DOI:
10.1039/D4TA02974G
(Paper)
J. Mater. Chem. A, 2024,
12, 16129-16142
Two-dimensional Janus antimony chalcohalides for efficient energy conversion applications†
Received
29th April 2024
, Accepted 22nd May 2024
First published on 4th June 2024
Abstract
Specific functionalities at the nanoscale can emerge from the broken inversion symmetry in two-dimensional (2D) Janus monolayers. In this work, we employed the first-principles theory to systematically investigate novel 2D Janus SbYZ (Y = S and Se and Z = Cl, Br, and I) monolayers and study their applications in various energy conversion fields such as piezoelectric, thermoelectric, photovoltaic solar cell, and photocatalytic water splitting. The positive phonon spectra and ab initio molecular dynamics (AIMD) simulation plots suggest that these monolayers are dynamically and thermally stable. Our findings demonstrate that these monolayers have extremely low lattice thermal conductivity and excellent electronic transport properties. The computed thermoelectric performances (ZT) of the monolayers range from 0.15 to 1.66 at 800 K. The inspection of the piezoelectric stress and strain coefficients demonstrates strong out-of-plane piezoelectricity. These monolayers also exhibit characteristics such as semiconductor nature, high carrier mobility, and visible light absorption. The proposed heterostructures of these monolayers show high power conversion efficiencies, up to 19% in the case of SbSeBr/AsTeI heterostructures. We have demonstrated the photocatalytic properties of Janus SbSI and SbSeBr monolayers, as the band alignments of these monolayers are appropriate for photocatalytic water splitting. The HER process can occur without an external potential in pH 0 medium for SbSeBr monolayer. The high solar-to-hydrogen (STH) conversion efficiency (up to 18%) and relatively larger electron–hole recombination rates (1.02 ns and 1.80 ns for the Janus SbSI and SbSeBr monolayers, respectively) demonstrated via NAMD simulations indicated that these monolayers are potential materials for efficient photocatalytic water splitting. Our study suggests that these monolayers have the potential for various energy conversion applications.
1. Introduction
After the successful fabrication of single-layer graphene,1 various 2D monolayers were prepared including germanene,2 silicene,3 antimonene,4 and bismuthene.5 Recently, novel 2D material derivatives known as Janus materials have attracted considerable scientific interest due to their distinctive qualities.6 The out-of-plane asymmetry of Janus materials induces an intrinsic electric field (EF).7 The asymmetric nature of Janus monolayers makes them potential candidates for various applications including thermoelectric,8 piezoelectric,9 photovoltaic,10 photocatalytic water splitting,11 and ultrafast charge transfer dynamics in van der Waals heterostructures (vdWHs).12
After the successful fabrication of Janus graphene,13 Lu et al. synthesized Janus MoSSe14 and Zheng et al. synthesized Janus WSSe.15 These experimental syntheses led to the theoretical investigations of numerous ternary Janus monolayers based on titanium chalcogenides,16 group-V transition metal trichalcogenides,17 group-IV(A) dichalcogenides,18 antimony-based chalco-halides,19 and transition metal dichalcogenide oxides.20 The thermoelectric properties of 2D Janus monolayers are characterized by a dimensionless quantity called the figure of merit (ZT). A small lattice thermal conductivity and high electrical conductivity are necessary conditions for a high thermoelectric performance. Ternary Janus monolayers such as BiTeBr,21 PdSX (X = Se and Te),22 BiOCl,23 WSX (X = Se and Te),24 AsTeX (Cl, Br and I),25 and BiTeCl (0.43)26 show moderate thermoelectric performance at room temperature (ZT ∼ up to 0.75).
Due to the absence of inversion symmetry, 2D Janus monolayers have an additional degree of freedom to tune piezoelectric properties.27 Piezoelectric materials have broad prospects such as human pulse diagnosis that can measure the weak vibration pattern of the human radial artery28 and detection of pressure distribution.29 The piezoelectric properties of 2D monolayers are characterized by in-plane and out-of-plane piezoelectric coefficients, i.e., d11 and d31. The piezoelectric properties of various Janus monolayers based on bismuth chalcogen halides,30 bismuth oxyhalide,31 group-IV monochalcogenides,9 PtXO (X = S, Se),27 γ-Ge2XX′ (X, X′ = S, Se, and Te),32 SnSSe,33 MoSTe,34 WXO (X = S, Se, and Te),20 X2PAs (X = Si, Ge, and Sn),35 MXY (M = Sb and As; X = Te and Se; Y = Br and I)36 and TiXY (X, Y = Cl, Br, and I)37 have been reported.
Due to the exciting properties of Janus monolayers, researchers further explored their application in excitonic solar cells.38–40 These solar cells feature a type-II band alignment and are centered on the donor–acceptor mechanism between two materials that form a heterostructure.41 Very recently, Janus monolayers were shown to possess good power conversion efficiency (PCE) in photovoltaic solar cells such as Pb2SSe/SnSe (20.02%),42 Pb2SSe/GeSe (19.28%),42 β-Te2S/α-Te2S (21.13%),10 ZrICl (15.91%),43 SbTeBr/SbSI44 and ZrIBr (14.13%).43
Apart from solar cells, Janus structures also show the potential for application in photocatalytic water splitting, which produces clean hydrogen energy using sunlight. For cost-effective commercial application, a photocatalyst's solar-to-hydrogen (STH) conversion efficiency should exceed 10%.45 Various Janus monolayers such as WSSe,46 MoSSe,47 PtSSe,48 WSeTe,49 AsTeX (X = Cl, Br),25 AsXY (X = Se, Te; Y = Br, I),50 and AlXY (X = S, Se; Y = Cl, Br and I)51 possess high STH conversion efficiency. The structural asymmetry-induced intrinsic electric field in Janus monolayers is favourable for hindering charge recombination.46
In addition to various energy conversion applications of 2D materials, the lifetimes of photogenerated charge carriers in these materials play a pivotal role in the performance of energy conversion devices. The suitable carrier lifetimes of 2D monolayers, i.e., black phosphorene (2.4 ns),52 TiO2 (31.1 ns),53 ReS2 (40.1 ps),54 ReSe2 (28.7 ps),54 XMMX′ (X = S, Se; M = Ga, In; X′ = Te) (39.9–72.8 ns),55 LiAlTe2 (1.69 ns),56 PtSe2 (>10 ns),57 MoSi2N4 (489 ps)58 and Cu2O (10.10 ps)59 make them potential candidates for various energy conversion fields.
Inspired by the experimental fabrication of 2D Janus BiTeX (X = Cl, Br and I) compounds,60,61 we systematically explored their analogous novel Janus monolayers SbYZ (Y = S, Se; Z = Cl, Br and I). The dynamical and thermal stability of Janus SbYZ was confirmed by performing phonon and AIMD simulations. We found that 2D SbYZ exhibits very low lattice thermal conductivity with high thermoelectric performance by including various elastic and inelastic scattering mechanisms. Next, we examined the mechanical response and the piezoelectric stress and strain coefficients. The effects of an electric field and magnetic field on free charges were evaluated in terms of Hall conductivity and Hall factor. These monolayers also showed high carrier mobility, computed using Matthiessen's rule, which included various scattering effects, i.e., acoustic, piezoelectric and polar-optical scattering. We also investigated the optical absorption spectrum using the GW+BSE level of theory. Type-II heterojunctions with other monolayers are designed to demonstrate the potential applications of 2D Janus monolayers in solar cells. The high STH conversion efficiency (>10%) with appropriate band edge positions was also obtained for Janus SbSI and SbSeBr monolayers. We also performed ab initio non-adiabatic molecular dynamics (NAMD) simulations on SbSI and SbSeBr Janus monolayers to evaluate the interfacial electron–hole recombination rate and charge transfer dynamics.
2. Computational details
We implemented density functional theory confined in the VASP package62,63 to perform calculations. To describe electron–ion interactions, projected augmented-wave (PAW)64 potentials were employed, while GGA parameterized by PBE65 was implemented to calculate exchange–correlation functionals. To certify the well-founded calculation of electronic properties, the screened hybrid HSE06 method was implemented by mixing nonlocal Fock exchange (25%) with PBE exchange (75%).66 We relaxed the structure until the forces on each atom decreased below 0.01 eV Å−1. The energy convergence criteria between sequential steps for structural relaxation were set to 10−5 eV. For the sampling of the Brillouin zone, the Γ-centred k-mesh of 16 × 16 × 1 (ref. 67) was implemented. A perpendicular cell length of 16 Å was used to eliminate the interactions between the adjoining layers along the z-direction.
The phonon dispersion spectra calculations were performed using the DFPT method with a q-mesh of 8 × 8 × 1 and convergence threshold of 10−16 Ry, as incorporated with the Quantum Espresso package.68 The thermal stability of 2D SbYZ Janus monolayers was validated by AIMD simulations at room temperature. To control the temperature in AIMD simulations, the Nose–Hoover thermostat69,70 was employed under the NVT ensemble. The finite difference method confined in VASP was implemented to demonstrate the elastic constants and piezoelectric stress coefficients by integrating the ionic and electronic contributions for the relaxed ions with a 16 × 16 × 1 k-point grid.
The magneto-transport properties were investigated using the theory implemented in the AMMCR package.71 The electrical transport properties calculated by the constant relaxation time approach do not include any scattering mechanisms, which affects the experimental validation of the theoretically anticipated results. Therefore, for more steadiness of theoretically predicted results, we include various kinds of momentum scattering mechanisms as follows:
| | 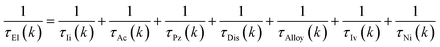 | (1) |
The subscripts Ii, Ac, Pz, Dis, Alloy, Iv and Ni in the above equation indicate ionized impurity scattering, polar optical phonon scattering, dislocation scattering, alloy scattering, intra-valley scattering, piezoelectric scattering, acoustic deformation scattering and neutral impurity scattering, respectively.
The lattice transport properties of Janus monolayers were calculated using the Phono3py code.72 The ab initio NAMD simulations were carried out within the Hefei-NAMD code encoded in VASP.73 To obtain charge transfer dynamics, we implemented the (NVT) microcanonical ensemble to perform MD simulation at 300 K with a supercell of size 3 × 3 × 1.
3. Results and discussion
3.1. Geometric structure and energetics
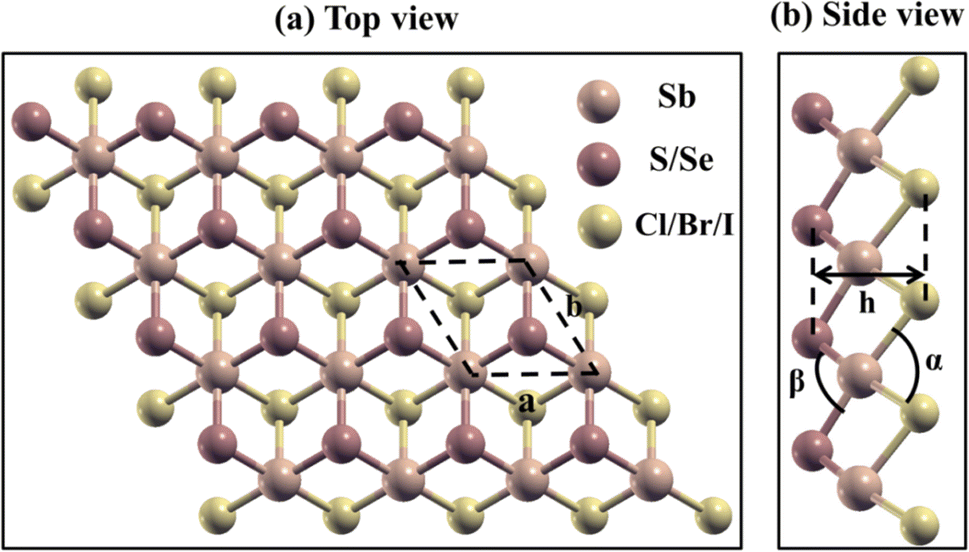
The schematic representation of the geometric optimized structure of 2D Janus SbYZ monolayers is depicted in Fig. 1. Janus SbYZ monolayers exhibit a trigonal structure with a three-atom hexagonal unit cell having a Sb atom sandwiched between Y and Z atoms. These monolayers exhibit a P3m1 space group with a C3v point group. The SbSCl possesses the smallest lattice constant due to the presence of the lightest atoms in it as compared to other SbYZ monolayers. The lattice constant of these monolayers is comparable with previous reports.19,30,36
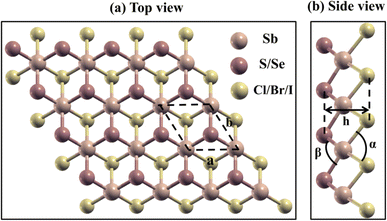 |
| | Fig. 1 (a) Top and (b) side views of Janus SbYZ monolayers. | |
The thickness of Janus monolayers increases with the elongation of bonds, i.e., Sb–Y and Sb–Z, due to the reduction of attractive forces among the layers. On the other hand, the bond angle α (Y–Sb–Y; Y = S, Se and Te) increases while the bond angle β (Z–Sb–Z; Z = Cl, Br and I) shows an opposite trend as given in Table 1. When the electronegativity difference between chalcogen (Y) and halogens (Z) increases, more electrons gather around the more electronegative atoms, which leads to the widening of the bond angle (Table 1).
Table 1 The optimized parameters: lattice constants (a, b), thickness (h), bond angle (α for Y–Sb–Y, β for Z–Sb–Z), bond length (dSb–Y and dSb–Z), amount of charge transfer ΔρSb–Y (charge transfer from Sb to Y) and ΔρSb–Z (charge transfer from Sb to Z), the difference between the work function (ΔΦ) of the chalcogen and halogen sides, and the cohesive energy (EC) of the SbYZ monolayers
| 2D monolayer |
(a = b) (Å) |
h (Å) |
α (°) |
β (°) |
d
Sb–Y (Å) |
d
Sb–Z (Å) |
ΔρSb–Y |
ΔρSb–Z |
ΔΦ (eV) |
E
C (eV per atom) |
| SbSCl |
3.94 |
3.89 |
86.61 |
94.74 |
2.75 |
2.95 |
0.76 |
0.40 |
0.25 |
3.84 |
| SbSBr |
3.99 |
4.02 |
86.83 |
94.71 |
2.83 |
3.01 |
0.60 |
0.76 |
0.23 |
3.56 |
| SbSI |
4.07 |
4.21 |
87.20 |
94.57 |
2.99 |
3.15 |
0.61 |
0.59 |
0.21 |
3.41 |
| SbSeCl |
4.03 |
3.99 |
88.02 |
97.08 |
3.03 |
3.07 |
0.42 |
0.59 |
0.29 |
3.48 |
| SbSeBr |
4.09 |
4.15 |
88.26 |
97.75 |
3.14 |
3.17 |
0.41 |
0.52 |
0.27 |
3.37 |
| SbSeI |
4.12 |
4.23 |
88.98 |
97.99 |
3.22 |
3.25 |
0.38 |
0.48 |
0.24 |
3.21 |
In Bader charge analysis, ΔρSb–Y (charge transfer from Sb to chalcogens) decreases as the atomic mass corresponding to the chalcogen increases but in the case of ΔρSb–Y (charge transfer from Sb to halogens), an irregular trend is observed. A similar kind of irregular charge transfer behaviour for the case of halogens is observed in the case of MXY (M = Sb, As; X = Te, Se; Y = Br, I)36 and γ-Ge2XX′ (X, X′ = S, Se and Te)32 monolayers. The bond lengths dSb–Y and dSb–Z of SbYZ monolayers increase as the lattice constant “a” increases as we move down the chalcogen and halogen groups. The unbalanced charge between the Y and Z generates an electric field between the Sb-atoms layer and the Y(Z)-atom layer, the direction of which points from Sb to Y(Z). The planar average potential of the SbY/SbZ sides and the potential difference between the two sides, i.e. Y and Z, are also shown in Fig. S1, ESI.† The charge differences between the work function (ΔΦ) of the Y and Z sides are listed in Table 1.
The energetic stability of novel 2D Janus monolayers was quantified in terms of cohesive energy (EC), which is formulated as follows:
| | 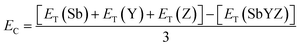 | (2) |
where
ET(Sb),
ET(Y), and
ET(Z) are the energies of single atoms corresponding to Sb, Y and Z, respectively, and
ET(SbYZ) is the total energy of SbYZ Janus monolayers. All the computed results of cohesive energy are listed in
Table 1. The cohesive energy decreases as ‘
a’ increases, which is correlated with the bond weakening. The high cohesive energy corresponds to the strong binding between the constituent atoms, which confirms the energetic stability of the Janus SbYZ monolayers. Accordingly, SbSCl and SbSeI are the most and least stable materials amidst the SbYZ Janus monolayers. The
EC of SbYZ is comparable with the BiXY (X = S, Se and Te; Y = F, Cl, Br and I) (2.46–3.58 eV per atom)
30 and Ge
2XX′ (X/X′ = S, Se and Te) (3.14–3.68 eV per atom)
32 monolayers.
3.2. Dynamical, thermal and mechanical stability
Next, we examine the dynamical and thermal stability of Janus SbYZ monolayers. The dynamical stability of Janus SbYZ monolayers is confirmed by computing phonon dispersion spectra. The phonon dispersion spectra of SbYZ monolayers have no imaginary frequencies, which indicates that these monolayers are dynamically stable, as depicted in Fig. 2. The phonon dispersion spectra of SbYZ monolayers incorporate nine vibrational modes (three acoustic phonon (AP) modes and six optical phonon (OP) modes). Three acoustic phonon modes are characterized by a flexural acoustic (ZA) branch that exhibits quadratic dispersion (p2), a transverse acoustic (TA) branch, and a longitudinal acoustic (LA) branch that possesses linear dispersion near the Γ-point.
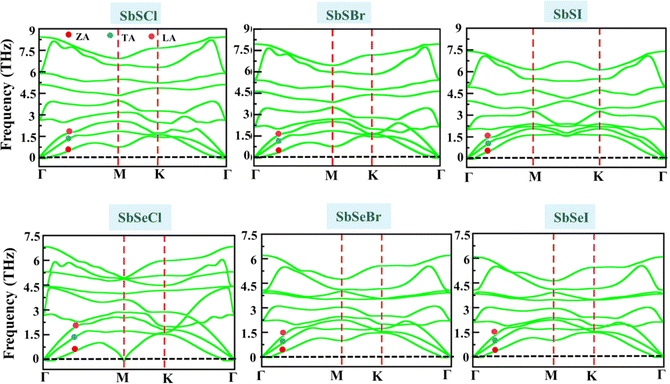 |
| | Fig. 2 Phonon dispersion spectra of 2D SbYZ monolayers. | |
The phonon modes of SbYZ monolayers shifted to the lower frequencies, and phonon dispersion spectra increased as the atomic mass of constituent atoms increased, as depicted in Fig. 2. In the case of SbSCl, a gap appeared between the AP and OP modes due to the large difference in the atomic masses and the electronegativity differences between the constituent atoms.30 Due to the overlapping of AP and OP modes, strong optical-acoustic scattering arises in SbSBr, SbSI, SbSeCl, SbSeBr and SbSeI. Note that the narrow band dispersion leads to the suppression of their group velocity.32
To confirm the thermal stability of the SbYZ monolayers, we performed AIMD simulations at 300 K (800 K) for 5 ps with a supercell size of 4 × 4 × 1, as shown in Fig. S2, ESI (Fig. S3, ESI†). The variation of temperature with time and the snapshots of final structures show that the crystalline form of Janus SbYZ was maintained at 300 K and 800 K.
After confirming the dynamical and thermal stability, the mechanical stability of the suggested structures was quantified by computing Young's modulus (Y2D), Poisson ratio (ν), and the nonzero elastic constants (Cij), listed in Table 2. All the Janus SbYZ monolayers satisfy the Born and Huang requirements (C11 > |C12| and C66 = (C11 − C12)/2 > 0) for the hexagonal lattice, confirming their mechanical stability.74 The mechanical properties of the 2D SbYZ monolayers were computed by calculating Young's modulus (Y2D) and the Poisson ratio (ν). Young's modulus assesses the stiffness or stretchability of 2D monolayers. The Y2D of 2D Janus SbYZ was computed as follows:
| | 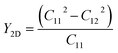 | (3) |
Table 2 Elastic coefficients (C11, C12), Young's modulus (Y2D) and Poisson's ratio (ν) of 2D Janus SbYZ
| 2D monolayers |
C
11 (N m−1) |
C
12 (N m−1) |
Y
2D (N m−1) |
(ν) |
| SbSCl |
35.56 |
11.53 |
31.91 |
0.31 |
| SbSBr |
33.83 |
11.96 |
29.60 |
0.34 |
| SbSI |
33.86 |
13.20 |
28.71 |
0.38 |
| SbSeCl |
36.16 |
12.74 |
31.67 |
0.34 |
| SbSeBr |
30.62 |
9.47 |
27.69 |
0.31 |
| SbSeI |
28.07 |
8.05 |
25.76 |
0.28 |
As we move down the halogen group, Y2D decreases, which indicates the elongation of the bonds (dSb–Z). Consequently, SbSeCl and SbSeI yielded the largest and smallest Y2D, respectively. Note that Y2D for SbSeBr was calculated to be 31.14 N m−1 in a previous report.36 Along the stretching direction, the fraction of the transverse strain to the axial strain delivers the Poisson ratio (ν = C12/C11). The computed ν values are in the range of 0.28–0.38, as shown in Table 2. According to the Frantsevich rule,75 the Poisson's ratio of the Janus SbSCl, SbSeBr and SbSeI monolayers are less than 1/3, which implies that these monolayers possess a brittle nature. Note that the Poisson's ratio in previous reports for SbSeBr36 and SbSeI19 was calculated to be 0.32 and 0.23, respectively.
3.3. Piezoelectric properties
The concept of “piezoelectricity” signifies the association of mechanical stress and electrical polarisation that occurs when external stress is applied and induces electric dipole moments in non-centrosymmetric materials, and vice versa. For 2D materials, the z-axis lattice parameter needs to be used to renormalize the piezoelectric stress coefficients, i.e., e11 and e31. Compared to their 3D counterparts, 2D materials exhibit improved piezoelectric constants, according to recent experimental research and theoretical predictions.76 The relaxed-ion piezoelectric tensor (eij) is calculated using a density functional perturbation theory (DFPT) based method, which is formulated as follows:where Ckj is the elastic tensor of the SbYZ monolayers. The piezoelectric tensor coefficients (eij and dij) depend on the symmetry of the crystal structures. The piezoelectric tensor of 2D SbYZ monolayers is characterized as e11/d11 for in-plane piezoelectricity and e31/d31 for out-of-plane piezoelectricity due to the C3v symmetry of the structure. The 2D materials' out-of-plane piezoelectric constants are influenced by the charge density difference between two surfaces and out-of-plane asymmetry. The Voigt notation of Janus SbYZ is formulated as follows:| | 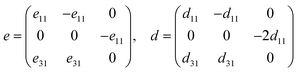 | (5) |
The relations between piezoelectric coefficients for Janus SbYZ are given as34,77
| | 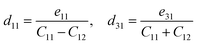 | (6) |
The calculated values of piezoelectric coefficients are tabulated in Table 3. The piezoelectric coefficient (d31) of SbSI and SbSeI, is higher as compared to the analogous Janus monolayers such as the MoSSe (0.29 pm V−1),77 MoSTe (0.4 pm V−1),34 1H-WSO (0.4 pm V−1),20 BiSF (−0.33 pm V−1),30 and BiSBr (−0.41 pm V−1)30 monolayers. Such high piezoelectric coefficient values make Janus SbSI and SbSeI monolayers potential candidates for piezoelectric devices. Note that the calculated value of d11 for SbSeBr (21.9 pm V−1) is comparable with the reported value of 22.84 pm V−1 in the literature.36
Table 3 Piezoelectric stress coefficients (e11 and e31) and the corresponding piezoelectric strain coefficients (d11 and d31) of 2D Janus SbYZ monolayers
| 2D monolayers |
e
11 (pC m−1) |
e
31 (pC m−1) |
d
11 (pm V−1) |
d
31 (pm V−1) |
| SbSCl |
7.52 |
−0.06 |
31.3 |
−0.12 |
| SbSBr |
8.16 |
−0.11 |
37.3 |
−0.26 |
| SbSI |
10.56 |
−0.23 |
51.1 |
−0.48 |
| SbSeCl |
3.68 |
−0.006 |
15.7 |
−0.01 |
| SbSeBr |
4.64 |
−0.1 |
21.9 |
−0.25 |
| SbSeI |
4.16 |
−0.22 |
20.8 |
−0.60 |
3.4. Thermoelectric properties
3.4.1. Lattice thermal conductivity.
The phonon transport properties play an important role in the calculation of lattice thermal conductivity (κl). The lattice thermal conductivity (κl) of Janus SbYZ monolayers is characterized in terms of phonon transport variables such as phonon group velocities (vg) and Gruneisen parameters (γ), etc. The (κl) at temperature T is formulated as follows:| | | κl,j = ∑∑Cpvg,j2(λ,q′)τ(λ,q′) | (7) |
where Cp represents specific heat capacity, λ is the phonon mode and q′ is the wave vector.
Janus SbYZ monolayers exhibit a decrease in lattice thermal conductivity as temperature increases, indicating Umklapp anharmonic interactions in these monolayers. The calculated values of κl at 300 K range from 0.61 W m−1 K−1 to 1.83 W m−1 K−1 for 2D Janus SbYZ (Fig. 3). The comparison of κl of 2D Janus SbYZ with the other similar monolayer at 300 K is given in Table 4.
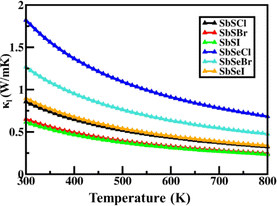 |
| | Fig. 3 The lattice thermal conductivity of 2D Janus SbYZ monolayers as a function of temperature. | |
Table 4 The lattice thermal conductivity (κl) of SbYZ monolayers and other similar 2D Janus monolayers at 300 K
| 2D monolayers |
κ
l (W m−1 K−1) |
Reference |
| SbSCl, SbSBr, SbSI, SbSeCl, SbSeBr, SbSeI |
0.86, 0.64, 0.61, 1.83, 1.27, 0.91 |
This study |
| BiOCl |
3.0 |
78
|
| AsTeCl, AsTeBr, AsTeI |
0.92, 2.02, 3.36 |
25
|
| PdSTe, PdSeTe |
5.45, 4.02 |
22
|
| BiTeCl |
1.46 |
26
|
| BiTeBr |
1.47 |
21
|
| In2SO, In2SeO |
0.42, 0.27 |
8
|
| 1T-ISbTe, 2H-ISbTe |
3.55, 1.63 |
79
|
Next, we computed the phonon's group velocities for different modes as follows:
| | 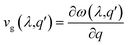 | (8) |
where
ω(
λ,
q′) is the phonon frequency. The maximum values of the LA, ZA and TA modes reached 25 km s
−1. SbSI monolayers show minimum contributions and Janus SbSeCl monolayers show maximum contributions corresponding to acoustic phonon modes (Fig. S4, ESI
†). It results in the lowest value of the lattice thermal conductivity corresponding to the SbSI monolayer and the highest lattice thermal contribution to the SbSeCl monolayer among all the Janus SbYZ monolayers. This happens because the
κl exhibits a lower value if the phonon group velocity is less or
vice versa.
κ
l can also be expressed in terms of a dimensionless quantity γ as follows:
| | 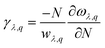 | (9) |
γ describes the anharmonic interaction in the crystal structure. A greater value of γ leads to a lower phonon relaxation time due to a higher phonon–phonon scattering rate. The calculated value of γ reached up to ∼45, corresponding to the Janus SbYZ monolayer. The ZA mode in γ contributes more than the TA and LA modes. It was observed that the contribution of optical modes is much less than acoustic modes (Fig. S5, ESI†), which indicates that κl majorly arises due to the anharmonic acoustic scattering of phonons.80 The high value of the Gruneisen parameter and the low value of the group velocity are responsible for the low lattice thermal conductivity in the case of the SbSI monolayer. The higher γ and lower vg are adequate conditions for a monolayer to exhibit low κl.
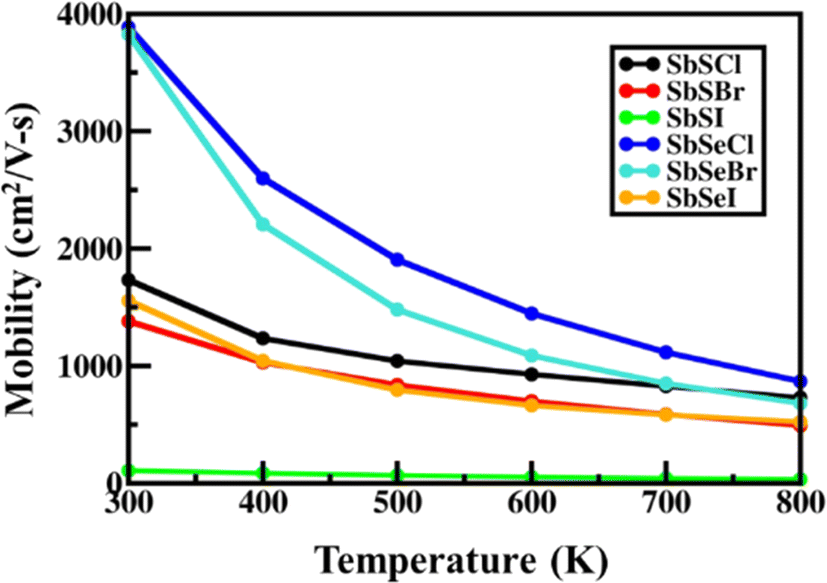
3.4.2. Carrier mobility and magneto-transport properties.
Next, we calculated the carrier mobility of the SbXY monolayer by including various scattering mechanisms. We implemented Matthiessen's rule for the calculation of temperature-dependent carrier mobility (μ), which is formulated as follows:| | 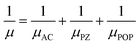 | (10) |
where μAC, μPZ and μPOP represent the carrier's mobility arising due to the contributions from the acoustic, piezoelectric and polar-optical mechanisms, respectively. Fig. 4, illustrates the carrier mobility of SbYZ monolayers w.r.t. temperature (T). Here, we incorporate only three types of scattering because in the case of Janus monolayers, mainly these scatterings play an important role. AC scattering arises due to the interaction of electrons with an acoustic phonon, piezoelectric scattering in Janus monolayer arises due to the presence of an in-built electric field (EF),27 and POP scattering arises due to high-temperature systems. The carrier mobility of these monolayers decreases with T because as T increases, the electrons scatter more and thus, their mobility decreases.
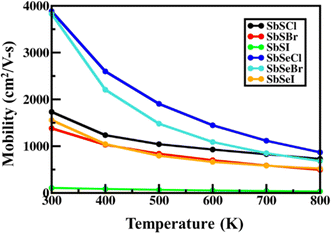 |
| | Fig. 4 Carrier mobility of 2D Janus SbYZ monolayers. | |
At room temperature, the carrier mobility of SbSeCl and SbSeBr reached 4000 cm2 V−1 s−1. We also studied the effect of magnetic field (B) on charge carriers. The magneto-transport properties of SbYZ monolayers are characterized in terms of the Hall conductivity (σ) and Hall factor (r). The Hall conductivity of SbYZ monolayers is expressed in the form of components of perturbations h(ε) and g(ε). This perturbation arises due to the B. The Hall conductivity in terms of perturbation is given as follows:81
| | 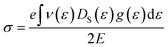 | (11) |
where
E is the electric field. The Hall conductivity (
σ) of the SbXY monolayer is illustrated in Fig. S6(a), ESI.
† In general, the Hall factor (
r) depends on
σ, which is formulated as
81| | 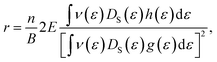 | (12) |
where
n is the number of charge carriers,
E is the electric field and
B is the magnetic field. In this formalism, the distribution functions
g(
e) and
h(
e) depend on the temperature-dependent scattering rate. The Hall factor of the SbYZ monolayer is illustrated in Fig. S6(b), ESI.
† Note that the calculated value of the Hall factor of SbYZ monolayers is less than 1. If the Hall factor is equal to one, then the carrier mobility and Hall mobility are the same; in that case, we can deal with one system with the constant relaxation time approach.
81
3.4.3. Electronic structure.
We have implemented different levels of theories, i.e., GGA-PBE, HSE06, and G0W0, for the computation of the electronic band structure of Janus SbYZ monolayers. Bandgap plays a crucial role in the calculation of electronic transport properties. Band structure, which resides in the reciprocal space of the periodic lattice, directly determines the electronic transport properties of the materials.82 If the bandgap of the material is large, it leads to a lack of electronic states of the charge carriers and causes poor electrical conductivity. The degeneracy in bands effectively optimizes the power factor by enhancing the Seebeck coefficient.83 The bandgaps obtained using different methods are tabulated in Table S1, ESI.† The HSE06 bandgap of these monolayers is smaller as compared to the analogous Janus monolayers, i.e., AsTeCl (2.32 eV),25 WSSe (2.15 eV),24 and comparable with WSTe (1.71 eV),24 AsTeBr (2.02 eV) and AsTeI (1.78 eV)25 monolayers. The calculated value of the HSE06 (GGA-PBE) bandgap of SbSeBr is correlated with previously reported literature, i.e., 1.79 eV (1.25 eV).19 The electronic band structure of Janus SbYZ indicates an indirect bandgap nature with semiconducting behavior, as depicted in Fig. 5.
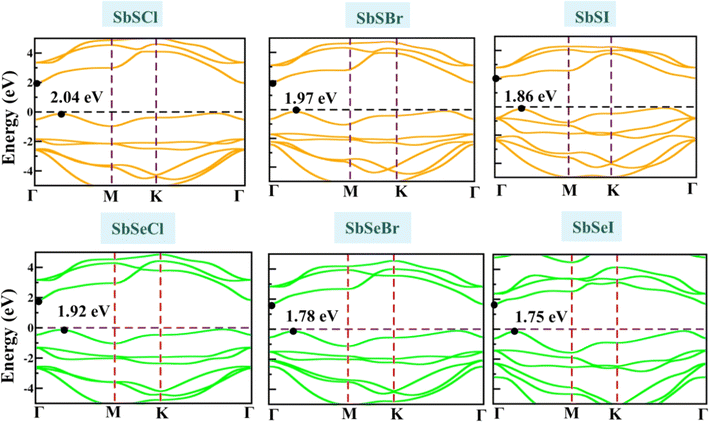 |
| | Fig. 5 The electronic band structure of 2D Janus SbYZ monolayers using the HSE06 method. | |
3.4.4. Electronic transport properties and thermoelectric performance.
Due to the limitation of the CRTA (constant relaxation time approach), we implemented Rode's algorithm71 to include eight different kinds of scattering mechanism. A detailed discussion of these scattering equations is provided in the ESI.† Out of these eight scattering mechanisms, τPOP, τAc and τPz play the most significant roles. POP (polar optical polar) scattering predominates in regions with higher temperatures or near room temperature. The coupling of electrons with non-polar acoustic phonons gives rise to acoustic deformation potential scattering. The contribution of piezoelectric scattering in Janus monolayers arises due to the presence of an in-built intrinsic electric field. We have taken the piezoelectric coefficient from Table 3 in the calculations of piezoelectric scattering. In Fig. S7, ESI,† the scattering rates of the SbYZ monolayers show almost the same trend. The inclusion of these scattering effects provides more consistency with experimental results.71 In calculating the electronic transport properties, the relaxation time is incorporated with various scattering mechanisms rather than the constant relaxation time approach. For the SbYZ monolayers, the computed value of relaxation time lies in the range of 0.005–0.03 (10−14 s).
We implemented the momentum-dependent scattering mechanism and semi-classical Boltzmann transport theory within the BoltzTraP code for the calculation of electronic transport properties.84 The Seebeck coefficient (S), electronic thermal conductivity (κe), electrical conductivity (σ), and power factor (P) of Janus SbYZ monolayers play an essential role in the calculation of (ZT). The electronic transport parameters, i.e., S, σ and κe are expressed as follows:84
| | 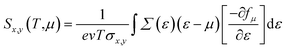 | (13) |
| | 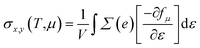 | (14) |
| | 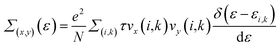 | (15) |
In the above equations,
x and
y correspond to the cartesian indices,
N is the number of
k-point sampling and
Σ(ε) is the transport distribution function. We have calculated the Seebeck coefficient (
S) in the temperature range of 300 K to 800 K (Fig. S8(a)
†). As the temperature rises to 800 K,
S decreases due to the reduction of the slope,
σ, around the valence band minima and conduction band maxima. The
σ of these monolayers also increases with temperature (Fig. S8(b)
†).
For the computation of κe, we used the Wiedemann–Franz law as follows:
where
L is Lorentz's number. Using
L = 1.5 + exp(−|
S|/116), different values of the Lorenz number were obtained, corresponding to the different values of the Seebeck coefficient (
S has the unit μV K
−1). The maximum calculated value of electronic thermal conductivity reached 50 W m
−1 K
−1 for the SbSeCl monolayer at 800 K, as depicted in Fig. S8(c).
† By the integrated effects of
σ and
S, we calculated the power factor (PF) as follows:
where
S is the Seebeck coefficient. The higher the value of the Seebeck coefficient and electronic conductivity, the higher will be the power factor. The computed values of the power factor were in the range of 0.0119–0.2351 W m
−1 K
−2 for the SbYZ monolayers in the temperature range of 300–800 K. Among all the Janus monolayers SbSeI possessed the highest power factor,
i.e., 0.2351 W m
−1 K
−2, which led to the high thermoelectric performance corresponding to this monolayer.
By the integrated effect of κl and PF, the thermoelectric performance of Janus SbYZ monolayers is formulated as follows:86
| | 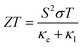 | (18) |
The thermoelectric performance gradually increased with temperature (Fig. 6) because of the reduction of the lattice thermal conductivity with temperature. The calculated values of ZT for Janus SbYZ monolayers and their comparison with the other 2D monolayers are listed in Table 5. From this comparison, it is clear that the SbYZ monolayer can act as a potential candidate in the field of thermoelectrics at higher temperatures.
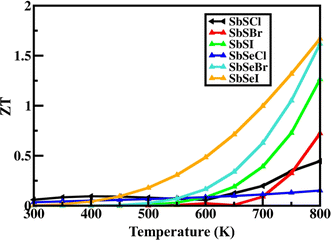 |
| | Fig. 6 The thermoelectric figure of merit (ZT) of 2D Janus SbYZ monolayers as a function of temperature. | |
Table 5 The thermoelectric performance (ZT) of SbYZ monolayers and other similar 2D Janus monolayers at high temperatures (800–900 K)
| 2D monolayers |
ZT
|
References |
| SbSCl, SbSBr, SbSI, SbSeCl, SbSeBr, SbSeI |
0.45, 0.72, 1.26, 0.15, 1.61, 1.66 |
This study |
| BiOCl |
1.1 |
23
|
| AsSBr |
0.91 |
85
|
| PdSSe, PtSSe, PtSTe, PtSeTe |
0.49, 0.28, 0.34, 0.52 |
22
|
3.5. Optical and photovoltaic solar cell properties
3.5.1. Optical properties.
To provide a clear picture of the SbYZ monolayers' capacity to harvest light, we computed their optical absorbance spectra using the G0W0-BSE87 level of theory, which takes into account the excitonic effect. To evaluate the optical absorbance spectra (A(ω)), we used the imaginary part of the dielectric function (εi(ω)),88| | 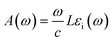 | (19) |
L in the above formula represents the length of the cell in the z-direction.
The optical absorbance spectra of SbYZ monolayers lie in the UV-visible region, with the first most prominent peak being in the visible region, as depicted in Fig. 7. This indicates that these monolayers have excellent visible light harvesting capabilities. The photoexcitation of photogenerated charge carriers is depicted in terms of excitonic binding energy. The excitonic binding energy (Eb) of SbYZ monolayers is as follows:
where
EQP and
EOPT represent the direct quasi-particle bandgap and energy corresponding to the most prominent optical absorption peak. The calculated value of the excitonic binding energy corresponding to the SbYZ monolayers is given in Table S1, ESI.
†
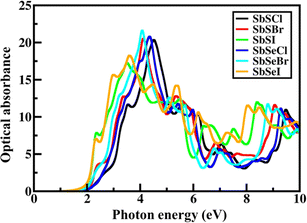 |
| | Fig. 7 Optical absorbance of 2D Janus SbYZ (Y = S, Se and Z = Cl, Br & I) monolayers using GW-BSE theory. | |
This excitonic binding energy is lower than other 2D monolayers, i.e., MgO (2.49 eV),89 BeN2 (1.07 eV),90 MoS2 (1.1 eV)91 and β-Te (0.84 eV).92 This signifies that 2D Janus SbYZ monolayers have good potential in the energy conversion fields.
3.5.2. Photovoltaic solar cell properties.
We then explored SbYZ monolayers in the context of heterojunction solar cells due to their semiconducting behaviour, high carrier mobility and excellent visible light absorption spectra. We proposed 31 type-II van der Waals heterostructures (vdWHs) of SbYZ monolayers with their similar Janus monolayers, which are tabulated in Table S2, ESI.† The inherent optoelectronic characteristics of distinct monolayers are retained within the heterojunctions. Thus, to calculate the conduction band offsets (ΔEC), we used the band edge alignment of SbYZ heterostructures and implemented the Anderson method.93 As a result, band bending was caused by the variation in electrostatic potential caused by the varied surface atoms.
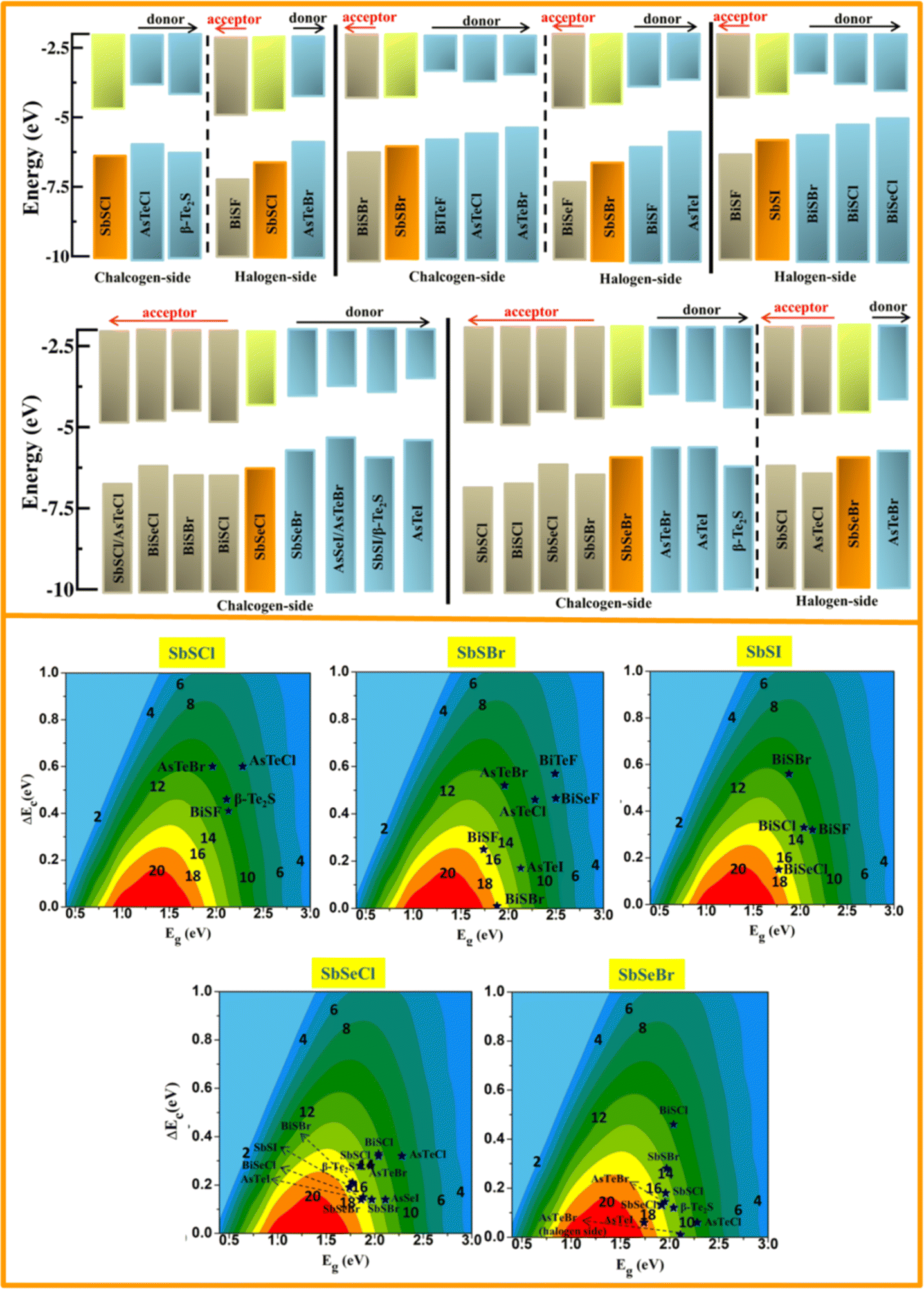
The band alignment of SbYZ monolayers w.r.t. to vacuum level is shown in Fig. 8 (upper panel). We aligned the 2D Janus monolayers corresponding to the chalcogen and halogen sides of SbYZ monolayers with minimum conduction band offset. The appropriate bandgap and smaller ΔEC led to a higher value of PCE. In the case of heterojunction solar cells, the material with the high value of CBM acts as a donor, and other materials act as acceptors.
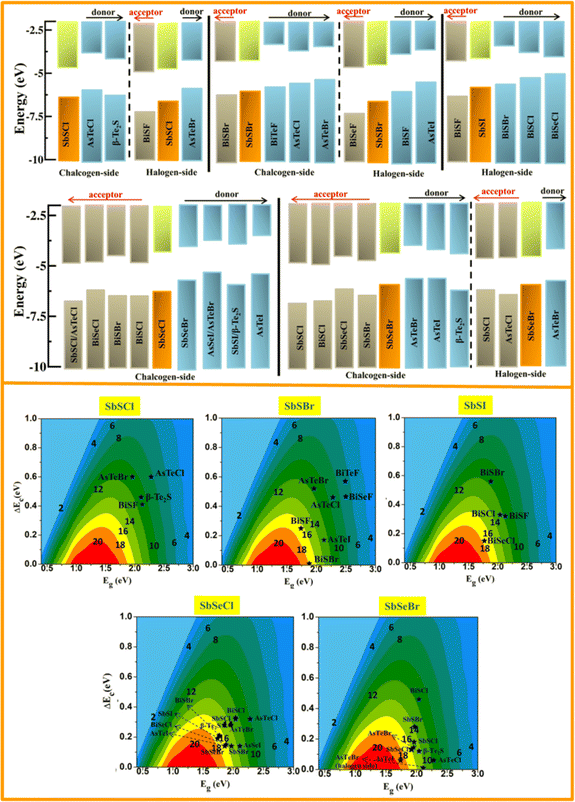 |
| | Fig. 8 The band alignments of SbSCl, SbSBr, SbSI, SbSeCl and SbSeBr monolayers w.r.t. their similar 2D monolayers (upper panel). The vacuum level is fixed at zero eV. The orange and green bars represent the VBM and CBM of SbYZ monolayers. The grey and sky-blue bars correspond to the acceptor and donor monolayers w.r.t. to SbYZ monolayers. The lower panel depicts the power conversion efficiencies of photovoltaic solar cells of 2D Janus SbYZ monolayers. | |
We implemented the Scharber et al. formula for calculating the power conversion efficiency of the proposed heterostructures.94 PCE can be calculated as follows:
| | 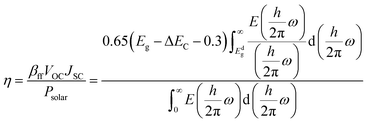 | (21) |
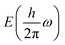
is the total solar energy. The estimated value of
βff is 0.65, which was calculated using the Shockley–Queisser limit. The power conversion efficiency of the proposed heterostructures as a function of donor band gap and conduction band offset is shown in
Fig. 8 (lower panel). The PCE value of the Janus SbYZ monolayer and other similar 2D Janus monolayers are tabulated in Table S3, ESI.
† The PCE value of SbSeBr/AsTeI heterostructure reached up to 19%, which makes them a potential candidate for photovoltaic solar cells.
3.6. Photocatalytic water-splitting properties
The photocatalytic properties were then computed. Among the different Janus SbYZ monolayers, it was discovered that only SbSI and SbSeBr monolayers had the appropriate band alignments for photocatalytic water splitting. As shown in Fig. 9 (left panel), the valence band position from the halogen side and conduction band position from the chalcogen side properly engulfed the redox potential of water, which made the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) feasible for both the Janus SbSI and SbSeBr monolayers. The photocatalyst stability in aqueous solution was demonstrated by calculating the reduction and oxidation potentials by implementing the method proposed by Chen and Wang.95 A detailed discussion of the calculation of these potentials is given in the ESI.†
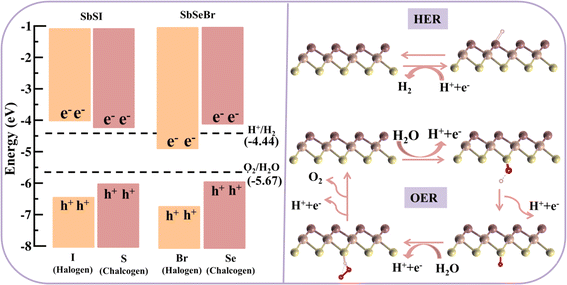 |
| | Fig. 9 The band alignments of 2D Janus SbSI and SbSeBr monolayers with respect to the vacuum level in the HSE06 scheme (left panel). The configuration of the adsorption of species for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is shown in the right panel. | |
3.6.1. Gibbs free energy profile.
For a detailed study of the HER and OER mechanism, we further explored the Gibbs free energy profiles. The parameters such as the zero-point energy and entropy contribution are presented in Table S4, ESI.† Note that the HER mechanism is a two-step process and the OER is a four-step process (Fig. 9). The calculated photogenerated electron potentials are 0.38 eV and 0.08 eV corresponding to SbSI and SbSeBr monolayers, respectively, at pH = 0, which is less than standard reduction potential value 1.23 eV of water. HER is feasible for SbSeBr without providing any external potential, while 0.56 eV external potential is required to make HER process spontaneous in SbSeI monolayer (Fig. 10). On the other hand, the computed values of the potentials of photogenerated holes are 2.01 eV and 2.18 eV, corresponding to the SbSI and SbSeBr monolayers, which can reduce the external potential to 0.76 eV and 0.17 eV, respectively (Fig. S9†). Various methods such as strain engineering96 and defect engineering97 play important roles in making the OER feasible for monolayer photocatalysts.
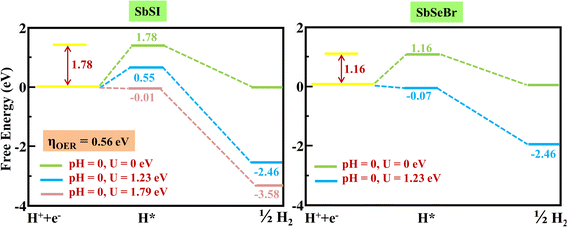 |
| | Fig. 10 The free energy change pathways for the hydrogen evolution reaction (HER) for the SbSI and SbSeBr monolayers, respectively. The green and sky-blue lines represent the presence and absence of light irradiation, respectively. | |
3.6.2. Solar-to-hydrogen conversion efficiency.
The solar-to-hydrogen conversion efficiency (ηSTH) is evaluated by the product of the light absorption efficiency (ηABS) and carrier utilization efficiency (ηCU). The ηABS is formulated as follows:| | 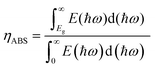 | (22) |
where E(ℏω) indicates the AM1.5G solar energy flux at the photon energy. The numerator and denominator indicate the photon density absorbed by the Janus monolayer and the total photon density of the reference sunlight spectra (AM1.5G), respectively.
η
CU is formulated as follows:
| | 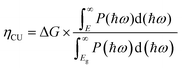 | (23) |
where Δ
G = 1.23 eV is the potential and
E is the photon energy used for the photocatalytic mechanism. The appropriate band alignments in Janus SbSI and SbSeBr monolayers suggest that they can act as efficient photocatalysts for the photocatalytic water-splitting mechanism. The aim of the water splitting mechanism is to maximize STH efficiency, which is based on the overpotentials of the hydrogen evolution reaction and the oxygen evolution mechanism. The solar energy conversion efficiency of the Janus SbSI and SbSeBr monolayers was examined by
ηABS,
ηCU and
ηSTH.
98 The calculated values of
ηCU and
ηABS for Janus SbSI (SbSeBr) monolayers are 53.83% (49.33%) and 35.58% (23.75%), respectively. The presence of an in-built intrinsic electric field in the SbSI and SbSeBr monolayers facilitates the flow of photogenerated charge carriers and is considered in the form of potential difference (Δ
V). The corrected solar-to-hydrogen efficiency
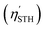
is as follows:
| | 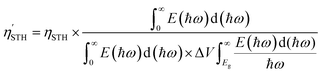 | (24) |
The 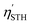 of the SbSI and SbSeBr monolayers fulfilled the 10% efficiency target for the commercial production of hydrogen. The power conversion efficiencies of SbSI and SbSeBr monolayers and other Janus monolayers are listed in Table S5, ESI.†
of the SbSI and SbSeBr monolayers fulfilled the 10% efficiency target for the commercial production of hydrogen. The power conversion efficiencies of SbSI and SbSeBr monolayers and other Janus monolayers are listed in Table S5, ESI.†
3.6.3. Charge transfer dynamics.
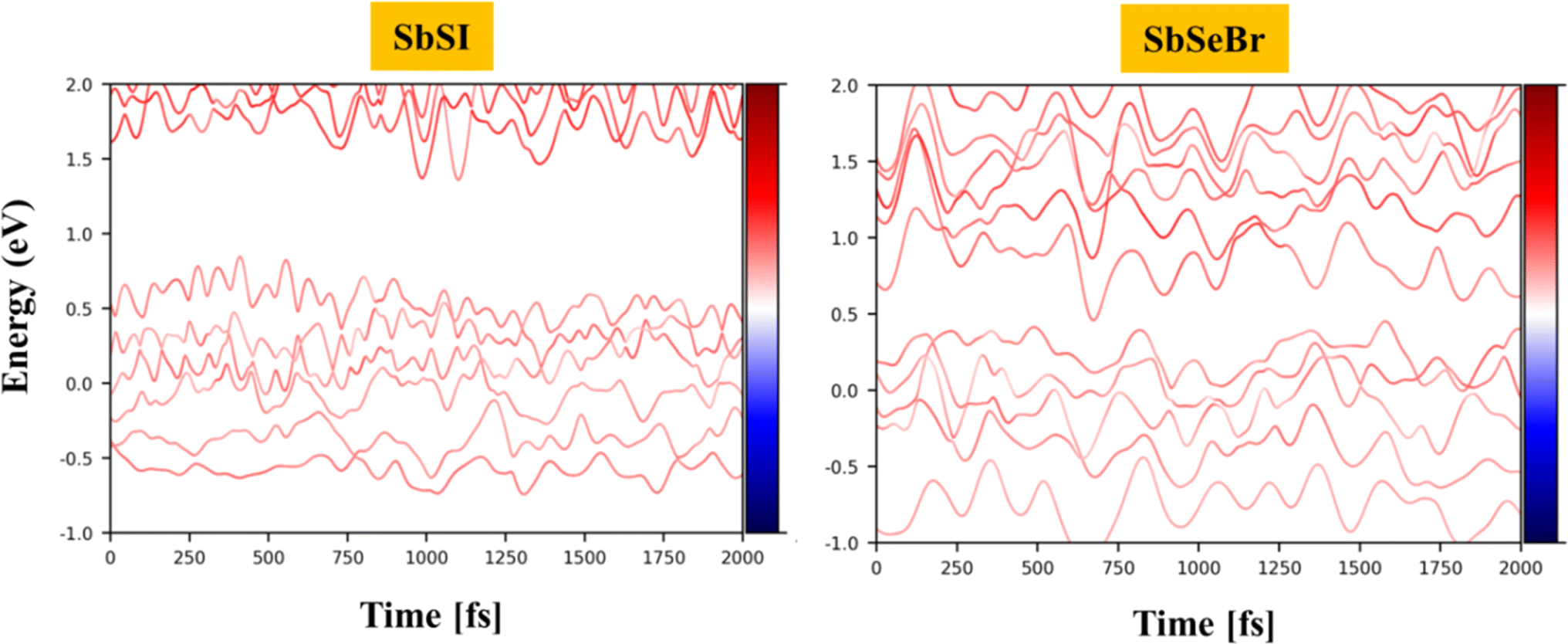
The carrier lifetime is an important aspect of the charge transfer dynamics. Apart from proper band alignment, the lifetime of photogenerated electrons and holes is another deciding factor for a material to act as an efficient photocatalyst. We employed density functional theory for the effective computation of the charge transfer and electron–hole pair recombination rate. The time evolution of the Kohn–Sham (KS) states of Janus SbSI and SbSeBr monolayers at 300 K within NAMD formulism is depicted in Fig. 11.
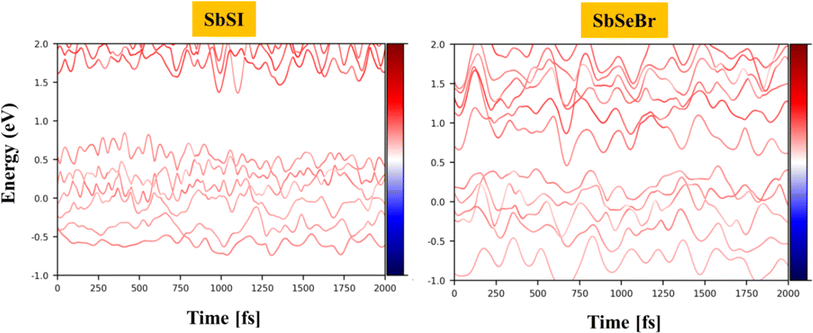 |
| | Fig. 11 The energy evolution of the Kohn–Sham (KS) states of SbSI and SbSeBr monolayers near the Fermi level at 300 K. | |
The time-dependent Kohn–Sham orbitals ψx(r,t) are expended in terms of KS orbitals ϕj(r′,R′(t)) as follows:73
| | 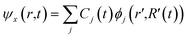 | (25) |
Photon excitation leads to the production of photogenerated charge carriers, electrons (e) and holes (h), in the CBM and VBM, which play a vital role in the recombination process. In the non-radiative electron–hole recombination process, the recombination time of electrons and holes depends on electron–phonon coupling. The recombination rate is determined by non-adiabatic coupling (NAC) and decoherence time.73 The smaller lifetime corresponds to smaller nonadiabatic coupling. The non-adiabatic coupling and decoherence time are related to phonon excitation and electron–phonon coupling. The NAC is given as follows:
| | 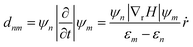 | (26) |
where
ṙ is the velocity of the nuclei. NAC is calculated from electron–phonon coupling elements (
ψn|∇
rH|
ψm), the energy gap (
εm −
εn) and (
ṙ). The computed value of non-adiabatic coupling corresponding to the SbSI and SbSeBr monolayers is 3.25 meV and 3.91 meV, respectively. The exponential decay recombination time (
τ) of photogenerated electrons and holes and electron (hole) dynamics were fitted according to the following:
99| | 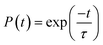 | (27) |
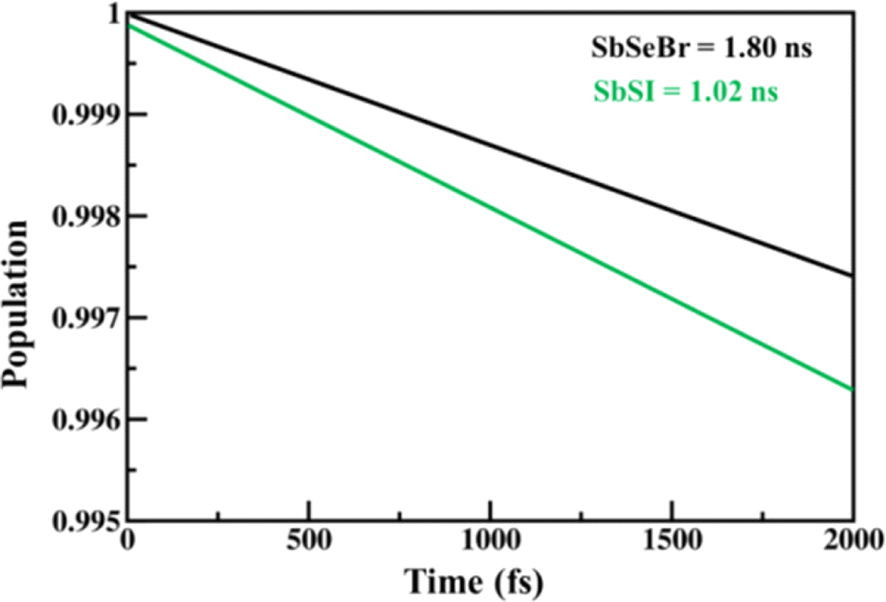
The calculated values of the electron (hole) transfer rate for SbSI and SbSeBr monolayers are 0.08 ps (11.73 ps) and 5.66 ps (20.96 ps) (Fig. S10, ESI†). The values of τ for the SbSI and SbSeBr monolayers were calculated as 1.02 ns and 1.80 ns, respectively (Fig. 12), which are higher as compared to the other monolayers such as pristine black phosphorene (0.29 ns),52 TiO2 (31.1 ns),53 GaSe (48.6 ns),55 GaS (39.9 ns),55 Ga2SeTe (51.5 ns),55 In2SeTe (61.1 ns),55 In2STe (72.8 ns),55 and PtSe2 (>10 ns).57
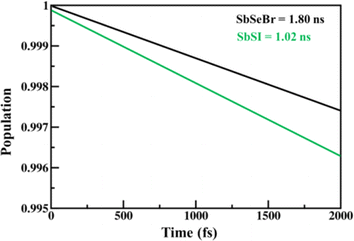 |
| | Fig. 12 The electron–hole recombination dynamics of 2D Janus SbSI and SbSeBr monolayers. | |
4. Conclusions
In this work, we investigated the thermoelectric, piezoelectric, magneto-transport, optoelectronic, photocatalytic, photovoltaic and charge transfer dynamics properties of Janus SbYZ monolayers. The phonon spectra, AIMD simulations and elastic tensor analysis confirmed their dynamical, thermal, and mechanical stability. These monolayers possess low lattice thermal conductivity ranging from 0.61 to 1.83 W m−1 K−1. We incorporated various kinds of elastic and inelastic scattering mechanisms to deal with the electronic transport properties. By the integrated effect of lattice thermal conductivity and electronic transport properties, the computed values of the thermoelectric performance (ZT) of Janus SbYZ monolayers can reach up to ∼1.6 at 800 K. The piezoelectric coefficients were calculated and a large out-of-plane piezoelectric constant was observed in these monolayers. The carrier mobility of these monolayers was calculated using Matthiessen's rule, including the effect of acoustic, piezoelectric and polar-optical scattering; the SbSeCl monolayer carrier mobility reached 4000 cm2 V−1 s−1. To demonstrate the effects of an electric field and magnetic field on free charges, we studied the magneto-transport properties of Janus SbYZ monolayers and these properties were characterized in terms of the Hall conductivity (σ) and Hall factor (r). The most prominent peak of the optical absorbance spectra of SbXY monolayers lies in the visible region. The proposed type-II heterojunction solar cells based on Janus SbYZ monolayers possess high power conversion efficiencies (PCEs), especially in the case of the SbSeBr/AsTeI heterostructures marked up to ∼19%. We also looked into the photocatalytic capabilities of Janus SbSI and SbSeBr monolayers; the redox potential of water lies in the band edge alignment w.r.t to the different surface vacuum potentials of these monolayers. At zero pH, SbSeBr monolayer can be potential candidates for hydrogen generation. Janus SbSI and SbSeBr monolayers showed 12% and 18% STH conversion efficiency, respectively. The charge transfer dynamics properties were studied in the context of photocatalytic water splitting. The calculated prolonged electron–hole recombination rates of SbSI and SbSeBr monolayers were calculated as 1.02 ns and 1.82 ns, respectively. Our study indicates that these monolayers can act as efficient contenders for various energy harvesting fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
PC & JS thanks the CSIR for providing financial support in the form of a senior research fellowship (SRF). The results presented in this work were obtained using the computational resources at the Department of Physics at the Central University of Punjab.
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D.-e. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Dávila, L. Xian, S. Cahangirov, A. Rubio and G. Le Lay, New J. Phys., 2014, 16, 095002 CrossRef.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef PubMed.
- E. Martínez-Periñán, M. P. Down, C. Gibaja, E. Lorenzo, F. Zamora and C. E. Banks, Adv. Energy Mater., 2018, 8, 1702606 CrossRef.
- F. Reis, G. Li, L. Dudy, M. Bauernfeind, S. Glass, W. Hanke, R. Thomale, J. Schäfer and R. Claessen, Science, 2017, 357, 287–290 CrossRef CAS PubMed.
- L. Zhang, Z. Yang, T. Gong, R. Pan, H. Wang, Z. Guo, H. Zhang and X. Fu, J. Mater. Chem. A, 2020, 8, 8813–8830 RSC.
- J. Zhang, S. Jia, I. Kholmanov, L. Dong, D. Er, W. Chen, H. Guo, Z. Jin, V. B. Shenoy and L. Shi, ACS Nano, 2017, 11, 8192–8198 CrossRef CAS PubMed.
- T. V. Vu, C. V. Nguyen, H. V. Phuc, A. Lavrentyev, O. Khyzhun, N. V. Hieu, M. Obeid, D. Rai, H. D. Tong and N. N. Hieu, Phys. Rev. B, 2021, 103, 085422 CrossRef CAS.
- N. Ghobadi, S. G. Rudi and S. Soleimani-Amiri, Phys. Rev. B, 2023, 107, 075443 CrossRef CAS.
- J. Singh and A. Kumar, J. Mater. Chem. C, 2023, 11, 1173–1183 RSC.
- D. Ozbey, M. Kilic and E. Durgun, Phys. Rev. Appl., 2022, 17, 034043 CrossRef CAS.
- B. Cai, J. Tan, L. Zhang, D. Xu, J. Dong and G. Ouyang, Phys. Rev. B, 2023, 108, 045416 CrossRef CAS.
- L. Zhang, J. Yu, M. Yang, Q. Xie, H. Peng and Z. Liu, Nat. Commun., 2013, 4, 1443 CrossRef PubMed.
- A.-Y. Lu, H. Zhu, J. Xiao, C.-P. Chuu, Y. Han, M.-H. Chiu, C.-C. Cheng, C.-W. Yang, K.-H. Wei and Y. Yang, Nat. Nanotechnol., 2017, 12, 744–749 CrossRef CAS PubMed.
- B. Zheng, C. Ma, D. Li, J. Lan, Z. Zhang, X. Sun, W. Zheng, T. Yang, C. Zhu and G. Ouyang, J. Am. Chem. Soc., 2018, 140, 11193–11197 CrossRef CAS PubMed.
- Y. Wang, W. Wei, H. Wang, N. Mao, F. Li, B. Huang and Y. Dai, J. Phys. Chem. Lett., 2019, 10, 7426–7432 CrossRef CAS PubMed.
- R. Ahammed, N. Jena, A. Rawat, M. K. Mohanta, Dimple and A. De Sarkar, J. Phys. Chem. C, 2020, 124, 21250–21260 CrossRef CAS.
- P. Nandi, A. Rawat, R. Ahammed, N. Jena and A. De Sarkar, Nanoscale, 2021, 13, 5460–5478 RSC.
- A. Bafekry, M. Faraji, M. Fadlallah, D. Hoat, H. Jappor, I. A. Sarsari, M. Ghergherehchi and S. Feghhi, Phys. Chem. Chem. Phys., 2021, 23, 25866–25876 RSC.
- M. J. Varjovi, M. Yagmurcukardes, F. M. Peeters and E. Durgun, Phys. Rev. B, 2021, 103, 195438 CrossRef CAS.
- S.-D. Guo and H.-C. Li, Comput. Mater. Sci., 2017, 139, 361–367 CrossRef CAS.
- W.-L. Tao, J.-Q. Lan, C.-E. Hu, Y. Cheng, J. Zhu and H.-Y. Geng, J. Appl. Phys., 2020, 127, 035101 CrossRef CAS.
- C.-W. Wu, X. Ren, G. Xie, W.-X. Zhou, G. Zhang and K.-Q. Chen, Phys. Rev. Appl., 2022, 18, 014053 CrossRef CAS.
- A. Patel, D. Singh, Y. Sonvane, P. Thakor and R. Ahuja, ACS Appl. Mater. Interfaces, 2020, 12, 46212–46219 CrossRef CAS PubMed.
- P. Chauhan, J. Singh and A. Kumar, J. Mater. Chem. A, 2023, 11, 10413–10424 RSC.
- P. Chauhan, J. Singh and A. Kumar, J. Phys. Chem. Solids, 2022, 167, 110758 CrossRef CAS.
- W. Zhang and W. Ji, Phys. Rev. B, 2023, 108, 035411 CrossRef CAS.
- Y. Chu, J. Zhong, H. Liu, Y. Ma, N. Liu, Y. Song, J. Liang, Z. Shao, Y. Sun and Y. Dong, Adv. Funct. Mater., 2018, 28, 1803413 CrossRef.
- Y. Cheng, Y. Ma, L. Li, M. Zhu, Y. Yue, W. Liu, L. Wang, S. Jia, C. Li and T. Qi, ACS Nano, 2020, 14, 2145–2155 CrossRef CAS PubMed.
- M. J. Varjovi and E. Durgun, Phys. Rev. Mater., 2021, 5, 104001 CrossRef CAS.
- X. Li, J. Qiu, X. Chen, F. Zhang, Z. Chen and J. Yu, Mater. Lett., 2022, 325, 132867 CrossRef CAS.
- M. J. Varjovi, S. Ershadrad and B. Sanyal, Phys. Rev. B, 2023, 107, 195421 CrossRef CAS.
- S.-D. Guo, X.-S. Guo, R.-Y. Han and Y. Deng, Phys. Chem. Chem. Phys., 2019, 21, 24620–24628 RSC.
- M. Yagmurcukardes, C. Sevik and F. Peeters, Phys. Rev. B, 2019, 100, 045415 CrossRef CAS.
- Y. Wu, C.-H. Yang, H.-N. Zhang, L.-H. Zhu, X.-Y. Wang, Y.-Q. Li, S.-Y. Zhu and X.-C. Wang, Appl. Surf. Sci., 2022, 589, 152999 CrossRef CAS.
- H.-Q. Xu, G. Xiao, W.-Z. Xiao and L.-L. Wang, Micro Nanostruct., 2022, 170, 207396 CrossRef CAS.
- Q. Yang, T. Zhang, C.-E. Hu, X.-R. Chen and H.-Y. Geng, Phys. Chem. Chem. Phys., 2023, 25, 274–285 RSC.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem.
Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Gong, K. Sumathy, Q. Qiao and Z. Zhou, Renewable Sustainable Energy Rev., 2017, 68, 234–246 CrossRef CAS.
- J. Dai and X. C. Zeng, J. Phys. Chem. Lett., 2014, 5, 1289–1293 CrossRef CAS PubMed.
- D. Jariwala, A. R. Davoyan, J. Wong and H. A. Atwater, ACS Photonics, 2017, 4, 2962–2970 CrossRef CAS.
- F. Zhang, J. Qiu, H. Guo, L. Wu, B. Zhu, K. Zheng, H. Li, Z. Wang, X. Chen and J. Yu, Nanoscale, 2021, 13, 15611–15623 RSC.
- Y. Naik, D. Mehta, P. Parmar and P. Thakor, Phys. B, 2023, 415499 Search PubMed.
- H.-y. Liu, H. Yang and Y. Zheng, Phys. Chem. Chem. Phys., 2024, 26, 6228–6234 RSC.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
- L. Ju, M. Bie, X. Tang, J. Shang and L. Kou, ACS Appl. Mater. Interfaces, 2020, 12, 29335–29343 CAS.
- L. Ju, M. Bie, J. Shang, X. Tang and L. Kou, J. Phys.: Mater., 2020, 3, 022004 CAS.
- R. Peng, Y. Ma, B. Huang and Y. Dai, J. Mater. Chem. A, 2019, 7, 603–610 RSC.
- P. Jamdagni, R. Pandey and K. Tankeshwar, Nanotechnology, 2021, 33, 025703 CrossRef PubMed.
- J. Wang, J. Lu, X. Zhao, G. Hu, X. Yuan and J. Ren, Eur. Phys. J. B, 2023, 96, 17 CrossRef CAS.
- L. Pan, T. Zhang, C.-E. Hu, X.-R. Chen and H.-Y. Geng, J. Mater. Chem. A, 2022, 10, 22676–22685 RSC.
- H. Guo, W. Chu, Q. Zheng and J. Zhao, J. Phys. Chem. Lett., 2020, 11, 4662–4667 CrossRef CAS PubMed.
- C. Gao, L. Zhang, Q. Zheng and J. Zhao, J. Phys. Chem. C, 2021, 125, 27275–27282 CrossRef CAS.
- W. Dou, L. Zhang, B. Song, C. Hua, M. Wu, T. Niu and M. Zhou, J. Phys. Chem. Lett., 2022, 13, 10656–10665 CrossRef CAS PubMed.
- B. Zhang, A. Li, J. Lin and W. Liang, Phys. Chem. Chem. Phys., 2022, 24, 23437–23446 RSC.
- H. Dong, J. Zhao, H. Yang and Y. Zheng, Phys. Rev. Mater., 2022, 6, 104001 CrossRef CAS.
- H. Huang, J. Peng, Z. Li, H. Dong, L. Huang, M. Wen and F. Wu, J. Phys. Chem. Lett., 2022, 13, 10988–10993 CrossRef CAS PubMed.
- J. Zhao, Y. Zhao, H. He, P. Zhou, Y. Liang and T. Frauenheim, J. Phys. Chem. Lett., 2021, 12, 10190–10196 CrossRef CAS PubMed.
- J. Su, J. Zhang, Y. Wang, C. Wang, Q. Niu, R. Sun and W. Zhang, ACS Omega, 2023, 8, 28846–28850 CrossRef CAS PubMed.
- D. Hajra, R. Sailus, M. Blei, K. Yumigeta, Y. Shen and S. Tongay, ACS Nano, 2020, 14, 15626–15632 CrossRef CAS PubMed.
- G. Bianca, C. Trovatello, A. Zilli, M. I. Zappia, S. Bellani, N. Curreli, I. Conticello, J. Buha, M. Piccinni and M. Ghini, ACS Appl. Mater. Interfaces, 2022, 14, 34963–34974 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef PubMed.
- A. K. Mandia, B. Muralidharan, J.-H. Choi, S.-C. Lee and S. Bhattacharjee, Comput. Phys. Commun., 2021, 259, 107697 CrossRef CAS.
- A. Togo, L. Chaput and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 094306 CrossRef.
- Q. Zheng, W. Chu, C. Zhao, L. Zhang, H. Guo, Y. Wang, X. Jiang and J. Zhao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1411 CAS.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
-
I. N. Frantsevich, Reference Book, 1982 Search PubMed.
- L. C. Gomes, A. Carvalho and A. C. Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 214103 CrossRef.
- L. Dong, J. Lou and V. B. Shenoy, ACS Nano, 2017, 11, 8242–8248 CrossRef CAS PubMed.
- Y. Gan, C.-W. Wu, Z.-X. Xie, Y.-X. Deng, Y. Zhang, W.-X. Zhou and X.-K. Chen, Langmuir, 2022, 38, 7733–7739 CrossRef CAS PubMed.
- V.-H. Chu, T.-H. Le, T.-T. Pham and D.-L. Nguyen, RSC Adv., 2023, 13, 4202–4210 RSC.
- D. Broido, A. Ward and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 014308 CrossRef.
- A. K. Mandia, N. A. Koshi, B. Muralidharan, S.-C. Lee and S. Bhattacharjee, J. Mater. Chem. C, 2022, 10, 9062–9072 RSC.
- T. Fang, X. Zhao and T. Zhu, Materials, 2018, 11, 847 CrossRef PubMed.
- N. Wang, M. Li, H. Xiao, Z. Gao, Z. Liu, X. Zu, S. Li and L. Qiao, npj Comput. Mater., 2021, 7, 18 CrossRef CAS.
- G. K. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67–71 CrossRef CAS.
- M. Liu, S.-B. Chen, C.-E. Hu, Y. Cheng and H.-Y. Geng, Solid State Commun., 2022, 342, 114612 CrossRef CAS.
- J. Yang, L. Xi, W. Qiu, L. Wu, X. Shi, L. Chen, J. Yang, W. Zhang, C. Uher and D. J. Singh, npj Comput. Mater., 2016, 2, 1–17 CrossRef.
- M. Rohlfing and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 4927 CrossRef CAS.
- L. Matthes, O. Pulci and F. Bechstedt, J. Phys.: Condens. Matter, 2013, 25, 395305 CrossRef PubMed.
- Y.-m. Ding, X. Nie and Y. Li, Phys. Rev. Mater., 2021, 5, 074005 CrossRef CAS.
- Y.-m. Ding, Y. Ji, H. Dong, N. Rujisamphan and Y. Li, Nanotechnology, 2019, 30, 465202 CrossRef CAS PubMed.
- H.-P. Komsa and A. V. Krasheninnikov, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 241201 CrossRef.
- J. Min, C. Zhao, Z. Zeng, Y. Jia and Z. Du, Phys. Rev. B, 2019, 100, 085402 CrossRef CAS.
- R. Anderson, IBM J. Res. Dev., 1960, 4, 283–287 CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- Z. Zafar, S. Yi, J. Li, C. Li, Y. Zhu, A. Zada, W. Yao, Z. Liu and X. Yue, Energy Environ. Mater., 2022, 5, 68–114 CrossRef CAS.
- X. Zhao, X. Yang, D. Singh, P. K. Panda, W. Luo, Y. Li and R. Ahuja, J. Phys. Chem. C, 2020, 124, 7884–7892 CrossRef CAS.
- C.-F. Fu, J. Sun, Q. Luo, X. Li, W. Hu and J. Yang, Nano Lett., 2018, 18, 6312–6317 CrossRef CAS PubMed.
- S. Wang, Q. Luo, W.-H. Fang and R. Long, J. Phys. Chem. Lett., 2019, 10, 1234–1241 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*

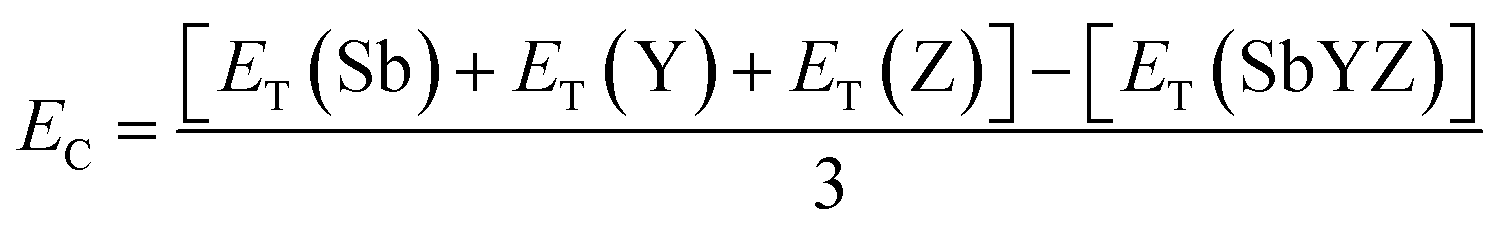

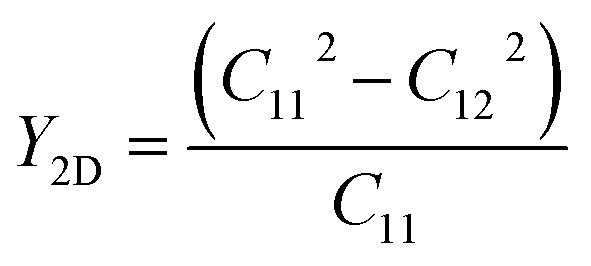












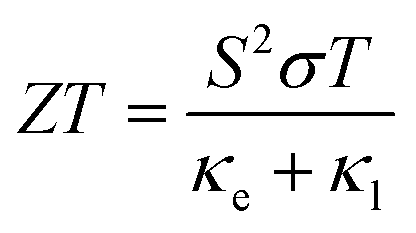




 is the total solar energy. The estimated value of βff is 0.65, which was calculated using the Shockley–Queisser limit. The power conversion efficiency of the proposed heterostructures as a function of donor band gap and conduction band offset is shown in Fig. 8 (lower panel). The PCE value of the Janus SbYZ monolayer and other similar 2D Janus monolayers are tabulated in Table S3, ESI.† The PCE value of SbSeBr/AsTeI heterostructure reached up to 19%, which makes them a potential candidate for photovoltaic solar cells.
is the total solar energy. The estimated value of βff is 0.65, which was calculated using the Shockley–Queisser limit. The power conversion efficiency of the proposed heterostructures as a function of donor band gap and conduction band offset is shown in Fig. 8 (lower panel). The PCE value of the Janus SbYZ monolayer and other similar 2D Janus monolayers are tabulated in Table S3, ESI.† The PCE value of SbSeBr/AsTeI heterostructure reached up to 19%, which makes them a potential candidate for photovoltaic solar cells.




 is as follows:
is as follows:
 of the SbSI and SbSeBr monolayers fulfilled the 10% efficiency target for the commercial production of hydrogen. The power conversion efficiencies of SbSI and SbSeBr monolayers and other Janus monolayers are listed in Table S5, ESI.†
of the SbSI and SbSeBr monolayers fulfilled the 10% efficiency target for the commercial production of hydrogen. The power conversion efficiencies of SbSI and SbSeBr monolayers and other Janus monolayers are listed in Table S5, ESI.†