
Metal-free bi-functional cooperative catalysis: amine and quaternary amine-functionalized dendritic fibrous nanosilica as heterogeneous catalysts for the Henry reaction and CO2 conversion†
Sanjay
Yadav‡
*ac,
Hanuman G.
Kachgunde‡
bc,
Nishu
Choudhary
ac,
Gopal H.
Wanole
bc,
Krishnan
Ravi
 *bc,
Ankush V.
Biradar
*bc and
Alok Ranjan
Paital
*ac
*bc,
Ankush V.
Biradar
*bc and
Alok Ranjan
Paital
*ac
aSalt and Marine Chemicals Division, CSIR-Central Salt & Marine Chemicals Research Institute, G. B. Marg, Bhavnagar-364002, Gujarat, India. E-mail: arpaital@csmcri.res.in; sychem00700@gmail.com
bInorganic Materials and Catalysis Division, CSIR-Central Salt & Marine Chemicals Research Institute, G. B. Marg, Bhavnagar-364002, Gujarat, India. E-mail: ankush@csmcri.res.in; krishkicha545@gmail.com
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 16th August 2024
Abstract
This study explores amine-functionalized dendritic fibrous nanosilica (DFNS) as a highly effective, base-free heterogeneous catalyst for nitro-aldol (Henry) condensation and additive-free CO2 utilization. Initially, high surface area DFNS was synthesized using a novel template in an emulsion biphasic system and chemically processed to create DFNS@NH2 for the Henry reaction and further quaternized to form DFNS@TBAB for CO2 utilization. DFNS is expected to outperform conventional mesoporous materials (SBA-15 and MCM-41) due to its dendritic morphology, higher surface area, superior mass transfer, ease of functionalization, and greater active site exposure, enhancing its catalytic efficiency and versatility. In addition, the total basicity values for DFNS@NH2 and DFNS@TBAB were determined to be 0.48 and 0.124 mmol g−1, respectively. Furthermore, FeSEM, TEM, and HR-TEM analyses evidenced the dendritic morphology of the solid substrate with a fibrous structure termed DFNS. Amine-functionalized DFNS achieved complete conversion of benzaldehyde to β-nitrostyrene with over 99% selectivity at 50 °C in 12 h using nitromethane. It successfully catalyzed a range of substrates in the Henry reaction, including electron-donating and electron-withdrawing aromatic aldehydes and biologically derived aldehydes such as furfural and 5-methyl furfural. Electron-withdrawing groups on aromatic rings showed reduced catalytic activity. Furthermore, DFNS functionalized with n-butyl bromide produced a quaternary amine catalyst capable of converting CO2 and styrene oxide to styrene carbonate without external additives, achieving 94% conversion and 98% selectivity. This dual functionality highlights DFNS's potential in carbon–carbon bond formation and CO2 utilization, offering a promising route for CO2 emission mitigation through chemical conversion.
1 Introduction
Heterogeneous catalysts with cooperative catalytic actions are vital for sustainability, selectivity, and reduced environmental impact in the chemical industry.1–3 Hybrid organic–inorganic substrates enhance catalytic activity and selectivity and are preferred over homogeneous catalysts due to their easy recovery and reusability.4–6 Effective cooperativity relies on the spatial separation and organized arrangement of acidic and basic active sites, whether they are chemically compatible or incompatible. Strategically placed, these sites can activate reactants and lower activation energy through dual or sequential processes. The proximity of acid and base sites significantly influences reactant activation, which is affected by factors such as active site distance, electronic effects, surface properties, and solvent effects.7,8 Mesoporous inorganic substrates such as SBA-15 and MCM-41 are ideal for catalytic performance due to their unique structural properties and their ability to immobilize organic ligands, creating dense active sites. Silanol groups on silica act as Lewis acid sites, and their interaction with Lewis bases enhances catalytic activity.9–12 Hybrid systems are created by functionalizing mesoporous silica with graftable alkoxysilanes, allowing customization of organic moieties for specific reactions.12–16 Functional groups are introduced either during silica synthesis (co-condensation) or by post-synthetic grafting with organosilanes. The effectiveness of these catalysts depends on the nature, extent, and pore structure of surface functionalization.6 However, achieving high active site loading while maintaining structural order is challenging. SBA-15 and MCM-41 have limited accessibility due to their small cylindrical pore channels, which restrict mass transport. High catalyst loading can block these pores, further reducing internal surface availability. For example, nitriding MCM-41 and SBA-15 with ammonia at high temperatures to develop solid bases led to restricted access to amine sites and structural distortion, limiting their catalytic performance and thermal stability.17–19Regarding the catalytic template, the dendritic fibrous nanosilica (DFNS) is considered superior to conventional mesoporous materials with its unique features of dendritic fibrous morphology, tuneable surface area, radially accessible pores, and enhanced stability.20–22 DFNS surpasses MCM-41 and SBA-15 in surface area accessibility, facilitating better diffusion of reactants and increased loading of active sites without pore blockage. The fibrous morphology of DFNS creates nanoscopic gaps between fibers, resulting in a substantial surface area and increased pore volume, which can be easily functionalized with active moieties.23 Owing to its high surface area and the availability of nanoscopic space for functionalization with active moieties, DFNS has found widespread application as an inorganic solid support material for hydrogenizing homogeneous catalysts.24,25 Previous studies have reported many metal-based catalysts for the Henry reaction.26 In addition, various acidic and basic catalytically active moieties have been immobilized on DFNS to produce efficient and recoverable catalysts for a range of organic transformations, including nitroarene reduction, Suzuki–Miyaura cross-coupling, and Suzuki coupling reactions.23 However, in the Henry reaction27 and CO2 utilization28,29 metal-free DFNS materials have not been extensively studied. The Asefa research group has investigated a range of amine-functionalized silica materials for their effectiveness in controlling and improving the selectivity of the nitroaldol condensation reaction. Their results show that the type of amine functionality within the silica materials has a significant effect on the outcome of the reaction. Specifically, they observed that silica materials functionalized with primary amines exhibited selectivity towards α and β-unsaturated nitro compounds, whereas those with secondary amine functionality enhanced the formation of Michael addition products.30 In addition to this, the research group is working intensively on the co-operative catalysis of the nitro-aldol condensation reaction between aldehydes and nitroalkanes.31 A recent development has been the introduction of a bifunctionalized boronic acid–amine cooperative catalyst for the Henry reaction. This innovative catalyst combines the advantages of amine-functionalized silica with a quaternary ammonium salt and has demonstrated efficacy in the cycloaddition of CO2 with epoxides. In addition, the incorporation of boronic acid into primary amine-functionalized silica has been shown to enhance the catalytic activity in nitroaldol condensation reactions.32 Furthermore, Collier et al. reported acid–base bifunctional amino-silica catalysts for nitroaldol condensation between p-hydroxybenzaldehyde and nitromethane. Interestingly, the presence of trimethylsilyl functionality on dendritic silica materials was found to decrease the catalytic activity. The silanol group (–OH) on silica materials showed weak acidic properties. Notably, the combination of primary amine and silanol groups was observed to synergistically enhance the catalytic activity of nitro-aldol condensation.33 Furthermore, the combination of primary amines with quaternary ammonium salt facilitates the synthesis of cyclic carbonates from epoxides and CO2. It has been observed that simple amine-functionalized materials alone are not sufficiently effective in activating CO2. Therefore, the use of additional co-catalysts such as quaternary ammonium salts and phosphonium salts is necessary for epoxide and CO2 activation.34 Numerous studies have been conducted on the synthesis of cyclic carbonates from epoxides using amine-functionalized silica, with quaternary ammonium salt being employed as an external additive.35–38 The amine functionality present on the solid substrate serves as a precursor for quaternary ammonium salts. Our group recently developed quaternary ammonium-functionalized sugarcane bagasse as a catalyst for additive-free CO2 cycloaddition with epoxides, showing moderate catalytic activity.38 Nevertheless, it's worth noting that the reported catalyst necessitates high CO2 pressure and raises concerns regarding thermal stability for commercial viability. Hence, there is a pressing need for metal-free catalyst systems that offer thermal stability and high conversion rates, ensuring efficiency in CO2 cycloaddition with epoxides under mild conditions, thereby promoting sustainable methodologies.
Herein, we present a metal-free cooperative catalyst comprising amine-functionalized dendritic fibrous nanosilica (DFNS@NH2) for the Henry reaction, operating under mild conditions and the further quaternized product DFNS@TBAB for CO2 conversion. Initially, the dendritic fibrous nanosilica (DFNS) was synthesized via an emulsion biphasic stratification method using a templating micellar approach yielding a high surface area. The use of tetraphenylphosphonium bromide as a template leads to the formation of highly uniform, discrete dendritic fibrous nanosilica (DFNS) with a high surface area, which has not been previously utilized. The dendritic structure of the material enables efficient access and encapsulation of organic ligands within the pore channels, facilitating the creation of densely active sites. These active sites play a crucial role in determining the catalytic performance efficiency of the materials. Post-synthesis of DFNS, it was covalently functionalized with 3-APTES to produce the DFNS@NH2 material and further functionalized with butyl bromide to yield the quaternized product, DFNS@TBAB. The catalyst, DFNS@NH2, demonstrates efficacy in promoting nitroaldol condensation between p-hydroxybenzaldehyde and nitromethane at room temperature. It also catalyzes a range of substrates in the Henry reaction, including electron-donating and electron-withdrawing aromatic aldehydes, as well as biologically derived aldehydes such as furfural and 5-methyl furfural. In addition, the quaternary ammonium-functionalized material, DFNS@TBAB, serves as a metal-free, solvent-free, and co-catalyst-free heterogeneous catalyst for the cycloaddition of CO2 with epoxides under mild conditions (using balloon CO2 pressure) achieving 94% conversion and 98% selectivity in the conversion of styrene oxide and CO2 to styrene carbonate. Therefore, this catalyst minimizes resource utilization and offers economic benefits without causing secondary pollution. Additionally, as a heterogeneous catalyst, it can be recycled easily, further contributing to sustainability. The excellent results are due to the bifunctional nature of the materials offering cooperative catalysis. The cooperative action of acid silanol groups and amine sites provides complementary catalytic effects, enhancing the overall efficiency and selectivity of the CO2 cycloaddition reaction with epoxides. The acid–base bifunctional nature of the catalyst system allows for synergistic interactions between the acidic and basic sites, leading to improved catalytic performance under mild conditions. To the best of our knowledge, this study marks the first instance of amine-functionalized dendritic fibrous nanosilica serving as a single catalyst for both the Henry reaction and CO2 conversion without the need for metals, co-catalysts, or solvents.
2 Results & discussion
2.1 Synthesis and characterization
The synthesis (Scheme 1) represents the biphasic stratification approach for the synthesis of the dendritic fibrous nanosilica (DFNS) substrate material. For the first time tetraphenylphosphonium bromide was used as a template, tetraethyl orthosilicate as a silica source, urea as a hydrolysis medium and p-xylene–H2O as a solution system, as detailed in the Experimental section.39,40 The nucleation phase was supported by cosolvent propanol, followed by the circular reassembly of the micellar seed phase at a mild temperature. The subsequent refluxing process resulted in a dendritic structure of the material with a high surface area and high pore volume of 1005 m2 g−1 and 1.899 cm3 g−1 compared to the original KCC-1 material.20 This enhanced structure is ideal for multifunctional payloads, creating effective dense sites for catalytic actions.20 After the synthesis of DFNS, it was then successfully functionalized with 3-aminopropyltriethoxysilane (3-APTES) creating the DFNS@NH2 material and a subsequent quaternization reaction with butyl bromide affording the final material as DFNS@TBAB.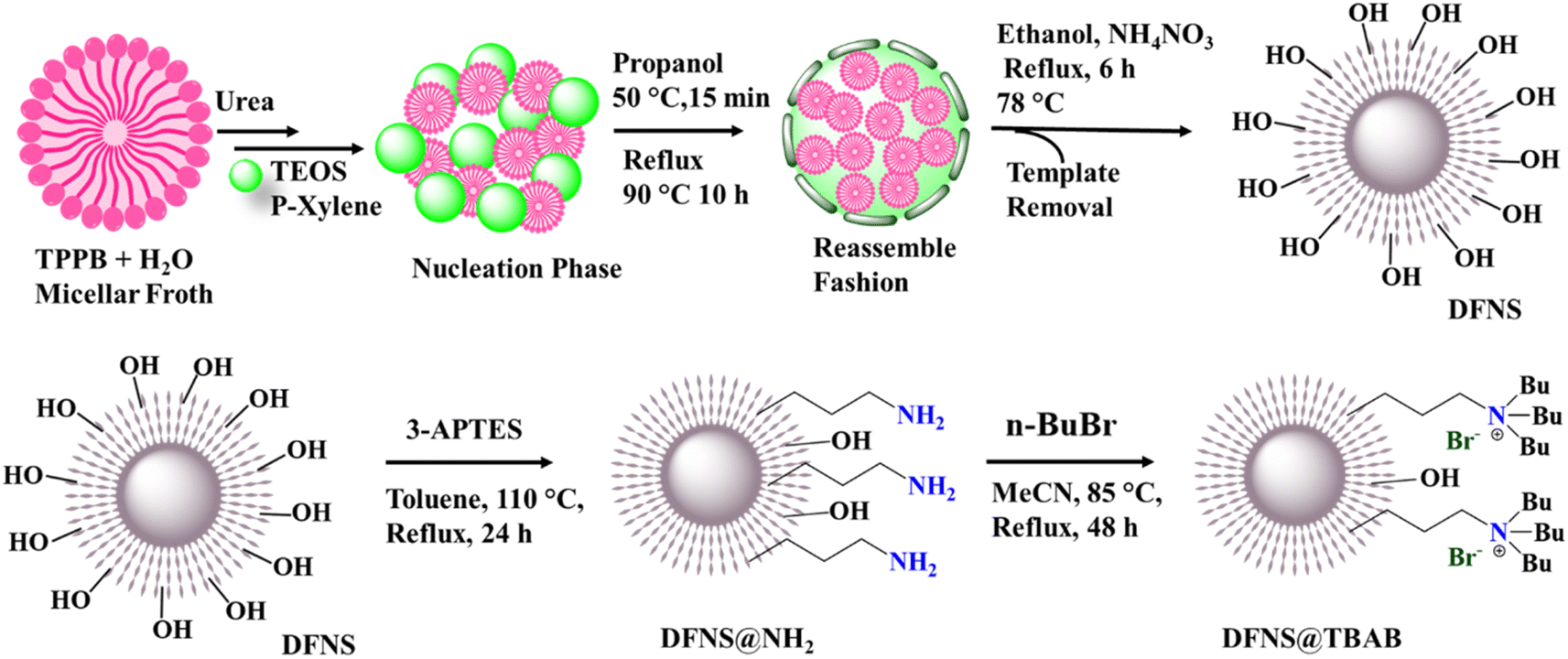 | ||
| Scheme 1 The schematic synthetic procedure of the dendritic fibrous nanosilica (DFNS), amine-functionalized DFNS (DFNS@NH2), and quaternary amine-functionalized DFNS (DFNS@TBAB). | ||
The synthesized materials were thoroughly characterized to determine the structural properties, functionalities, stability, nature and surface states of the materials. The morphological characteristics of the materials were revealed through SEM images, depicting the formation of spherical silica particles with an approximate size of 320–340 nm (Fig. 1A–F). A detailed examination of the FeSEM images unveiled 3-D dimensional fibrous arrangements, forming dendrimer-like structures. The silica framework of the synthesized was confirmed by EDX analysis and elemental mapping showing signals for silicon (Si) and oxygen (O) elements (Fig. S1†).
