
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-supported antimony tin oxide anode with Sb segregation promoted atrazine removal†
Xue
Wang
a,
Jia-Fang
Xie
 *ab,
Quan-Bao
Zhao
*ab and
Qian
Sun
ab
*ab,
Quan-Bao
Zhao
*ab and
Qian
Sun
ab
aCAS Key Laboratory of Urban Pollutant Conversion, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen, 361021, China. E-mail: jfxie@iue.ac.cn; qbzhao@iue.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 13th September 2024
Abstract
Electrochemical oxidation is a sustainable approach to remove persistent organic pollutants, although it suffers from slow reaction transfer. Herein, a robust self-supported antimony tin oxide anode was developed by compaction–sintering process with engineering fiber as the pore-forming reagent and second binder. The obtained EF-ATO anode presented a Sb segregation-enhanced built-in electric field (BIEF), realizing 95% removal efficiency of atrazine (ATZ) in 30 min. Kinetics, Kelvin probe force microscopy, and in situ EPR analysis revealed that BIEF accelerated the reaction charge transfer for the co-generation of three reactive oxygen species, contributing to the highly efficient ATZ removal. Furthermore, the robust EF-ATO exhibited low energy consumption, high durability over 10 times cycling tests, and wide applicability in water pH and pollutant types. Thus, EF-ATO possessed high potential as a promising candidate for the eco-removal of persistent organic pollutants. This work provides an approach for designing robust self-supported metal oxide-based anodes and reveals an electrochemical oxidation process promoted by doping metal segregation-induced BIEF.
 Jia-Fang Xie | Jia-Fang Xie received her B.S. degree (2011) in Chemistry and PhD degree (2016) in Applied Chemistry from the University of Science and Technology of China. Since 2020, she has served as an associate professor at the Institute of Urban Environment, Chinese Academy of Sciences. She is now a member of the Youth Innovation Promotion Association CAS. Her research interest focuses on developing advanced electrocatalytic nanomaterials and technologies for sustainable environment and energy applications, including energy storage, green synthesis, and water treatment. |
Introduction
Clean water is vital for the survival of all living organisms and the health of ecosystems. However, it is threatened by persistent organic pollutants such as pesticides. Atrazine (ATZ), an extensively used pesticide, has been found to be widely dispersed in multiple bodies of water.1,2 However, traditional biological treatment and physical methods cannot effectively remove this pollutant.Several technologies have been developed to remove ATZ-including persistent organic pollutants, among which electrochemical oxidation (EO) has attracted increasing attention.3 EO has the advantages of high controllability and efficiency,4 minimal secondary pollution and aeration, and wide applicability to diversified persistent organic pollutants.5,6 Reactive oxygen species (ROS), such as hydroxyl radicals (˙OH), superoxide radicals (˙O2−), and singlet oxygen (1O2), generated in the EO process can effectively remove and even mineralize persistent organic pollutants due to their high oxidation potentials.7 The ROS generation performance heavily depends on the anode.8 However, for widely studied metal oxide-based anodes, a sluggish charge transfer in the metal oxides often limited the ROS generation and pollutant removal efficiency.9
There are two strategies to prepare EO anodes with metal oxide catalyst powder: (1) coating a catalyst-containing ink onto the planar-structured substrate layer-by-layer, and repeated annealing to obtain a planar catalyst-coated anode;10 and (2) compacting a catalyst-containing powder mixture in a mould, and performing one time sintering to obtain 3D self-supported porous anodes.11 The former one uses less catalyst material but requires an interlayer (which usually contains Pt, Ru, and Ir) to distribute and stabilize the catalyst powder on the anodes.12,13 The latter strategy requires the addition of pore-forming (polymethacrylate, carbon fiber (CF), starch, etc.)11,14,15 and binder reagents (paraffin oil, polyacrylic acid, chitosan, etc.)16–18 to a greater amount of catalyst to prepare the EO anode. After sintering, the self-supported anodes gain a uniform composition and porous structure because the pore-forming and binder reagents decompose during sintering. Freestanding SnOx/La–Sb anode and Ti4O7 anodes19,20 prepared by this compaction–sintering strategy showed the effective and rapid removal of moxifloxacin and 4-chlorophenol, respectively. Despite these great achievements, the removal efficiency and kinetics have still been limited by the sluggish charge transfer in metal oxides. Although a second metal has been doped to increase the conductivity, there have been few studies on how doping metals can improve the reaction charge transfer.
