
Unveiling the solar-driven synergistic production of a cyclic fuel-additive and carbon-free solar fuel from biogenic furfural: mediated by a metal-free organic semiconductor†
Shivali
Dhingra
,
Arpna
Jaryal
,
Deepak Kumar
Chauhan
and
Kamalakannan
Kailasam
 *
*
Advanced Functional Nanomaterials Group, Institute of Nano Science and Technology (INST), Knowledge City, Sector 81, SAS Nagar, Manauli PO, Mohali, 140306, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
First published on 6th November 2024
Abstract
Photocatalytic biomass upgradation to fine chemicals and fuels offers a promising strategy to address the current energy crisis and presents a prominent step towards carbon neutrality. Despite several reports in recent years, biomass valorization is still facing a lot of challenges including poor selectivity and inefficient conversion. Notably, photooxidation of biomass results in inefficient utilization of charge carriers which hampers the overall efficiency of the photocatalytic process. In the ongoing quest for effective biomass upgradation, here, we present a metal-free urea-derived carbon-nitride for the photocatalytic acetalization of furfural (Ffal) with ethylene glycol (EG) to generate a cyclic acetal i.e. 2-furyl-1,3-dioxolane (FD), a promising bio-fuel additive integrated with H2O2 production under visible light for the first time. Importantly, an 85% cyclic acetal yield is achieved in 6 h with 99% selectivity along with 162 µmol g−1 of H2O2 production. Under natural sunlight, an exceptionally high yield of FD has been achieved, reaching 70% yield, presenting the practicality of the UCN photocatalyst for the large-scale production of cyclic acetals. In situ EPR analysis, photoluminescence spectroscopy, and photo-electrochemical studies along with various control experiments elucidated the charge transfer mechanism involved in the photoredox process. Thus, the current study offers an encouraging approach for harnessing a metal-free photocatalyst to generate solar fuel coupled with biomass upgradation to fuel additives, thereby presenting a viable pathway for the sustainable production of fuels and fine chemicals.
1. Introduction
The expeditious consumption of fossil fuel reserves has led to profound concern regarding the future availability of energy resources.1 Additionally, the excessive utilization of fossil fuels causes the relentless release of greenhouse gases which contribute substantially to the environmental crisis.2 To tackle the above-stated issues, developing sustainable alternative energy resources has become imperative. In this regard, biomass is emerging as a promising alternative to fossil fuel due to its renewable and abundant nature, offering the potential for sustainable fuel production and production of value-added chemicals. However, solar-driven biomass valorization to fuel and value-added chemicals has gained significant attention as it is carried out under environmentally benign conditions and does not necessitate any additional energy input. Lignocellulosic biomass (non-edible) consists of cellulose, hemicellulose, and lignin which can be depolymerized to generate platform chemicals.3,4 In this line, furfural (Ffal) has gained remarkable recognition as an important biomass-derived platform chemical that can be obtained from pentose sugar present in agricultural residues.5 Furfural has remarkable reactivity and can undergo reactions such as hydrogenation, oxidation, esterification, and acetalization to generate value-added chemicals and biofuels.6,7In recent years, acetalization of furfural has garnered significant attention as it results in cyclic acetals on reaction with diol or triols, which serve as promising bio-fuel additives. Utilizing biomass-derived substrates in the acetalization process makes it a sustainable approach for synthesizing bio-fuel additives.8 The environmental impact of the fuel can be reduced by blending it with fuel additives such as cyclic acetal and acetal as they act as oxygenated additives in fuels. They have the potential to increase the octane number to improve the fuel's lubricity, reduce sulfur content, and further improve the viscosity index of the fuel. In addition to their application in the fuel industry, acetal is also utilized by the food industry as a flavoring additive, as a plasticizer in the polymer industry, and as an aroma enhancer in the cosmetic industry. Moreover, acetal formation is considered a powerful tool for protecting the carbonyl group in multistep organic synthesis.9,10
The conventional synthesis of acetals employs the use of strong acids such as HCl and H2SO4 which are exceedingly corrosive in nature.11 In addition to this, various other homogeneous catalysts have been reported including ionic liquids,12 NBS,13 and N,N′– bis [3,5-bis (trifluoromethyl) phenyl] thiourea.14 Apart from homogeneous catalysts, the heterogeneous catalyst for acetalization has been studied owing to its numerous advantages over homogeneous catalysis. Various heterogeneous catalysts have been reported for the acetalization including zeolites, sulfonic acid-functionalized carbons, and metal-supported mesoporous silica.15,16 However, it's worth mentioning that all the above-mentioned catalytic systems suffer from various challenges such as rigorous reaction conditions, utilizing strong acids, and high temperature which makes this process environmentally unsustainable. The broad applicability of acetalization has directed the development of strategies that require milder reaction conditions. Consequently, harnessing abundant sunlight to drive the acetalization of aldehydes will enhance the sustainability of the process.
In recent years, the following photocatalytic systems have been reported for the acetalization of aldehydes which include Eosin Y,17 thioxanthenone,18 and (N,N′-bis[3,5-bis(trifluoromethyl)-phenyl]-thiourea),19 whereas there are seldom reports available for the photocatalytic acetalization of biomass-derived precursors. Wang et al.20 utilized a phosphated TiO2 photocatalyst for the acetalization of furfural. Bhagat et al.21 reported metal-free porphyrin-based photocatalysts for the acetalization of furfural. However, all the above-mentioned photocatalytic systems have certain limitations such as dependency on metal-based systems, utilization of UV light (only 4% in the solar spectrum), cost-ineffectiveness, and poor stability. Importantly, all the above-stated photocatalysts result in inefficient utilization of charge carriers which hampers the overall efficiency of the photocatalytic process. In view of these drawbacks, it becomes indispensable for the synergistic utilization of holes and electrons for the sustainable production of fuels and fine chemicals, thus enhancing the effectiveness of the photocatalytic system.
In this line, the generation of fuel additives coupled with H2O2 production by synergistically utilizing photogenerated holes and electrons presents a remarkable strategy. H2O2 is an industrially essential chemical used in a variety of applications such as organic synthesis, wastewater treatment, pollutant treatment, medical disinfection, the paper industry, etc.22 In recent years, H2O2 has gained substantial attention as a promising carbon-free fuel for fuel cells compared to H2. H2O2 transport is notably more convenient compared to H2 due to its high solubility, making it a promising energy carrier for the next-generation fuel sector. Currently, the industry predominantly relies on the anthraquinone oxidation process for the production of H2O2. However, this process is not environmentally friendly as it uses external H2 and toxic solvents and generates a large amount of liquid waste. Certainly, employing photocatalysis for the production of H2O2 is an environmentally benign process that produces H2O2 from O2 reduction or H2O oxidation by using photogenerated electrons at its conduction band or holes at its valence band, respectively.23,24 Therefore, coupling furfural acetalization with H2O2 production will maximize the overall energy utilization of photocatalysts to produce fuel and bio-fuel additives. To date, there is no report in the literature for the simultaneous production of bio-fuel additives coupled with H2O2 production. Therefore, the development of a cost-effective and metal-free photocatalyst for the sustainable production of bio-fuel additives coupled with fuel production is a matter of paramount importance.
In this line, graphitic carbon nitride (g-C3N4) has gained remarkable attention in the catalysis community as a metal-free semiconductor with an optimum band gap (2.7 eV) in the visible region. Furthermore, g-C3N4 is highly cost-effective and easy to synthesize, exhibits high photostability, and can be synthesized from earth-abundant precursors such as urea, melamine, thiourea, and cyanamide making it a promising candidate for photocatalysis.25 In 2009, Wang et al.26 utilized for the first time g-C3N4 as a metal-free photocatalyst for H2 production. In particular, after this research, g-C3N4 has gained remarkable attention. Subsequently, g-C3N4 has been extensively employed for various photocatalytic applications such as CO2 reduction, H2O2 production, organic transformation, and biomass valorization. These studies highlighted the potential of g-C3N4 as a versatile and efficient candidate for numerous photocatalytic applications.26–28
Motivated by the above-mentioned previous studies, herein, for the first time we have employed a g-C3N4 photocatalyst derived from a urea precursor (UCN) for the acetalization of furfural (Ffal) with ethylene glycol (EG),29 a biomass-derived chemical, coupled with H2O2 production without utilizing any external acids and under ambient conditions. This study provides us with an excellent approach to utilize both the electrons and holes for the synergistic production of H2O2 as fuel and 2-furyl-1,3-dioxolane (FD) as a bio-fuel additive, respectively. The versatility of the UCN photocatalyst for photocatalytic acetalization was investigated with various biomass substrates and different alcohols. The state-of-the-art UCN has been compared to the existing thermal catalysts and the available photocatalysts for Ffal acetalization with EG (Table S1†). We believe that this study broadens the application domain, particularly towards the upgradation of biomass for sustainable production of value-added chemicals and fuels, thereby playing a significant role in advancing the bio-chemical economy.
2. Results and discussion
2.1 Characterization of the catalyst
Carbon nitride (g-C3N4) was synthesized from a urea precursor, named UCN, and detailed synthesis is given in the ESI†. MCN (carbon nitride synthesized from a melamine precursor) was also synthesized (detailed synthesis given in the ESI†) to compare the photocatalytic activity with that of UCN. Initially, Fourier transform infrared spectroscopy (FTIR) measurement was carried out for MCN and UCN (Fig. S1†). Similar FTIR spectra were obtained for UCN and MCN as the structural moieties remained the same. FTIR spectra of UCN and MCN exhibit a broad peak at around 3000 cm−1 to 3200 cm−1 which is attributed to the stretching vibrations of N–H groups. The peak at around 1200 cm−1 to 1700 cm−1 can be attributed to the bending vibration and stretching vibration of the C–N heterocyclic ring. The peak that arises at 807 cm−1 is due to the breathing mode of the tri-s-triazine core unit. XRD patterns of both UCN and MCN (Fig. S2a†) exhibited a weak diffraction peak at 13.1°, corresponding to the (100) diffraction plane resulting from in-plane ordering and a strong diffraction peak at 27.3°, corresponding to the (002) diffraction planes that resulted from interplanar stacking of conjugated tri-s-triazine sheets.30 The N2 adsorption–desorption isotherms of both UCN and MCN shown in Fig. S2b† are identified as type IV isotherms with a H3 hysteresis loop, which indicates the mesoporous structure of the material. The surface area analysis of UCN and MCN was carried out using the Brunauer–Emmett–Teller (BET) method and the surface area was estimated to be 85 and 18 m2 g−1, respectively. XPS analysis was carried out to determine the elemental state and chemical composition of carbon nitride (Fig. S3†). XPS survey scan of UCN (Fig. S3a†) exhibits three peaks that correspond to C1s, N1s, and O1s binding energies. The high-resolution C 1s XPS spectrum (Fig. S3b†) can be deconvoluted into three peaks with binding energy at 284.8, 286.6, and 288.1 eV. The peak that appeared at 284.8 eV is due to the presence of adventitious carbon (C–C) and the peaks observed at 286.6 eV and 288.1 eV were due to the presence of sp2 hybridized carbon C–NHx and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the tri-s-triazine unit of UCN, respectively. The high-resolution N 1s XPS spectrum (Fig. S3c†) can be deconvoluted into four peaks. The intense peak at binding energy 398.4 eV indicates the presence of nitrogen atoms in the sp2-hybridization state attached to the carbon atom and peaks at binding energy 399.6 and 401 eV correspond to the tertiary N–(C)3 and –NHx functional groups present in the tri-s-triazine unit of the carbon nitride core, respectively. A weak intense peak at 403.9 eV in N 1s spectra was caused by the π-excitations.31 The high-resolution O 1s spectra (Fig. S3d†) show a peak at 532.2 eV which corresponds to the absorbed moisture.32 Notably, MCN exhibits similar C 1s and N 1s XPS spectra to those observed for UCN.
N in the tri-s-triazine unit of UCN, respectively. The high-resolution N 1s XPS spectrum (Fig. S3c†) can be deconvoluted into four peaks. The intense peak at binding energy 398.4 eV indicates the presence of nitrogen atoms in the sp2-hybridization state attached to the carbon atom and peaks at binding energy 399.6 and 401 eV correspond to the tertiary N–(C)3 and –NHx functional groups present in the tri-s-triazine unit of the carbon nitride core, respectively. A weak intense peak at 403.9 eV in N 1s spectra was caused by the π-excitations.31 The high-resolution O 1s spectra (Fig. S3d†) show a peak at 532.2 eV which corresponds to the absorbed moisture.32 Notably, MCN exhibits similar C 1s and N 1s XPS spectra to those observed for UCN.
The FE-SEM micrograph (Fig. S4a and b†) represents significant agglomeration in UCN and MCN. The TEM image (Fig. S4c†) displays the 2D sheet-like structure of UCN and a similar observation was noted for MCN (Fig. S4d†). Furthermore, AFM analysis was carried out to determine the thickness of UCN nanosheets. The AFM demonstrates (Fig. S5†) that the thickness of UCN nanosheets is ≈11 nm. DR UV-vis (diffuse reflectance ultraviolet-visible) spectra of UCN (Fig. 1a) show strong absorption in the visible region at 450 nm. Similarly, MCN exhibits absorption in the visible region but with a slight red shift compared to UCN (Fig. S6†). The absorption of UCN in the visible region makes it an effective catalyst to harness the visible range of the solar spectrum. The optical band gap of the photocatalyst is measured by using a Tauc plot, as shown in Fig. 1b. The band gap of UCN was found to be 2.75 eV. The valence band potential (EVB) of UCN is calculated by using the valence band XPS as shown in Fig. 1c, which is 1.72 eV. Furthermore, based on the optical band gap and from the valence band position, the conduction band of UCN is calculated by using the relation ECB = EVB − Eg.33 Therefore, the ECB of the UCN is estimated to be −1.03 eV vs. NHE. Fig. 1d displays the band energy diagram of UCN with favorable conduction band potential to reduce O2 to reactive oxygen species.
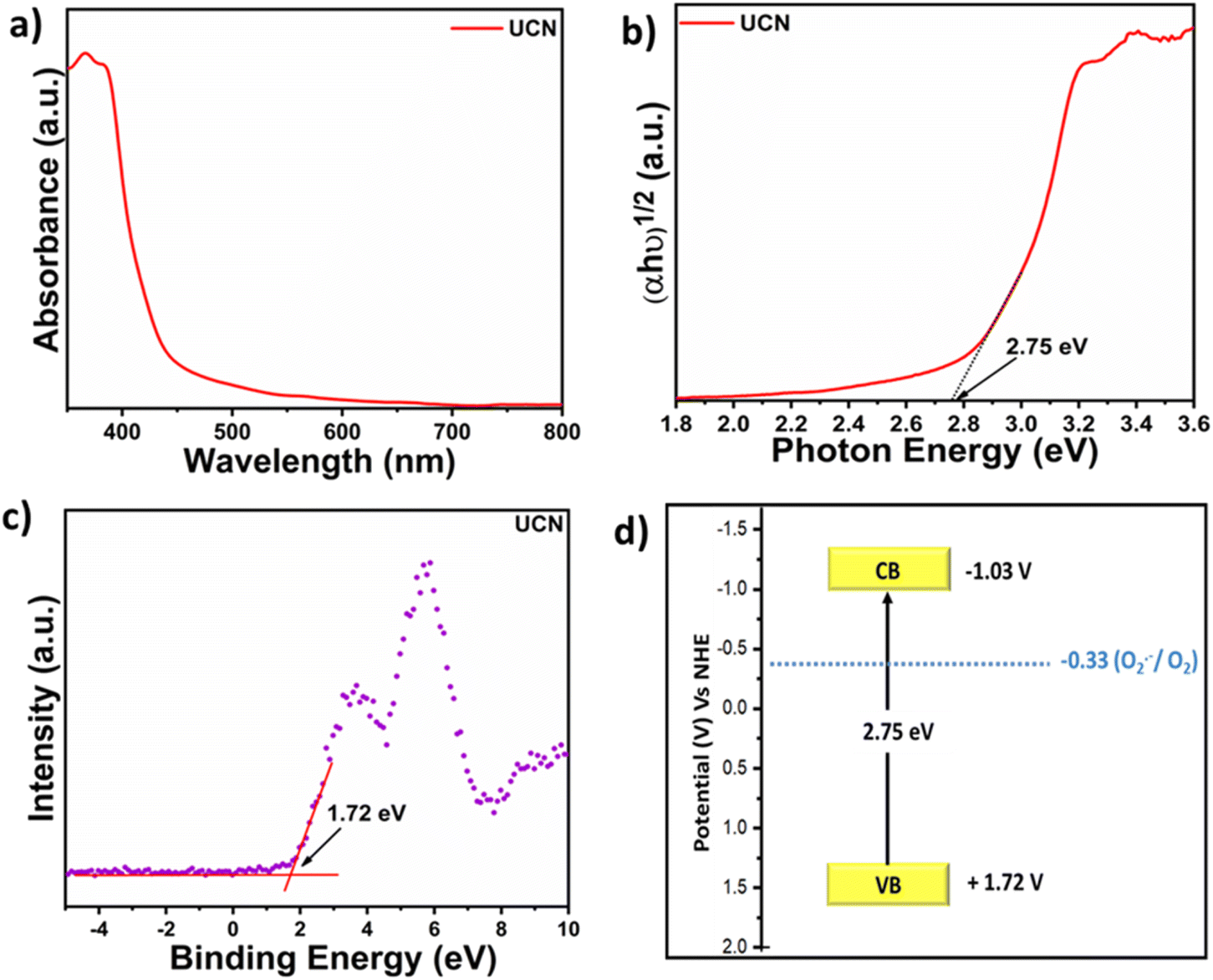 | ||
| Fig. 1 (a) DR UV-vis spectra of UCN; (b) Tauc plot of UCN; (c) valence band XPS of UCN; and (d) band energy diagram. | ||
2.2 Photocatalytic activity
Upon successfully characterizing the catalyst, UCN was tested for photocatalytic acetalization of biomass-derived Ffal with EG under an O2 atmosphere to synthesize cyclic acetal coupled with H2O2 generation. The photocatalytic reaction setup is given in Scheme S1.† The photocatalytic acetalization of Ffal performed under different conditions is summarized in Table 1. It should be noted that the photocatalytic activity of UCN was compared with that of melamine-derived carbon nitride (MCN). In the case of UCN, the yield of FD obtained is 85% and the H2O2 yield is about 162 µmol g−1 (Table 1, entry 1). Under the same reaction conditions, MCN has shown less activity compared to UCN as shown in Table 1, entries 1 and 2. The better activity in the case of UCN can be attributed to the higher surface area (Fig. S2b†) and better charge transfer kinetics (Fig. S7†) of UCN compared to MCN. The photocatalytic reaction was found to be dependent on certain essential parameters such as light and catalyst as shown in Table 1 (entries 4 and 5). From the experiment (Table 1, entry 3), it can be depicted that acetalization can be carried out effectively in ambient air, resulting in a significant amount of the desired product.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Light | Atmosphere | FD yieldb (%) | FD selectivityc (%) | H2O2 µmol g−1 |
| a Reaction conditions: photocatalyst (5 mg), substrate (Ffal 0.1 mmol), solvent (EG 2 mL), O2 atmosphere, and light source: 400 W xenon lamp (100 mW cm−2) (>420 nm). b FD yield determined by GC-MS. c FD selectivity determined by GC-MS. d Photocatalyst (MCN). e Without a catalyst. f Without light, and ND = not detected. | ||||||
| 1 | UCN | + | O2 | 85 | 99 | 162 |
| 2d | MCN | + | O2 | 60 | 99 | 131 |
| 3 | UCN | + | Air | 70 | 99 | 140 |
| 4e | + | O2 | 5 | 99 | ND | |
| 5f | UCN | — | O2 | 10 | 99 | ND |
Based on the above results, we conclude that UCN significantly promoted the synergistic formation of FD with a good selectivity of 99% with a significant amount of H2O2. The obtained product FD was analyzed by GC-MS (Fig. S8 and S9†) whereas H2O2 formed was quantified by the iodometry method (Fig. S10a–c†). For broad substrate scope, photocatalytic experiments were conducted for different aldehyde-functionalized biomass-derived substrates and alcohols as shown in Table S2† under similar reaction conditions. It was shown that EG with 5-hydroxymethylfurfural (HMF) produced cyclic acetal ([5-(1,3-dioxolan-2-yl)-2-furyl] methanol) with good catalytic yield i.e. 73% (Table S2,† entry 2). In the case of 2,5-diformylfuran (DFF) which has two aldehyde groups, a reaction with EG leads to two products. One with two cyclic acetals 2,2′-(2,5-furandiyl)bis(1,3,-dioxolane) with 36% yield whereas the other with one cyclic acetal 5-(1,3-dioxolan-2-yl)-2-furfural with 18% yield (Table S2,† entry 3). Furthermore, the acetalization of Ffal was also tested with methanol and ethanol (Table S2,† entries (4 and 5)).
From the catalytic results, it was concluded that EG exhibits better activity compared to methanol and ethanol in the formation of acetals. This can be associated with greater stability of the hemiacetal intermediate formed in the case of cyclic acetal as compared to open chain acetals.34 Furthermore, the acetalization of Ffal with EG was carried out at different time intervals under the optimized conditions (Fig. 2a and b). With increasing reaction time, an increase in the conversion of Ffal along with the generation of H2O2 was observed. Interestingly, the selectivity of the product was maintained throughout each preceding hour of the reaction.
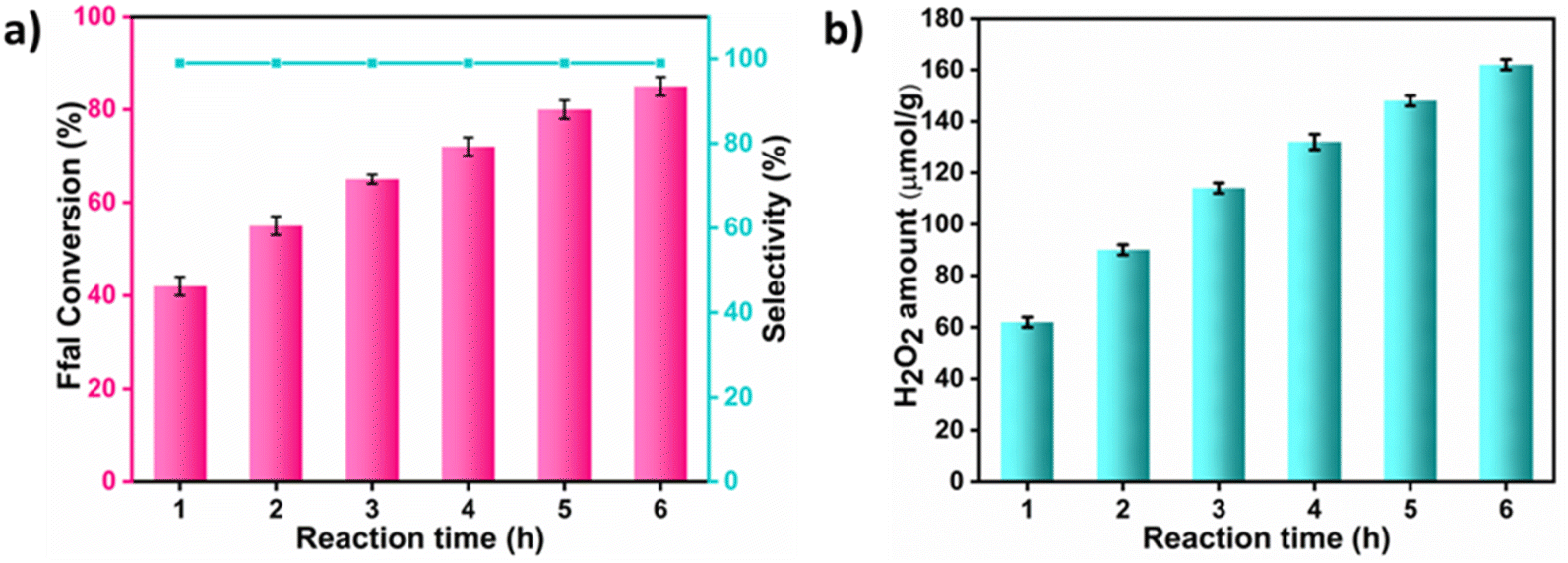 | ||
| Fig. 2 (a and b) Time study of acetalization of Ffal with EG. Reaction conditions: Ffal (0.1 mmol), UCN (5 mg), EG (2 mL), O2 pressure (1 atm), 25 °C, and light source: a 400 W xenon lamp (100 mW cm−2) (>420 nm). | ||
2.3 Mechanistic studies
Various control experiments were performed to get insight into the mechanism behind this photocatalytic acetalization process (Table S3†). Introducing TEMPO (radical scavenger) in the reaction mixture led to a remarkable reduction in the cyclic acetal yield, indicating that the reaction proceeds via a radical mechanism (Table S3,† entry 2). Different scavengers were utilized to find the role of photogenerated charge carriers such as holes and electrons. Ammonium formate (NH4HCO2) was used as a hole scavenger (Table S3,† entry 3) to determine the role of holes in the reaction initiation. A negligible amount of product was formed, indicating the crucial role of holes in the catalysis. After adding an electron scavenger i.e. (AgNO3), the product yield increased from 85% to 87% in 6 h (Table S3,† entry 4). The observed increase in the activity can be attributed to the greater availability of photogenerated holes which was achieved by adding an electron scavenger. This elucidates the role of molecular oxygen (O2) as an electron-scavenging agent which turns into another industrially important product i.e. hydrogen peroxide (H2O2). To further validate the role of photogenerated holes in Ffal acetalization, the reaction was carried out under an argon atmosphere (Table S3,† entry 5). Interestingly, 36% of the product was formed in 6 h by efficiently harnessing the potential of photogenerated holes. Moreover, adding a superoxide scavenger (1,4-benzoquinone) into the reaction mixture exhibited no significant change in the yield of the product (Table S3,† entry 6), thus ruling out the role of superoxide in photocatalytic acetalization. Based on the results obtained in the above studies, it can be concluded that oxygen acts as an electron scavenger that undergoes reduction reaction to produce H2O2 and the photocatalytic acetalization reaction is mediated by the photogenerated holes.Photoluminescence (PL) study (Fig. 3a) was carried out to gain deeper insight into the charge transfer mechanism underlying the synergistic photocatalytic process catalyzed by UCN. Appreciable quenching in the PL intensity of UCN was observed upon adding EG to UCN and a subsequent additional drop in PL intensity was observed when Ffal was added to UCN in the presence of EG. This indicates the lower rate of recombination of photogenerated charge carriers, thereby corroborating facile charge transfer occurring during the photocatalytic process. Furthermore, time-resolved photoluminescence (TRPL) decay was employed to study the lifetime of photogenerated excitons in UCN via the time-correlated single photon counting (TCSPC) technique which was recorded at 390 nm wavelength (Fig. S11†). From this, the average lifetime of photogenerated electrons in UCN was calculated to be 3.91 ns which lies within the timescale to initiate the chemical reaction.35 The photogenerated holes left behind in the valence band after exciting electrons towards the conduction band were then consumed for the oxidative acetalization of Ffal with EG. Also, the photogenerated electrons were utilized for the reduction of O2 to H2O2. Thus, the long-lived excitons generated in UCN resulted in improved charge separation and consequent migration during the photocatalytic acetalization coupled with the H2O2 production process.
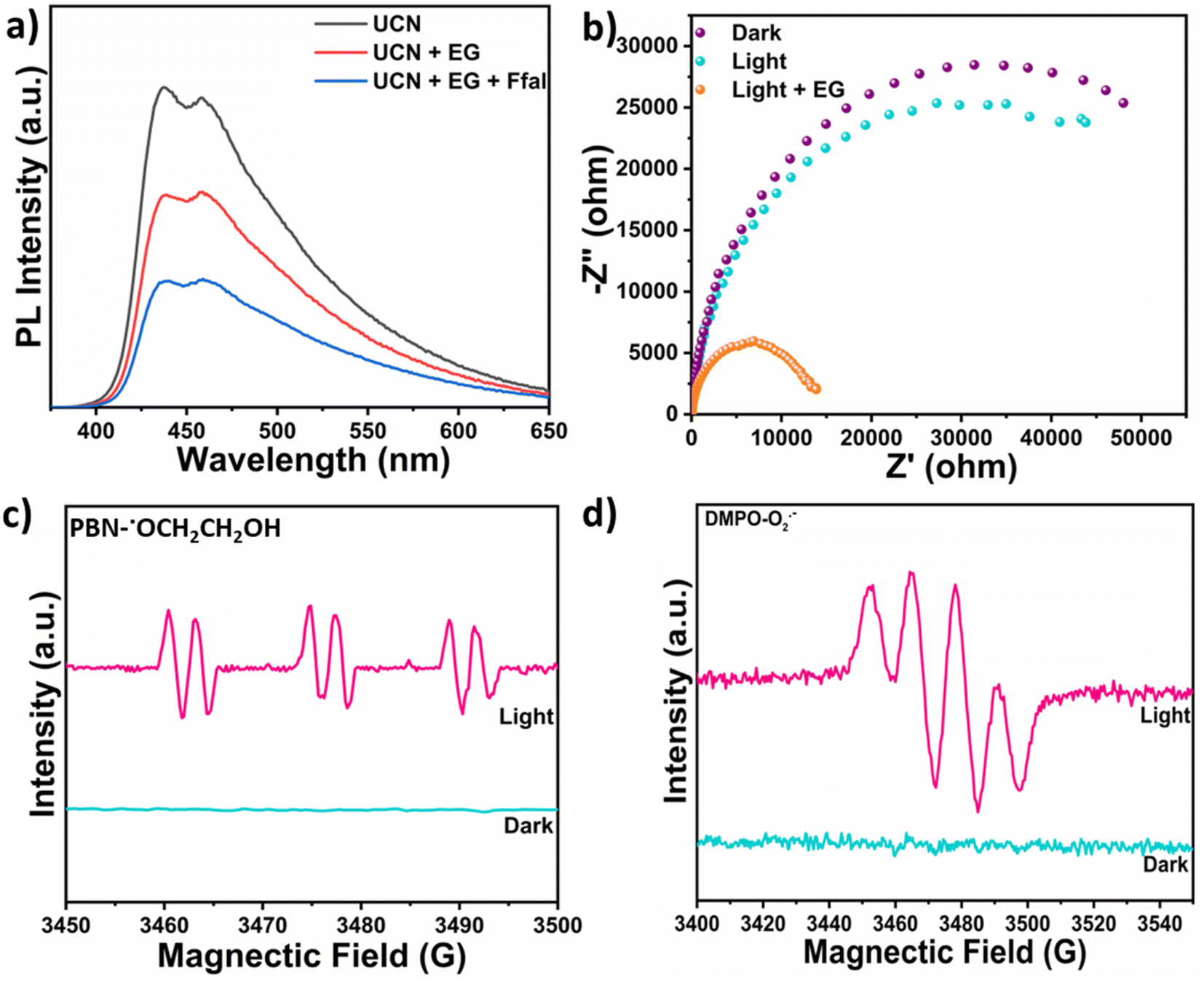 | ||
| Fig. 3 (a)PL spectra under different conditions; (b) Nyquist plot under different conditions; (c) EPR spectra of PBN-˙OCH2CH2OH in EG; and (d) EPR spectra of DMPO-O2˙− for superoxide. | ||
Photo-electrochemical studies were conducted to investigate the facile charge transfer occurring during the Ffal acetalization. The EIS-Nyquist plot of UCN was recorded (Fig. 3b) at 1.86 V vs. RHE bias potential. The arc radius in the Nyquist plot is correlated with the charge transfer resistance between the working electrode (coated with UCN) and the electrolyte interface.36 Upon light illumination, a notable reduction in the arc radius was observed, corroborating the lower charge transfer resistance (i.e. improved charge transfer kinetics) compared to dark conditions. Surprisingly, the arc radius further decreased significantly with the addition of EG under light. This strongly suggests the capability of UCN as a photocatalyst for EG oxidation, which subsequently drives the acetalization of Ffal.37 Furthermore, LSV (linear sweep voltammetry) (Fig. S12†) was also performed to evaluate the photocatalytic activity of UCN. The addition of ethylene glycol results in an increased anodic current density to 0.40 mA cm−2 with a decrease in onset potential (1.46 V vs. RHE).38 This supports the facile oxidation of EG over UCN, which further initiates the acetalization of Ffal. Invariably, all the above studies corroborate facile charge transfer between UCN and the substrates which facilitates Ffal acetalization with EG.
The mechanistic pathway for the synergistic acetalization of the Ffal and H2O2 production is illustrated in Scheme 1 based on the scavenging experiment, PL and photo-electrochemical studies. First, strong absorption of substrates i.e. Ffal and EG occurs over the surface of the UCN photocatalyst. Under visible light illumination, charge carriers (holes and electrons) were generated on the surface of the photocatalyst. The holes in the valence band of UCN facilitate the oxidization of EG through interfacial charge transfer which is well aligned with PL and photo-electrochemical studies. The photo-oxidation of EG results in the generation of H+ and an alpha hydroxy ethoxy radical (CH2OHCH2O., intermediate 1) which is in good agreement with significant quenching in the photocatalytic acetalization reaction on adding TEMPO as a radical scavenger in the reaction mixture (Table S3,† entry 2). Furthermore, the formation of radical intermediate 1 was confirmed by in situ EPR analysis under light irradiation by using N-tert-butyl-a-phenylnitrone (PBN) as a spin-trapping agent in EG solution. After adding PBN, six hyperfine splitting signals were seen which corresponded to PBN-˙OCH2CH2OH (Fig. 3c).39 Additionally, corroboration of intermediate 1 provides indisputable evidence, to substantiate the formation of H+. Concurrently, the electrons in the conduction band reduce molecular O2 to generate a superoxide radical (O2˙−). Furthermore, EPR studies were employed to trap the O2˙− by using DMPO (5,5-dimethyl-1-pyrroline N-oxide) as a spin-trapping agent which exhibits the four hyperfine peaks as shown in Fig. 3d that correspond to characteristic peaks of the DMPO-O2˙− adduct.40 Furthermore, the formed O2˙− then combined with in situ generated H+, formed during oxidation of EG through holes, to produce H2O2. On the other hand, in situ generated H+ exhibited greater affinity towards electron-rich oxygen present in Ffal, thereby activating Ffal, resulting in the formation of intermediate 2. Subsequently, intermediate 2 undergoes a reaction with intermediate 1 leading to the formation of hemiacetal intermediate 3. Finally, the hemiacetal intermediate 3 will lose water molecules and undergo cyclization to generate the desired cyclic acetal product.41
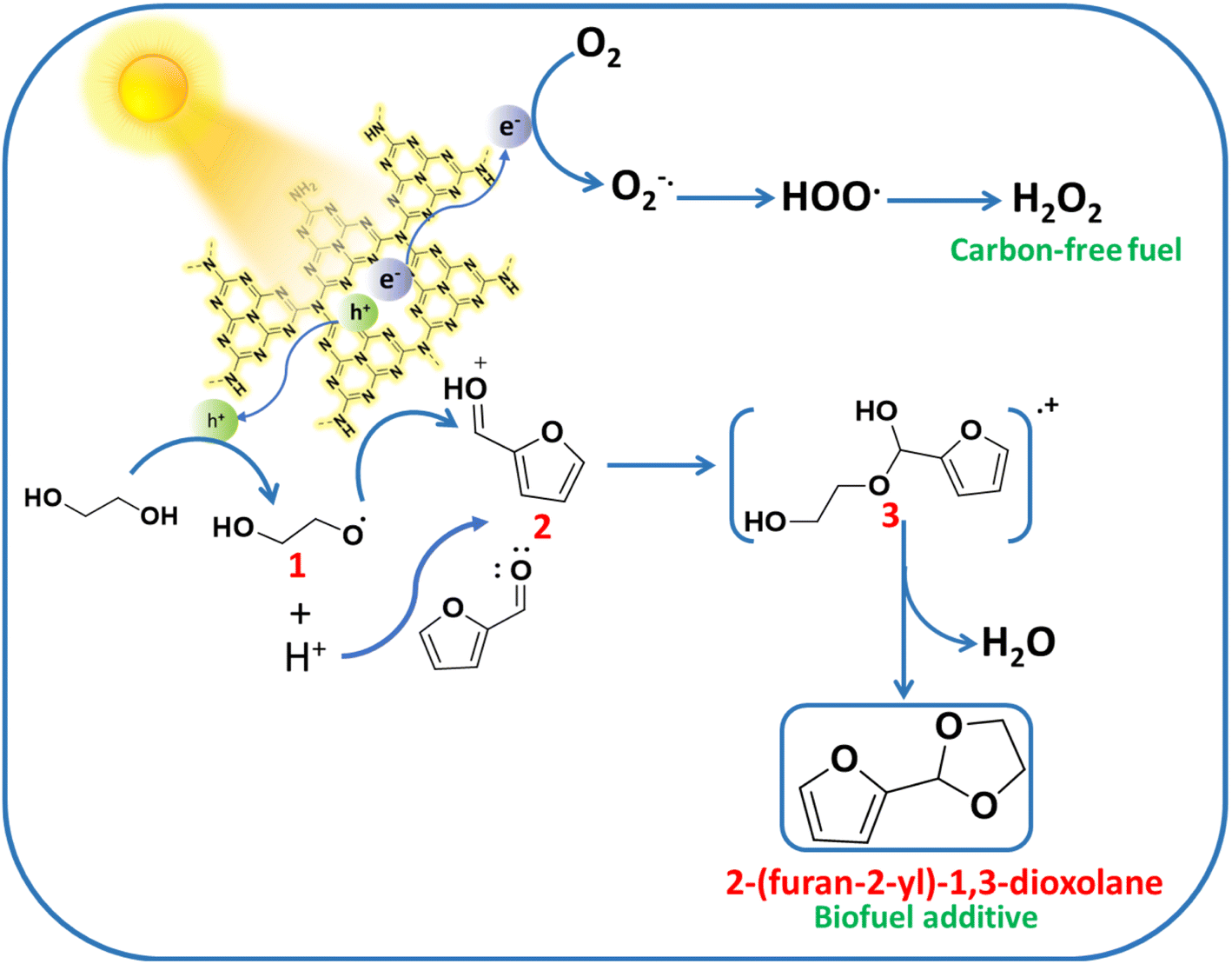 | ||
| Scheme 1 Mechanistic illustration of acetalization of Ffal with EG on UCN under visible light. | ||
2.4 Stability studies
The effectiveness of the photocatalyst depends upon its reusability. Recycling experiments were carried out for four consecutive cycles to determine the photocatalyst's robustness under these conditions. As exhibited in Fig. 4, UCN showed only a minimal decrease in the product (FD) yield (81%) and H2O2 yield 158 µmol g−1 after the 4th cycle. PXRD spectra of UCN after recyclability exhibited a similar pattern to fresh UCN as shown in Fig. S13a.† To examine the morphological aspect of the recycled catalyst, FESEM analysis was carried out as shown in Fig. S13b,† and no significant change was observed in the morphology of the catalyst. Furthermore, the surface area of the recovered catalyst was measured and it slightly decreased to 72 m2 g−1 (Fig. S13c†) which might be due to the pore blockage. Importantly, the slight decrease in surface area might be the reason for the minimal decrease in photocatalytic activity in the 4th cycle.42 Ultimately, from the recyclability studies, the reusability and robustness of the photocatalyst for the acetalization of Ffal can be concluded.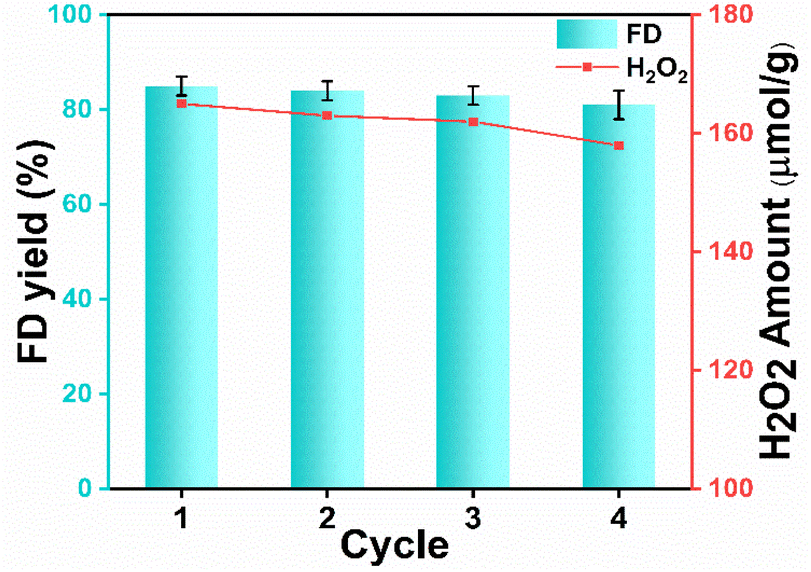 | ||
| Fig. 4 Recyclability study of the UCN photocatalyst. Reaction conditions: Ffal (1 mmol), UCN (50 mg), EG (20 mL), reaction time (6 h), O2 pressure (1 atm), 25 °C, and visible light. | ||
In regard to the large-scale production of cyclic acetal coupled with H2O2 production, the utilization of natural sunlight rather than artificial light sources represents a prominent step for an economically viable process. Thus, we have carried out the reaction under natural sunlight for 6 h as shown in Fig. S14a.† Under natural sunlight, we have achieved 70% cyclic acetal (FD) yield accompanied by 150 µmol g−1 H2O2 after 6 h of reaction time. The intensity of sunlight and temperature during the reaction is presented in Fig. S14b.† The liquid product was analyzed by GC-MS (Fig. S14c†) whereas H2O2 was quantified using iodometry (Fig. S14d†). The outstanding results obtained under natural sunlight for the simultaneous production of cyclic acetals and H2O2 by employing a metal-free photocatalyst signifies the practicality of this work and offers a viable pathway for sustainable and economically feasible large-scale production of commodity chemicals through biomass valorization.
3. Conclusion
This present study reports the effective utilization of metal-free photocatalysts for the acetalization of Ffal to synthesize bio-fuel additives coupled with H2O2 production under visible light for the first time. Notably, conventional acetalization synthesis often entails the utilization of strong acids and high temperatures, resulting in environmentally unfriendly reaction conditions. However, our approach of photocatalytic acetalization of biomass-derived substrates eliminates the exigency for external acid and can be accomplished under ambient conditions. Importantly, the metal-free photocatalyst i.e. UCN has shown exceptional performance yielding 85% of FD (bio-fuel additive) along with a significant amount of H2O2 in 6 h of reaction time. Moreover, the remarkably high yield of cyclic acetal under natural sunlight exhibits the practicality of the UCN photocatalyst for possible large-scale production. Photoluminescence and photo-electrochemical studies provide insight into the charge transfer mechanism underlying the photocatalytic acetalization of Ffal. The mechanism for acetalization has been illustrated through in situ EPR studies under light irradiation and experiments performed under controlled reaction conditions. Overall, this study offers a viable pathway for developing metal-free photocatalysts for efficiently harnessing sunlight for sustainable production of solar fuel integrated with biomass upgradation to value-added products.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Shivali Dhingra thanks INST Mohali for a research fellowship. K. Kailasam would like to thank the Anusandhan National Research Foundation (ANRF) for the project Science & Engineering Research Board (SERB), Core Research Grant Scheme (Sanction order No. “CRG/2023/001875”) for the financial support. Arpna Jaryal thanked CSIR, New Delhi, for a fellowship under File No. 09/1129(0010)/2017-EMR-I. Deepak Kumar Chauhan thanks INST Mohali for a research fellowship. The author thanks CRF IIT Delhi Sonipat campus for providing the EPR facility.References
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- M. Wang, J. Ma, H. Liu, N. Luo, Z. Zhao and F. Wang, ACS Catal., 2018, 8, 2129–2165 CrossRef CAS.
- C. Li, J. Li, L. Qin, P. Yang and D. G. Vlachos, ACS Catal., 2021, 11, 11336–11359 CrossRef CAS.
- D. K. Chauhan, V. R. Battula, A. Giri, A. Patra and K. Kailasam, Catal. Sci. Technol., 2022, 12, 144–153 RSC.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- D. S. Desai and G. D. Yadav, Ind. Eng. Chem. Res., 2019, 58, 16096–16105 CrossRef CAS.
- J. He, Q. Qiang, L. Bai, W. Su, H. Yu, S. Liu and C. Li, Ind. Chem. Mater., 2024, 2, 30–56 RSC.
- S. Kirchhecker, A. Dell'Acqua, A. Angenvoort, A. Spannenberg, K. Ito, S. Tin, A. Taden and J. G. de Vries, Green Chem., 2021, 23, 957–965 RSC.
- K. Kumar, S. Pathak and S. Upadhyayula, Renewable Energy, 2021, 167, 282–293 CrossRef CAS.
- J. L. Dong, L. S. H. Yu and J. W. Xie, ACS Omega, 2018, 3, 4974–4985 CrossRef CAS PubMed.
- D. Li, F. Shi, J. Peng, S. Guo and Y. Deng, J. Org. Chem., 2004, 69, 3582–3585 CrossRef CAS PubMed.
- B. Karimi, H. Hazarkhani and J. Maleki, Synthesis, 2005, 279–285 CrossRef CAS.
- M. Kotke and P. R. Schreiner, Tetrahedron, 2006, 62, 434–439 CrossRef CAS.
- H. Song, F. Jin, Q. Liu and H. Liu, Mol. Catal., 2021, 513, 111752 CrossRef CAS.
- M. Iwamoto, Y. Tanaka, N. Sawamura and S. Namba, J. Am. Chem. Soc., 2003, 125, 13032–13033 CrossRef CAS PubMed.
- H. Yi, L. Niu, S. Wang, T. Liu, A. K. Singh and A. Lei, Org. Lett., 2017, 19, 122–125 CrossRef CAS PubMed.
- N. F. Nikitas, I. Triandafillidi and C. G. Kokotos, Green Chem., 2019, 21, 669–674 RSC.
- N. Spiliopoulou, N. F. Nikitas and C. G. Kokotos, Green Chem., 2020, 22, 3539–3545 RSC.
- X. Liu, Y. Zhou, D. Zeng, H. Wang, S. Qiao, L. Zhang and W. Wang, ChemistrySelect, 2021, 6, 8074–8079 CrossRef CAS.
- S. U. Raut and P. R. Bhagat, Biomass Convers. Biorefin., 2023, 13, 7737–7754 CrossRef CAS.
- J. Chang, J. Shi, Q. Li, S. Li, Y. Wang, Y. Chen, F. Yu, S. Li and Y. Lan, Angew. Chem., 2023, 135, e202303606 CrossRef.
- H. Liang, A. Wang, R. Cheng, X. Tian, S. Jing and P. Tsiakaras, Small, 2023, 19, 2303813 CrossRef CAS PubMed.
- Q. Hu, Y. Huang, X. Yu, S. Gong, Y. Wen, Y. Liu, G. Li, Q. Zhang, R. Ye and X. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 42611–42621 CrossRef CAS PubMed.
- W. Ong, L. Tan, Y. H. Ng, S. Yong and S. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- M. Abdullah Khan, I. F. Teixeira, M. M. J. Li, Y. Koito and S. C. E. Tsang, Chem. Commun., 2016, 52, 2772–2775 RSC.
- G. Xu, A. Wang, J. Pang, X. Zhao, J. Xu, N. Lei, J. Wang, M. Zheng, J. Yin and T. Zhang, ChemSusChem, 2017, 10, 1390–1394 CrossRef CAS PubMed.
- M. Yang, R. Lian, X. Zhang, C. Wang, J. Cheng and X. Wang, Nat. Commun., 2022, 13, 4900 CrossRef CAS PubMed.
- A. Kumar, P. K. Prajapati, M. S. Aathira, A. Bansiwal, R. Boukherroub and S. L. Jain, J. Colloid Interface Sci., 2019, 543, 201–213 CrossRef CAS PubMed.
- V. R. Battula, B. Rawat and K. Kailasam, ACS Appl. Polym. Mater., 2023, 5, 1989–1997 CrossRef CAS.
- A. Jaryal, V. R. Battula and K. Kailasam, ACS Appl. Energy Mater., 2020, 3, 4669–4676 CrossRef CAS.
- K. Kumar, S. Pathak and S. Upadhyayula, Renew. Energ, 2021, 167, 282–293 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 735–758 CrossRef CAS.
- X. T. Xu, L. Pan, X. Zhang, L. Wang and J. J. Zou, Adv. Sci., 2019, 6, 1801505 CrossRef PubMed.
- S. Q. Liu, M. R. Gao, S. Wu, R. Feng, Y. Wang, L. Cui, Y. Guo, X. Z. Fu and J. L. Luo, Energy Environ. Sci., 2023, 16, 5305–5314 RSC.
- D. Liu, J.-C. Liu, W. Cai, J. Ma, H. Bin Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef PubMed.
- J. B. G. Filho, R. D. F. Rios, C. G. O. Bruziquesi, D. C. Ferreira, H. F. V. Victória, K. Krambrock, M. C. Pereira and L. C. A. Oliveira, Appl. Catal., B, 2021, 285, 119814 CrossRef CAS.
- X. Nie, Y. Zhao, W. Gao, W. Liu, X. Cheng, Y. Gao, N. Shang, S. Gao and C. Wang, Chem.–Eur. J., 2023, 29, e202203607 CrossRef CAS PubMed.
- C. H. Rao, S. Ma, J. W. Cui, M. Z. Jia, X. R. Yao and J. Zhang, Green Chem., 2022, 24, 5862–5868 RSC.
- D. Feng, Y. Cheng, J. He, L. Zheng, D. Shao, W. Wang, W. Wang, F. Lu, H. Dong, H. Liu, R. Zheng and H. Liu, Carbon, 2017, 125, 454–463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03730h |
| This journal is © The Royal Society of Chemistry 2024 |