 | ||
| Fig. 1 (A and D) The FeSEM images of the dendritic fibrous nanosilica (DFNS); (B and E) the FeSEM images of the DFNS@NH2 material; (C and F) the FeSEM images of the DFNS@TBAB material; (G and J) the TEM & HR-TEM images of the material DFNS; (H and K) the TEM images of the aminated material DFNS@NH2; (I and L) the TEM images of the quaternized material DFNS@TBAB. | ||
The fibrous nature was further affirmed by TEM images & HR-TEM images, illustrating fiber-like structures that appeared denser after functionalization (Fig. 1G–L). The DFNS's fibrous structure, coupled with a high surface area, facilitates 3-D accessibility of pores for ligand functionalization creating active dense sites. Sorption studies using SBET N2 (Fig. 2A and B) revealed a typical type IV isotherm with a capillary condensation maximum for DFNS and DFNS@NH2, transitioning to a type II isotherm with negligible capillary condensation for the final material DFNS@TBAB (Fig. 3A). The DFNS exhibited a high surface area/pore volume/pore diameter (1005 m2 g−1/1.89 cm3 g−1/6.2 nm), which sequentially decreased with DFNS@NH2 (225 m2 g−1/0.6347 cm3 g−1/4.2 nm) and DFNS@TBAB (28.35 m2 g−1/0.192 cm3 g−1/1.8 nm). The progressive reduction in surface area, pore volume, and pore diameter from DFNS to DFNS@TBAB (Table 1) signifies the surface functionalization of radially available pore channels with the organic moieties. Despite the chemical processing, the amorphous nature of the DFNS and the quaternized material DFNS@TBAB remains unchanged, as evidenced by broad peaks at 23.56 and 23.01 for DFNS (Fig. 2C). The thermogravimetric profiles of DFNS@NH2 and DFNS@TBAB were also studied to determine the stability, nature, and loading of organic functionalities in the materials (Fig. 2D). In the initial region of both materials (0 to 200 °C), there is a loss of physically adsorbed water. The subsequent region (200–550 °C) indicates the loss of organic functionalities, while the third region (550–800 °C) represents the dehydroxylation of surface silanol groups. The TGA profile aligns with the progressive functionalization reactions in DFNS@NH2 and DFNS@TBAB, resulting in a greater organic loss of −33.18% in the latter.39
 | ||
| Fig. 2 (A and B) The SBET N2 sorption isotherms and pore size distribution of the synthesized materials DFNS, DFNS@NH2 & DFNS@TBAB; (C and D) the high-angle PXRD and TGA profiles of the synthesized materials DFNS, DFNS@NH2 & and DFNS@TBAB. | ||
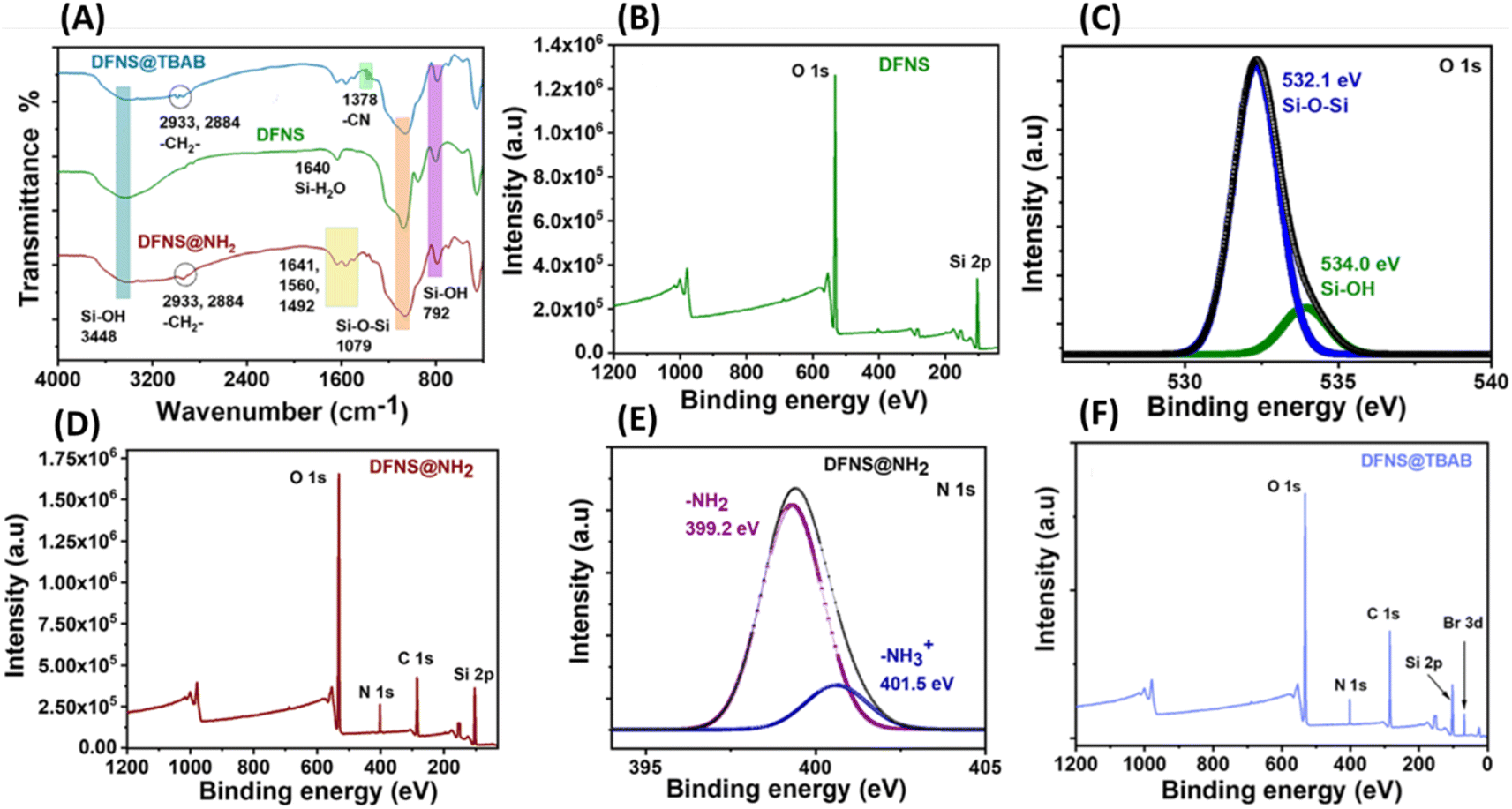 | ||
| Fig. 3 (A) The comparison of the FTIR spectra of the synthesized DFNS, DFNS@NH2 and the final material DFNS@TBAB; (B and C) the XPS full scan and O 1s core–shell spectrum of the DFNS material; (D and E) the XPS full scan and N 1s core–shell spectrum of the DFNS@NH2 material; (F) the XPS full scan spectrum of the material DFNS@TBAB. | ||
| Samples | Surface area [m2 g−1] | Pore volume [cm3 g−1] | Pore width [nm] |
|---|---|---|---|
| DFNS | 1005 | 1.89 | 6.2 |
| DFNS@NH2 | 225 | 0.634 | 4.2 |
| DFNS@TBAB | 28.35 | 0.192 | 1.8 |
The information on covalent functionalization was further analyzed through FT-IR & XPS spectra analyses (Fig. 3). The characteristic peaks at 3448 cm−1 for Si–OH and 1079 cm−1 for Si–O–Si linkage and additional peaks at 1641, and 1560 cm−1 are attributed to –NH bending and stretching vibrations in the DFNS@NH2 material. Peaks at 2933, 2884, and 1492 cm−1 were associated with alkyl –CH stretching of the APTES group, confirming the successful immobilization of amine functionality in the DFNS@NH2 material.39,41 The quaternization reaction was confirmed by the intense alkyl stretching (–CH2–) and C–N peaks (1378 cm−1) in the final material DFNS@TBAB with an intact silica framework supported by FT-IR (Fig. 3A) and EDX analysis, which show peaks for elemental Br (Fig. S2B†).
The chemical and surface states of the material were also evaluated by X-ray photoelectron spectroscopy analysis (XPS). The synthesis of the DFNS material was confirmed by the XPS full scan spectrum, which gave signals for silicon Si 2p (Si) and oxygen 1s (O) supported by EDX analysis & elemental colour mapping (Fig. 3B and S1†). The Si–O–Si framework peak at 532.1 eV and Si–OH groups (534.0 eV) were evident from core–shell O 1s spectra of the DFNS material (Fig. 3C). The covalent attachment of the APTES group was evident from the full scan XPS spectrum of the DFNS@NH2 material, which revealed the presence of oxygen, nitrogen, carbon, and silicon elements. This was further confirmed by the EDX analysis (Fig. 3D and S2A†). The core–shell C 1s spectrum showed peaks for C–Si, C–C and C–N functionalities at 283.5, 284.5 and 286.5 eV. Also, the core–shell N 1s of the DFNS material exhibited peaks at 399.2 and 401.5 eV for the NH2 and NH3+ groups (Fig. 3E and S3A†).35 The quaternization reaction of the DFNS@NH2 material to afford the DFNS@TBAB material was confirmed by the full scan XPS analysis showing increased intensity of the C 1s and the presence of bromide ions (Fig. 3F and S2, S3B†). To confirm further, the Raman spectra of the DFNS and DFNS@NH2 were recorded and compared (Fig. S4A†). The fingerprint region of the Si–O–Si network of the DFNS ranges from 400 to 1100 cm−1 corresponding to bending, symmetric, and asymmetric stretching vibrations, whereas the broad intense band at around 3100 cm−1 is due to Si–OH stretching and the band at 850 cm−1 cm corresponds to the bending vibrations. Apart from these characteristic peaks, DFNS@NH2 exhibited extra peaks at 1065, 1295, 1443, 1412, 1534, 1600, 3424, and 3341 cm−1 corresponding to C–C, alkyl (–CH2–, wag & bending), Si–C, C–N and NH bending and stretching frequency vibrations respectively. Furthermore, the UV-vis spectral analysis was performed for the synthesized DFNS, DFNS@NH2, and DFNS@TBAB materials (Fig. S4B†). Although the materials displayed no significant absorption peaks, the DFNS@NH2 & DFNS@TBAB materials showed a slightly broad absorption band at 282 nm indicating a functionalization process. The acidity of the amine-modified DFNS and DFNS modified with quaternary ammonium salt was analyzed through CO2-TPD. The amine-modified DFNS demonstrated robust acidity at 500 °C, whereas the DFNS modified with quaternary ammonium salt displayed moderate to strong acidity at temperatures of 300 °C and 500 °C. The overall basicity values for DFNS@NH2 and DFNS@TBAB were found to be 0.48 and 0.124 mmol g−1, respectively (Fig. S5†).
2.2 Catalytic activity
α,β-Unsaturated nitroalkenes are considered one of the most important intermediates in the pharmaceutical industry and are used as building blocks in bioactive compounds. They are synthesized through the condensation of carbonyl compounds with nitroalkanes in the presence of a basic catalyst (Scheme 2). | ||
| Scheme 2 Synthesis of β-nitrostyrene using a heterogeneous catalyst. | ||
In this study, benzaldehyde and nitromethane were used as model substrates for the synthesis of α,β-unsaturated nitroalkenes, and the results are depicted in Fig. 4A. In the absence of a catalyst, fumed silica showed no catalytic activity, highlighting the necessity of a basic environment for the condensation reaction. Amine-functionalized dendritic mesoporous silica nanoparticles showed 87% conversion of benzaldehyde with good selectivity for β-nitrostyrene. Achieving selective production of α,β-unsaturated nitroalkenes over other nitro alcohols and Michael addition products is a challenging task in the Henry reactions. Previous work by Bedasso et al. reported a 65% selectivity of an α,β-unsaturated nitroalkene (2-(2-nitrovinyl)furan) in the reaction between furfural and nitromethane using an amino group-functionalized pitch-based carbocatalyst.42 The lower selectivity of α,β-unsaturated nitroalkenes in carbocatalysts may be attributed to the fewer basic sites present in the carbocatalyst. Furthermore, metal catalysts such as Zn-MOF by Gu et al. claimed to achieve size-dependent conversion rates, where smaller nitroalkanes yield better conversion than larger ones, indicating molecular size-dependent conversion rates.43 Similarly, in another report, an 80% yield of 2-nitro alcohol was achieved using a calcined eggshell (CES) (CaO and Ca(OH)2) as a natural catalyst.44 When MgO is used as the catalyst, the reaction achieves only a 51% conversion rate but retains a remarkable 99% selectivity for the desired product. However, introducing amine functionalization to MgO enhances catalytic activity while diminishing the selectivity for the desired α,β-unsaturated nitroalkenes. As further support, when SiO2 (fumed) and Ba(OH)2 were used as catalysts under similar conditions no catalytic activity was observed (Table S1,† entries 3 and 5). Also, to evaluate the effect of porosity on catalytic performance, the amine-functionalized DFNS was found to show better catalytic activity compared to aminated fumed silica. This improved performance can be attributed to the unique dendritic porous structure of the DFNS with a higher surface area and greater accessibility to the active sites (Table S1,† entries 4 and 6).
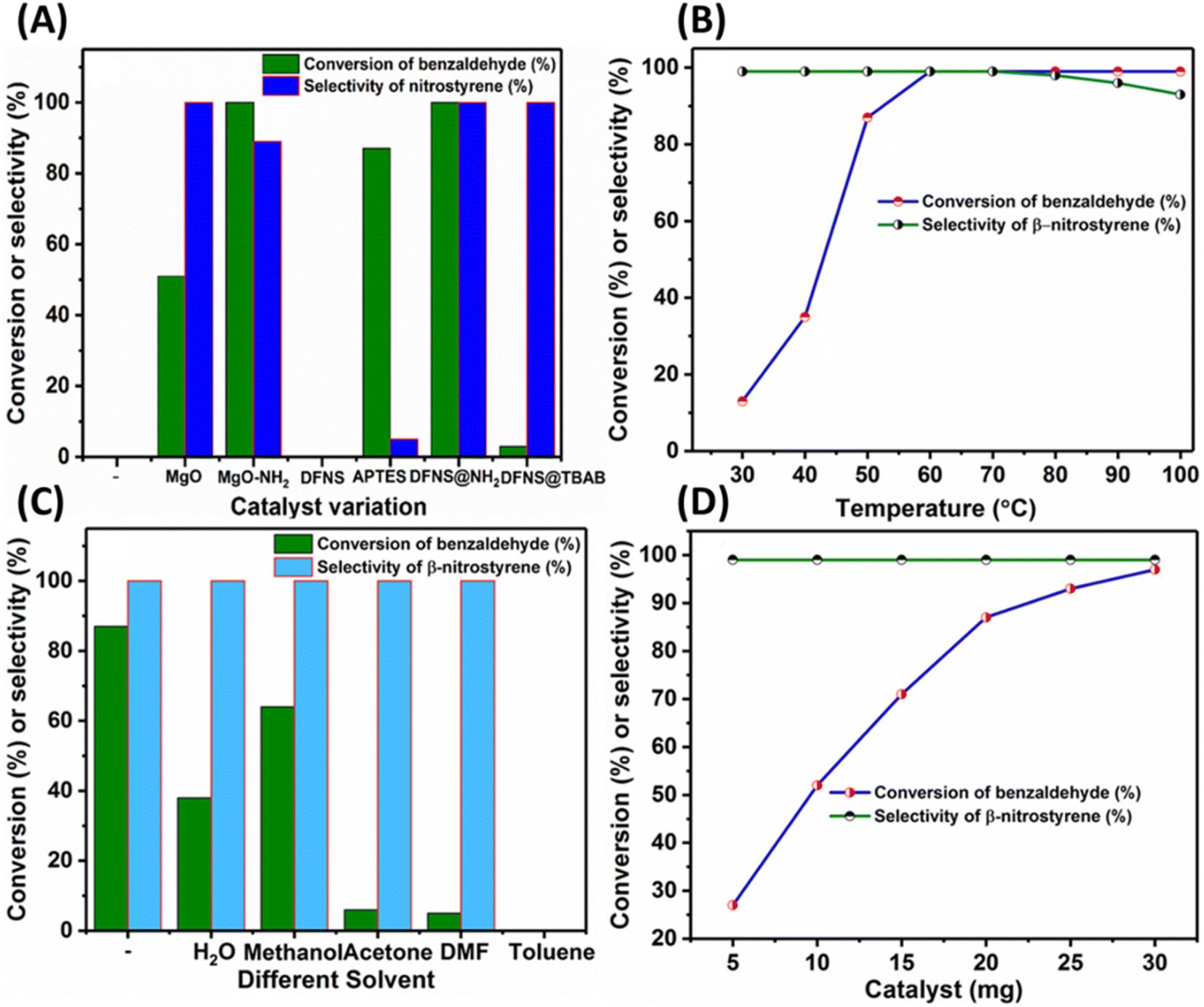 | ||
| Fig. 4 Optimization studies. Reaction conditions: (A) benzaldehyde: 2 mmol, nitromethane: 11.2 mmol, catalyst; 20 mg, temperature; 60 °C, time: 6 h, (B) benzaldehyde: 2 mmol, nitromethane: 11.2 mmol, DFNS@NH2; 20 mg, temperature; (30–100 °C), 6 h, (C) benzaldehyde: 2 mmol, nitromethane: 11.2 mmol, DFNS@NH2; 20 mg, solvent: 1 mL, temperature; 50 °C, time: 6 h, and (D) benzaldehyde: 2 mmol, nitromethane: 11.2 mmol, DFNS@NH2; (5–30 mg), temperature; 50 °C, time: 6 h. | ||
The impact of reaction temperature on the selective synthesis of β-nitrostyrene from benzaldehyde and nitromethane was examined, as depicted in . As the temperature increased, the conversion of benzaldehyde also increased. At 50 °C, an 87% conversion rate of benzaldehyde with over 99% selectivity for β-nitrostyrene was attained. However, at 100 °C, the selectivity for β-nitrostyrene decreased to 93% due to the formation of the Michael addition product. The influence of solvent on the nitro-aldol condensation between benzaldehyde and nitromethane was examined using various solvents at 50 °C for 6 h, as depicted in . Under solvent-free conditions, benzaldehyde achieved an 87% conversion rate with 99% selectivity for the desired β-nitrostyrene product. Polar protic solvents such as H2O and methanol gave 38% and 64% conversion rates for benzaldehyde, respectively, with 99% selectivity for β-nitrostyrene. Conversely, polar aprotic solvents such as DMF and acetone exhibited only 5% and 6% conversion rates for benzaldehyde, respectively, with 99% selectivity for β-nitrostyrene. Toluene, on the other hand, showed no formation of the nitro-aldol product, indicating that it may not be suitable as a solvent for the nitro-aldol condensation reaction. These results suggest that solvent-free conditions are effective in promoting the nitro-aldol condensation reaction.
To understand the influence of the catalyst amount on maximizing benzaldehyde conversion, the catalyst amount was varied from 5 wt% to 30 wt% at 50 °C for 6 h (Fig. 4D). The conversion of benzaldehyde increased with the catalyst amount, reaching a plateau beyond 20 wt% without affecting the selectivity of β-nitrostyrene. Hence, it was concluded that a 20 wt% catalyst is sufficient for achieving maximum conversion of benzaldehyde. In the final screening experiments, time variation studies were conducted from 2 to 12 h. These studies revealed that prolonging the reaction time from 2 to 12 h increased the conversion of benzaldehyde, while the selectivity of β-nitrostyrene remained consistently high (Fig. S6†). The absence of the Michael addition product even after 12 h suggests that the selectivity of the desired product is not negatively impacted by prolonged reaction times.
2.3 Substrate scope
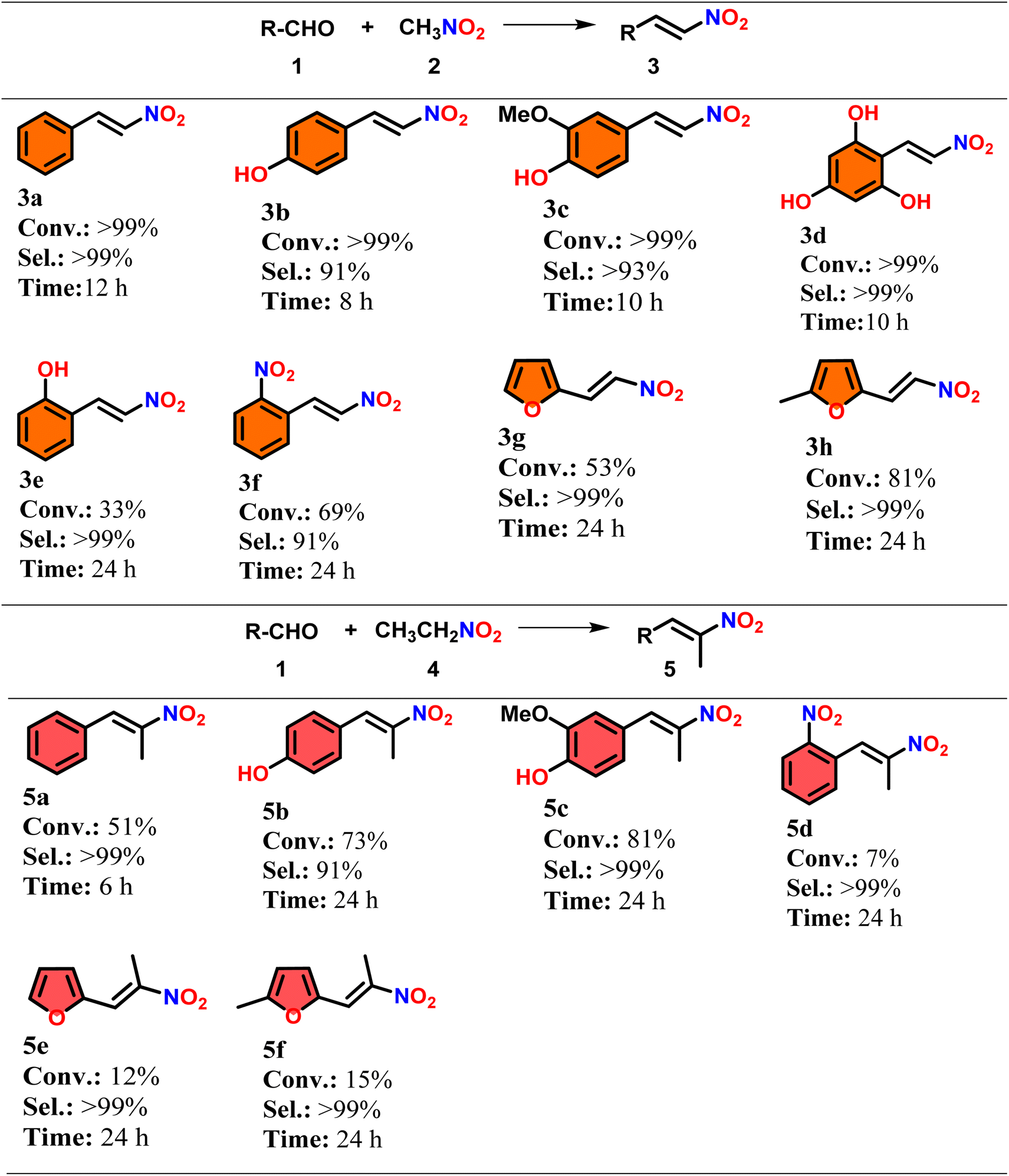
Our synthesized DFNS@NH2 was tested for its efficiency across a broad range of substrates, including electron-withdrawing and electron-donating aromatic substituted benzaldehydes and furanic aldehydes and the results are shown in Table 2. Initially, our investigation focused on p-hydroxyl substituted aromatic aldehydes (1b–1d), which, when reacted with nitromethane (2), yielded 91–99% selectivity for the desired p-hydroxyl substituted α,β-unsaturated nitrostyrene derivatives (3b–3d). However, only a 33% yield was observed for 2-hydroxy benzaldehyde (1e), resulting in sole product formation (3e). Moving on to 2-nitro benzaldehyde (1f), a 69% conversion rate was achieved with 91% selectivity for the desired product (3f). Furanic aldehydes (1g and 1h) smoothly transformed into substituted 2-(2-nitrovinyl) furan derivatives (3g and 3h), indicating the good conversion and selectivity of the desired product with nitromethane when using amine-functionalized DFNS. The electronic properties of aldehydes were found to significantly affect catalytic activity, with aromatic aldehydes containing electron-donating groups exhibiting higher conversion rates compared to those with electron-withdrawing groups. Additionally, reactions proceeded more rapidly when the hydroxyl group was situated in the para position of aromatic aldehydes compared to the ortho position, likely due to steric hindrance. Furthermore, DFNS@NH2 was applied to the nitroaldol condensation between various carbonyl compounds and nitroethane, resulting in lower conversion rates for all aromatic carbonyl compounds. For instance, the reaction between benzaldehyde (1a) and nitroethane (4) yielded only a 59% conversion rate with sole product formation (5a). In contrast, 4-hydroxy and 3-methoxy substituted benzaldehydes (1b and 1c) exhibited conversion rates of 73% and 81%, respectively, with 91% and 93% selectivity for the desired products (5c and 5d). The substituted furanic aldehydes showed lower conversion rates but maintained good selectivity for the desired products. This discrepancy in conversion rates between carbonyl compounds and nitromethane suggests a steric hindrance effect caused by the alkyl group in nitroethane.37| a Reaction conditions (3a–h): substrate: 2 mmol, nitromethane: 11.2 mmol [for 3b, c, d, f for solid substrate – (1 mL nitromethane)], DFNS@NH2; 10 wt% to substrate, temperature; 50 °C, 3d, 3e, 3f, 3g, and 3h [90 °C]. Reaction conditions (5af): substrate: 2 mmol, nitroethane: 8.4 mmol [for solid substrate 5b, c, d – (1 mL nitroethane)], DFNS@NH2; 10 wt% to substrate, temperature; 70 °C, 5d, e, f [90 °C]. For solid substrate solvent: 1 mL 3b, 3c, 3d, and 3f. |
|---|
|
2.4 Recyclability studies
The recyclability of the synthesized amine-functionalized DFNS was demonstrated in the nitro-aldol condensation reaction of benzaldehyde (2 mmol) with nitromethane (11.2 mmol) at 50 °C for 6 h. After each reaction, the amine-functionalized DFNS was recovered from the mixture by centrifugation, washed three times with ethyl acetate, and dried overnight at 80 °C. The catalyst's activity was reevaluated for up to five cycles of the nitro-aldol condensation reaction. Over these cycles, the conversion of benzaldehyde decreased gradually from 86% to 60%, while the selectivity for β-nitrostyrene remained unaffected (Fig. S7†). FESEM analysis of the recycled catalyst confirmed no morphological changes after the fourth cycle (Fig. S8†). The literature performance with other catalysts is also compared for Henry's reaction between benzaldehyde and nitromethane (Table S3†).2.5 Plausible mechanism
The proposed mechanism for the nitro-aldol condensation of benzaldehyde with nitromethane, shown in Scheme 3, highlights the critical role of a cooperative catalyst system.45 In this mechanism, the DFNS@NH2 catalyst, functionalized with both amine and hydroxyl groups, demonstrates the importance of cooperative catalysis in improving reaction efficiency and selectivity. The amine functionality on the DFNS@NH2 catalyst abstracts a proton from nitromethane to form a nitronate anion (II).46 At the same time, a proton from a hydroxyl group on DFNS@NH2 forms a partial hydrogen bond with the oxygen of the benzaldehyde carbonyl group (III). This partial hydrogen bond facilitates the activation of benzaldehyde towards nucleophilic attack by enhancing the electrophilic character of the carbonyl carbon. The importance of cooperative catalysis is evident in this step, where the combined action of the amine and hydroxyl groups on DFNS@NH2 enhances the reactivity of both reactants. Control experiments likely confirm the essential role of both the amine and hydroxyl functionalities on DFNS@NH2 in catalyzing the nitro-aldol and subsequent reactions (Scheme S1†). The nitronate anion (II) then attacks the activated carbonyl of the benzaldehyde (III), leading to the formation of a hydroxyl alcohol intermediate (IV). The amine functionality of DFNS@NH2 abstracts another proton from the hydroxyl alcohol intermediate (IV), resulting in the formation of β-nitrostyrene (V). This step involves the elimination of water and the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The presence of excess nitromethane induces a Michael addition reaction with the newly formed β-nitrostyrene (V), resulting in the formation of a Michael addition product (VI). In addition, the quaternary ammonium functionalized version of DFNS@NH2 is used for the cycloaddition of CO2, potentially leading to further product formation and value addition. This demonstrates the versatility and broad applicability of the cooperative catalyst system.
C bond. The presence of excess nitromethane induces a Michael addition reaction with the newly formed β-nitrostyrene (V), resulting in the formation of a Michael addition product (VI). In addition, the quaternary ammonium functionalized version of DFNS@NH2 is used for the cycloaddition of CO2, potentially leading to further product formation and value addition. This demonstrates the versatility and broad applicability of the cooperative catalyst system.
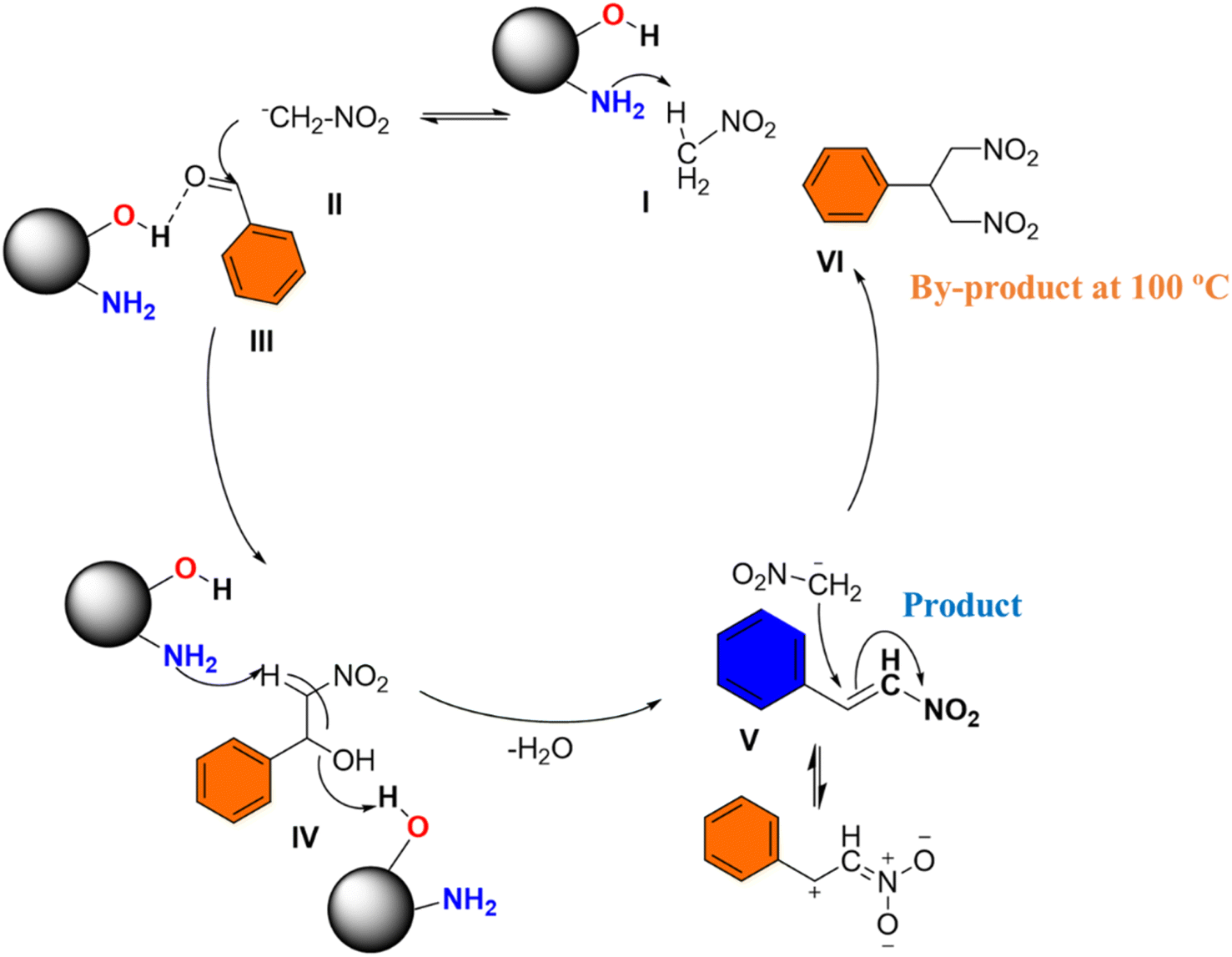 | ||
| Scheme 3 Plausible mechanism for the production of β-nitrostyrene from benzaldehyde. | ||
2.6 CO2 utilization
In the future, achieving carbon neutrality will play a crucial role in mitigating global warming. A significant amount of CO2 is emitted during industrial processes. This emitted CO2 can be harnessed for the synthesis of highly valuable chemicals such as formic acid, methanol, and cyclic carbonates.28,29 Using CO2, a major greenhouse gas, for the synthesis of valuable chemicals such as cyclic carbonates presents a promising approach to achieving carbon neutrality. Cyclic carbonates, such as styrene carbonate, are of particular importance due to their applications as bio-solvents and intermediates in organic synthesis. Of particular importance is the exploration of the catalytic activity of the quaternized product (DFNS@TBAB) for the synthesis of styrene carbonate from styrene oxide and CO2 (Scheme 4). | ||
| Scheme 4 Cycloaddition of styrene oxide with CO2. | ||
In the absence of a catalyst or with amine-functionalized DFNS alone, there is no significant catalytic activity, highlighting the complexity of activating CO2 for chemical transformations (Table S2,† entries 1 and 2). The improved conversion and selectivity achieved by introducing halogenated additives (CTAB and KI) in addition to amine-functionalized DFNS indicates the crucial role of halides in the cycloaddition process (Table S2,† entries 3 and 4). This suggests a synergistic effect, where the halides may facilitate the activation of epoxide (ring opening and formation of reactive alkoxide intermediates)47,48 and the amine sites activate CO2 (forming a carbamate or increasing its electrophilicity), thus making the reaction with styrene oxide more favorable. The specific combination of amine-functionalized DFNS with n-butyl bromide leading to a 46% conversion of styrene oxide with 94% selectivity for styrene carbonate is particularly compelling (Table S2,† entry 5). It not only demonstrates the potential of this system in CO2 utilization but also highlights the significant role of n-butyl bromide in the catalytic process. The comparison with the performance of n-butyl bromide alone, which showed much lower conversion, further emphasizes the necessity of both the amine-functionalized DFNS (nitrogen sites) and the halide for effective catalysis (Table S2,† entry 6). These results underline the necessity of the cooperative influence of halides and nitrogen sites playing a significant role in the cycloaddition reaction, providing valuable insights into the design of catalytic systems for CO2 utilization.
The observation that both the conversion of styrene oxide and the selectivity for styrene carbonate increase with an increased amount of catalyst is indicative of the catalytic process being dependent on the availability of active sites (Table S2,† entries 8–11). This suggests that the reaction is surface-limited, and increasing the catalyst amount provides more sites for the reactants to interact, leading to higher conversion rates and potentially more controlled selectivity towards styrene carbonate. The temperature-dependence study reveals a direct correlation between reaction temperature and catalytic activity, with no activity observed at 60 °C and a gradual increase in activity peaking at 120 °C (Table S2,† entries 12–15). The fact that maximum conversion (88%) was observed at 120 °C indicates that the activation energy for the reaction is sufficiently overcome at 120 °C, facilitating the efficient conversion of styrene oxide to styrene carbonate. Concluding the investigation, time variation studies were carried out for the synthesis of styrene carbonate using the DFNS@TBAB material. After 3 h, only a 10% conversion of styrene oxide was observed. However, extending the reaction time to 6 h resulted in a conversion increase to 34%. With further increments in reaction time, both conversion and selectivity continue to increase. Notably, 94% conversion of styrene oxide was achieved within 15 h at 120 °C with 98% selectivity and 2% 1-phenylethane-1,2-diol (Table S2,† entries 16–20). The overall optimization studies for the cycloaddition reaction between styrene oxide and CO2 using DFNS@TBAB highlight a significant advancement in CO2 utilization. It's excellent that the catalyst allows the reaction to proceed without additional alkali halides or organic quaternary ammonium salts, such as KI and CTAB, which simplifies the reaction system and potentially reduces costs and environmental impact. Future studies could explore the use of DFNS@TBAB in other types of condensation and cycloaddition reactions or fine chemical production. Also, we have compared other metal-free catalysts highlighting their performance in the CO2 cycloaddition reaction (Table S4†). The probable mechanistic pathway for the formation of styrene carbonate from styrene oxide is outlined below in Scheme 5.38,47 For effective catalysis to promote the cycloaddition reaction of CO2, the activation and ring opening of epoxides is critically significant as it is the rate-determining step. Polarized active sites (N+ and Br−) and hydrogen bond donors (HBDs) can cooperatively influence the dual activation of epoxides thereby stabilizing the intermediates and promoting efficiency during the cyclic catalytic process.47–49 The catalytic material DFNS@TBAB has intrinsically polarized units as quaternary nitrogen with Br−, silanol groups as HBDs (Si–OH) and a unique dendritic morphology with 3-D accessibility of pore channels. The porous nature and dendritic morphology of the DFNS are ideal for greater and effective mass diffusion, which will assist in the enrichment of CO2 and diffusion of substrates. This combination of the dual activation strategy using polarized units along with HBDs accounts for high catalytic activity. At first, the hydroxyl group (–OH) of DFNS@TBAB interacts with the oxygen atom of styrene oxide, facilitating the activation of the epoxide ring by polarizing the C–O bond (step I). Simultaneously, the nearby Br− anion acting as a nucleophile attacks the β-carbon of the strained three-membered ring of the activated epoxide forming a bromoalkoxide intermediate (step II), that is stabilized by the Si–OH bonds (silanol) present in the material. Subsequently, this intermediate reacts with the electrophilic carbon center (Cδ+) of the inserted CO2 to form an alkyl carbonate intermediate. Finally, intramolecular cyclization of the alkyl carbonate anion occurs, resulting in the desired cyclic styrene carbonate product. The material DFNS@TBAB harbors the optimum desired catalytic microenvironment rich in nucleophilic Br− ions with quaternary nitrogen (N+) and HBD silanol groups accelerating the synergistic effects of activation promoting efficiency under mild conditions.
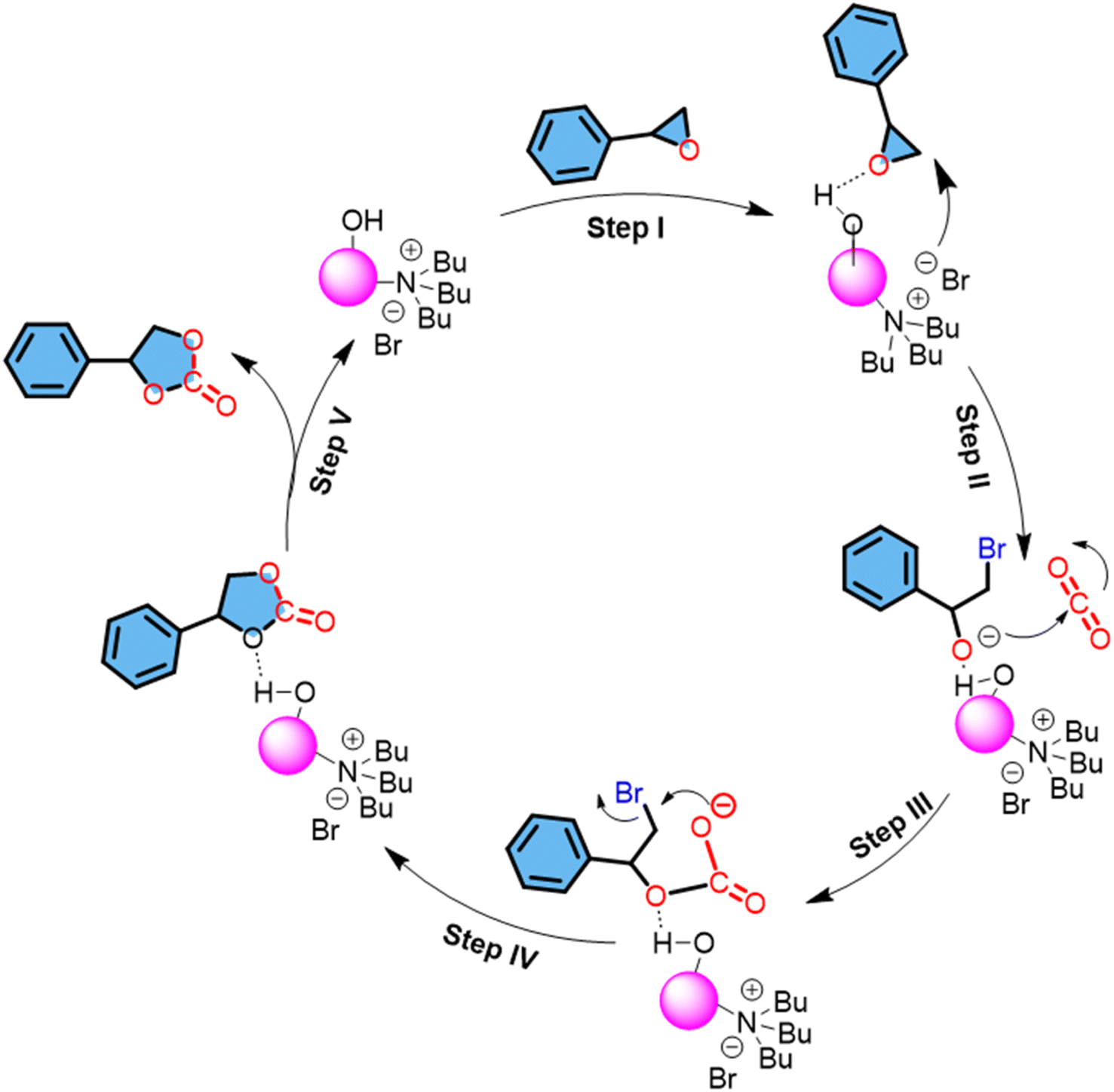 | ||
| Scheme 5 The plausible mechanism for the cycloaddition of CO2 with styrene oxide. | ||
3 Experimental section
3.1 Synthesis of dendritic fibrous nanosilica (DFNS)
For the synthesis of dendritic fibrous nanosilica, typically, 1.5 g (3.57 mmol) of tetraphenylphosphonium bromide (TPPB) and 3 g (49.95 mmol) of urea were added in 150 mL of Milli-Q water and stirred for 20 min. Following this, a solution containing 5 mL (22.39 mmol) of TEOS in 30 mL of p-xylene was slowly introduced into the reaction mixture, along with 2 mL of propanol as a mineralizing agent. The reaction temperature was then raised to 50 °C and maintained for 15 min under continuous stirring at 550 rpm. Subsequently, the reaction mixture was refluxed at 90 °C for 10 h, resulting in the formation of white precipitates. These precipitates were separated by centrifugation, thoroughly washed with water, methanol, and chloroform (3 times each), and finally dried overnight in an oven at 70 °C. To remove the template, the material underwent multiple solvent extractions using an NH4NO3/ethanol solution for 6 h, repeated twice, followed by thorough washing with water and methanol (twice each). The resulting product, denoted as DFNS, weighed 4.8 g and appeared as a pure white substance after drying in the oven for 10 h.3.2 Synthesis of aminated catalyst DFNS (DFNS@NH2)
For the functionalization of DFNS with 3-APTES, typically, 3 g of the above synthesized DFNS was refluxed with 7 mL (29.91 mmol) of 3-APTES in 100 mL toluene at 110 °C for 24 h. After the reaction times, the reaction mixture was cooled down and white precipitates were separated by centrifugation followed by thorough washing with chloroform and methanol 3 times and finally dried in an oven at 50 °C for 8 h giving 3.14 g of the pure aminated product as the DFNS@NH2 material.3.3 Synthesis of the quaternized catalyst (DFNS@TBAB)
For the quaternization of the DFNS@NH2 material, typically 2 g of the DFNS@NH2 material was refluxed with 10 mL (92.69 mmol) of the n-butyl bromide in acetonitrile at 85 °C for 48 h. The reaction mixture was allowed to cool and finally centrifuged and washed with acetonitrile and methanol 3 times and finally dried in an oven for 8 h at 60 °C to afford the product DFNS@TBAB.3.3.1.1 Henry reaction between benzaldehyde and nitromethane. To evaluate the catalytic properties of synthesized amine functionalized dendritic fibrous silica used for the Henry reaction between benzaldehyde and nitromethane, the Henry reaction was typically conducted in a 30 mL glass tube containing benzaldehyde (0.250 g) dissolved in 2 mL of nitromethane, followed by the addition of the catalyst. The reaction mixture was then heated at varying temperatures (30–100 °C). Periodic samples were taken and analyzed using GC-MS (Shimadzu, QP-2010) (Fig. S9–S26†). The resulting α,β-unsaturated nitroarenes were also identified using 1H and 13C NMR spectroscopy (Fig. S27–S31†).
3.3.1.2 Cycloaddition of CO2 with styrene oxide. The cycloaddition of epoxides and CO2 was performed in a glass tube (10 mL capacity) with constant stirring. For each reaction, styrene oxide (0.25 g) and the catalyst (0.02 g) were added to the tube, which was then pressurized with 1 atm of CO2 at room temperature. The reaction mixture was heated to the desired temperature (60–120 °C) while stirring at 600 rpm. After the reaction, the tube was cooled to room temperature and the catalyst was separated from the mixture by centrifugation. The recovered catalyst was washed twice with ethyl acetate and dried, while the solution was analyzed using GC-MS (Shimadzu, QP-2010).
4 Conclusion
The versatility of DFNS demonstrated for nitro-aldol condensation and the cycloaddition of styrene oxide with CO2 underline the potential of engineered nanomaterials in sustainable chemistry. In nitro-aldol condensation, the presence of both hydroxyl and amine functionalities within the DFNS structure plays a crucial role in the activation of aldehydes and nitroalkanes. These functional groups facilitate the formation of intermediates through hydrogen bonding or proton transfer mechanisms, thereby increasing the efficiency of the reaction at a relatively mild temperature of 50 °C. The subsequent modification of amine-functionalized DFNS through quaternization with n-butyl bromide to produce a quaternary ammonium-functionalized DFNS (DFNS@TBAB) represents a strategic pivot towards cycloaddition of CO2 with epoxides. This quaternary ammonium-functionalized DFNS can facilitate the complete conversion of styrene oxide to the desired styrene carbonate, with 98% selectivity and without the need for additional additives at 120 °C for 15 h. The results demonstrate the catalyst's efficiency in activating and incorporating CO2 into organic substrates, in line with carbon capture and utilization (CCU) objectives.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was collaboratively written by all authors, and all have approved the final version.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
A. R. P., S. Y., and N. C. acknowledge the Council of Scientific and Industrial Research (CSIR), Govt. of India, for financial support. S. Y. & N. C. acknowledge CSIR & AcSIR for the fellowship & PhD degree. HGK is grateful to the CSIR Govt. of India for the SRF Fellowship. GHW acknowledges the UGC, Govt. of India, for the JRF Fellowship. AB acknowledges the project MLP0077. KR thanks the CSIR, Govt. of India, for the Senior Research Fellowship (CSIR Grant No. 31/028(11360)/2021-EMR-I). All authors acknowledge the Analytical and Environmental Science Division and Centralized Instrumentation Facility of CSIR-Central Salt & Marine Chemicals and Research Institute, Bhavnagar, for providing all the required instrumental analysis. A CSIR-CSMCRI communication number 128/2024.References
- C. M. Friend and B. Xu, Acc. Chem. Res., 2017, 50, 517–521 CrossRef CAS PubMed
.
- F. Zaera, Chem. Rev., 2022, 122, 8594–8757 CrossRef CAS PubMed
- S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC
- A. West, Nat. Rev. Chem, 2018, 2, 0140 CrossRef
- E. D. Goodman, C. Zhou and M. Cargnello, ACS Cent. Sci., 2020, 6, 1916–1937 CrossRef CAS PubMed
- C. Khoury, C. Gadipelly, S. Pappuru, D. Shpasser and O. M. Gazit, Adv. Funct. Mater., 2020, 30, 1901385 CrossRef CAS
- P. Chandra, A. M. Jonas and A. E. Fernandes, ACS Catal., 2018, 8, 6006–6011 CrossRef CAS
- N. A. Brunelli, S. A. Didas, K. Venkatasubbaiah and C. W. Jones, J. Am. Chem. Soc., 2012, 134, 13950–13953 CrossRef CAS PubMed
- S. Singh, A. Kumar, L. Nebhani and C. K. Hazra, JACS Au, 2023, 3, 3400–3411 CrossRef CAS PubMed
- C. Li, W. Xiong, T. Zhao, F. Liu, H. Cai, P. Chen and X. Hu, Appl. Catal., B, 2023, 324, 122217 CrossRef CAS
- C. Khoury, S. Holton, D. Shpasser, E. Hallo, A. Kulkarni, F. C. Jentoft and O. M. Gazit, ACS Catal., 2022, 12, 9846–9856 CrossRef CAS
- T. Himiyama, M. Waki, Y. Maegawa and S. Inagaki, Angew. Chem., 2019, 131, 9248–9252 CrossRef
- S. Huh, H. Chen, J. W. Wiench, M. Pruski and V. S. -Y. Lin, Angew. Chem., 2005, 117, 1860–1864 CrossRef
- S. Khoonsap, L. Buengkitcharoen, S. Amnuaypanich, N. Thongnoppakhun, N. Patdhanagul, S. Suthirakun, C. Sukpattanacharoen and S. Amnuaypanich, Chem. Eng. J., 2023, 477, 147074 CrossRef CAS
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118 RSC
- A. E. Fernandes and A. M. Jonas, Catal. Today, 2019, 334, 173–186 CrossRef CAS
- T. Asefa, M. Kruk, N. Coombs, H. Grondey, M. J. MacLachlan, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 11662–11673 CrossRef CAS PubMed
- K. Sugino, N. Oya, N. Yoshie and M. Ogura, J. Am. Chem. Soc., 2011, 133, 20030–20032 CrossRef CAS PubMed
- M. Bouhrara, C. Ranga, A. Fihri, R. R. Shaikh, P. Sarawade, A.-H. Emwas, M. N. Hedhili and V. Polshettiwar, ACS Sustain. Chem. Eng., 2013, 1, 1192–1199 CrossRef CAS
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS PubMed
- V. Polshettiwar, Acc. Chem. Res., 2022, 55, 1395–1410 CrossRef CAS PubMed
- Y. Wang, X. Du, Z. Liu, S. Shi and H. Lv, J. Mater. Chem. A, 2019, 7, 5111–5152 RSC
- A. Maity and V. Polshettiwar, ChemSusChem, 2017, 10, 3866–3913 CrossRef CAS PubMed
- A. Maity, R. Belgamwar and V. Polshettiwar, Nat. Protoc., 2019, 14, 2177–2204 CrossRef CAS PubMed
- R. Belgamwar, R. Verma, T. Das, S. Chakraborty, P. Sarawade and V. Polshettiwar, J. Am. Chem. Soc., 2023, 15, 8634–8646 CrossRef PubMed
- F. G. Cirujano, R. Luque and A. Dhakshinamoorthy, Molecules, 2021, 26, 1445 CrossRef CAS PubMed
- G. B. B. Varadwaj, S. Rana, K. Parida and B. B. Nayak, J. Mater. Chem. A, 2014, 2, 7526 RSC
- Y. Shao and H. C. Zeng, J. Mater. Chem. A, 2023, 11, 2698–2710 RSC
- R. Das, T. Ezhil, A. S. Palakkal, D. Muthukumar, R. S. Pillai and C. M. Nagaraja, J. Mater. Chem. A, 2021, 9, 23127–23139 RSC
- A. V. Biradar, K. K. Sharma and T. Asefa, Appl. Catal., A, 2010, 389, 19–26 CrossRef CAS
- K. K. Sharma, A. V. Biradar and T. Asefa, ChemCatChem, 2010, 2, 61–66 CrossRef CAS
- H. Zhang, H. Zhang, H. Du, J. Xiao, M. Tao, N. Ma and W. Zhang, ACS Catal., 2023, 13, 7844–7856 CrossRef CAS
- V. E. Collier, N. C. Ellebracht, G. I. Lindy, E. G. Moschetta and C. W. Jones, ACS Catal., 2016, 6, 460–468 CrossRef CAS
- D. Kim, S. Subramanian, D. Thirion, Y. Song, A. Jamal, M. S. Otaibi and C. T. Yavuz, Catal. Today, 2020, 356, 527–534 CrossRef CAS
- F. D. Bobbink, W. Gruszka, M. Hulla, S. Das and P. J. Dyson, Chem. Commun., 2016, 52, 10787–10790 RSC
- F. Lagarde, H. Srour, N. Berthet, N. Oueslati, B. Bousquet, A. Nunes, A. Martinez and V. Dufaud, J. CO2 Util., 2019, 34, 34–39 CrossRef CAS
- Y. Ye, D. Li, P. Xu and J. Sun, Inorg. Chem. Front., 2020, 7, 3636–3645 RSC
- K. Ravi, J.-L. K. GBE, S. Mehra, S. Tothadi, A. Kumar and A. V. Biradar, J. Environ. Chem. Eng., 2023, 11, 109737 CrossRef CAS
- S. Yadav, N. Choudhary, M. Ranjan Dash and A. Ranjan Paital, Chem. Eng. J., 2022, 450, 138042 CrossRef CAS
- S. Yadav, N. Choudhary, V. Sonpal and A. Ranjan Paital, Chem. Eng. J., 2023, 471, 144715 CrossRef CAS
- S. Yadav, N. Choudhary and A. R. Paital, Carbon, 2023, 205, 527–539 CrossRef CAS
- G. D. Bedasso, D.-L. M. Tzou and P.-W. Chung, J. Taiwan Inst. Chem. Eng., 2024, 158, 104801 CrossRef CAS
- J.-M. Gu, W.-S. Kim and S. Huh, Dalton Trans., 2011, 40, 10826 RSC
- S. D. Jadhav, R. C. Patil, A. A. Jagdale and S. S. Patil, Res. Chem. Intermed., 2022, 48, 593–606 CrossRef CAS
- K. Motokura, M. Tada and Y. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 9230–9235 CrossRef CAS PubMed
- G. Demicheli, R. Maggi, A. Mazzacani, P. Righi, G. Sartori and F. Bigi, Tetrahedron Lett., 2001, 42, 2401–2403 CrossRef CAS
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS
- M. Yin, L. Wang and S. Tang, ACS Catal., 2023, 13, 13021–13033 CrossRef CAS
- L. Zhu, P. Cheng, Z. Xiao, C. Lu, B. Li, X. Jiang, Z. Shen, N. Qian, W. Zhong and Y. He, Chem. Eng. J., 2024, 481, 148359 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03980g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |