
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of washing on nickel-rich NCMs: methodology to quantify the lithium/proton-exchange kinetics, the inserted protons, and the structural changes†
Stefan
Oswald‡§
 *a,
Rebecca
Wilhelm‡
a,
Tim
Kratky
b,
László
Szentmiklósi
c,
Boglárka
Maróti
c,
Ildikó
Harsányi¶
c,
Sebastian A.
Hallweger
d,
Gregor
Kieslich
d,
Sebastian
Günther
b and
Hubert A.
Gasteiger
a
*a,
Rebecca
Wilhelm‡
a,
Tim
Kratky
b,
László
Szentmiklósi
c,
Boglárka
Maróti
c,
Ildikó
Harsányi¶
c,
Sebastian A.
Hallweger
d,
Gregor
Kieslich
d,
Sebastian
Günther
b and
Hubert A.
Gasteiger
a
aTechnical University of Munich, TUM School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Chair of Technical Electrochemistry, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: Stefan.Oswald@tum.de
bTechnical University of Munich, TUM School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Associate Professorship of Physical Chemistry with Focus on Catalysis, Lichtenbergstraße 4, 85748 Garching, Germany
cHUN-REN Centre for Energy Research, Nuclear Analysis and Radiography Department, Konkoly-Thege Miklós út 29-33, Budapest, 1121, Hungary
dTechnical University of Munich, TUM School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Chair of Inorganic and Metal-Organic Chemistry, Lichtenbergstraße 4, 85748 Garching, Germany
First published on 23rd August 2024
Abstract
The washing of cathode active materials (CAMs) for lithium-ion batteries (LIBs) to remove unwanted contaminants is of high importance in industrial CAM manufacturing. Nickel-rich NCMs (LiMO2, M = Ni, Co, Mn), a family of commercial high-energy CAMs, are known for their detrimental reactivity with liquid/gaseous water; yet, insights on the kinetics of the occurring lithium/proton (Li+/H+) exchange are still lacking. To assess the Li+/H+ exchange, a set of five independent techniques is developed and employed to quantify its extent for a nickel-rich NCM (LiNi0.83Co0.12Mn0.05O2) as a function of the washing parameters. Using titration, prompt-gamma activation analysis, thermogravimetric analysis, temperature-dependent X-ray diffraction, and X-ray photoelectron spectroscopy, the proton content of washed NCM samples is determined, exploring the impact of washing time and water-to-CAM mass ratio. Furthermore, a highly protonated NCM (H0.25Li0.75Ni0.83Co0.12Mn0.05O2) is used to investigate the bulk properties, e.g., the change of lattice parameters and the spatial proton distribution, as well as the thermal decomposition of the protonated phase. The gained knowledge on the mechanism and the kinetics of the Li+/H+ exchange as well as on the decomposition mechanism of the protonated NCM phase is essential for the successful aqueous processing of nickel-rich CAMs, advancing the sustainable production of LIBs.
Introduction
Layered lithium transition metal oxides (LiMO2) are the most widely used cathode active materials (CAMs) in commercial lithium-ion batteries (LIBs).1,2 NCMs (M = Ni, Co, Mn; LiNiaCobMncO2, with a+b+c = 1) are the best-known representative, providing a high theoretical capacity of 275 mAh g−1. Due to structural instabilities of the NCM structure at high degrees of delithiation, however, the utilized capacity has to be limited;3–8 otherwise, the release of reactive oxygen from the NCM structure at high state of charge oxidizes the electrolyte chemically,4,9–12 the formed oxygen-depleted layer on the NCM surface increases the resistance of the CAM,3,13–16 and the conventionally used polycrystalline particles expose their internal surface area due to particle cracking.17–20 Fortunately, nickel-rich NCMs, being cost-efficient due to the reduced cobalt content,21 provide a large share of up to 85% (or 234 mAh g−1) of their theoretical capacity in their stable operation window.8 Even though the use of nickel-rich NCMs is essential to enable higher specific capacities and thus energy densities for future LIBs, their successful application is challenged by their tendency to form unwanted surface species, which increases with the nickel content.22,23In nickel-rich NCMs, these surface species originate from: (i) residual lithium salts (such as lithium carbonate) which remain from the slight lithium excess (of few mol%) required for the synthesis, especially for CAMs with a high nickel content,24,25 (ii) the use of flux agents (such as NaCl, KCl, LiOH, LiNO3, or Li3BO3), which are beneficial for the synthesis of single-crystalline NCM particles,26–29 and (iii) the formation of surface contaminants such as lithium and transition-metal hydroxides, carbonates, and bicarbonates when the CAM is stored in humid/ambient atmosphere containing water vapor (and carbon dioxide (CO2)).23,30–34 Especially the surface contaminants are detrimental to electrode manufacturing, cell performance, and battery safety, as they: (i) increase the pH value of the slurry due to their basicity, resulting in the cross-linking of polymer chains of the polyvinylidene-fluoride (PVDF) binder and thus the gelation of the slurry during mixing;35–37 (ii) result in an alkaline slurry which may corrode the aluminum current collector and evolve hydrogen during electrode casting;38 (iii) evolve CO2 upon the chemical decomposition of the carbonate species during the first few cycles;23,39,40 and/or, (iv) compromise the cycle life of a battery.23,30–32
To counter these contaminant-induced detriments and remove any unwanted surface species prior to electrode manufacturing, CAMs are typically washed in H2O or in alcohols to remove any surface species.41–44 Due to their high solubility in H2O, the surface can be cleared efficiently from the present salts.42,45–47 Even though this washing step induces additional cost during the manufacturing of the CAM, it is crucial for electrode manufacturing, reduces gassing during cell operation, and improves the cycle life of a battery. On the downside, the washing with water is accompanied by an unwanted lithium/proton (Li+/H+) cation exchange, yielding a partially protonated NCM (HxLi1−xMO2) and lithium hydroxide (LiOH) dissolved in the washing solution:46
LiMO2 + x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) H2O → HxLi1−xMO2 + xLiOH H2O → HxLi1−xMO2 + xLiOH | (1) |
Based on the current mechanistic understanding, the Li+/H+ exchange occurs within the near-surface region of the NCM particles, both during washing46 and storage in humid atmosphere.34 This cation exchange decreases the lithium inventory of the CAM and thus of the cell, while the intercalation of protons results in resistance build-up and deteriorates the cycle life.44–46 The implications of this exchange reaction were first investigated by Pritzl et al. for a nickel-rich NCM (with a nickel content of 85 mol%),46 which is known for its accelerated reaction with H2O.23,32,34,48,49 Their study revealed that the protonated material exhibits a compromised thermal stability as it releases H2O and molecular oxygen (O2) upon heating,33,46 resulting in an oxygen-depleted surface layer, similar to what was observed for partially delithiated NCMs.22,50,51 Consequently, the release of oxygen from the NCM structure at high degrees of delithiation, investigated by on-line electrochemical mass spectrometry (OEMS), is mitigated – or even eliminated – for a washed NCM that was dried at 180 or 300 °C prior to electrode preparation, reducing the extent of unwanted electrolyte oxidation reactions.46 Unfortunately, the thus formed oxygen-depleted surface layer also leads to a roughly 10-fold higher impedance as compared to the unwashed NCM, thereby diminishing its overall electrochemical performance.46 Therefore, it is essential to understand and optimize the washing parameters – such as the time, the concentration (i.e., the water-to-CAM mass ratio), the temperature as well as the drying conditions – with the aim to maximize the removal of surface species (enhanced for extended washing) while suppressing the Li+/H+ exchange (reduced for minimized washing). Typically, this optimization is performed empirically by evaluating the effect of the washing parameters on the amount of remaining surface species, the extent of gas evolution, as well as the electrochemical cell performance and cycle life. Particularly, the latter two should depend on the proton content of the washed CAM, independent of the exact washing conditions; however, the proton content after washing has not yet been assessed.
In this study, the extent and the kinetics of the Li+/H+ cation exchange, i.e., the proton content of a nickel-rich CAM (LiNi0.83Co0.12Mn0.05O2, further on referred to as NCM831205 or simply NCM) is investigated as a function of the washing parameters. A set of five independent methods is employed to conclusively quantify the proton content of differently washed NCM samples in terms of the mole fraction of protons ion-exchanged into the NCM material (xH+, in units of mol%, defined by HxLi1−xMO2): acid–base titration, prompt-gamma activation analysis (PGAA), thermogravimetric analysis coupled with mass spectrometry (TGA-MS), in situ and ex situ X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). This allows to determine the dependence of the proton content on the washing time and the water-to-CAM mass ratio. Furthermore, a highly protonated NCM831205 sample is prepared to investigate the bulk properties, e.g., the change of lattice parameters and the spatial proton distribution, as well as the decomposition mechanism of the protonated phase. Nickel-rich NCA (LiNiaCobAlcO2) materials are equivalent to nickel-rich NCMs (as both have a nickel content between 80 and 95 mol%, while, for NCAs, 5 mol% of manganese are replaced by 5 mol% of aluminum); thus, it is expected that the findings of this study apply to both NCMs and NCAs for similar nickel contents on the order of 83 mol%.
Results and discussion
Acid–base titration
So far, mainly the titration technique has been utilized to determine the amount of surface contaminants, often referred to as Warder's method.52,53 During washing, the surface contaminants such as lithium carbonate (Li2CO3) as well as transition-metal (TM) hydroxides and carbonates, which may remain from the synthesis and/or from improper storage in humid atmosphere, are expected to get dissolved, which can then be quantified from the titration curve of the washing solution. Upon contact of NCM with either gaseous or liquid water (H2O), however, lithium ions (Li+) from the NCM host structure are exchanged by protons (H+), resulting in the formation of lithium hydroxide (LiOH), as depicted in eqn (1).31,34,46,54 While LiOH remains on the NCM surface upon the reaction with H2O vapor,23,30,34 it gets dissolved in the solution upon washing.34,46,54 In this work, we extend this approach, aiming to quantify the protons exchanged during washing from the LiOH concentration of the washing solution.Previous studies have assigned the detected LiOH in the washing solution to hydroxide species which stem solely from surface contaminants of the CAM.22,33 The here investigated as-received NCM material (see Fig. S1a and Table S1† for particle morphology) has been stored for over one year in an argon-filled glovebox that contained no H2O (<0.1 ppm) vapor but had CO2 (several hundred ppm). Therefore, we expect that most surface hydroxides have been transformed to their respective carbonate analogue; the impact of the initial hydroxide content is discussed in more detail in the ESI.† This implies that the hydroxide ions found in the washing solution are expected to stem from the Li+/H+ exchange; in this case, the amount of H+ intercalated into the NCM structure from the washing solution is equal to the amount of deintercalated Li+ and thus of LiOH. Furthermore, deionized and degassed water has a pH value of 7, which rises to values above 10 within less than a minute when H2O is mixed with nickel-rich NCMs.46,48 Additionally, it has to be noted that no dissolution of TMs from the NCM is expected since this process mainly occurs with acidic solutions and is absent for a pH value above 10.48,54 Thus, most of the detected hydroxides and carbonates in the following experiments are expected to be lithium salts, which are summarized for simplicity as LiOH and Li2CO3, respectively.
To quantify the amount of LiOH, an acid–base titration of the washing solution is performed using 10 mM HCl (aq.) as titrant. Upon addition of HCl, the monoprotic base LiOH is neutralized, while the diprotic base Li2CO3 gets fully protonated in two steps. Here, the second equivalence point of Li2CO3 overlaps with the one of LiOH (see eqn (2) to (4)). A typical titration curve based on these three processes is exemplarily shown for a washing solution obtained for the 1h-5:1 sample (the first number giving the washing time of 1 h and the second the water-to-CAM mass ratio of 5:1, see Methods section) depicted in Fig. 1a, showing the pH value as a function of the added titrant volume VHCl (see black curve, plotted against the left y-axis). The titration curve of 0.5 ml of the washing solution diluted by 20 ml of H2O starts at a pH value of 10.9, before it reaches the final target pH value of 3.5 after the addition of 3.1 ml of HCl. Here, it has to be noted that, while the dilution does not affect the quantification of the lithium salts, it does affect the observed pH value; thus, the initial pH value of 10.9 of the diluted solution corresponds to a pH value of 12.5 for the undiluted washing solution. Up to a VHCl of 1.2 ml, the pH decreases gradually to a value of 10 before dropping stepwise to a pH of 4 after 2.1 ml HCl were added; upon further HCl addition, the pH gradually decreases to 3.5.
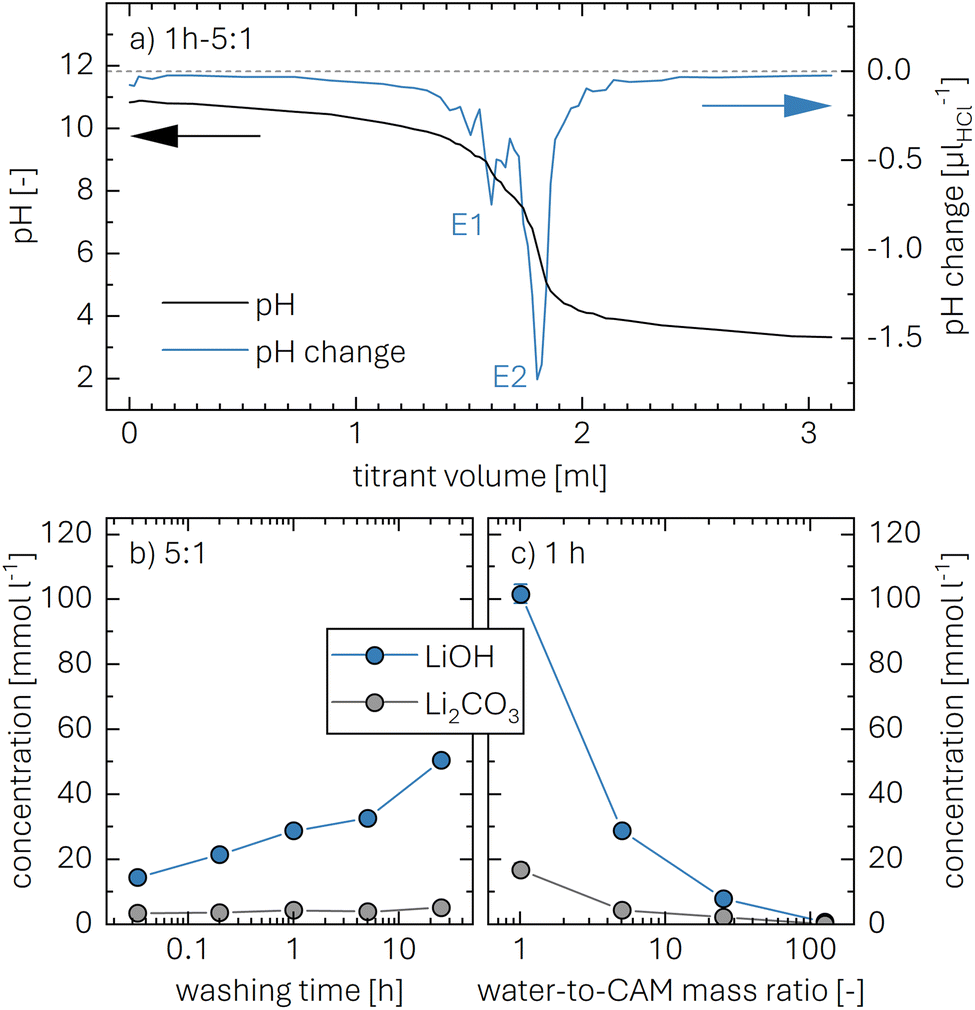 | ||
| Fig. 1 Acid–base titration of the washing solutions which were collected after the washing of the NCM with different washing procedures. 0.5 ml of washing solution were diluted by 20 ml of H2O and titrated with 10 mM HCl (aq.) until a pH value of 3.5 was reached. (a) Exemplary titration curve for the solution obtained for the 1h-5:1 sample (1 h at a water-to-CAM mass ratio of 5:1), showing the pH value (black, depicted on the left y-axis) and the derivative of the pH with respect to the added HCl volume in units of μlHCl−1 (blue, depicted on the right y-axis), both as a function of the added HCl volume. The position of the two equivalence points E1 and E2 are marked in the figure (the gray dashed line marks a dpH/dVHCl of 0). The bottom panels show the concentrations of both LiOH (blue) and Li2CO3 (gray) in units of mmol l−1 in the washing solution: (b) as a function of the washing time for a water-to-CAM mass ratio of 5:1; or, (c) as a function of the water-to-CAM mass ratio for a washing time of 1 h, both in a logarithmic representation. | ||
To identify the equivalence points of the three acid–base reactions, being the points with the highest change of the pH value as a function of VHCl, the derivative of the pH with VHCl (dpH/dVHCl, in units of μlHCl−1) is computed (see blue curve in Fig. 1a, depicted on the right y-axis). For 1h-5:1, the two equivalence points E1 and E2 appear for an added titrant volume VE1HCl of 1.6 ml and VE2HCl of 1.8 ml, respectively. E1, occurring at a pH value of 9, stems from the neutralization of LiOH to lithium chloride (LiCl) as well as the protonation of Li2CO3 to lithium bicarbonate (LiHCO3):55
| LiOH + HCl → LiCl + H2O | (2) |
| Li2CO3 + HCl → LiHCO3 + LiCl | (3) |
E2, where the protonation of LiHCO3 to carbonic acid (H2CO3) occurs, is observed at a pH value of 6:55
| LiHCO3 + HCl → H2CO3 + LiCl | (4) |
Since between E1 and E2 only the protonation of LiHCO3 occurs, the titrant volume 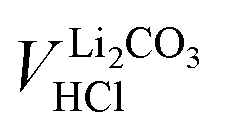 required for the protonation LiHCO3 can be calculated by the difference of VE1HCl and VE2HCl:56
required for the protonation LiHCO3 can be calculated by the difference of VE1HCl and VE2HCl:56
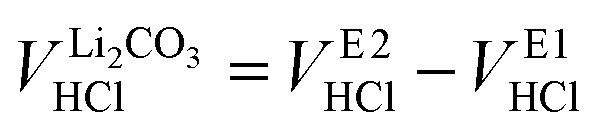 | (5) |
Even though the neutralization of LiOH is convoluted with the first protonation step of Li2CO3,54 the titrant amount VLiOHHCl required to neutralize LiOH can be calculated by subtracting 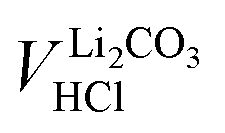 from VE1HCl since the titrant volume of the two protonation steps of Li2CO3 is identical:
from VE1HCl since the titrant volume of the two protonation steps of Li2CO3 is identical:
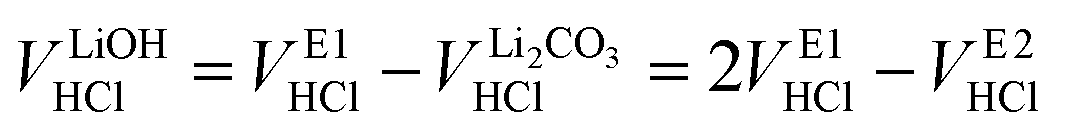 | (6) |
By taking into account the concentration cHCl of 10 mM of the titrant and the sample volume Vsamplewashing of 0.5 ml of the washing solution used for the titration, the concentration of each anion can be determined, yielding the following value for CO32−, i.e., for Li2CO3:
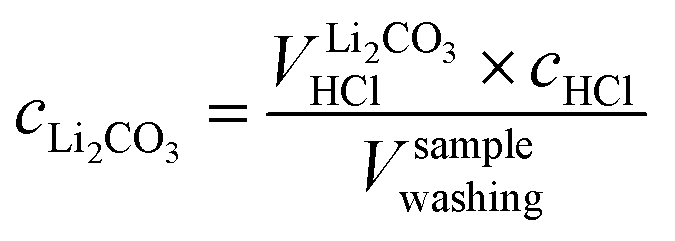 | (7) |
The concentration of Li2CO3 in the washing solution is depicted for a fixed water-to-CAM mass ratio of 5:1 as a function of the washing time (see gray symbols in Fig. 1b). For the exemplary data of 1h-5:1 shown in Fig. 1a, the experimental values of VE1HCl and VE2HCl result in 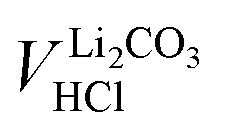 of 0.2 ml, corresponding to cLi2CO3 of 4.2 mM. Even though the washing times differ by almost three orders of magnitude, cLi2CO3 remains within 3.7 to 5.4 mM, indicating that Li2CO3 surface species are readily dissolved within minutes for a water-to-CAM mass ratio of 5:1. The weight percentage of Li2CO3 in the NCM material can be determined by taking into account the molar mass of Li2CO3 (MLi2CO3) of 73.9 g mol−1, the mass of the washed NCM (mNCM), and the total volume of the washing solution Vtotalwashing:
of 0.2 ml, corresponding to cLi2CO3 of 4.2 mM. Even though the washing times differ by almost three orders of magnitude, cLi2CO3 remains within 3.7 to 5.4 mM, indicating that Li2CO3 surface species are readily dissolved within minutes for a water-to-CAM mass ratio of 5:1. The weight percentage of Li2CO3 in the NCM material can be determined by taking into account the molar mass of Li2CO3 (MLi2CO3) of 73.9 g mol−1, the mass of the washed NCM (mNCM), and the total volume of the washing solution Vtotalwashing:
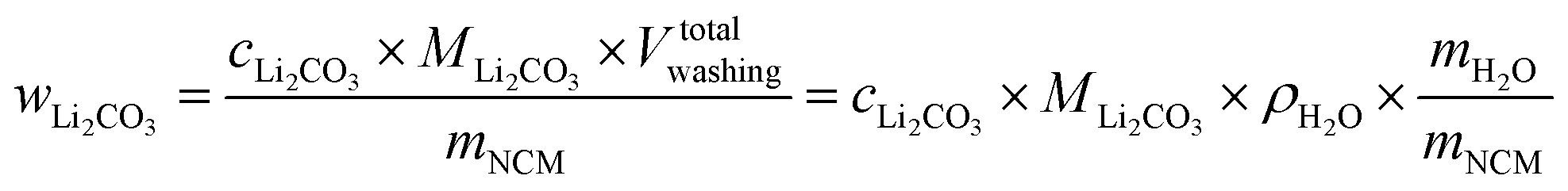 | (8) |
As a next step, to assess the amount of LiOH in the solution, its concentration is determined by considering VLiOHHCl (see eqn (6)), cHCl of 10 mM, and Vsamplewashing:
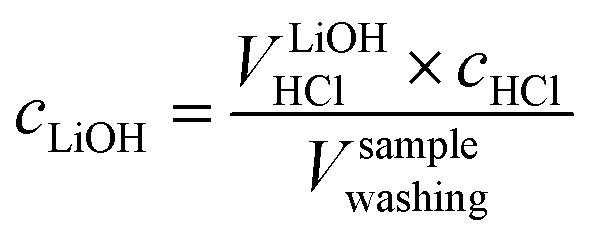 | (9) |
The concentration of LiOH in the washing solution is depicted for a fixed water-to-CAM mass ratio of 5:1 as a function of the washing time (see blue data in Fig. 1b). For the exemplary data of 1h-5:1 in Fig. 1a, VE1HCl and VE2HCl result in VLiOHHCl of 1.4 ml, corresponding to cLiOH of 29.0 mM. This concentration of hydroxide ions translates to an approximate pH value of 12.5 (i.e., pH ≈ 14 − log(0.029), ignoring the activity coefficient of the hydroxide species for this rough estimate), which was observed for the undiluted washing solution (see discussion above). For the variation of the washing time at a water-to-CAM mass ratio of 5:1, cLiOH increases roughly proportionally with the logarithm of the washing time (see Fig. 1b), indicating that either the dissolution of LiOH is very slow or that LiOH is produced by the Li+/H+ exchange of the CAM in contact with water. In the experiment where the water-to-CAM mass ratio is varied for a fixed washing time of 1 h (see blue data in Fig. 1c), cLiOH decreases with increasing water-to-CAM mass ratio. Assuming that the measured amounts of LiOH are due to the Li+/H+ exchange of the CAM, the obtained cLiOH values (see Table 1) can be converted into the proton content of the NCM in terms of the mole fraction of protons ion-exchanged into the NCM material (xH+, in units of mol%, defined by HxLi1−xMO2) by considering Vtotalwashing, mNCM, and the molar mass MNCM of the NCM of 97.5 g mol−1:
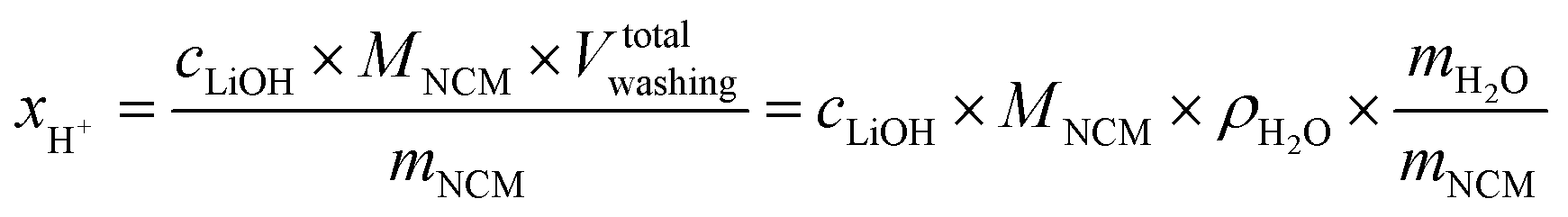 | (10) |
| Sample | c LiOH (in mM) | x H+ (in mol%) |
|---|---|---|
| 2min-5:1 | 14.7 | 0.72 |
| 12min-5:1 | 21.6 | 1.05 |
| 1h-5:1 | 29.0 | 1.41 |
| 5h-5:1 | 32.9 | 1.60 |
| 25h-5:1 | 50.7 | 2.47 |
| 1h-1:1 | 101.7 | 0.99 |
| 1h-5:1 | 29.0 | 1.41 |
| 1h-25:1 | 8.1 | 1.98 |
| 1h-125:1 | 1.0 | 1.17* |
| 55h-25:1-80°C | 54.4 | 13.33* |
| 2min-1:1-0°C | 48.5 | 0.47 |
The determined xH+ is summarized in Table 1 (also, later in Table 6 and Fig. 8), both for varying washing times and varying water-to-CAM mass ratios. For example, for 1h-5:1, xH+ equals to 1.41 mol%, corresponding to the amount of H+ ion-exchanged into the NCM (equivalent to the Li+ exchange from the NCM into the washing solution). When increasing the washing time at a constant water-to-CAM mass ratio of 5:1 (rows 1 to 5), xH+ increases roughly linearly with the logarithm of the washing time (see green circles in Fig. 8a). Similarly, when increasing the water-to-CAM mass ratio for a fixed washing time of 1 h (rows 6 to 9), the xH+ value increases roughly linearly with the logarithm of the water-to-CAM mass ratio (see green circles in Fig. 8b). Only for the 1h-125:1 sample, xH+ does not follow this trend, as it is lower than that of the 1h-25:1 sample; this is likely due to the comparably low cLiOH of the washing solution of only 1.0 mM (see Table 1), so that small amounts of CO2, that may be absorbed by the solution during the titration under ambient air, can substantially alter the result. Additionally, for the 55h-25:1-80°C washed sample (row 10) that was prepared at a washing temperature of 80 °C to obtain an NCM with a very high xH+ for XRD analysis, a very high Li2CO3 content of 1.5 wt% was obtained, which we ascribe to the intrusion of CO2 from ambient air, permeating over the long washing time; since intruding CO2 would also affect the LiOH concentration, the calculated xH+ value is not trustworthy. Finally, the sample washed under the most mild conditions, namely for 2 min at a water-to-CAM mass ratio of 1:1 and at 0 °C (row 11), the Li2CO3 weight percentage of 0.21 wt% is essentially identical to that of all other measurements, while its calculated proton content is still 0.47 mol%. The latter implies that either a perfect exclusion of the Li+/H+ exchange during washing with water is impossible and/or that the CAM contains approx. 0.12 wt% of LiOH surface contaminants; the latter is unlikely since the CAM was stored for more than a year under a glove-box atmosphere with several hundred ppm of CO2, so that any residual LiOH surface contaminants should have reacted to Li2CO3.
Alternative methods to the above quantification of xH+via analysis of the washing solution are based on the direct analysis of the washed NCM powders, which will be discussed in the following sections.
Prompt-gamma activation analysis (PGAA)
PGAA is used to determine the bulk elemental composition of samples.57,58 It is based on the radiative neutron-capture process, where characteristic gamma radiation is emitted promptly after a nucleus captures a neutron. PGAA is particularly suited for detecting protons due to their large neutron-capture cross-sections upon irradiation with cold neutrons, resulting in the emission of a photon with an energy of 2223.3 keV:59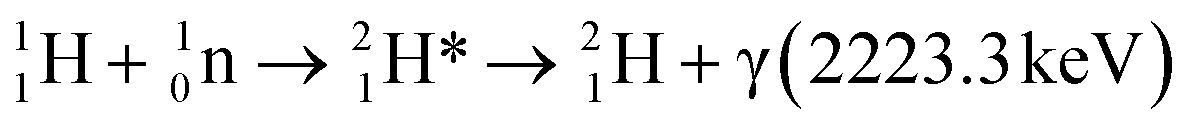 | (11) |
Similar processes occur for other elements and their isotopes, including lithium, nickel, cobalt, and manganese. The emitted photons, having nuclide-specific energies, enable the quantification of isotopic constituents in a sample from the intensity of the entire PGAA spectrum, even for trace amounts. The mass m of the respective element in the irradiated volume can be determined from the peak area, i.e., from the number of counts NP in one single peak, the analytical sensitivity S of each nuclide-specific photon energy, and the measurement time t:
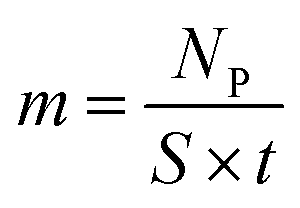 | (12) |
The analytical sensitivity S (in units of s−1 g−1) is calculated for protons, lithium, and the transition metals using the Avogadro constant NA, the molar mass M of the respective element, the thermal-equivalent neutron flux Φ0, the partial gamma-ray-production cross-section σγ, as well as the detector efficiency ε(Eγ) at the respective gamma energy Eγ:
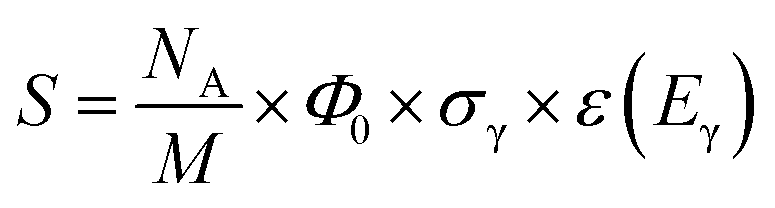 | (13) |
Previously, PGAA has already been used to quantify xH+ in chemically delithiated, cobalt-free CAMs.60,61 For typical NCM materials containing substantial amounts of cobalt to enhance structural stability and lithium-ion diffusion kinetics,8,62,63 it is more challenging to quantify xH+ from its single peak (at Eγ of 2223.3 keV) in the presence of cobalt (2221.6 keV) since the gamma-ray energies are only 2 keV apart, resulting in the overlap of the two peaks. Additionally, another spectroscopic interference occurs between lithium and cobalt at 2032.3 and 2032.8 keV, respectively (data not shown). Therefore, to accurately quantify the amount of lithium and hydrogen, a fit of selected regions within the entire PGAA spectrum is required, and an appropriate correction has to be applied, using the reference-peak method.58 On the other hand, the amount of the transition metals in each sample can be obtained from several interference-free peaks: at 282.9 and 8998.4 keV for nickel; at 277.2, 447.7, 556.0, and 6985.4 keV for cobalt; at 212.0, 271.2, and 6783.7 keV for manganese (data not shown).59 Unfortunately, the amount of oxygen could not be quantified due to the low analytical sensitivity of the recorded peak at 870 keV.
Based on eqn (12) and (13), the mass m of the elements of interest can be calculated from the respective peak areas, from which their molar amounts n are determined when taking into account the molar mass M of each element. Due to the spatial inhomogeneity of the neutron beam spot and the influence of self-shielding and/or due to a partial illumination of the sample, however, it is imprecise to quantify the absolute amounts of each element. When considering the typical mass of roughly 200 mg of an irradiated NCM sample and its molar mass MNCM of 97.5 g mol−1, an amount of 2 mmol of TMs would be expected; the here observed smaller amounts of 1 mmol detected by PGAA (data not shown) are attributed to self-shielding and partial illumination of the sample. Nevertheless, the molar ratios between the elements of interest can be determined precisely since they are independent of the analyzed fraction for any homogeneous samples, such as an NCM powder. The observed molar ratios of the transition metals nNi:nCo:nMn, which are presented in Table 2 and determined with a relative uncertainty below 4% for each TM signal, are close to the expected ratio of 83:12:05 for Ni:Co:Mn of the as-received NCM. Since the total amount of TMs is not expected to change upon washing in water, as they do not dissolve at a pH value above 9,46,54 the amount of lithium and protons can be referenced to either one and/or all of the TMs.
| Sample | Relative amounts (in mol%) | ||
|---|---|---|---|
|
n
Ni:nCo:nMn |
n Li/nTM ≡ xLi+ | n H/nTM ≡ xH+ | |
| as-received | 82.5:12.6:4.9 |
97 ± 4 | 0* |
| calcined | 82.5:12.7:4.9 |
99 ± 5 | 0* |
| 1h-1:1-[65°C] | 82.6:12.6:4.8 |
95 ± 5 | 0.9 ± 0.3 |
| 1h-5:1-[65°C] | 82.4:12.8:4.9 |
97 ± 5 | 1.7 ± 0.4 |
| 55h-25:1-80°C-[65°C] | 82.7:12.6:4.7 |
82 ± 3 | 25.5 ± 0.7 |
| 55h-25:1-80°C-[120°C] | 82.5:12.7:4.9 |
83 ± 3 | 26.6 ± 0.8 |
| 55h-25:1-80°C-[180°C] | 82.7:12.6:4.8 |
79 ± 3 | 16.5 ± 0.7 |
| 55h-25:1-80°C-[230°C] | 82.4:12.7:4.9 |
75 ± 3 | 8.1 ± 0.4 |
| 55h-25:1-80°C-[300°C] | 82.7:12.6:4.7 |
75 ± 5 | 3.2 ± 0.3 |
| 55h-25:1-80°C-[450°C] | 82.7:12.5:4.8 |
74 ± 7 | 4.1 ± 0.4 |
Based on the cobalt mass obtained from multiple interference-free peaks, the expected area of the peak of cobalt at 2032.8 keV is calculated using eqn (12). With this calculated intensity, the area of the lithium peak at 2032.3 keV can be quantitatively corrected for the cobalt contribution. The thus obtained molar amount nLi of lithium, which is a weighted average of the area of the lithium peaks at 980.6, 1051.8, and 2032.3 keV, can be converted into the degree of lithiation xLi+ (see third column of Table 2) by dividing nLi by the sum of the molar amount of all transition metals nTM:
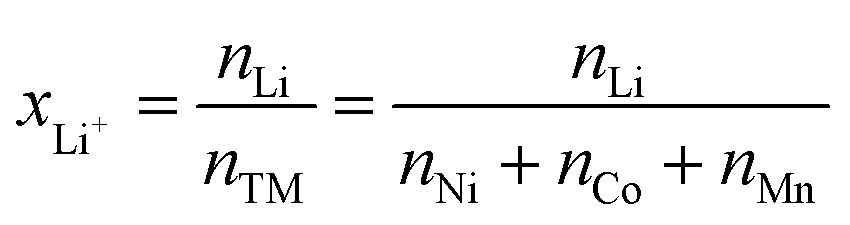 | (14) |
According to the PGAA data, the lithium content of the as-received and the calcined NCM sample of 97 ± 4 and 99 ± 5 mol%, respectively, agrees within the experimental error with the expected value of 100 mol%. For the mildly washed samples, i.e., for 1h-1:1-[65°C] and 1h-5:1-[65°C] (both dried at 65 °C under dynamic vacuum after washing), for which a proton content of only 1.4 to 1.6 mol% (see Table 1) or a lithium content of 98.6 to 98.4 mol% is expected, the experimental error of the xLi+ value derived from the PGAA analysis is too high to detect these minor changes in xLi+ (see row 3 and 4 in Table 2). Only for the harshly washed 55h-25:1-80°C-[65°C] sample with an expected xH+ value of more than 10 mol% (see Table 1), a clear decrease in xLi+ to 82 ± 3 mol% can be observed. Upon drying this sample at higher temperature (of 120 to 450 °C), xLi+ remains at similar values within the determined uncertainty. Even though smaller changes in the lithium content cannot be detected reliably due to the relatively high absolute uncertainties of up to 7 mol%, resulting from the low intensity of lithium due to its unfavorable nuclear properties, the removal of more than 10 mol% of lithium due to extensive washing (55 h at a water-to-CAM mass ratio of 25:1 and a temperature of 80 °C), which was indicated by titration (see Table 1), can be confirmed.
The data of the as-received NCM, presented in Fig. 2a, show the normalized counts (black symbols), from which the background signal (dark gray line) as well as the total fit (black line and gray highlighted area) are determined. The contribution of the cobalt (light green line/area) and of the protons (light blue line/area), both obtained from the fit of the peak depicted in Fig. 2a, illustrate the relative contribution of the two components.
 | ||
| Fig. 2 Spectroscopic data from PGAA analysis between 2217 and 2228 keV, depicting the recorded counts (circles) for different NCM samples: (a) as-received (black symbols); (b) as-received and calcined under O2 at 525 °C (gray symbols); (c) 1h-1:1-[65°C] (light blue symbols); (d) 1h-5:1-[65°C] (dark blue symbols); (e) 55h-25:1-80°C-[65°C] (dark red symbols); (f)–(j) 55h-25:1-80°C washed samples dried under dynamic vacuum at temperatures higher than 65 °C (viz., at 120, 180, 230, 300, or 450 °C; dark red symbols). The data were normalized by the fitted area of the cobalt peak. For each sample, the figure shows exemplary fits of the data (black line and gray area), the fitted background signal (gray line below each peak), and the computed contributions of cobalt (light green line/area, with Eγ = 2221.6 keV) and protons (light blue line/area, with Eγ = 2223.3 keV). | ||
For the as-received NCM calcined at 525 °C under O2, the PGAA spectrum between 2017 and 2028 keV shows no signal at a gamma-ray energy of 2223 keV, indicating the absence of protons in the calcined NCM material (see Fig. 2b). For the washed NCMs dried at 65 °C, protons are detected: For 1h-1:1-[65°C] (see Fig. 2c), a small but significant proton signal is observed, which increases with increasing water-to-CAM mass ratio, from a substantial signal for the 1h-5:1-[65°C] (see Fig. 2d) to a dominating signal for 55h-25:1-80°C-[65°C] (see Fig. 2e). When the latter sample is dried at increasingly higher temperatures, i.e., at 120, 180, 230, 300, and 450 °C (see Fig. 2f–j), the proton signal gradually decreases, particularly at temperatures of 180 °C or greater, indicating the temperature-induced removal of protons. It has to be noted, however, that the here shown peak fits of cobalt and protons are for illustrative purposes only, indicating the changes in the signal intensity of the two elements.
In order to quantify xH+ of the NCM samples, the area of the only proton peak with a maximum at 2223.3 keV is corrected by the expected area of the cobalt peak at 2221.6 keV (based on the four interference-free cobalt peaks), as it was also done for the above described quantification of the lithium amount, resulting in the contained amount of protons nH. Subsequently, the ratio of nH and of the sum of the transition metals nTM yield the value of xH+, according to eqn (15):
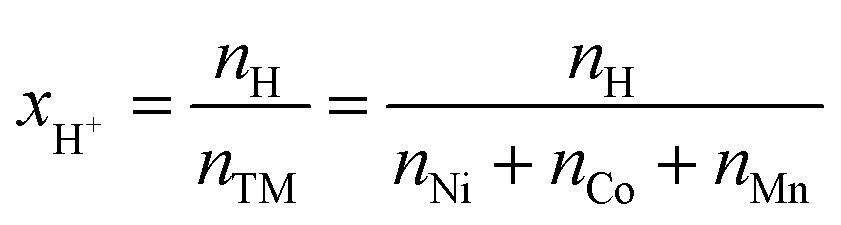 | (15) |
This analysis results in a relatively low xH+ of 0.6 mol% for the as-received NCM, with a 1σ standard uncertainty of 0.4 mol%. In the here used analysis protocol, the quantification limit of the method differs from the detection limit and is defined as the value for which the uncertainty from the counting statistics does not exceed 50%. As the uncertainty of xH+ of the as-received sample was found to be above this threshold, i.e., that the presence of the protons could be detected but not quantified reliably, xH+ was not stated quantitatively (marked as 0* in Table 2). For the calcined NCM (see Fig. 2b), no significant amount of protons could be detected. In all other cases, the proton signals could be quantified reliably.
The resulting xH+ is listed in Table 2 for each NCM sample and compared below to the results of the other methods (see also Table 6 and yellow symbols in Fig. 8). For the mildly washed samples 1h-1:1-[65°C] and 1h-1:1-[65°C], xH+ is 0.9 ± 0.3 and 1.7 ± 0.4 mol%, respectively. By increasing the washing time and temperature, xH+ even reaches 25.5 ± 0.7 mol% for 55h-25:1-80°C-[65°C]. Even though xH+ is only affected insignificantly when the washed material is dried at the higher temperature of 120 °C, showing a value of 26.6 ± 0.8 mol%, it continuously decreases with increasing drying temperature: xH+ is reduced to 16.5 ± 0.7 mol% at 180 °C, to 8.1 ± 0.4 mol% at 230 °C, and to similar values of 3.2 ± 0.3 and 4.1 ± 0.4 mol% at 300 and 450 °C, respectively, indicating the removal of most of the protons upon drying of the washed sample at temperatures of 300 °C or higher. As the protons require oxygen atoms to be released from the NCM structure in the form of H2O,46 and as the NCM particle surface region is oxygen-depleted due to the expected temperature-induced decomposition of the layered NCM structure to rock-salt/spinel phases (observed for partially delithiated NCMs),22,50 oxygen must diffuse to the surface where it can be released as H2O upon the reaction with the intercalated protons. Thus, the 3 to 4 mol% of protons remaining in 55h-25:1-80°C-[450°C] might be trapped due to a limited oxygen diffusivity; when increasing the temperature further, however, it is expected that the remaining protons are released. This suggests that, for the more mildly washed samples with a much lower xH+, no protons might remain after drying at 450 °C since less oxygen atoms are required to release all protons in the form of H2O.
Based on the Li+/H+-exchange reaction formulated in eqn (1), the amounts of protons and lithium are expected to add up to 100 mol%. Within the obtained uncertainties of the PGAA analysis, being relatively large for the determined lithium amount in the NCM samples, the PGAA data for 55h-25:1-80°C are consistent with a stoichiometric replacement of Li+ by H+, at least for the samples dried at 65 and 120 °C. This does confirm that the reaction with H2O does not induce the exclusive delithiation of the NCM but results in the Li+/H+ exchange. Beyond a heating temperature of 120 °C, xH+ rapidly decreases and the sum of xLi+ and xH+ becomes substantially less than 100 mol%. To elucidate the apparent removal of protons at temperatures above 120 °C, the thermal stability of protonated NCMs is further studied by thermogravimetric analysis.
Thermogravimetric analysis coupled with mass spectrometry (TGA-MS)
The thermal analysis of CAMs by TGA(-MS) has proven useful to identify the surface contaminants which form on the surface of CAM particles upon their exposure to ambient atmosphere, particularly on nickel-rich CAMs.30,33,34,64 So far, the mass loss as well as the evolving gasses have often been assigned solely to the thermal decomposition of the contaminants; during ambient air storage of CAMs, however, water vapor from humid air does not only form contaminants such as lithium hydroxide that gradually reacts further with CO2 to the more stable lithium carbonate,23,30 but, simultaneously, H+ is intercalated to replace the extracted Li+.34 This protonated NCM phase has been found to be thermally less stable than fully lithiated NCMs.34,46,54 Thus, for the thermal analysis of stored NCMs, the decomposition of the surface contaminants and of the protonated, thermally less stable NCM phase are convoluted since the temperature regions of the decomposition reactions overlap.46,65 However, after washing an NCM, only minor amounts of contaminants are expected to remain, which is why, in this case, the thermal decomposition of the protonated NCMs can be investigated without significant interference.Using a custom-made setup attached to an MS, Pritzl et al. showed that no oxygen is released from the NCM structure during the washing process;46 therefore, the layered NCM structure is expected to be maintained, at least at relatively low drying temperatures of 120 °C or below, as discussed further in the later XRD analysis. The thermal decomposition of washed and thus protonated NCMs has been investigated by Pritzl et al. and Hamam et al.: Both discussed a two-step decomposition reaction, first evolving H2O above 120 °C and then oxygen (O2) above 250 °C, resulting in the formation of a rock-salt-type phase (MO).46,54 Since the extent of this decomposition reaction is expected to scale with the extent of protonation of the NCM caused by washing, the relative mass loss upon heating allows to quantify xH+.
The thermal analysis of the as-received, the calcined, and the washed NCMs is depicted exemplarily in Fig. 3. The applied temperature profile (see Fig. 3a) consists of a temperature ramp (of 10 K min−1) to 450 °C under argon, with intermittent temperature holds at 25 °C (for 10 min) as well as at 120 and 450 °C (for 30 min each), marked by the gray areas. Up to 120 °C, the samples lose between 0.005 and 0.05 wt% of their initial mass (see Fig. 3b), which is attributed to the desorption of physisorbed H2O (see Fig. 3c). Since we expect to have removed all physisorbed water up to 120 °C, the mass loss m(T) of the NCM samples, depicted in Fig. 3b, is normalized by the value m(120 °C) at the end of the temperature hold at 120 °C (reached after 49.5 min); the thus normalized mass at the end of the 450 °C temperature hold is marked in the figure and is referred to as m(450 °C). The mass loss between 120 and 450 °C is only 0.09 wt% for the as-received NCM (black), consistent with previous reports;23,30,34 in contrast, this mass loss does not exceed 0.01 wt% for the calcined NCM (gray) since it was not exposed to ambient air after calcination at 525 °C under oxygen. The washed NCM 1h-1:1-[65°C] (light blue) exhibits an increased mass loss of 0.20 wt% at 450 °C, initiating at approx. 140 °C. For the sample washed at the higher water-to-CAM mass ratio of 1h-5:1-[65°C] (dark blue), which is expected to have a higher xH+ (see Fig. 1), the mass loss reaches 0.34 wt%, while 55h-25:1-80°C-[65°C] (dark red) even loses 4.42 wt% of its mass up to 450 °C (see also Fig. S3 in the ESI†). For all samples, the mass loss essentially ceases at 450 °C, indicating the completion of the thermal decomposition of the protonated NCM. Note that the here observed temperature range for the mass loss of the washed/protonated nickel-rich NCMs of 120 to 450 °C agrees well with the decomposition temperature of nickel oxyhydroxide (NiOOH, which can conceptually also be written as HNiO2),66,67 and, since HNiO2 is essentially the fully delithiated/protonated phase of nickel-only layered lithium nickel oxide (LiNiO2), which has a significantly higher decomposition temperature of >600 °C, similar to nickel-rich NCMs, the compromised thermal stability of a washed NCM is attributed to its protonation.
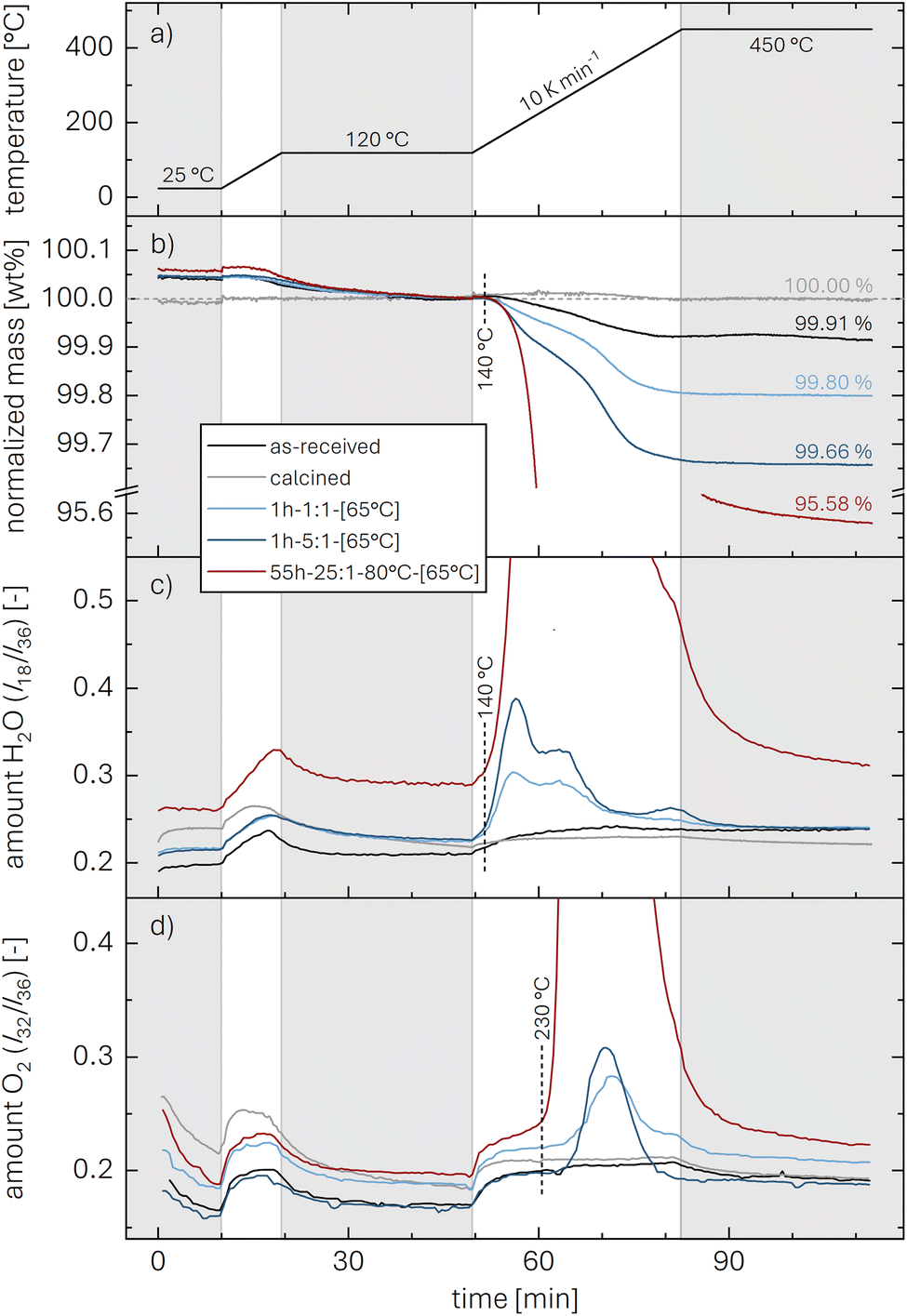 | ||
| Fig. 3 TGA-MS data recorded under argon (60 ml min−1) of as-received (black), calcined (at 525 °C under O2, gray), and washed NCM samples (dried at 65 °C). For the latter, exemplary data of 1h-1:1-[65°C] (light blue), 1h-5:1-[65°C] (dark blue), and 55h-25:1-80°C-[65°C] (dark red) are presented. The samples were heated to 450 °C at 10 K min−1 with intermittent temperature holds at 25 °C (for 10 min) as well as at 120 and 450 °C (for 30 min each), marked by the gray areas. (a) Temperature profile. (b) Relative NCM mass, normalized to the mass at the end of the temperature hold at 120 °C; the relative mass loss at 450 °C is marked in the figure (it is also given for all investigated samples in Table 3). MS ion currents of (c) H2O (m/z = 18) and (d) O2 (m/z = 32) are normalized by that of argon (m/z = 36). Due to the large difference in signal intensity, the complete data of 55h-25:1-80°C-[65°C] are not shown here but can be found in Fig. S3.† | ||
The mass loss of the washed NCMs is accompanied by the release of H2O (m/z = 18, see Fig. 3c) and O2 (m/z = 32, see Fig. 3d), which are both normalized by the argon ion current (m/z = 36). The release of H2O is detected together with the onset of the mass loss at 140 °C, while most of the H2O signal is detected below approx. 350 °C (for 55h-25:1-80°C-[65°C], see also Fig. S3†). In contrast, the onset of the O2 release occurs at approx. 230 °C and continues up to 450 °C. Note that the observed decrease of the O2 signal over the first 10 min during the initial hold at 25 °C (see Fig. 3d) as well as the increase of both the H2O and O2 signal during the temperature ramps of 10 K min−1 both from 25 to 120 °C and from 120 to 450 °C (see Fig. 3c and d), being present for all samples, do not originate from the NCM sample but are related to the background signal, which is discussed in more detail for Fig. S2 in the ESI.†
Pritzl et al. suggested that relatively small amounts of oxygen (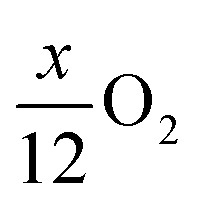 per HxLi1−xMO2) are released at the lower temperatures together with H2O upon the formation of a spinel phase (M3O4).46 However, since no O2 signal is observed below 230 °C for the here obtained TGA-MS data of the washed NCMs, not even for the highly protonated 55h-25:1-80°C-[65°C] that has very large mass loss and gas signals (see dark red data in Fig. 3d and S3d†), it is suggested that no O2 is released below 230 °C and no M3O4 is formed. Instead, an oxygen-depleted corundum phase (M2O3) could develop below 230 °C, which, according to eqn (16), would only release H2O and no O2 (note that the here suggested decomposition pathway will actually be disproven by the later discussed XRD analysis):
per HxLi1−xMO2) are released at the lower temperatures together with H2O upon the formation of a spinel phase (M3O4).46 However, since no O2 signal is observed below 230 °C for the here obtained TGA-MS data of the washed NCMs, not even for the highly protonated 55h-25:1-80°C-[65°C] that has very large mass loss and gas signals (see dark red data in Fig. 3d and S3d†), it is suggested that no O2 is released below 230 °C and no M3O4 is formed. Instead, an oxygen-depleted corundum phase (M2O3) could develop below 230 °C, which, according to eqn (16), would only release H2O and no O2 (note that the here suggested decomposition pathway will actually be disproven by the later discussed XRD analysis):
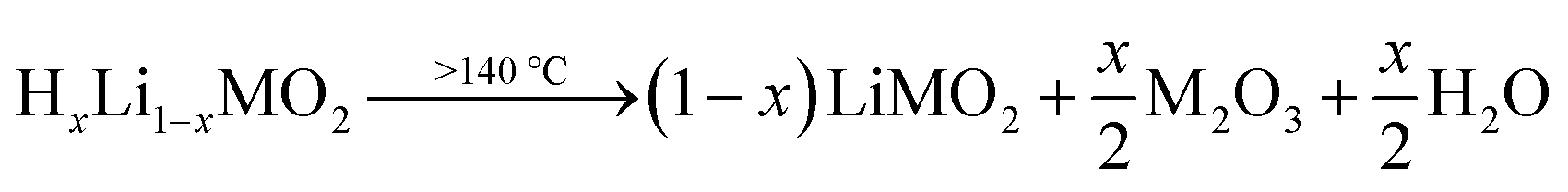 | (16) |
Similar to the thermal decomposition of (electro)chemically delithiated NCMs (Li1−xMO2),22,50,51 the release of O2 is observed at higher temperatures between 230 and 450 °C, eventually resulting in the formation of a rock-salt phase (MO) according to eqn (17):
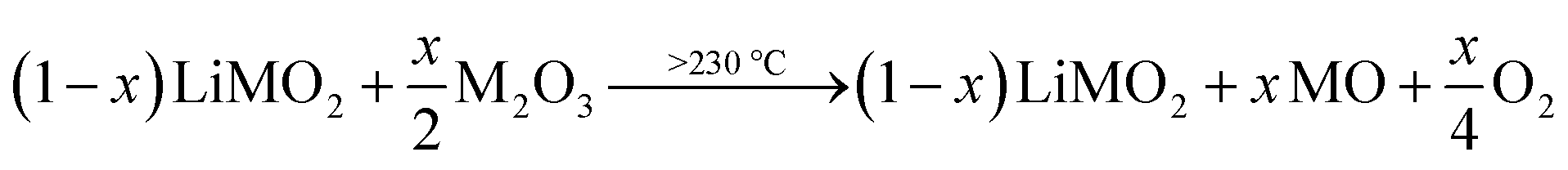 | (17) |
Since fully lithiated LiMO2 is stable up to >600 °C even for nickel-rich materials,23,33 only the protonated fraction of the HxLi1−xMO2 would be expected to be decomposed below 450 °C. In this case, the fraction of the HMO2 phase can be quantified by the ratio of the formed MO and the remaining LiMO2 phases (see eqn (16) and (17)). For a complete decomposition of the HMO2 phase, xH+ can be obtained from the mass values of the washed NCM samples at 120 °C (m(120 °C)), corresponding to a nominal mixture of (1−x) parts of LiMO2 and x parts of HMO2, and at 450 °C (m(450 °C)), corresponding to a nominal mixture of (1−x) parts of LiMO2 and x parts of MO, taking into account the molar masses of the various compounds (of 97.5, 91.5, and 74.5 g mol−1 for MLiMO2, MHMO2, and MMO, respectively); this results in eqn (18), being a simple mass balance that can be solved for x (corresponding to xH+), and, when solved for xH+, in eqn (19):
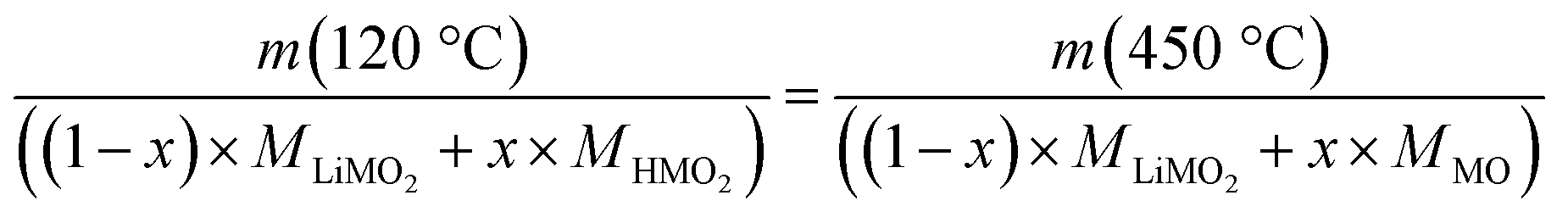 | (18) |
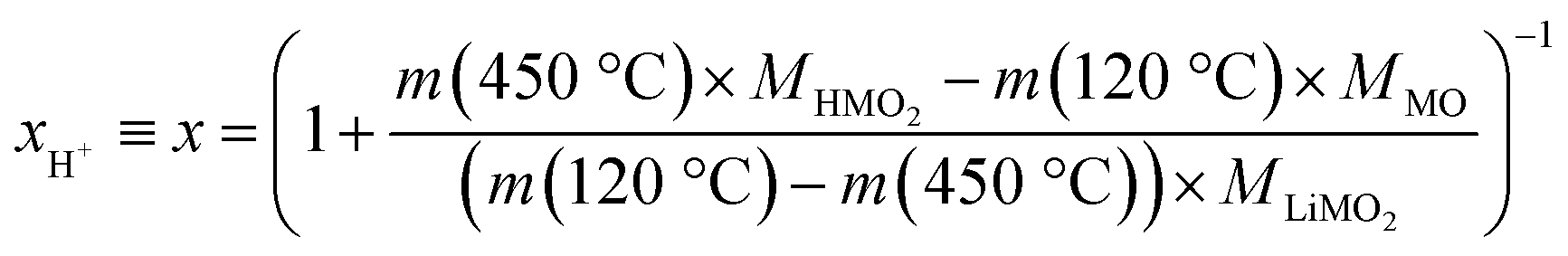 | (19) |
Note that for this quantification of xH+, it is irrelevant what the intermediate species are, as long as the only compounds present at 450 °C are the fully lithiated layered phase and the corresponding rock-salt phase.
For 1h-1:1-[65°C] and 1h-5:1-[65°C], shown in Fig. 3b, the values for the mass loss of 0.20 and 0.34 wt% between 120 and 450 °C equate to xH+ of 1.15 and 1.96 mol%. For the harshly washed 55h-25:1-80°C-[65°C] sample with an observed mass loss of 4.42 wt% (Fig. 3b), eqn (19) yields a value for xH+ of 25.0 mol%. For this high proton content, however, some protons potentially remain in the structure even at 450 °C, as discussed for the PGAA data (see Fig. 2), so that the actual xH+ value might be approximately 10 to 15% higher (for 3 to 4 mol% of protons remaining, see Table 2). While the mass loss of 0.003 wt% of the calcined NCM is <0.01 wt%, the larger mass loss of 0.09 wt% of the as-received NCM (see Table 3) may stem from the decomposition either of surface contaminants such as LiOH or TM carbonates and/or of a protonated phase formed by short-term ambient air exposure (e.g., during packaging). Additionally, for the most mildly washed 2min-1:1-0°C-[65°C] sample, a mass loss of 0.13 wt% is observed, corresponding to xH+ of 0.74 mol%. All results for m(450 °C) and xH+ obtained by TGA-MS are depicted in Table 3 and Fig. 8 (gray symbols), both for varying water-to-CAM mass ratios and washing times. To elucidate the assumed decomposition of HxLi1−xMO2 to MO, the structural changes of the highly protonated NCM sample upon heating will be investigated by XRD next.
| Sample | m(450 °C) (in wt%) | x H+ (in mol%) |
|---|---|---|
| as-received | 99.91 | 0.49 |
| calcined | 100.00 | 0.02 |
| 2min-5:1-[65°C] | 99.79 | 1.21 |
| 12min-5:1-[65°C] | 99.74 | 1.50 |
| 1h-5:1-[65°C] | 99.66 | 1.96 |
| 5h-5:1-[65°C] | 99.42 | 3.31 |
| 25h-5:1-[65°C] | 99.41 | 3.35 |
| 1h-0.2:1-[65°C] | 99.87 | 0.74 |
| 1h-1:1-[65°C] | 99.80 | 1.15 |
| 1h-5:1-[65°C] | 99.66 | 1.96 |
| 1h-25:1-[65°C] | 99.65 | 2.00 |
| 1h-125:1-[65°C] | 99.61 | 2.21 |
| 55h-25:1-80°C-[65°C] | 95.58 | 25.0 |
| 2min-1:1-0°C-[65°C] | 99.87 | 0.74 |
In situ temperature-resolved X-ray powder diffraction (TR-XRD)
The utilization of in situ TR-XRD allows to illuminate bulk structural changes of samples as a function of temperature. In the context of CAMs, it has been used, e.g., to elucidate their synthesis as well as their decomposition upon heating.68–70 Furthermore, Bak et al. employed TR-XRD to elucidate the thermal decomposition of delithiated NCMs to identify the resulting spinel- and rock-salt-type phases.50 For protonated NCMs, showing a similar structural (in)stability upon heating as delithiated NCMs,20,22,71 the thermal decomposition of the protonated phase (HMO2) is investigated here by TR-XRD for temperature steps of 5 K between 120 and 450 °C. Since (TR-)XRD is a bulk-sensitive analysis technique that requires a significant phase fraction to enable its detection, identification, and quantification, the highly protonated NCM sample 55h-25:1-80°C-[65°C] (with xH+ of 25.0 mol%) is investigated by TR-XRD and compared to the as-received NCM, being thermally stable far beyond 450 °C.33The obtained diffractograms are normalized by the reflections with the highest intensity, i.e., the ones at 8.5° (corresponding to the (003)L lattice plane, being perpendicular to the layers in the layered NCM structure (L)). The thus normalized diffractograms are depicted in Fig. S4a† for the as-received sample and in Fig. S4b† for the 55h-25:1-80°C-[65°C] sample as a function of temperature through a contour plot (where the x-axis represents the diffraction angle 2θ and the y-axis the temperature, while the intensity is given by the color). The visible reflections at 120 °C (i.e., at the bottom of each contour plot) agree with the reflections expected for a layered NCM phase (see ticks at the bottom of Fig. S4†), with the lattice parameters of 2.87 Å for aL and 14.2 Å for cL (see discussion for Fig. 5 and S5† as well as Table 4). At first sight, the two samples exhibit similar diffraction patterns, and newly occurring phases, both upon washing or heating, are not obvious from the full diffractograms; thus, changes in the already apparent reflections will be discussed for the zoomed-in regions of the contour plot shown in Fig. 4.
| Sample | I 8.5°/I20.0° | χ | a L (in Å) | c L (in Å) | c L/aL |
|---|---|---|---|---|---|
| as-received | 1.42 | 0.96 | 2.8711 | 14.1894 | 4.9422 |
| calcined | 1.69 | 0.92 | 2.8706 | 14.1904 | 4.9433 |
| 55h-25:1-80°C-[65°C] | 1.60 | 0.86 | 2.8694 | 14.2374 | 4.9618 |
| 55h-25:1-80°C-[120°C] | 1.40 | 0.98 | 2.8696 | 14.2408 | 4.9627 |
| 55h-25:1-80°C-[180°C] | 1.31 | 0.93 | 2.8696 | 14.2484 | 4.9654 |
| 55h-25:1-80°C-[230°C] | 1.17 | 1.15 | 2.8680 | 14.2248 | 4.9598 |
| 55h-25:1-80°C-[300°C] | 1.16 | 0.93 | 2.8738 | 14.1961 | 4.9398 |
| 55h-25:1-80°C-[450°C] | 1.17 | 0.87 | 2.8746 | 14.1952 | 4.9382 |
 | ||
| Fig. 4 Zoomed-in regions of in situ TR-XRD contour plots of the NCM samples acquired between 120 and 450 °C (at 5 K steps): (a)–(d) as-received NCM; (e)–(h) highly protonated 55h-25:1-80°C-[65°C] NCM. The data are depicted for selected regions of the Mo-Kα1 2θ angle, viz., for (a) and (e) between 7.9° and 8.9°, (b) and (f) between 16.1° and 17.6°, (c) and (g) between 19.3° and 20.3°, as well as (d) and (h) between 27.7° and 28.9°, all extracted from the complete data set in Fig. S4.† The white labels indicate the lattice planes of the layered (L) and the rock-salt (RS) phase; the dashed white line across the bottom panels marks the temperature of 230 °C. | ||
Fig. 4a–d present selected regions of the as-received NCM which were extracted from the XRD data of Fig. S4a.† The shown reflections do not expose any structural changes upon heating but exclusively exhibit a decrease of the diffraction angle with increasing temperature due to the thermal expansion of the lattice parameters, which becomes more pronounced for the reflections at higher diffraction angles. For the as-received sample, no increase in the diffraction background with temperature is observed, and no additional phases can be found.
The selected regions of the highly protonated NCM sample 55h-25:1-80°C-[65°C] are depicted in Fig. 4e–h, showing the reflections at similar angles as the as-received NCM. Based on this observation, the layered structure clearly remains intact during washing in water, and no new crystalline phase is observed at 120 °C; this finding agrees with the observed absence of any oxygen release during washing46 or of any release of oxygen from the NCM structure during heating to 120 °C (see Fig. 3). Since there is no additional phase to be found by XRD for 55h-25:1-80°C-[65°C], the protonated NCM does not form a separate crystalline phase. In contrast to the as-received sample, however, the 55h-25:1-80°C-[65°C] washed NCM exhibits additional features upon heating to 450 °C, which mainly appear at 230 °C (indicated by the white dashed line in the bottom panels of Fig. 4). Fig. 4e presents the region between 7.9° and 8.9°, which includes the reflection at 8.5° that corresponds to the (003)L lattice plane of the layered (L) phase (LiMO2) and is its most intense reflection. For the (003)L plane, in addition to the thermal expansion, a slight shift to greater diffraction angles as compared to the as-received NCM can be observed; this observation will be discussed in more details for Table 4 and Fig. S5.† In contrast, both the (101)L reflection at 16.6° as well as the (10![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) )L reflection at 17.4°, depicted in Fig. 4f (between 16.1° and 17.6°), exhibit a shift to smaller angles at 230 °C. Furthermore, in addition to the weak (006)L reflection at 17.2°, a more intense reflection occurs at 17.2° for temperatures above 230 °C, which stems presumably from the (111)RS lattice plane of a rock-salt (RS) phase (i.e., indicating the formation of an MO phase). Similarly, the (104)L reflection at 20.0°, which is shown in Fig. 4g (between 19.3 and 20.3°), is broadened towards smaller angles, likely induced by an overlapping reflection appearing at 19.8° above 230 °C; this reflection is attributed to the (200)RS plane, being the most intense one of MO. Furthermore, the (10
)L reflection at 17.4°, depicted in Fig. 4f (between 16.1° and 17.6°), exhibit a shift to smaller angles at 230 °C. Furthermore, in addition to the weak (006)L reflection at 17.2°, a more intense reflection occurs at 17.2° for temperatures above 230 °C, which stems presumably from the (111)RS lattice plane of a rock-salt (RS) phase (i.e., indicating the formation of an MO phase). Similarly, the (104)L reflection at 20.0°, which is shown in Fig. 4g (between 19.3 and 20.3°), is broadened towards smaller angles, likely induced by an overlapping reflection appearing at 19.8° above 230 °C; this reflection is attributed to the (200)RS plane, being the most intense one of MO. Furthermore, the (10![[8 with combining macron]](https://www.rsc.org/images/entities/char_0038_0304.gif) )L reflection at 28.3° and the (110)L reflection at 28.6°, which are depicted in Fig. 4h (between 27.7° and 28.9°), are separated and of similar intensity at 120 °C; above 230 °C, however, the two peaks partially merge and their intensity ratio changes, likely explained by an occurring reflection at 28.3°, which may originate from the (220)RS reflection of MO. Additionally, the increase of the background, which becomes most obvious in Fig. S4b† and is absent for the as-received sample, might emerge from amorphous contributions, i.e., the partial loss of crystallinity of some material. When analyzing the XRD data without normalizing them by the intensity of the (003)L reflection (not shown), a decrease in the (003)L reflection intensity is observed (see discussion below for Table 4), which decreases between 230 and 330 °C from approx. 750 to 550 counts (data not shown) and, consequently, increases the background signal upon normalization. This indicates the decomposition of the layered phase at 230 °C.
)L reflection at 28.3° and the (110)L reflection at 28.6°, which are depicted in Fig. 4h (between 27.7° and 28.9°), are separated and of similar intensity at 120 °C; above 230 °C, however, the two peaks partially merge and their intensity ratio changes, likely explained by an occurring reflection at 28.3°, which may originate from the (220)RS reflection of MO. Additionally, the increase of the background, which becomes most obvious in Fig. S4b† and is absent for the as-received sample, might emerge from amorphous contributions, i.e., the partial loss of crystallinity of some material. When analyzing the XRD data without normalizing them by the intensity of the (003)L reflection (not shown), a decrease in the (003)L reflection intensity is observed (see discussion below for Table 4), which decreases between 230 and 330 °C from approx. 750 to 550 counts (data not shown) and, consequently, increases the background signal upon normalization. This indicates the decomposition of the layered phase at 230 °C.
Since even nickel-rich LiMO2 phases are stable up to >600 °C,33 only the HMO2 fraction of the NCM is decomposed below 450 °C. The appearance of MO above 230 °C agrees with the release of O2, as it was shown by TGA-MS (see Fig. 3). Even though the release of oxygen from the NCM structure in the form of H2O occurs already at temperatures greater than 140 °C (and thus far below 230 °C), clearly no new crystalline phase is visible between 140 and 230 °C (see Fig. S4b†). The most intense reflections of the previously hypothesized intermediate spinel (S) phase (M3O4, suggested by Pritzl et al.46) or the corundum (C) phase (M2O3, see eqn (16)) would occur at 14.3° for the (220)S plane or at 15.2° for the (222)C plane, respectively;50,72–74 however, no additional reflections can be observed in that region between 120 and 450 °C. Thus, one can exclude the formation of M2O3 and M3O4 phases upon washing and heating of a protonated NCM831205 material.
In addition to the decomposition of the layered phase, a kink-like, sudden shift of the diffraction angles is observed for all five reflections in the temperature region of 230 °C: This shift also results in a change of the distance between the (10)L and the (110)L reflections (Fig. 4h), which decreases from 0.3° up to 230 °C to 0.2° above 280 °C. For the (electro)chemical (de)lithiation of the NCM structure, it is known that these two reflections move apart from each other when lithium ions are deintercalated.75 Based on this observation, this shift of the reflections at 230 °C is assigned to a lithiation of the layered phase, while, at 120 °C, the structure appears to be slightly delithiated as compared to the as-received NCM. This suggests that the partially protonated HxLi1−xMO2 phase decomposes between 120 and 230 °C into a slightly lithiated layered phase and a rock-salt phase, concomitant with the release of H2O:
 | (20) |
The onset temperature for this process is marked by the release of H2O in the TGA-MS experiments, which occurs at 140 °C (see Fig. S3c†). The partially lithiated layered phase in the bulk is then decomposed at higher temperatures, which is accompanied by the release of O2, initiating at 230 °C (see Fig. S3d†):
 | (21) |
Note that despite the different intermediate phases, the sum of eqn (20) and (21) results in the same overall reaction as the sum of eqn (16) and (17), so that the above proposed TGA-MS analysis of the proton content is valid (see eqn (19)), irrespective of the intermediate phases.
Therefore, instead of the hitherto hypothesized formation of an intermediate M2O3 phase upon heating protonated NCMs to a temperature between 140 and 230 °C (eqn (16)), the here presented XRD analysis suggests a thermal decomposition into an MO phase (likely in the surface-near region due to the slow diffusion of oxygen in the NCM structure) and a partially lithiated layered phase (likely in the bulk), as described by eqn (21). To verify this hypothesis and to quantify the amount of the MO phase, samples of the highly protonated NCM material heat-treated at different temperatures were analyzed by ex situ powder XRD, as this provides a higher signal-to-noise ratio and, through a Rietveld refinement, also the values of the lattice parameters a and c.
Ex situ X-ray powder diffraction (XRD)
As shown in Fig. 4 and S4,† the reflections of the rock-salt phase (MO) have a significant overlap with those of the layered phase (LiMO2), rendering the discrimination of the two phases challenging. The phase analysis is facilitated by: (i) high-quality diffraction patterns (high signal-to-noise ratio, small full width at half maximum of peak shapes), (ii) a large amount of the side phase after heating, here of the rock-salt phase, and (iii) a Rietveld refinement to quantify the contribution of the phases as well as their lattice parameters. Therefore, ex situ powder XRD data were collected for the highly protonated NCM sample 55h-25:1-80°C, which was heated beforehand to temperatures of 65, 120, 180, 230, 300, and 450 °C; these are then compared to the as-received as well as the calcined NCM. The measurements were performed at room temperature, thus avoiding contributions by thermal lattice expansion, in contrast to the in situ TR-XRD data presented in Fig. 4 and S4.†Exemplarily, the diffractogram of the highly protonated and subsequently at 450 °C heat-treated NCM (55h-25:1-80°C-[450°C]) is depicted in Fig. 5a (black data points). As suggested by Friedrich et al.,14 a Rietveld refinement based on the combination of a layered NCM phase with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group as well as a rock-salt phase with an Fmm space group should, in the absence of additional phases, fully account for all reflections between 7° and 48° using a Mo-Kα1 source (see red line). This is indeed the case, yielding a reasonably small difference of data and fit (see gray data in Fig. 5a). The difference can also be expressed by the goodness-of-fit parameter χ, as discussed in the ESI,† having values of χ between 0.86 and 1.15 and thus confirming a reliable fit.
m space group as well as a rock-salt phase with an Fmm space group should, in the absence of additional phases, fully account for all reflections between 7° and 48° using a Mo-Kα1 source (see red line). This is indeed the case, yielding a reasonably small difference of data and fit (see gray data in Fig. 5a). The difference can also be expressed by the goodness-of-fit parameter χ, as discussed in the ESI,† having values of χ between 0.86 and 1.15 and thus confirming a reliable fit.
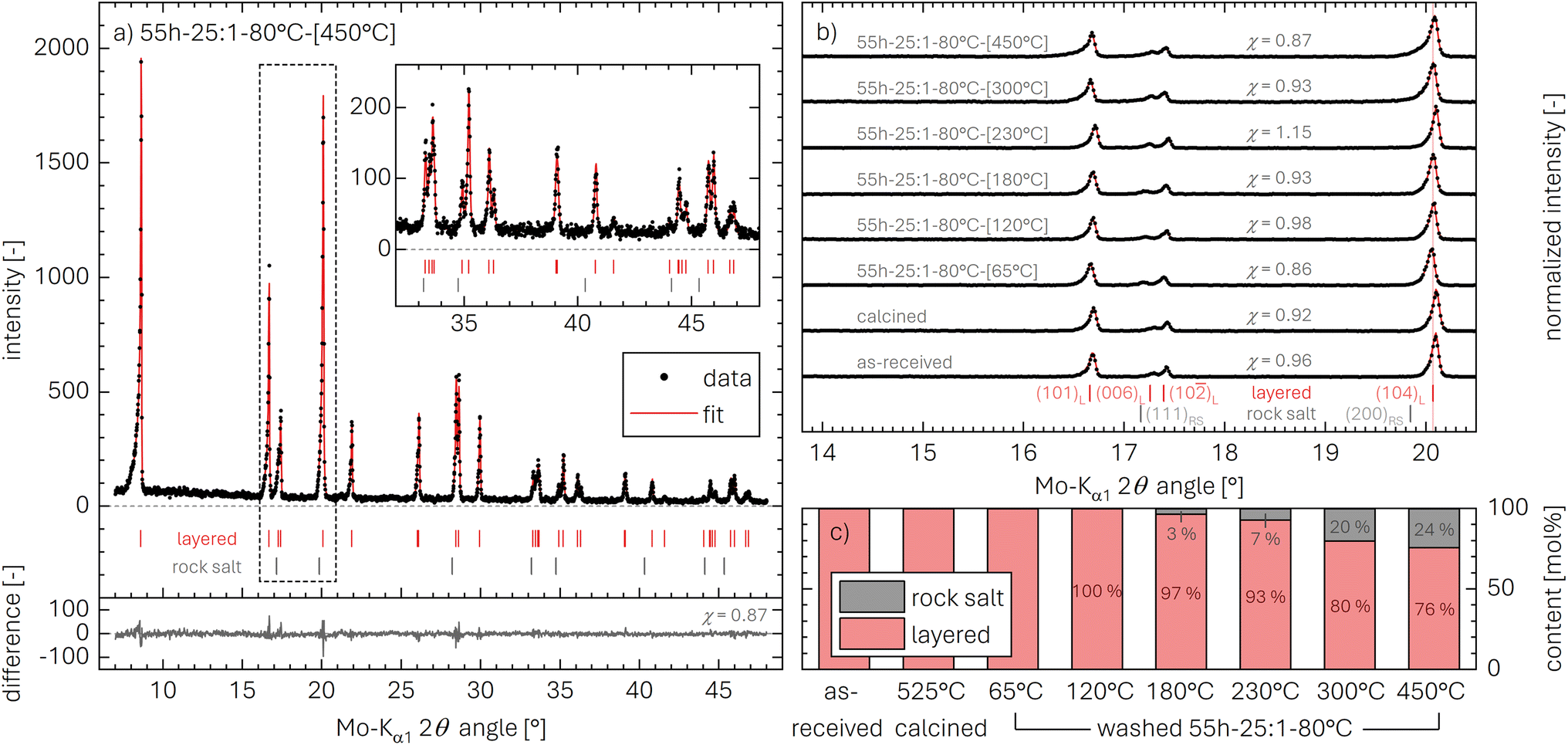 | ||
| Fig. 5 Ex situ powder X-ray diffractograms (black data points) and fit by Rietveld refinement (red lines) for various NCM samples: the XRD experiments were conducted in transmission mode using Mo-Kα1 radiation, taking one data point every 0.15° between 7° and 48°. The red and gray vertical bars indicate the expected position of the layered and the rock-salt phase, respectively. The goodness of fit χ is stated for each data set. (a) Exemplary diffraction pattern of the highly protonated NCM sample after heating at 450 °C, including the difference between data and fit (gray line). (b) Magnified diffraction patterns of all samples, showing the intensity normalized to the highest peak (i.e., the (003)L reflection at 8.5°) between 16.1° and 20.9° (see black dashed box in panel a). The red and gray labels indicate the lattice planes of the layered (L) and the rock-salt (RS) phase, respectively. (c) Relative molar fraction (in mol%) of the layered (red) and the rock-salt (gray) phase for each sample, extracted from the XRD data by the Rietveld refinement. | ||
To investigate the changes in the NCM structure upon washing and/or subsequent heating, an overview of all collected diffractograms is shown in Fig. 5b for the region between 16.1° and 20.9°. This region includes the (101)L, (006)L, (102)L, and (104)L reflections of the layered NCM structure and, if present, two reflections of the MO structure, viz., the (111)RS reflection at 17.2° and the (200)RS reflection at 19.8°. When comparing the diffraction pattern of the as-received and the calcined NCM, no changes are observed. In contrast, for the highly protonated sample heated to only 65 °C (55h-25:1-80°C-[65°C]), a shift of the reflections towards smaller angles occurs, which is discussed below for the evaluation of the lattice parameters aL and cL in Table 4 and Fig. S5.† Nevertheless, the NCM structure maintains its layered structure and no other crystalline phases are observed, despite the high proton content xH+ of 25.0 mol% (see Table 6), underlining that no oxygen is released from the NCM structure upon washing, consistent with the on-line mass spectrometry analysis by Pritzl et al.46 The formation of a separated phase for HMO2 with reflections at different diffraction angles is excluded by XRD, as there is no peak broadening observed upon protonation, evident when comparing the calcined and the 55h-25:1-80°C-[65°C] sample; furthermore, the protonated phase cannot be amorphous either, as this would result in the unchanged diffractogram of a pure LiMO2 phase, while the XRD data of 55h-25:1-80°C-[65°C] exhibit a clear shift of the (104)L reflection to lower diffraction angles.
With increasing drying temperature, only little changes are observed up to 180 °C, while, starting at 230 °C, a broadening of the (104)L reflection (at 20.1°, see Fig. 5b) towards lower diffraction angles can be observed. The broadening is explained by the appearance of the (200)RS reflection (at 19.8°), which is accompanied by the simultaneous development of the (111)RS reflection (at 17.2°), both challenging to observe by eye. The ratio I8.5°/I20.0° of the integrated intensities of the (003)L reflection (at 8.5°) as well as the (104)L and the (200)RS reflection (both at 20.0°), however, provides further insights: In the literature, this ratio (often referred to as I003/I104 value, with I104 including the intensity of the (200)RS reflection) is an indicator both for lithium-TM disorder as well as for rock-salt formation.14,76,77 The as-received NCM exhibits an I8.5°/I20.0° value of 1.42 (see Table 4), while it increases for the calcined NCM to 1.69, potentially due to the partial conversion of an MO phase present in the as-received material to a layered phase. Upon protonation, relatively small changes are observed (I8.5°/I20.0° = 1.60), while, upon heating of the highly protonated NCM, the I8.5°/I20.0° values decrease with increasing drying temperature (from 1.40 to 1.16, see Table 4), possibly indicating a partial decomposition of the layered phase to the MO phase. Similar to the TR-XRD results (see Fig. 4), no indication of an intermediate spinel (S) phase (M3O4) or a corundum (C) phase (M2O3) are observed for any heat-treatment temperature, as for these reflections at 14.3° for (220)S or at 15.2° for (222)C would be expected, both of which are absent for the ex situ XRD data as well.
From the Rietveld refinements of the diffractograms, the relative contributions of the layered LiMO2 and the rock-salt MO phase are extracted and presented in Fig. 5c (note that the relative mass fraction (in units of wt%) obtained by Rietveld refinement is stated here in the more meaningful molar fraction (in units of mol%), calculated from the mass fraction using the molar masses of LiMO2 (97.5 g mol−1) and MO (74.5 g mol−1)). Up to 120 °C, no MO can be determined. Starting at 180 °C, 3 mol% of MO are detected, increasing to 7 mol% at 230 °C and 20 mol% at 300 °C. At the highest temperature of 450 °C, where the TGA-MS experiments indicate the completion of the thermal decomposition of protonated NCMs (see Fig. 3 and S3†), a value of 24.1 mol% of MO is determined.
The observations that exclusively H2O is released up to 230 °C (see Fig. S3†) while the formation of MO is observed to start already at 180 °C, provide further evidence that the thermal decomposition of the protonated NCM between 140 and 230 °C proceeds viaeqn (20) (yielding a partially lithiated layered and a rock-salt phase), rather than via the previously proposed formation of a spinel phase46,78 or the potential formation of a corundum phase (eqn (16)). As the thermal decomposition is completed at 450 °C (see Fig. S3†), solely yielding a layered LiMO2 and a rock-salt MO phase (see Fig. 5), consistent with eqn (20) and (21), the combination of eqn (20) and (21) predicts that, at 450 °C, 1 mol of HxLi1−xMO2 should be converted to 1−x mol LiMO2 and x mol MO, so that the MO mole fraction corresponds to x, i.e., to xH+:
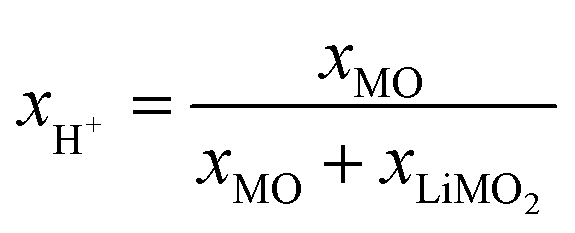 | (22) |
The thus obtained proton content xH+ of 24.1 mol% for the highly protonated 55h-25:1-80°C sample will be compared to those obtained by the other methods in Table 6. However, considering that the PGAA analysis indicated that protons remain in its structure even at 450 °C (see Fig. 2), the here obtained xH+ value might be somewhat underestimated (as in the case of the TGA-MS analysis).
The Rietveld refinement also allows to determine the lattice parameters aL, cL, and aRS for the highly protonated and heat-treated samples. In the literature, the change of the lattice parameters has already been investigated for washed and/or heated nickel-rich layered oxides;41,42,49,79–81 however, for the typical washing conditions which would result in a small Li+/H+ exchange on the order of 1 mol%, the effect on the lattice parameters would be minor. In this work, however, changes in the lattice parameters, listed in Table 4 and discussed for Fig. S5 in the ESI,† are rather pronounced due to the relatively high xH+ of 25 mol%.
As already observed for the TR-XRD experiments of the as-received NCM and the highly protonated 55h-25:1-80°C-[65°C] sample, the diffraction angles of certain reflections of the latter are slightly shifted as compared to the as-received NCM; additionally, this shift is reversed when the washed NCM is heated above 230 °C (see Fig. 4). These changes of the lattice parameters aL and cL were quantified for the layered phase via Rietveld refinement of the ex situ XRD data and are presented in Table 4: aL and cL of the as-received NCM show the expected values of 2.8711 and 14.1894 Å of an NCM851005, respectively, consistent with previous studies.14,75,82 Comparing the as-received NCM with the calcined one, the differences of aL and cL are minor. Upon protonation, however, aL changes by only 0.04% to 2.8694, while cL increases by 0.33% to 14.237 Å. Upon heating up to 180 °C, aL remains essentially constant (at 2.8696 Å), and cL also increases by only 0.08% to 14.248 Å. More significant changes are observed with the release of O2 starting at 230 °C (see also Fig. S3†), where the cL parameter again decreases, reaching 14.196 and 14.194 Å at 300 and 450 °C, respectively, which are within 0.04% of the value in the as-received material; at the same time, the aL parameter increases to 2.8738 and 2.8746 Å, which is 0.14% higher than in the as-received material. It must be noted that, even though both the as-received/calcined sample as well as the washed and heat-treated samples dried at 300 and 450 °C are expected to consist of a fully lithiated layered structure (and an additional rock-salt shell for the washed and heat-treated samples), their lattice parameter aL of the layered structure differs. This may be caused by the relatively thick rock-salt surface layer formed on the particles of the 55h-25:1-80°C-[300°C] and the 55h-25:1-80°C-[450°C] samples: For a molar fraction of the rock-salt phase of 24 mol% and a specific surface area of 1.67 m2 g−1 (corresponding to a particle diameter of 0.7 μm, see Table S1†), a spherical approximation would yield a thickness of 18 nm for the MO shell after washing and heat treatment; after washing without heat treatment, an HMO2 shell of 33 nm would be present (see detailed derivation of eqn (S12) and (S14) in the ESI†). This MO shell of 18 nm may then distort the lattice parameters of the layered phase in the core. The here described variation of the aL and cL values is also illustrated in Fig. S5.† The lattice parameter aRS of the rock-salt phase, appearing at 180 °C, exhibits a value of 4.11 ± 0.01 Å that remains constant also at higher temperatures; it is close to the one of nickel-rich rock-salt-type phases of TM oxides, e.g., of NiO.83,84 Based on the results of Iida et al. on the dissolution of lithium oxide in NiO, the relatively low value of aL of 4.11 Å might suggest that the rock-salt phase contains up to 25 mol% of lithium.85 Overall, however, the changes of the lattice parameters of at most 0.14% for aL and 0.42% for cL are relatively small in comparison to their changes upon (electro)chemical delithiation of up to 2% for aL and up to 5% for cL.75
Furthermore, the parameter cL/aL, being a descriptor for the distortion of the unit cell, has a very similar value of 4.9433 and 4.9422 for the as-received and the calcined NCM, respectively. It increases by 0.40% to 4.9618 for the highly protonated NCM with xH+ of 25.0 mol% (see Table 4); upon heating, the cL/aL values further increase, reaching a maximum of 4.9654 at 180 °C. This trend becomes reversed between 180 and 230 °C, and the cL/aL value decreases to 4.9382 after the heat treatment at 450 °C, a value that is within 0.08% of that of the as-received and calcined NCM. This finding confirms our hypothesis taken in eqn (21) that protonated NCMs heat-treated at 450 °C are composed of a rock-salt structure on the surface and a fully lithiated LiMO2 phase in the core.
As the protonation affects the lattice parameters and thus also changes the bulk properties of the NCM material, the distribution of protons and lithium ions within an NCM particle is of great interest. Since the here observed variations of the lattice parameters of a highly protonated and heat-treated NCM are significant but rather small, two possible structures of the washed NCM particles remain plausible: either (i) the protons mix with the lithium ions and distribute homogeneously within the NCM particle, or (ii) a layered material evolves with a delithiated, protonated NCM layer around an essentially unperturbed fully lithiated NCM core. To elucidate the spatial distribution of the protons within the NCM primary particles, X-ray photoelectron spectroscopy is applied in the following.
X-ray photoelectron spectroscopy (XPS)
Core-level spectra are acquired by XPS for the calcined NCM material as well as for samples washed with different washing times, temperatures, and water-to-CAM mass ratios. The obtained survey spectra with the assigned core levels and Auger transitions are depicted in Fig. S7.† Besides the expected core-level photoelectron and Auger emission from lithium, oxygen, and the transition metals, a C 1s signal of similar intensity is found at 284.5 eV for every sample, originating from the adventitious carbon, which is typical for sample surfaces that have not been pre-treated in ultra-high vacuum prior to XPS analysis.86 For the calcined sample, which is the only unwashed sample investigated by XPS, traces of sodium and sulfur are detected on the surface, which are impurities stemming from the NCM synthesis (see discussion of Fig. S7†).To study the chemical environment and the concentration of the lithium and TM species in the near-surface region of the NCM particles, high-resolution spectra of the TM 3p and Li 1s core levels are acquired (see Fig. 6a). As all respective core levels lie in a very narrow range with a binding energy between 45 and 85 eV, this region allows for a simplified and more accurate quantitative analysis (see below). Quantitative analysis of the TM 2p core levels is omitted since the respective peaks suffer from a complicated satellite structure and overlap with electron emission from Auger transitions, especially in case of the Mn 2p peak, which cannot be resolved at low concentration due to the overlap with the intense Ni L2M23M45 Auger transition peak. The spectra depicted in Fig. 6a are normalized by the total integrated Ni 3p intensity and corrected for sample charging by using the C 1s signal of adventitious carbon at 284.5 eV. The overall sample charging is low in all cases (<0.1 eV), except for the highly protonated 55h-25:1-80°C-[65°C] sample, which charges by 1.3 eV during the XPS measurements. This larger extent of sample charging goes in line with inhomogeneous (differential) charging effects leading to peak broadening, particularly visible for the Co 3p and Mn 3p peak. After subtraction of a Shirley background, the deconvolution of all peaks in the acquired spectra is accomplished by a global fit with ten components, of which four components are used for the Ni 3p peak and two components each for the Co 3p (blue area in Fig. 6b–g), the Li 1s (red), and the Mn 3p (brown) peaks. The two components for the Li 1s peaks aim at deconvoluting two spectroscopically resolved, chemically different Li 1s species (see below); all other peak components are solely introduced to mimic the complex peak shape and satellite structure of the TM 3p photoemission peaks in order to correctly determine their peak areas.
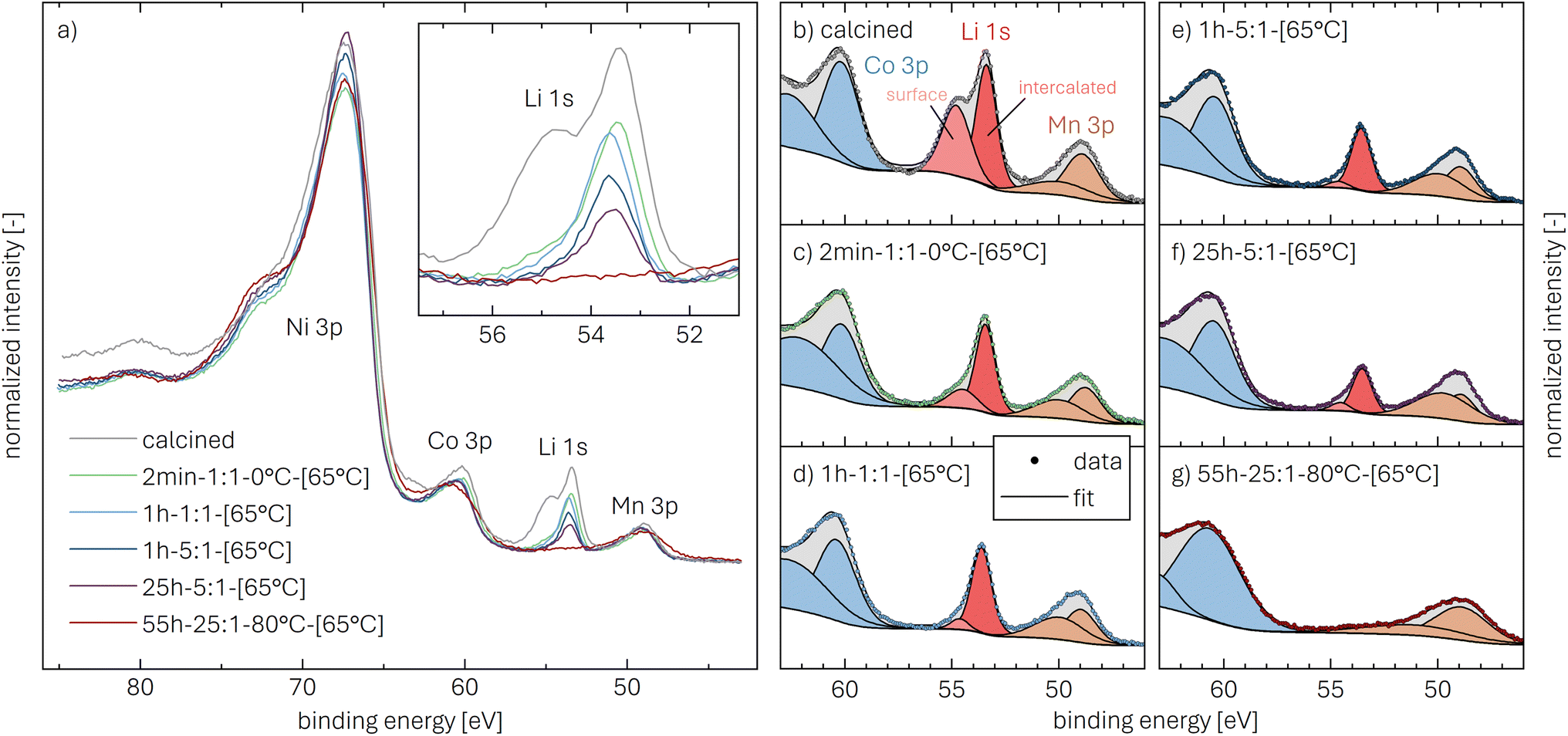 | ||
| Fig. 6 XPS data of the calcined NCM and of differently washed NCM samples heat-treated at 65 °C, which are normalized by the total integrated Ni 3p intensity and corrected for sample charging by using the C 1s signal of adventitious carbon at 284.5 eV. (a) Data overview. (b)–(g) Ten-component fit of the core levels of Ni 3p (68 eV, data not shown, four components), Co 3p (60 eV, blue, two components), Li 1s (54 eV, red, two components), and Mn 3p (49 eV, brown, two components) for each of the NCM samples. The ten-component fit aims at distinguishing two chemical Li 1s species, i.e., lithium-containing surface impurities such as Li2CO3 (54.8 eV, pale red) and intercalated lithium (53.4 eV, red); all other peak components are solely introduced to correctly mimic the complex peak shape and satellite structure of the TM 3p photoemission peaks, having no physical meaning but enabling an accurate determination of their respective peak areas. | ||
The binding energies of the Ni 3p, Co 3p, and Mn 3p core levels of 67, 60 and 49 eV, respectively, are similar for the calcined and all the washed NCMs, indicating that the protonation of the NCM does not significantly change the oxidation state or chemical environment of the TMs, as one would expect for a Li+/H+-exchange reaction. However, the Ni 3p peak shape is affected by the washing procedure: For the calcined sample, its peak shape resembles a single broad component that tails to the high binding energy side, whereas all washed samples clearly show a similar, more narrow peak shape with a resolved second component at 72 eV. This additional component reflects a satellite and does not originate from the spin–orbit splitting which is below 2 eV for Ni 3p.87 Mansour et al. found that the absence of this satellite is characteristic for LiNiO2 systems;86,88–90 lithium-free nickel compounds such as rock-salt NiO or nickel-oxide species formally containing Ni3+, such as NiOOH and Ni2O3, show pronounced satellites at similar binding energy. Thus, this satellite might be sensitive towards the chemical environment and the lithium content of an ordered NCM structure, which is influenced by replacement of lithium ions by protons. All other TM 3p signals remained essentially unaffected upon washing of the NCM.
The Li 1s peaks of the calcined sample are deconvoluted by two components: a sharp component at a binding energy of 53.4 eV (marked by a red background in Fig. 6b–g), referring to lithium intercalated in the layered-oxide structure of the NCM (further on referred to as [Li 1s]int.), and a broader component at 54.8 eV (pale red), indicative for typical surface contaminants such as Li2CO3,30,34,46,47 remaining from the NCM synthesis or produced during NCM exposure to ambient atmosphere (further on referred to as [Li 1s]surf.). For the calcined NCM, the [Li 1s]surf. component contributes 44% to the overall Li 1s intensity (see Fig. 6b), while, for the washed samples, the [Li 1s]surf. contribution decreases substantially. For 2min-1:1-0°C-[65°C], the surface contaminants contribute only 24% (see Fig. 6c), while all other washing conditions are sufficient to remove most surface contaminants, so that their contribution to the Li 1s signal is below 10%. Besides the [Li 1s]surf. intensity, the washing procedure also affects the intensity of the [Li 1s]int.. In accordance with titration and PGAA experiments, both an increased washing time as well as an increased water-to-CAM mass ratio reduces the [Li 1s]int. intensity, as intercalated lithium is replaced by protons. For the extreme washing conditions of 55h-25:1-80°C-[65°C], for which xH+ of 25 mol% has been found by PGAA, TGA-MS, and XRD (see Tables 2–4), the [Li 1s]int. signal is below the detection limit, suggesting the formation of an entirely delithiated and thus fully protonated near-surface region (see Fig. 6g).
In order to study the effect of the lithium/proton intermixing within the lithium layer of the NCM structure, the intensity changes of the [Li 1s]int. signal are analyzed using a model for the NCM surface composition based on a quantitative peak intensity analysis. First, the intensity IA of a core level A is given by:91
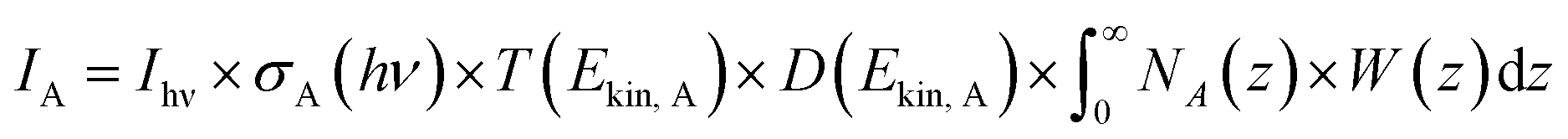 | (23) |
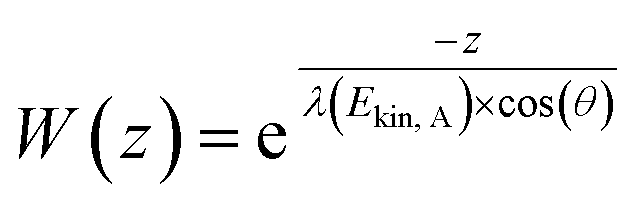 | (24) |
This limits the probed surface layer to typically 3λ(Ekin, A) or approximately 2 to 10 atomic layers, making XPS a surface-sensitive technique. For a solid with a homogeneous distribution of element A, NA is not dependent on z and the integral in eqn (23) results in:
 | (25) |
In the case of the NCM material, the TM atoms are assumed to be homogeneously distributed, which is not expected to change during washing, as the TMs do not dissolve above a pH value of 7;48,54 furthermore, the layered structure is not altered by the Li+/H+ exchange, as observed by XRD (see Fig. 5). Considering an intensity ratio of two elements within the same data set, the X-ray intensity Ihv and the detection efficiency D are identical and thus cancel out. As the kinetic energy of the TM 3p core levels varies within a very narrow range of 35 eV compared to the absolute kinetic energy of more than 1400 eV (for the here used Al-Kα radiation with an energy of 1486.6 eV), the changes in the analyzer transmission T(Ekin) as well as the signal attenuation length λ(Ekin) can be neglected, so that these contributions cancel out when calculating an intensity ratio. Hence, the atomic ratio of two elements A and B is dependent on the core level intensity ratio and the photoionization cross sections only:
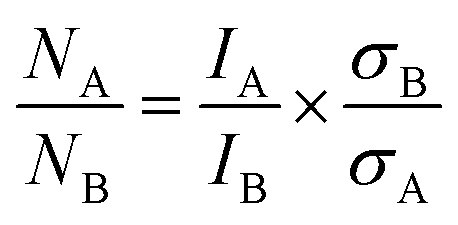 | (26) |
Following this approach, the molar ratios of the transition metals in the investigated NCM samples can be determined and are summarized in Table 5. For the calcined and all the washed samples, the obtained molar ratios of the TMs match the bulk composition of the NCM831205 material as well as the PGAA results (see Table 2). Thus, no differences between surface and bulk stoichiometry of the TMs in the NCM particles are observed, and changes of the NCM stoichiometry upon washing can be ruled out.
 is normalized by the one of the calcined sample
is normalized by the one of the calcined sample 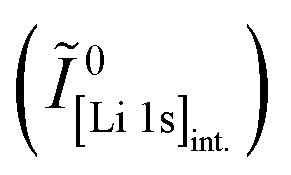 . The latter is indicated by 1* since, for the calcined sample, it is assumed that no protons are present, indicated by a layer thickness of 0*. For 55h-25:1-80°C-[65°C], the normalized intensity of intercalated lithium is below the detection limit, indicated by <0.05, which results in a lower limit of l, indicated by >8.1
. The latter is indicated by 1* since, for the calcined sample, it is assumed that no protons are present, indicated by a layer thickness of 0*. For 55h-25:1-80°C-[65°C], the normalized intensity of intercalated lithium is below the detection limit, indicated by <0.05, which results in a lower limit of l, indicated by >8.1
| Sample |
N
Ni:NCo:NMn (in mol%) |
|
l (in nm) |
|---|---|---|---|
| calcined | 84.4:10.4:5.2 |
1* | 0* |
| 2min-1:1-0°C-[65°C] | 85.0:10.8:4.3 |
0.86 | 0.4 |
| 1h-1:1-[65°C] | 84.5:10.9:4.6 |
0.77 | 0.7 |
| 1h-5:1-[65°C] | 84.3:11.1:4.6 |
0.57 | 1.5 |
| 25h-5:1-[65°C] | 83.8:11.5:4.8 |
0.38 | 2.6 |
| 55h-25:1-80°C-[65°C] | 83.4:10.7:6.0 |
<0.05 | >8.1 |
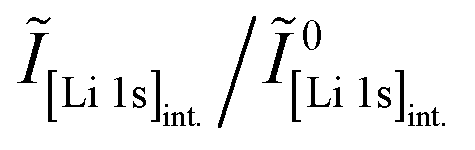
In contrast to the TMs, the distribution of Li+ within the NCM host structure might be affected by washing, for which different scenarios can be imagined:
• As lithium ions are removed by the washing solution from the near-surface region of the NCM particles and simultaneously replaced by protons, a protonated near-surface layer may form around a lithiated NCM core without significant intermixing of the protonated and lithiated phases. This could either be caused by an extremely slow interdiffusion (i.e., kinetic limitations) or by a thermodynamically favored phase separation (i.e., intercalated lithium ions prefer to be surrounded by lithium ions rather than by protons and vice versa).
• In case of a thermodynamically favored and relatively fast lithium/proton intermixing within the NCM structure (i.e., fast interdiffusion), a partially protonated/lithiated NCM phase with a homogeneous distribution of both lithium ions and protons throughout the entire NCM particle would be obtained.
These two hypotheses can be tested with the NCM material subjected to the harshest washing procedure (55h-25:1-80°C-[65°C]), for which PGAA, TGA-MS, and XRD suggest that approx. 25 mol% of the intercalated lithium ions are exchanged by protons. Assuming complete intermixing of Li+ and H+, this would result in a decrease of the [Li 1s]int. intensity by approx. 25% compared to the value obtained for the calcined NCM, for which a homogeneous Li+ distribution without a delithiated surface layer can be assumed. However, the [Li 1s]int. peak intensity measured for 55h-25:1-80°C-[65°C] is not decreased by only 25%, but its signal drops below the detection limit. Thus, no intercalated lithium is present within the probed surface layer (of 3λ, corresponding to 8 nm) of the highly protonated NCM particles, indicating that the lithium/proton intermixing within the NCM structure must either be extremely slow or thermodynamically unfavorable. The latter is implied by an additional experiment, as the protonated surface layer is maintained even during a temperature hold at 120 °C for 18 h (data not shown), which will be discussed in a future publication.
To quantify the thickness of this surface layer, in a first step, a flat NCM surface consisting of a protonated, lithium-depleted near-surface layer above an unaffected, fully-lithiated substrate is assumed. For this geometry, the [Li 1s]int. intensity originating from the substrate is attenuated by the protonated layer. Compared to the [Li 1s]int. intensity of the calcined sample with a homogeneous lithium distribution 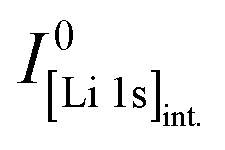 , an exponential decay of the [Li 1s]int. intensity of a washed sample
, an exponential decay of the [Li 1s]int. intensity of a washed sample 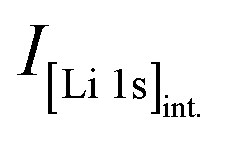 as a function of the protonated layer thickness l is expected, which scales with the signal attenuation length λ(Ekin, Li 1s) and the emission angle θ:
as a function of the protonated layer thickness l is expected, which scales with the signal attenuation length λ(Ekin, Li 1s) and the emission angle θ:
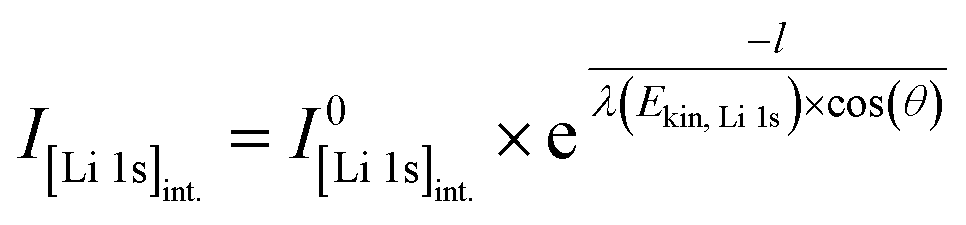 | (27) |
Thus, the thickness of the protonated layer can be derived from the [Li 1s]int. intensity ratio of a washed NCM compared to the calcined NCM (note that here the attenuation by surface contaminants of the calcined NCM is expected to be small and thus neglected). However, the intensities of a single photoemission line acquired from different samples might suffer from deviations in X-ray intensity or detection efficiency (see eqn (23)) and require normalization. As the TMs remain unchanged during the washing procedure, a TM 3p intensity can serve for the normalization. Due to the complicated satellite structure of Ni 3p and the overlap between Ni 3p and Co 3p, the Mn 3p intensity was used to normalize the [Li 1s]int. intensity, i.e., Ĩ[Li 1s]int. = I[Li 1s]int./IMn 3p. As noted above, the signal attenuation length is often approximated by the IMFP. According to Seah et al., the IMFP of an electron can be estimated by λ = CE0.5, with E being the energy of the photoelectron (in units of eV) and C being a material constant, which is typically 0.096 nm for inorganic compounds.32,92 At the kinetic energy of 1430 eV for Li 1s photoelectrons when using an aluminum X-ray source, λ in the NCM solid amounts to 3.6 nm. Considering that the NCM material does not provide a flat surface but approximately spherical particles in the micrometer range, the photoelectron emission angle between the surface normal and the analyzer changes depending on the position on the particle. According to Kuipers et al., this can be accounted for by a correction of the IMFP by a factor of 0.75, independent of the particle geometry and size,93 resulting in an effective attenuation length λeff = 0.75λ = 2.7 nm for a kinetic energy of 1430 eV. For this geometry, the thickness of the delithiated shell l can be calculated by eqn (28) (note that the data given for 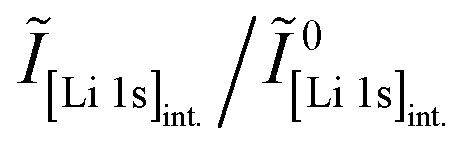 in Table 5 is the inverse of the value required here):
in Table 5 is the inverse of the value required here):
 | (28) |
The calculated layer thickness, summarized in Table 5, varies between 0.4 nm for the material washed for the shortest time at the lowest temperature (2min-1:1-0°C-[65°C]) and 2.6 nm for the 25h-5:1-[65°C] sample. Note that, obviously, no layer thickness can be derived when no [Li 1s]int. intensity is observed, as it is the case for the highly protonated 55h-25:1-80°C-[65°C] sample; however, the lower limit of its layer thickness can be estimated by 3λeff, for which most signal of the lithium in the bulk is attenuated (i.e., 95% of 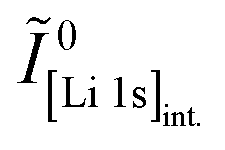 ), resulting in a layer thickness of more than 8.1 nm.
), resulting in a layer thickness of more than 8.1 nm.
In general, the proton contents of the washed NCM materials can be derived based on the thickness of the protonated surface layer determined by XPS. The proton content reflects the contribution of the protonated surface layer to the total particle volume. However, the total particle volume is a bulk-related value that is inaccessible by surface-sensitive XPS. Hence, additional knowledge of the particle size distribution is required, which must be addressed by other analytical techniques. A common approach considers the specific surface area of the materials: Assuming all particles are spheres of the same size, an effective particle diameter can be extracted (see Table S1†) and related to the thickness of the protonated surface layer obtained by XPS. The absolute proton content determined by this approach is on the correct order of magnitude and differs by a factor of 2 from the values obtained by the other techniques, as discussed in more detail in Table S2.† Considering that the actual particle size distribution of the NCM materials is very broad and not monodisperse (see Fig. S1†), the deviations of the proton contents are within the expected error. Nonetheless, calculating the protonated layer thickness via XPS provides plausible results.
Thus, for a semi-quantitative interpretation of the XPS data, the thickness of the protonated layer obtained by XPS (see Table 5) is plotted in Fig. 7 as a function of the proton content obtained from titration (green symbols in Fig. 7, see Table 1), PGAA (yellow, Table 2), and TGA-MS (gray, Table 3), both in units of λeff (equal to 2.7 nm, left y-axis) as well as nm (right y-axis). For the calcined sample (see black symbol), the layer thickness and the proton content is defined to be zero since, based on the TGA-MS and PGAA data, the absence of protons has been confirmed. With increasing intensity of the washing conditions (i.e., with prolonged washing time, higher water-to-CAM mass ratio, and higher temperature), the thickness of the protonated layer increases and is directly proportional to the proton content determined by the other techniques. Due to this linear relationship of layer thickness and proton content (see gray line), it is concluded that the washing of NCM materials results in the formation of a delithiated but protonated surface layer, in which lithium ions are fully replaced by protons while the bulk remains lithiated. The intermixing of H+ and Li+ was not observed, thus it does not take place to a significant extent.
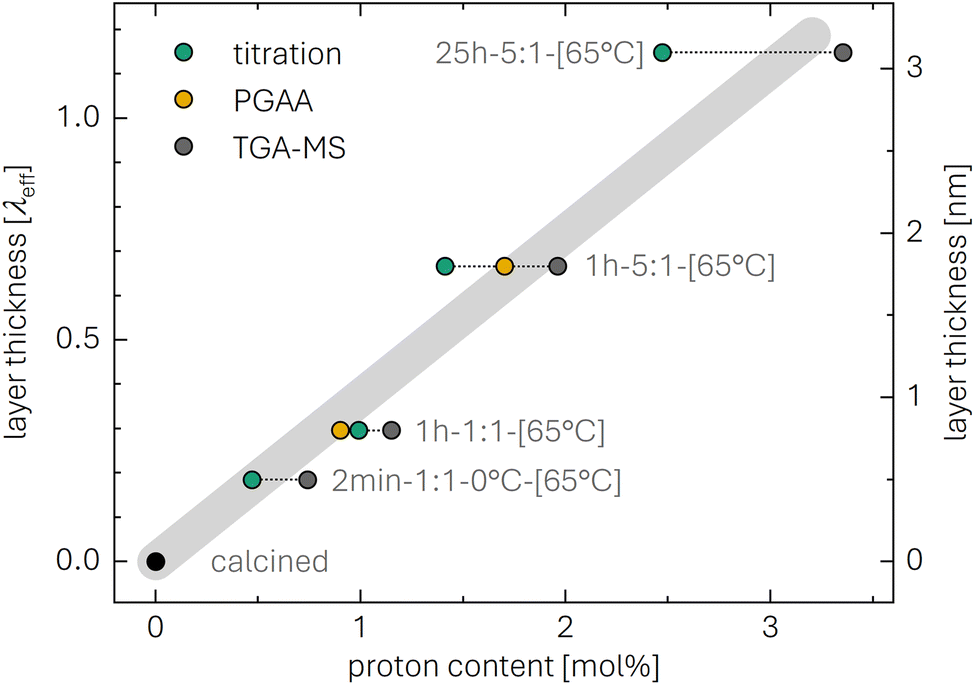 | ||
| Fig. 7 Thickness of the protonated surface layer l (in units of λeff and nm on the left and right y-axis, respectively), extracted from XPS (see Table 5), as a function of the proton content xH+ (in mol%) obtained by titration (green symbols, see Table 1), by PGAA (yellow symbols, see Table 2), and TGA-MS (gray symbols, see Table 3). The gray line was calculated by linear regression of the data points (while, for the calculation, a proton content of 0 mol% was defined to correspond to a layer thickness of 0 nm) and serves as guide to the eye. | ||
At first sight, it seems contradictory that the XRD data (see Fig. 4 and 5) indicate changes of the lattice parameters in the bulk, while XPS indicates the presence of a protonated surface layer with no/negligible intermixing of the protons with the intercalated lithium from the NCM bulk (see Fig. 6). In the case of a core–shell particle comprising two phases with slightly different lattice parameters, a tiny difference (e.g., of 1%) in the layer spacing would likely accumulate over tens of layers within an NCM particle. As the bending of the layers to such an extent is energetically unfavorable, the induced stress/strain may result either in (i) plane gliding or stacking faults of the structure, or (ii) in a distortion of the lattice parameters within the entire particle. From the collected XRD data, the formation of stacking faults between the protonated and the lithiated phase cannot be excluded. The XPS data, however, indicate the formation of core–shell particles, consisting of a protonated surface layer and a lithiated bulk without significant intermixing, whereby the intrinsically different lattice parameters of the surface layer appear to slightly distort the lattice parameters of the entire particle. Such distortions are observed for epitaxially grown surface films on single-crystalline substrates in surface science, e.g., for gold on alumina, niobium-germanium alloys on silicon, silver on sapphire, gallium-nitride semiconductor on aluminate or silicon carbide, and vanadium oxide on sapphire.94–96 However, even though the XPS measurements provide unequivocal evidence for the core–shell nature of protonated NCM particles, it cannot be excluded that a (relatively small) fraction of the protons diffuses into the lithiated phase, what might contribute to the overall change of the lattice parameters, as observed by XRD (see Table 4 and Fig. S5†).
Comparative evaluation of the methodology
The methods employed in this study assess the extent of the Li+/H+ exchange of NCM CAM powder upon washing, quantifying: (i) the amount of extracted lithium by titration; (ii) the amount of inserted protons by PGAA; (iii) the mass loss of the protonated, thermally unstable NCM phase by TGA-MS; and (iv) the thermally decomposed phase by XRD for the harshly washed sample. Even though the four methods illuminated xH+ by completely independent approaches, their results show good agreement, as presented in Fig. 8 and Table 6. In addition, the thickness of the protonated/lithium-depleted surface layer was determined by XPS. | ||
| Fig. 8 Summary of the obtained proton content xH+ for the two measurement series: (a) with a water-to-CAM mass ratio of 5:1 as a function of washing time, and (b) for a washing time of 1 h as a function of the water-to-CAM mass ratio. Note that the x-axes are plotted in a logarithmic representation. The data are extracted from Table 1 for the titration technique (green circles), from Table 2 for PGAA (yellow symbols), and from Table 3 for TGA-MS (gray symbols). | ||
| Method | Quantity | Units | as-received | 1h-1:1 | 1h-5:1 | 55h-25:1-80°C |
|---|---|---|---|---|---|---|
| Titration | LiOH concentration | mmol l−1 | — | 99 | 29 | 55 |
| proton content | mol% | — | 0.97 | 1.42 | 13.4 | |
| PGAA | proton-to-TM molar ratio | — | 0* | 0.9 | 1.7 | 25.5 |
| proton content | mol% | 0* | 0.9 | 1.7 | 25.5 | |
| TGA-MS | mass loss (at 450 °C) | wt% | 0.09 | 0.20 | 0.34 | 4.42 |
| proton content | mol% | 0.49 | 1.15 | 1.96 | 25.0 | |
| XRD | rock-salt content (at 450 °C) | mol% | 0* | — | — | 24.1 |
| proton content | mol% | 0* | — | — | 24.1 | |
| XPS | intensity ratio | — | — | 0.77 | 0.57 | <0.05 |
| protonated-layer thickness | nm | — | 0.7 | 1.5 | >8.1 |
For example, when studied by three independent techniques (i.e., titration, PGAA, and TGA-MS), the 1h-1:1 and 1h-5:1 washed NCM samples exhibit values for xH+ between 0.9 and 1.2 mol% as well as between 1.4 and 2.0 mol%, respectively. For the harshly washed 55h-25:1-80°C sample, the highest xH+ values between 24.1 and 25.5 mol% are observed, as presented in Table 6. Furthermore, the XPS-derived thickness of the essentially fully delithiated/protonated near-surface layer is 0.7 nm for 1h-1:1 and 1.5 nm for 1h-5:1. Since the Li+/H+ exchange is assumed to occur on various exposed crystal facets, for simplicity, a cubic NCM unit cell with an edge length of 3.2 Å is assumed (corresponding to the actual cell volume of 33.7 Å3 per LiMO2 unit75), as done in a previous work.16 For the 1h-1:1 and the 1h-5:1 samples, the values for the layer thickness equate to the exchange of approximately 2 and 5 monolayers, respectively. It has to be noted that, for the quantification by titration, xH+ of only 13.4 mol% is observed for 55h-25:1-80°C (see Table 6), which is also demonstrated in repetition measurements (data not shown); while the origin of this deviation remains unclear, it should be noted that this washing condition is much harsher than what is relevant for practical applications. For the samples washed at 25 °C, the titration-based xH+ values are in good agreement with those obtained by the other techniques (see Fig. 8). Note that a summary and comparison of the five methods regarding effort of experiment and analysis, accessibility, accuracy, required sample amount, advantages as well as disadvantages/limitations is presented in Table S3 of the ESI.†
Based on the measurements of the pH value of the washing solution as a function of time, previous studies have observed that the Li+/H+ exchange increases with washing time, whereby this increase appears to cease after usually 5 to 10 h, depending on the used CAM and the washing parameters.42,46,54,97 As the pH value is a logarithmic representation of the proton concentration in the washing solution, it is challenging to detect (not to mention quantify) smaller changes of the proton concentration reliably, especially for the typical, relatively high pH values of 11 to 13 during washing. Within the precision of the here used techniques, it is shown that the rate of the Li+/H+ exchange at room temperature decreases for extended washing times; however, the exchange reaction does not stop but continues for at least 25 h at an albeit decreasing rate, as depicted in Fig. 8a for a water-to-CAM mass ratio of 5:1. Independent of the measurement method, at a constant water-to-CAM mass ratio, xH+ appears to increase roughly linearly with the logarithm of time for practically relevant washing times t of up to several hours. As shown for the harshly washed 55h-25:1-80°C sample (see Table 6), the extent of the Li+/H+ exchange can be driven to relatively high values at the elevated washing temperature, which may be due to either faster Li+/H+-exchange kinetics or different exchange equilibrium conditions.
Phenomenologically, Fig. 8b shows that xH+ increases roughly linearly with the water-to-CAM mass ratio for a washing time of 1 h. Here, for a given extent of the Li+/H+ exchange, the increasing amount of water in the washing solution with increasing water-to-CAM mass ratio results in lower LiOH concentrations and thus lower solution pH, corresponding to a higher proton activity in the washing solution at any given xH+ value. This higher proton activity is apparently able to further drive the Li+/H+-exchange reaction, resulting in the apparent logarithmic relationship between xH+ and the water-to-CAM mass ratio. Thus, combining the observations from Fig. 8a and b, the relationship of the xH+ value obtained under practical washing conditions (i.e., up to several hours at room temperature) can be described by its logarithmic dependence on the water-to-CAM mass ratio and the washing time t:
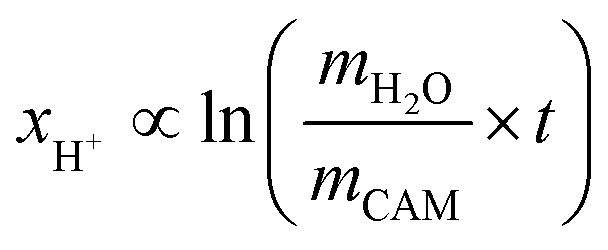 | (29) |
To extend this relation by other relevant washing parameters, future studies should also quantify the dependence of the Li+/H+-exchange kinetics on the applied washing temperature, the effect of other protic solvents than water (e.g., alcohols),43,44,79,98 the nickel content of the CAM, as well as the specific surface area of the CAM powder.
Conclusions
Five independent techniques were developed and employed to assess the extent of the Li+/H+ exchange of a nickel-rich cathode-active-material (CAM) powder upon washing in water (H2O), expressed in terms of the mole fraction of protons xH+. These independent methods allowed to reliably quantify: (i) the amount of extracted lithium by titration; (ii) the amount of inserted protons by prompt-gamma activation analysis (PGAA); (iii) the mass loss of the protonated, thermally unstable NCM phase by thermogravimetric analysis coupled with mass spectrometry (TGA-MS); (iv) the thermally decomposed phase by in situ and ex situ X-ray diffraction (XRD); and (v) the thickness of the protonated/lithium-depleted near-surface layer of NCM particles by X-ray photoelectron spectroscopy. Even though the five methods illuminated xH+ by completely independent approaches, their results show good agreement. Washing at room temperature under conditions relevant to application resulted in xH+ values between 0.7 and 3.6 mol%, depending on the washing parameters. This variation of the washing parameters exposed a linear dependence of xH+ on the logarithms of both the washing time and the water-to-CAM mass ratio.By washing at elevated temperature, a model material with a relatively large xH+ of approx. 25 mol% was prepared, as quantified by PGAA and TGA-MS. This enabled a reasonably accurate XRD analysis of the initially formed protonated phase and of the temperature-dependent formation of various phases that are produced upon heating of the protonated NCM material. A Rietveld refinement of the XRD data exposed that the protonation of the surface alters the lattice parameters a and c of the lithiated core. Upon heating of the washed NCM, both PGAA and TGA-MS indicated the removal of the ion-exchanged protons, and the effect on the lattice parameters was reversed. The thermal decomposition of the protonated phase (HMO2) initiates at 140 °C and is accompanied by the release of H2O only (i.e., without the release of O2) as well as the formation of an oxygen-depleted rock-salt-type surface layer (MO) and a partially delithiated layered structure (Li1−xMO2). Above 230 °C, the Li1−xMO2 phase is decomposed by the release of O2, resulting in an even thicker MO layer.
Using XPS, it was verified that the surface contaminants were successfully removed from the surface, even for the shortest washing time of 2 min. More importantly, however, it was shown that the ion-exchanged protons in the NCM phase do not mix with the intercalated lithium ions, i.e., they do not distribute homogeneously within the bulk but rather form a protonated near-surface layer. Finally, the key findings of this work are summarized graphically in Scheme 1.
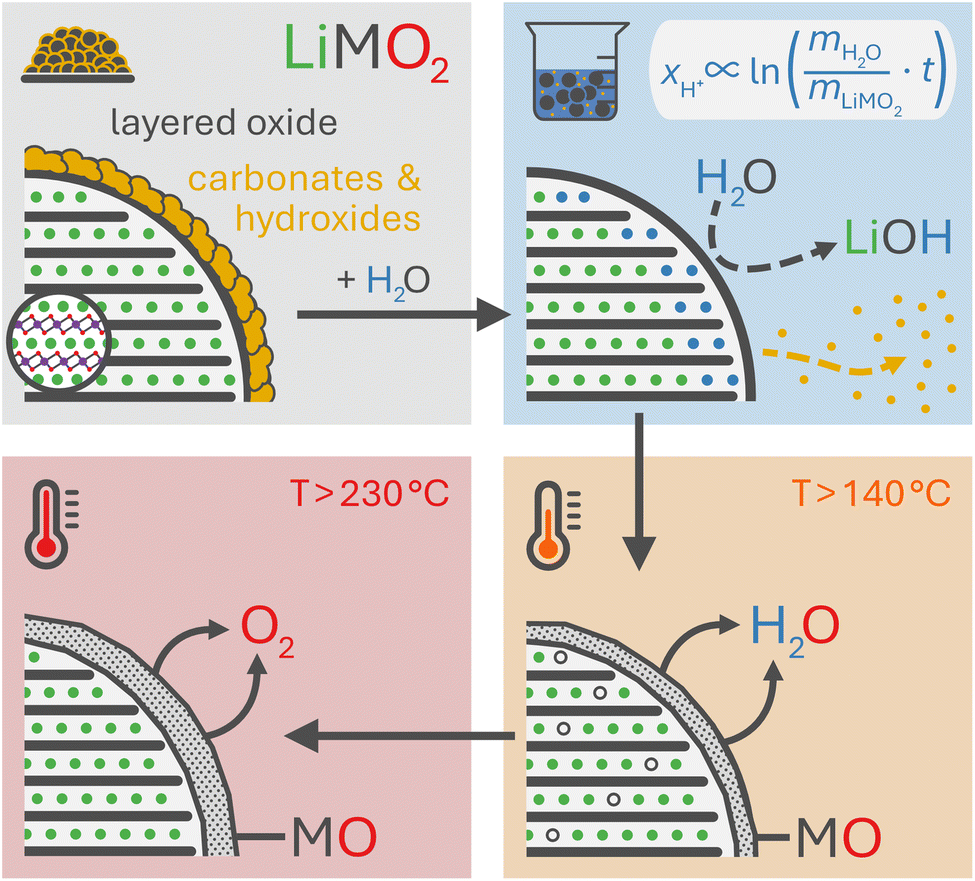 | ||
| Scheme 1 Graphical summary of the key findings of this work. The as-received NCM powder (top left) with its layered crystal structure also contains contaminants on its surface (yellow). Upon washing in H2O (top right), the surface contaminants are dissolved while simultaneously the lithium (green) in the NCM is exchanged by protons (blue), resulting in a protonated phase in the near-surface regions as well as LiOH in the washing solution. Phenomenologically, the proton content xH+ of the washed NCM depends on the water-to-CAM mass ratio and on the washing time in a logarithmic manner. Upon heating above 140 °C (bottom right), H2O is released from the structure of the washed NCM, leaving behind an oxygen-depleted surface layer and a partially delithiated layered phase (open circles indicate vacant lithium sites). Above 230 °C (bottom left), O2 (red) is released, decomposing the partially delithiated structure and forming an even thicker rock-salt-type phase in the near-surface regions. | ||
To elucidate the effect of the Li+/H+ exchange on the gas evolution and on the electrochemical performance of differently washed NCM831205 materials, on-line electrochemical mass spectrometry (OEMS) as well as cycling experiments in half- and full-cells of the here prepared protonated NCMs with a known proton content will be presented in the second part of this study.
Methods
Washing and sample preparation
Nickel-rich LiNi0.83Co0.12Mn0.05O2 (NCM831205, referred to as as-received, BASF SE, Germany, 0.62 mBET2 gNCM−1, see Fig. S1a and Table S1† for the particle morphology), which was not washed, coated, or doped in the synthesis, was mixed in a high-density-polyethylene (HDPE) bottle with deionized and degassed water (H2O, 18 MΩ cm−1, Merck MilliPore Milli-Q, USA, additionally degassed by bubbling nitrogen for at least 48 h), and stirred at 500 rpm using a magnetic stirring bar. To exclude the unwanted dissolution of carbon dioxide from ambient air and its effect on the carbonate concentration in the suspension of CAM and H2O, the gas atmosphere in the HDPE bottle was exchanged with argon before the bottle was closed and placed inside a closed argon-filled glass bottle (wide-mouth Duran bottle).In the first set of experiments, water-to-CAM mass ratios of 1:1, 5:1, 25:1, and 125:1 (H2O:CAM w/w) were chosen, while all suspensions were stirred at room temperature for 1 h. For example, for an water-to-CAM mass ratio of 1:1, 5 g of CAM were mixed with 5 ml of H2O. For 125:1, only 2 g of CAM were mixed with 250 ml of H2O to reduce the total volume of the suspension. For 0.2:1, 2 g of NCM were mixed with 0.4 g of H2O and 1.6 g of N-methyl-2-pyrrolidone (NMP, anhydrous, 99.5%, Sigma-Aldrich) to form a suspension with a processable viscosity while reducing the water-to-CAM mass ratio. In a second set of experiments, a suspension with a water-to-CAM mass ratio of 5:1 was stirred for 2, 12, 60, 300, or 1500 min (or 0.033, 0.2, 1, 5, or 25 h, respectively) at room temperature. To generate a highly protonated CAM, a suspension with a water-to-CAM mass ratio of 25:1 was stirred for 55 h at 80 °C, while a suspension of 1:1 was stirred for 2 min at 0 °C to remove surface contaminants but minimize the exchange of protons. After the washing, the washing solution was removed by vacuum filtration using a membrane filter (cellulose acetate, 0.2 μm pore size, Whatman, United Kingdom), collected in an HDPE bottle, which was filled with argon, and stored in an argon-filled glovebox (MBraun, Germany, <1 ppm O2 and H2O). All NCM powders were dried in a glass oven (Büchi, Switzerland) under dynamic vacuum at 65 °C for at least 6 h and transferred to the argon-filled glovebox without exposure to ambient air.
Additional subsequential heat treatments at 120, 180, 230, or 300 °C were conducted under dynamic vacuum in the Büchi glass oven for at least 6 h; furthermore, to obtain samples heat-treated at 450 °C, the NCM powders investigated by TGA-MS under argon atmosphere up to 450 °C (see below) were used. The chosen nomenclature for the washed and heat-treated NCM samples, e.g., 1h-1:1-[65°C], describes a sample washed in a suspension with a water-to-CAM mass ratio of 1:1 for 1 h at room temperature, while the drying temperature of 65 °C is shown in square brackets; consistently, the sample heated during washing is labeled by 55h-25:1-80°C-[65°C], while the one cooled during washing is labeled by 2min-1:1-0°C-[65°C]. Additionally, as-received NCM was also calcined in a tube furnace under oxygen atmosphere (1 l min−1) at 525 °C for 1 h before the sample was transferred to an argon-filled glovebox without exposure to ambient air (referred to as calcined).
Scanning electron microscopy (SEM)
The morphology of the NCM powder samples was investigated via top-view SEM using a MIRA3 FEG-SEM (TESCAN, Czech Republic) in secondary electron mode, applying an acceleration voltage of 5 kV.Surface-area analysis (Kr-BET)
The surface area of the NCM powders was determined by krypton-gas-physisorption measurements (also referred to as Kr-BET)99 at 77 K (adapted and reprinted here from previous publications7,14–16,19,20 for the sake of completeness), measuring at 13 points between 0.01 ≤ p/p0 ≤ 0.30, using an autosorb iQ (Quantachrome Instruments, USA). Prior to Kr-BET measurements, the NCM samples were dried at 120 °C under vacuum for 6 h.Titration
For the titration, 0.5 ml of the collected washing solutions were pipetted into a polyethylene beaker and diluted with 20 ml of H2O. The solution was stirred with a magnetic stirring bar and only exposed to ambient atmosphere for the relatively short duration of the measurement (of approx. 5 min). To obtain the titration curve, an acidic titrant solution of 10 mM hydrochloric acid (HCl, Sigma-Aldrich) in H2O was added stepwise with a resolution dVHCl of the added titrant volume of 0.1 ml using an automated titrator (TitroLine 7000, SI Analytics, Xylem Inc., USA) until a pH value of 3.5 was reached, recording the pH value as a function of time and added acid volume. The NCM sample washed with a water-to-CAM mass ratio of 0.2:1 was not titrated, as the washing solution consisted of H2O and NMP.Prompt-gamma activation analysis (PGAA)
The experiments were performed at the PGAA station of the Budapest Neutron Centre.100 Portions of ∼200 mg of NCM powder were weighed into 200 μl aluminum crucibles, closed with an aluminum lid, and heat-sealed into a polytetrafluoroethylene (PTFE) bag filled with nitrogen gas. These samples were irradiated with a white beam of cold neutrons (with a wavelength between 1 and 7 Å) at room temperature and under ambient conditions. The thermal equivalent neutron flux at the sample position was about 7.7 × 107 cm−2 s−1. The size of the neutron-collimator aperture was set to 20 mm × 20 mm; therefore, the entire volume of each sample was irradiated. The measurement times varied typically between 3 and 18 hours. The beam time was allocated considering the mass of the samples and the count rates of the lithium and hydrogen peaks, as, due to the higher analytical sensitivities, the peaks of nickel, cobalt, and manganese were always of adequate statistical precision (note that, by this strategy, a 1σ-uncertainty of 1.7 to 4.0% was achieved for all components of interest). The prompt-gamma spectra were recorded using an Ortec DSPEC 502A digital gamma spectrometer in 64k histogram channels between 45 keV and 12 MeV and in Compton-suppressed mode. The detector had been calibrated for efficiency and for non-linearity of the energy response using sealed radioactive sources and lines of the radiative neutron capture process of nitrogen and chlorine.The in-house software PeakFit was used to evaluate the spectra.101 This program fits asymmetric Gaussian-like peak-shape functions and a smooth baseline to the spectrum counts in selected regions, using the weighted nonlinear least-squares method. Special background and peak shape features of the PGAA spectra are also implemented. While the element hydrogen has only one line at 2223.3 keV, all other constituents have several analytical peaks in the PGAA spectrum. The element identification is based on matching the experimentally determined peak energies to entries in the nuclear data library of Révay et al.,59 while the formulae for the concentration calculation were presented by Révay.102 The nuclear data of each prompt gamma peak includes the natural abundance of the specific isotope, and natural isotopic composition is assumed for all the elements. The quantity of an element is obtained as the weighted average of masses from up to 25 analytical lines; this approach reduces the risk of a bias due to interferences and offers an inherent way to check the consistency of the reported results. The spectroscopic background, which originated from the structural and shielding materials of the facility, was corrected for.
Thermogravimetric analysis coupled with mass spectrometry (TGA-MS)
For TGA-MS, portions of roughly 100 mg of CAM powder were filled into an aluminum-oxide crucible (150 μl, calcined beforehand for 1 h at 1000 °C in oxygen), which was transferred to a TGA system (TGA/DSC 1, Mettler Toledo, Switzerland) with minimized air exposure. The TGA device was flushed using argon at a flowrate of 200 ml min−1 at room temperature for 10 min (data not shown) before the flowrate was reduced to 60 ml min−1. After an additional 10 min at 25 °C, the sample was heated from 25 °C to 120 °C at 10 K min−1, where it was held for 30 min, followed by an additional heating step at 10 K min−1 to 450 °C, where the temperature was also held constant for 30 min. Part of the evolving gases was diverted through a capillary and detected in a mass spectrometer (ThermoStar GSD320, Pfeiffer Vacuum, Germany). The monitored sample mass is normalized to the last measurement point of the temperature hold at 120 °C, while the ion currents of the evolved water (m/z = 18) and oxygen (m/z = 32) are normalized by the one of the argon-36 isotope (m/z = 36). To provide a baseline for the sample mass as well as the evolving gases, a measurement with only an empty crucible, i.e., without sample, was run separately with the same experimental conditions.In situ temperature-resolved X-ray powder diffraction (TR-XRD)
For in situ TR-XRD, variable-temperature measurements were performed on a STADI P Dual (STOE, Germany) using Mo-Kα1 radiation (λ = 0.7093 Å, Ge monochromator). The calcined NCM sample as well as the highly protonated one (55h-25:1-80°C-[65°C]) were filled into a quartz capillary (with a diameter of 1 mm), placed onto the sample stage of an INSITU HT furnace (STOE, Germany), and measured in Debye–Scherrer geometry with a Mo-Kα1 source over a 2θ range of angle of 6.0° and 48.0° in temperature steps of 5 K from 120 to 450 °C. Between each temperature step, a temperature ramp of 10 K min−1 was applied. Each spectrum was recorded after an equilibration time of 1 min at each temperature step while each temperature was held for 36 min in total.Ex situ X-ray powder diffraction (XRD)
For ex situ powder XRD, the NCM powder samples were filled into 0.3 mm glass capillaries (Hilgenberg, Germany), which were sealed in an argon-filled glovebox. The experiments were conducted at room temperature at a STADI P diffractometer (STOE, Germany) in transmission mode using Mo-Kα1 radiation (0.7093 Å, 50 kV, 40 mA) and a Mythen 1 K detector with one data point every 0.15° from 7° to 48°, resulting in a total acquisition time of roughly 45 min. For the determination of instrumental broadening, a silicon standard material (NIST SRM 640c) was used. Rietveld refinement was performed using the Topas software package;103 the fitting procedure is based on the publication of Friedrich and Strehle et al., where all relevant details on the Rietveld refinement are presented.14 During the refinement, the data was simultaneously fitted for both a layered oxide (LiMO2, with the space group Rm) as well as a rock-salt phase (MO, Fmm). In a similar manner, the fitting of LiMO2 in combination with either a corundum phase (M2O3) or a spinel phase (M3O4) was attempted, but neither of the two phases could be detected in any sample.
X-ray photoelectron spectroscopy (XPS)
For XPS measurements, the NCM powder samples were pressed into cavities of a stainless-steel stub in the argon-filled glovebox and transferred into the spectrometer (Axis Supra, Kratos, UK) without exposure to ambient air using a portable inert-transfer suitcase (Kratos, UK). All spectra were recorded at a pressure below 10−8 torr using monochromatized Al-Kα radiation with an energy of 1486.6 eV. The concentric hemispherical analyzer was operated at a constant pass energy of 40 eV for the core-level spectra. Since the powders were mounted conductively, no active charge neutralization was applied.The region of interest, containing the core levels of Ni 3p, Co 3p, Li 1s, and Mn 3p, was investigated in the binding-energy (BE) range between 35 and 85 eV using 10 sweeps with a step size of 0.1 eV and a dwell time of 1000 ms. Additionally, a survey spectrum (for BEs between 1202.5 and 0 eV with a step size of 0.5 eV, a dwell time of 100 ms, and a pass energy of 160 eV) as well as a spectrum of the regions containing the C 1s, O 1s, Co 2p, and Ni 2p core levels (with a step size of 0.1 eV and a dwell time of 200 ms, data not shown) were recorded using two sweeps, all without active charge neutralization. The energy scale of the spectra was corrected for sample charging by using the adventitious-carbon peak at a BE of 284.5 eV. After a Shirley background was subtracted, the core level spectra were deconvoluted by Voigt functions.
Data availability
The data supporting the findings of this study have been included as part of the ESI† and are also available from the authors upon reasonable request.Author contributions
Stefan Oswald: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, visualization, writing – original draft. Rebecca Wilhelm: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft. Tim Kratky: formal analysis, investigation, methodology, writing – original draft. László Szentmiklósi: formal analysis, investigation, resources, writing – review & editing. Boglárka Maróti: formal analysis, investigation, writing – review & editing. Ildikó Harsányi: investigation, writing – review & editing. Sebastian A. Hallweger: investigation, writing – review & editing. Gregor Kieslich: resources, writing – review & editing. Sebastian Günther: formal analysis, writing – review & editing. Hubert A. Gasteiger: conceptualization, funding acquisition, methodology, resources, writing – review & editing.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
Stefan Oswald and Rebecca Wilhelm gratefully acknowledge the BASF Battery Research Network and the BMBF (Federal Ministry for Education and Research, Germany) within the AQua-POp project (grant no. 03XP0329B) for their financial support. László Szentmiklósi and Boglárka Maróti thank Zsolt Révay (TUM/FRM II) for providing the latest version of the prompt-gamma spectroscopy library, containing refined data on lithium. The PGAA measurements at the Budapest Neutron Centre were part of Project No. RRF-2.3.1-21-2022-00009 (National Laboratory for Renewable Energy), which is implemented with the support provided by the Recovery and Resilience Facility of the European Union within the framework of Programme Széchenyi Plan Plus. Gregor Kieslich and Sebastian A. Hallweger acknowledge support from the DFG (project number 524525093 and 493871295). BASF SE (Germany) and Steve Zhang (BASF SE, Shanghai, China) are kindly acknowledged for providing the active materials. The authors acknowledge Clare P. Grey and the Yusuf Hamied Department of Chemistry at the University of Cambridge for the access to the scanning electron microscope. The authors also kindly thank Felix Riewald and Moritz Bock (both TUM/BASF SE), Corbinian Grön, Benjamin Strehle, and Michele Piana (all TUM) for fruitful discussions. The authors acknowledge Philip Rapp (TUM) for creating the scheme.References
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- J. U. Choi, N. Voronina, Y.-K. Sun and S.-T. Myung, Adv. Energy Mater., 2020, 10, 2002027 CrossRef CAS.
- J. Choi and A. Manthiram, J. Electrochem. Soc., 2005, 152, A1714–A1718 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- A. O. Kondrakov, H. Geßwein, K. Galdina, L. de Biasi, V. Meded, E. O. Filatova, G. Schumacher, W. Wenzel, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 24381–24388 CrossRef CAS.
- H. Li, A. Liu, N. Zhang, Y. Wang, S. Yin, H. Wu and J. R. Dahn, Chem. Mater., 2019, 31, 7574–7583 CrossRef CAS.
- S. Oswald, D. Pritzl, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 100511 CrossRef CAS.
- S. Oswald and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 030506 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS PubMed.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- A. T. S. Freiberg, M. K. Roos, J. Wandt, R. de Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828–8839 CrossRef CAS PubMed.
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS.
- R. Jung, P. Strobl, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A2869–A2879 CrossRef CAS.
- F. Friedrich, B. Strehle, A. T. S. Freiberg, K. Kleiner, S. J. Day, C. Erk, M. Piana and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A3760–A3774 CrossRef CAS.
- B. Strehle, F. Friedrich and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 050512 CrossRef CAS.
- S. Oswald, M. Bock and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 090505 CrossRef.
- J.-H. Kim, H.-H. Ryu, S. J. Kim, C. S. Yoon and Y.-K. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 30936–30942 CrossRef CAS PubMed.
- E. Trevisanello, R. Ruess, G. Conforto, F. H. Richter and J. Janek, Adv. Energy Mater., 2021, 11, 2003400 CrossRef CAS.
- S. Oswald, D. Pritzl, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 120501 CrossRef CAS.
- S. Oswald, M. Bock and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 050501 CrossRef CAS.
- S. Lee and A. Manthiram, ACS Energy Lett., 2022, 7, 3058–3063 CrossRef CAS.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- J. Sicklinger, M. Metzger, H. Beyer, D. Pritzl and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A2322–A2335 CrossRef CAS.
- S. T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- F. Schipper, E. M. Erickson, C. Erk, J.-Y. Shin, F. F. Chesneau and D. Aurbach, J. Electrochem. Soc., 2017, 164, A6220–A6228 CrossRef CAS.
- Y. Kim, ACS Appl. Mater. Interfaces, 2012, 4, 2329–2333 CrossRef CAS PubMed.
- Y. Zong, Z. Guo, T. Xu, C. Liu, Y. Li and G. Yang, Int. J. Energy Res., 2020, 44, 8532–8541 CrossRef CAS.
- J. Langdon and A. Manthiram, Energy Storage Mater., 2021, 37, 143–160 CrossRef.
- K. Shishino, T. Yamada, K. Fujisawa, M. Motoi, T. Hatakeyama and K. Teshima, ACS Appl. Energy Mater., 2022, 5, 2747–2757 CrossRef CAS.
- N. V. Faenza, L. Bruce, Z. W. Lebens-Higgins, I. Plitz, N. Pereira, L. F. J. Piper and G. G. Amatucci, J. Electrochem. Soc., 2017, 164, A3727–A3741 CrossRef CAS.
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. T. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489–A1498 CrossRef CAS.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- A. C. Martinez, S. Grugeon, D. Cailleu, M. Courty, P. Tran-Van, B. Delobel and S. Laruelle, J. Power Sources, 2020, 468, 228204 CrossRef CAS.
- L. Hartmann, D. Pritzl, H. Beyer and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 070507 CrossRef CAS.
- G. J. Ross, J. F. Watts, M. P. Hill and P. Morrissey, Polymer, 2000, 41, 1685–1696 CrossRef CAS.
- W. M. Seong, K.-H. Cho, J. Park, H. Park, D. Eum, M. H. Lee, I.-S. S. Kim, J. Lim and K. Kang, Angew. Chem., Int. Ed., 2020, 59, 18662–18669 CrossRef CAS PubMed.
- Y. Bi, Q. Li, R. Yi and J. Xiao, J. Electrochem. Soc., 2022, 169, 020521 CrossRef CAS.
- E. Deltombe and M. Pourbaix, Corrosion, 1958, 14, 16–20 CrossRef.
- S. E. Renfrew and B. D. McCloskey, J. Am. Chem. Soc., 2017, 139, 17853–17860 CrossRef CAS PubMed.
- A. T. S. Freiberg, J. Sicklinger, S. Solchenbach and H. A. Gasteiger, Electrochim. Acta, 2020, 346, 136271 CrossRef CAS.
- R. Moshtev, P. Zlatilova, S. Vasilev, I. Bakalova and A. Kozawa, J. Power Sources, 1999, 81–82, 434–441 CrossRef CAS.
- J. Kim, Y. Hong, K. S. Ryu, M. G. Kim and J. Cho, Electrochem. Solid-State Lett., 2006, 9, A19–A23 CrossRef CAS.
- D. Weber, Đ. Tripković, K. Kretschmer, M. Bianchini and T. Brezesinski, Eur. J. Inorg. Chem., 2020, 2020, 3117–3130 CrossRef CAS.
- Y. Zhou, Z. Hu, Y. Huang, Y. Wu and Z. Hong, J. Alloys Compd., 2021, 888, 161584 CrossRef CAS.
- J.-H. Park, B. Choi, Y.-S. Kang, S. Y. Park, D. J. Yun, I. Park, J. H. Shim, J.-H. Park, H. N. Han and K. Park, Energy Technol., 2018, 6, 1361–1369 CrossRef CAS.
- D. Pritzl, T. Teufl, A. T. S. Freiberg, B. Strehle, J. Sicklinger, H. Sommer, P. Hartmann and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A4056–A4066 CrossRef CAS.
- Y. Kato, A. Nagahara, N. Gerile, S. Fujinaka, N. Hamamoto, H. Nishimura and H. Nakai, J. Electrochem. Soc., 2022, 169, 060543 CrossRef CAS.
- M. Hofmann, M. Kapuschinski, U. Guntow and G. A. Giffin, J. Electrochem. Soc., 2020, 167, 140512 CrossRef CAS.
- M. Wood, J. Li, R. E. Ruther, Z. Du, E. C. Self, H. M. Meyer, C. Daniel, I. Belharouak and D. L. Wood, Energy Storage Mater., 2020, 24, 188–197 CrossRef.
- S.-M. Bak, E. Hu, Y. Zhou, X. Yu, S. D. Senanayake, S.-J. Cho, K.-B. Kim, K. Y. Chung, X.-Q. Yang and K.-W. Nam, ACS Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef CAS PubMed.
- S. Qian, A. T. S. Freiberg, F. Friedrich and H. A. Gasteiger, in preparation.
- R. B. Warder, Chem. News, 1881, 43, 228 Search PubMed.
- A. A. Benedetti-Pichler, M. Cefola and B. Waldman, Ind. Eng. Chem., Anal. Ed., 1939, 11, 327–332 CrossRef CAS.
- I. Hamam, N. Zhang, A. Liu, M. B. Johnson and J. R. Dahn, J. Electrochem. Soc., 2020, 167, 130521 CrossRef CAS.
- W. M. Seong, Y. Kim and A. Manthiram, Chem. Mater., 2020, 32, 9479–9489 CrossRef CAS.
- M. Toledo, Titration Appl. Note, M828 v1.0 Search PubMed.
- R. L. Paul, Analyst, 1997, 122, 35R–41R RSC.
- Z. Révay and T. Belgya, in Handbook of Prompt Gamma Activation Analysis with Neutron Beams, ed. G. L. Molnár, Springer, New York, 2004, pp. 1–30 Search PubMed.
- Z. Révay, R. B. Firestone, T. Belgya and G. L. Molnár, in Handbook of Prompt Gamma Activation Analysis with Neutron Beams, ed. G. L. Molnár, Springer, New York, 2004, pp. 173–364 Search PubMed.
- S. K. Aghara, S. Venkatraman, A. Manthiram and E. Alvarez, J. Radioanal. Nucl. Chem., 2005, 265, 321–328 CrossRef CAS.
- J. Choi, E. Alvarez, T. A. Arunkumar and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A241–A244 CrossRef CAS.
- K.-K. Lee and K.-B. Kim, J. Electrochem. Soc., 2000, 147, 1709–1717 CrossRef CAS.
- D. D. MacNeil, Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1332–A1336 CrossRef CAS.
- J. Sicklinger, H. Beyer, L. Hartmann, F. Riewald, C. Sedlmeier and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 130507 CrossRef CAS.
- H. Beyer, S. Meini, N. Tsiouvaras, M. Piana and H. A. Gasteiger, Phys. Chem. Chem. Phys., 2013, 15, 11025–11037 RSC.
- A. Van der Ven, D. Morgan, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2006, 153, A210 CrossRef CAS.
- X. Z. Fu, X. Wang, Q. C. Xu, J. Li, J. Q. Xu, J. D. Lin and D. W. Liao, Electrochim. Acta, 2007, 52, 2109–2115 CrossRef CAS.
- J. Zhao, W. Zhang, A. Huq, S. T. Misture, B. Zhang, S. Guo, L. Wu, Y. Zhu, Z. Chen, K. Amine, F. Pan, J. Bai and F. Wang, Adv. Energy Mater., 2017, 7, 1601266 CrossRef.
- M. Bianchini, F. Fauth, P. Hartmann, T. Brezesinski and J. Janek, J. Mater. Chem. A, 2020, 8, 1808–1820 RSC.
- W.-N. Wang, D. Meng, G. Qian, S. Xie, Y. Shen, L. Chen, X. Li, Q. Rao, H. Che, J. Liu, Y.-S. He, Z.-F. Ma and L. Li, J. Phys. Chem. C, 2020, 124, 27937–27945 CrossRef CAS.
- S. Albrecht, J. Kümpers, M. Kruft, S. Malcus, C. Vogler, M. Wahl and M. Wohlfahrt-Mehrens, J. Power Sources, 2003, 119–121, 178–183 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- M. H. Rossouw and M. M. Thackeray, Mater. Res. Bull., 1991, 26, 463–473 CrossRef CAS.
- N. M. Vuong, N. M. Hieu, H. N. Hieu, H. Yi, D. Kim, Y. S. Han and M. Kim, Sens. Actuators, B, 2014, 192, 327–333 CrossRef.
- L. de Biasi, A. O. Kondrakov, H. Geßwein, T. Brezesinski, P. Hartmann and J. Janek, J. Phys. Chem. C, 2017, 121, 26163–26171 CrossRef CAS.
- Z. Chen, J. Wang, D. Chao, T. Baikie, L. Bai, S. Chen, Y. Zhao, T. C. Sum, J. Lin and Z. Shen, Sci. Rep., 2016, 6, 25771 CrossRef CAS PubMed.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef CAS PubMed.
- S. Radloff, R. G. Scurtu, G. Carbonari, M. Hölzle, T. Diemant, M. Bozorgchenani, F. Klein and M. Wohlfahrt-Mehrens, J. Power Sources, 2023, 580, 233314 CrossRef CAS.
- L. Azhari, X. Zhou, B. Sousa, Z. Yang, G. Gao and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 57963–57974 CrossRef CAS PubMed.
- W. Lee, S. Lee, E. Lee, M. Choi, R. Thangavel, Y. Lee and W.-S. Yoon, Energy Storage Mater., 2022, 44, 441–451 CrossRef.
- S. Xu, X. Wang, W. Zhang, K. Xu, X. Zhou, Y. Zhang, H. Wang and J. Zhao, Solid State Ionics, 2019, 334, 105–110 CrossRef CAS.
- Y.-K. Sun, D. J. Lee, Y. J. Lee, Z. Chen and S.-T. Myung, ACS Appl. Mater. Interfaces, 2013, 5, 11434–11440 CrossRef CAS PubMed.
- F. Fiévet, P. Germi, F. de Bergevin and M. Figlarz, J. Appl. Crystallogr., 1979, 12, 387–394 CrossRef.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- Y. Iida, J. Am. Ceram. Soc., 1960, 43, 225 CrossRef CAS.
- A. N. Mansour and C. A. Melendres, Surf. Sci. Spectra, 1994, 3, 263–270 CrossRef CAS.
- F. U. Hillebrecht, J. C. Fuggle, P. A. Bennett and Z. Zolnierek, Phys. Rev. B, 1983, 27, 2179–2193 CrossRef CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 279–286 CrossRef CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 231–238 CrossRef CAS.
- A. N. Mansour and C. A. Melendres, Surf. Sci. Spectra, 1994, 3, 271–278 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical Surface Analysis: Auger and X-Ray Photoelectron Spectroscopy, Wiley, New York, 1990 Search PubMed.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 2–11 CrossRef CAS.
- H. P. C. E. Kuipers, H. C. E. van Leuven and W. M. Visser, Surf. Interface Anal., 1986, 8, 262 CrossRef.
- E. L. Haase, Thin Solid Films, 1985, 124, 283–291 CrossRef CAS.
- C. Kisielowski, J. Krüger, S. Ruvimov, T. Suski, J. W. Ager, E. Jones, Z. Liliental-Weber, M. Rubin, E. R. Weber, M. D. Bremser and R. F. Davis, Phys. Rev. B, 1996, 54, 17745–17753 CrossRef CAS PubMed.
- H. Schuler, S. Klimm, G. Weissmann, C. Renner and S. Horn, Thin Solid Films, 1997, 299, 119–124 CrossRef CAS.
- X. Xiong, Z. Wang, P. Yue, H. Guo, F. Wu, J. Wang and X. Li, J. Power Sources, 2013, 222, 318–325 CrossRef CAS.
- R. Fantin, E. Trevisanello, R. Ruess, A. Pokle, G. Conforto, F. H. Richter, K. Volz and J. Janek, Chem. Mater., 2021, 33, 2624–2634 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- L. Szentmiklósi, T. Belgya, Z. Révay and Z. Kis, J. Radioanal. Nucl. Chem., 2010, 286, 501–505 CrossRef.
- L. Szentmiklósi, J. Radioanal. Nucl. Chem., 2018, 315, 663–670 CrossRef.
- Z. Révay, Anal. Chem., 2009, 81, 6851–6859 CrossRef PubMed.
- A. Coelho, TOPAS-academic V6, Coelho Software, Brisbane, Australia, 2015 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03319a |
| ‡ Equal contribution. |
| § Current affiliation: Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, United Kingdom. |
| ¶ Current affiliation: HUN-REN Wigner Research Centre for Physics, Budapest, Hungary. |
| This journal is © The Royal Society of Chemistry 2024 |