In this work, we take Sb-doped SnO2 (ATO), a widely studied metal oxide EO catalyst, as a model catalyst to develop a robust and effective self-supported ATO anode by a double binder strategy. Polypropylene engineering fiber (EF) was chosen as a pore-forming reagent because of its additional binder function and beneficial qualities, including low cost and density, easy decomposition without residue under low temperature, and hard-elastic properties. EFs are widely applied in the construction industry because it forms a scrambled support system in cement. The structural properties of the obtained EF-ATO anode were studied with the focus on Sb segregation driven by 1000 °C sintering. The EO performance of the EF-ATO anode for ATZ removal was investigated. Under optimized conditions, the durability and applicability of the EF-ATO anode were evaluated. The effect of Sb segregation on the excellent EO performance was revealed with multiple techniques. Finally, the intermediates of ATZ degradation with the EF-ATO anode and their ecotoxicity were analysed.
Results and discussion
By compaction and 1000 °C sintering with EF, some micron-level cavities derived from EF could be observed without residue in the EF-ATO anode (Fig. 1a). High-resolution scanning electron microscope studies revealed that the size of the ATO particles grew from about 20 nm (Fig. S1†) to nearly 80 nm in EF-ATO (Fig. 1a). X-ray diffraction (XRD) patterns of these two materials shared the same peaks located at 26.58°, 33.91°, 37.95°, 51.80°, and 54.76° (Fig. 1c), in good agreement with the (110), (101), (200), (211) and (220) crystal planes of the tin oxide phase (PDF no. 01-088-2348), respectively. The higher and narrower peaks of EF-ATO proved its superior crystal structure after sintering, which would facilitate charge transfer. Meanwhile, the specific surface area was reduced by nearly 80%, and the micropores and small mesopores almost disappeared (Fig. S2 and Table S1†), indicating the smoother surface of the larger ATO particles in EF-ATO. Notably, the EF-ATO anode possessed higher integrity and mechanical strength over the ATO anode with CF as the pore-forming reagent (CF-ATO, Fig. S3 and Table S2†), proving the advantage of EF with the extra binder function. This feature would allow the self-supported EF-ATO to adjust to various device configurations and tough operation conditions for complicated applications. Additionally, although CF-ATO showed a similar N2 adsorption–desorption isotherm with EF-ATO, a carbon fiber residue could be observed (Fig. S4†). This is possibly due to the incomplete decomposition of CF with a much higher decomposition temperature than EF. | ||
Fig. 1 The structural properties of EF-ATO. (a and b) SEM images. (c) XRD patterns. (d) N2 adsorption–desorption isotherms. The initial ATO powder is shown for comparison. (e) Surface Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sb atomic ratio and in-depth Sn:Sb from XPS. (f) Illustration of heat-driven Sb segregation in EF-ATO. :Sb atomic ratio and in-depth Sn:Sb from XPS. (f) Illustration of heat-driven Sb segregation in EF-ATO. | ||
The survey X-ray photoelectron spectroscopy (XPS) spectra of the EF-ATO anode showed the presence of Sn, Sb, and O, in addition to C from the adsorbed CO2 (Fig. S5a†). C 1s was calibrated to 284.8 eV (Fig. S5b†) before fitting the high-resolution XPS spectra of Sn, Sb, and O. Based on the fitting results, the surface atomic ratio of Sn:Sb was 2.0 for EF-ATO (Fig. 1e). This ratio is apparently smaller than that for the initial ATO powder (5.2) and similar with the CF-ATO anode (2.2), indicating that the 1000 °C sintering is driving this change. The further depth-profiling XPS spectra of EF-ATO presented the gradually concentrated Sb from the inner part to the surface, proving the Sb segregation driven by 1000 °C sintering. Moreover, the relative content of oxygen vacancy was firstly evaluated by calculating the area ratio of the adsorbed oxygen (Oad) to lattice oxygen (OL) species.21 The oxygen vacancy content for EF-ATO was calculated to be three times higher than that of the initial ATO, nearly opposite to the surface Sn:Sb ratio, implying that the 1000 °C sintering step generated the oxygen vacancy on ATO.22 The EPR results further confirmed the enhanced oxygen vacancy content in EF-ATO over the initial ATO powder (Fig. S6†). The oxygen vacancy would tune the metal–O bonds by offering unsaturated coordination and optimize the interactions between ATO and water-including oxygen-containing species, benefiting the EO process.23,24 Therefore, the EF-ATO anode prepared by 1000 °C sintering with EF as the pore-forming reagent possessed Sb segregation and a more crystalline structure.
The EO properties of the EF-ATO anode were investigated with ATZ as a typical persistent organic pollutant. Commercial boron doped diamond (BDD) and Ti mesh-loaded ATO particles (ATO@Ti) were also examined for comparison. As shown in Fig. 2a, the EF-ATO anode presented the highest removal efficiency of about 95% at 30 min and 99.3% at 60 min. As comparison, the commercial BDD and ATO@Ti anodes showed a much lower removal efficiency of 79% and 61% at 60 min for ATZ, respectively. Correspondingly, EF-ATO held the higher value of ATZ removal rate constant as 0.086 min−1 (Fig. 2b) over BDD (0.026 min−1) and the ATO@Ti anode (0.016 min−1). Notably, this high-rate removal of ATZ on the EF-ATO anode exceeds most recent anodes (Table S3†). Furthermore, the energy consumption of the EF-ATO anode for ATZ removal was only 3.08 kW h m−3, much lower than that of BDD and commercial ATO@Ti. These results demonstrated the advantages of the self-supported EF-ATO over the powder-coated ATO@Ti, which has also been reported by many other electrochemical reactions.25
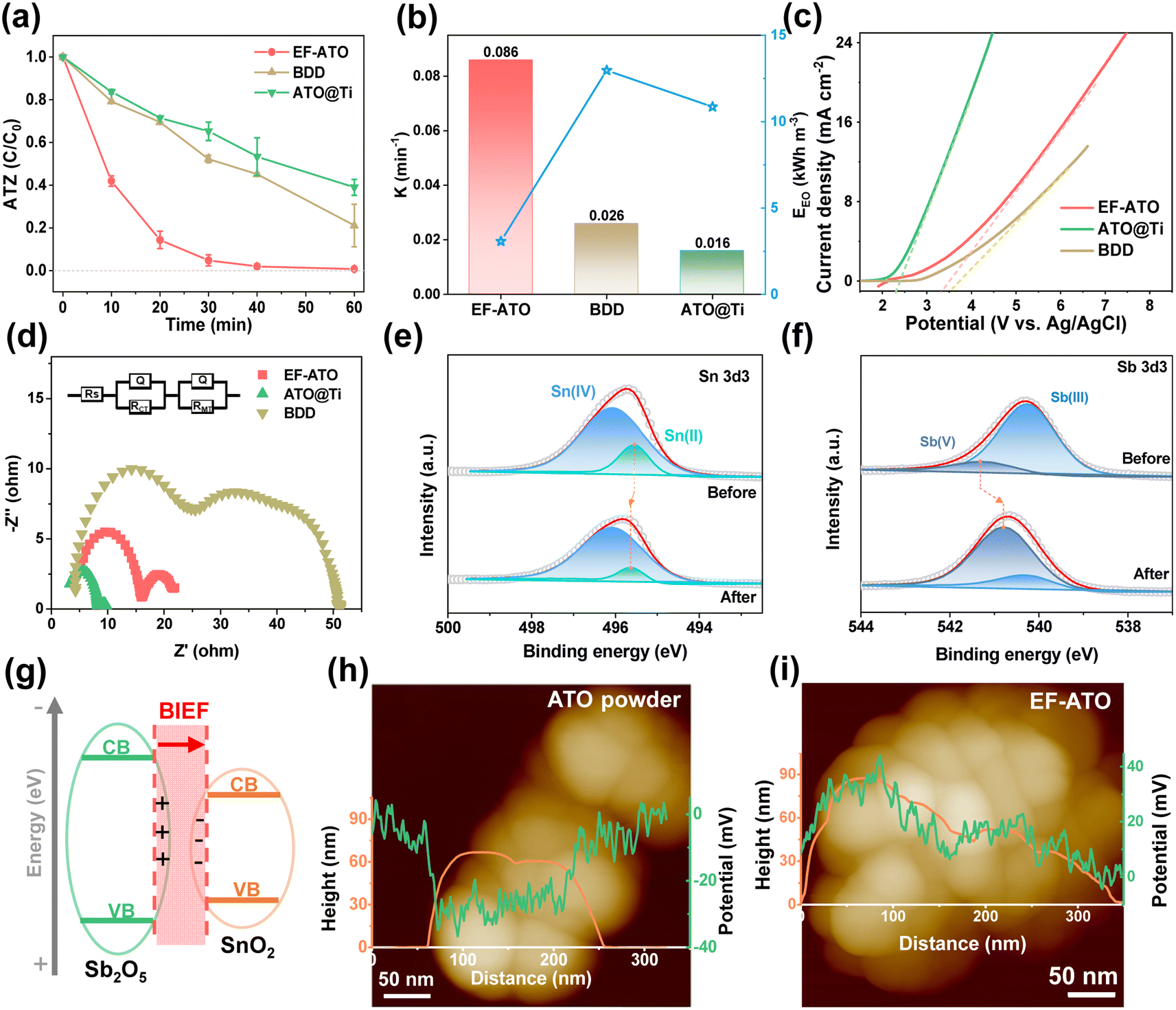 | ||
| Fig. 2 EO performance for ATZ removal on EF-ATO. (a) ATZ removal rate, (b) the corresponding removal rate constant and energy consumption. (c) LSV curve in 0.1 M Na2SO4. (d) EIS in 0.1 M Na2SO4. Commercial BDD and ATO@Ti ware also shown as comparison. High-resolution XPS of (e) Sn 3d3 and (f) Sb 3d3. (g) The illustration of BIEF between Sb2O5 and SnO2. KPFM image with linear height and surface potential distribution for the (h) initial ATO powder and (i) EF-ATO. If not specified, experimental conditions: C0 = 20 mg L−1, current density = 10 mA cm−2, pH = 6, and Na2SO4 concentration = 0.1 M. | ||
To understand the electro-kinetics of the EF-ATO anode, linear scanning voltammetry (LSV) was conducted to evaluate the oxygen evolution potential (OEP). The high OEP can significantly reduce the side reactions of the oxygen evolution, and thus enhance the generation of ROS, benefiting the ATZ removal capacity and energy efficiency.26 As shown in Fig. 2c, the OEP of the EF-ATO anode was 3.36 V, which is very close to that of the BDD (3.50 V) and far beyond ATO@Ti (2.35 V). Thus, EF-ATO is suggested to exhibit excellent ROS generating performance similar to that of BDD, and exceeding that of ATO@Ti. Furthermore, the electrochemical impedance spectroscopy (EIS) study can reveal the impedance properties of the electrodes, especially the reaction charge transfer resistance (RCT) that is highly related to the EO performance. According to the EIS curves, EF-ATO possessed an apparently smaller semicircle than BDD (Fig. 2d). Further fitting analysis showed that the RCT of EF-ATO was 6.78 Ω, lower than that of BDD (30.12 Ω) and close to the RCT of ATO@Ti (5.78 Ω) (Table S4†).
XPS characterization of EF-ATO after the EO process was carried out to investigate the possible variations of the atomic ratio and valence of metal elements. After C 1s was calibrated to 284.8 eV, the high-resolution spectra of Sn 3d and Sb 3d & O 1s were fitted (Fig. S7†). The surface atom ratio of Sn:Sb was slightly increased to 2.5, suggesting the stable existence of Sb segregation in EF-ATO. Interestingly, the location of the Sn(II) peak moved to the higher binding energy. Meanwhile, the Sb(V) peak moved negatively to lower binding energy, suggesting an electron transfer from Sn(II) to Sb(V) during the EO process. The electron transfer increased the ratio of Sb(V) in Sb and Sn(IV) in Sn, which would benefit the ROS generation and ATZ removal by the EO process with EF-ATO (Fig. S8†). Moreover, the oxygen vacancy slightly increased after the EO process (Fig. S9†), suggesting its stable existence. Moreover, Sb segregation induced the concentrated Sb2O5–SnO2 heterojunction structure on the surface over the inner part. The Sb2O5–SnO2 heterojunction would generate the built-in electric field (BIEF) between them (Fig. 2g), facilitating the charge transfer. Kelvin probe force microscopy (KPFM) was further applied to measure the surface potential of the ATO particles, which is highly related to the built-in electric field (BIEF) strength.27 According to the KPFM results, pure tin oxide without Sb doping had the weakest BIEF strength (Fig. S10†) and the initial ATO with uniform Sb doping had a stronger BIEF than pure tin oxide (Fig. 2h). After 1000 °C sintering, EF-ATO with Sb segregation showed the strongest BIEF strength (Fig. 2i). By accelerating the charge transfer and tuning the intermediate adsorption for electrochemical reactions, the enhanced BIEF would promote ROS generation in the EO process on the EF-ATO anode.22,28
Influence factors on the EO process with EF-ATO were further studied. As shown in Fig. S11a,† four binder contents during the EF-ATO preparation made a slight difference in the ATZ removal efficiency. A 5% binder content, achieving the highest removal rate constant of 0.086 min−1 (Fig. S11b†), was thus fixed for the following investigation of other influence factors. The electrolyte concentration affected the ATZ removal efficiency a bit when it was as low as 0.02 mol L−1 (Fig. 3a). When the electrolyte concentration was gradually increased from 0.05 to 0.15 mol L−1, the ATZ removal efficiencies at 60 min were nearly the same. Meanwhile, the removal rate constant achieved the highest value of 0.086 min−1 with 0.1 M electrolyte concentration (Fig. S11c†). Thus, 0.1 M electrolyte would provide sufficient conductivity for this EO system with the EF-ATO anode.29 Increasing the initial pH of the electrolyte from 3 to 11 showed a slight influence on the ATZ removal efficiency by the EO process with EF-ATO (Fig. 3b), but played an obviously negative role in the removal rate constant (Fig. S11d†). This could be possibly ascribed to the enhancement of the ROS generation in acid solution, as reported by previous works.30 Taking the cost and removal efficiency into consideration together, the initial pH of 6 was chosen as the optimized one. The current density was gradually increased from 5 to 15 mA cm−2, which resulted in a tiny improvement of the ATZ removal efficiency (Fig. S11e†). The removal rate constant grew from 0.054 min−1 to 0.125 min−1 as the current density increased (Fig. 3c). Up to 96.9% ATZ could be removed in 30 min electrolysis at the current density of 15 mA cm−2. However, when the energy consumption was also taken into consideration, the current density of 10 mA cm−2 was considered to be the better choice with the lowest energy consumption to obtain one of the highest removal efficiencies in 60 min. Thus, 5% binder content, 0.1 M Na2SO4 solution with pH of 6, current density of 10 mA cm−2, and 60 min electrolysis were chosen as the optimized conditions for the following experiments.
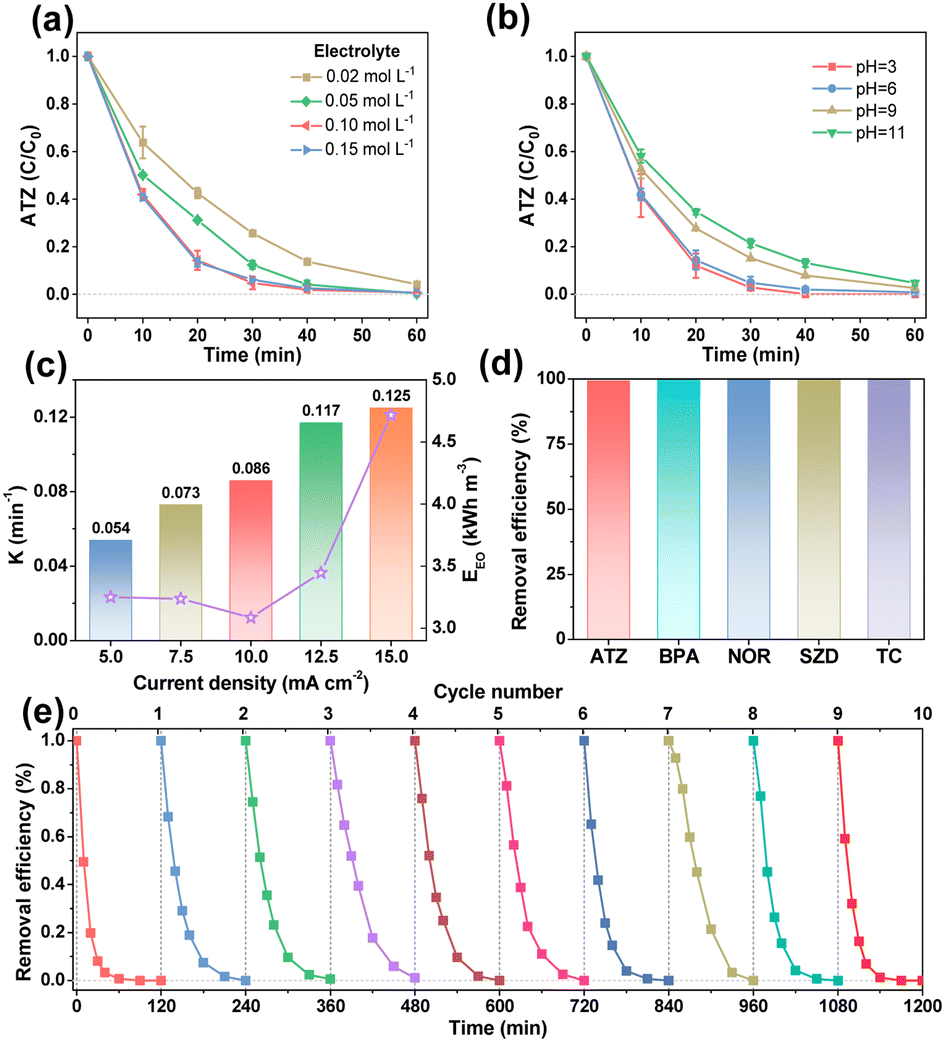 | ||
| Fig. 3 Influence factors of ATZ removal on EF-ATO anode. Removal efficiency under a series of electrolyte (a) concentration and (b) pH. (c) Removal rate constant and EEO at a series of current densities. (d) Removal efficiency of different pollutants. (e) Recyclability of EF-ATO anode for ATZ removal. If not specified, experimental conditions: C0 = 20 mg L−1, j = 10 mA cm−2, pH = 6, and Na2SO4 concentration = 0.1 M. | ||
The EO properties of EF-ATO for more kinds of persistent organic pollutants were also evaluated. The EO system with EF-ATO presented excellent performance of removing bisphenol A (BPA), levofloxacin (LFX), sulfadiazine (SDZ), and tetracycline (TC) (Fig. 3d and S11f†). These results suggest the promising application of EF-ATO in the efficient removal of other kinds of persistent organic pollutants. Finally, ten times consecutive removal experiment showed highly stable removal efficiency of ATZ throughout the ten times experiments (Fig. 3e). Therefore, the robust EF-ATO could remove persistent organic pollutants with high rate, high durability, low energy consumption, and wide applicability in different pH environments and pollutant types.
To explore the possible ROS in the EO process on EF-ATO, quenching experiments with three concentrations of quenching reagent were carried out. tert-Butanol alcohol (TBA), furfuryl alcohol (FFA), and para-benzoquinone (p-BQ), respectively, were added into the electrolyte before the EO process took place (Fig. S12†).31 As depicted in Fig. 4a, the ATZ removal efficiency was the most sensitive to the concentrated addition of FFA, as 50 mM FFA addition resulted in the negligible removal of ATZ. TBA and p-BQ additions also decreased the ATZ removal efficiency up to around 50%. Their sum exceeded 100%, possibly owing to the complicated interconversion of ROS.32In situ EPR further confirmed the characteristic signals of DMPO–˙OH, DMPO–˙O2−, and TEMP-1O2, and their intensities improved as the electrolysis time continued (Fig. 4b–d). The relative contributions of the three ROS to the ATZ removal were evaluated with quenching experiments based on the equations in ESI.† Accordingly, ˙OH, ˙O2−, and 1O2 contributed 30.8%, 48.7%, and 20.5%, respectively, to ATZ removal. These results demonstrated the co-generation of ˙OH, ˙O2−, and 1O2 in the EO process. This could be ascribed to the effective removal of multiple persistent organic pollutants, as different ROS molecules have their respective dominant reaction activity to differed typical pollutant structures.
 | ||
| Fig. 4 ROS determination. (a) Quenching tests with TBA, FFA, and p-BQ, respectively. In situ EPR spectra of (b) ˙OH, (c) ˙O2−, and (d) 1O2. (e) Illustration of EF-ATO with Sb segregation enhanced EO process for persistent organic pollutants removal. | ||
Finally, based on 19 intermediates of ATZ degradation identified by UPLC-MS/MS system with triple quadrupole mass spectrometer (Fig. S13 and Table S5†), the possible 3 main pathways of ATZ removal in the EO process with EF-ATO were proposed (Fig. S14†). There were 6 reactions involved, including dechlorohydroxylation, dealkylation, dehydroxylation, alkyl hydroxylation, and hydroxylation reactions. To comprehensively evaluate the potential ecotoxicity of these intermediate products, the QSAR method combined with T.E.S.T was applied. According to the evaluation of acute toxicity, bioaccumulation factor, developmental toxicity, and mutagenicity of ATZ and 19 degradation intermediates (Fig. S15†), the ecotoxicity of ATZ could be efficiently decreased by the EO process with EF-ATO. Thus, the mechanism of EF-ATO with Sb segregation enhancing the eco-removal of persistent organic pollutants by EO process was illustrated in Fig. 4e. On robust EF-ATO, Sb segregation induced the BIEF-enhanced reaction electron transfer in the electrochemical water oxidation for ROS generation. The ˙OH, ˙O2−, and 1O2 molecules all played an important role in the robust eco-removal of persistent organic pollutants.
Conclusions
This work describes a self-supported EF-ATO anode with Sb segregation synthesized using a double binder strategy for persistent organic pollutant removal by EO process. EF-ATO possessed an enhanced build-in electric field driven by Sb segregation, facilitating charge transfer and promoting the generation of ˙OH, ˙O2−, and 1O2 in EO. Accordingly, the removal efficiency of ATZ reached 95% in 30 min and 99.3% at 60 min with a low energy consumption of 3.08 kW h m−3. Accompanied with high durability and wide applicability in pH and pollutant type, EF-ATO heightens its potential as a promising candidate for practical application in removing persistent organic pollutants. These results extend the understanding of segregation-induced BIEF promoting EO process and strategies of designing robust self-supported electrodes for energy and environment application.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Xue Wang: investigation, formal analysis, writing – original draft. Jia-Fang Xie: conceptualization, formal analysis, visualization, writing – review & editing, supervision. Quan-Bao Zhao: supervision, funding acquisition, writing – review & editing. Qian Sun: resources, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Key Research & Development Program of China (2021YFA1202700), National Natural Science Foundation of China (52027815), the Youth Innovation Promotion Association CAS (2022308) to J. F. Xie. We thank Professor Xing Zhang from Anhui University for helping heterojunction construction and analysis.References
- S. H. Wu, H. J. He, X. Li, C. P. Yang, G. M. Zeng, B. Wu, S. Y. He and L. Lu, Chem. Eng. J., 2018, 341, 126e136 Search PubMed.
- X. Yu, X. Jin, N. Wang, Q. Zheng, Y. Yu, J. J. Tang, L. Wang, R. Zhou, J. Sun and L. Zhu, Environ. Int., 2022, 166, 107377 CrossRef CAS PubMed.
- D. Seibert, C. F. Zorzo, F. H. Borba, R. M. Souza, H. B. Quesada, R. Bergamasco and J. J. Inticher, Sci. Total Environ., 2020, 748, 141527 CrossRef CAS PubMed.
- G. R. P. Malpass, D. W. Miwa, S. A. S. Machado, P. Olivi and A. J. Motheo, J. Hazard. Mater., 2006, 137, 565–572 CrossRef CAS PubMed.
- B. P. Chaplin, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 RSC.
- S. Komtchou, A. Dirany, P. Drogui, N. Delegan, M. A. El Khakani, D. Robert and P. Lafrance, Chemosphere, 2016, 157, 79–88 CrossRef CAS PubMed.
- G. Lofrano, R. Pedrazzani, G. Libralato and M. Carotenuto, Curr. Org. Chem., 2017, 21, 1054–1067 CrossRef CAS.
- E. Mousset, N. Oturan, E. D. van Hullebusch, G. Guibaud, G. Esposito and M. A. Oturan, Water Res., 2014, 48, 306–316 CrossRef CAS PubMed.
- P. Zhang, L. Yu and X. W. Lou, Angew. Chem., Int. Ed. Engl., 2018, 57, 15076–15080 CrossRef CAS PubMed.
- Y. Wang, C. Shen, M. Zhang, B.-T. Zhang and Y.-G. Yu, Chem. Eng. J., 2016, 296, 79–89 CrossRef CAS.
- C. Yang, Y. Fan, P. Li, Q. Gu and X.-y. Li, Chem. Eng. J., 2021, 422, 130032 CrossRef CAS.
- X. Li, D. Shao, H. Xu, W. Lv and W. Yan, Chem. Eng. J., 2016, 285, 1–10 CrossRef CAS.
- D. Santos, M. J. Pacheco, A. Gomes, A. Lopes and L. Ciríaco, J. Appl. Electrochem., 2013, 43, 407–416 CrossRef CAS.
- S. Mestre, A. Gozalbo, M. M. Lorente-Ayza and E. Sánchez, J. Eur. Ceram. Soc., 2019, 39, 3392–3407 CrossRef CAS.
- G. Wang, Y. Liu, Y. Duan, J. Ye and Z. Lin, Ceram. Int., 2023, 49, 15357–15364 CrossRef CAS.
- P. Parikh, M. Sina, A. Banerjee, X. Wang, M. S. D'Souza, J.-M. Doux, E. A. Wu, O. Y. Trieu, Y. Gong, Q. Zhou, K. Snyder and Y. S. Meng, Chem. Mater., 2019, 31, 2535–2544 CrossRef CAS.
- Y. Jing, S. Almassi, S. Mehraeen, R. J. LeSuer and B. P. Chaplin, J. Mater. Chem. A, 2018, 6, 23828–23839 RSC.
- L. Chai, Q. Qu, L. Zhang, M. Shen, L. Zhang and H. Zheng, Electrochim. Acta, 2013, 105, 378–383 CrossRef CAS.
- C. Yang, S. Shang and X.-y. Li, J. Hazard. Mater., 2022, 436, 129212 CrossRef CAS PubMed.
- Z. Zhao, J. Zhang, J. Yao and S. You, Environ. Res., 2022, 210, 113004 CrossRef CAS PubMed.
- X. Wang, P. Ren, H. Tian, H. Fan, C. Cai and W. Liu, J. Alloys Compd., 2016, 669, 29–37 CrossRef CAS.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS PubMed.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2021, 31, 2009070 CrossRef CAS.
- M.-C. Han, J.-H. Zhang, C.-Y. Yu, J.-C. Yu, Y.-X. Wang, Z.-G. Jiang, M. Yao, G. Xie, Z.-Z. Yu and J. Qu, Angew. Chem., Int. Ed. Engl., 2024, 63, e202403695 CrossRef CAS PubMed.
- X. Sun, S. Wang, Y. Hou, X. F. Lu, J. Zhang and X. Wang, J. Mater. Chem. A, 2023, 11, 13089–13106 RSC.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- Y. Li, D. Zhang, P. Wang, J. Qu and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2407012121 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- S. Periyasamy and M. Muthuchamy, J. Environ., 2018, 6, 7358–7367 CAS.
- X. Sun, H. Qi and Z. Sun, Chemosphere, 2022, 286, 131972 CrossRef CAS PubMed.
- C. Yang, S. Shang, L. Lin, P. Wang, Z. Ye, Y. Wang, K. Shih, L. Sun and X.-y. Li, Nat. Water, 2024, 2, 793–802 CrossRef.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03129f |
| This journal is © The Royal Society of Chemistry 2024 |