
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into the activity of Ni-based thermal catalysts for dry reforming of methane
Ziquan
Wang
a,
Ziyu
Mei
a,
Luyuan
Wang
a,
Qilong
Wu
b,
Changlei
Xia
c,
Song
Li
*a,
Tianyi
Wang
*a and
Chuangwei
Liu
 *ad
*ad
aKey Laboratory of Anisotropy and Texture of Materials, School of Materials Science and Engineering, Northeastern University, Shenyang, 110819, China. E-mail: lis@atm.neu.edu.cn; tianyiwang@mail.neu.edu.cn
bIntelligent Polymer Research Institute, Australian Institute for Innovative Materials, Innovation Campus, University of Wollongong, Squires Way, North Wollongong, NSW 2500, Australia
cCollege of Materials Science and Engineering, Nanjing Forestry University, Nanjing, Jiangsu 210037, China
dState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Science, Dalian, 116023, China. E-mail: cwliu@dicp.ac.cn
First published on 29th August 2024
Abstract
Dry reforming of methane (DRM) is a sustainable technology that converts methane and carbon dioxide into syngas, achieving value-added utilization of greenhouse gases and reducing carbon emissions in the energy industry. Ni-based catalysts are preferred for promoting the DRM reaction due to their high activity and low price compared to other active metals. However, the long-term catalytic stability of Ni-based catalysts is limited by carbon deposition and metal sintering. In this review, the mechanism of the formation and elimination of carbon deposition in the DRM reaction is proposed first. Then, a comprehensive summary of the current development of Ni-based catalysts for the DRM reaction is presented. The summary highlights various aspects from an experimental perspective that affect the activity and stability of catalysts, including synthesis methods and conditions, selection of supports, doping of promoters and second active metals, and regulation of reaction conditions. Additionally, it also covers the practical application conditions of catalysts, aiming to promote the industrial application of DRM reaction.
 Ziquan Wang | Ziquan Wang obtained his Bachelor's degree in 2022 from Shenyang University of Technology and is currently pursuing a Master's degree at Key Laboratory of Anisotropy and Texture of Materials, School of Materials Science and Engineering, Northeastern University, Shenyang, China. His research focuses on the design and application of novel electric-heating-integrated catalysts. |
 Song Li | Song Li is a professor of Materials Science and Engineering at the Northeastern University, China. He received his PhD in materials physics from University of Lorraine in Metz and Northeastern University in Shenyang (2009). He was a visiting scholar at Boston College, from 2015 to 2016. Li's recent research focuses on energy conversion and industrial decarbonization by developing novel functional materials. |
 Tianyi Wang | Tianyi Wang received her PhD in 2022 in the School of Science, Computing and Engineering Technologies, Swinburne University of Technology. She is currently a postdoctoral fellow at Northeast University. Her interests focus on computer-aid design electrochemical catalysts for nitrogen reduction reaction. |
 Chuangwei Liu | Chuangwei Liu received his PhD in 2019 in the School of Chemistry, Monash University, Australia. After a stay as a postdoctoral fellow in the Technical University of Denmark, he joined Northeastern University as an associate professor in the School of Materials Science and Engineering. His research interest is molecular modeling and the design of innovative materials for clean energy, catalysts, and environment through computation methods. |
1. Introduction
In recent years, the substantial growth in worldwide energy demand has led to extensive utilization of conventional fossil fuels and excessive emission of greenhouse gases. In particular, from 1960 to 2020, global carbon dioxide emissions increased by over 30%, surpassing 40 billion tonnes annually,1,2 resulting in global environmental degradation, as well as more extreme weather events and climate fluctuations.1,3–5 Various studies have demonstrated the significant correlation between rising global temperatures and greenhouse gas concentrations including methane (CH4) and carbon dioxide (CO2).6 Notably, CH4 exhibits worse influences as a greenhouse gas relative to CO2, despite its lower atmospheric concentration.7,8 Internationally, the Paris Agreement, ratified by 178 nations in 2016, aims to raise global awareness to combat global climate change. Moreover, the International Conference on Carbon Dioxide Utilization (ICCDU) offers a platform for researchers to share advancements in technology for capturing, storing, and converting carbon dioxide.9 The target of China is to peak carbon emissions by 2030 and achieve carbon neutrality by 2060. Thus, mitigating CH4 and CO2 emissions is an efficient strategy, and the reasonable utilization of CH4 and CO2 is also crucial for addressing the challenges generated by global warming.10–13 Hydrogen energy emerges as a promising energy alternative devoid of combustion byproducts, thereby increasing global annual hydrogen consumption.14 Presently, approximately 80–85% of hydrogen production is derived from the conversion of natural gas. Therefore, converting CH4 and CO2 greenhouse gases into H2 presents a mutually beneficial approach towards environmental stewardship and the promotion of sustainable resource utilization.15 Dry reforming of methane (DRM), steam reforming of methane (SRM), and partial oxidation of methane (POM) are mainstream methods for converting CH4 into CO and H2, followed by eqn (1)–(3).8,16 Currently, SRM is the dominant technique for industrial synthesis gas production. However, high production costs and high H2O/CH4 ratio impede its widespread application.8 In contrast, DRM is a cost-effective approach to effectively mitigate both CO2 and CH4 emissions concurrently, which is favorable for regions abundant in natural gas but limited in water resources.17 In addition, DRM is particularly suitable for the Fischer–Tropsch process to facilitate industrial long-chain hydrocarbon synthesis, attributing to the neat unity of H2/CO.16,18–21 The flow chart of carbon cycling achieved through dry reforming of methane reaction is shown in Fig. 1(a).| DRM: CH4(g) + CO2(g) → 2CO(g) + 2H2(g) ΔH298K = 247 kJ mol−1 | (1) |
| SRM: CH4(g) + H2O(g) → CO(g) + 3H2(g) ΔH298K = 206 kJ mol−1 | (2) |
| POM: CH4(g) + 1/2O2(g) → CO(g) + 2H2(g) ΔH298K = −36 kJ mol−1 | (3) |
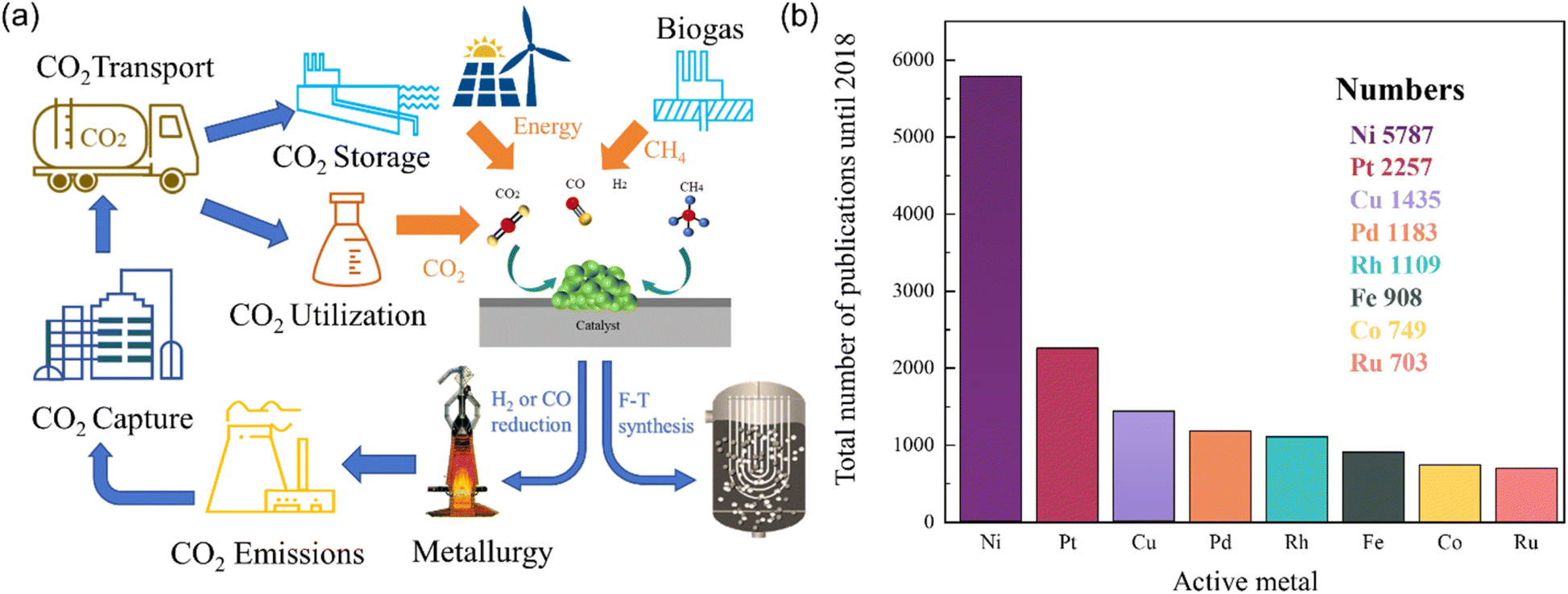 | ||
| Fig. 1 (a) Carbon cycling achieved through dry reforming of methane reaction. (b) Comparison of the number of scientific studies using several transition metals for reforming. | ||
However, DRM is characterized by a significant endothermic nature within a reaction temperature range of 650–1000 °C. When the reaction temperature is too high, the conversion of reactants does not increase significantly. High heat input is pivotal for reactant dissociation while limiting the increase in conversion rate. Moreover, the limitation of reverse water gas shift (RWGS) reaction alongside DRM is another challenge due to the decrease in H2/CO ratio.22 Therefore, exploring high-performance catalysts is critical to enhancing conversion efficiency and reducing energy input. Precious metals (Ru, Rh, Pt) and transition metals (Fe, Co, Ni) serve as primary active components of thermal catalyst candidates for DRM application.8,23 Yu et al.23 constructed Gibbs free energy diagrams of eight transition metals and concluded that Ni, Rh, andIr are the most active elemental metals in DRM. Rh, Ir-based catalysts exhibit higher activity and stability,24 but their expensive preparation costs hinder the large-scale application. In contrast, Ni-based catalysts offer a cost-effective alternative with outstanding catalytic activity. Fig. 1(b) illustrates the increasing research interest of Ni-based catalysts in reforming reactions in recent years. However, it is noteworthy that Ni-based catalysts are susceptible to sintering at high temperatures, and their activity sites may be obstructed by carbon deposition resulting from CH4 dissociation. These factors engender a decline in stability over extended periods, thereby significantly constraining their industrial application.25 Currently, the CALCOR process, which involves the production of high-purity carbon monoxide by reforming carbon dioxide, and the sulfur passive reforming process (SPARG), which mitigates carbon deposition by sulfur coverage on the catalyst surface, offer technical assistance in maintaining the stability of Ni-based catalysts in DRM.26 However, these technologies come at the cost of sacrificing the required H2/CO ratio and catalytic activity. Therefore, maintaining the activity and selectivity of Ni-based catalysts while improving stability is crucial for the industrial application of DRM. Hussien et al.27 provided a comprehensive overview of the principal challenges associated with employing Ni-based catalysts for DRM, focusing on the fundamental mechanisms of DRM reaction and the design of Ni-based catalysts. Fundamental mechanisms of DRM reaction focus on investigating the CH4 activation mechanism, thermodynamic and kinetic analysis, together with mitigating side reactions. The target of the design of Ni-based catalysts is to improve the anti-poisoning capability, activity, and selectivity of the catalyst. This review will primarily focus on the recent development of Ni-based catalysts with anti-sintering and anti-coking properties.
2. Catalytic performance and deactivation of Ni-based catalysts
2.1 Evaluation of catalytic performance of Ni-based catalysts
The catalytic performance of Ni-based catalysts in the DRM reaction can be evaluated based on activity, selectivity, and stability. Activity and selectivity determine the catalytic efficiency of the catalyst, while stability affects the practical application of catalysts. Traditionally, enhancing both the stability and activity of single Ni-based catalysts is challenging. Increased activity leads to more methane decomposition at active sites, resulting in increased carbon deposition. To enhance stability, hydrogen sulfide can be selectively added to poison the catalyst surface. While this may slightly reduce activity, the higher toxicity of sulfur atoms towards graphite formation can significantly improve catalyst stability.28 The thermodynamic conditions, kinetic conditions, catalyst properties, and catalytic pathway all play a role in determining the catalytic performance of methane dry reforming.From a thermodynamic standpoint, it is observed that as temperature increases, the conversion rate of reactants also increases due to more negative Gibbs free energy (Fig. 2(a)). There is an optimal operational temperature for both DRM and various side reactions.27 Due to the non-overlapping optimal operational temperature range of carbon deposition reaction (557–700 °C) and DRM (870–1040 °C),27 DRM has a lower conversion rate and more carbon deposition at low temperatures. At high temperatures, as the conversion rate of DRM increases, carbon deposition also decreases, and sintering becomes the main reason for catalyst deactivation. Furthermore, the equilibrium of the reaction can be influenced by gas composition (see Section 7) and side reactions, leading to alterations in the relative content and composition of the products (Fig. 2(b)). Typically, CO2 conversion exceeds CH4 conversion because of the reverse water-gas shift reaction (CO2 + H2 ↔ CO + H2O) at high temperatures.25
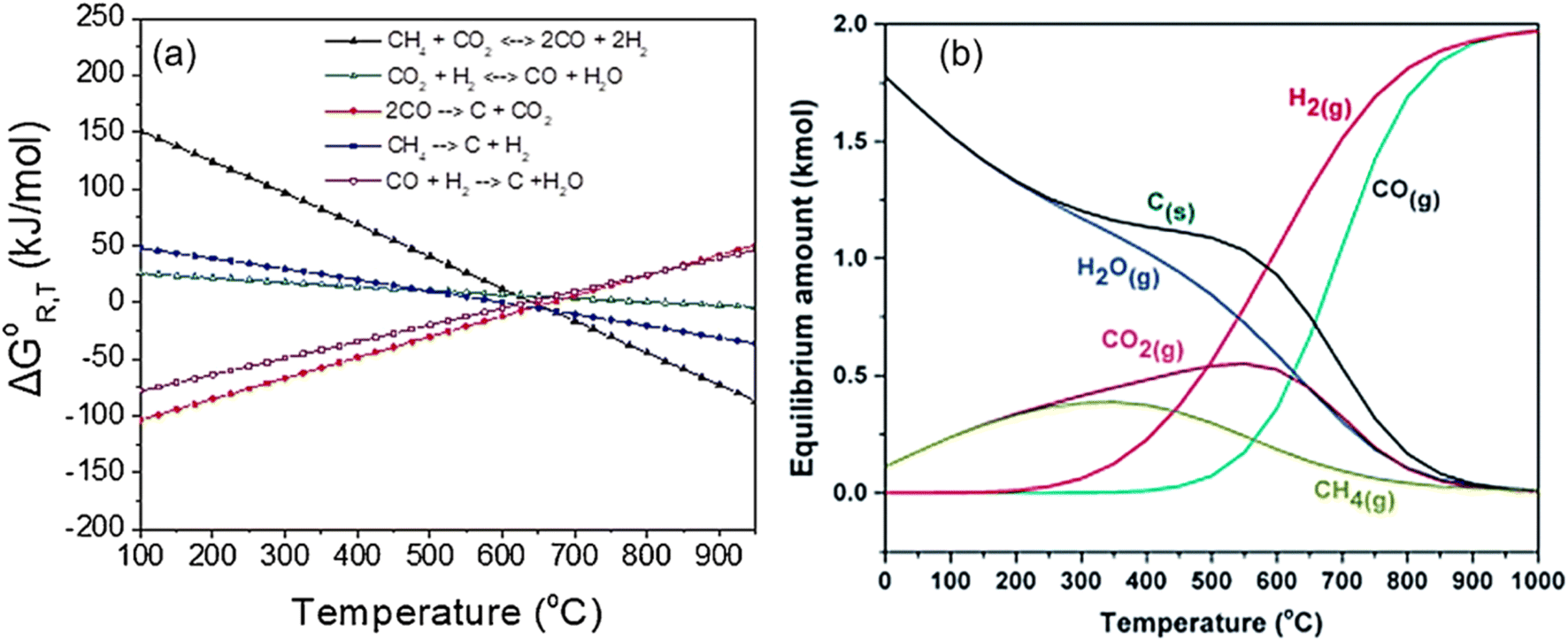 | ||
| Fig. 2 Thermodynamic analysis at equilibrium calculated as a function of temperature. (a) ΔG0R,T of all the reactions involved in the DRM process at 1 bar. Reprinted with permission from ref. 16 Copyright 2021, Elsevier. (b) Equilibrium data of chemical compounds for DRM. Reprinted with permission from ref. 29 Copyright 2016, The Royal Society of Chemistry. | ||
The establishment of a large-scale reactor model for DRM and the selection of operating conditions (mass transfer, heat transfer, etc.) are largely influenced by kinetic conditions. However, this will lead to overly complex dynamic models that are not suitable for ideal conditions in the laboratory. In terms of kinetics, Wei et al.30 proposed that the forward reaction rate of CH4 is directly proportional to the pressure of CH4, while it remains unaffected by the pressure of CO2, as shown in Fig. 3(a). This phenomenon can be attributed to the fact that the reaction rate of CH4 is primarily determined by the activation of C–H bonds and is not influenced by the characteristics or concentration of the reactants. However, many studies have shown different understandings of the rate-determining step in DRM.32–35 Hussien et al.27 provided a synthesis of prior studies concerning the rate-controlling step in the process of DRM. Their analysis indicated that the dissociation of CH4 is not the rate-determining step. The interaction between adsorbed carbon and oxygen dissociated from active Ni sites, leading to the production of CO, serves as the rate-determining step.
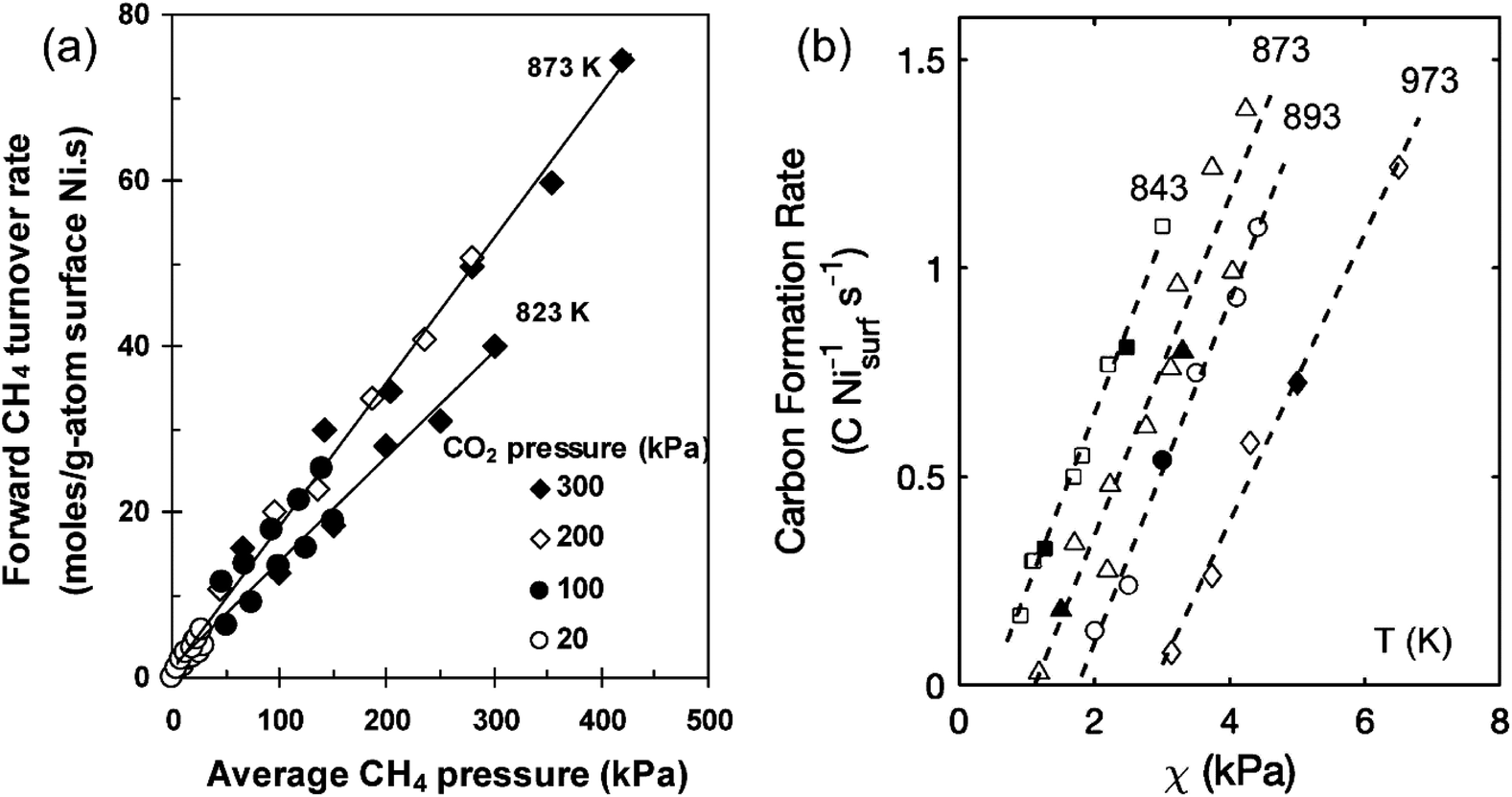 | ||
| Fig. 3 The kinetic mechanism of reactant decomposition and carbon deposition in reforming reactions. (a) Effects of CH4 pressure on forward CH4 reaction rate for DRM on Ni/MgO. Reprinted with permission from ref. 30 Copyright 2004, Elsevier. (b) χ (PCH4PCO/PCO2) on carbon formation rates for DRM on Ni/MgO (open symbol). Reprinted with permission from ref. 31 Copyright 2020, American Chemical Society. | ||
From the perspective of catalytic processes, competitive adsorption of reactants and the formation/decomposition of intermediates are crucial factors of catalytic performance.16,36 Therefore, maintaining a balance between the adsorption and dissociation of CO2 and CH4 is the key to improving catalytic stability and activity.
From the perspective of catalysts, catalysts are composed of active metals and support, and the factors influencing catalytic performance can be categorized into two types:
(1) Factors of active metals: particle size and distribution, metal dispersion, active sites with different coordination, and the interaction between multiple metals.37,38
(2) Factors of support: the redox ability of the support, surface acidity or basicity, oxygen vacancy, oxygen transfer rate, interaction between the metal and support, and metal–oxide interface area.20,39 In the following chapters, we will investigate the influence of synthesis methods, supports, second active metals, and promoters on the catalyst structure, surface properties, interface structure, and Ni electronic properties from an experimental perspective. Furthermore, the consequential effects of such factors on the overall catalytic performance of catalysts will be discussed. The scope of this review is illustrated in Fig. 4.
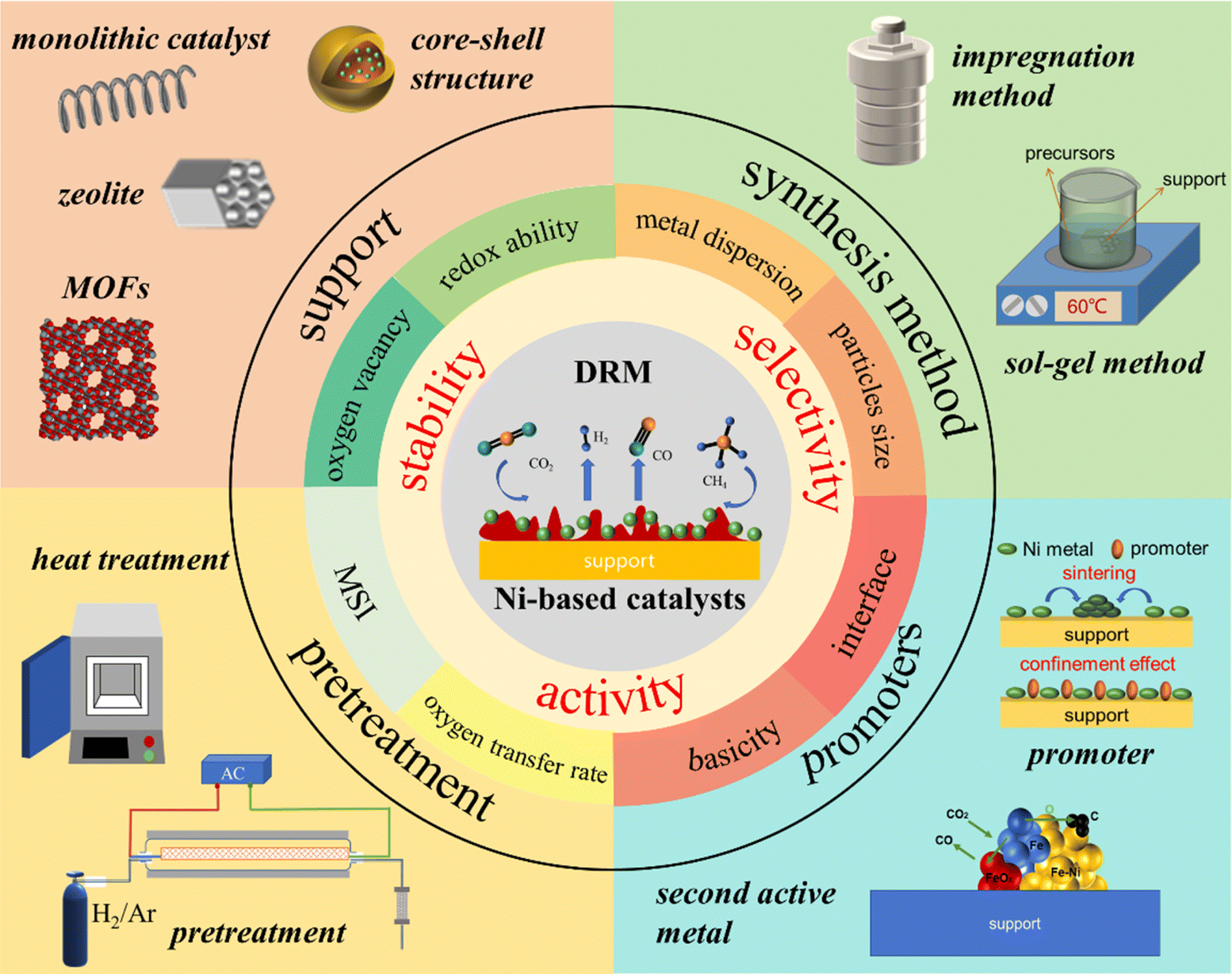 | ||
| Fig. 4 Graphical illustration of the scope of this review. | ||
2.2 Formation and elimination of carbon deposition
DRM is frequently conducted at high temperatures to achieve the required production output. At elevated temperatures, carbon deposition primarily results from the complete breakdown of CH4, rather than from the Boudouard reaction or CO reduction, owing to the highly endothermic nature of CH4 decomposition1,27 (Fig. 2(a)). Therefore, a viable approach to mitigate carbon deposition is to promote the formation of intermediate CH4 to curb its complete dehydrogenation. The primary side reactions in DRM are summarized in Table 1.16,27
| Side reaction | Chemical equation | ΔH298Kθ | Operational temperature |
|---|---|---|---|
| Boudouard reaction | 2CO(g) ↔ C(s) + CO2(g) | −173 kJ mol−1 | <700 °C |
| Decomposition of CH4 | CH4(g) ↔ C(s) + 2H2(g) | 75 kJ mol−1 | >557 °C |
| RWGS | CO2(g) + H2(g) ↔ CO(g) + H2O(g) | 41 kJ mol−1 | 870–1040 °C |
| CO reduction | CO(g) + H2(g) ↔ C(s) + H2O(g) | −131 kJ mol−1 | — |
To promote incomplete dissociation of CH4, it is necessary to understand the dissociation mechanism of the reactants. In the last decades, first-principles calculation was employed to investigate DRM reaction mechanism and carbon deposition on Ni-based catalysts. Lian et al.40 proposed a four-step mechanism for the DRM reaction on Ni/CeO2, including CO2 activation, CH4 dehydrogenation, CHx oxidation, and CHxO dehydrogenation. In addition, the adsorption sites of CH4 and CO2 were explored as well. The unfilled d-orbital of metallic Ni is prone to accepting C–H bonds σ, and then promoting CH4 adsorption and dissociation.41 The oxygen vacancies and basic sites are mainly active sites for CO2 adsorption, which will also be discussed in the review. Moreover, the adsorption of CO2 or CH4 is structure-sensitive, which is more likely to occur at low coordinated sites.42,43 Yu et al.23 discovered that the stepped (211) surface demonstrated higher reactivity compared to the close-packed (111) surface. Fig. 5 illustrates DRM reaction steps and molecular conversion on the catalyst surface.
 | ||
| Fig. 5 (a) Principal steps of DRM reaction. (b) Active sites and molecular conversion on the catalyst surface in DRM reaction, which shows the dissociation of CH4 and CO2, formation of OH* and CHxO*, decomposition of CHxO*, and release of CO and H2. | ||
From the comprehensive perspective of active sites and reaction mechanisms, DRM reaction mechanism can be divided into:44,45
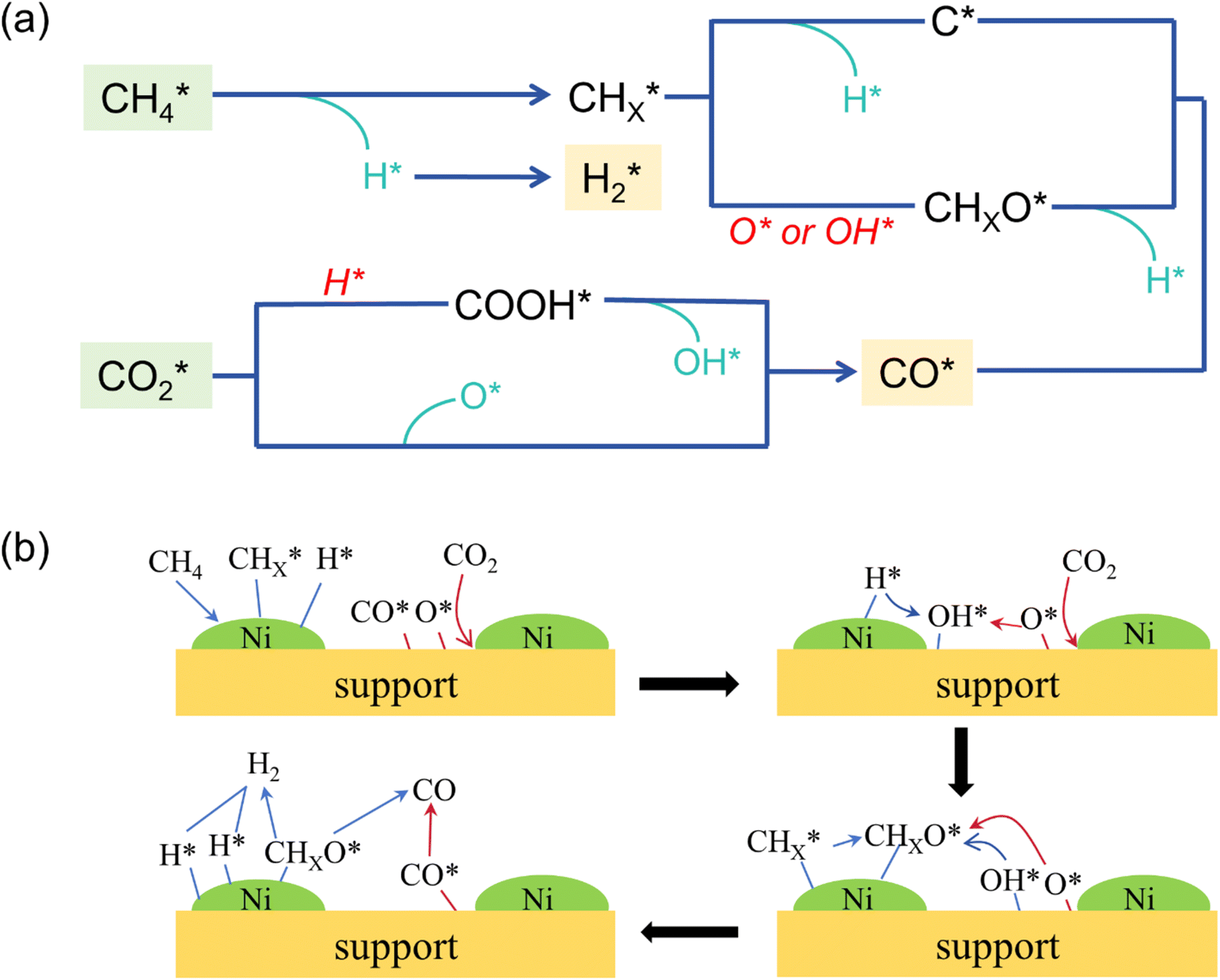
(1) The adsorption and continuous dehydrogenative dissociation of CH4 on the active metal Ni to form the methyl-like species 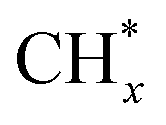 (0 ≤ x < 4) and different amounts of H*.
(0 ≤ x < 4) and different amounts of H*.
(2) The adsorption and dissociation of CO2 on basic sites or interface oxygen vacancies, including direct dissociation 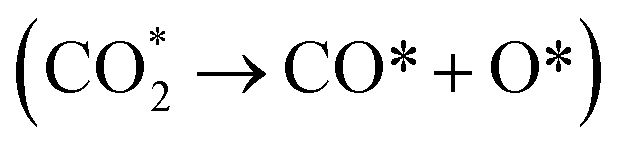 and H-assisted dissociation
and H-assisted dissociation  .
.
(3) 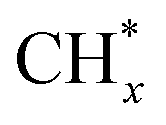 reacts with O* or OH* to form the formate intermediate CHxO*.
reacts with O* or OH* to form the formate intermediate CHxO*.
(4) CHxO* decomposes to generate CO* and H*, and H* combines with each other to form 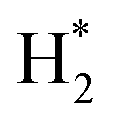 .
.
(5) Release of adsorbed CO and H2, which are rate-determining steps.
Thermal sintering and carbon deposition are commonly observed phenomena on Ni-based catalysts, and their occurrence is intricately linked. Initially, the sintering-induced growth of Ni particles resulted in increased particle sizes and decreased surface areas, accelerating the rate of carbon formation. This is attributed to carbon molecules diffusing through the crystalline structure of Ni particles, while large particles provide more pathways to facilitate such diffusion phenomena.46 Meanwhile, carbon formation and subsequent gasification contribute to the migration of Ni particles, consequently altering their size and shape.47,48 Owing to the low H/C ratio in the reactants, carbon deposition is often unavoidable in DRM. Hence, promoting intermediate formation and inhibiting complete CH4 dehydrogenation are viable strategies for mitigating carbon deposition. In addition, Rao et al.49 posited that the active site plays a significant role in determining the reaction pathway and generation of intermediate species. Specifically, 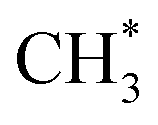 prefers to react with oxygen to produce CH3O* on the Ni–O site, whereas
prefers to react with oxygen to produce CH3O* on the Ni–O site, whereas 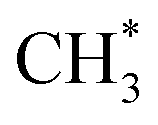 species are prone to forming
species are prone to forming 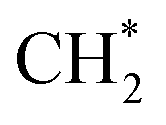 and H* on the Ni–Ni site. The carbon deposition can be classified into two categories:50,51 deactivated carbon (polymeric amorphous films, bulk Ni carbide, graphitic platelets or films, filament carbon) and non-deactivated carbon (surface adsorbed atomic carbon, vermicular filaments).
and H* on the Ni–Ni site. The carbon deposition can be classified into two categories:50,51 deactivated carbon (polymeric amorphous films, bulk Ni carbide, graphitic platelets or films, filament carbon) and non-deactivated carbon (surface adsorbed atomic carbon, vermicular filaments).
Non-deactivated carbon does not influence its activity and can be easily eliminated by oxidation.44 In contrast, deactivated carbon encapsulates the active metal Ni, blocks pores, and even physically damages the catalyst structure. Furthermore, the deactivation attributed to deactivated carbon is irreversible.52
Among all deactivated carbon, filament carbon is a major contributor to pore damage and structure collapse due to its high strength. For filament carbon formation, two mechanisms have been mentioned, the bulk dissolution mechanism and the surface mechanism (Fig. 6).
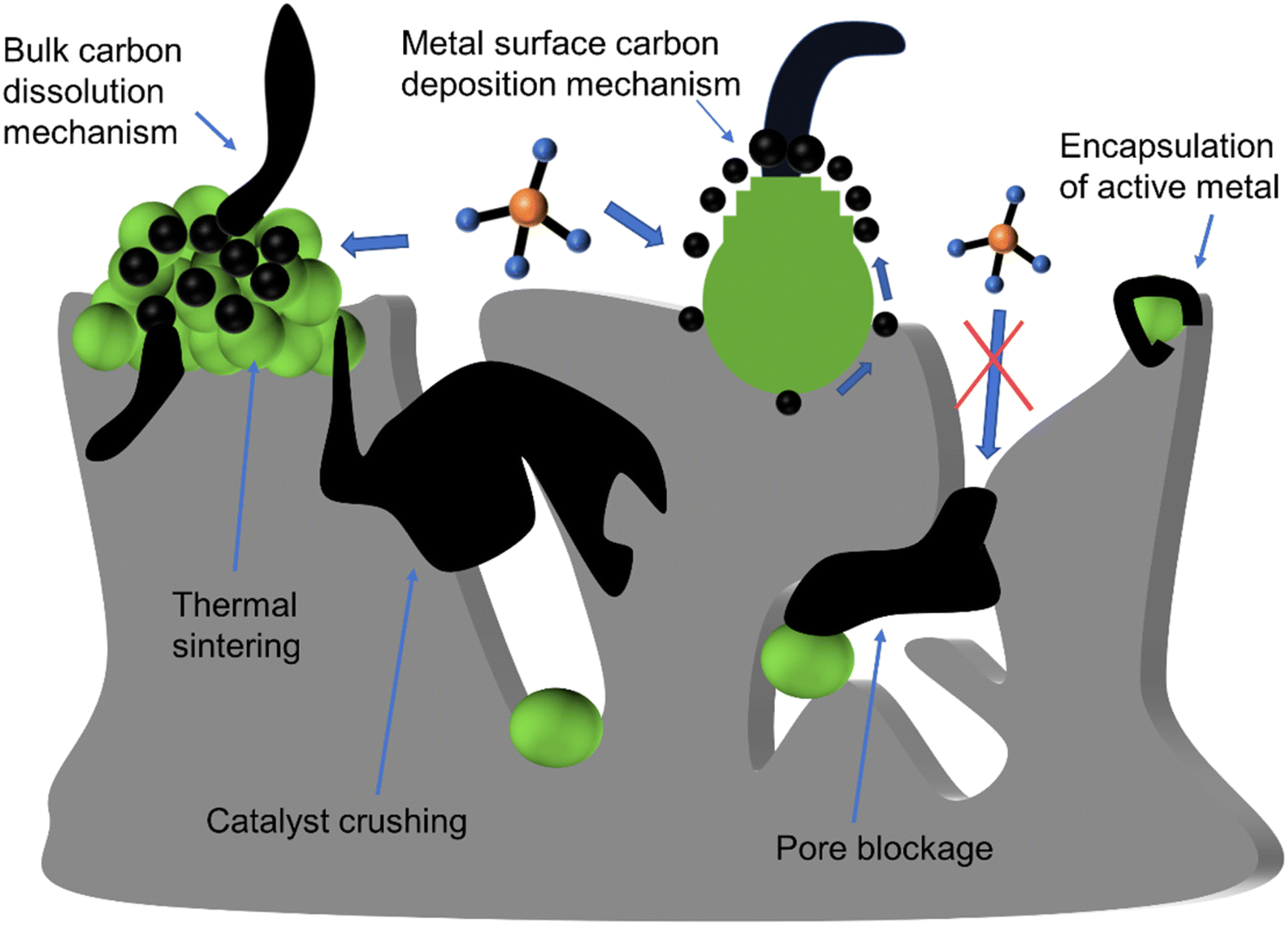 | ||
| Fig. 6 Schematic of chemical or mechanical deactivation of Ni-based catalysts. | ||
In the bulk dissolution mechanism, carbon-containing gas molecules decompose to yield carbon adatoms that dissolve in the active metal Ni. Subsequently, these carbon atoms undergo diffusion and segregation on the appropriate growth plane, promoting filament carbon growth.53 The carbon atoms diffusion process was proposed to be the rate-determining step in coke formation.54,55 After establishing the location of the growth of filament carbon, the filament carbon achieves a stable growth state along the plane as a result of the ongoing migration of carbon atoms. Larsen et al.56 posit that the growth of filament carbon is influenced by the disparity in rates between the creation of surface-adsorbed atoms and the migration of carbon atoms within the bulk phase. If the carbon atom generation rate reaction rate exceeds the bulk diffusion rate of carbon atoms, the bulk diffusion process becomes the limiting factor, potentially leading to catalyst poisoning by an excess of surface carbon atoms. Conversely, if the carbon atom supply rate is slower than bulk diffusion, filament carbon growth may persist indefinitely until the carbon fiber breaks.
In the surface mechanism, carbon nucleation at the step sites and the ensemble size effect are critical for the formation of filament carbon. Under-coordinated sites, such as Ni step edge sites, play a critical role in the nucleation and expansion of filament carbon due to the stronger binding of carbon to these sites compared to sites at the close-packed facets of Ni. The step sites on the Ni surface provide more favorable active sites for carbon atom adsorption, thus becoming the initial sites for the growth of filament carbon. Specifically, Ni nanoclusters experience cycles of elongation and contraction induced by reforming reactions.57 During this process, due to the higher reactivity of carbon atoms at the gas/catalyst interface compared to the catalyst/graphite layer interface,24 Ni step edges spontaneously form at the interface between graphene and Ni, and move towards the end of the Ni aggregates. This promotes the migration of carbon atoms at the interface and the formation of filament carbon sites.43 Additionally, the ensemble size effect starts to influence the process when carbon atoms aggregate at the step sites on the Ni surface. As the number of carbon atoms increases, the size of the aggregates grows, altering their physical and chemical properties, which in turn impacts the growth behavior of filament carbon. For example, larger aggregates may exhibit higher reactivity and lower growth barriers, which is beneficial for the growth of filament carbon. If the crystal surface or boundary is of insufficient size, nucleation is hindered, further inhibiting the formation of carbon fiber. The growth of filament carbon ceases once carbon atoms completely encapsulate the Ni nanoclusters.
Various factors influence both the type of coke and growth mechanism, such as the surface chemical properties of Ni, the precursor containing Ni2+, grain size, pretreatment gas, and reaction conditions (such as temperature, pressure, and gas composition).44 Carbon formation rate is dependent on the thermodynamic carbon activity and is impacted by the pressure ratio (PCH4PCO/PCO2), reaction temperature, and the size of Ni particles.30,31 When the levels of PCH4PCO/PCO2 are elevated, multiple sites for carbon formation initiate simultaneously, increasing carbon formation rate, leading to the encapsulation of Ni nanoparticles by a carbon layer, as shown in Fig. 3(b). Additionally, the rate of carbon formation decreases with a reduction in the size of Ni particles, as carbon filaments formed on smaller Ni particles exhibit lower stability. In addition, an increase in temperature can also lead to a decrease in carbon deposition in Fig. 2(b). By selecting appropriate synthesis methods, supports, and reaction conditions, it is feasible to inhibit the transformation of activated carbon into non-activated carbon. In addition to carbon deposition, the other deactivation types and mechanisms of Ni-based catalysts are shown in Fig. 6 and Table 2.42,44,45
| Mechanism | Type | Description |
|---|---|---|
| Thermal sintering | Mechanical deactivation | Metal accumulation leads to the blockage of the active site |
| Pore blockage | Mechanical deactivation | The carbon growing around the pores blocks the channels, restricting contact between the reactants and the catalyst |
| Metal encapsulation | Chemical deactivation | The carbon deposition on the metal surface encapsulates the entire metal particle, leading to deactivation |
| Catalyst crushing | Mechanical deactivation | The growth of strong carbon filaments compresses catalyst pores, leading to support fracturing and degradation |
| Inactive phase | Chemical deactivation | The interaction between side reaction products and catalyst components leads to the generation of inactive phases |
The oxygen atoms generated by the decomposition of CO2 participate in the gasification of surface carbon deposition. Therefore, the slow dissociation of CO2 may not hinder carbon deposition which can cover the active metal and impede its further involvement in the reaction process. On the other hand, excessively rapid CO2 activation can lead to the oxidation of active sites, resulting in the formation of the inactive phase. Moreover, the abundant oxygen atoms produced from the dissociation of CO2 can react with H2, compromising the selectivity of H2. Therefore, it is crucial to achieve effective CO2 dissociation and sustained participation in the reaction. The adsorption and activation of CO2 are influenced by the concentration of oxygen vacancies and surface basic sites, which will be further discussed in the subsequent sections.60
The hydroxylation reaction initiates upon contact between the oxide support and water on the surface.62 The nature of hydroxyl groups, whether acidic or basic, depends on the property of the oxide support. The basic sites on the support facilitate acidic gas adsorption like CO2, while the number and intensity of basic sites affect catalytic activity, selectivity, and stability.63 The research underscores that moderate intensity of basic sites is beneficial for CO2 adsorption and dissociation, increasing the oxygen transfer rate and stabilizing the carbonate intermediates.64 Meanwhile, basic sites with appropriate intensity can prevent excessive CO2 adsorption, thereby promoting catalytic efficiency and H2/CO ratio due to limitations in the RWGS reaction. In contrast, weak basic sites limit efficacy in CO2 adsorption, while excessively strong basic sites heighten adsorption ability and impede chemical bond cleavage, thereby attenuating CO2 reactivity.44,65 The generation of basic sites can be facilitated through the selection of suitable basic supports or promoters. Additionally, pretreatment processes that remove H2, CO2, and O2 from the surface of basic oxides can induce the formation of basic sites.66 Different pretreatment temperatures affect the degree of desorption of these gases.67 Therefore, the optimal pretreatment temperatures yield an ideal intensity of basic sites to maximize catalytic activity. However, protracted pretreatment duration may induce the rearrangement of catalyst atoms, altering the number and properties of surface basic sites.
In addition to basic sites, the number of oxygen vacancies is also a crucial factor in eliminating carbon deposition. CO2 exhibits the capacity to modify its structure by incorporating oxygen atoms into vacancies, thereby facilitating the dissociation process. Oxygen vacancies can arise through various mechanisms:
(1) Charge imbalance and lattice distortion are caused by the partial substitution of atoms within the lattice, thereby increasing oxygen vacancies.68
(2) The interaction between supports with different oxygen concentrations induces the migration of oxygen atoms, generating oxygen vacancies.69
An optimal number of oxygen vacancies enhances CO2 dissociation and facilitates oxygen atom migration, thereby promoting the oxygen transfer rate.44 However, excessive oxygen vacancies blocks active metal sites, limiting access between reactants and metal surfaces, and reducing CH4 and CO2 conversion in DRM reactions.44 The modification of catalysts to improve CO2 adsorption can be achieved by introducing oxide supports and promoters. This strategy will be discussed in the following sections.
3. Synthesis method
The primary synthesis methods of DRM are summarized in Table 3. Contescu et al.81 conducted a review of the catalyst preparation technologies and categorized them into two main categories. The first category involves transforming precursors containing the active metal, promoter, and support in the liquid phase into solid-phase catalysts through precipitation or chemical decomposition. The second category involves introducing and fixing the active metals in the prepared solid phase supports using the impregnation method. The method of synthesis significantly influences the specific surface area, dispersion of active metals, and particle size of catalysts. External factors, such as heat and pressure, are frequently utilized to enhance the interaction between active metals and supports. For example, the conventional hydrothermal technique involves introducing the salt solution containing active metals and supports into a sealed reactor, where they combine mutually under conditions of elevated pressure and temperature. Subsequently, a catalyst with strong metal–support interaction is formed through specific procedures (e.g. drying and calcination). This review primarily introduces five common approaches for synthesizing Ni-based catalysts: the impregnation method, precipitation method, sol–gel method, solution combustion method, and gasification-deposition method. Additionally, many physical or chemical auxiliary techniques utilized to enhance active metal dispersion and promote metal–support interaction were discussed as well.| Catalyst | Synthesis method | T [°C] | GHSVb [ml g−1 h−1] | Timec [h] | CH4 conversion [%] | CO2 conversion [%] |
|---|---|---|---|---|---|---|
| a K = °C + 273.15. b Gas hourly space velocity. c It is demonstrated by the duration for which the catalyst remains active. | ||||||
| 10Ni/10Al2O3–10ZrO2 (ref. 70) | Impregnation with plasma treatment | 850 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
24 | 92 | 95 |
| Ni/fumed silica71 | Pressure dilution | 600 | 1440000 |
19 | 88 | 95 |
| Ni/Al2O3 (ref. 72) | Impregnation with EDF technique | 800 | 120000 |
— | 58.8 | 77.3 |
| Ni/SiO2-0.7G73 | Glycine-assisted impregnation process | 600 | 60000 |
20 | 38.3 | 52 |
| Ni/SBA-15 (ref. 74) | Homogeneous precipitation | 700 | 36000 |
100 | 79 | 74 |
| Ni/palygorskite72 | Ultrasonic-assisted hydrothermal precipitation | 800 | 90000 |
6 | 75.62 | 79.58 |
| Ni/Ce0.75Zr0.25O75 | Surfactant-assisted co-precipitation | 850 | 12000 |
9 | 92 | 94 |
| 5NiO/MgO76 | Sol–gel | 750 | 30000 |
50 | 72 | 88 |
| Ni/4La2O3–1CeO2 (ref. 77) | Ammonia solution-assisted sol–gel | 800 | — | 24 | 94.0 | 97.5 |
| Ni/ZrO2–Al2O3 (ref. 78) | Modified pechini sol–gel | 700 | 25000 |
70 | 50 | 55 |
| Ni–Mg/La2O3 (ref. 79) | Solution combustion | 700 | 30000 |
100 | 83.2 | 90.8 |
| Ni/La2O3 (ref. 80) | Combustion synthesis of nitrogen-rich precursors | 800 | 20000 |
24 | 73 | 84 |
3.1 Impregnation method
The impregnation method commonly entails submerging porous supports in nitrate solutions containing active Ni components, wherein capillary force regulates solution penetration into the pores, followed by water removal through evaporation. The resultant catalytic performance is notably affected by the support structure with the technique renowned for its simplicity and cost-effectiveness. Rao et al.49 use the impregnation method to synthesize Ni/CeO2 catalysts with distinct Ni–O or Ni–Ni coordination by adjusting the Ni-containing precursor solution. Zhang et al.82 prepared a series of Ni/MSS (monodisperse dendritic mesoporous SiO2 spheres) using three impregnation methods: conventional impregnation, glycine-assisted impregnation, and ethylene glycol-assisted impregnation. The results showed that glycine-assisted impregnation and ethylene glycol-assisted impregnation effectively improved the dispersion of Ni while maintaining the unique pore structure of the MSS support and producing smaller Ni particles compared to conventional impregnation. As a result, the catalyst exhibited excellent activity and stability.The morphology and performance of catalysts are greatly influenced by treatment and activation methods. The impregnation method lacks the necessary force to achieve proper contact between metal particles and the support, and it is difficult to control metal particle size and maintain activity. Consequently, various physical or chemical assistant technologies are employed to enhance the interaction between metals and supports, thereby facilitating the loading process. For example, Ren et al.83 utilized the ultrasonic impregnation method to prepare Ni catalysts on SiO2/TiO2 composite oxides with varying molar ratios of silicon, which is applied for CO methanation with improved interaction through physical interaction.
In recent years, many non-thermal physical assisted methods have been applied in the synthesis of catalysts, which can effectively improve the dispersion of active metals and the metal–support interaction, and even change the reaction pathway. Odedairo et al.84 prepared Ni/CeO2 catalysts by impregnation method and used microwave plasma-assisted method instead of conventional heat treatment, which can effectively improve the conversion of reactants. Rahemi et al.70 employed synthesized impregnation and non-thermal plasma treatment to synthesize Ni/Al2O3–ZrO2 nanocatalysts. Plasma treatment can enhance the interaction between the metal and support, which also prevents support particle aggregation and collapse, resulting in uniform morphology and smaller particle formation. After plasma treatment, the catalytic performance is superior to untreated counterparts, with remarkable CH4 and CO2 conversions reaching 93% and 94%, respectively. Khoja et al.63 fabricated Ni/γ-Al2O3–MgO nanocomposite catalysts via cold plasma assisted incipient wetness impregnation method in a dielectric barrier discharge (DBD) plasma reactor, assessing the influence of specific input energy (SIE) on catalytic performance. Their findings indicated that the selectivity of H2 and CO has a positive correlation with the SIE value, accompanied by a decrease in by-products C2H6. Thus, increasing SIE is an efficient way to alter the reaction pathways and enhance H2 yield, possibly through the suppression of CH3 and H recombination or facilitating the breakdown of CH3. In addition, Zhu et al.85 investigated the effect of argon discharge plasma treatment on the structure and reactivity of Ni/Al2O3 catalyst for DRM. The DRIFT spectra analysis reveals that the sample prepared with the plasma treatment (NiAl-PC) maintains Ni particle size but exhibits variations in morphology compared with untreated counterparts (NiAl–C). The flatter morphology and larger metal–support interface of NiAl-PC restrict CH4 dissociation, resulting in a better carbon formation-gasification balance. Furthermore, the plasma-treated catalyst contains a high concentration of close-packed planes, leading to the improvement of Ni dispersion and Ni–Al2O3 interaction, thereby enhancing catalytic activity and resisting filamentous carbon formations. Moreover, Danghyan et al.71 utilized pressure dilution combined with wet impregnation to prepare Ni/fumed silica catalyst for DRM. They mixed catalyst powder prepared by the traditional impregnation method with additional fumed SiO2, and catalyst nanoparticles formed with excellent dispersion (61%). The higher dispersion leads to higher catalyst activity and stability, allowing carbon deposition nanotubes to disperse more freely with reduced entanglement.
The principle of chemical auxiliary techniques is to cross-link metal atoms onto organic radicals by adding certain special organic solvents, avoiding partial segregation of metals caused by the interaction between metal ions in the solution.
Zheng et al.86 used acetic acid to selectively etch the inner layer of HNTs to preserve the original nanotube-like morphology, while expanding the inner diameter from 12.9 nm to 17.9 nm. Meanwhile, the inner surface was hydroxylated, promoting the introduction and adsorption of the metal precursor. Currently, a slightly modified equilibrium deposition filtration (EDF) technique is often used to prepare Ni/Al2O3 catalysts.72 During the traditional impregnation process, uncontrolled precipitation of metal particles on the support generated larger metal particle sizes. In comparison, species of the catalyst using EDF are deposited not only by physical adsorption but also via reaction with the receptor sites, such as surface oxygens and hydroxyls, which developed on the support surface throughout the whole process. The continuous deposition during the mixing process between the precursor and support resulted in high metal dispersion and imparted resistance to sintering. Yang et al.73 proposed that in the conventional impregnation method for preparing Ni/SiO2 catalysts, the NiO particles are mainly distributed on the outer surface of SiO2. The XPS results (Fig. 7(c)) indicate the presence of a Ni 2p3/2 peak at 854.1 eV in the Ni/SiO2 catalysts. This peak is attributed to the weak interaction between larger NiO particles and the support, thereby facilitating carbon deposition and catalyst sintering. To address this issue, a series of Ni/SiO2-XG catalysts (X represents the molar ratio of glycine to nitrate) were prepared via the glycine-assisted impregnation method. By adjusting the glycine content, the Ni precursors were highly dispersed in SiO2 pores, resulting in smaller NiO particle sizes (Fig. 7(a and b)) and stronger interaction between NiO and SiO2. Experimental results revealed that the Ni/SiO2-0.7G exhibited optimal catalytic activity, stability, and resistance to carbon deposition (Fig. 7(d)) during low-temperature DRM reaction. Li et al.87 used the L-arginine ligand-assisted incipient wetness impregnation (LA-IWI) approach to synthesize a highly dispersed Ni/ZrO2 catalyst. This catalyst demonstrated superior catalytic activity and stability for the steam-CO2 dual reforming of methane compared to the Ni/ZrO2 catalyst synthesized using the IWI method. Experiments further confirmed that complexing agents can enhance the interaction between the metal and supports, increase oxygen vacancies, and promote the replacement of Zr4+ with Ni2+ in the ZrO2 lattice. This substitution enhances the stability of both the Ni2+ cation and the thermodynamically unstable tetragonal ZrO2 phase. Zhang et al.88 prepared a series of Ni/SBA-15 catalysts via the chelating ligand-assisted impregnation method. Their results indicated that the presence of the chelating ligand with electron-pair donating atoms and the high viscosity effectively inhibits the aggregation of Ni species, which is beneficial to improving dispersion.
 | ||
| Fig. 7 TEM images of reduced catalysts of (a) Ni/SiO2-0G and (b) Ni/SiO2-0.7G. (c) Ni 2p X-ray photoelectron spectra of fresh catalysts. (d) TGA profiles for spent catalysts. Reprinted with permission from ref. 73 Copyright 2022, Elsevier. | ||
In addition to using auxiliary methods, the catalytic performance can also be affected by adopting an appropriate impregnation sequence. Zhen et al.89 prepared Ni–Ru/γ-Al2O3 using co-impregnation and sequential impregnation methods, demonstrating controllability of Ni and Ru through adjustment of preparation steps. For catalysts prepared by the co-impregnation method, Ru segregation occurs on the metal surface, which has the potential to alter the chemical state of surface Ru species to favor metallic state reduction. Yao et al.90 compared the catalytic activity of Zr and Mn co-promoted Ni/SiO2 catalysts (Ni–MnOx–ZrOx/SiO2, MnOx/Ni–ZrOx/SiO2, and ZrOx/Ni–MnOx/SiO2 catalysts) via co-impregnation and sequential impregnation synthesis methods. Among these catalysts, due to the strong ability to absorb CO2 and form intermediate, ZrOx/Ni–MnOx/SiO2 performed higher activity at low temperatures. By adjusting the reduction temperature to 550 °C, the ZrOx/Ni–MnOx/SiO2 exhibited smaller Ni species particles with a narrow particle size distribution (5–6 nm).
3.2 Precipitation method
The precipitation method serves as a prominent technique for catalyst preparation, with factors such as pH, temperature, reagents nature, impurities, and precipitation method impacting catalyst particle size and pore structure. High supersaturation promotes the nucleation rate of solid particles from small particles.81 However, controlling the growth and aggregation of particles of the precipitation method is a challenge due to the difficulties in achieving supersaturation for extended periods and the occurrence of Ostwald ripening.The co-precipitation method is commonly used to synthesize catalysts that contain various active phases. This is due to the solubility difference between each component, which allows for a change in the precipitation order of different active phases. Albarazi et al.91 prepared Ce and Zr doped Ni/SBA-15 catalysts using co-precipitation and impregnation methods for DRM. Compared with the impregnation method, the co-precipitation method can significantly improve catalyst stability, but catalytic activity is slightly lower. This attributed to the co-precipitation method effectively inhibits the direct CH4 dissociation and minimizes carbon formation. Simultaneously, the existence of CH4 will decrease its activity. Zhang et al.74 also compared the DRM catalytic performance of Ni/SBA-15 prepared by homogeneous precipitation method (Ni-HP) and impregnation method (Ni-IM). H2-TPR revealed that catalysts prepared by the homogeneous precipitation method have stronger metal–support interaction and higher stability, owing to the type of coke mainly being disordered filamentous carbon without impact on catalytic performance. After 100 hours of reaction at 700 °C, the CH4 conversion of Ni-IM catalyst dropped from 61.7% to 37.3%, while the CH4 conversion of Ni-HP catalyst slightly decreased from 74.5% to 73.8%, ascribed to its excellent anti-sintering property. Li et al.92 prepared the palygorskite-supported Ni catalysts (Ni/pal) using five methods (deposition precipitation, impregnation, ultrasonic-assisted impregnation, hydrothermal precipitation, and ultrasonic-assisted hydrothermal precipitation). The experimental results indicated that the catalyst prepared by ultrasonic-assisted hydrothermal precipitation exhibited the homogeneous dispersion of active nanoparticles, a large surface area and pore diameter, strong interaction between the Ni component and palygorskite (Fig. 8(a)), and favorable basic sites (Fig. 8(b)), resulting highest DRM catalytic activity with reactant conversions of 75.62% (CH4) and 79.58% (CO2), respectively (Fig. 8(c and d)).
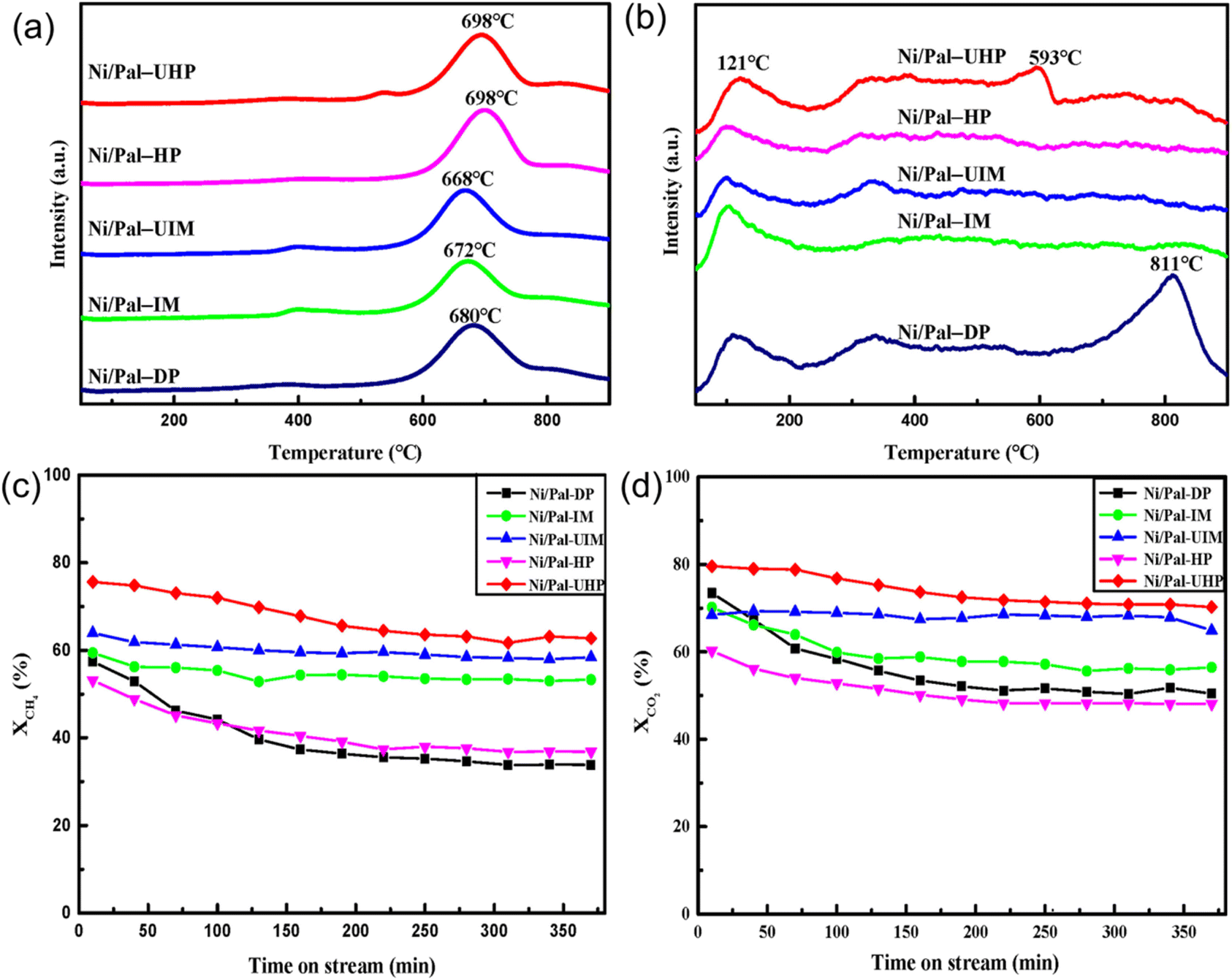 | ||
| Fig. 8 (a) H2-TPR profiles, (b) CO2-TPD profiles, (c) CH4 conversion, and (d) CO2 conversion for Ni/Pal with different preparation methods. Reprinted with permission from ref. 92 Copyright 2022, Elsevier. | ||
Additionally, adding precipitation agents or surfactants could reduce the solubility of the precipitate. However, extra addition is possible to generate excessive concentration in certain solution areas with uneven active component distribution. Chen et al.75 prepared Ni/Ce0.75Zr0.25O2 catalysts using co-precipitation with the surfactant (CTAB) assistance for DRM, combining the advantages of Zr and Ce. The surfactant-assisted process encourages Ni2+ to enter the lattice of solid solution and interact with Ce0.75Zr0.25O2, and then reduce the interfacial energy and the surface tension of water existing in the sample pores, thereby obtaining a larger surface area and pore size. Experimental results suggest that the CH4 and CO2 conversions were respectively about 92% and 94% under the conditions of 0.1 MPa, 1123 K, and gas hourly space velocity of 12000 ml g−1 h−1. Zanganeh et al.93 prepared Ni0.1Mg0.9O powder using the surfactant (PEG-PPG-PEG) assisted co-precipitation method for DRM, and then investigated the effects of several process parameters on the structural properties of the powders. Their findings demonstrated that NiO–MgO samples have a higher specific surface area and smaller NiO–MgO solid solution crystallite size under the conditions of high refluxing time (>20 h), high refluxing temperature (>80 °C), and low calcination temperature (<600 °C).
Moreover, the co-precipitation method is widely applied for preparing ternary oxides, particularly solid solutions. Xie et al.94 used a modified one-pot strategy by combining co-precipitation with a sol–gel process to prepare SiO2 supported Ni0.2Mn0.8O ternary metal oxide catalysts for DRM. In this method, the metal salt solutions are added to an aqueous sodium silicate solution, resulting in simultaneous precipitation of the metal alongside the gelation of the silica precursor. After heat treatment, dispersed metal oxide nanoparticles are formed on porous SiO2, exhibiting a relatively smaller particle size compared to bulk Ni0.2Mn0.8O prepared by the conventional co-precipitation method, enhancing activity and stability in DRM low-temperature reaction (<550 °C).
3.3 Sol–gel method
The sol–gel method is related to the co-precipitation method, which forms the catalyst through the transition from the liquid phase into the solid phase. The rapid gel formation of the sol–gel preparation process results in the incorporation of sol molecules within the gel framework, thereby inhibiting particle agglomeration and enhancing catalyst stability.95 However, the sol–gel method demands a long operation duration and emits substantial harmful gases.The sol–gel method is usually used to prepare composite materials and multi-functional materials by mixing varying material components. Jafarbegloo et al.76 prepared NiO–MgO solid solution using the sol–gel method, achieving lower calcination temperature than that of the impregnation method and precipitation method. Rad et al.96 conducted a comparative study between the sol–gel method and the impregnation method for preparing Ni/Al2O3 catalyst, and results indicated that the sol–gel method facilitated NiAl2O4 spinel formation with enhanced metal–support interaction, which also contains higher metal dispersion, higher reducibility, and larger surface area. Venezia et al.97 investigated the effects of the sol–gel method and grafting method on Ni/SiO2 with ZrO2 and TiO2 as promoters. The Ni/SiO2 with TiO2 prepared by the sol–gel method exhibits higher structural stability and chemical stability, resulting in stable CH4 and CO2 conversion (the deactivation rate is only 7% within 24 hours at 650 °C).
Researchers also try to improve the sol–gel method to further enhance the structural stability of the catalyst. Chen et al.98 prepared xerogel and aerogel Ni/CeO2–Al2O3 by sol–gel method combined with two drying methods (conventional drying and supercritical drying). Experiments demonstrated that the aerogel preparation method has a higher specific surface area (282.9 vs. 195.9 m2 g−1), larger pore diameter (0.24 vs. 0.18 cm3 g−1), smaller bulk density (0.44 vs. 0.74 g ml−1) and smaller particle size (10.6 vs. 17.8 nm) than xerogel. Grabchenko et al.77 utilized the sol–gel method to synthesize CeO2 and binary La2O3–CeO2 oxides with different La/Ce atomic ratios (1:4; 1:1; 4:1). The citric acid (C6H8O7·H2O) and ammonia (NH4OH) serve as complexing agent and pH agent for preparing Ni/La2O3–CeO2 catalysts. Their results prove that citric acid and ammonium hydroxide can effectively promote the formation of gel and La2−xCe2xO3−δ. Peak shift towards high temperature in H2-TPR indicated that solid solution formation enhances the interaction between the metal and supports, improving the stability of Ni/La2O3–CeO2 catalysts. Shin et al.78 prepared Ni/ZrO2–Al2O3 catalyst by modified Pechini sol–gel method, urea hydrolysis method, and physical mixing method. Their work indicated that the Pechini sol–gel method obtained higher crystallinity to improve the reducibility and chemical adsorption ability of the catalyst surface.
3.4 Solution combustion method
As a redox-based reaction, the solution combustion method predominantly occurs in a homogeneous aqueous solution containing oxidizing agents (metal nitrates) and reducing agents (fuels). This reaction releases heat without external heat sources, facilitating rapid combustion waves and depositing the active phase onto a solid support to enhance metal–support interaction. The designed components and structures of the catalysts are obtained via rapid automatic combustion waves in the self-sustaining reaction.For the solution combustion method, controlling the heat release and transfer rate is utilized to control the structure, and the appropriate fuel is a key point. Xu et al.99 employed the impregnation combustion method to synthesize a series of Ni/CeO2/SiO2 catalysts. During the calcination process, the fuels NH3 and N2O generated from the raw materials interact to promote the combustion reaction. Due to the instantaneous and high exothermic combustion reaction, Ni nanoparticles rapidly nucleate with highly dispersed Ni particles (6 nm), elevating catalytic activity. Cross et al.100 prepared a Ni/fumed SiO2 catalyst using glycine and ammonium nitrate as fuel. The mixed solution formed a gel during the drying and preheating process, and then the gel burned through ignition to form highly dispersed Ni nanoparticles. The molecular-level precursor mixture in the active aqueous solution used in the solution combustion method can produce catalyst particles with a higher surface area compared to the co-precipitation method. The experimental results demonstrated that the specific surface area of the catalyst prepared by the solution combustion method reached 155 m2 g−1, and the size of the Ni nanoparticles was less than 5 nm. Ahmad et al.79 employed solution combustion synthesis to synthesize Ni/La2O3, which is promoted by alkali metals or alkaline earth metals (K, Na, Cs, Li, and Mg) and applied in DRM reaction. Their experimental results indicated that Ni–Mg/La2O3 has the highest specific surface area and pore content. At 700 °C, the CH4 and CO2 conversions can reach 83.2% and 90.8%, respectively, which also maintains prolonged stability after over 100 hours of reaction time. Danghyan et al.101 employed the improved cellulose-assisted solution combustion method (PACS) to synthesize NiO–MgO solid solution, comparing them with catalysts prepared by a single-step solution combustion method without cellulose. The catalyst prepared by PACS has a high specific surface area of 139 m2 g−1, owing to the template effect of cellulose paper.
In addition, cellulose acts as an additional fuel, intensifying combustion reactions and augmenting gas release during the combustion process at elevated temperatures. This mechanism effectively prevents sintering and forms smaller grain sizes. Sorcar et al.80 synthesized Ni/La2O3 catalysts via combustion synthesis of Ni and La complexes derived from nitrogen-rich precursors and the influence of precursor structure on catalytic performance was also analyzed. Their findings proved that nitrogen-rich precursors demonstrated superior efficiency in improving crystallinity and metal–support interaction. The ability of the fuel to release heat and nitrogen gas significantly affects the crystallization of catalysts and metal–support interaction. Among the various nitrogen-rich precursors, Ni-BT ([Ni(H2BT)2][NO3]2) generates the highest gas pressure during its combustion, resulting in the catalyst with enhanced reducibility and the highest dispersion of Ni nanoparticles. Synthesis, structures, and appearance of different nitrogen-rich Ni-precursors with different molecular structures of their ligands are shown in Fig. 9.
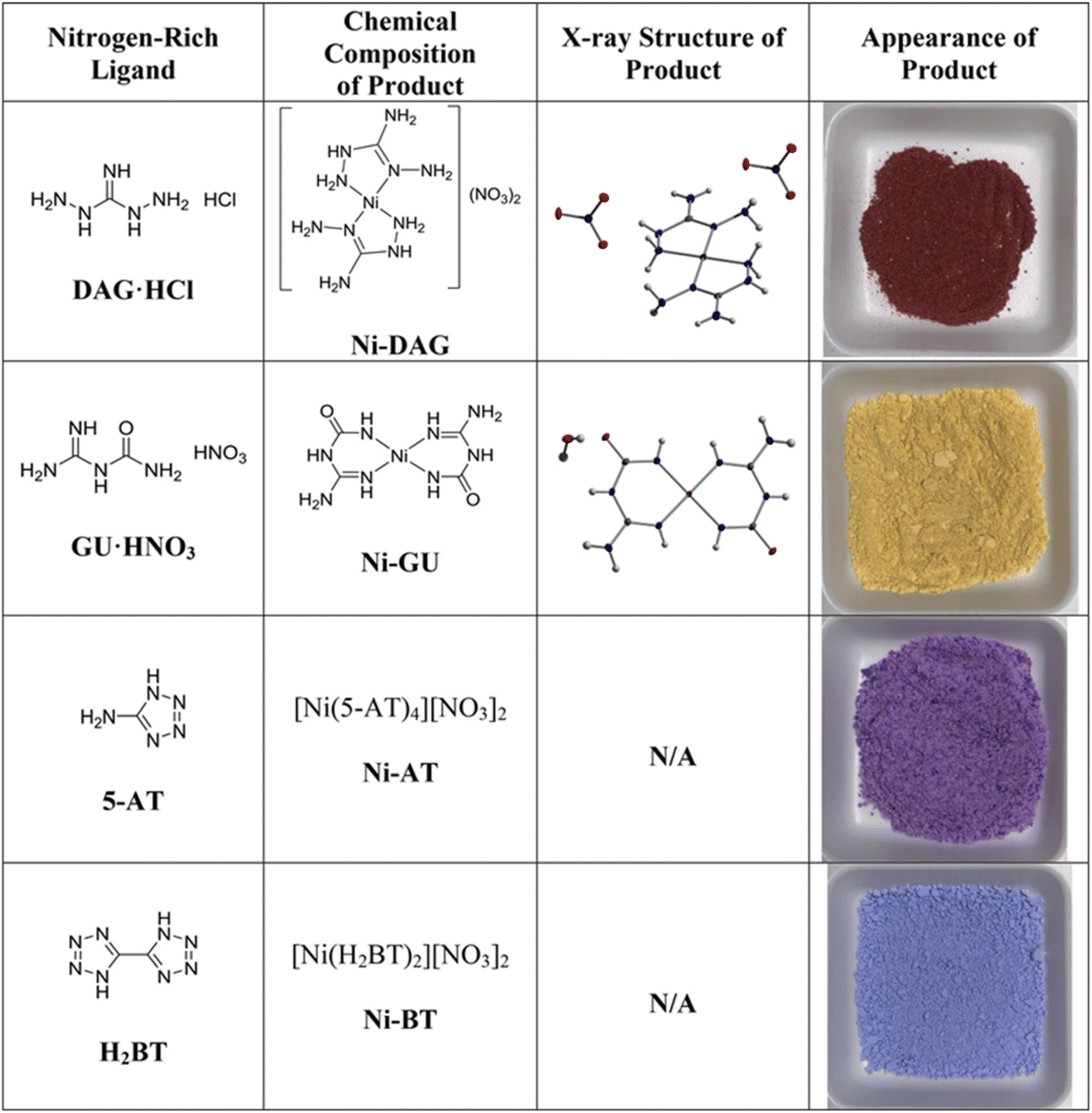 | ||
| Fig. 9 Synthesis, structures, and appearance of different nitrogen-rich Ni-precursors with different molecular structures of their ligands. Reproduced with permission from ref. 80 Copyright 2022, Elsevier. | ||
3.5 Gasification-deposition method
Porous supports can improve metal dispersion attributed to the confinement effect, which is crucial for anti-sintering and inhibition of carbon deposition growth. The gasification-deposition method, which includes evaporation and sublimation, is an effective technique for preparing porous catalysts. Bian et al.102 used ammonia evaporation to generate multiple Ni nanoparticles (diameter: 6 nm) supported on the inner silica core. Compared to other core–shell structure construction methods, this synthesis method has simple experimental steps without toxic chemicals. Wang et al.103 utilized the evaporation-induced self-assembly (EISA) method to synthesize a series of ordered mesoporous Ni–Ce–Al composite oxides with different cerium contents and applied them in DRM reaction. Specifically, hydrophobic Ni precursors are directly incorporated into the hydrophobic core of surfactant micelles, thereby stabilizing highly dispersed Ni nanoparticles in the mesoporous channels of the alumina matrix. Ma et al.104 prepared bimodal mesoporous-macroporous alumina supports using the evaporation-induced self-assembly (EISA) method, which was further employed for the preparation of Ni/Al2O3 catalysts. These catalysts have higher stability due to their non-macroporous structure. These studies demonstrated the deposition of active substances on the support surface through aqueous solution evaporation. In addition to the evaporation method, Xie et al.105 used a sublimation-deposition strategy to prepare Ni–Ce/SiO2 catalysts. Specifically, they placed Ni-containing compounds and modified porous silica solids separately in a quartz tube, and Ni-containing compounds were sublimated by heating and deposited on the support surface to form a mixture. Thereafter, porous Ni–Ce/SiO2 catalysts were formed through centrifugation and reduction, which have superior Ni dispersion compared to conventional impregnation methods.Simple and cost-effective preparation methods often lack sufficient load driving force, so physical means or promoters are needed to improve the dispersion and uniformity of catalyst particles. However, this strategy can lead to an escalation in both the time and expenses involved in catalyst preparation. This trade-off is one of the important factors to consider when choosing a synthesis method. Table 4 summarizes the advantages and disadvantages of common synthesis methods.
| Synthesis method | Advantages | Disadvantages |
|---|---|---|
| Impregnation | The preparation process is simple and the step of catalyst molding can be omitted. The utilization rate of catalytic components is high. The properties of catalysts depend on the physical structural characteristics of the support | Requiring multiple impregnations to meet the required impregnation amount and the binding force between the active component and the support is insufficient |
| Precipitation | It is suitable for depositing active metals on metal substrates and easy to prepare nanopowder materials with small particle size and uniform distribution | The precipitation process cannot be controlled and the precipitants may cause uneven particle size distribution |
| Sol–gel | Compared to the co-precipitation method, the active components have better uniformity and the required temperature is relatively low | The raw materials are relatively expensive and prone to producing harmful gases, and it need long preparation process time |
| Solution combustion | The reaction can proceed without the need for an external heat input, the process can be controlled by changing the type of fuel and the required preparation time is extremely short | Compared to the solution method, choosing the appropriate fuel is quite difficult and the reaction requires a higher temperature |
4 Support
4.1 Effect of support on catalytic performance
The structure and physicochemical properties of supports have a significant influence on catalyst structure stability, active metals loading, activity, and stability (coking resistance and sintering resistance). It is essential to consider various properties to select suitable supports. For example, supports with a high specific surface area but weak metal–support interaction may cause catalyst deactivation, and it is critical to evaluate performance from multiple influencing factors simultaneously. Herein, we will explore the impact of supports on catalytic performance from three aspects: activity, coking resistance, and sintering resistance.The basic site strength supports reducibility (oxygen storage capacity),39,106 and the oxygen transfer rate are important indicators of the catalyst's stability. Notably, the capability of the catalyst to clean surface carbon is the key to stability, rather than the carbon deposition amount. The duration of catalyst activity maintenance is determined by the relative magnitude of the oxygen transfer rate and carbon deposition rate.39 CH4 decomposes on the metal surface and produces deposited carbonaceous species, which subsequently react with surface oxygen on the support. After that, oxygen atoms on the support could be compensated through CO2 dissociation continuously. Noronha et al.39 investigated the relationship between the reducibility of Pt/CexZr1−xO2 catalyst support and catalytic performance. The maximized amount of Ce3+ performs optimal activity and stability simultaneously. The presence of CeO2 with variable valence oxide greatly increases the oxygen transfer rate, allowing the carbon deposition on the metal surface to be oxidized timely.
Metal–support interaction (MSI) affects the dispersion and size of metal particles, which is important for maintaining high activity and resistance to sintering. In the H2-TPR experiment, a higher reduction temperature indicates a stronger interaction between the metal and support, making it more difficult to break the chemical bond between them. It has been demonstrated that excessive interaction can result in a reduction in metal dispersion, due to the incorporation of Ni into the support, and a subsequent reduction in the number of active sites. Conversely, weak interaction can facilitate the occurrence of metal sintering during the early stages of the reaction. Thus, moderating optimal MSI intensity may be the most suitable. However, the difficult-to-reduce Ni2+ may serve as a slow-release agent to continuously supply Ni particles for deactivated catalyst through a slow reduction process.104 Additionally, for bimetallic-based catalysts, choosing inert supports to reduce MSI can strengthen the interaction between two active metals, fostering the synergistic effect of bimetallic materials. Currently, various studies emphasize the importance of MSI. Zhang et al.106 prepared Ni-based catalysts using SiO2 and Al2O3 as supports. The results indicate that the strong MSI between Ni and Al2O3 generates small Ni particles and little carbon deposition on the Ni/Al2O3 catalyst. Xu et al.107 proved that the low mobility of “bound” Ni species at high temperatures is a key factor in maintaining metal dispersion and high catalyst stability after reduction.
MSI affects active metal configurations in the catalyst. Upon calcination or reduction, a variety of Ni species emerge on the support. Moreover, different Ni species positions (support surface or metal–support interface) have different effects on the DRM reaction. Yang et al.108 assumed that the Ni species that existed in the reduced Ni/γ-Al2O3 is characterized as three types: Ni2+ in spinel, the bounded Ni exsoluted from spinel, and free state Ni formed by NiO reduction. Various studies were conducted to investigate their impacts on the catalytic performance of DRM. For Ni/Al2O3 catalyst, Rogers et al.109 proposed that the four coordinated Ni2+ in the NiAl2O4 spinel structure are highly active in DRM, while Foppa et al.110 emphasizes the importance of the metal–support interface, considering the metallic Ni as the active site in the DRM reaction. Thus, divergence is expressed about the influence of Ni species on catalytic activity.
In addition, varying factors of support have influences on active metal migration and aggregation, such as specific surface area and pores. Li et al.111 prepared hydroxyapatite (HAP) supported Ni catalysts for DRM, and the proportions of mesopores/macropores directly affected the distribution of Ni particles. A high proportion of macropores led to large-size Ni nanoparticles, while a high proportion of small-size mesopores resulted in complete blockage of the pore structure, which has adverse effects on improving the interaction between the metal and support.
Supports with large specific surface areas and high pore content are commonly selected to construct a core–shell structure, which limits the range of active metals. The confinement effect is typically employed to enhance the dispersion of active metals, thereby facilitating greater contact with reactant gases and effectively preventing the sintering of active metals. Meanwhile, such an effect also inhibits carbon deposition and blocks filamentous carbon growth.112 Common shell materials include SiO2, Al2O3, zeolite, etc. Except for materials selection, adjusting the pore size and shell thickness,84 and inventing more advanced core–shell structures (such as sandwich structures,113 yolk–shell structures,114etc.) are efficient strategies. The principal obstacle to the construction of a core–shell structure is the transfer of mass and heat. Shell structures may create gas wall blockage holes at high flow rates to prevent reactants from encountering internal metals, thereby reducing activity. Meanwhile, shell structures with poor heat resistance also hinder heat transfer.114
The support morphology also affects catalytic performance. Wang et al.115 synthesized Ni/CeO2 with different morphologies (nanorods, nanotubes, nano-octahedral, and nanoparticles), as shown in Fig. 10(a–h). The study demonstrated that the nanorod Ni/CeO2 catalyst exhibited the highest catalytic activity and stability in the DRM reaction, attributed to its smallest crystalline size and highest value of oxygen vacancies (Fig. 10(i–l)).
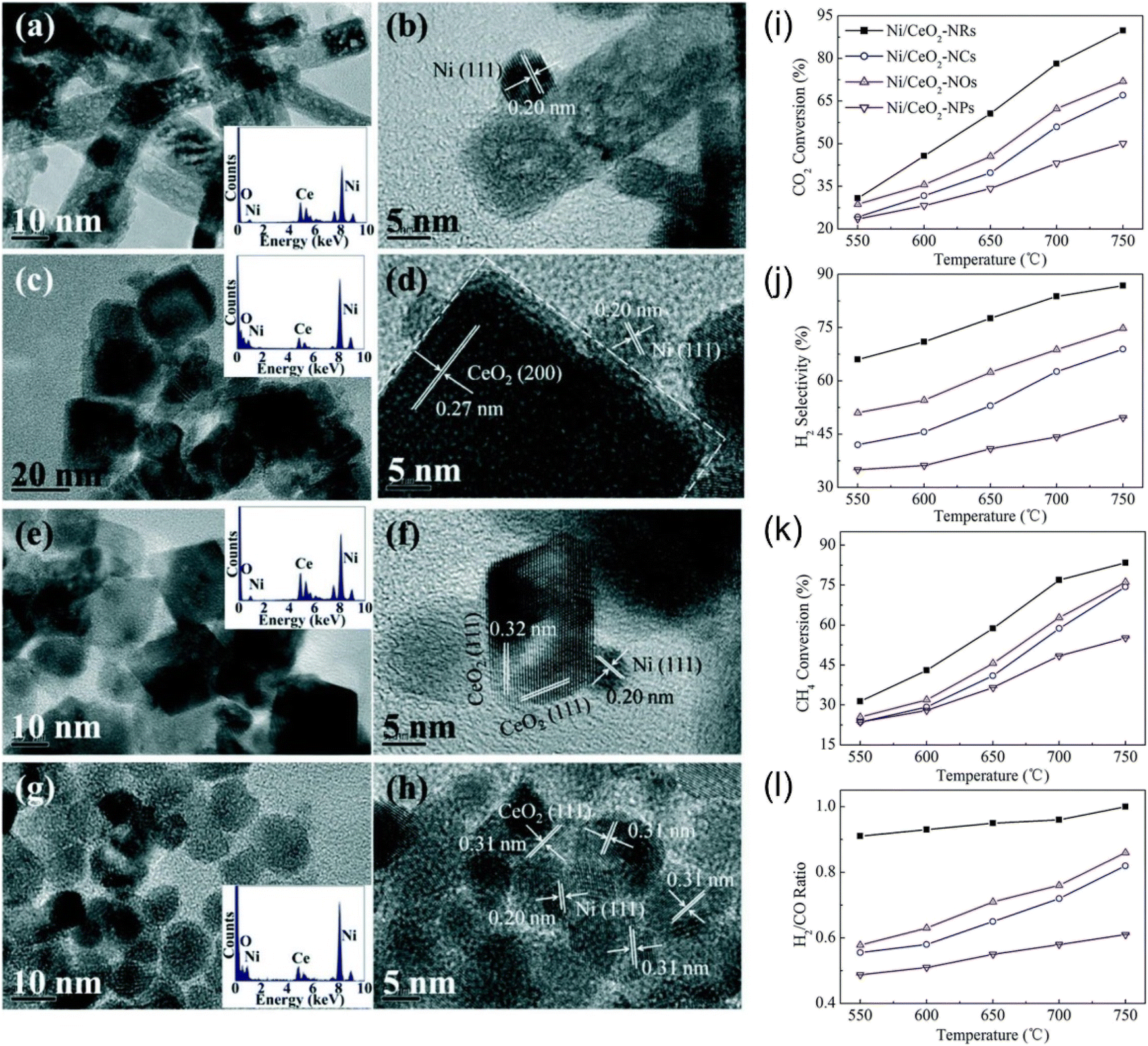 | ||
| Fig. 10 TEM, HRTEM, SAED (inset) images of CeO2: (a and b) rods, (c and d) cubes, (e and f) octahedrons, and (g and h) particles. (i–l) Catalytic performance with different CeO2 morphology over different temperatures. Reprinted with permission from ref. 115 Copyright 2016, The Royal Society of Chemistry. | ||
4.2 Effects of different types of supports
Popular supports used in Ni-based catalysts are summarized in Table 5. Due to their varying surface and structural properties, the interaction of these supports with metallic Ni differs, ultimately impacting their catalytic performance. Kumar et al.126 prepared a series of Ni-based catalysts with different supports for DRM, SRM, and POM triple reforming reactions, such as Al2O3, ZrO2, TiO2, SBA-15, MgO, and CeO2–ZrO2. Among these supports, Ni/Al2O3 has stronger metal–support interaction due to the formation of spinel NiAl2O4, resulting in dispersed Ni particles and the highest initial conversion of reactants. The hexagonal porous network of zeolite SBA-15 confines Ni particles, which prevents metal particles from sintering. Ni/ZrO2 resists re-oxidation of metallic Ni owing to its oxophilic properties. After adding CeO2 to form Ni/CeO2–ZrO2, the basic site concentration decreases and the interaction between the metal and supports weakens, resulting in poor CH4, CO2, and H2O conversions. Zhang et al.106 studied the effect of different supports (ZrO2, TiO2, SiO2, MgO, Mg–Al solid solution, etc.) on the catalytic performance of Ni-based catalysts for DRM. The Mg–Al solid solution has a strong metal–support interaction with superior catalytic performance even at a high gas hourly space velocity. However, SiO2, TiO2, and ZrO2 as supports have weak interaction with NiO, which is beneficial for the reduction of corresponding catalysts but aggravates active metal aggregation. Han et al.127 applied the sol–gel method to create various metal oxide coatings on the surface of 5.2 nm nickel nanoparticles fixed on silica spheres. They investigated the impact of the support on the CH4 turnover frequency and stability for DRM. The results revealed that only Ni covered by TiO2 underwent severe Ni aggregation before the reaction (Fig. 11(a–h)), and its reaction activity was the lowest (Fig. 11(i)). After 20 hours of DRM reaction, Ni supported on TiO2 catalyst produced the most graphite carbon (Fig. 11(j)).| Catalyst | Synthesis method | T [°C] | GHSV [ml g−1 h−1] | Time [h] | CH4 conversion [%] | CO2 conversion [%] |
|---|---|---|---|---|---|---|
| Ni/mesoporous-Al2O3 (ref. 116) | Incipient impregnation | 700 | 120000 |
20 | 77.6 | 85.4 |
| Ni/COMA117 | One-pot synthesis | 850 | 18000 |
240 | 99 | 96 |
| Ni@SiO2–S1 (ref. 118) | Seed-directed method | 750 | 750000 |
28 | 73 | 80 |
| NiCo@SiO2 (ref. 119) | Microemulsion | 800 | 600000 |
150 | 90 | 93 |
| Ni–Mg PSNTS@silica120 | Hydrothermal | 750 | 60000 |
75 | 86 | 89 |
| Ni/BNf121 | Impregnation | 750 | 108000 |
100 | 63 | — |
| Defect confined Ni/BN122 | Sonication-assisted alcoholysis and chemical peeling | 750 | 25000 |
125 | 86 | 94 |
| Cube LaNiO3 (ref. 123) | Hydrothermal and chemical precipitation | 650 | 125000 |
100 | 70 | 80 |
| Ce0.70La0.20Ni0.10O2−δ750 (ref. 124) | Combustion synthesis | 750 | 26400 |
50 | 88 | 89 |
| 1.5Ni–0.5Pt@Hol S-1 (ref. 125) | Incipient-wetness impregnation | 800 | 72000 |
6 | 80 | 85 |
 | ||
| Fig. 11 (a–d) TEM images of the catalysts with different metal oxide overlayers after reduction and stabilization. (e–h) The overlapped elemental mapping by HAADF-STEM for the catalysts after reduction and stabilization (green: Al, Mg, Zr, or Ti; red: Ni). (i) Dependence of CH4 turnover frequency on the catalysts with different metal oxide overlayers. (j) TPO results after running the DRM reaction at 800 °C for 20 h. Reprinted with permission from ref. 127 Copyright 2017, Elsevier. | ||
γ-Al2O3 can enhance metal–support interaction by forming NiAl2O3 with high thermal stability (eqn (4)), which was widely studied as a support for DRM reactions.128 The intrinsic acidity of Al2O3 is beneficial for CH4 decomposition but weakens CO2 adsorption. Consequently, the imbalance of conversion of two reactants results in the C* produced by CH4 decomposition cannot be oxidized by O* timely, leading to catalyst deactivation.26 Hence, several modification strategies should be employed to enhance the anti-coking property of Ni/Al2O3 including the control of morphology and the reduction of surface acidity.
| Ni + Al2O3 + CO2 → NiAl2O4 + CO | (4) |
The pore structure of mesoporous alumina enables the confinement of metals, thereby promoting metal dispersion and inhibiting coke growth. This results in superior catalytic performance compared to non-porous alumina. Bian et al.116 prepared mesoporous alumina supported Ni by impregnation method, and then produced a NiAl2O4 spinel structure at an appropriate calcination temperature of 700 °C. While maintaining a high metal–support interaction, the size of the Ni crystals was only about 5 nm. At 700 °C, the conversions of CH4 and CO2 were 77.6% and 85.4%, respectively. The amount of carbon deposition was only 3.8% after a long-term reaction of 20 hours. Xu et al.129 prepared Ni/Al2O3 catalysts using layered double hydroxides (LDH) as precursors via an in situ growth method. The LDH structure can enhance metal–support interaction and Ni dispersion, which is advantageous for improving activity. Additionally, the layered structure of LDH provides a larger interface area, which is more favorable for CO2 activation and significantly reduces carbon deposition.
The pore structure, pore size, and specific surface area of Al2O3 have a significant impact on the DRM catalytic performance of Ni/Al2O3 catalysts. Hwang et al.130 studied the effect of micropore and mesoporous content on CH4 conversion and found that mesopores can effectively disperse active sites without affecting the contact between reactants and metals, which is beneficial to improving the initial CH4 conversion. Gholizadeh et al.117 prepared cubic ordered mesoporous alumina (COMA) via employing the one-pot and dispersed Ni uniformly into the alumina support by adjusting the pH to neutral. The cage-like pores are connected to provide a confinement effect for the active metal Ni and prevent the formation and growth of whisker coke, maintaining efficient mass transport between interconnected cages. At 850 °C, the conversions of CH4 and CO2 can reach 99% and 97%, respectively. Ma et al.104 constructed a macroporous–mesoporous structure Al2O3 using polystyrene latex spheres as sacrificial templates. The experiments showed that the macroporous structure can promote mass transfer and maintain high stability at a higher gas hourly space velocity (115 L g−1 h−1), due to its tolerance to graphitic carbon. Chica et al.131 supported Ni–Ce bimetallic materials on nanofibers and nanoparticles γ-Al2O3. The experiment found that the nanofibers γ-Al2O3 have a higher mesoporous content, which reduces Ni particle size and improves dispersion, achieving CO2 conversion of 95% at 750 °C. Additionally, the nanofibers exhibit higher sintering resistance and carbon resistance compared to nanoparticles.
The atomic deposition method (ALD) is an effective method for constructing layered catalysts, as it can control the thickness of the deposition layer for optimal performance. Zhao et al.113 synthesized the Al2O3/Ni/Al2O3 sandwich catalyst using ALD. First, the Ni nanoparticles were deposited onto Al2O3 support to form Ni/Al2O3. Subsequently, the Al2O3/Ni/Al2O3 sandwich catalyst was prepared by coating the Al2O3-supported Ni nanoparticles with a porous Al2O3 thin film using ALD. The double interactions of Ni with both the γ-Al2O3 support and the Al2O3 film effectively suppress the aggregation of Ni at high temperatures. The thickness of the film significantly impacts the activity and stability of catalyst. Experimental results show that the catalyst with 80 layers of Al2O3 film exhibits superior stability at 800 °C (400 hours) compared to the catalyst with 40 layers of Al2O3 film. Increased thickness of the Al2O3 film effectively protects the catalyst from damage at high temperatures. Additionally, the high thermal conductivity of Al2O3 film rapidly dissipates heat from the active Ni sites, further preventing Ni agglomeration and ensuring greater stability than thinner films. However, further increasing the Al2O3 film thickness is detrimental, as the encapsulated Ni active sites are less able to adsorb and activate CO2 and CH4, leading to reduced activity or selectivity. However, the author did not provide a specific analysis on the catalytic performance of the catalyst with thicker Al2O3 layer. Baktash et al.132 extensively investigated the influence of the number of Al2O3 layers on catalytic activity and stability in ALD technology. They found that catalytic activity and stability were highest with five ALD cycles of Al2O3, while activity gradually decreased with additional layers, indicating that excessive thickness of the Al2O3 layer affects the contact between gas and active substances.
SiO2 has a high specific surface area, which promotes high Ni dispersion. Furthermore, the high thermal stability of SiO2 prevents structural collapse and carbon deposition at high temperatures, and it is commonly used as a shell material. However, its inertness cannot generate stronger metal–support interaction, resulting in metal sintering. In addition, supports containing SiO2 may incur support reactions under reforming conditions, resulting in the formation of gas-phase impurities and causing impure composition of the synthesis gas (eqn (5) and (6)).21
| SiO2 + 2H2 → SiH4(g) + 2H2O | (5) |
| SiO2 + 2H2O → Si(OH)4(g) | (6) |
Zhang et al.106 compared the effects of different supports on the performance of Ni-based catalysts in the DRM. They found that SiO2 has the highest specific surface area (239 m2 g−1) and pore volume (0.76 cm3 g−1), which achieves the highest Ni dispersion (10.3%). Based on the above advantages, Ni/SiO2 showed a very high initial CH4 conversion. However, over a long reaction duration, the weak metal–support interaction led to metal sintering. Fortunately, SiO2 can act as a shell to form a core–shell structure (Fig. 12), which helps to prevent Ni particle sintering. Currently, the mainstream research directions for core–shell catalysts include constructing multi-core or multi-shell structures and optimizing the structural and preparation parameters.112 Xu et al.118 used a seed-directed method to fix Ni nanoparticles with a small particle size (2.9 nm) in the silicalite-1 zeolite framework. The strong interaction between the metal and support hinders the migration and aggregation of Ni particles, resulting in stable CO2 and CH4 conversions of 80% and 73% respectively within 28 hours. Additionally, the authors compared various synthesis methods for Ni nanoparticles encapsulated in silicon, including the post-treatment method, direct hydrothermal method, and seed-directed method. The results indicated that the post-treatment method and direct hydrothermal method exhibited incomplete encapsulation due to the co-existence of the Ni phase on the internal and external surfaces, resulting in severe carbon deposition and Ni sintering. Zhao et al.119 synthesized a multi-core Ni–Co/SiO2 catalyst via the microemulsion method, which exhibits high activity and stability in DRM at 800 °C. Moreover, it remains stable activity after a 1000 h reaction, which is superior to Ni/SiO2 and Co/SiO2 catalysts with a single metal as the core. Except for using Ni–Co as the core metal, multi-core structures composed of Ni–Mg, Ni–In120,133 and sandwich shell structures composed of SiO2, Al2O3, ZrO2, and MgO are utilized to prepare core–shell catalysts.127 Li et al.134 designed Ni@Ni phyllosilicate@SiO2 core–shell hollow spheres (Ni@NiPhy@SiO2-HS) based on Ni@Ni phyllosilicate construction, resistant to carbon deposition at low temperatures during DRM below 600 °C. TPO-MS, H2-TPR, XPS, and TEM characterizations showed that the confinement effect of SiO2 and the strong interaction between Ni and NiPhy hindered the detachment of Ni from the NiPhy surface, thereby eliminating carbon accumulation and the formation of carbon nanotubes. Dou et al.135 coated Ni@SiO2 with a porous ZrO2 shell, which exhibited a CH4 conversion six times higher than that of Ni@SiO2 catalysts for DRM at 700 °C. The calculation results illustrated that the existence of ZrO2 can lower the dissociation energy barrier of CH4 and CO2, which is key to increasing activity on the SiO2@Ni@ZrO2 catalyst.
 | ||
| Fig. 12 Common core–shell structures. (a) Nanotube, (b) spherical structure, (c) sandwich structure with low and high content of shell materials. | ||
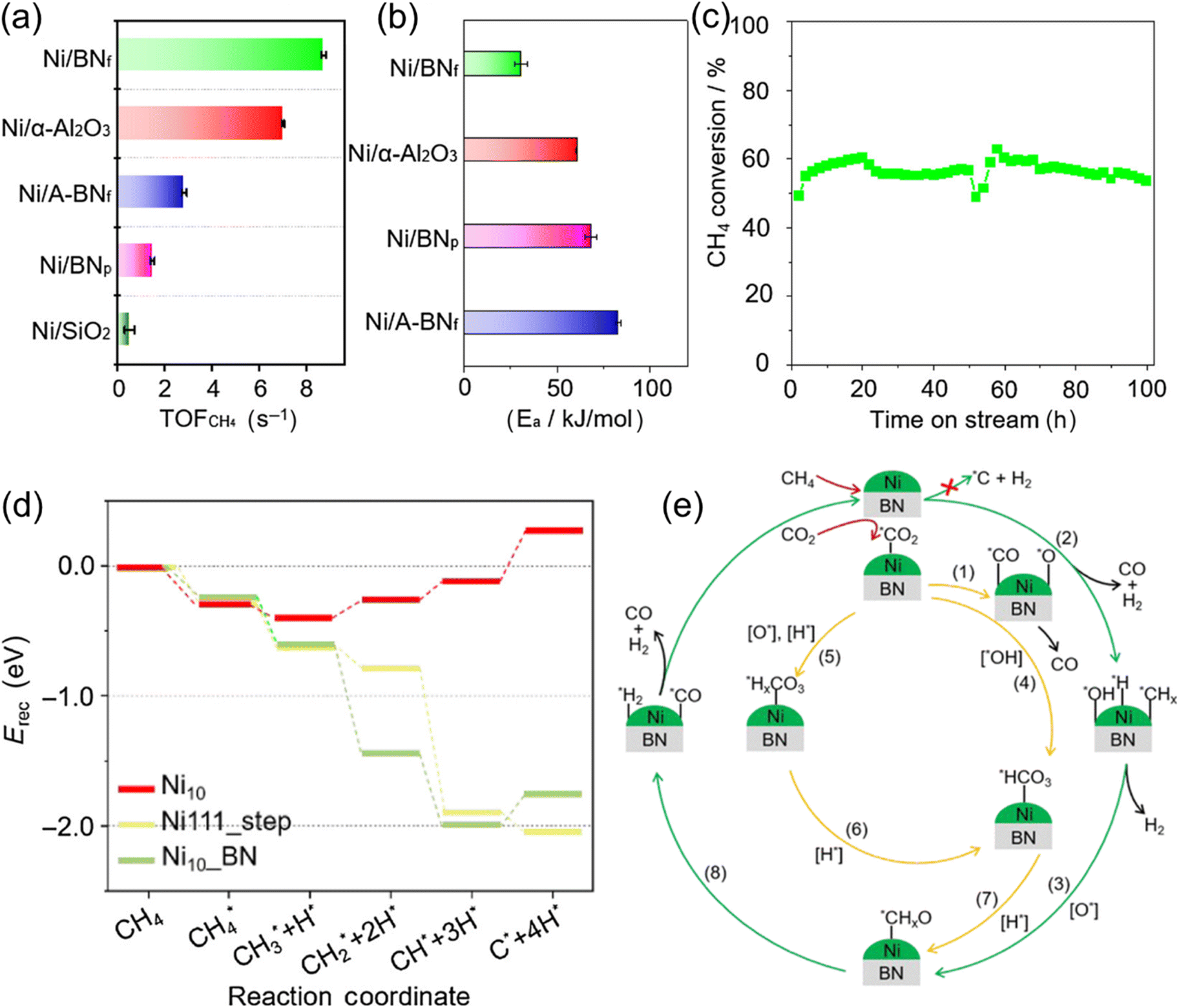 | ||
| Fig. 13 (a) Turnover frequency of CH4 and (b) activation energy calculated based on the conversion of CH4 over various catalysts. (c) CH4 conversion of Ni/BNf catalyst in which the catalyst was cooled after the first 50 h and then restarted for another 50 h. (d) The energy profiles for CH4 decomposition on Ni111_step, Ni10_BN, and Ni10 catalyst. (e) The reaction pathway over Ni/BNf catalyst. Reprinted with permission from ref. 121 Copyright 2023, Chinese Chemical Society. | ||
Various strategies are employed to prevent sintering, such as introducing atomic defects of B or N, or a second promoter to enhance the interaction between the metal and the support. Cao et al.136 prepared core–shell Ni/BN@mSiO2 to prevent the sintering of hexagonal boron nitride-supported catalysts (Ni/h-BN). Utilizing the synergistic effect of SiO2 and BN, the catalyst has both anti-sintering and anti-coking capabilities, that is, the porous nature of SiO2 limited the sintering of Ni, and the BN provides more active sites to form B–OH, which promotes CHx oxidation and dehydrogenation, thereby effectively accelerating product formation and the elimination of carbon deposition. Li et al.137 supported CeO2 and NiO on BN through a two-step method. The strong interaction between Ni and CeO2 was utilized to enhance the anti-sintering property of the Ni/BN. Their experimental results indicated that the specific activity of the ceria-upgraded boron nitride exceeded that of boron nitride-supported Ni by 3 times. Bu et al.122 dispersed BN in isopropanol and then broke B–N by the sonication-assisted alcoholysis and chemical peeling techniques to form the B-rich defective nanosheets BN supported Ni-based catalyst (Ni/d-BN). Ni was embedded in the defect sites of BN, which improved Ni dispersion and enhanced metal–support interactions according to the FTIR, XPS, H2-TPR, and EELS analysis. In addition, the presence of B-site defect also increased the basicity of the catalyst, making the active species O* and H* in reaction rapidly binding to B terminal sites to reform B–OH species. The B–OH can be consumed and regenerated during the catalytic process, which effectively reduces coke deposition from CH4 dissociation.
Adjusting the BN surface charge number is also a pathway to alter CO2 adsorption capacity, assisting in the elimination of deposited carbon. Sun et al.138 found that BN nanomaterials with negative charges can enhance the interaction between CO2 and the support. The results demonstrate that CO2 adsorption/desorption can be controlled by modifying the charge states of BN nanomaterials, thereby modulating CO2 conversion.
Carbon-based support, as another type of non-oxide material, is also used as support for Ni-based catalysts due to their porous structure, chemical inertness, and lower cost.17 In contrast to BN, carbon materials have various types with distinct structures and surface chemical states, such as carbon nanotubes with cylindrical structures,139,140 and activated carbon with multiple micropores and a large specific surface area,141,142etc. Figueira et al.139 studied multi-walled carbon nanotubes (MWCNTs) as the support for DRM to enhance mass transfer. Ma et al.140 found that the Ni nanoparticles inside CNTs exhibited higher catalytic activity and stability than the Ni nanoparticles exposed outside the CNT. Wang et al.141 explored the influence of reduction temperature on the catalytic performance of Ni/activated carbon, and further investigated the effect of non-thermal plasma on catalytic performance. Tan et al.142 compared the pore structure and reaction activity of different activated carbons, and found that wood-derived activated carbon (WAC) exhibits a CH4 conversion four times higher than coal-derived activated carbon (CAC).
 | ||
| Fig. 14 Structural properties of different mesoporous silica supports. (a) Ni/SBA-15, (b) Ni/TMS, (c) Ni/KIT-6, (d) Ni/MCM-41. Reproduced with permission from ref. 143 Copyright 2020, MPDI. | ||
In addition to the porous silica zeolites, metal–organic frameworks (MOFs) have been extensively studied as promising materials in DRM due to their structural adjustability, control of surface chemistry, and composite synthesis capability.147,148 The coordination of metal ions or clusters with ligands is accomplished via various methods, such as solvothermal synthesis,149 microwave-assisted synthesis,150 and electrochemical deposition synthesis,151etc., which are effective for porous structure formation in MOFs. Several strategies are investigated to enhance the CO2 capture capability and adaptability of MOFs to extreme environments, such as the incorporation of open metal sites,152 the introduction of different polar functional groups,147 changes in external stimuli,153etc. Due to the poor thermal stability of MOFs at high temperatures, they cannot be directly used as supports for DRM catalysts.147 However, they can be employed as precursors for highly active catalysts in DRM reactions. Researchers have attempted to synthesize MOF-derived Ni/CeO2 catalysts using organic ligands with different chain lengths, with notable success in improving their catalytic performance.148 Shao et al.154 have synthesized Ni/ZrO2 using Zr-MOFs as sacrificial templates. The experimental results demonstrated that the construction of the metal–oxide interface and the strengthening of MSI through the MOFs template sacrificial strategy were effective, resulting in a significant improvement in catalytic performance.
 | ||
| Fig. 15 In situ XRD profiles of (a) Ni/CeZrO2 and (b) Ni/CeO2 samples under different DRM reaction temperatures. (c) The H2 selectivity and (d) reactants conversion for the DRM reaction over Ni/CeO2 and Ni/CeZrO2 samples. Reprinted with permission from ref. 157 Copyright 2020, American Chemical Society. | ||
The performance of perovskite catalysts in the DRM reaction has been widely explored by replacing the A sites (Mg, Ca, Sr, Ba, etc.) or B sites (Ni, Ru, Co, Ti, Zr, etc.).158 After the reaction, Ni2+ can occupy the B site of the perovskite and form uniformly distributed Ni particles with a strong metal–support interaction. However, the synthesis process of perovskite structures requires multi-step heat treatment, which results in a low specific surface area. Therefore, it is necessary to use the template casting method to prepare porous or hollow structures, or directly prepare bulk perovskite crystalline oxides containing homogeneous active metal.158 Nair et al.159 utilized the nano-casting method to synthesize high surface area LaNiO3 perovskite precursors using ordered mesoporous silica SBA-15 as a hard template. After H2 reduction, the perovskite structure was gradually destroyed, and Ni2+ migrated to the support surface to form uniformly distributed particles. Compared with the citrate synthesis method, the nanocasting method can obtain a higher specific surface area of the catalyst (150 m2 g−1vs. 50 m2 g−1). Singh et al.123 prepared cubes, spheres, and rods of LaNiO3 by modified hydrothermal and precipitation routes. Spherical and rod-shaped perovskite structures have more stacking faults in the TEM, which change the reduction pathway from the parent perovskite nanoparticle to the final catalyst phase.
Currently, it is a common strategy for DRM to control active metal nanoparticle formation and growth via utilizing the reversible co-exsolution mechanism of perovskite oxides.160,161 Nanoparticle exsolution or impregnation in the solid solution can be controlled by regulating synthesis and pretreatment conditions (A/B cation ratio, composition of the perovskite precursors, reducing gas and temperature, etc.), thereby improving particle distribution and size, and impurity resistance of the catalyst. This is a new approach to catalytic performance optimization.162 Under reducing conditions, B cations migrate from the ABO3 matrix through grain boundaries or oxygen vacancies and then nucleate on the surface in zero-valent states, forming metal particles with strong metal–support interaction.161,163 In contrast, nanoparticles move within the perovskite matrix when exposed to an oxidative environment, facilitating their regeneration.164 Najimu et al.163 discovered a correlation between modifying the La:Fe ratio of La(Fe, Ni)O3 solid precursors and the alteration of cation and anion point defects, which regulated the size and composition of exsolved NiFe nanoparticles. Furthermore, it was demonstrated that the composition and size of the alloy nanoparticles can undergo alterations in response to alterations in the reduction time. Shah et al.160 found that in the LaFe0.8Ni0.2O3 catalyst, Ni and Fe would exsolve on the surface of the support at different reduction temperatures and form alloy particles. While in the DRM reaction, most Fe would be oxidized by CO2 and re-enter the perovskite structure in the form of LaFeO3, while Ni would still exist on the surface in the metallic state to participate in the reaction.
5. Addition of active metals and promoters
After supporting Ni on a suitable support, the electronic structure or surface environment of the catalyst can be improved by adding second active metals or promoters,160 thereby changing the dissociation barrier of reactants and the desorption ability of products. The active metals and promoters involved in this section are illustrated in Table 6.| Catalyst | Synthesis method | T [°C] | GHSV [ml g−1 h−1] | Time [h] | CH4 conversion [%] | CO2 conversion [%] |
|---|---|---|---|---|---|---|
| Ni–Pd/SiO2 (ref. 165) | Oleic acid-assisted preparation | 700 | 24000 |
24 | 65 | — |
| Ni–Mo/MgO166 | Polyol-mediated reductive growth | 800 | 300000 |
500 | 100 | 99 |
| Ni–Sn/CeO2–Al2O3 (ref. 167) | Sequential impregnation | 700 | 60000 |
21 | 80 | 95 |
| 6Ni–6Cu/MgAlOx168 | Hydrothermal crystallization | 700 | 40000 |
70 | 85 | 90 |
| La2O3–NiO–Al2O3 (ref. 169) | Hydrolysis-deposition | 800 | 14400 |
8 | 94 | 97 |
| Ni/5MgO–Al2O3 (ref. 170) | Wet impregnation | 750 | 30000 |
4 | 94 | 96 |
| 5Ni/OMA–5CaO171 | Evaporation induced self-assembly | 700 | 15000 |
100 | 82 | 82 |
| In0.5Ni@SiO2 (ref. 133) | One-pot synthesis | 800 | 18000 |
20 | 92 | 98 |
5.1 Effects of the second active metals
Yu et al.23 utilized thermodynamic analysis, descriptor-based microkinetic analysis, and the unsupervised clustering learning technique to screen 23 binary intermetallic catalysts (A3B1 and A1B1) for DRM, based on their anti-coking, anti-sintering, anti-oxidation, catalytic activity, and cost, including Ni–In, Ni–Ge, Ni–Ga, Ni–Zn, Ni–Fe, Ni–Sb, and others. These findings offer a theoretical foundation for developing bimetallic catalysts in experimental settings.In addition, precious metals are more effective in inhibiting side reactions compared to Ni. Foppa et al.35 discovered that the free energy span of the RWGS reaction is lower for Ni (228 kJ mol−1) than for Pd and Pt (253 and 257 kJ mol−1, respectively), indicating that using Ni as an active metal is more conducive to the RWGS reaction.
The “dilution effect” caused by the addition of precious metals keeps different active metals separated from each other, effectively preventing catalyst sintering. Pan et al.165 synthesized a bimetallic Ni–Pd/SiO2 catalyst, which exhibited excellent catalytic performance (the H2/CO ratio was close to 1, and the H2 selectivity reached 98.4% at 700 °C). It was found that Pd was conducive to the reduction and distribution of the active metal Ni on the catalyst surface, thereby increasing the proportion of active Ni0 species, which significantly improved anti-sintering properties.
Precious metals addition can effectively prevent catalyst carbon deposition. Theoretical studies have shown that the carbon deposition methods of precious metals and Ni are different,35 precious metals are more thermodynamically advantageous for methane cracking to form carbon species compared to Ni. Hussien et al.27 believe that Ni-precious metal bimetallic catalysts can promote carbon species gasification formed from methane cracking, which exhibits high coke resistance compared to single Ni-based catalysts. Dai et al.125 utilized hollow silicalite-1 (Hol S-1) as the shell material to encapsulate Ni–Pt bimetallic particles, which demonstrated superior anti-sintering and anti-coking properties than single-metal catalysts and catalysts without core–shell structure. It was observed that the platinum content in 1.5NiO–0.5Pt@Hol S-1 was greater than in 0.5Pt@Hol S-1, indicating that Ni effectively inhibited platinum aggregation and loss after encapsulation pretreatment. In addition, compared with other catalysts, the synergistic effect of the protective shell and alloying resulted in almost no filament carbon on the 1.5Ni–0.5Pt@Hol S-1 catalyst after 6 hours of reaction (Fig. 16 (a–d)). After reduction at 800 °C, the average size of the Ni–Pt bimetallic particles was 4.40 nm, exhibiting excellent anti-sintering capability and catalytic stability (Fig. 16(e)).
 | ||
| Fig. 16 TEM images of (a) 1.5Ni/S-1, (b) 1.5Ni@Hol S-1, (c) 1.5Ni–0.5Pt/S-1, (d) 1.5Ni–0.5Pt@Hol S-1. (e) The stability test of the four catalysts. Reprinted with permission from ref. 125 Copyright 2015, The Royal Society of Chemistry. | ||
Reducing the surface energy of Ni by adding a second active metal with a higher electron cloud density than Ni has been proven by many studies. Liu et al.133 prepared In–Ni/SiO2 catalysts and demonstrated that the addition of In can reduce carbon deposition and activity. The analysis also suggests that the smaller electronegativity of In leads to electron transfer from Into the Ni surface, increasing the electron cloud density of metallic Ni, which weakens C–H bond dissociation, thereby reducing carbon deposition and CH4 conversion. Besenbacher et al.173 suggest that alloying Au atoms onto the Ni surface increases the electron density of adjacent Ni atoms. Although Ni–Au alloys exhibit lower activity compared to pure Ni catalysts, they have a higher capacity for adsorbing unstable carbon atoms on the surface, which facilitates their oxidation.
Zhou et al.174 designed a Ni–Ru bimetallic catalyst and confirmed that Ru can increase the activation barrier for the rate-determining CH4 dissociation step, decreasing the carbon deposition rate. Moreover, Ru can change the type of carbon deposition from a recalcitrant graphitic one that can only be gasified by O2 to a soft type that can be facilely gasified by CO2. Mozammel et al.175 added Ni, Rh, and Co to mesoporous alumina (MAl) to form bimetallic Rh–Ni/MAl and Ni–Co/MAl catalysts. It was found through research that the nature of these two alloys is different (Ni forms a homogeneous alloy phase with Co, while it forms a heterogeneous bimetallic phase with Rh). In Ni–Co homogeneous alloy, Co preferentially interacted with the support, which was beneficial to inhibit Ni0 oxidation, while Ni–Rh heterogeneous alloy enhanced hydrogen spillover and promoted the gasification of carbon deposition.
Yao et al.177 successfully designed Ni–Si/ZrO2 and Ni–Zr/SiO2 bimetallic catalysts. Compared to Ni–Zr/SiO2, the active Ni particles formed by the Ni–Si/ZrO2 catalyst featured a smaller size (6–9 nm) and stronger electron donor ability, which keeps Ni in a metallic state and forms C1-type coke. Such coke formation is easily removed at 400 °C. However, the metallic Ni on the Ni–Zr/SiO2 catalyst will be re-oxidized into NiO at 400 °C, leading to its deactivation. Lu et al.178 found that adding a small amount of V (Ni:V = 10:1) to Ni–Mg–Al layered double hydroxide can significantly reduce the activation energy of CH4 cracking to form CH3 and H species (72.1 kJ mol−1 on pure Ni (111) and 37.1 kJ mol−1 on V0.50Ni (111)). In situ CO2-IR analysis reveals that the introduction of the V promoter can significantly enhance the presence of monodentate carbonate species, which are intermediates in CO2 activation. The appropriate addition of V enhanced Ni site dispersion and Ni particle size reduction. Furthermore, V atoms infiltrate the lattice of Ni particles in the reduction process, interacting with the Ni atoms and increasing the electron cloud density of Ni active sites. Nevertheless, an excessive V amount can disrupt the LDH structure, resulting in activity decline. Song et al.166 supported Ni–Mo bimetallic alloy on MgO using a polyol-mediated reductive growth method, and PVP was added to control the particle size at 2.9 nm. After 850 hours of reaction, the conversions of CH4 and CO2 remained at 100%, and H2/CO also approached 1. By comparing experimental methods, raw material sources, and other factors, a “nanocatalysts on single crystal edges” technique was proposed. It is believed that the diffusion and aggregation of Ni–Mo nanoparticles towards the high-energy step edges of the crystalline MgO (111) form a stable and sustained average size of 17 nm particles, which is the main reason for preventing further sintering and significantly prolonging the catalyst lifetime. However, its industrial application is constrained by the requirement to sustain reaction conditions at elevated temperatures of up to 800 °C for extended durations. Zheng et al.86 found that the introduction of Mo in Ni–Mo/halloysite nanotube catalysts has a positive effect on inhibiting particle sintering and carbon deposition, slowing down H2S poisoning of Ni sites, and promoting the recovery of H2S poisoned catalysts. According to XPS, Mo can compete with Ni to adsorb H2S and convert it into high valence sulfides, alleviating the poisoning of Ni0 sites. However, the specific mechanism and optimal Ni–Mo ratio were not further investigated in the study. Stroud et al.167 found that Sn can improve the catalytic activity and stability of Ni–Sn/Al2O3 and Ni–Sn/CeO2–Al2O3 catalysts. Sn atoms can occupy the step sites where carbon nucleates because the valence shell of Sn is isoelectronic to C. However, excessive Sn will cover Ni active sites, thereby reducing CH4 and CO2 conversions. Nikolla et al.179 discovered that Sn displaces Ni atoms from under-coordinated sites on Ni particles and shifts the rate-controlling CH4 activation step to more abundant, well-coordinated terrace sites. The presence of Sn atoms raises the activation barrier for methane dissociation and weakens the binding between carbon atoms and low-coordination sites. On Sn–Ni alloy catalysts, the rate of C–C bond formation is lower compared to C–O bond formation, which leads to the preferential oxidation of C atoms and CHx fragments, thereby enhancing the stability of the catalyst. Theofanidis et al.180 found that Fe partially separates from the Ni–Fe alloy during DRM reaction to form FeOx according to EDX element mapping, thereby promoting CO2 reduction (Fig. 17(a and b)). The surface carbon undergoes gasification through interaction with FeOx lattice oxygen (Fig. 17(c)). The experiment shows that the catalyst has the highest activity and stability when the molar ratio of Fe/Ni is 0.7.
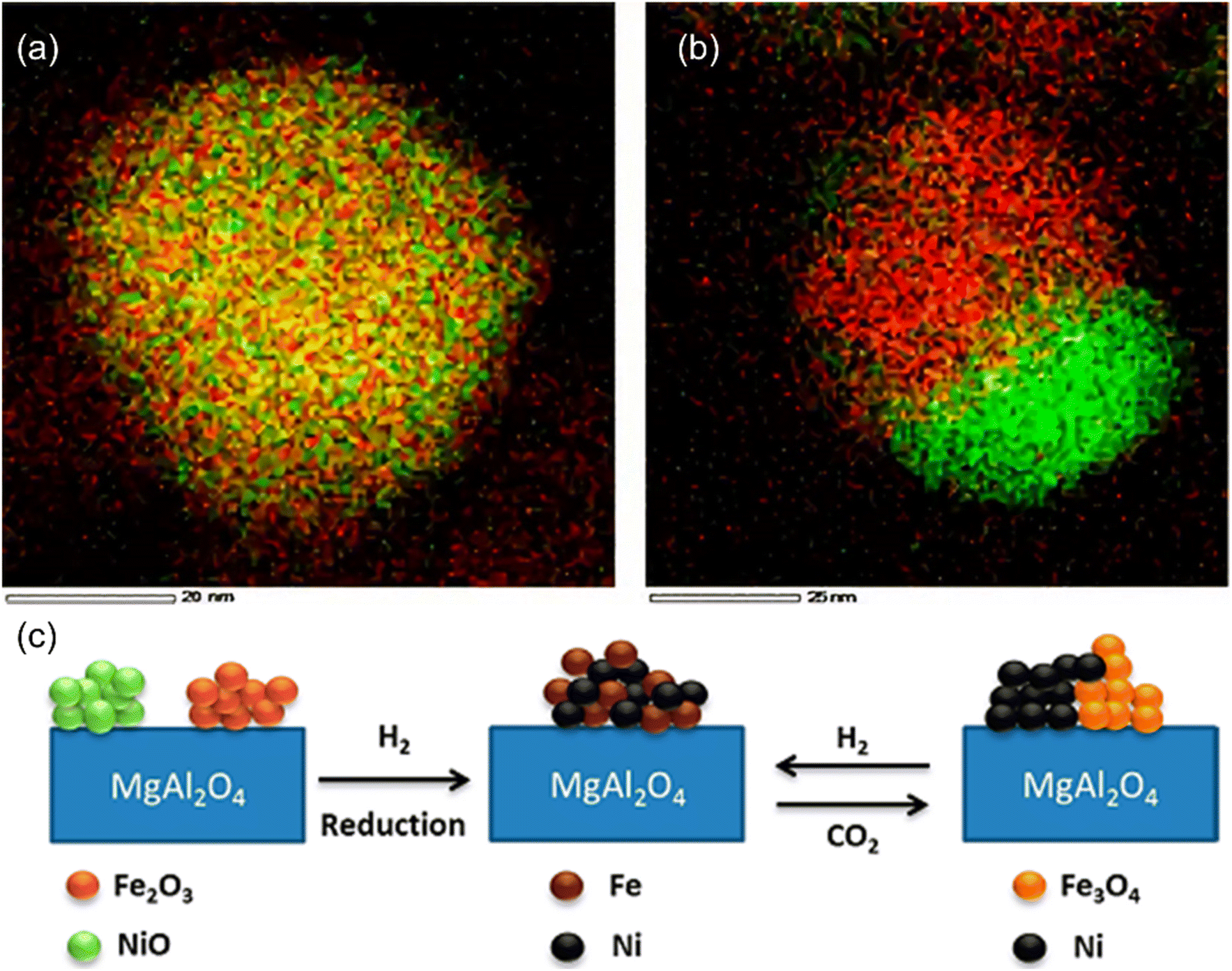 | ||
| Fig. 17 EDX element mapping of 0.7-Fe/Ni (a) after H2 reduction and (b) after CO2 oxidation during DRM reaction. (c) Schematic diagram of the effect of Fe–Ni alloy on CO2 activation and coke elimination. Reprinted with permission from ref. 180 Copyright 2015, American Chemical Society. | ||
The surface reconstruction phenomenon on bimetallic catalysts is of great significance for constructing core–shell structures and changing the reactant dissociation pathway. Bian et al.172 believed that temperature or adsorbents are the reasons for inducing surface reconstruction of bimetallic particles. Xiao et al.168 supported Ni–Cu alloy on periclase-phase MgAlOx nanosheets using the hydrothermal method. The experiment showed that Ni–Cu alloy nanoparticles undergo reconstruction during the reaction, forming Cu atoms that segregate on the surface of the alloy, reducing the adsorption energy of C*. When the Cu and Ni content reaches 6 wt%, the conversion of CH4 and CO2 remains above 85% and 90% respectively, even after a long-term reaction at 700 °C for 70 hours.
5.2 Effects of oxide promoters
The imbalance of CH4 and CO2 dissociation is the main reason for carbon deposition, and the dissociate rate of reactants is largely influenced by the degree to which the reactants are adsorbed by the support. Carbon dioxide, as an acidic substance, is easily adsorbed on the basic sites of the supports.44 Currently, studies primarily focus on improving the activity and stability of catalysts by adding a small amount of alkaline earth oxides or rare earth oxides such as MgO, CaO, CeO2, La2O3, etc. Alkaline earth metal oxides provide basic sites to improve CO2 conversion, while rare earth oxides promote the elimination of carbon deposition through their oxygen storage capacity. Therefore, the rational utilization of the advantages of both simultaneously improves the activity and stability of the catalyst.To further mitigate the issue of carbon deposition on the catalyst surface, Jin et al.181 introduced MgO to the Ni/Al2O3 catalyst. The authors posited that the introduction of MgO is expected to enhance the generation of oxygen vacancies and OH groups. Furthermore, the introduction of Mg2+ into the lattice of Al2O3 or NiAl2O4 would result in surface defects creation, causing the adjacent surface oxygen ions to become coordinately unsaturated through charge compensation. These oxygen ions with low coordination or protruded at vertices, steps, or kinks have been observed to readily interact with the empty orbitals of CO2, leading to the binding of CO2 molecules and the formation of various carbonate structures. Chatla et al.182 conducted a study to enhance the basicity of Ni/MgO catalysts by incorporating Zr. The addition of Zr resulted in the formation of ZrO2, which was achieved by dissolving Zr in the NiO–MgO solid solution. This formation of ZrO2 had a positive impact on the interaction between metal and support, improving reducibility and increasing the dispersion of Ni. Additionally, Zr reduces the dissociation energy of CH4 and facilitates a better match between the conversion of CH4 and CO2. However, it is important to note that excessive Zr content can cause pore blockage in the support and reduce the specific surface area of the catalyst. Navarro et al.183 compared the effects of different promoters on the hydrogen production ability of Ni/Al2O3. The results showed that the order of the metal dispersion trend was as follows: La2O3–Al2O3 > MgO–Al2O3 > CeO2–Al2O3 > Al2O3 > ZrO2–Al2O3. Different promoters can affect the amount and type of coke on the catalyst surface. Among them, La2O3 and CeO2 can prevent the formation of carbon filaments on the Ni surface and affect selectivity, while MgO and ZrO2 can reduce surface acidity, forming an active metal–promoter–support interface, thereby promoting CO2 activation. Additionally, the metal dispersion of catalysts caused by the promoters is completely different (Ni/CeO2–Al2O3: 6.9% vs. Ni/ZrO2–Al2O3: 5.3%), which can be attributed to the different intensities of the metal–support interaction. Compared to ZrO2–Al2O3, the addition of Ce to Al2O3 support leads to moderate intensity of metal–support interaction and Ni phases with moderate dispersion. The extremely high oxygen storage capacity of CeO2 can accelerate CO2 dissociation to remove carbon deposition and is often added as a promoter in catalysts. The promoting effect of CeO2 is generally believed to have the following two points.39 Firstly, CeO2 exhibits a notable oxygen exchange capability, facilitating the absorption or release of oxygen atoms via surface interactions with reducing agents like CO, H2, or hydrocarbons. This process leads to the reversible conversion of cerium atoms into the Ce4+ and Ce3+ oxidation states, enabling the accumulation or release of oxygen atoms and promoting carbon deposition removal.184,185 Secondly, CeO2 can provide strong metal–support interaction, preventing the thermal sintering of metal particles. This effect is not only beneficial for increasing the surface area of the active metal but also for maintaining a large interface area between the metal and support, which is the key to effectively removing carbon on the surface of metal particles. DRM over CeO2 can be presented as the following eqn (7) and (8).
| CeO2 + kCH4 → CeO2−k + kCO + 2kH2 | (7) |
| CeO2−k + kCO2 → CeO2 + kCO | (8) |
Recent studies have also found that La2O3 also has oxygen storage capacity like CeO2. La2O3 can enhance the surface basicity and maintain strong metal–support interaction (Fig. 18(a)). Thus, CO2 can chemically adsorb on the La2O3 surface to generate oxidized carbonate (La2O2CO3), eliminating carbon deposition on the support surface,186,187 according to the following eqn (9) and (10). Mo et al.169 added different amounts of La to the Ni/Al2O3 catalyst and found that La can effectively avoid the contact of Ni particles with other particles due to the “confinement effect” of lanthanum species (see Fig. 18(d), this effect indicates that lanthanum species dilute the Ni active metals, preventing their aggregation and sintering), thereby improving the anti-sintering ability. The results showed that the Ni/Al2O3 catalyst with La content of 0.95 wt% had a smaller average Ni particle size and exhibited good catalytic performance at 800 °C (Fig. 18(b and c)). The Khoja et al.186 incorporated web structured-like La2O3 into MgAl2O4, changing the irregular structure of MgAl2O4 into flakes, which improved Ni dispersion and Ni-support interaction. Farooqi et al.188 synthesized Ni/Al2O3–La2O3 and Ni/Al2O3–CeO2 using the sol–gel method and found that CeO2 promoter can increase reducibility and dispersion of Ni, while La2O3 can inhibit the formation of non-active phase, which plays a significant role in the stability of the catalysts during the 8 hours reaction.
| La2O3 + CO2 → La2O2CO3 | (9) |
| La2O2CO3 + C → La2O3 + 2CO | (10) |
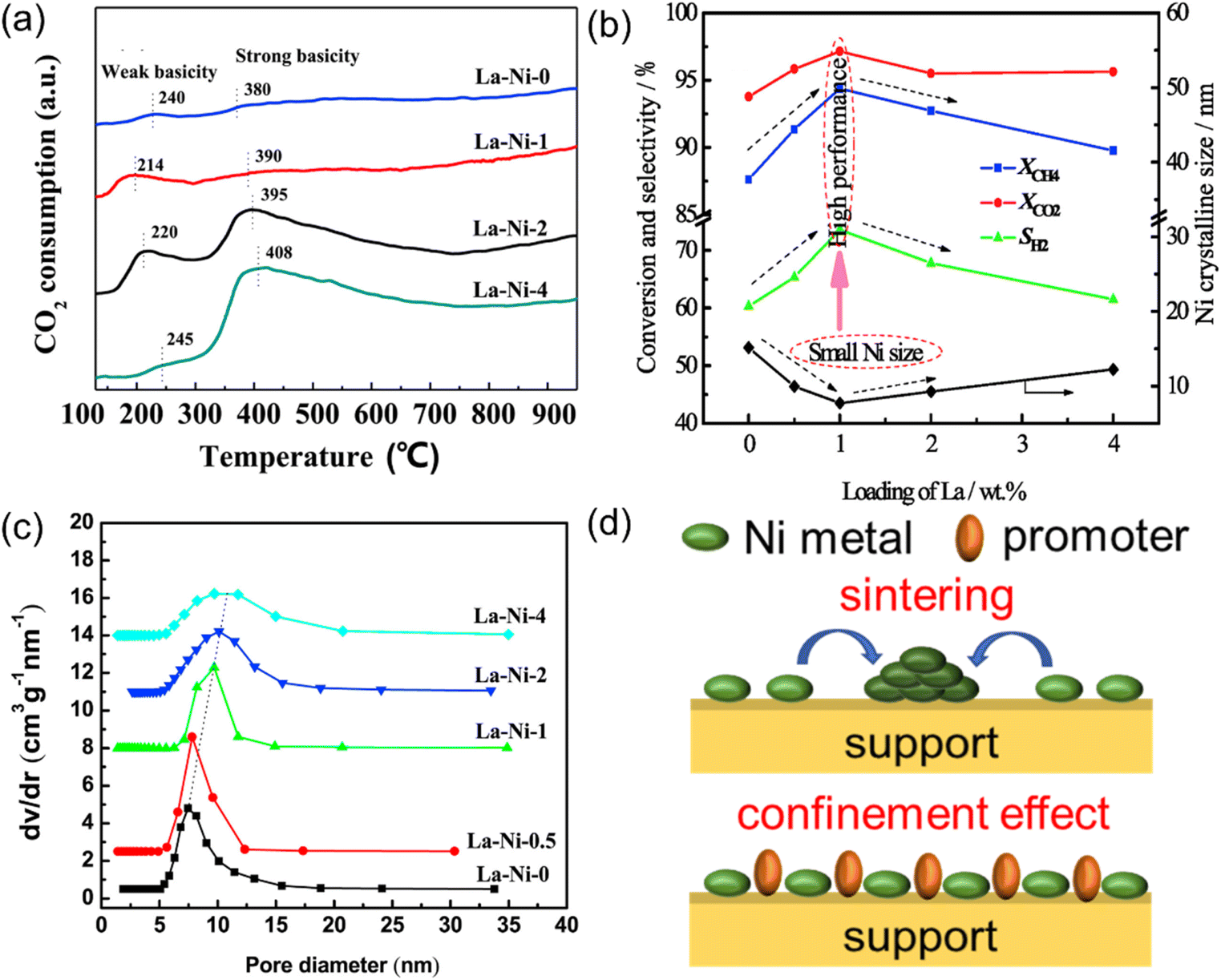 | ||
| Fig. 18 The effect of La2O3 promoters. (a) CO2-TPD profiles with different La loading. (b) The catalytic performance and Ni crystalline size with different La loading (wt%). (c) Pore diameter distributions of catalysts with different La loading. Reprinted with permission from ref. 169 Copyright 2019, Elsevier. (d) Schematic diagram of the anti-sintering mechanism of promoters. | ||
The amount of promoters added has an impact on the activity and stability of the catalyst. Bach et al.170 prepared Ni/Al2O3 catalysts doped with different contents (3, 5, 10 wt%) of MgO. The results showed that MgO can greatly improve the dispersion and the number of active sites, and the catalyst with 5 wt% MgO content has the best carbon deposition resistance, with the H2/CO ratio close to 1. Excessive promoters may cause the active sites to be covered, resulting in a decrease in catalyst activity. Bellido et al.189 discovered that CaO in Ni–ZrO2 catalysts has a significant impact on oxygen ionic conductivity, consequently influencing oxygen vacancy concentration which can facilitate CO2 activation. Their study showed that at the CaO concentration of 8 mol%, the oxygen ionic conductivity reached its maximum, resulting in the highest CO2 conversion. However, as the CaO concentration exceeded the optimal level, the aggregation of oxygen vacancies and the emergence of oxygen-vacancy dopant pairs caused a decline in conductivity, subsequently resulting in a reduction in CO2 conversion. Adding multiple promoters may be another way to improve stability. Singha et al.190 compared the catalytic performance of CeO2 and MgO-promoted Ni/ZnO2 catalysts for DRM, and found that the Ni–MgO/ZnO2 catalyst had better low-temperature activity than the Ni–CeO2/ZnO2 catalyst. Hence, the synergistic effect of the high basicity of MgO and the redox performance of CeO2 can be utilized to increase the Ni dispersion, forming a strong metal–support interaction, and effectively reducing carbon deposition on the catalyst surface.
The synergistic effect of promoters and support structures or synthesis methods can improve the catalytic performance of the catalyst. Xu et al.171 synthesized CaO-promoted ordered mesoporous Al2O3via an improved evaporation-induced self-assembly strategy to construct Ni-based catalysts. In a 100 hours long-term stability test, ordered mesoporous structures (Ni–CaO/ordered mesoporous alumina) maintain more stable catalytic performance and activity compared to disordered mesoporous structures (Ni–CaO/non-mesoporous alumina). Wang et al.191 used the one-pot method to coat MgO on SBA-15 mesoporous silica and found that compared with Ni/MgO-SBA-15 prepared only by the impregnation method, the MgO-coated catalyst has a better order in the mesostructure and medium basic sites, which effectively improves the initial activity.
6. Effects of preparation and reaction conditions
Preparation conditions such as Ni content, reduction temperature, pretreatment gas, pH of the solution, and calcination temperature, can significantly affect the structural properties of the catalyst. In addition, reaction conditions such as CO2/CH4 ratio, gas hourly space velocity (GHSV), and reaction temperature, can affect the thermodynamic equilibrium of DRM. Considering these factors according to experimental results and thermodynamic analysis is of great significance for optimizing the process parameters of DRM.6.1 Pretreatment process
The reduction process has a significant impact on the size of active metal particles and the metal–support interaction.192 Since unreduced Ni-based catalysts typically exhibit oxide phases, a reduction process is required to obtain the active phase of metallic Ni before the reaction proceeds. Different supports require different reducing gases. Gao et al.192 summarized the influence of different pretreatment gases on the properties of the catalyst, which were as follows:(1) Decrease particle size: oxygen free radicals lead to a decrease in nucleation, while reductive gases can react with oxygen free radicals to form inert gases such as water, thereby increasing nucleation and reducing the size of Ni metal particles. Meanwhile, reductive gases can lower the decomposition temperature of Ni precursors, promote the diffusion of Ni species, and improve Ni dispersion.
(2) Retain surface oxygen: when using CeO2 as a support, inert gas needs to be selected to ensure the presence of surface oxygen. Compared with inert gases like Ar, the use of oxidative gases (CO2 and O2) will reduce the mobility and reactivity of surface oxygen, while the use of reductive gases (H2, NO, N2O, and CO) will reduce the oxygen storage capacity of CeO2.
(3) Wet the metals: for Ni/TiO2, oxidative gases can react with Ni to form NiOx which encapsulates Ni particles. Due to the lower interfacial energy of the oxide–oxide interface than that of oxide–metal, the bonding between TiO2 and NiOx was easier, which is beneficial to reducing the growth rate of Ni particles.
(4) Improves the degree of reduction: reductive gases in Ni/Al2O3 moderately reduce metal–support interaction and improve the reduction degree by reducing NiAl2O4 formation, which increases the amount of Ni0.
(5) Change the metal–support interaction: different supports can regulate MSI by using different pretreatment gases. Reductive gas is usually used on Al2O3 to reduce MSI, while when using SiO2 as the support, it is suitable to use air as the pretreatment gas to promote silicate formation, thereby improving MSI. For CeO2, using inert gas is the best choice to improve MSI.
Yao et al.90 discovered that the catalytic activity of ZrOx/Ni-MnOx/SiO2 changes at different reduction temperatures due to the varying intensities of interaction between the metal and support, caused by the aggregation and migration of Ni particles. Caballero et al.193 studied the effects of the reduction process with CO or H2 on Ni particle size in the Ni/ZrO2 catalyst for DRM (Fig. 19(a)). The appearance of a band centered at 2067 cm−1 in the FTIR spectra (Fig. 19(b)) indicates that Ni atoms react with CO to form Ni(CO)4 complexes after partial reduction of NiO by CO at 400 °C, corroding the Ni particles and reducing particle size, thereby exhibiting higher catalytic stability than catalysts reducing with H2 (Fig. 19(c)). Lovell et al.194 utilized a simple reduction-oxidation-reduction method for the pretreatment of Ni/SiO2 catalysts. The samples were first reduced in hydrogen, then oxidized in oxygen, and finally re-reduced in hydrogen. This method can reduce the size of metal particles and increase the interaction between Ni and SiO2 without the need for expensive precursors and complex synthesis techniques to control particle deposition. As a result, CH4 conversion increased from 57% to 69% at 800 °C, and the H2/CO ratio became closer to unity. Chen et al.195 compared the effects of different reduction temperatures on the particle size of Ni-based catalysts. The results showed that although high-temperature reduction increased catalyst sintering and particle size, Ni nanoparticles are also encapsulated by the ZrO2 support during the reaction process, resulting in a smaller particle area exposed to the atmosphere, effectively reducing carbon deposition during the reaction process.
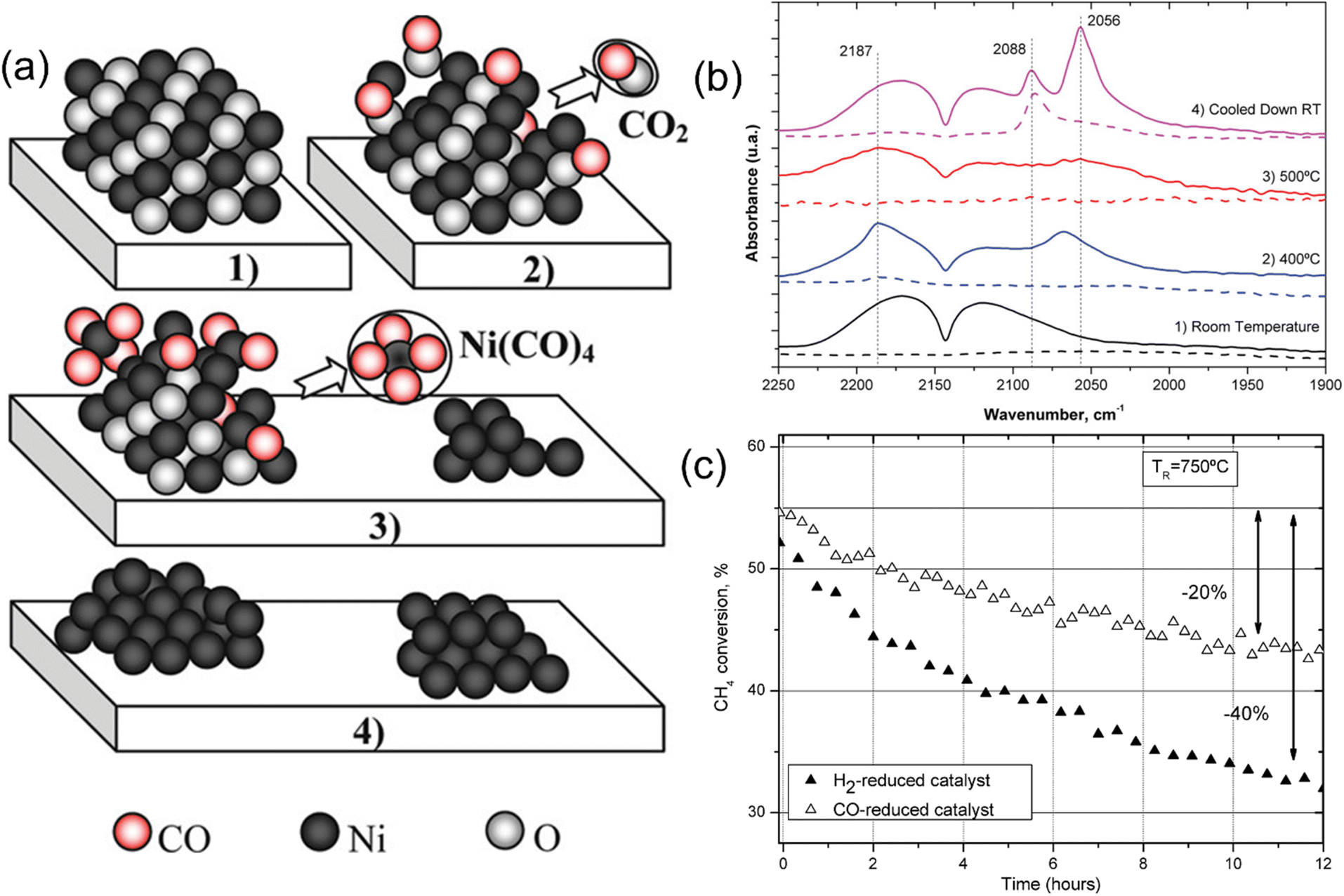 | ||
| Fig. 19 (a) Schematic diagram of the reaction between NiO and CO, which represents (1) NiO particle, (2) NiO reacts with CO to form CO2 and Ni, (3) CO reacts with Ni to form Ni(CO)4, and (4) Ni(CO)4 transfers to the surrounding regions on the zirconia support to form smaller Ni particles than NiO particles, respectively. (b) FTIR spectra of the Ni/ZrO2 sample during reduction treatment with CO. (c) CH4 conversion in the reaction of DRM over Ni/ZrO2 catalyst after different reduction treatments. Reprinted with permission from ref. 193 Copyright 2011, American Chemical Society. | ||
6.2 Heat treatment process
The catalyst needs to undergo washing, drying, and calcination processes after forming a solid phase. Zanganeh et al.93 studied the catalytic performance of Ni0.1Mg0.9O catalyst for DRM at different calcination temperatures. The results show that increasing the calcination temperature negatively affects the catalytic activity, and the catalyst calcined at 600 °C had the highest CH4 and CO2 conversion. This is because the calcination temperature can affect the pore size distribution. As the calcination temperature increases, the pore size subsequently increases, leading to the particle size increase (13.31 nm at 600 °C and 30.9 nm at 800 °C) and the specific surface areas decrease (115.9 m2 g−1 at 600 °C and 49.81 m2 g−1 at 800 °C). Yang et al.108 made precise adjustments to the type of active Ni by controlling the Ni loadings and annealing temperature, and the influence of bound Ni and free Ni on catalytic performance in Ni/γ-Al2O3 has been experimentally demonstrated. The bound Ni precipitated from the spinel structure has better CH4 and CO2 dissociation ability, as well as higher CO desorption ability, solving the contradiction that activity and stability cannot be improved simultaneously.6.3 Ni loadings
The different Ni loadings have a direct relationship with the active metal dispersion. Shamskar et al.196 synthesized mesoporous nanocrystalline NiAl2O3 powders with different Ni loadings (5 wt% to 35 wt%) using the ultrasonic-assisted co-precipitation method and applied them in DRM. The experiment showed that increasing Ni loadings from 15 wt% to 33 wt% decreased the crystallite size and improved catalyst reducibility. An ideal amount of Ni enhances the surface area, while an additional increase in Ni loadings results in a detrimental impact on the surface area. Meanwhile, increasing Ni loadings generates an opposite trend in activity and stability, that is, an increase in Ni loadings will enhance activity but increase carbon deposition content with weaker stability. Consequently, it is necessary to explore the optimal Ni content and they found that NiAl2O3 with 25 wt% Ni loadings had the best catalytic performance. Xu et al.129 used layered double hydroxides (hydrotalcite, LDH) as precursors to prepare Ni/Al2O3 catalysts by a facile in situ growth method. By changing the amount of urea, the Ni loading was controlled. When the urea content was insufficient, Ni2+ and Al2O3 could not be effectively bonded through chemical bonds, resulting in a decrease in Ni loading during the washing process. However, when the urea content is too high, excessive ammonia will form [Ni(NH3)6]6+ with Ni2+, leading to a turbostratic disorder of the LDH layer and crystallographic LDH structure deformation. Asencios et al.197 combined POM and DRM reactions to design a NiO–MgO–ZrO2 catalyst. Due to a good balance between active sites and oxygen acancies produced by the MgO–ZrO2 solid solution, the activity and selectivity of the catalyst loaded with 20 wt% Ni is maximized, while decreased above this loading.6.4 Thermodynamic reaction conditions
Various studies use thermodynamic methods combined with experimental results to study the effect of reaction conditions on reactant conversion and product yield. Akbari et al.198 prepared mesoporous nanocrystalline Ni–MgO–Al2O3 catalysts with a high specific surface area using a co-precipitation method and compared the effects of CO2/CH4 intake ratio (Fig. 20(a and b)), Ni loadings (Fig. 20(c)) and GHSV (Fig. 20(d)) on the catalytic performance of DRM. Under higher GHSV conditions, the decrease in contact time between the catalyst surface and reactants leads to a decrease in reactant conversions. In addition, insufficient contact time between the reactant and the catalyst surface can lead to the reaction being far from thermodynamic equilibrium, thereby diminishing reactant conversion. Moreover, the CH4 conversion increased while CO2 conversion decreased with the increase in CO2/CH4 ratio. Bach et al.170 studied the effects of temperature, pressure, and feed composition through thermodynamics analysis and concluded that DRM reactions require high temperature, low pressure, and low CH4/CO2 molar ratio to achieve minimum Gibbs free energy.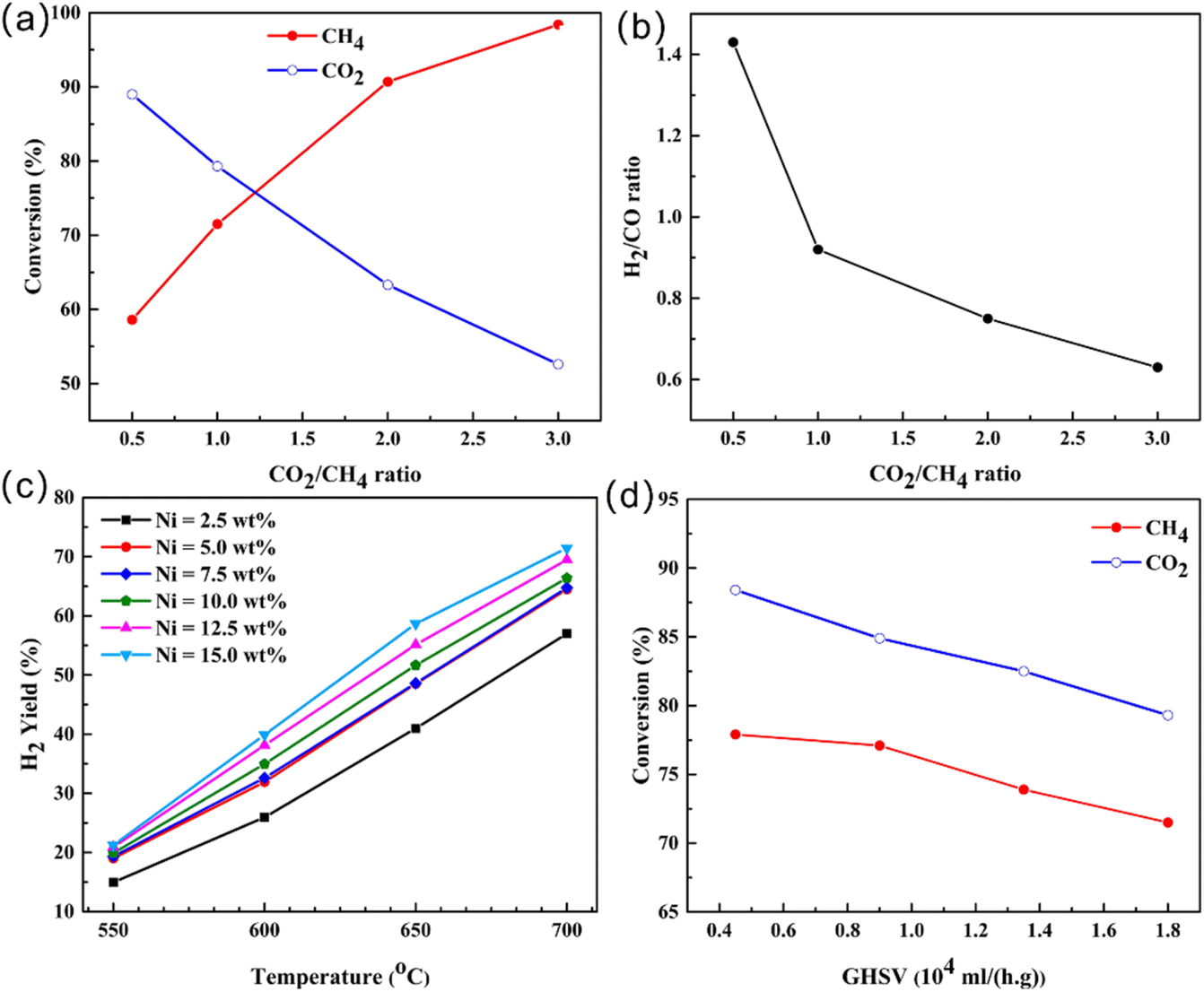 | ||
| Fig. 20 The influence of reaction conditions on the catalytic performance of the 12.5 wt% Ni–MgO–Al2O3 catalyst at 700 °C. (a) Effect of CO2/CH4 ratio on the CH4 and CO2 conversions. (b) Effect of CO2/CH4 ratio on H2/CO molar ratio. (c)Effect of Ni content and temperature on H2 yield. (d) Effect of GHSV on the catalytic activity at CO2/CH4 = 1. Reprinted with permission from ref. 198 Copyright 2017, Elsevier. | ||
Fig. 21 provides an overview of the catalytic performance of various common catalysts, demonstrating that the composition of the catalyst, particularly the choice of support, plays a crucial role in influencing its catalytic performance.199–207 This phenomenon is attributed to the confinement or interface effect facilitated by the support, which significantly influences active sites distribution and reaction pathways. Various synthesis techniques are employed to load individual or multiple active metals onto specific supports to improve the catalytic performance of Ni-based catalysts further. Additionally, the addition of promoters and the construction of specialized support structures can be utilized, combined with optimizing the pretreatment process and reaction conditions, to achieve a maximum balance of activity and stability. Once a highly stable Ni-based catalyst has been designed, it is imperative to consider the practical circumstances (pressure, reactor, gas composition) to facilitate the industrial application of the DRM reaction and progress toward an economically and environmentally sustainable industrialization process.
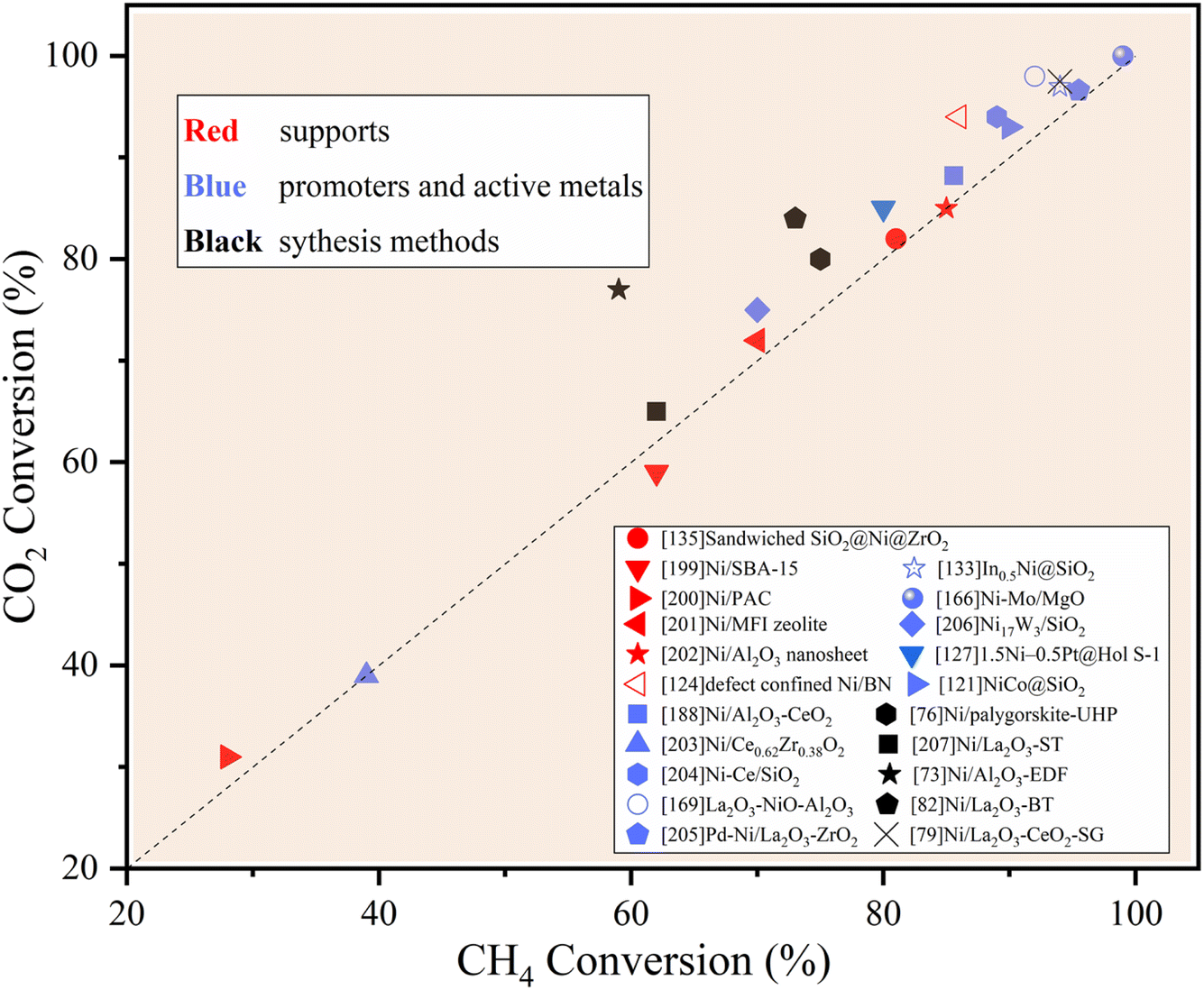 | ||
| Fig. 21 The comparison diagram of CH4 and CO2 conversion of different catalysts at 800 °C. | ||
7. Conclusions and perspectives
Currently, the lack of comprehensive understanding of the general mechanism underlying catalytic reactions hinders substantial enhancement of the selectivity of DRM. The characteristic of Ni-based catalysts being prone to deactivation makes it challenging to meet the high catalyst lifespan requirements in the industry. Researchers are encouraged to conduct comprehensive investigations into catalytic mechanisms, enhance catalyst designs for improved efficiency, and refine process conditions. Moreover, it is imperative to account for industrialization demands by thoroughly evaluating factors like cost-effectiveness, stability, and sustainability to guarantee the viability and applicability of novel Ni-based catalysts in industrial settings. The practical application of DRM requires a comprehensive understanding of the differences between laboratory scale and industrial production scale. Furthermore, the incorporation of catalysts and reactors into integrated systems is essential to meet the needs of diverse sectors such as automotive, energy storage, and other industrial applications. The following summarizes the research directions (Fig. 22) that need further efforts and attention for Ni-based catalysts, and provides prospects: | ||
| Fig. 22 Research directions of Ni-based catalysts for DRM reaction. | ||
(1) Development of characterization tools and computational methods: the utilization of advanced characterization tools, such as steady-state isotope transient kinetic analysis (SSITKA), extended X-ray absorption fine structure (EXAFS) and in situ infrared spectroscopy (In Situ FTIR), scanning tunneling microscopy (STM), and atomic force microscopy (AFM), facilitates the observation of surface transient reaction phenomena and exploration of various factors, such as the mobility of lattice oxygen, electronic states, and abundance of active metals on the surface catalysis of DRM.27 A comprehensive study can be conducted on the DRM reaction mechanism and catalyst deactivation mechanism by combining DFT computational results.
(2) Improvement of synthesis process: to achieve uniform and stable metal dispersion, as well as optimal utilization of active sites, more advanced auxiliary methods and suitable active metal precursors should be adopted. These auxiliary methods mainly refer to the promotion of contact and interaction between substances through mechanical and heating methods. Recently, such as non-thermal glow discharge plasma treatment,70 electrodeposition method,208 and improved multi-bubble sonoluminescence method,209 have been employed to improve the MSI, producing highly dispersed Ni particles, thereby improving stability and activity. In addition, developing an integrated catalyst based on metal and modifying the substrate surface using techniques like the micro-arc oxidation method to enhance the distribution of active metals is a crucial strategy for advancing industrial decarbonization in the future.210
(3) Adjustment of catalyst composition: adjusting the catalyst reaction composition is also crucial for designing Ni-based catalysts with superior performance. Two-dimensional layered double (MOFs),147 hollow zeolites, and other structural supports27,211 are hotspots as templates for adjusting catalyst structure and synthesizing new Ni-based catalysts with high metal dispersion and MSI. Another effective approach involves integrating an active metal into a thermally durable crystalline oxide, creating a well-dispersed metal–support catalyst through the exsolution of the active metal from the oxide structure during reduction. This results in catalysts with reduced particle size, enhanced dispersion, and improved stability compared to traditional supported metal oxide catalysts. The objective of adjusting the catalyst composition is to establish a quantitative correlation between surface properties (such as the additional amount of second active metals and promoters, basic sites, and oxygen vacancies), reactant adsorption and dissociation mechanism, and catalytic performance.
(4) Development of new reactor types and reaction process: inventing a new reactor to maximize the utilization of energy and reaction gas is an important technology to break the bottleneck of traditional catalyst design. Baharudin et al.44 summarized various new DRM reactors, such as ionization-driven reactors and microwave-assisted reactors that provide new heating methods, photocatalytic reactors that improve resource utilization efficiency, etc. Recently, the technology of chemical cycle dry reforming of methane has emerged (Fig. 23(a)).212 This technology promotes the reduction and regeneration of catalysts during the reaction process by enabling cyclic contact between catalysts and reagents, thereby enhancing the yield and selectivity of synthesis gas.213 This review introduces an electrothermal catalysis internal model using sustainable energy power to drive DRM, which achieves a more uniform temperature distribution than traditional external heating hydroxides (LDHs),three-dimensional metal–organic frameworks modes (Fig. 23(b)).214–216 For traditional powder catalysts, it is necessary to ensure uniform heat distribution between the tubes in the tubular reformer and the catalyst bed in the adiabatic reformer. By comparison, integral catalysts have natural advantages in terms of heat distribution uniformity. In addition, the pulse heating method uses the programmable current to quickly heat and quench, which confirms that the reaction stops in a local equilibrium state in a multi-step reaction promptly, enhancing selectivity and stability.217 In the future, this method could be attempted to be applied to DRM reactions.
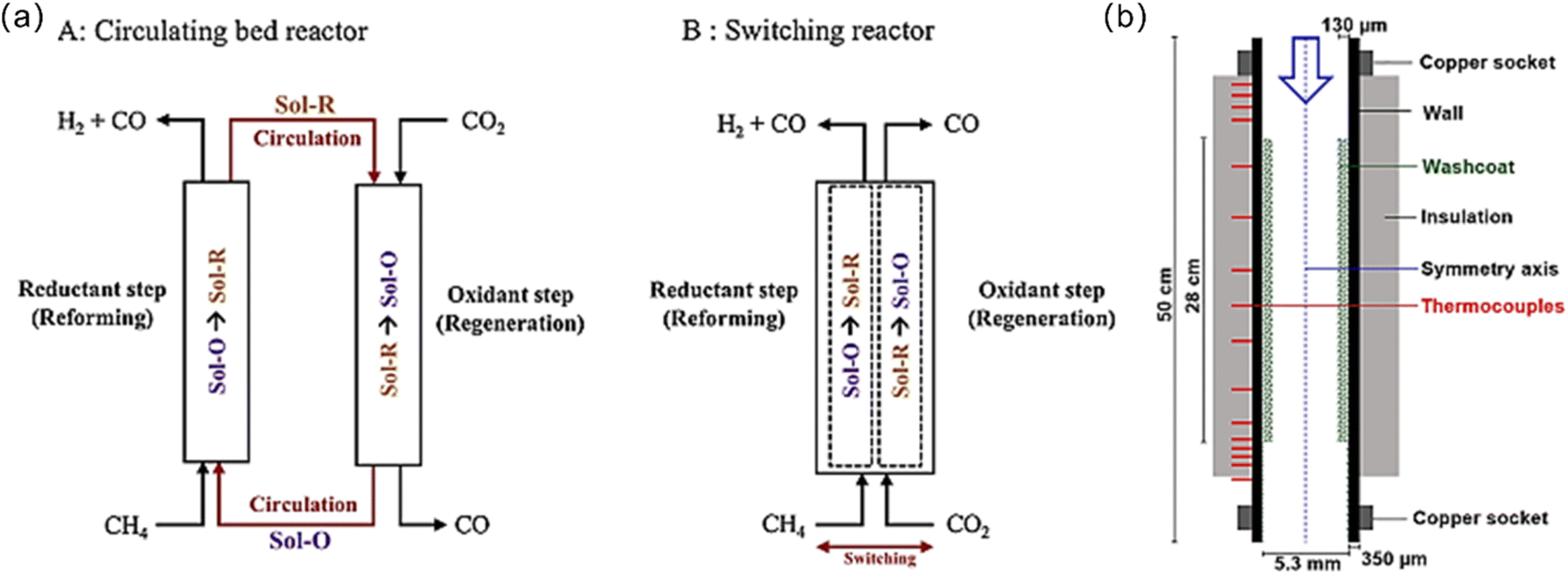 | ||
| Fig. 23 (a) Schematic representation of the chemical looping dry reforming of methane (CLDRM) process using a (A) circulating bed reactor or (B) switching feed reactor. Reproduced with permission from ref. 212 Copyright 2019, Elsevier. (b) Cross section of ohmic resistance heating reactor. Reproduced with permission from ref. 214 Copyright 2021, Elsevier. | ||
(5) Progress towards industrialization: scaling up DRM technology to an industrial level necessitates a thorough understanding of reaction engineering principles about catalysts, heat transfer, pressure dynamics, and fluid flow in challenging environments commonly encountered in DRM. Adjusting the feed ratio of CO2/H2O/O2 and combining different reforming reactions (DRM, POM, SRM) not only effectively reduces carbon deposition but also prevents damage caused by hot spot formation.197 Additionally, the gases utilized in practical DRM reactions (natural gas, flue gas, or biogas) contain other impurities, such as sulfides, chlorides, etc. These impurities may cause catalyst poisoning and deactivation. Investigating the effect of impurities on catalyst deactivation properties is a critical indicator to verify the stability under actual conditions for achieving the industrial application of DRM. In industrial processes, DRM reactions are typically conducted at high pressure to optimize thermodynamic equilibrium and reaction kinetics, enhancing efficiency and yield while lowering energy consumption. Addressing the issue of heightened carbon deposition when operating under high pressure represents a significant obstacle in advancing the industrialization of DRM.
Data availability
All data analyzed in this review article are derived from previously published research articles, which are duly cited within this manuscript.Author contributions
Chuangwei Liu, PhD (supervision: lead) Tianyi Wang (writing – review & editing: lead) Ziquan Wang (data curation: lead; writing – original draft: lead) Qilong Wu (investigation: equal) Luyuan Wang (formal analysis: equal) Song Li (supervision: supporting).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The project was financially supported by the Foundation of China's National Key R&D Programme (grant no. 2023YFB3810600), and S. Li acknowledges the China BaoWu Low Carbon Metallurgical Innovation Foundation (no. BWLCF202113). The authors thank the Beijing PARATERA Tech Co., Ltd. for providing HPC resources.Notes and references
- M. Yusuf, A. S. Farooqi, L. K. Keong, K. Hellgardt and B. Abdullah, Chem. Eng. Sci., 2021, 229, 116072 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- E. R. Harrould-Kolieb, Clim. Pol., 2019, 19, 1225–1238 CrossRef.
- R. Waheed, D. Chang, S. Sarwar and W. Chen, J. Clean. Prod., 2018, 172, 4231–4238 CrossRef.
- A. M. Ranjekar and G. D. Yadav, J. Indian Chem. Soc., 2021, 98, 100002 CrossRef CAS.
- C. D. Elvidge, M. D. Bazilian, M. Zhizhin, T. Ghosh, K. Baugh and F.-C. Hsu, Energy Strat. Rev., 2018, 20, 156–162 CrossRef.
- J. J. Torrez-Herrera, S. A. Korili and A. Gil, Catal. Rev., 2021, 65, 1300–1357 CrossRef.
- I. A. Karimi and S. Kawi, Ind. Eng. Chem. Res., 2016, 55, 7839–7841 CrossRef CAS.
- L. Wang, X. Zhao, D. Lv, C. Liu, W. Lai, C. Sun, Z. Su, X. Xu, W. Hao, S. X. Dou and Y. Du, Adv. Mater., 2020, 32, 2004311 CrossRef CAS PubMed.
- L. Li, X. Zhang, C. Liu, V. S. S. Mosali, J. Chen, A. M. Bond, Q. Gu and J. Zhang, Appl. Catal., B, 2023, 331, 122597 CrossRef CAS.
- Z. Guo, Y. Yu, C. Li, E. C. dos Santos, T. Wang, H. Li, J. Xu, C. Liu and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202319913 CrossRef CAS PubMed.
- X. Zhang, C. Liu, Y. Zhao, L. Li, Y. Chen, F. Raziq, L. Qiao, S.-X. Guo, C. Wang, G. G. Wallace, A. M. Bond and J. Zhang, Appl. Catal., B, 2021, 291, 120030 CrossRef CAS.
- X. Gao, S. Yang, L. Hu, S. Cai, L. Wu and S. Kawi, Carbon Capture Sci. Technol., 2022, 3, 100039 CrossRef CAS.
- F. Fayaz, H. T. Danh, N. H. Chinh, K. B. Vu, B. Abdullah and D.-V. N. Vo, Procedia Eng., 2016, 148, 646–653 CrossRef CAS.
- I. V. Yentekakis, P. Panagiotopoulou and G. Artemakis, Appl. Catal., B, 2021, 296, 120210 CrossRef CAS.
- X. Wu, L. Xu, M. Chen, C. Lv, X. Wen, Y. Cui, C. E. Wu, B. Yang, Z. Miao and X. Hu, Front. Chem., 2020, 8, 581923 CrossRef CAS PubMed.
- S. Posada-Pérez, F. Viñes, J. A. Rodriguez and F. Illas, Top. Catal., 2014, 58, 159–173 CrossRef.
- C. Liu, A. Tian, H. Yang and X. Xue, Mater. Lett., 2013, 106, 1–4 CrossRef CAS.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556–3575 CrossRef CAS PubMed.
- J. Rostrup-Nielsen and L. J. Christiansen, Concepts in Syngas Manufacture, Imperial College Press, London, 2011, vo. 1 Search PubMed.
- G. Zhang, J. Liu, Y. Xu and Y. Sun, Int. J. Hydrog. Energy, 2018, 43, 15030–15054 CrossRef CAS.
- Y.-X. Yu, J. Yang, K. K. Zhu, Z. J. Sui, D. Chen, Y. A. Zhu and X. G. Zhou, ACS Catal., 2021, 11, 8881–8894 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev., 1999, 41, 1–42 CrossRef CAS.
- X. Gao, Z. Ge, G. Zhu, Z. Wang, J. Ashok and S. Kawi, Catalysts, 2021, 11, 1003 CrossRef CAS.
- K. Wittich, M. Krämer, N. Bottke and S. A. Schunk, ChemCatChem, 2020, 12, 2130–2147 CrossRef CAS.
- A. G. S. Hussien and K. Polychronopoulou, Nanomaterials, 2022, 12, 3400 CrossRef CAS PubMed.
- N. T. Andersen, F. Topsøe, I. Alstrup and J. R. Rostrup-Nielsen, J. Catal., 1987, 104, 454–465 CrossRef CAS.
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668–108688 RSC.
- J. Wei and E. Iglesia, J. Catal., 2004, 224, 370–383 CrossRef CAS.
- S. L. Leung, J. Wei, W. L. Holstein, M. Avalos-Borja and E. Iglesia, J. Phys. Chem. C, 2020, 124, 20143–20160 CrossRef CAS.
- T. Osaki and T. Mori, J. Catal., 2001, 204, 89–97 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renew. Sustain. Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- S. A. Theofanidis, R. Batchu, V. V. Galvita, H. Poelman and G. B. Marin, Appl. Catal., B, 2016, 185, 42–55 CrossRef CAS.
- L. Foppa, M.-C. Silaghi, K. Larmier and A. Comas-Vives, J. Catal., 2016, 343, 196–207 CrossRef CAS.
- Y. Liu, C. Liu, H. Zhou, G. Qin and S. Li, Colloids Surf., A, 2023, 667, 131392 CrossRef CAS.
- Y. Wang, L. Yao, S. Wang, D. Mao and C. Hu, Fuel Process. Technol., 2018, 169, 199–206 CrossRef CAS.
- Z. Li, Q. Lin, M. Li, J. Cao, F. Liu, H. Pan, Z. Wang and S. Kawi, Renew. Sustain. Energy Rev., 2020, 134, 110312 CrossRef CAS.
- F. B. Noronha, E. C. Fendley, R. R. Soares, W. E. Alvarez and D. E. Resasco, Chem. Eng. J., 2001, 82, 21–31 CrossRef CAS.
- Z. Lian, S. O. Olanrele, C. Si, M. Yang and B. Li, J. Phys. Chem. C, 2020, 124, 5118–5124 CrossRef CAS.
- O. Omoregbe, H. T. Danh, C. Nguyen-Huy, H. D. Setiabudi, S. Z. Abidin, Q. D. Truong and D.-V. N. Vo, Int. J. Hydrog. Energy, 2017, 42, 11283–11294 CrossRef CAS.
- J. S. Bitters, T. He, E. Nestler, S. D. Senanayake, J. G. Chen and C. Zhang, J. Energy Chem., 2022, 68, 124–142 CrossRef.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- L. Baharudin, N. Rahmat, N. H. Othman, N. Shah and S. S. A. Syed-Hassan, J. CO2 Util., 2022, 61, 102050 CrossRef CAS.
- Y. Kathiraser, U. Oemar, E. T. Saw, Z. Li and S. Kawi, Chem. Eng. J., 2015, 278, 62–78 CrossRef CAS.
- N. D. Charisiou, G. Siakavelas, L. Tzounis, V. Sebastian, A. Monzon, M. A. Baker, S. J. Hinder, K. Polychronopoulou, I. V. Yentekakis and M. A. Goula, Int. J. Hydrog. Energy, 2018, 43, 18955–18976 CrossRef CAS.
- Y. Li, B. Zhang, X. Xie, J. Liu, Y. Xu and W. Shen, J. Catal., 2006, 238, 412–424 CrossRef CAS.
- C. Vogt, J. Kranenborg, M. Monai and B. M. Weckhuysen, ACS Catal., 2019, 10, 1428–1438 CrossRef.
- Z. Rao, K. Wang, Y. Cao, Y. Feng, Z. Huang, Y. Chen, S. Wei, L. Liu, Z. Gong, Y. Cui, L. Li, X. Tu, D. Ma and Y. Zhou, J. Am. Chem. Soc., 2023, 145, 24625 CAS.
- N. A. K. Aramouni, J. G. Touma, B. A. Tarboush, J. Zeaiter and M. N. Ahmad, Renew. Sustain. Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- J. Grams and A. M. Ruppert, Catalysts, 2021, 11, 265 CrossRef CAS.
- L. S. Lobo, J. L. Figueiredo and C. A. Bernardo, Catal. Today, 2011, 178, 110–116 CrossRef CAS.
- N. M. Rodriguez, J. Mater. Res., 1993, 8, 3233–3250 CrossRef CAS.
- J. Yoo, Y. Bang, S. J. Han, S. Park, J. H. Song and I. K. Song, J. Mol. Catal. A Chem., 2015, 410, 74–80 CrossRef CAS.
- J. H. Larsen and I. Chorkendorff, Surf. Sci. Rep., 1999, 35, 163–222 CrossRef CAS.
- A. Ohtomo and H. Y. Hwang, Nature, 2004, 427, 423–426 CrossRef CAS PubMed.
- B. AlSabban, L. Falivene, S. M. Kozlov, A. Aguilar-Tapia, S. Ould-Chikh, J.-L. Hazemann, L. Cavallo, J.-M. Basset and K. Takanabe, Appl. Catal., B, 2017, 213, 177–189 CrossRef CAS.
- C. Wan, K. Song, J. Pan, M. Huang, R. Luo, D. Li and L. Jiang, Int. J. Hydrog. Energy, 2020, 45, 33574–33585 CrossRef CAS.
- X. Gao, P. Cai, Z. Wang, X. Lv and S. Kawi, Top. Catal., 2022, 66, 299–325 CrossRef.
- U. J. Etim, C. Zhang and Z. Zhong, Nanomaterials, 2021, 11, 3265 CrossRef CAS PubMed.
- H. P. Boehm, Discuss. Faraday Soc., 1971, 52, 264–275 RSC.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Fuel Process. Technol., 2018, 178, 166–179 CrossRef CAS.
- T. S. Phan, A. R. Sane, B. R. de Vasconcelos, A. Nzihou, P. Sharrock, D. Grouset and D. Pham Minh, Appl. Catal., B, 2018, 224, 310–321 CrossRef CAS.
- H. Yang, L. Xu, M. Chen, C. Lv, Y. Cui, X. Wen, C.-e. Wu, B. Yang, Z. Miao, X. Hu and Q. Shou, Micropor. Mesopor. Mat., 2020, 302, 110250 CrossRef CAS.
- H. Hattori, Mater. Chem. Phys., 1988, 18, 533–552 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2001, 222, 247–259 CrossRef CAS.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- A. W. Budiman, S.-H. Song, T.-S. Chang, C.-H. Shin and M.-J. Choi, Catal. Surv. Asia, 2012, 16, 183–197 CrossRef CAS.
- N. Rahemi, M. Haghighi, A. A. Babaluo, M. F. Jafari and P. Estifaee, J. Ind. Eng. Chem., 2013, 19, 1566–1576 CrossRef CAS.
- V. Danghyan, S. C. Novoa, A. Mukasyan and E. E. Wolf, Appl. Catal., B, 2018, 234, 178–186 CrossRef CAS.
- M. A. Goula, N. D. Charisiou, K. N. Papageridis, A. Delimitis, E. Pachatouridou and E. F. Iliopoulou, Int. J. Hydrog. Energy, 2015, 40, 9183–9200 CrossRef CAS.
- J. Yang, X. Lu, C. Han, H. Liu, D. Gong, L. Mo, Q. Wei, H. Tao, S. Cui and L. Wang, Int. J. Hydrog. Energy, 2022, 47, 32071–32080 CrossRef CAS.
- Q. Zhang, J. Wang, P. Ning, T. Zhang, M. Wang, K. Long and J. Huang, Korean J. Chem. Eng., 2017, 34, 2823–2831 CrossRef CAS.
- J. Chen, Q. Wu, J. Zhang and J. Zhang, Fuel, 2008, 87, 2901–2907 CrossRef CAS.
- M. Jafarbegloo, A. Tarlani, A. W. Mesbah, J. Muzart and S. Sahebdelfar, Catal. Lett., 2015, 146, 238–248 CrossRef.
- M. Grabchenko, G. Pantaleo, F. Puleo, T. S. Kharlamova, V. I. Zaikovskii, O. Vodyankina and L. F. Liotta, Catal. Today, 2021, 382, 71–81 CrossRef CAS.
- S. A. Shin, Y. S. Noh, G. H. Hong, J. I. Park, H. T. Song, K.-Y. Lee and D. J. Moon, J. Taiwan Inst. Chem. Eng., 2018, 90, 25–32 CrossRef CAS.
- Y. H. Ahmad, A. T. Mohamed, A. Kumar and S. Y. Al-Qaradawi, RSC Adv., 2021, 11, 33734–33743 RSC.
- S. Sorcar, J. Das, E. P. Komarala, L. Fadeev, B. A. Rosen and M. Gozin, Mater. Today Chem., 2022, 24, 100765 CrossRef CAS.
- C. Contescu, A. Contescu and J. A. Schwarz, Chem. Rev., 1995, 95, 477–510 CrossRef.
- Y. Zhang, G. Zhang, J. Liu, T. Li, X. Zhang, Y. Wang, Y. Zhao, G. Li and Y. Zhang, Fuel Process. Technol., 2023, 250, 107891 CrossRef CAS.
- J. Ren, H. Li, Y. Jin, J. Zhu, S. Liu, J. Lin and Z. Li, Appl. Catal., B, 2017, 201, 561–572 CrossRef CAS.
- T. Odedairo, J. Chen and Z. Zhu, Catal. Commun., 2013, 31, 25–31 CrossRef CAS.
- X. Zhu, P. Huo, Y.-p. Zhang, D.-g. Cheng and C.-j. Liu, Appl. Catal., B, 2008, 81, 132–140 CrossRef CAS.
- J. Zheng, S. Impeng, J. Liu, J. Deng and D. Zhang, Appl. Catal., B, 2024, 342, 123369 CrossRef CAS.
- W. Li, Z. Zhao, F. Ding, X. Guo and G. Wang, ACS Sustainable Chem. Eng., 2015, 3, 3461–3476 CrossRef CAS.
- Q. Zhang, K. Long, J. Wang, T. Zhang, Z. Song and Q. Lin, Int. J. Hydrog. Energy, 2017, 42, 14103–14114 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 16472–16479 RSC.
- L. Yao, Y. Wang, J. Shi, H. Xu, W. Shen and C. Hu, Catal. Today, 2017, 281, 259–267 CrossRef CAS.
- A. Albarazi, M. E. Gálvez and P. Da Costa, Catal. Commun., 2015, 59, 108–112 CrossRef CAS.
- Y. Li, Y. Wang, X. Zhang, X. Ding, Z. Liu, R. Zhu, L. Wu and L. Zheng, Int. J. Hydrog. Energy, 2022, 47, 20851–20866 CrossRef CAS.
- R. Zanganeh, M. Rezaei and A. Zamaniyan, Adv. Powder Technol., 2014, 25, 1111–1117 CrossRef CAS.
- X. Xie, T. Otremba, P. Littlewood, R. Schomäcker and A. Thomas, ACS Catal., 2013, 3, 224–229 CrossRef CAS.
- R. D. Gonzalez, T. Lopez and R. Gomez, Catal. Today, 1997, 35, 293–317 CrossRef CAS.
- S. J. H. Rad, M. Haghighi, A. A. Eslami, F. Rahmani and N. Rahemi, Int. J. Hydrog. Energy, 2016, 41, 5335–5350 CrossRef.
- V. La Parola, L. F. Liotta, G. Pantaleo, M. L. Testa and A. M. Venezia, Appl. Catal., A, 2022, 642, 118704 CrossRef CAS.
- J. Chen, R. Wang, J. Zhang, F. He and S. Han, J. Mol. Catal. A Chem., 2005, 235, 302–310 CrossRef CAS.
- Y. Xu, H. Wan, X. Du, B. Yao, S. Wei, Y. Chen, W. Zhuang, H. Yang, L. Sun, X. Tao and P. Wang, Fuel Process. Technol., 2022, 236, 107418 CrossRef CAS.
- A. Cross, S. Roslyakov, K. V. Manukyan, S. Rouvimov, A. S. Rogachev, D. Kovalev, E. E. Wolf and A. S. Mukasyan, J. Phys. Chem. C, 2014, 118, 26191–26198 CrossRef CAS.
- V. Danghyan, A. Kumar, A. Mukasyan and E. E. Wolf, Appl. Catal., B, 2020, 273, 119056 CrossRef CAS.
- Z. Bian and S. Kawi, ChemCatChem, 2018, 10, 320–328 CrossRef CAS.
- N. Wang, K. Shen, L. Huang, X. Yu, W. Qian and W. Chu, ACS Catal., 2013, 3, 1638–1651 CrossRef CAS.
- Q. Ma, Y. Han, Q. Wei, S. Makpal, X. Gao, J. Zhang and T.-s. Zhao, J. CO2 Util., 2020, 35, 288–297 CrossRef CAS.
- T. Xie, X. Zhao, J. Zhang, L. Shi and D. Zhang, Int. J. Hydrog. Energy, 2015, 40, 9685–9695 CrossRef CAS.
- R.-j. Zhang, G.-f. Xia, M.-f. Li, Y. Wu, H. Nie and D.-d. Li, J. Fuel Chem. Technol., 2015, 43, 1359–1365 CrossRef CAS.
- Z. Xu, Y. Li, J. Zhang, L. Chang, R. Zhou and Z. T. Duan, Appl. Catal., A, 2001, 210, 45–53 CrossRef CAS.
- B. Yang, J. Deng, H. Li, T. Yan, J. Zhang and D. Zhang, iScience, 2021, 24, 102747 CrossRef CAS PubMed.
- J. L. Rogers, M. C. Mangarella, A. D. D'Amico, J. R. Gallagher, M. R. Dutzer, E. Stavitski, J. T. Miller and C. Sievers, ACS Catal., 2016, 6, 5873–5886 CrossRef CAS.
- L. Foppa, T. Margossian, S. M. Kim, C. Müller, C. Copéret, K. Larmier and A. Comas-Vives, J. Am. Chem. Soc., 2017, 139, 17128 CrossRef CAS PubMed.
- B. Li, X. Yuan, B. Li and X. Wang, Fuel Process. Technol., 2020, 202, 106359 CrossRef CAS.
- D. S. B. Zhoufeng, Chem. Ind. Eng. Prog., 2023, 42, 247–254 Search PubMed.
- Y. Zhao, Y. Kang, H. Li and H. Li, Green Chem., 2018, 20, 2781–2787 RSC.
- C. Wang, H. Wu, X. Jie, X. Zhang, Y. Zhao, B. Yao and T. Xiao, ACS Appl. Mater. Interfaces, 2021, 13, 31699–31709 CrossRef CAS PubMed.
- N. Wang, W. Qian, W. Chu and F. Wei, Catal. Sci. Technol., 2016, 6, 3594–3605 RSC.
- Z. Bian, W. Zhong, Y. Yu, Z. Wang, B. Jiang and S. Kawi, Int. J. Hydrog. Energy, 2021, 46, 31041–31053 CrossRef CAS.
- F. Gholizadeh, A. Izadbakhsh, J. Huang and Y. Zi-Feng, Micropor. Mesopor. Mat., 2021, 310, 110616 CrossRef CAS.
- S. Xu, T. J. A. Slater, H. Huang, Y. Zhou, Y. Jiao, C. M. A. Parlett, S. Guan, S. Chansai, S. Xu, X. Wang, C. Hardacre and X. Fan, Chem. Eng. J., 2022, 446, 137439 CrossRef CAS.
- Y. Zhao, H. Li and H. Li, Nano Energy, 2018, 45, 101–108 CrossRef CAS.
- Z. Bian, I. Y. Suryawinata and S. Kawi, Appl. Catal., B, 2016, 195, 1–8 CrossRef CAS.
- J. Deng, M. Gao, J.-y. Hasegawa, X. Zhang, A. Wang, A. Chen and D. Zhang, CCS Chem., 2022, 5, 2111–2124 CrossRef.
- K. Bu, J. Deng, X. Zhang, S. Kuboon, T. Yan, H. Li, L. Shi and D. Zhang, Appl. Catal., B, 2020, 267, 118692 CrossRef CAS.
- S. Singh, D. Zubenko and B. A. Rosen, ACS Catal., 2016, 6, 4199–4205 CrossRef CAS.
- L. Pino, C. Italiano, A. Vita, M. Laganà and V. Recupero, Appl. Catal., B, 2017, 218, 779–792 CrossRef CAS.
- C. Dai, S. Zhang, A. Zhang, C. Song, C. Shi and X. Guo, J. Mater. Chem. A, 2015, 3, 16461–16468 RSC.
- R. Kumar, K. Kumar, N. V. Choudary and K. K. Pant, Fuel Process. Technol., 2019, 186, 40–52 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- Y. Xu, X.-h. Du, J. Li, P. Wang, J. Zhu, F.-j. Ge, J. Zhou, M. Song and W.-y. Zhu, J. Fuel Chem. Technol., 2019, 47, 199–208 CrossRef CAS.
- Y. Xu, X. Du, L. Shi, T. Chen, H. Wan, P. Wang, S. Wei, B. Yao, J. Zhu and M. Song, Int. J. Hydrog. Energy, 2021, 46, 14301–14310 CrossRef CAS.
- K.-S. Hwang, H. Y. Zhu and G. Q. Lu, Catal. Today, 2001, 38, 183–190 CrossRef.
- J. J. González, J. F. Da Costa-Serra and A. Chica, Int. J. Hydrog. Energy, 2020, 45, 20568–20581 CrossRef.
- E. Baktash, P. Littlewood, R. Schomäcker, A. Thomas and P. C. Stair, Appl. Catal., B, 2015, 179, 122–127 CrossRef CAS.
- W. Liu, L. Li, S. Lin, Y. Luo, Z. Bao, Y. Mao, K. Li, D. Wu and H. Peng, J. Energy Chem., 2022, 65, 34–47 CrossRef CAS.
- Z. Li, B. Jiang, Z. Wang and S. Kawi, J. CO2 Util., 2018, 27, 238–246 CrossRef CAS.
- J. Dou, R. Zhang, X. Hao, Z. Bao, T. Wu, B. Wang and F. Yu, Appl. Catal., B, 2019, 254, 612–623 CrossRef CAS.
- Y. Cao, M. Lu, J. Fang, L. Shi and D. Zhang, Chem. Commun., 2017, 53, 7549–7552 RSC.
- X. Li, M. Phornphimon, X. Zhang, J. Deng and D. Zhang, Chem. Asian J., 2022, 17, e202101428 CrossRef CAS PubMed.
- Q. Sun, Z. Li, D. J. Searles, Y. Chen, G. M. Lu and A. Du, J. Am. Chem. Soc., 2013, 135, 8246–8253 CrossRef CAS PubMed.
- C. E. Figueira, P. F. Moreira, R. Giudici, R. M. B. Alves and M. Schmal, Appl. Catal., A, 2018, 550, 297–307 CrossRef CAS.
- Q. X. Ma, D. Wang, M. B. Wu, T. S. Zhao, Y. Yoneyama and N. Tsubaki, Fuel, 2013, 108, 430–438 CrossRef CAS.
- H. Wang, J. Han, Z. Bo, L. Qin, Y. Wang and F. Yu, Mol. Catal., 2019, 475, 110486 CrossRef CAS.
- Y. Tan, S. Wang, L. Li, B. Meng, J. Chen, Z. Yang, K. Yan and X. Qin, Chem. Eng. Process., 2019, 145, 107662 CrossRef CAS.
- M. H. Amin, Catalysts, 2020, 10, 51 CrossRef CAS.
- Q. Zhu, H. Zhou, L. Wang, L. Wang, C. Wang, H. Wang, W. Fang, M. He, Q. Wu and F.-S. Xiao, Nat. Catal., 2022, 5, 1030–1037 CrossRef CAS.
- S. Wang, Y. Wang and C. Hu, Int. J. Hydrog. Energy, 2018, 43, 13921–13930 CrossRef CAS.
- X. Gao, S. Deng and S. Kawi, iScience, 2022, 25, 105343 CrossRef CAS PubMed.
- J. Gandara-Loe, L. Pastor-Perez, L. F. Bobadilla, J. A. Odriozola and T. R. Reina, React. Chem. Eng., 2021, 6, 787–814 RSC.
- R. D. Alli and N. Mahinpey, Fuel, 2024, 361, 130756 CrossRef CAS.
- X. Zhu, J. Gu, Y. Wang, B. Li, Y. Li, W. Zhao and J. Shi, Chem. Commun., 2014, 50, 8779–8782 RSC.
- S. Tai, W. Zhang, J. Zhang, G. Luo, Y. Jia, M. Deng and Y. Ling, Micropor. Mesopor. Mat., 2016, 220, 148–154 CrossRef CAS.
- I. Stassen, M. Styles, T. V. Assche, N. Campagnol, J. Fransaer, J. Denayer, J.-C. Tan, P. Falcaro, D. De Vos and R. Ameloot, Chem. Mater., 2015, 27, 1801–1807 CrossRef CAS.
- Z. Zhang, Q. Zhang, J. Shi, Y. S. Chu, X. Yu, K. Xu, M. Ge, H. Yan, W. Li, L. Gu, Y. S. Hu, H. Li, X. Q. Yang, L. Chen and X. Huang, Adv. Energy Mater., 2016, 7, 1601196 CrossRef.
- S. Bureekaew, S. Shimomura and S. Kitagawa, Sci. Technol. Adv. Mater., 2008, 9, 014108 CrossRef PubMed.
- J. Shao, C. Li, Z. Fei, Y. Liu, J. Zhang and L. Li, Mol. Catal., 2024, 558, 114028 CrossRef CAS.
- E. Mamontov, T. Egami, R. Brezny, M. Koranne and S. Tyagi, J. Phys. Chem. B, 2000, 104, 11110–11116 CrossRef CAS.
- I. C. Sophiana, F. Iskandar, H. Devianto, N. Nishiyama and Y. W. Budhi, Nanomaterials, 2022, 12, 1556 CrossRef CAS.
- F. Zhang, Z. Liu, X. Chen, N. Rui, L. E. Betancourt, L. Lin, W. Xu, C.-j. Sun, A. M. M. Abeykoon, J. A. Rodriguez, J. Teržan, K. Lorber, P. Djinović and S. D. Senanayake, ACS Catal., 2020, 10, 3274–3284 Search PubMed.
- Z. Bian, Z. Wang, B. Jiang, P. Hongmanorom, W. Zhong and S. Kawi, Renew. Sustain. Energy Rev., 2020, 134, 110291 CrossRef CAS.
- M. M. Nair, S. Kaliaguine and F. Kleitz, ACS Catal., 2014, 4, 3837–3846 CrossRef CAS.
- S. Shah, J. Hong, L. Cruz, S. Wasantwisut, S. R. Bare and K. L. Gilliard-AbdulAziz, ACS Catal., 2023, 13, 3990–4002 CrossRef CAS.
- J. Wang, A. Kumar, J. L. Wardini, Z. Zhang, H. Zhou, E. J. Crumlin, J. T. Sadowski, K. B. Woller, W. J. Bowman, J. M. LeBeau and B. Yildiz, Nano Lett., 2022, 22, 5401–5408 CrossRef CAS PubMed.
- B. Zhao, B. Yan, S. Yao, Z. Xie, Q. Wu, R. Ran, D. Weng, C. Zhang and J. G. Chen, J. Catal., 2018, 358, 168–178 CrossRef CAS.
- M. Najimu, S. Jo and K. L. Gilliard-AbdulAziz, Acc. Chem. Res., 2023, 108, 430–438 Search PubMed.
- H. Tanaka, M. Taniguchi, M. Uenishi, N. Kajita, I. Tan, Y. Nishihata, J. Mizuki, K. Narita, M. Kimura and K. Kaneko, Angew. Chem., Int. Ed., 2006, 45, 5998–6002 CrossRef CAS PubMed.
- C. Pan, Z. Guo, H. Dai, R. Ren and W. Chu, Int. J. Hydrog. Energy, 2020, 45, 16133–16143 CrossRef CAS.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS PubMed.
- T. Stroud, T. J. Smith, E. Le Saché, J. L. Santos, M. A. Centeno, H. Arellano-Garcia, J. A. Odriozola and T. R. Reina, Appl. Catal., B, 2018, 224, 125–135 CrossRef CAS.
- Z. Xiao, F. Hou, J. Zhang, Q. Zheng, J. Xu, L. Pan, L. Wang, J. Zou, X. Zhang and G. Li, ACS Appl. Mater. Interfaces, 2021, 13, 48838–48854 CrossRef CAS PubMed.
- W. Mo, F. Ma, Y. Ma and X. Fan, Int. J. Hydrog. Energy, 2019, 44, 24510–24524 CrossRef CAS.
- V. R. Bach, A. C. de Camargo, T. L. de Souza, L. Cardozo-Filho and H. J. Alves, Int. J. Hydrog. Energy, 2020, 45, 5252–5263 CrossRef CAS.
- L. Xu, Z. Miao, H. Song, W. Chen and L. Chou, Catal. Sci. Technol., 2014, 4, 1759–1770 RSC.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Nøskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS.
- H. Zhou, T. Zhang, Z. Sui, Y.-A. Zhu, C. Han, K. Zhu and X. Zhou, Appl. Catal., B, 2018, 233, 143–159 CrossRef CAS.
- T. Mozammel, D. Dumbre, R. Hubesch, G. D. Yadav, P. R. Selvakannan and S. K. Bhargava, Energy Fuels, 2020, 34, 16433–16444 CrossRef CAS.
- H. Wang, J. T. Miller, M. Shakouri, C. Xi, T. Wu, H. Zhao and M. C. Akatay, Catal. Today, 2013, 207, 3–12 CrossRef CAS.
- Y. Wang, L. Yao, Y. Wang, S. Wang, Q. Zhao, D. Mao and C. Hu, ACS Catal., 2018, 8, 6495–6506 CrossRef CAS.
- Y. Lu, L. Kang, D. Guo, Y. Zhao, Y. Zhao, S. Wang and X. Ma, ACS Catal., 2021, 11, 8749–8765 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, J. Catal., 2009, 263, 220–227 CrossRef CAS.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- B. Jin, S. Li and X. Liang, Fuel, 2021, 284, 119082 CrossRef CAS.
- A. Chatla, I. W. Almanassra, P. Kallem, M. A. Atieh, H. Alawadhi, V. Akula and F. Banat, J. CO2 Util., 2022, 62, 102082 CrossRef CAS.
- M. Sanchezsanchez, R. Navarro and J. Fierro, Int. J. Hydrog. Energy, 2007, 32, 1462–1471 CrossRef CAS.
- A. Tian, T. Ma, X. Shi, D. Wang, W. Wu, C. Liu and W. Pei, Coatings, 2021, 11, 1095 CrossRef CAS.
- A. Tian, Z. Mei, L. Wang, G. Liu, Z. Liu, G. Kong, W. Tang and C. Liu, Sustain. Energy Fuels, 2024, 8, 1405–1411 RSC.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Energy Fuels, 2019, 33, 11630–11647 CrossRef CAS.
- J. Wang, Y. Mao, L. Zhang, Y. Li, W. Liu, Q. Ma, D. Wu and H. Peng, Fuel, 2022, 315, 123167 CrossRef CAS.
- A. S. Farooqi, B. M. Al-Swai, F. H. Ruslan, N. A. M. Zabidi, R. Saidur, S. A. F. a. Syed Muhammad and B. Abdullah, Arab. J. Chem., 2020, 13, 5740–5749 CrossRef CAS.
- J. D. A. Bellido, J. E. De Souza, J. C. M'Peko and E. M. Assaf, Appl. Catal., A, 2009, 358, 215–223 CrossRef CAS.
- R. K. Singha, A. Yadav, A. Shukla, Z. Iqbal, C. Pendem, K. Sivakumar and R. Bal, ChemistrySelect, 2016, 1, 3075–3085 CrossRef CAS.
- N. Wang, X. Yu, K. Shen, W. Chu and W. Qian, Int. J. Hydrog. Energy, 2013, 38, 9718–9731 CrossRef CAS.
- X. Gao, J. Ashok and S. Kawi, Catal. Today, 2022, 397–399, 581–591 CrossRef CAS.
- V. M. Gonzalez-Delacruz, R. Pereñiguez, F. Ternero, J. P. Holgado and A. Caballero, ACS Catal., 2011, 1, 82–88 CrossRef CAS.
- E. C. Lovell, A. Fuller, J. Scott and R. Amal, Appl. Catal., B, 2016, 199, 155–165 CrossRef CAS.
- Q. Chen, J. Zhang, B. Pan, W. Kong, Y. Chen, W. Zhang and Y. Sun, Chem. Eng. J., 2017, 320, 63–73 CrossRef CAS.
- F. R. Shamskar, M. Rezaei and F. Meshkani, Int. J. Hydrog. Energy, 2017, 42, 4155–4164 CrossRef.
- Y. J. O. Asencios and E. M. Assaf, Fuel Process. Technol., 2013, 106, 247–252 CrossRef CAS.
- E. Akbari, S. M. Alavi and M. Rezaei, Fuel, 2017, 194, 171–179 CrossRef CAS.
- S. Singh, M. B. Bahari, B. Abdullah, P. T. T. Phuong, Q. D. Truong, D.-V. N. Vo and A. A. Adesina, Int. J. Hydrog. Energy, 2018, 43, 17230–17243 CrossRef CAS.
- R. K. Liew, M. Y. Chong, O. U. Osazuwa, W. L. Nam, X. Y. Phang, M. H. Su, C. K. Cheng, C. T. Chong and S. S. Lam, Res. Chem. Intermed., 2018, 44, 3849–3865 CrossRef CAS.
- H. U. Hambali, A. A. Jalil, A. A. Abdulrasheed, T. J. Siang and D.-V. N. Vo, J. Energy Inst., 2020, 93, 1535–1543 CrossRef.
- D. Y. Shen, M. M. Huo, L. Li, S. Lyu, J. H. Wang, X. Y. Wang, Y. H. Zhang and J. L. Li, Catal. Sci. Technol., 2020, 10, 510–516 RSC.
- M. Radlik, M. Adamowska-Teyssier, A. Krzton, K. Koziel, W. Krajewski, W. Turek and P. Da Costa, C. R. Chim., 2015, 18, 1242–1249 CrossRef CAS.
- J. Q. Zhu, X. X. Peng, L. Yao, X. J. Deng, H. Y. Dong, D. M. Tong and C. W. Hu, Int. J. Hydrog. Energy, 2013, 38, 117–126 CrossRef CAS.
- H. W. Cheng, S. H. Feng, W. Tao, X. G. Lu, W. L. Yao, G. S. Li and Z. F. Zhou, Int. J. Hydrog. Energy, 2014, 39, 12604–12612 CrossRef CAS.
- S. H. Zhang, C. Shi, B. B. Chen, Y. L. Zhang and J. S. Qiu, Catal. Commun., 2015, 69, 123–128 CrossRef CAS.
- Y. H. Ahmad, A. T. Mohamed, H. A. El-Sayed, A. Kumar and S. Y. Al-Qaradawi, Int. J. Hydrog. Energy, 2022, 47, 41294–41309 CrossRef CAS.
- Q. Xu, Y. Liu, C. Liu, A. Tian, X. Shi, C. Dong, Y. Zhou and H. Zhou, RSC Adv., 2015, 5, 14458–14464 RSC.
- K.-M. Kang, H.-W. Kim, I.-W. Shim and H.-Y. Kwak, Fuel Process. Technol., 2011, 92, 1236–1243 CrossRef CAS.
- G. P. Thiel and A. K. Stark, Joule, 2021, 5, 531–550 CrossRef CAS.
- C. Liu, A. Tian, Q. Li, T. Wang, G. Qin, S. Li and C. Sun, Adv. Funct. Mater., 2022, 33, 2210759 CrossRef.
- J. Guerrero-Caballero, T. Kane, N. Haidar, L. Jalowiecki-Duhamel and A. Löfberg, Catal. Today, 2019, 333, 251–258 CrossRef CAS.
- A. Löfberg, T. Kane, J. Guerrero-Caballero and L. Jalowiecki-Duhamel, Chem. Eng. Process., 2017, 122, 523–529 CrossRef.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, W. L. Eriksen, C. Frandsen, P. M. Mortensen and I. Chorkendorff, Chem. Eng. J., 2021, 425, 131509 CrossRef CAS.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364, 756–759 CrossRef CAS PubMed.
- L. Dou, C. Yan, L. Zhong, D. Zhang, J. Zhang, X. Li and L. Xiao, Chem. Commun., 2019, 56, 205–208 RSC.
- Q. Dong, Y. Yao, S. Cheng, K. Alexopoulos, J. Gao, S. Srinivas, Y. Wang, Y. Pei, C. Zheng, A. H. Brozena, H. Zhao, X. Wang, H. E. Toraman, B. Yang, I. G. Kevrekidis, Y. Ju, D. G. Vlachos, D. Liu and L. Hu, Nature, 2022, 605, 470–476 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |