
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali-promoted indium oxide as a highly active and selective catalyst for photo-thermal CO2 hydrogenation†
Xinhuilan
Wang
,
Alejandra
Rendón-Patiño
,
Jean Marcel R.
Gallo
,
Diego
Mateo
 * and
Jorge
Gascon
*
* and
Jorge
Gascon
*
Advanced Catalytic Materials (ACM), KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: diego.mateo@kaust.edu.sa; jorge.gascon@kaust.edu.sa
First published on 26th July 2024
Abstract
Photo-thermal carbon dioxide (CO2) reduction has recently gained significant attention as a strategy to harness solar energy and address environmental challenges. Among other photo-thermal catalysts, indium oxide (In2O3) has emerged as a promising candidate for the CO2 hydrogenation reaction. However, owing to its wide band gap semiconductor nature and relatively low CO2 adsorption capacity, modifications are imperative to facilitate efficient light absorption and CO2 activation. In this study, we report alkali-promoted In2O3 catalysts for efficient and selective photo-thermal CO2 hydrogenation to carbon monoxide (CO). By virtue of the enhanced CO2 adsorption capacity, Cs-promoted In2O3 demonstrated superior catalytic performance with a CO production rate of 28 mmol g−1 h−1 and 100% selectivity under full solar spectrum irradiation and without external heating, which is more than 3 times higher compared to that of pure In2O3. Mechanistic studies indicated that non-thermal effects dominate the reaction pathway, particularly at low reaction temperatures and high light intensities, accompanied by minor pure thermal effects. Additional experiments revealed that the in situ formation of defective sites on the indium oxide catalyst surface under illumination could enhance the light absorption ability, thereby significantly enhancing CO production. Diffuse-reflectance infrared Fourier transform spectroscopy (DRIFTS) demonstrated that light irradiation could stabilize key reaction intermediates and accelerate the CO2 hydrogenation reaction at low temperatures, ultimately boosting CO production compared to dark conditions.
1. Introduction
The global demand for energy and environmental concerns resulting from the consumption of fossil fuels have reinforced the urgent need for alternative clean energy solutions. In this regard, the direct utilization of carbon dioxide (CO2) as a feedstock for the production of fuels or chemicals entails a double benefit, as it does not only allow for the mitigation of greenhouse gas emissions but also enables the valorization of CO2 as a raw material. In particular, the conversion of CO2 to carbon monoxide (CO) through the reverse water gas shift (RWGS) reaction is an attractive approach because the obtained CO can serve as a direct feedstock for the Fischer–Tropsch process or methanol synthesis.1–4 However, according to thermodynamics, CO is the major product only at temperatures above 700 °C, thus imposing a considerable penalty in terms of energy efficiency and catalyst stability.5–7 For these reasons, there is an increasing interest in the development of novel processes and catalysts to soften the operating conditions of the RWGS reaction.8,9In this context, photo-thermal catalysis, based on the synergistic combination of light-induced thermal and non-thermal effects, has recently emerged as a promising strategy to perform chemical reactions using sunlight as a unique energy source.10,11 On the one hand, thermal effects derived from the non-radiative relaxation of charge carriers favor localized heating at the active sites, thus improving the overall efficiency of the process.12 On the other hand, non-thermal effects can reduce the thermodynamic barrier of certain reaction steps or even activate specific reaction pathways.13,14 Therefore, the integration of thermochemistry and photochemistry in a single process circumvents the energy constraints and low catalytic performances associated with traditional thermal catalysis and pure photocatalytic systems, respectively, representing a viable approach to produce solar fuels or chemicals such as methane (CH4), methanol (CH3OH) and, of more relevance to the scope of this work, carbon monoxide.15–18
Among other metal oxides, indium oxide (In2O3) has been widely investigated as a catalyst for the thermal hydrogenation of CO2.19–23 Nevertheless, its wide bandgap (3.2 eV) and limited CO2 adsorption properties hinder its direct application in photo-thermal systems for CO2 reduction.24–26 Consequently, many efforts have been devoted to improve the photo-thermal properties of In2O3.27–31 For instance, previous reports have demonstrated that it was possible to enhance the light-harvesting properties of indium oxide through a pre-treatment with hydrogen at high temperatures.31 In a related precedent, Ye and co-workers showed that the loading of In2O3 with precious metals such as Ru, Rh, or Pd could enhance the catalytic activity of indium oxide in the photo-thermal CO2 hydrogenation to methanol.32 Authors attributed the improved catalytic performance to a more effective activation of CO2 and an effective light-to-heat transformation by noble metals.33 However, despite these advances, both high-temperature pre-treatment steps and the incorporation of precious metals still constitute major drawbacks for the wide implementation of indium oxide in photo-thermal catalysis.
Alkali metals have gained significant attention as cost-efficient promoters to enhance both the catalytic activity and selectivity in a wide number of thermal processes such as CO2 hydrogenation,34–36 ammonia decomposition37 and ammonia synthesis.37–39 Surprisingly, the impact of the addition of small quantities of alkali metals on the performance of photo-thermal catalysts remains as an almost unexplored field of research. In this work, we present for the first time the use of alkali-promoted indium oxide as an effective and selective photo-catalyst for the photo-thermal CO2 reduction to CO. Among all the catalysts, Cs-promoted indium oxide displayed the highest catalytic activity, achieving remarkable CO production rates in the order of 28 mmol g−1 h−1 with a 100% selectivity in the absence of external heating. Experimental data suggest that the introduction of alkali promoters favors the CO2 adsorption capacity of the catalyst. Furthermore, it has been demonstrated that light induces the in situ formation of surface defects on the indium oxide structure, in the absence of any pre-treatment step, thus accelerating the reaction at lower temperatures compared to dark conditions. Mechanistically, non-thermal effects dominate the reaction pathway, particularly at low temperatures and high light intensities, in combination with pure thermal effects. Overall, these results indicate that alkali-promotion is a straightforward and cost-efficient strategy to enhance the photo-thermal catalytic activity of indium oxide.
2. Materials and methods
2.1. Catalyst synthesis
As-synthesized indium oxide was prepared through calcination of In(OH)3. Typically, 6 g of In(NO3)3·H2O (Sigma-Aldrich, 99.9%) was dissolved in 100 mL of deionized water, followed by the addition of NH4OH (Sigma-Aldrich, 28.0–30.0% NH3 basis) with a pH-value adjusted to 8. After centrifugation, the precipitate was collected and washed with deionized water three times. In(OH)3 was obtained after drying at 80 °C overnight. After that, a variety of alkali metals (Na, K, Rb, and Cs) were loaded on the In(OH)3via wet-impregnation method with the loading amount of metal designed at 2 wt%. Finally, alkali-promoted In2O3 was obtained from calcination under air at 350 °C for 3 h.2.2. Photo-thermal catalytic experiments
The photo-thermal reverse water gas shift reaction (RWGS) was evaluated using a commercial flow reactor equipped with a quartz window (Harrick, HVC-MRA-5) and a 300 W Xe lamp as the irradiation source. In a typical experiment, 40 mg of catalyst was loaded on the reactor and a thermocouple to monitor the temperature was located ca. 1 mm below the catalyst's surface. The reaction gas mixture (H2/CO2 molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was introduced through the reactor with a total flow rate of 10 mL min−1 (GSHV = 15000 mL h−1 g−1). Nitrogen was used as an internal standard. The outlet of the reactor was directly connected to the inlet of the gas chromatograph (SRA instruments) equipped with two modules and a TCD detector to determine the concentration of gases. One of the modules has a MS5A column and analyzes H2, CH4, and CO using Ar as carrier gas. The second module has a PPU column and analyzes CO2 and up to C2+ hydrocarbons using He as carrier gas.
:1) was introduced through the reactor with a total flow rate of 10 mL min−1 (GSHV = 15000 mL h−1 g−1). Nitrogen was used as an internal standard. The outlet of the reactor was directly connected to the inlet of the gas chromatograph (SRA instruments) equipped with two modules and a TCD detector to determine the concentration of gases. One of the modules has a MS5A column and analyzes H2, CH4, and CO using Ar as carrier gas. The second module has a PPU column and analyzes CO2 and up to C2+ hydrocarbons using He as carrier gas.
CO2 conversion (XCO2, %) and CO selectivity (SCO, %) were calculated according to the following equations:
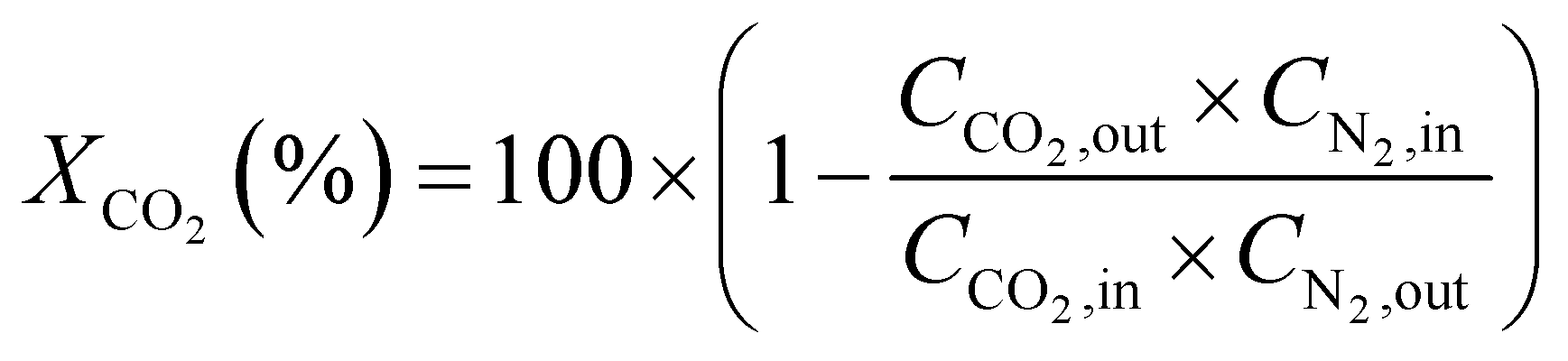
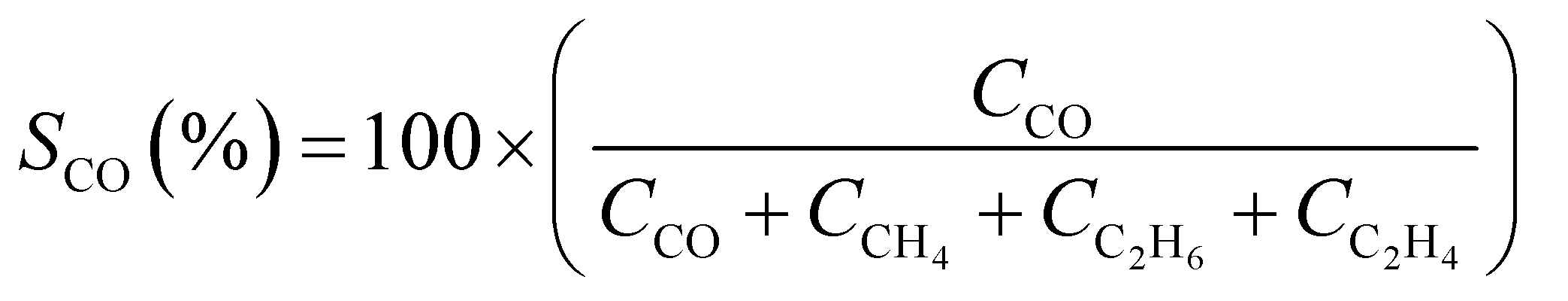
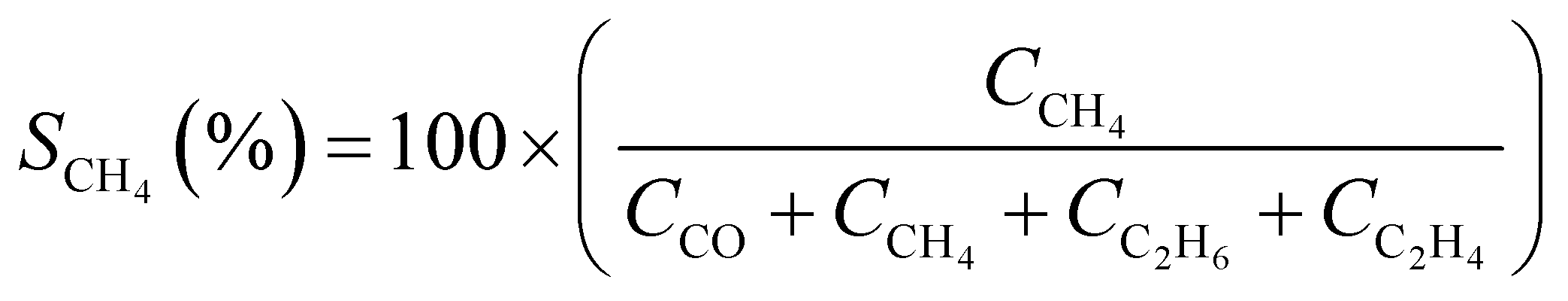
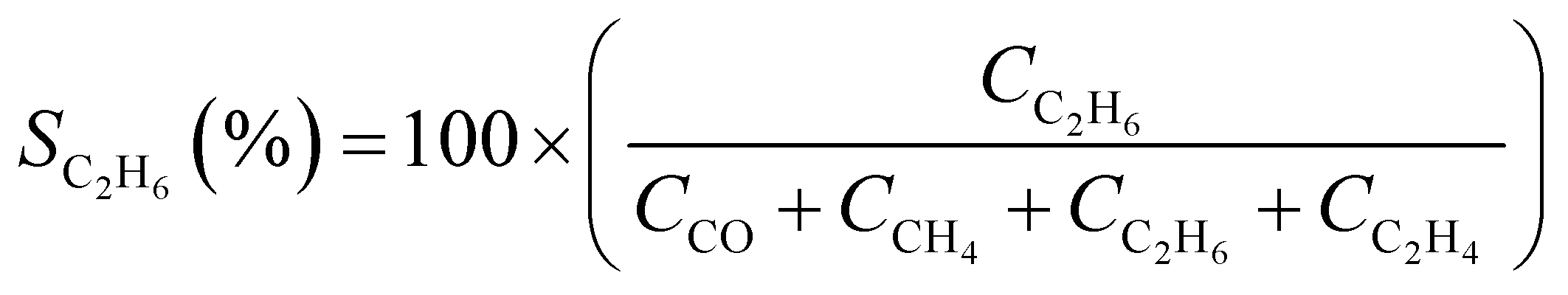
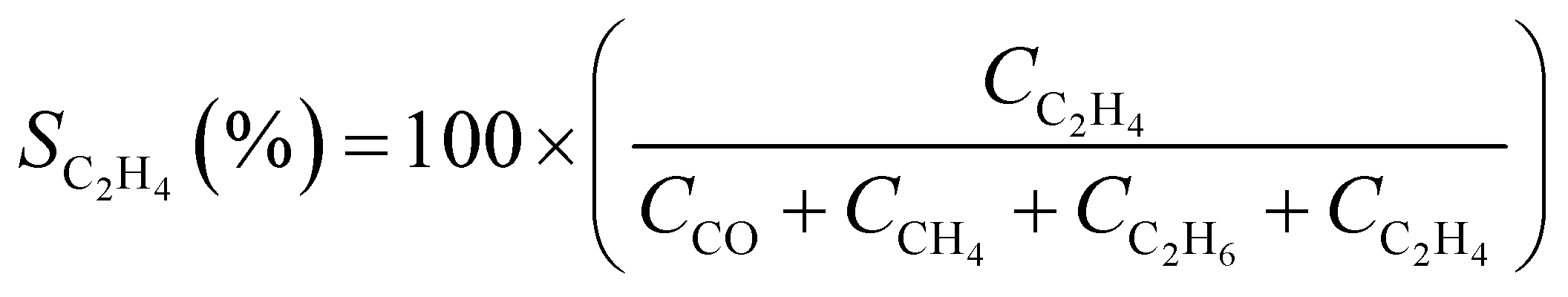
3. Results and discussion
On the basis of literature review, we selected 4 potential promoters (Na, K, Rb and Cs) to impregnate the In2O3 obtained from the calcination of In(OH)3 precursor, as described above. Fig. S1† shows the PXRD patterns of as-synthesized pure and alkali-promoted In2O3 samples. The characteristic peaks of all In2O3 samples appear at 2θ values of 21.5°, 30.6°, 35.5°, 51° and 60.7°, which are ascribed to the diffractions of (211), (222), (400), (440) and (622) facets of c-In2O3 (JCPDS 06-0416). No additional peaks associated with the alkali metals are observed in the XRD patterns, indicating its well dispersion in In2O3.As shown in Fig. S2,† the thermal decomposition of In(OH)3 occurs at approximately 250 °C in air. In fact, the absence of a correlated diffraction peak of In(OH)3 (Fig. S1†) and the negligible weight loss of In2O3, demonstrated the complete conversion of the precursor to In2O3 during the calcination process.
The UV-Vis-NIR absorption spectra of pure and alkali-promoted In2O3 were also measured to determine their light harvesting properties (Fig. S4†). Both pure and alkali-promoted In2O3 exhibited similar absorption peaks around 300 nm, displaying calculated bandgaps of 3.06, 2.93, 2.96, 3.06, 2.97 eV for pure, Na-promoted, K-promoted, Rb-promoted, and Cs-promoted In2O3, respectively (Fig. S4b†).40
N2 adsorption measurements were performed in order to determine the specific surface area of the samples. As shown in Fig. S3a,† pure In2O3 exhibited a type IV isotherm, indicating the presence of mesopores in the material, with a calculated surface area of 67 m2 g−1 (Table S1†).41 However, for the alkali-promoted samples, the surface area was considerably reduced, probably due to the incorporation of the promoter into the existing pores of indium oxide, as it can be seen for Cs-promoted In2O3 (Fig. S3b and Table S1†).
STEM images revealed that the pure In2O3 sample consisted in the aggregation of small In2O3 nanoparticles with an average particle size distribution of 12.2 ± 4.5 nm (Fig. S5†). The STEM analysis of alkali-promoted In2O3 samples showed that the alkali promotion did not alter significantly the size of indium oxide. For instance, in the case of Cs-promoted In2O3 the average particle size distribution was 13.8 ± 5.2 nm (Fig. 1b). High magnification images of the nanoparticles allowed to measure a lattice distance of 0.29 nm attributable to the (222) plane of In2O3, in good agreement with the main peak observed in XRD (Fig. 1c and S1†). Furthermore, STEM coupled with EDX revealed a homogeneous distribution of In, O and Cs in the samples and further confirmed the presence of In2O3 (Fig. S6†).
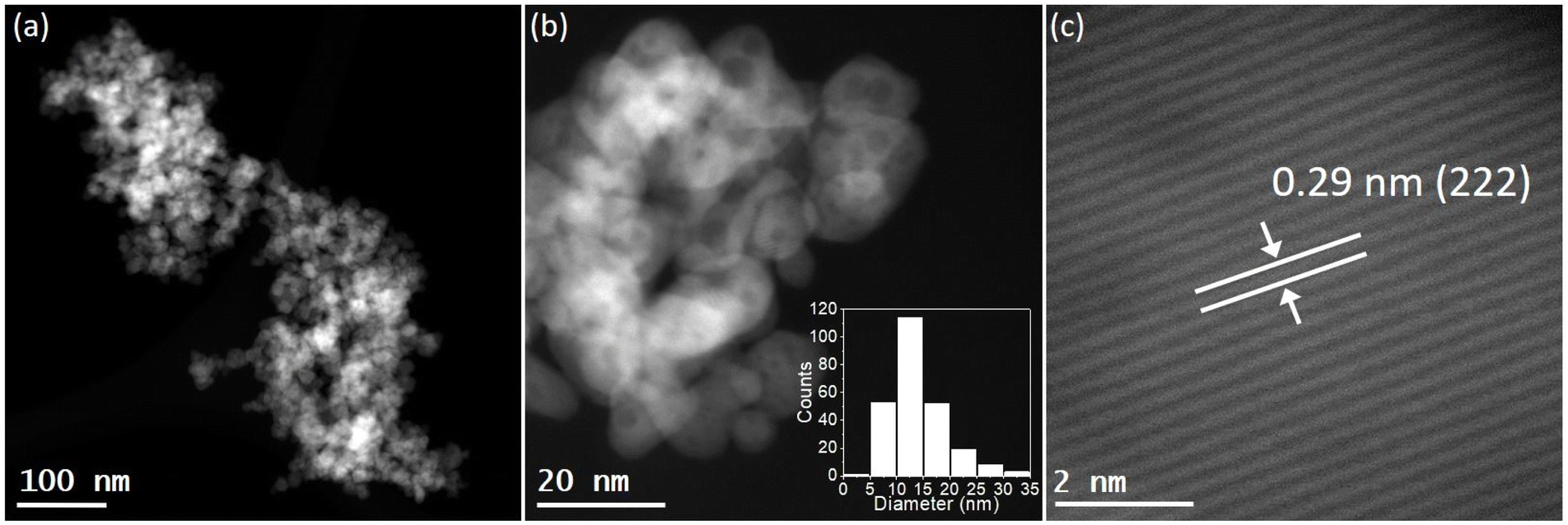 | ||
| Fig. 1 STEM images of Cs-promoted In2O3 at (a) low and (b) high magnification. Inset in (b) corresponds to the particle size distribution. (c) Interplanar distance corresponding to the lattice distance of (222) plane of In2O3. | ||
The catalytic performance of alkali-promoted In2O3 samples towards the RWGS reaction was studied using a 300 W Xe lamp at 3.3 W cm−2 light intensity under continuous flow configuration. As it can be seen in Fig. 2a, the surface temperature of the catalysts rapidly increased upon irradiation, reaching equilibrium in around 15 minutes. It is worth noting that potassium, rubidium and cesium-promoted In2O3 catalysts exhibited more than 30 °C higher surface temperature (ca. 225–230 °C) compared to pure In2O3 under the same conditions, suggesting that the presence of these alkali metals improves the photo-thermal performance of indium oxide. In general, the photo-thermocatalytic efficiency of alkali-promoted samples decreased upon increasing promoter electronegativity. Thus, Cs-promoted In2O3 displayed the best performance, achieving a 25% CO2 conversion and 28 mmol g−1 h−1 CO production rate, while Na-promoted In2O3 showed the lowest catalytic activity, even below pristine indium oxide (Fig. 2b). Interestingly, for all samples, independently on the presence or the nature of the promoter, the selectivity towards CO was 100%. To the best of our knowledge, these results outperform the current state-of-the-art of indium-based catalysts for photo-thermal CO2 reduction under continuous flow (Table S4†).
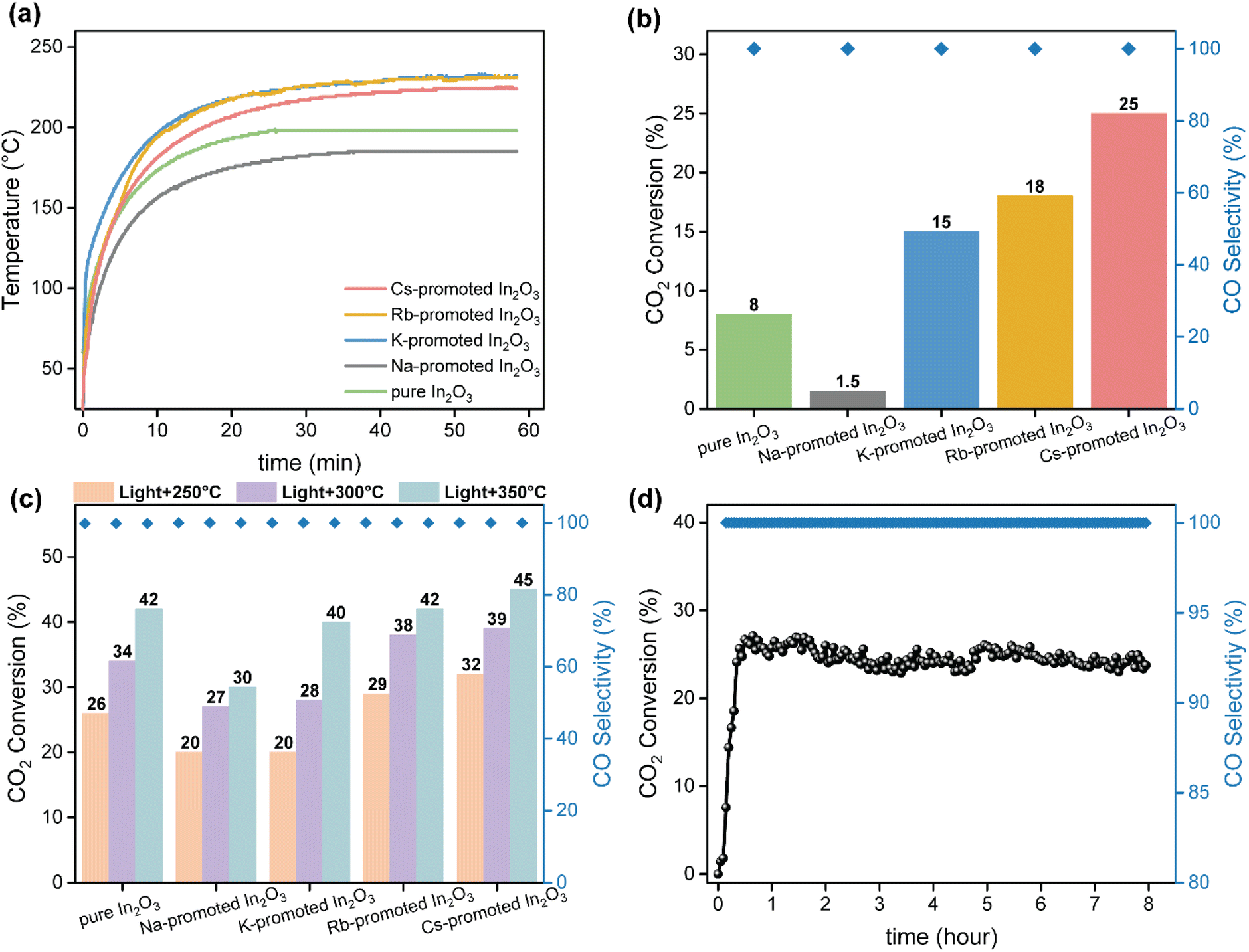 | ||
| Fig. 2 (a) Temperature evolution over pure and alkali-promoted catalysts under 3.3 W cm−2 light irradiation. (b) CO2 conversion and CO selectivity over pure and alkali-promoted In2O3 without external heating under irradiation of 3.3 W cm−2. Reaction conditions: GSHV = 15000 mL h−1 g−1, 1 bar. (c) CO2 conversion and CO selectivity over pure and alkali promoted In2O3 with external heating at 250, 300 and 350 °C under irradiation of 3.3 W cm−2. Reaction conditions: GSHV = 15000 mL h−1 g−1, 1 bar. Stability test of Cs-promoted In2O3 with external heating at 250 °C under irradiation of 3 W cm−2. Reaction conditions: GSHV = 15000 mL h−1 g−1, 1 bar. | ||
Previous reports have observed that the higher the difference in electronegativity between the alkali promoter and the metallic active site, the stronger the interaction between the CO2 molecule and the catalyst.34 Our results are very much in line with these findings and we hypothesize that the enhanced catalytic activity of Cs-promoted indium oxide derives from intense electronic effects that, ultimately, improve the CO2 adsorption capacity and the strength of the interaction between CO2 and our catalyst. In order to support this hypothesis, CO2-TPD measurements were performed to evaluate the ability of CO2 adsorption/desorption on the catalyst surface (Fig. S7†). The small desorption peak in Region I (around 100 °C) is ascribed to physically adsorbed CO2 while the broad peak at about 250 °C in Region II is assigned to thermal-induced oxygen vacancies.42 It is worth noting that the stronger peak intensities and the higher desorption temperature of Cs-promoted In2O3 compared to pristine In2O3 indicate a higher CO2 adsorption ability and a more intense interaction between CO2 molecule and indium oxide in the presence of the alkali promoter, particularly in the high-temperature region (Region III), attributed to the decomposition of bicarbonate (HCO3−) and carbonate (CO32−) species.43
To further understand the transformation of bicarbonate/carbonate species during the CO2 adsorption/desorption process and their decomposition behavior, CO2-TPD experiments of Cs-promoted and pure In2O3 were monitored by in situ DRIFTS (Fig. S8 and S9†). The IR spectra were collected at elevated temperatures (50–600 °C) under helium flow after CO2 adsorption at 50 °C for 60 minutes. As shown in Fig. S8a,† multiple peaks related to different surface species are identified in the range between 1900 cm−1 and 1150 cm−1, including bicarbonate (HCO3−), monodentate carbonate (m-CO32−), bidentate carbonate (b-CO32−), polydentate carbonate (p-CO32−) and carboxylate (CO2δ−).43–47 The predominant adsorbed species on the surface of Cs-promoted In2O3 after CO2 adsorption at room temperature are identified as bicarbonate (HCO3−, 1625 cm−1) and carboxylate species (CO2δ−, 1598 cm−1) (Fig. S8a†). Upon increasing the temperature, the adsorbed carboxylate and bicarbonate species start to decompose, which is consistent with the physical CO2 desorption at around 100 °C in the corresponding MS results (Fig. S8b†) and the CO2-TPD profile (Fig. S7†). Upon further increased temperature, the signal at 1505 cm−1 is attributed to the carboxylate species (CO2δ−) adsorbed on oxygen vacancies, observed at about 250 °C. This is confirmed by a clear CO peak (m/z = 28) in Fig. S8b† at approximately 250 °C, indicating that the oxygen vacancies on the Cs-promoted In2O3 catalyst improve the dissociation of CO2 to CO.48 Moreover, the bicarbonate signal at 1625 cm−1 exhibits a monotonic decrease as the temperature steadily rises to 600 °C. Meanwhile, the characteristic feature associated with bidentate carbonate (b-CO32−) at 1270 cm−1 intensifies until reaching 400 °C, then the peak subsequently declines with further increasing temperature. These results suggest that the bicarbonate species on the catalyst surface decompose to water and bidentate carbonate (b-CO32−) during the desorption process, aligning with the observed water signal (m/z = 18) from MS results in Fig. S8b.† Additionally, the negative peaks in the range between 1400–1300 cm−1 demonstrate the progressive decomposition of monodentate carbonate (m-CO32−) and polydentate carbonate (p-CO32−) with elevating temperature, in accordance with the CO2-TPD results (Fig. S7†).
Conversely, in the case of pure In2O3 no obvious positive peaks are observed after CO2 adsorption, thus indicating a limited CO2 adsorption capacity for this sample (Fig. S9†). Interestingly, when the temperature rises to 600 °C, all carbonates and bicarbonates on pure In2O3 are completely decomposed. However, the positive peak at 1270 cm−1 of Cs-promoted In2O3 persists due to the remaining bidentate carbonate. Overall, in situ DRIFTS-MS experiments reveal that the introduction of Cs induces a strong CO2 adsorption capacity that boosts the catalytic performance towards the RWGS reaction, as evidenced by the catalytic results presented above.
As the RWGS reaction is favored at high reaction temperatures due to its endothermic nature, we further explored the catalytic performance of alkali-promoted indium oxide samples in the temperature range from 250 to 350 °C. Given that the maximum temperature displayed by the catalyst is limited by the maximum irradiance of the lamp, we used an external heater positioned at the bottom of the catalyst bed to achieve the desired temperature. As shown in Fig. 2c, increasing the external heating from 250 °C to 350 °C led to a progressive improvement in catalytic performance. Both at 250 °C and 300 °C, the Cs-promoted In2O3 consistently demonstrated superior catalytic activity, in line with the results in the absence of external heating. At 350 °C, however, all catalysts, except for Na-promoted In2O3, showed a comparable performance. This indicates that, upon increasing the reaction temperature the above-mentioned electronic effects derived from alkali promoters start to vanish, thus equilibrating the activity of all samples. Interestingly, in terms of CO selectivity, all catalysts exhibited an outstanding CO selectivity for all reaction temperatures. In view of the excellent catalytic properties of Cs-promoted indium oxide, particularly at low temperatures, we assessed the long-term stability of this catalyst under reaction conditions. As shown in Fig. 2d, no noticeable CO2 conversion or CO selectivity drop is observed after 8 hours of continuous reaction, thus demonstrating the robustness of the catalyst.
When coupled to Fischer–Tropsch process or methanol synthesis, performing the RWGS reaction under moderate pressures features notable advantages, particularly the absence of compression stages between the different reactors. For this reason, besides the effect of reaction temperature, we also studied the effect of pressure on the catalytic activity of alkali-promoted indium oxide samples. Fig. S10† illustrates the photo-thermal CO2 conversion and CO selectivity of Cs-promoted In2O3 at different pressures in the range of 250–350 °C. Although the pressure could not affect the thermodynamic equilibrium for RWGS reaction, the methanation reaction is more favorable under high pressure and H2-rich environment. Surprisingly, the photo-thermal performance at 10 bar increases significantly compared to ambient conditions, displaying a near unity selectivity towards CO and without any evident sign of methane formation. For instance, remarkable CO production rates in the order of 67 and 74 mmol g−1 h−1 were achieved at 250 °C and 300 °C, respectively, far exceeding the productivity at ambient conditions and establishing a new benchmark for the photo-thermal RWGS reaction by indium-based catalysts under continuous flow configuration (Table S4†). This enhancement could be explained by a better surface coverage of the catalyst at 10 bar, thereby fostering more favorable reaction kinetics. However, upon further increasing the pressure to 15 bar, no substantial improvement in catalytic performance is observed, probably due to a complete coverage of the catalyst surface. In addition to this, the selectivity towards CO at 15 bar started to decrease due to the formation of CH4 as by-product as a result of the partial reduction of indium oxide, as we describe below.
Interestingly, the analysis of the spent samples by XRD confirmed that metallic indium was present in the samples tested at 10 and 15 bar at 350 °C (Fig. S11†). However, the reduction temperature of In2O3 to metallic indium under hydrogen atmosphere occurs at temperatures above 600 °C.49,50 Therefore, these results suggested that light could somehow facilitate the formation of metallic indium at considerably lower temperatures. In order to investigate in more detail the role of light in the reduction of indium oxide, we carried out H2-TPR experiments over the Cs-promoted In2O3 sample both under dark and light conditions. As shown in Fig. S12a,† the first intense peak centered at 234 and 236 °C for Cs-promoted In2O3 under light and dark conditions, respectively, is assigned to the reduction of surface In2O3. In the high-temperature region, the H2 consumption peaks are associated with the reduction of bulk In2O3 to metallic indium.50,51 Notably, under light illumination, the reduction temperature of bulk In2O3 to metallic indium occurs at 518 °C, which is approximately 70 °C lower than its dark counterpart (587 °C). The presence of metallic indium in the irradiated sample was further confirmed through XRD measurements, however, no characteristic diffraction of metallic indium was detected in the sample reduced under dark conditions (Fig. S12b†). Altogether, H2-TPR experiments demonstrated that light irradiation effectively lowers the reduction temperature of indium oxide to metallic indium, inducing the formation of defects in the lattice of indium oxide.
In view of this, additional XPS measurements of the fresh and spent Cs-promoted In2O3 samples were performed to prove the formation of metallic indium on the surface of the catalyst after reaction. In the fresh sample, the In3d core level exhibited two symmetric peaks of In3d5/2 and In3d3/2 spin orbits located at 443.7 and 451.3 eV, respectively, thus matching the peak-to-peak energy gap of 7.6 eV characteristic of In2O3 (Fig. S13a†).52 The O1s core level was deconvoluted into two peaks centered at 529.2 and 531.1 eV corresponding to lattice oxygen and surface –OH species, respectively (Fig. S13b†).53 Interestingly, the Cs3d core level spectrum showed two peaks located at 724.1 and 738.2 eV, attributable to CsOH, indicating a positive charge on Cs (Fig. S13c†).54 This observation explains the negative shift (∼0.7 eV) in the binding energy of both indium and oxygen components, owing to the electron donation activity of Cs and the increase of electron density of In and O atoms. In contrast, the In3d core level spectrum of the spent sample showed the presence of both metallic and In3+ components at 442.9 and 443.5 eV, respectively, thereby indicating the partial reduction of surface In3+ (Fig. S13d†). Furthermore, the O1s spectrum showed a new component at 529.1 eV attributable to defect-related oxygen species, thus decreasing the fraction of lattice oxygen in the spent Cs-promoted In2O3 sample from 65.9 to 35.4% (Fig. S13e†). In general, a negative shift in all the peaks both in In3d and O1s regions could be observed, probably due to an increase in the electron density in the In–O bonding upon the creation of defects in the indium oxide lattice.55 Overall, XPS data demonstrated that, under moderate pressure and temperature conditions, light could induce the reduction of In3+ to metallic indium and the creation of surface defects in the photocatalyst during irradiation.
In an effort to further confirm the introduction of defects in the indium oxide lattice, Raman measurements were performed in both fresh and spent Cs-promoted In2O3 catalysts. As shown in Fig. S14a,† the observed peaks at 130 and 307 cm−1 are associated with the In–O vibration of the InO6 octahedra, while the peak at 366 cm−1 is attributed to the stretching vibration of the In–O–In bond, reflecting the oxygen vacancy in the In2O3 structure.56 As it can be seen, compared to the fresh catalyst, a noticeable redshift is evident in the spent catalysts. In particular, the peak at 307.8 cm−1 in the fresh sample shifted to 303.2 cm−1 after the reaction at 10 bar. This significant shift indicates the distortion of the In2O3 structure during the photo-thermal reaction. Moreover, the relative peak intensity ratio between unsaturated InO6−x (366 cm−1) and saturated InO6 (307 cm−1) is presented in Fig. S14b.† The increased ratio in the spent sample compared to fresh Cs-promoted In2O3 suggests an enhanced oxygen vacancy concentration after the photo-thermal process, in good agreement with previous H2-TPR and XPS results.
In general, the formation of surface oxygen vacancies in semiconductor oxides is attributed to a self-structural transformation of the material under illumination. In our case, we hypothesize that the mechanism for the light-induced oxygen vacancy generation in the indium oxide begins with the photo-generation of electrons and the self-reduction of surface In3+ to In(3−x)+. Secondly, the photo-generated holes are trapped in the lattice oxygen ions to form free oxygen radical species (O˙). In a last step, the combination of two oxygen radical species releases an oxygen molecule and creates the oxygen vacancies on the surface.57–59 To confirm this hypothesis, Cs-promoted In2O3 samples were subjected to illumination under argon atmosphere without external heating and also at 250 °C in order to induce the formation of oxygen vacancies. After this treatment, samples were analyzed by UV-Vis absorption spectroscopy and TGA under air (Fig. S15a and c†). As it can be seen, samples subjected to illumination exhibited an enhanced absorption in the visible-NIR region compared to the fresh sample, clearly indicating the formation of mid-gap defect states in the bandgap of indium oxide associated with oxygen vacancies.31 When it comes to TG analysis, both samples showed a weight increase upon increasing the temperature, thus demonstrating the reoxidation and refilling of the light-induced oxygen vacancies. Furthermore, mass spectroscopy measurements confirmed that molecular oxygen was released during the light-induced formation of the surface oxygen vacancies under light irradiation (Fig. S15d†), therefore validating our initial hypothesis. In view of these results, we suggest that the effect of light, combined with the elevated temperatures and the presence of hydrogen favored the formation of defects in the structure of In2O3. Apart from the improvement in the light-harvesting properties, previous studies have also revealed that oxygen vacancies can improve the photo-thermal performance towards CO2 hydrogenation because they are able to trap photo-generated electrons, activate CO2 molecules and stabilize the reaction intermediates under light illumination.30 For these reasons, we hypothesize that the remarkable catalytic activity of Cs-promoted In2O3 derives not only from an enhanced interaction between CO2 and the catalyst due to alkali promotion, but also from the in situ formation of surface defects under radiation. It is worth reminding, however, that an excessive degree of defects in the indium oxide structure can alter the selectivity patterns, for instance promoting the generation of undesirable CH4, as we observed in the case of the experiments performed at 15 bar. For these reasons, it is crucial to finely control the reaction conditions to achieve a perfect balance between surface defects and structural integrity in the indium oxide catalyst.
4. Mechanistic study
As commented above, photo-thermal catalysis arises from the interplay between thermal and non-thermal effects of light. A straightforward way to isolate the contribution of each effect consists in performing the reaction both under dark and light conditions at the same temperature. However, this approach is too simplistic as it overlooks the presence of potential thermal gradients within the catalyst bed that can point to misleading conclusions. To minimize this issue, we employed indirect illumination experiments to extract the pure photo-chemical contribution, following the approach recently reported by Liu and Everitt.60 In these experiments, a 1 mm thin layer of Ti2O3 (a material with excellent properties to transform light into heat but inactive for RWGS reaction) was applied on top of the catalyst bed to ensure that the underlying catalyst is not directly illuminated but maintains the same temperature profile as in the direct photo-thermal reaction. Ultimately, the comparison between the direct and indirect illumination experiments gives a good approximation on the relative contribution of pure photo-chemical effects to the overall catalytic activity.Fig. 3a shows the CO2 conversion and CO selectivity of direct and indirect illumination experiments over Cs-promoted In2O3 at 1 bar. As shown in Table S2,† all surface temperatures of the catalyst covered with Ti2O3 are in good agreement with those obtained under direct illumination, suggesting that this thin layer provides comparable photo-thermal heating while blocking pure photo-chemical effects. As for without external heating, the CO2 conversion of indirect illumination exhibits only 3.8%, which is more than 6 times lower than direct illumination, indicating the prevalence of non-thermal effects. However, the difference in CO2 conversion between direct and indirect illumination is reduced from 84.8 to 34.4% upon increasing the external heating up to 300 °C (Table S2†), suggesting that the contribution of hot carriers derived from the photo-excitation of In2O3 gradually decreases upon increasing reaction temperature due to charge recombination.
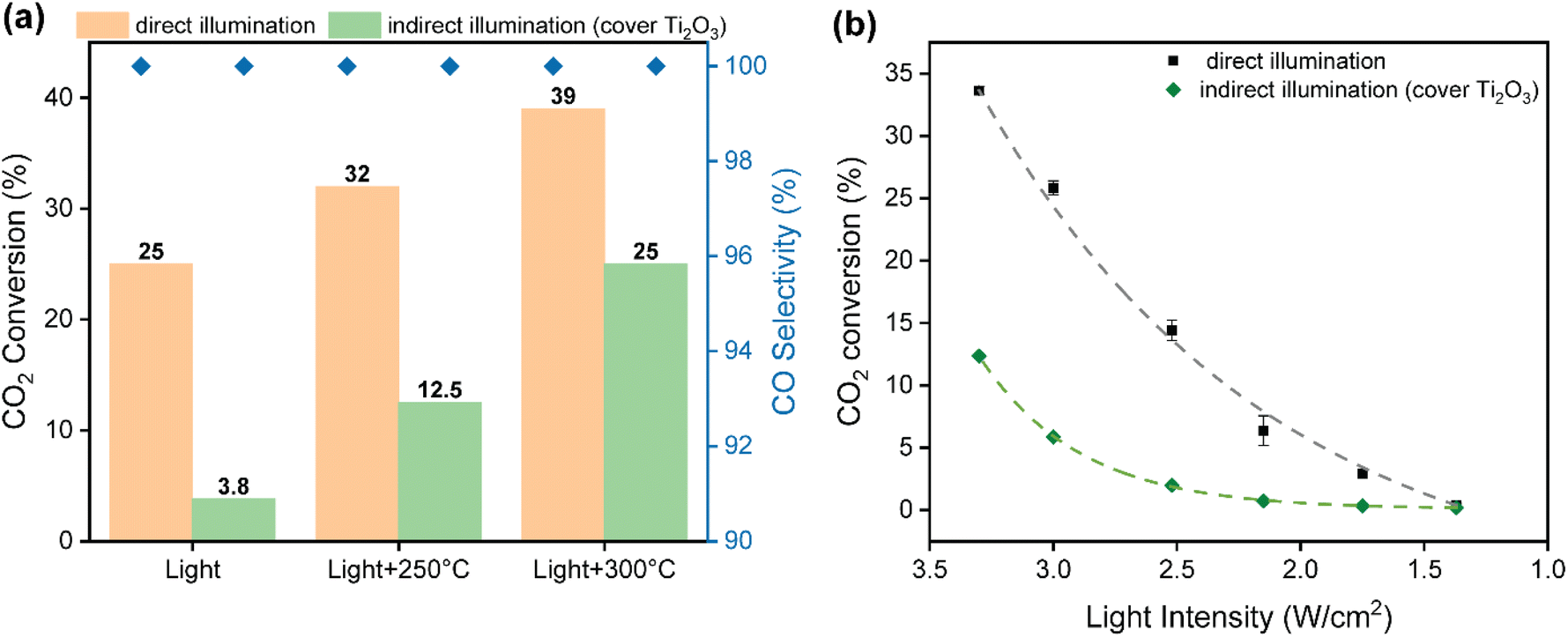 | ||
| Fig. 3 (a) CO2 conversion and CO selectivity over Cs-promoted In2O3 under direct and indirect illumination of 3.3 W cm−2 without and with external heating. Reaction conditions: GSHV = 15000 mL h−1 g−1, 1 bar. (b) Study of the effect of light intensity on the CO2 conversion over Cs-promoted In2O3 catalyst under direct and indirect illumination. Reaction conditions: GSHV = 15000 mL h−1 g−1, 1 bar, 250 °C. | ||
To further investigate the role of thermal and non-thermal effects on the reaction mechanism, we evaluated the catalytic activity at different light intensities with and without the Ti2O3 layer (Fig. 3b). As it can be seen, the difference in CO2 conversion values between dark and light conditions augmented upon increasing the irradiance, even displaying a similar surface temperature (Table S3†). These observations therefore suggest that non-thermal effects are dominant at high light intensities. Finally, the apparent activation energy was calculated by the Arrhenius equation according to the CO production rate and surface temperature in the light intensity tests. As shown in Fig. S16,† the apparent activation energy of Cs-promoted In2O3 in the dark was 119 kJ mol−1, while this value was reduced to 105 kJ mol−1 under light irradiation. Overall, these results provide solid evidences of the participation of light-induced charge carriers in the reaction enhancement, particularly at low temperatures and high light intensities, in combination with pure thermal effects. Interestingly, as it can be seen in Fig. 3a, CO selectivity was independent on the presence of direct or indirect illumination, thus indicating that in this case hot carriers were not altering the overall reaction pathway.
The reaction orders of CO2 and H2 were also investigated to deeply analyze the kinetic behavior under both light illumination and dark conditions, with reaction temperature being an independent parameter. As shown in Fig. S17,† the reaction orders of CO2 under light and dark conditions were 0.41 and 0.47, respectively. This suggests that light irradiation could accelerate to some extent activation of CO2. Interestingly, the reaction order of H2 in the dark was −0.75. This negative value indicates that adsorbed H2 (presumably in the form of indium hydride species or hydroxyl groups) could block the active sites of the catalyst, competing with CO2 for adsorption and activation.61–63 As for under light irradiation, the H2 reaction order increased from −0.75 to −0.13. These observations suggest that light weakens H2 adsorption, therefore minimizing the H2 poisoning effect. Altogether, these kinetic data further confirm the presence of non-thermal effects in our system, in good agreement with our previous mechanistic studies.
To have a better insight on these observations, in situ DRIFTS measurements were conducted to investigate the reaction pathway involved in the photo-thermal RWGS both under dark and light reaction conditions (Fig. 4). As shown in Fig. 4a, after the exposure of Cs-promoted In2O3 to the reaction mixture (H2/CO2 = 4:1) at room temperature, a small band appeared at 1625 cm−1, corresponding to the surface bicarbonate species.43,44 Surprisingly, at a temperature as low as 50 °C, several peaks are observed in the range between 2200 cm−1 and 800 cm−1. The bands at 1575 and 1370 cm−1 correspond to the symmetric and asymmetric OCO stretching of bidentate formate.64,65 The peak at 1445 cm−1 is assigned to the polydentate carbonate (p-CO32−).43 Moreover, the signal at 1235 cm−1 is assigned to the C–O stretching of COOH* intermediate.66 Meanwhile, a peak at around 2078 cm−1 appeared and increased, corresponding to the formation of CO adsorbed on the catalyst surface. In stark contrast, under dark conditions, only a minor band attributed to bicarbonate species is discernible until reaching 120 °C (Fig. 4b). Furthermore, the characteristic bands associated with bidentate formate, p-CO32−, adsorbed CO* and COOH* species are only observed at a significantly higher temperature of 150 °C in the absence of light.
 | ||
| Fig. 4
In situ DRIFTS spectra of Cs-promoted In2O3 under H2 and CO2 (4:1) at 10 bar with increased temperature upon (a) light and (b) dark conditions. IR peak intensities of COOH* and bidentate formate species as a function of temperature under (c) light and (d) dark conditions. | ||
To better understand the formation rate of key intermediates in the photo-thermal RWGS reaction, the IR peak intensities of COOH* and bidentate formate both under light and dark in the range of 25–250 °C are illustrated in Fig. 4c and d. Under light illumination, both COOH* and b-formate increase significantly in intensity and reach their maximum at 100 °C. Conversely, these intermediates begin their formation at 120 °C under dark conditions and reach their maximum at 160 °C. In addition, the intensity of b-formate decreases above 200 °C in the dark, while it remains stable up to 250 °C under illumination. Overall, DRIFTS measurements not only reveal that bidentate formate and COOH* species serve as active intermediates in the RWGS reaction but also indicate that the presence of light promotes the formation and stabilization of key intermediates at lower reaction temperatures, thus enhancing the reaction activity compared to dark conditions.
Our proposed mechanism for the photo-thermal RWGS reaction over Cs-promoted In2O3 is illustrated in Fig. 5. Firstly, owing to the improved CO2 adsorption properties derived from the alkali promotion, the CO2 molecules are adsorbed on the catalyst surface in the form of bicarbonate/carbonate species. In a further step, the bicarbonate/carbonate intermediates react with adsorbed H* to generate formate and COOH* species that, eventually, decompose to generate CO, as demonstrated by DRIFTS measurements.
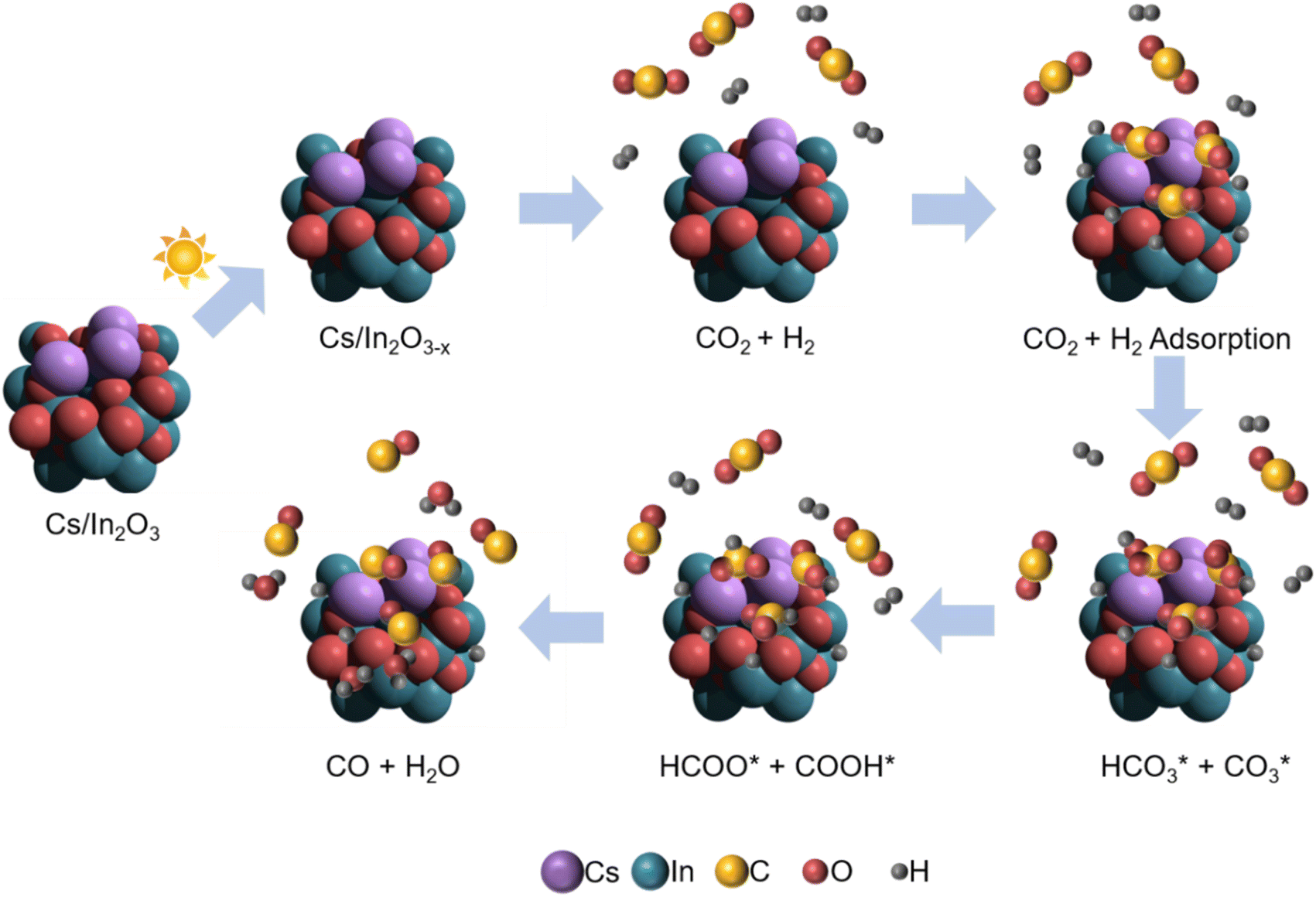 | ||
| Fig. 5 Schematic illustration for the proposed reaction mechanism of the photo-thermal CO2 hydrogenation over Cs-promoted In2O3. | ||
5. Conclusions
In summary, we have successfully developed an efficient and selective alkali-promoted In2O3 catalyst for the photo-thermal hydrogenation of CO2. The introduction of cesium significantly enhanced the CO2 adsorption capacity of indium oxide, thus improving the catalytic activity with a CO production rate of 28 mmol g−1 h−1 in the absence of external heating. Notably, this catalyst exhibited exceptional performance under mild pressure conditions, achieving a CO yield of 74 mmol g−1 h−1 at 300 °C and 10 bar. Mechanistic studies revealed that the reaction follows a non-thermal pathway, particularly at low reaction temperatures and high light intensities, in combination with thermal contributions. Experimental results showed that light illumination facilitated the in situ formation of defect sites on the In2O3 surface, thereby enhancing light-harvesting properties during the photo-thermal reaction. Furthermore, DRIFTS measurements indicated that light could stabilize crucial intermediates (COOH* and formate species) and accelerate the reaction at lower temperatures compared to dark conditions. Overall, this work demonstrates that the incorporation of alkali promoters is a promising and cost-efficient approach to improve the photo-thermal activity of indium oxide in the CO2 hydrogenation reaction.Data availability
Data from this study have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from King Abdullah University of Science and Technology (KAUST).References
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC
.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, React. Chem. Eng., 2021, 6, 954–976 RSC
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC
- T. A. Atsbha, T. Yoon, P. Seongho and C.-J. Lee, J. CO2 Util., 2021, 44, 101413 CrossRef CAS
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC
- A. Wolf, A. Jess and C. Kern, Chem. Eng. Technol., 2016, 39, 1040–1048 CrossRef CAS
- M. Zhu, Q. Ge and X. Zhu, Trans. Tianjin Univ., 2020, 26, 172–187 CrossRef CAS
- C. Lv, X. Bai, S. Ning, C. Song, Q. Guan, B. Liu, Y. Li and J. Ye, ACS Nano, 2023, 17, 1725–1738 CrossRef CAS
- R. Ma, J. Sun, D. H. Li and J. J. Wei, Int. J. Hydrogen Energy, 2020, 45, 30288–30324 CrossRef CAS
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 Search PubMed
- J. Wang, Y. Li, L. Deng, N. Wei, Y. Weng, S. Dong, D. Qi, J. Qiu, X. Chen and T. Wu, Adv. Mater., 2017, 29, 1603730 CrossRef
- I. S. Khan, L. Garzon-Tovar, D. Mateo and J. Gascon, Eur. J. Inorg. Chem., 2022, 28, e202200316 CrossRef
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 CrossRef CAS PubMed
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC
- I. S. Khan, D. Mateo, G. Shterk, T. Shoinkhorova, D. Poloneeva, L. Garzón-Tovar and J. Gascon, Angew. Chem., 2021, 133, 26680–26686 CrossRef
- D. Mateo, N. Morlanes, P. Maity, G. Shterk, O. F. Mohammed and J. Gascon, Adv. Funct. Mater., 2021, 31, 2008244 Search PubMed
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., 2016, 128, 6369–6373 CrossRef
- S. Dang, B. Qin, Y. Yang, H. Wang, J. Cai, Y. Han, S. Li, P. Gao and Y. Sun, Sci. Adv., 2020, 6, eaaz2060 CrossRef CAS PubMed
- M. S. Frei, M. Capdevila-Cortada, R. García-Muelas, C. Mondelli, N. López, J. A. Stewart, D. C. Ferré and J. Pérez-Ramírez, J. Catal., 2018, 361, 313–321 CrossRef CAS
- M. S. Frei, C. Mondelli, R. García-Muelas, J. Morales-Vidal, M. Philipp, O. V. Safonova, N. López, J. A. Stewart, D. C. Ferré and J. Pérez-Ramírez, Nat. Commun., 2021, 12, 1960 CrossRef CAS
- J. Guo, Z. Wang, T. Gao and Z. Wang, Chem. Eng. J., 2024, 149370 CrossRef CAS
- Y. Dong, K. K. Ghuman, R. Popescu, P. N. Duchesne, W. Zhou, J. Y. Loh, F. M. Ali, J. Jia, D. Wang and X. Mu, Advanced Science, 2018, 5, 1700732 CrossRef
- L. He, T. E. Wood, B. Wu, Y. Dong, L. B. Hoch, L. M. Reyes, D. Wang, C. Kubel, C. Qian, J. Jia, K. Liao, P. G. O'Brien, A. Sandhel, J. Y. Loh, P. Szymanski, N. P. Kherani, T. C. Sum, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 5578–5586 CrossRef CAS PubMed
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS
- L. He, T. E. Wood, B. Wu, Y. Dong, L. B. Hoch, L. M. Reyes, D. Wang, C. Kübel, C. Qian and J. Jia, ACS Nano, 2016, 10, 5578–5586 CrossRef CAS PubMed
- L. B. Hoch, P. G. O'Brien, F. M. Ali, A. Sandhel, D. D. Perovic, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 9017–9025 CrossRef CAS PubMed
- Y. Qi, J. Jiang, X. Liang, S. Ouyang, W. Mi, S. Ning, L. Zhao and J. Ye, Adv. Funct. Mater., 2021, 31, 2100908 CrossRef CAS
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu and E. E. Storey, Nat. Commun., 2020, 11, 2432 CrossRef CAS
- B. Deng, H. Song, Q. Wang, J. Hong, S. Song, Y. Zhang, K. Peng, H. Zhang, T. Kako and J. Ye, Appl. Catal., B, 2023, 327, 122471 CrossRef CAS
- M. Gao, J. Zhang, P. Zhu, X. Liu and Z. Zheng, Appl. Catal., B, 2022, 314, 121476 CrossRef CAS
- Q. Yang, V. A. Kondratenko, S. A. Petrov, D. E. Doronkin, E. Saraçi, H. Lund, A. Arinchtein, R. Kraehnert, A. S. Skrypnik and A. A. Matvienko, Angew. Chem., Int. Ed., 2022, 61, e202116517 CrossRef CAS
- A. Ramirez, L. Gevers, A. Bavykina, S. Ould-Chikh and J. Gascon, ACS Catal., 2018, 8, 9174–9182 CrossRef CAS
- G. Varvoutis, M. Lykaki, E. Papista, S. A. Carabineiro, A. C. Psarras, G. E. Marnellos and M. Konsolakis, J. CO2 Util., 2021, 44, 101408 CrossRef CAS
- L. A. Parker, J. H. Carter, N. F. Dummer, N. Richards, D. J. Morgan, S. E. Golunski and G. J. Hutchings, Catal. Lett., 2020, 150, 3369–3376 CrossRef CAS
- Q. Wang, J. Guo and P. Chen, Chem, 2021, 7, 3203–3220 CAS
- Y. Peng, J. Albero, A. Franconetti, P. Concepción and H. García, ACS Catal., 2022, 12, 4938–4946 CrossRef CAS
- S. Kulkarni and D. Patil, J. Mater. Sci.: Mater. Electron., 2016, 27, 3731–3735 CrossRef CAS
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res, 2018, 3, 34–50 Search PubMed
- T. Yan, N. Li, L. Wang, W. Ran, P. N. Duchesne, L. Wan, N. T. Nguyen, L. Wang, M. Xia and G. A. Ozin, Nat. Commun., 2020, 11, 6095 CrossRef CAS PubMed
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS
- E.-M. Köck, M. Kogler, T. Bielz, B. Klötzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666–17673 CrossRef
- Z. Zhou, B. Qin, S. Li and Y. Sun, Phys. Chem. Chem. Phys., 2021, 23, 1888–1895 RSC
- Y. Wang, L. Zhu, Y. Liu, E. I. Vovk, J. Lang, Z. Zhou, P. Gao, S. Li and Y. Yang, Appl. Surf. Sci., 2023, 631, 157534 CrossRef CAS
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed
- Y. Yang, Y.-X. Pan, X. Tu and C.-j. Liu, Nano Energy, 2022, 101, 107613 CrossRef CAS
- P. W. Park, C. S. Ragle, C. L. Boyer, M. L. Balmer, M. Engelhard and D. McCready, J. Catal., 2002, 210, 97–105 CrossRef CAS
- S. Tan, L. B. Gil, N. Subramanian, D. S. Sholl, S. Nair, C. W. Jones, J. S. Moore, Y. Liu, R. S. Dixit and J. G. Pendergast, Appl. Catal., A, 2015, 498, 167–175 CrossRef CAS
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C.-j. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS
- I. N. Reddy, C. V. Reddy, M. Cho, J. Shim and D. Kim, Mater. Res. Express, 2017, 4, 086406 CrossRef
- L. Sun, R. Li, W. Zhan, Y. Yuan, X. Wang, X. Han and Y. Zhao, Nat. Commun., 2019, 10, 2270 CrossRef PubMed
- Y. V. Larichev, I. Prosvirin, D. Shlyapin, N. Shitova, P. Tsyrul’nikov and V. Bukhtiyarov, Kinet. Catal., 2005, 46, 597–602 CrossRef CAS
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef
- J. Gan, X. Lu, J. Wu, S. Xie, T. Zhai, M. Yu, Z. Zhang, Y. Mao, S. C. I. Wang and Y. Shen, Sci. Rep., 2013, 3, 1021 CrossRef
- B. Wang, X. Wang, L. Lu, C. Zhou, Z. Xin, J. Wang, X.-k. Ke, G. Sheng, S. Yan and Z. Zou, ACS Catal., 2018, 8, 516–525 CrossRef CAS
- C. Xu, Y. Zhang, J. Chen, J. Lin, X. Zhang, Z. Wang and J. Zhou, Appl. Catal., B, 2017, 204, 324–334 CrossRef CAS
- C. Xu and J.-L. Luo, JPhys Energy, 2023, 5, 011001 CrossRef
- X. Li, H. O. Everitt and J. Liu, Nano Res., 2019, 12, 1906–1911 CrossRef CAS
- Y. Peng, H. Szalad, P. Nikacevic, G. Gorni, S. Goberna, L. Simonelli, J. Albero, N. López and H. García, Appl. Catal., B, 2023, 333, 122790 CrossRef CAS
- Y. Peng, A. Melillo, R. Shi, A. Forneli, A. Franconetti, J. Albero and H. García, Appl. Catal., B, 2023, 339, 123143 CrossRef CAS
- A. I. Serykh, J. Phys. Chem. C, 2016, 120, 21436–21440 CrossRef CAS
- B. Deng, H. Song, K. Peng, Q. Li and J. Ye, Appl. Catal., B, 2021, 298, 120519 CrossRef CAS
- Y. Wang, Y. Liu, L. Tan, X. Lin, Y. Fang, X. F. Lu, Y. Hou, G. Zhang and S. Wang, J. Mater. Chem. A, 2023, 11, 26804–26811 RSC
- S. Ning, H. Ou, Y. Li, C. Lv, S. Wang, D. Wang and J. Ye, Angew. Chem., 2023, 135, e202302253 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04387a |
| This journal is © The Royal Society of Chemistry 2024 |