
Scalable, low-cost synthesis of high volumetric capacity LiMn0.5Fe0.5PO4 cathode for lithium-ion batteries†
Seth
Reed
,
Kevin
Scanlan
and
Arumugam
Manthiram
 *
*
Materials Science and Engineering Program, Texas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: manth@austin.utexas.edu
First published on 16th July 2024
Abstract
LiMnxFe1−xPO4 (LMFP) cathodes in lithium-ion batteries (LIBs) can improve the energy density by up to 15% compared to LiFePO4. However, the smaller primary particle size needed to overcome sluggish Mn2+/3+ redox kinetics results in lower electrode density, limiting the practical energy density of LMFP cells. We present here a scalable, wastewater-free synthesis of high energy density LMFP cathodes by employing a spinel LiMnFeO4 precursor obtained by a facile solid-state reaction. The LMFP synthesized with LiMnFeO4, referred to as S-LMFP, has uniform secondary particles formed via spray drying and greater tap density than LMFP synthesized with a conventional co-precipitation route, referred to as P-LMFP. While S-LMFP offers a lower specific capacity (147 mA h g−1) than P-LMFP (153 mA h g−1) at C/5 rate due to inferior Li+ diffusion kinetics during Mn2+/3+ redox, it exhibits a ∼15% higher electrode-level energy density due to its exceptional electrode packing density (2.6 g cm−3). Rate capability testing of LMFP cathodes with different electrode densities reveals that at low rates (<1C), electrode density is more important than specific capacity in determining the energy density of LMFP cathodes. Overall, this work highlights the complex tradeoff between Mn2+/3+ redox kinetics and electrode density, which dictates the practical energy density of LMFP cathodes. With further optimization of composition, particle size, and synthesis conditions, LMFP synthesized from LiMnFeO4 is promising to improve the energy density of the next generation of LIBs.
Introduction
The projected exponential increase in electric vehicle (EV) production over the next decade will require a commensurate increase in the production of Li-ion batteries (LIB) that power them. To ensure the cost competitiveness of EVs, a significant reduction in the cost of LIBs (i.e., dollars per kW h) is needed, which demands both further improvement in energy density and reduction in materials cost.1–3 LiNixMnyCozO2 (NMC) has been the dominant cathode chemistry in EV batteries due to its superior energy density, but its dependence on critical minerals – namely, nickel and cobalt – presents serious issues for cost, sustainability, and supply chain security.4–7 Due to recent advances in cell and pack design, LiFePO4 (LFP) has emerged as a leading cathode material for a sustainable production of low-cost LIBs.8,9 At approximately one third of the material cost of NMC due to the use of abundant and inexpensive iron and phosphorus, LFP can mitigate the economic and supply chain risks associated with NMC cathodes.10,11 LFP also has remarkable electrochemical and thermal stability, resulting in LIB cells with much better safety and lifetime than NMC-based cells.12,13 However, with a maximum practical capacity of 160 mA h g−1 at an average discharge voltage of 3.4 V vs. Li/Li+, LFP offers much lower specific energy compared to NMC.5,12,13 Additionally, due to the lower bulk density of LFP along with the sub-micron particle size required to achieve full capacity, the maximum packing density of LFP electrodes (e.g., <2.5 g cm−3) is much lower than that of NMC (e.g., >3.4 g cm−3), which further reduces practical energy density. To increase the energy density of LFP, Mn can be substituted for Fe in the olivine structure to form LiMnxFe1−xPO4 (LMFP).8,14–19 Since the Mn2+/3+ redox in LMFP occurs at a higher voltage of 4.0 V vs. Li/Li+, the average operating voltage – and thus energy density – can theoretically be increased by about 15% compared to LFP.8,14However, once the Mn content in LMFP surpasses 40%, kinetic limitations arise during Mn2+/3+ redox, which can limit the accessible capacity and decrease the average discharge voltage.14,19 The root cause of these kinetic limitations is the Jann–Teller distortion caused by the high-spin 3d4 electronic configuration of Mn3+. This phenomenon results in an anisotropic lattice mismatch between the lithiated (Mn2+ containing) and delithiated (Mn3+ containing) phases which hinders the Li+ diffusion kinetics along the phase boundary.18–20 For example, the apparent Li+ diffusion coefficient calculated from the Atlung Method for Intercalant Diffusion was shown to be orders of magnitude higher during Fe2+/3+ redox compared to Mn2+/3+ redox for LMFP with 50% Mn content.17 These kinetic issues can be alleviated through various strategies, but each approach has detrimental effects towards energy density. Inclusion of electrochemically inactive dopants, such as Mg, can mitigate the lattice strain and improve the Li+ diffusion kinetics along both the phase boundary and particle surface but with a penalty to theoretical specific capacity.14,21 Reducing the primary particle size of LMFP (i.e., to <100 nm) decreases the Li+ diffusion path length and greatly improves the Mn2+/3+ redox kinetics.14,15 However, excessively small particle size typically results in a low press density of the electrode, which reduces the cell-level energy density. For example, the tap density of high-Mn LMFP powders is often <1.0 g cm−3, which results in an electrode press density of <2.0 g cm−3, while the tap density of LFP can be >1.5 g cm−3, yielding a press density of >2.5 g cm−3.21–23 As the Mn content in LMFP is increased past 60%, the kinetic limitations of Mn2+/3+ redox become severe, requiring large amounts of inactive dopants (e.g., 5%) combined with very small primary particle sizes (e.g., 50 nm) to achieve full theoretical capacity.17,18 As a result, it is highly challenging for LMFP cathodes to achieve more than a 10% improvement in cell-level volumetric energy density compared to LFP.
For LMFP to achieve a reduced cost per kW h at the cell-level compared to LFP, the cost of the LMFP powder must be equivalent to that of LFP. While the raw material costs of Mn and Fe are similar, the more complex methods often employed to synthesize high-performance LMFP (e.g., hydrothermal) can add significant processing costs. Generally, commercial LFP is manufactured by one of two methods: (i) a precipitation reaction of FeSO4, H3PO4, and H2O2 to form FePO4, followed by solid-state reaction with Li2CO3, or (ii) a direct reaction of Li2CO3 and H3PO4 with a Fe source such as Fe2O3.24,25 Spray drying is often employed, either before or after the synthesis of the olivine phase, to densely agglomerate the primary particles into spherical secondary particles, which significantly improves the tap density.25 Due to the low synthesis temperature (i.e., 650 °C) and short time (i.e., <2 h) required to prevent sintering of the LMFP primary particles, intimate mixing of Mn and Fe at the atomic/molecular level in the LMFP precursor is necessary. If not, there is the possibility of the formation of Mn-rich particles which would experience more severe kinetic limitations. Co-precipitation of a phosphate precursor, such as NH4MnxFe1−xPO4·H2O, is a viable route to ensure atomic level mixing of Mn and Fe and can produce LMFP cathodes with high specific capacity.21,26–29 However, co-precipitation synthesis produces large amounts of wastewater, which is expensive and energy intensive to treat, and typically results in LMFP powders with low tap density. The direct synthesis of LFP with Fe2O3 has been shown to yield very high density LFP electrodes while avoiding waste generation, but such an approach has not been studied widely for LMFP. This is because achieving a sufficiently intimate atomic scale mixing of separate oxide precursors (e.g., Mn2O3 and Fe2O3) by scalable processes, such as ball milling, is extremely challenging.
In this work, LiMnFeO4 (LMFO), a spinel structure in which Mn and Fe are mixed at an atomic level, is employed as a precursor for the synthesis of LiMn0.5Fe0.5PO4 cathode, which is hereafter referred to as S-LMFP. The material properties and electrochemical performance of S-LMFP are compared with those of a LMFP cathode with the same composition that is synthesized with a conventional co-precipitation method, which is hereafter referred to as P-LMFP. Through rate performance testing of LMFP electrodes with various press densities, the maximum achievable electrode-level volumetric energy density of the two materials is compared. It is shown that despite its lower specific capacity, S-LMFP greatly outperforms P-LMFP in volumetric energy density due to its much higher achievable electrode density. By electrochemical testing at different temperatures, the lower capacity of S-LMFP is found to be a result of sluggish Li+ diffusion kinetics during Mn2+/3+ redox, which are caused by the larger primary particle size. Overall, it is suggested that achieving a maximum energy density with LMFP cathodes requires a careful balancing between the electrode density and Mn2+/3+ redox kinetics through a careful selection of precursors and optimization of synthesis conditions.
Experimental
Materials preparation
To synthesize the LMFO precursor, first 21.60 g of Fe2O3 and 23.52 g of MnO2 (both 98%, Thermo Scientific) were mixed with approximately 125 mL of deionized water in a 250 mL high-density polyethylene bottle, and the mixture was milled on a roller jar mill (U.S. Stoneware) with 3–10 mm zirconia media for 72 h. After drying the milled mixture for 12 h at 110 °C, a stoichiometric amount of LiOH·H2O (99.9%, FMC Corporation) was mixed with the metal oxides by grinding with a mortar and pestle to give nominal compositions ranging from Li0.90MnFeO4 to Li1.10MnFeO4. A composition of Li0.95MnFeO4 was used for the synthesis of LMFP. The mixture was calcined in a box furnace at 500 °C for 3 h, followed by 900 °C for 6 h, with a heating rate of 5 °C per min.The phosphate precursor, NH4Mn0.5Fe0.5PO4·H2O, was synthesized by a co-precipitation reaction. The metal-ion solution (250 mL) contained 0.50 M FeSO4·7H2O (98%, Thermo Scientific), 0.50 M MnSO4·H2O (99+%, Thermo Scientific), and 1.00 M H3PO4 (ACS reagent, Sigma-Aldrich). The base solution (100 mL) contained 5.00 M NH4OH (Certified ACS Plus, Fisher Chemical). A 2 L reaction vessel was initially filled with 350 mL of a solution containing 0.07 M H3PO4 and 0.07 M NH4OH. The metal-ion and base solutions were added dropwise to the reaction vessel with a peristaltic pump at flow rates of, respectively, 3.63 and 1.43 mL min−1. A temperature of 80 °C, a pH of 8, and continuous stirring at 450 rpm were maintained during the reaction. After cooling, the precipitated product was filtered, rinsed thoroughly with deionized water, and dried overnight at 110 °C under vacuum.
Three separate LMFP samples with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 molar ratio of Mn:Fe were prepared for this study: LMFP from LMFO (S-LMFP), LMFP from NH4Mn0.5Fe0.5PO4·H2O (P-LMFP), and LMFP from MnO2 and Fe2O3 (O-LMFP). Each sample had a nominal composition of Li1.06Mn0.475Fe0.475V0.02Mg0.02Co0.01(PO4)1.025 and a nominal carbon content of ∼3 wt%. A two-step carbon coating synthesis process was used for all three LMFP samples.21 Specifically, Fig. S-1† depicts this process for S-LMFP synthesis. For this process, LMFO was combined with Li3PO4 (Thermo Scientific, extra pure) or Li2CO3 (Sigma-Aldrich, ACS reagent), NH4H2PO4 (Sigma-Aldrich, 98.5%), CoC2O4·2H2O (Thermo Scientific, reagent grade), MgC2O4·2H2O (Alfa Aesar, 98.5%), and NH4VO3 (Acros Organics, 98%). 50% of the total carbon in the synthesis was added during the first milling step in the form of a 1:1 weight ratio between citric acid monohydrate (Sigma-Aldrich, >99.0%) and glucose (MP Biomedicals, molecular biology grade).
:50 molar ratio of Mn:Fe were prepared for this study: LMFP from LMFO (S-LMFP), LMFP from NH4Mn0.5Fe0.5PO4·H2O (P-LMFP), and LMFP from MnO2 and Fe2O3 (O-LMFP). Each sample had a nominal composition of Li1.06Mn0.475Fe0.475V0.02Mg0.02Co0.01(PO4)1.025 and a nominal carbon content of ∼3 wt%. A two-step carbon coating synthesis process was used for all three LMFP samples.21 Specifically, Fig. S-1† depicts this process for S-LMFP synthesis. For this process, LMFO was combined with Li3PO4 (Thermo Scientific, extra pure) or Li2CO3 (Sigma-Aldrich, ACS reagent), NH4H2PO4 (Sigma-Aldrich, 98.5%), CoC2O4·2H2O (Thermo Scientific, reagent grade), MgC2O4·2H2O (Alfa Aesar, 98.5%), and NH4VO3 (Acros Organics, 98%). 50% of the total carbon in the synthesis was added during the first milling step in the form of a 1:1 weight ratio between citric acid monohydrate (Sigma-Aldrich, >99.0%) and glucose (MP Biomedicals, molecular biology grade).
For S-LMFP, 0.060 mol of LMFO was combined with 0.024 mol of Li3PO4, 0.104 mol of NH4H2PO4, 1.25 mmol. of CoC2O4·2H2O, 2.50 mmol of MgC2O4·2H2O, 2.50 mmol of NH4VO3, 1.44 g of citric acid, 0.88 g of glucose, and 13 mL of deionized water. For P-LMFP, 0.057 mol of NH4Mn0.5Fe0.5PO4·H2O was combined with 0.032 mol of Li2CO3, 4.23 mmol of NH4H2PO4, 0.60 mmol of CoC2O4·2H2O, 1.21 mmol of MgC2O4·2H2O, 1.21 mmol of NH4VO3, 0.55 g of citric acid, 0.33 g of glucose, and 14 g of deionized H2O. For O-LMFP, 0.028 mol of Fe2O3 and 0.056 mol of MnO2 were combined with 0.042 mol of Li3PO4, 0.040 mol of NH4H2PO4, 1.19 mmol of CoC2O4·2H2O, 2.38 mmol of MgC2O4·2H2O, 2.38 mmol of NH4VO3, 1.37 g of citric acid, 0.84 g of glucose, and 13 mL of deionized water.
The precursors and carbon sources were milled in solution in an 80 mL stainless steel ball mill jar with 70 g of 5 mm diameter stainless steel media using a Fritsch Pulverisette 6 planetary ball mill for 6 h at 450 rpm. The viscosity of this solution was high to ensure effective milling and a uniform distribution of precursors. The milled product was dried under vacuum at 110 °C to evaporate all the water and then calcined at 550 °C for 3 h under Ar flow to produce an intermediate product. This powder was combined with the remaining 50% of carbon in the form of lactose (ChemCenter, reagent grade) with a mass of 1.85 g, 0.70 g, and 1.76 g, respectively, for S-LMFP, P-LMFP, and O-LMFP. 5.25 mg carboxymethyl cellulose (Nippon Paper, Sunrose MAC Series) was added to the solution as a dispersant. After milling for 1 h at 450 rpm in deionized water, the resultant solution was fed into a Labfreez benchtop spray dryer at a rate of 0.63 mL min−1 and a solid loading of 52%. The spray gun was pressurized to 0.15 MPa with air, and the air flow rate through the spray dryer was set at 18 scfm. The inlet temperature was maintained at 130 °C, and the resultant exhaust temperature remained around 65 °C. The collected spray dried product was calcined at 650 °C for 2 h under Ar flow to form LMFP.
Materials characterization
Structural properties of the various LMFO and LMFP samples were examined with X-ray diffraction (XRD) with Cu Kα radiation (Rigaku Miniflex). XRD was conducted at a scan rate of 2° per min. Rietveld refinement analysis was conducted with Rigaku PDXL integrated powder XRD software. The morphologies of the spray dried LMFP samples were examined with scanning electron microscopy (SEM). SEM was paired with energy dispersive X-ray spectroscopy (EDS) to examine the metal-ion distribution in the precursors, and SEM-EDS images were captured on a Tescan Vega3 SEM. SEM-EDS was conducted at a beam voltage of 10 kV. SEM was also paired with a focused ion beam (FIB) to investigate the secondary particle pore structure of the calcined LMFP samples, and the FIB-SEM images were captured on a Scios 2 DualBeam FIB SEM. FIB-SEM was conducted at a beam voltage of 2 kV. Tap density measurement was conducted with a Quantachrome Autotap Tap Density Analyzer. Approximately 7 g of LMFP was massed into a 10 mL graduated cylinder and allowed to tap for 1 h at a rate of 3 taps per min. Final volume measurements were taken with ±0.01 cm3 precision. Thermogravimetric analysis (TGA) was conducted with a Netzsch STA 449 F3 Jupiter Thermal Analyzer. The LMFP samples were heated to 700 °C at a heating rate of 5 °C min−1 under air flow and held at 700 °C for 30 min to obtain a steady-state value at this final temperature. The carbon content was measured assuming a 2.5 wt% increase in weight from the oxidation of LMFP. Brunauer–Emmett–Teller (BET) surface area analysis based on nitrogen physisorption isotherms was performed at 77 K with a Micromeritics TriStar II Plus. The LMFP samples were dried under vacuum overnight at 120 °C prior to BET measurements.Electrochemical characterization
All the LMFP samples were formed into cathodes via a slurry casting method. LMFP was dispersed in N-methyl-2-pyrrolidone (Sigma-Aldrich, 99.5%) with a conductive carbon mixture of Super C65 (Timcal) and vapor-grown carbon fibres (Resonac) in a 4:1 ratio by weight and polyvinylidene fluoride (Kynar HSV1800) binder. The compositions of the active material, conductive carbon, and binder were, respectively, 95:2.5:2.5 by weight. Slurry mixing was carried out in an ARV-310 Thinky mixer. The slurry was cast on to a carbon-coated Al foil at an aerial loading of 1.0–1.6 mA h cm−2 depending on the test conducted on the cathode. After casting, the cathode was dried at 110 °C to evaporate all the solvent and hot calendared at 110 °C between stainless steel shims to a specified press density. The cathodes were then stored under vacuum at 110 °C overnight to completely dry the electrode.
The LMFP cathodes prepared were punched into 1/2 in (1.26 cm) diameter discs and tested in 2032-type coin cells. Assembly of the coin cell was carried out inside an argon-filled glovebox. Half cells were composed with LMFP as the cathode, 50 μL of LP57 electrolyte with 2% vinylene carbonate by weight, 3/4 in diameter Celgard 2325 separator, and a 5/8 in diameter lithium-metal anode. The LP57 electrolyte refers to 1.0 M LiPF6 in ethylene carbonate (EC)/ethyl methyl carbonate (EMC) (3:7 by weight). LMFP half cells were cycled from 2.0 to 4.3 V vs. Li/Li+ at a C/5 rate. A constant current/constant voltage charge procedure was used; C/5 rate current was applied until 4.3 V was reached, followed by a voltage hold at 4.3 V until the current dropped below C/25 rate. The discharge procedure did not include a constant voltage discharge step. Discharge rate testing was conducted with a C/10 charge rate, followed by the specified discharge rate. All cycling and rate testing were conducted on an Arbin LBT-20084.
Electrochemical impedance spectroscopy (EIS) was conducted with a VMP3 Biologic potentiostat. A symmetric cell was built with pristine LMFP as both the cathode and anode (5/8 in diameter electrodes). The cell was flooded with 200 μL of a blocking electrolyte (BE): 1 M tetraethylammonium tetrafluoroborate (Sigma-Aldrich, 99%) in acetonitrile (Fisher Chemical, ACS grade). This electrolyte is commonly used in electric double-layer capacitors, where no chemical reactions occur.30 Therefore, BE does not allow intercalation of Li+ ions into either electrode, leaving contact resistance as the sole variable investigated. In this study, contact resistance refers to the resistance between the active material and the current collector, conductive carbon matrix/electronic pathway in the cathode, and/or other active material particles. EIS was operated within the frequency range of 500 kHz to 100 mHz.
Results and discussion
Initially, multiple oxide phases that contain both Fe and Mn were tested as precursors for high-density LMFP. A rock salt phase, Fe0.5Mn0.5O, was successfully synthesized by heating a mixture of Mn2O3, Fe2O3, and activated carbon under Ar flow at 800 °C.31 However, when mixed with an aqueous solution of NH4H2PO4, the Fe0.5Mn0.5O powder immediately reacted to form a viscous paste (Fig. S-2†). Mn2+ dissolution from Fe0.5Mn0.5O occurs rapidly under the mildly acidic pH (∼4.5) of the NH4H2PO4 solution and the dissolved Mn2+ reacts with oxygen to form MnOOH and Mn3O4.32,33 The costly synthesis conditions of Fe0.5Mn0.5O, combined with this unfavorable reaction during mixing make this phase unsuitable as a precursor for scalable synthesis of LMFP. Two other ternary Mn/Fe oxides were also investigated as precursors: spinel Fe1.5Mn1.5O4 and ilmenite FeMnO3. The Fe1.5Mn1.5O4 phase could not be synthesized with conventional solid-state methods due to the miscibility gap between Fe3O4 and Mn3O4 below 1160 °C. Forming Fe1.5Mn1.5O4 would require quenching from high temperature to make a metastable phase.34 FeMnO3 can successfully be synthesized by a sol–gel or hydrothermal synthesis, but it exhibited the same Mn dissolution issues as Fe0.5Mn0.5O.35,36 The electronic instability (Jann–Teller distortion) associated with high-spin Mn3+ (3d4) caused a disproportionation reaction in acidic solutions to form Mn4+ and soluble Mn2+.37 To avoid these dissolution issues, an oxide phase containing Mn4+ oxide could be employed, but there are no known ternary Mn/Fe oxides with Mn4+. For example, since Fe4+ is not chemically stable, the (Fe, Mn)O2 phase could not be formed. Considering that Li is also a necessary precursor for LMFP synthesis, we next investigated the quaternary Li/Mn/Fe oxide system. The synthesis of spinel LiMnFeO4 by solid-state reaction has been previously reported.38 The atomic scale mixing of Li, Mn, and Fe in the spinel structure, ease of synthesis, and greater resistance to dissolution in acidic solutions due to the absence of Mn3+ together make LMFO an ideal oxide precursor for LMFP synthesis.SEM imaging shows that the LMFO phase forms truncated octahedral crystals with a size of 1–5 μm, while the EDS mapping reveals a uniform distribution of Mn and Fe throughout the bulk of the spinel phase (Fig. 1a–c). For comparison, a NH4Mn0.5Fe0.5PO4·H2O precursor synthesized by a co-precipitation reaction, showing a uniform distribution of Mn and Fe according to EDS, is given in Fig. S-3.† The phase purity of LMFO was investigated with XRD as a function of synthesis temperature between 500 and 900 °C (Fig. 1d). At 500 °C, the two main phases present are LiMn2O4 and Fe2O3. The preferential lithiation of MnO2 by LiOH can be attributed to the lower decomposition temperature of MnO2 compared to Fe2O3.39,40 The gradual disappearance of Fe2O3 as the synthesis temperature increases shows that Fe3+ diffusion into the spinel structure is the rate limiting step in the formation of LMFO. At 800 °C, the powder retains a slight red tint after calcination, indicating residual amounts of Fe2O3. Therefore, 900 °C was found to be the optimal synthesis temperature for LMFO.
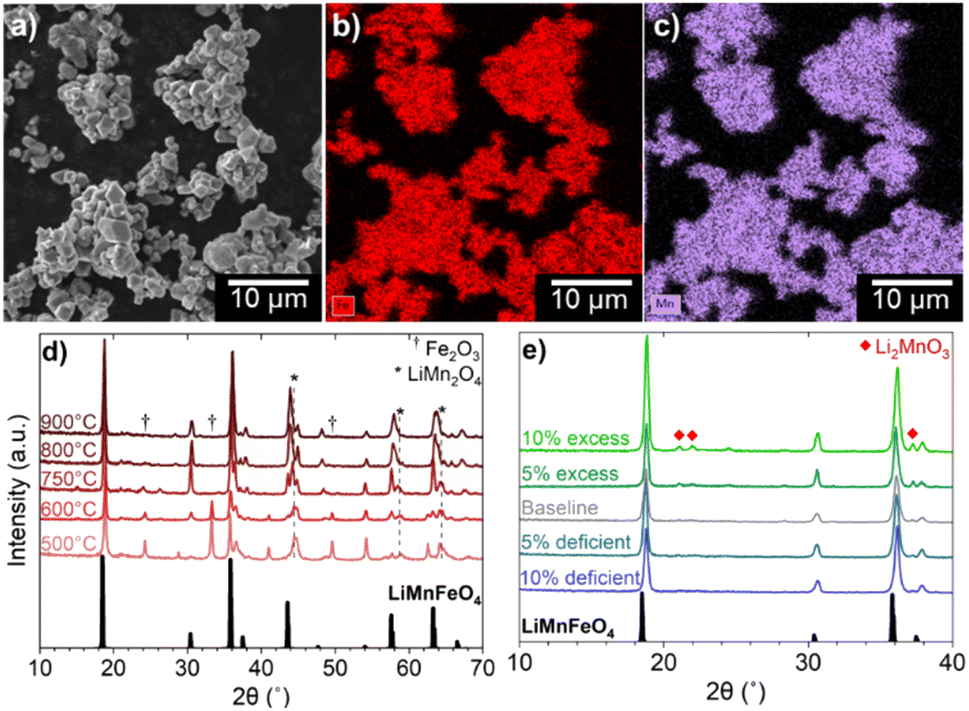 | ||
| Fig. 1 (a) SEM image of LMFO spinel precursor with EDS mapping of (b) Fe and (c) Mn. XRD patterns of LMFO (d) heated from 500 to 900 °C and (e) with adjusted Li amounts, from 10% deficient Li to 10% excess Li based on molar ratios. JCPDS #04-008-1660, #00-035-0782, #00-039-1346, and #00-027-1252 are used to identify the phases as, respectively, LMFO, LiMn2O4, Fe2O3, and Li2MnO3. | ||
The stoichiometry of Li also plays a large role in controlling the phase purity of LMFO. LMFO was synthesized with varying Li amounts ranging from 10 mol% deficient to 10 mol% excess (Fig. 1e), and the phase purity was assessed with XRD. A trace amount of Li2MnO3 impurity is detected when an equi-molar stoichiometric amount of Li is used, and the peak intensity of this impurity increases when excess Li is added. The reason for Li2MnO3 formation is rooted in Fe3+ with a high-spin 3d5 configuration. High-spin Fe3+ has a zero octahedral-site stabilization energy (OSSE), resulting in no preference for either the octahedral or tetrahedral sites of the spinel lattice.41 Therefore, some Fe3+ ions tend to reside in the tetrahedral sites of LMFO rather than being completely in the octahedral sites, displacing out some Li+ and Mn4+ from the spinel structure. According to XRD, the Li2MnO3 impurity could be completely eliminated by simply using 5 mol% deficient Li in the synthesis. Notably, many of the dopants commonly employed for LMFP can be incorporated into the LMFO precursor as well. For example, LMFO synthesized with 1 mol% of both Mg and Co is phase-pure according to XRD (Fig. S-4†). However, the stoichiometry of Li may need to be adjusted according to the site preference and amount of the dopants. For example, Mg2+ generally tends to go into the tetrahedral sites as known in the spinel MgAl2O4 mineral, while Co3+ will tend to go into the octahedral sites due to its high OSSE. Most other common dopants for LMFP, such as Al, Zn, Ti, V, Cr, Ni, Zn, and Nb, are also expected to be soluble in the LMFO spinel lattice. Employing LMFO spinel as an oxide precursor for LMFP offers the unique advantage of accommodating a variety of dopants at an atomic scale along with Mn and Fe. Additionally, the scalable, wastewater-free synthesis of LMFO presents economic viability and chemical versatility for LMFP manufacturing.
After optimizing the precursor synthesis parameters, LMFP was synthesized either from LMFO, NH4Mn0.5Fe0.5PO4·H2O, or separate oxide precursors via the two-step carbon coating and spray drying procedure. Fig. 2a presents the XRD patterns of all three LMFP samples. S-LMFP and P-LMFP are both phase pure, while O-LMFP shows impurity peaks at 43.5° and 62.4°, which can be indexed to metallic Fe. The decompositions of Fe2O3 and MnO2 may occur at different rates during the synthesis of O-LMFP, leading to phase separation in the final product. Notably, there was no difference in milling procedures among the three samples. While it is possible that more aggressive milling conditions could eliminate this impurity, the absence of impurities in the LMFP synthesized from the spinel LMFO emphasizes the advantage of an atomically mixed Mn/Fe precursor; the Mn/Fe distribution is entirely insensitive to milling conditions, which is beneficial for manufacturing scalability. The physical and electrochemical properties of S-LMFP and P-LMFP are compared throughout this work and are summarized in Table 1. The lattice parameters of both S-LMFP and P-LMFP are within ±0.02 Å for a, b, and c with Rwp values of 10%, indicating no differences in the bulk crystal structure (Table S-1†). Additionally, S-LMFP and P-LMFP both have very similar BET surface area (∼27 m2 g−1) and carbon content (∼3 wt%) and exhibit minimal electronic resistance within the porous electrode (Fig. 2b–d). Therefore, S-LMFP and P-LMFP can be assumed to be very similar cathode materials chemically, which is further seen in the SEM micrographs (Fig. 3). Spray drying causes the primary particles to aggregate into porous, spherical secondary particles, which have a diameter of 3–15 μm for all samples (Fig. 3a–c). A low magnification image of each cathode shows the size distribution of the secondary particles (Fig. S-5†). It is observed that S-LMFP and P-LMFP both have larger primary particles compared to O-LMFP where impurities could have altered the surface morphology of the material (Fig. 3d–f). With minimal differences in the primary particle size and surface morphology of S-LMFP and P-LMFP, further investigation is needed to determine the physical differences between the cathode materials. The internal morphology of the secondary particles was characterized with SEM after preparing cross sections of the secondary particles with a focused ion beam. S-LMFP and O-LMFP both contain a much larger number of internal voids compared to P-LMFP (Fig. 3g–i), which are likely caused by the formation of steam bubbles during the spray drying process.42 Tuning the spray drying parameters, such as the solid loading of the dispersion and the inlet temperature, may be effective at reducing the internal voids within the secondary particles and optimizing the packing density of primary particles within the spray dried secondary particles.
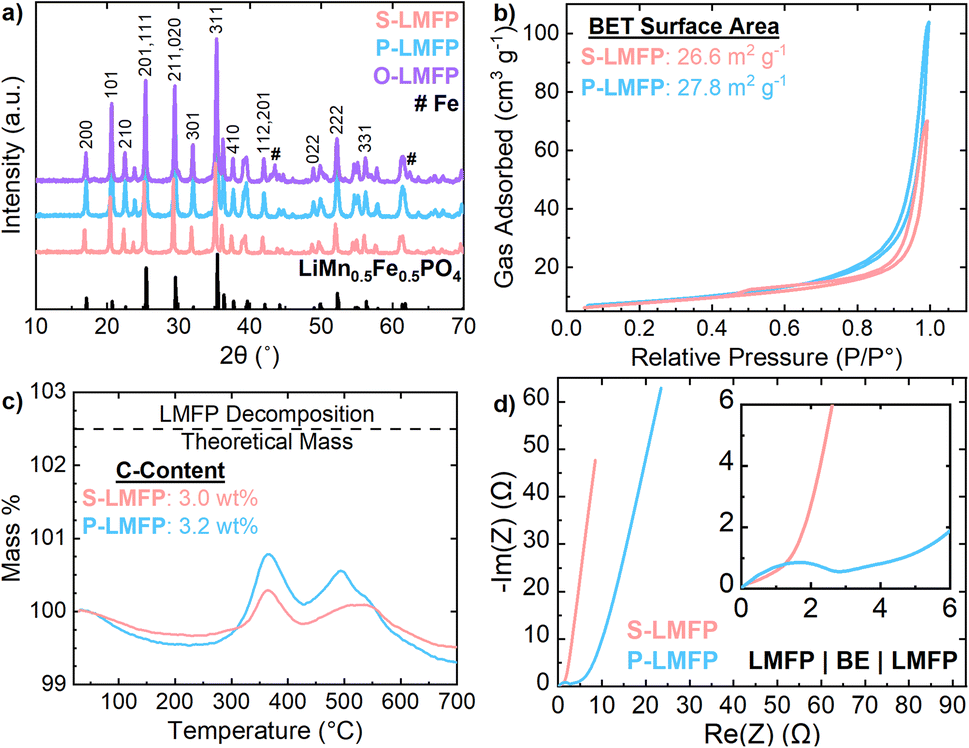 | ||
| Fig. 2 (a) XRD patterns of all three calcined LMFP samples synthesized with various precursors. JCPDS #04-013-7460 and #00-006-0696 are used to identify the phases of, respectively, LMFP and Fe. (b) Nitrogen adsorption isotherms for S-LMFP and P-LMFP with calculated surface area values presented. (c) TGA of carbon coated LMFP samples with calculated carbon amounts presented based on subtraction from the theoretical decomposition mass of LMFP. (d) EIS of a symmetric cell with a blocking electrolyte. | ||
| Sample | Tap density (g cm−3) | Press density (g cm−3) | Discharge capacity (mA h g−1) | Average discharge voltage (V) | Volumetric energy density (W h L−1) |
|---|---|---|---|---|---|
| S-LMFP | 1.21 | 2.2–2.6 | 145 | 3.61 | 1210 |
| P-LMFP | 1.00 | 1.8–2.2 | 151 | 3.64 | 1060 |
 | ||
| Fig. 3 SEM images of LMFP samples synthesized with (a, d and g) LMFO, (b, e and h) NH4Fe0.5Mn0.5PO4, and (c, f and i) separate oxide precursors. Spray dried secondary particles (a–c) in their entirety and (d–f) zoomed into the surface of the secondary particle to investigate the primary particles. (g–i) FIB-SEM images of the cross-section of the secondary particles. | ||
Despite the more porous appearance under FIB-SEM, S-LMFP has a significantly higher tap density of 1.2 g cm−3 than P-LMFP (1.0 g cm−3), which indicates that the primary particles within S-LMFP are much more densely aggregated than those in P-LMFP. Upon calendaring the electrodes with 95 wt% active material content, the maximum press density achieved before delamination of the electrode coating is much higher for S-LMFP (>2.6 g cm−3) than for P-LMFP (<2.2 g cm−3), which can be attributed to the higher packing density of primary particles. It is likely that the large internal voids present within the S-LMFP secondary particles collapse during intense calendaring, so improvements in the spray drying process may further improve the tap density of the powder, but likely not the press density of the electrode.
The cycle life of all LMFP samples is excellent, showing little or no capacity loss after 100 cycles in half cells at a C/5 rate (Fig. 4a). O-LMFP has poor electrochemical performance by comparison, achieving less than 130 mA h g−1 at a C/5 rate due to a metallic Fe impurity (Fig. S-6a†). On a gravimetric basis, the electrochemical performance of P-LMFP is superior to that of S-LMFP, offering both a higher discharge capacity and improved capacity retention at high rates (Fig. 4b). The discharge capacity of P-LMFP is 153 and 149 mA h g−1 at rates of, respectively, C/10 and C/3, compared to 147 and 141 mA h g−1 for S-LMFP. The discharge rate performance is slightly better for P-LMFP, which retains 86.6% of the C/10 rate capacity when discharged at 5C rate, while S-LMFP retains 83.2%. From the discharge voltage profiles at various rates, it is evident that the reduced capacity and lower average voltage of both samples at higher discharge rates is mainly due to the kinetic limitations of Mn2+/3+ redox (Fig. 4c and d). The inferior kinetics of Mn2+/3+ redox can be generally attributed to solid-state Li+ transport limitations that stem from the 1-D diffusion pathways of Li+ ions.
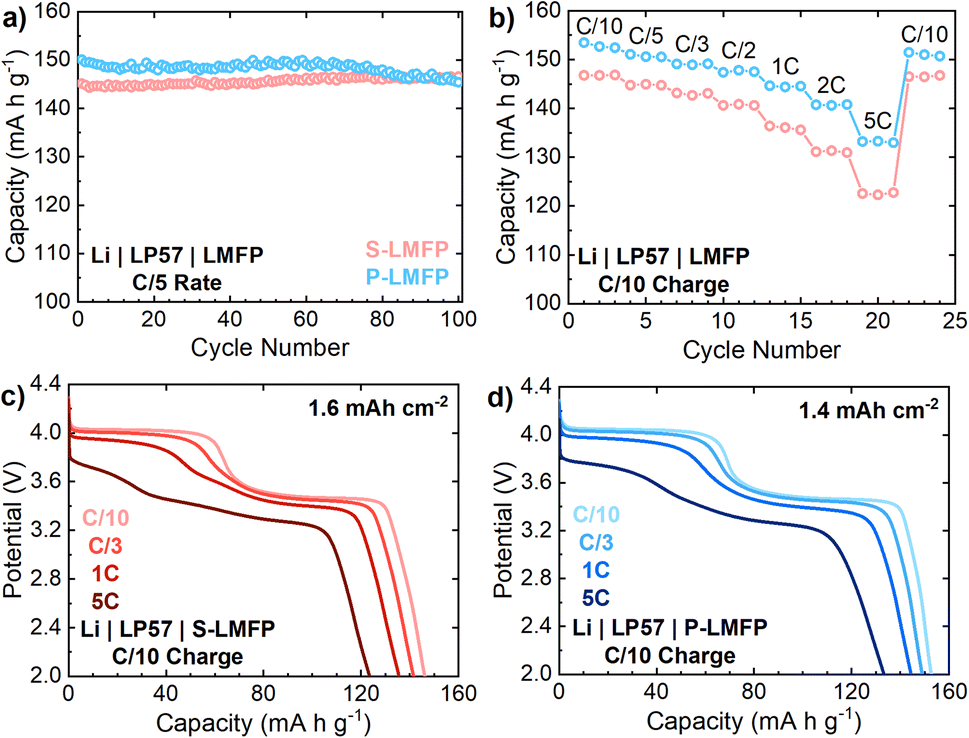 | ||
| Fig. 4 Electrochemical performances of S-LMFP and P-LMFP samples on a gravimetric basis. (a) Half-cell cycling performance of LMFP samples over 100 cycles at C/5 charge and discharge rate, (b) discharge rate testing from C/10 to 5C rate with a constant C/10 charge rate, and the voltage profiles presented at various discharge rates for (c) S-LMFP and (d) P-LMFP. Aerial capacities are indicated, and press densities for S-LMFP and P-LMFP are, respectively, 2.31 and 1.93 g cm−3. | ||
As mentioned previously, the apparent Li+ diffusion coefficient of LMFP is an order of magnitude lower for Mn2+/3+ redox than for Fe2+/3+ redox.16 The diffusion limitations for Mn2+/3+ redox manifest as increased voltage polarization and lower accessible capacity of the 4.0 V plateau at high rates. It appears that some additional capacity from Mn2+/3+ redox is obtained between 3.7 to 3.5 V, where Fe2+/3+ redox becomes active, which is expected to improve the Li+ diffusion kinetics. The kinetic limitations of Mn2+/3+ redox are also evident at C/5 rate: S-LMFP achieves a charge capacity of 147 mA h g−1 with 10 mA h g−1 coming from the CV hold at 4.3 V, while P-LMFP achieves a first charge capacity of 153 mA h g−1 with only 3 mA h g−1 coming from the CV hold (Fig. S-6b†). Compared to P-LMFP, there is a larger portion of the capacity from S-LMFP which can only be accessed at low rates (i.e., <C/10). Considering that both samples have the same composition and exhibit low electronic resistance, it is likely that this difference in apparent diffusion kinetics is caused by the denser particles of S-LMFP. Additionally, it is important to note that carbothermal reduction will occur during S-LMFP synthesis. Fe2+ and Mn2+ are present in LMFP, while the S-LMFP precursor LMFO contains Fe3+ and Mn4+. Carbothermal reduction will not occur during P-LMFP synthesis since the precursor already contains Fe2+ and Mn2+. Despite the carbon amount being similar for both cathode materials, the carbon in S-LMFP will help reduce Mn and Fe, leading to possible variances in the carbon coating structure and uniformity when compared to P-LMFP.43 Carbon coating variance may present a root cause for the differing kinetics between S-LMFP and P-LMFP.
While the electrochemical performance of LIB active materials is typically reported on a gravimetric basis, in many applications, such as electric vehicles, the volumetric energy density of the battery is much more important than the specific energy.44 The volumetric energy density is determined by the product of specific capacity, average discharge voltage, and electrode density. In practical electrode compositions with 95 wt% active material, P-LMFP can only be compacted to a maximum press density of 2.2 g cm−3 before delamination of the electrode coating from the current collector, while S-LMFP can be compacted to 2.6 g cm−3. Notably, the press density of S-LMFP meets or exceeds that of state-of-the art LFP active materials, which typically have a press density of ∼2.5 g cm−3.45 As a result, despite the lower specific capacity of S-LMFP, it can achieve an energy density of 1210 W h L−1 on an electrode-level basis at a C/5 rate, compared to 1060 W h L−1 for P-LMFP, which is a 14% improvement. However, the lower rate performance of S-LMFP compared to that of P-LMFP would be expected to reduce the energy density at higher rates. The lower porosity of electrodes at high press densities, like that in S-LMFP, can cause electrolyte transport limitations which also limit the rate performance. Accordingly, rate performance testing was conducted for both samples across a wide range of press densities to determine the optimal conditions for maximizing energy density (Fig. 5).
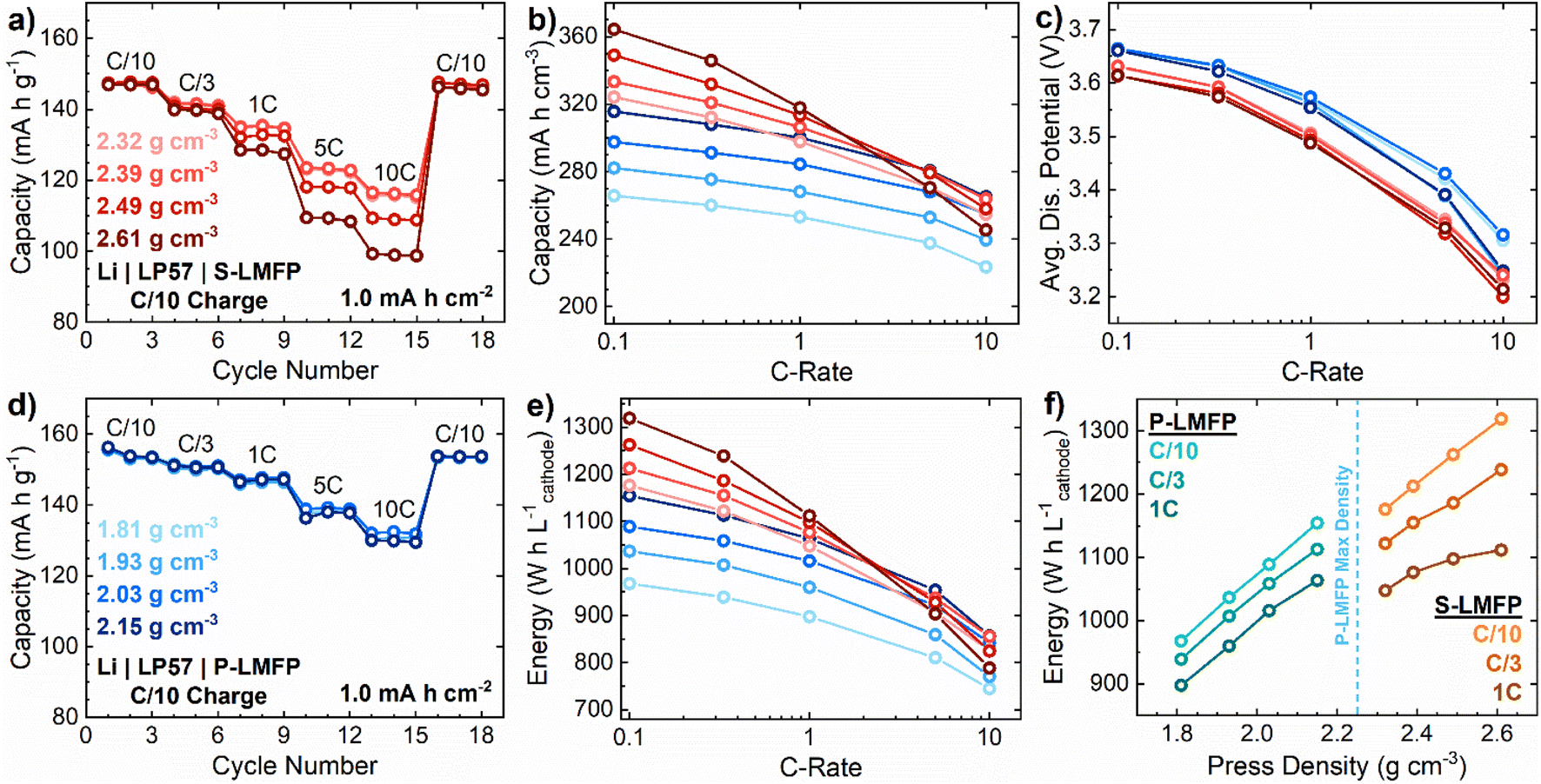 | ||
| Fig. 5 Discharge rate testing of (a) S-LMFP and (d) P-LMFP up to 10C rate when compressed to varying press densities with a C/10 charge rate. (b) Volumetric capacity, (c) average discharge potential, and (e) volumetric energy on a cathode level of all S-LMFP and P-LMFP samples tested. (f) Volumetric energy examined as a function of press density with the P-LMFP projected values until the critical press density is reached. | ||
For P-LMFP electrodes, with an active material loading of 1.0 mA h cm−2, there is essentially no difference in the rate performance between press densities of 1.81 to 2.15 g cm−3 (Fig. 5d). For S-LMFP electrodes with the same loading, the rate performance is similar between press densities of 2.32 to 2.39 g cm−3 but decreases at a press density of 2.49 g cm−3 or higher (Fig. 5a). Since volumetric capacity is directly proportional to press density, S-LMFP greatly outperforms P-LMFP in volumetric capacity at low rates when capacity retention is high (Fig. 5b). S-LMFP has about a 0.05 V lower average discharge voltage than P-LMFP, and both samples experience about a 0.4 V drop in average discharge voltage from a C/10 to a 10C rate. Overall, the press density does not appreciably affect the average discharge voltage (Fig. 5c). Therefore, the electrode-level volumetric energy follows essentially the same trend as volumetric capacity (Fig. 5e). At low rates (i.e., ≤C/3) where kinetic limitations are less severe, press density is clearly far more important than specific capacity or average discharge voltage in determining the energy density, and S-LMFP greatly outperforms P-LMFP. At intermediate rates (i.e., 1C), S-LMFP still outperforms P-LMFP in energy density, but by a smaller margin. At high rates (i.e., ≥5C), both materials exhibit essentially the same maximum energy density.
Specifically, at a 10C rate, the energy density of S-LMFP electrodes with a press density of 2.39 g cm−3 is equivalent to that of P-LMFP electrodes with a press density of 2.15 g cm−3, and S-LMFP electrodes with either a higher or lower press density exhibit inferior energy density. At high rates, electrolyte transport limitations within the porous electrode may become problematic in LMFP electrodes with high press density. These limitations will be exacerbated at higher electrode loadings of 3–4 mA h cm−2, which are typical of commercial LIB cells. The optimal press density of LMFP electrodes to maximize energy density will, therefore, depend on the specific application requirements and several aspects of cell design, including the electrode loading and electrolyte composition. The dependence of energy density on press density and discharge rate is summarized in Fig. 5f. In electric vehicle applications where LMFP is most likely to be used, the continuous discharge rate typically does not exceed C/3; in this case, it is expected that S-LMFP would greatly outperform P-LMFP in useable energy density. More generally, to achieve LIB cells with high practical energy density, it may be necessary to sacrifice the specific capacity and/or average voltage of the LMFP cathode in order to increase the press density of the electrode.
To achieve the maximum possible energy density, LMFP cathodes should ideally exhibit excellent Mn2+/3+ redox kinetics, which could enable both high press density and specific capacity. To understand the extent to which the energy density can be improved by mitigating the kinetic limitations of Mn2+/3+ redox, S-LMFP and P-LMFP half cells were tested at temperatures of 0 °C, 25 °C, and 55 °C (Fig. 6). Upon raising the temperature from 25 to 55 °C, the capacity of S-LMFP increases from 145 to 155 mA h g−1 at a C/5 rate, while the capacity of P-LMFP increases from 151 to 159 mA h g−1 (Fig. 6a and c). For both samples, this increase in capacity can be attributed mainly to an improvement in the Mn2+/3+ redox kinetics, as evidenced by the increased capacity from the 4.0 V plateau, as well as the decreased voltage hysteresis observed in the dQ/dV plots (Fig. 6b and d). Notably, for S-LMFP, a long CV hold is not needed to achieve the full capacity at 55 °C, and compared to room temperature, the electrode-level energy density increases by about 8% at 55 °C. Conversely, upon lowering the temperature from 25 to 0 °C, the capacity of S-LMFP and P-LMFP decreases to, respectively, 102 and 127 mA h g−1. The decrease in capacity at a lower temperature can likewise be attributed to a worsening of the Mn2+/3+ redox kinetics. However, the lower capacity from Fe2+/3+ redox in both samples at 0 °C indicates that there is also a contribution from electrolyte transport limitations due to its lower conductivity.
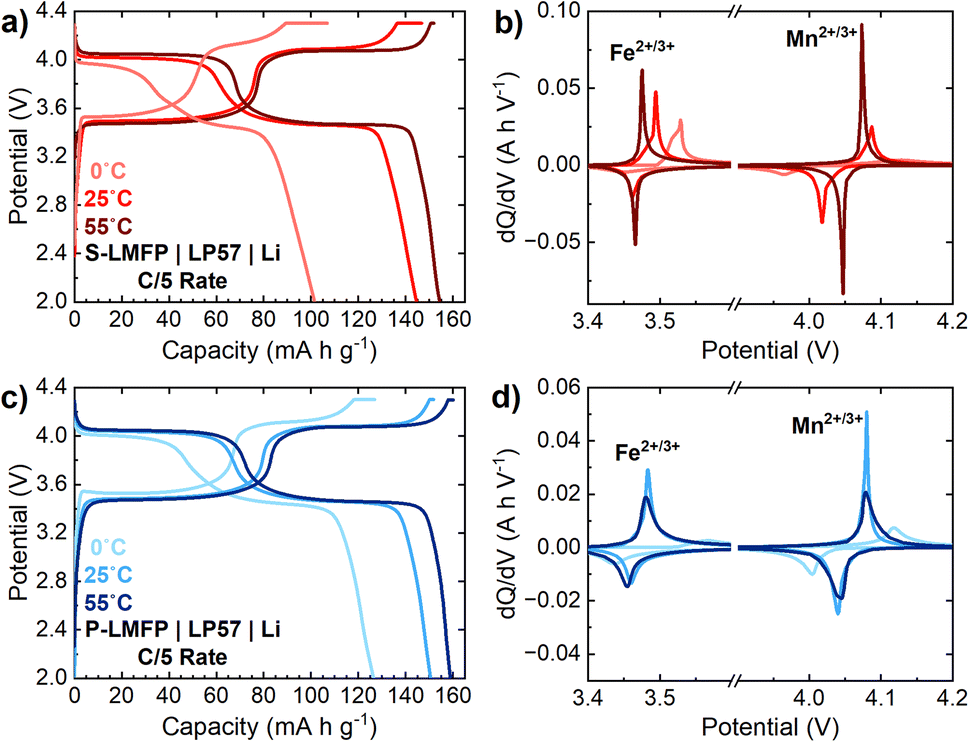 | ||
| Fig. 6 Electrochemical performances of S-LMFP (a and c) and P-LMFP (b and d) at varying temperatures during cycling. (a and b) Discharge voltage profiles and (c and d) dQ/dV plots at C/5 rate. All electrodes have an aerial capacity of 1.5 mA h cm−2. | ||
The temperature dependence of the Mn2+/3+ redox kinetics presents both challenges and opportunities for the development of high-energy-density LMFP cells. The capacity retention of S-LMFP at low temperatures is poor because of the combined effects of (i) restricted Mn2+/3+ redox kinetics due to denser primary particles and (ii) increased electrolyte transport limitations as a result of the lower porosity of the electrodes. Depending on the application requirements, the low-temperature performance of high-density LMFP may become problematic and must be considered in the overall performance evaluation. Conversely, high operating temperatures (40–50 °C) experienced during fast (dis)charging could help to overcome the kinetic limitations of LMFP, the high temperature lifetime of LMFP cells is much worse than LFP cells, likely due to elevated Mn2+ dissolution induced by the disproportionation of Jann–Teller active Mn3+ ions.46,47 Therefore, electrolyte design to improve transport properties and reduce Mn2+ dissolution will play a critical role in improving the practical volumetric energy density of LMFP cells. To further improve the energy density of S-LMFP at the material level, detailed electrochemical kinetic characterization is needed to better understand the reaction mechanisms during dis(charge) of LMFP. By understanding the limiting factors restricting the kinetics and capacity of Mn2+/3+ redox, strategies can be developed by altering the synthesis of either the LMFO precursor or LMFP cathode without sacrificing electrode density.
Conclusions
A scalable and wastewater-free synthesis process for high volumetric capacity LMFP cathodes has been developed based on a novel quaternary transition-metal oxide precursor, the LiMnFeO4 spinel, which is synthesized with a facile, low-cost solid-state reaction. The LiMnFeO4 precursor ensures a homogeneous mixing of Mn and Fe at the atomic scale, while improving the packing density of primary particles compared to LMFP synthesized from a NH4Mn0.5Fe0.5PO4·H2O precursor (P-LMFP). The LMFP synthesized with LiMnFeO4 (S-LMFP) achieves a specific capacity of 145 mA h g−1 at C/5 rate in electrodes with 95 wt% active material content and an exceptional press density of up to 2.6 g cm−3. The S-LMFP offers a slightly lower specific capacity and average discharge voltage than P-LMFP due to the kinetic limitations of Mn2+/3+ redox, arising from its larger primary particle size. Despite its lower specific energy, S-LMFP offers 15% higher electrode-level volumetric energy than P-LMFP due to its much higher electrode density. Rate performance testing of both materials with various electrode densities reveals that at low discharge rates (i.e., <1C), which are typical of electric vehicle batteries, the electrode density is much more important in determining the energy density than the specific capacity and average discharge voltage. Accordingly, in order to realize high practical energy density in LMFP cells, it is necessary to synthesize LMFP cathodes which can achieve comparable electrode density to state-of-the-art LFP (i.e., >2.5 g cm−3). The synthesis of LMFP cathodes from spinel LiMnFeO4 is a promising route to improve the density of LMFP electrodes; however, additional optimization of composition, particle size, and synthesis conditions is needed to overcome the sluggish Mn2+/3+ redox kinetics.Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to the ongoing process of filing a patent application, but are available from the authors on reasonable request.Author contributions
S. R. and K. S. conceptualized the project idea and developed the methodology. S. R. performed the material preparation, electrochemical testing, and structural and spectroscopic characterizations. All the authors wrote the manuscript. A. M. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U. S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under award number DE-SC0005397. One of the authors (S. R.) was supported by the National Science Foundation Graduate Fellowship.References
- C. Curry, Lithium-ion Battery Costs and Market, Bloomberg New Energy Finance, https://enerjiye.com/wp-content/uploads/2021/07/battery-market.pdf, (accessed 2024-04-10) Search PubMed.
- B. Nykvist and M. Nilsson, Nat. Clim. Change, 2015, 5, 329–332 CrossRef.
- B. E. Murdock, K. E. Toghill and N. Tapia-Ruiz, Adv. Energy Mater., 2021, 11, 39 CrossRef.
- C. Earl, I. H. Shah, S. Cook and C. R. Cheeseman, Sustainability, 2022, 14(7), 4124 CrossRef CAS.
- S. Lee and A. Manthiram, ACS Energy Lett., 2022, 7(9), 2058–3063 Search PubMed.
- LME Cobalt Official Prices, London Metals Exchange, https://www.lme.com/en/metals/ev/lme-cobalt, (accessed 2024-04-10) Search PubMed.
- LME Nickel Official Prices, London Metals Exchange, https://www.lme.com/en/metals/non-ferrous/lme-nickel, (accessed 2024-04-10) Search PubMed.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- F. M. N. U. Khan, M. G. Rasul, A. S. M. Sayem and N. K. Mandal, J. Energy Storage, 2023, 71, 108033 CrossRef.
- K. W. Knehr, J. J. Kubal, P. A. Nelson and S. Ahmed, Battery Performance and Cost Modeling for Electric-Drive Vehicles (A Manual for BatPaC v5.0), Argonne National Laboratory, 2022, ANL/CSE-22/1 Search PubMed.
- M. Fleischer, J. Chem. Educ., 1954, 31(9), 446 CrossRef CAS.
- A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 2001, 148, A224 CrossRef CAS.
- K. Zaghib, et al. , J. Power Sources, 2012, 219, 36–44 CrossRef CAS.
- T. Murallganth and A. Manthiram, J. Phys. Chem. C, 2010, 114, 15530–15540 CrossRef.
- D. B. Ravnsbæk, K. Xiang, W. Xing, O. J. Borkiewicz, K. M. Wiaderek, P. Gionet, K. W. Chapman, P. J. Chupas and Y. M. Chiang, Nano Lett., 2014, 14, 1484–1491 CrossRef.
- S. Wi, J. Park, S. Lee, J. Kim, B. Gil, A. J. Yun, Y. E. Sung, B. Park and C. Kim, Nano Energy, 2017, 39, 371–379 CrossRef CAS.
- E. Lyle, R. Vaeli, M. Cormier and M. Metzger, J. Electrochem. Soc., 2022, 169, 060527 CrossRef CAS.
- D. B. Ravnsbæk, K. Xiang, W. Xing, O. J. Borkiewicz, K. M. Wiaderek, P. Gionet, K. W. Chapman, P. J. Chupas, M. Ting and Y. M. Chiang, Nano Lett., 2016, 16(4), 2375–2380 CrossRef.
- A. Yamada, Y. Takei, H. Koizumi, N. Sonoyama, R. Kanno, K. Itoh, M. Yonemura and T. Kamiyama, Chem. Mater., 2006, 18(3), 804–813 CrossRef CAS.
- Z. X. Nie, C. Y. Ouyang, J. Z. Chen, Z. Y. Zhong, Y. L. Dua, D. S. Liu, S. Q. Shi and M. S. Lei, Solid State Commun., 2010, 150, 40–44 CrossRef CAS.
- W. Liu, P. Gao, Y. Mi, J. Chen, H. Zhou and X. Zhang, J. Mater. Chem. A, 2013, 1, 2411 RSC.
- L. Wu, S. H. Zhong, J. Q. Liu, F. Lv and K. Wan, Mater. Lett., 2012, 89, 32–35 CrossRef CAS.
- J.-K. Kim, CrystEngComm, 2014, 16, 2818 RSC.
- C. T. Hsieh, I. L. Chen, W. Y. Chen and J. P. Wang, Electrochim. Acta, 2012, 83, 202–208 CrossRef CAS.
- W. C. Chien, K. N. Liu, S. C. Chang and C. C. Yang, Thin Solid Films, 2018, 660, 931–937 CrossRef CAS.
- I. Seo, B. Senthilkumar, K. H. Kim, J. K. Kim, Y. Kim and J. H. Ahn, J. Power Sources, 2016, 320, 59–67 CrossRef CAS.
- J. Li, Y. Wang, J. Wu, H. Zhao, H. Wu, Y. Zhang and H. Liu, Chemelectrochem, 2017, 4, 175–182 CrossRef CAS.
- J. Li, M. Qiang, Y. Wang, J. Wu, H. Zhao and H. Liu, J. Mater. Chem. A, 2017, 5, 7952–7960 RSC.
- S. M. Oh, S. T. Myung, Y. S. Choi, K. H. Oh and Y. K. Sun, J. Mater. Chem., 2011, 21, 19363–19374 Search PubMed.
- P. Y. Yang, S. P. Ju, H. S. Hsieh and J. S. Lin, RSC Adv., 2017, 7, 55044–55050 RSC.
- L. Gao, Z. Liu, Z. Yang, L. Cao, C. Feng, M. Chu and J. Tang, Appl. Surf. Sci., 2020, 508, 145292 CrossRef CAS.
- J. D. Hem, Geochim. Cosmochim. Acta, 1981, 45, 1369–1374 CrossRef CAS.
- The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals, ed. M. J. O'Neil, Merck and Co., Inc., 2006, p. 88 Search PubMed.
- H. J. Van Hook and M. L. Keith, Am. Mineral., 1958, 43, 69–83 CAS.
- B. Saravanakumar, S. P. Ramachandran, G. Ravi, V. Ganesh, R. K. Guduru and R. Yuvakkumar, Mater. Res. Express, 2018, 5, 015504 CrossRef.
- S. Gowreesan and A. R. Kumar, Appl. Phys. A, 2017, 123, 689 CrossRef.
- T. Liu, et al. , Nat. Commun., 2019, 10, 4721 CrossRef CAS.
- M. Gracia, J. F. Marco, J. R. Gancedo, J. Ortiz, R. Pastene and J. L. Gautier, J. Phys. Chem. C, 2010, 114, 12792–12799 CrossRef CAS.
- D. M. Tinsley and J. H. Sharp, J. Therm. Anal., 1971, 3, 43–48 CrossRef CAS.
- Q. Fradet, M. Kurnatowska and U. Riedel, Thermochim. Acta, 2023, 726, 179552 CrossRef CAS.
- J. Darga, J. Lamb and A. Manthiram, Energy Technol., 2020, 8(12), 2000723 CrossRef CAS.
- D. E. Walton and C. J. Mumford, Trans IChemE, 1999, 77A, 442–460 CrossRef.
- X. Ren, et al. , Chem. Eng. J., 2023, 453, 139611 CrossRef CAS.
- X. Zeng, M. Li, D. A. El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- W. Li, H. Gu, H. Yang, Q. Li, X. Li, Y. Wang and G. Liang, J. Electrochem. Soc., 2024, 171, 02054 Search PubMed.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- C. H. Lu and S. W. Lin, J. Mater. Res., 2002, 17, 1476–1481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03438d |
| This journal is © The Royal Society of Chemistry 2024 |