
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient electrosynthesis of hydrogen peroxide in neutral media using boron and nitrogen doped carbon catalysts†
Brianna N.
Ruggiero
a,
Xiao Kun
Lu
a,
Bingzhang
Lu
a,
Adrien E.
Deberghes
a,
Dennis
Nordlund
b,
Justin M.
Notestein
 a and
Linsey C.
Seitz
*a
a and
Linsey C.
Seitz
*a
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois 60208-3113, USA. E-mail: linsey.seitz@northwestern.edu
bSLAC National Accelerator Laboratory, 2575 Sand Hill Rd, Menlo Park, California 94025, USA
First published on 16th August 2024
Abstract
The two-electron electrocatalytic oxygen reduction reaction (2e− ORR) is a promising, sustainable, and efficient route for on-site and small-scale production of hydrogen peroxide (H2O2) compared to the conventional anthraquinone oxidation process. Although heteroatom doped carbon materials are widely studied for the 2e− ORR, few studies have been conducted on the synergistic relationship between multiple doping elements on H2O2 performance. Moreover, there is a lack of efficient and stable catalysts for H2O2 electrosynthesis in neutral media. In this work, a series of boron and nitrogen co-doped carbon (BNC) catalysts are synthesized and assessed for the 2e− ORR in neutral pH electrolyte. Through X-ray photoelectron and absorption spectroscopy analyses, we characterize B–N–C moieties and correlate them with H2O2 production performance. In rotating ring disk electrode measurements, BNC catalysts pyrolyzed at 900 °C exhibit H2O2 Faradaic efficiency of ∼70% and demonstrate excellent long-term stability over 10 hours of continuous operation. In a three-electrode flow cell, this catalyst achieves ∼78% H2O2 Faradaic efficiency and 0.9 mmol cm−2 h−1 productivity at 0.20 V vs. RHE, surpassing most previously reported carbon-based catalysts in neutral media. This work contributes fundamental insights into the role of boron and nitrogen heteroatom dopants toward 2e− ORR and highlights the practical viability of these catalysts for real world applications.
 Linsey C. Seitz | Linsey Seitz is an Assistant Professor of Chemical and Biological Engineering at Northwestern University. Before joining Northwestern in 2018, she was a Helmholtz Postdoctoral Fellow at Karlsruhe Institute of Technology and completed her PhD at Stanford University. Prof. Seitz's research develops and applies diagnostic tools and experimental platforms at the interface of electrocatalysis and spectroscopy to investigate dynamic catalyst materials and reaction environments towards the sustainable production of fuels and chemicals. She is also passionate about outreach, teaching, and learning. Prof. Seitz has received the ACS Catalysis Early Career Award (2024) and the NSF CAREER Award (2022). |
1. Introduction
Hydrogen peroxide (H2O2) serves as an eco-friendly oxidant employed across diverse industries, including chemical synthesis, paper and pulp, semi-conductor, environmental remediation, and others.1–3 The global demand for H2O2 is rapidly growing, especially after the recent COVID-19 pandemic where it was used as a disinfectant in the healthcare industries.4,5 Current industrial production of H2O2 is achieved via the multi-step anthraquinone oxidation process, which is not only energy-intensive, but also environmentally hazardous, producing significant amounts of organic by-product waste.6 In addition, this process relies on a centralized infrastructure and requires expensive and hazardous transport and storage of concentrated H2O2 solutions.7 Thus, the development of a low-cost and decentralized H2O2 production method is highly desirable to reduce the costs and safety concerns associated with H2O2 synthesis, storage, and transportation.8At the forefront of a potential sustainable and efficient route to the on-site and small-scale production of H2O2 is the two-electron electrocatalytic oxygen reduction reaction (2e− ORR).9,10 At present, state-of-the-art 2e− ORR catalysts are based on noble metals and their alloys (e.g., Pd, Au, and Pt–Hg).8,11–14 However, such catalysts have considerable drawbacks, namely, scarcity and high cost, which impede their implementation on a large-scale. Recently, carbon-based materials emerged as promising alternatives for the 2e− ORR due to their relative abundance, facile preparation, low cost, and excellent stability.15–18 More importantly, their tunable surface and structural properties make it possible to modify the electrochemical performance of these carbon-based electrocatalysts.19 Among various modification methods, nitrogen doping has been reported to generate efficient active sites with favorable electrochemical properties.20,21 However, many of these studies with high selectivity toward 2e− ORR are performed in alkaline conditions, while much higher ORR overpotentials are reported in acidic or neutral pH conditions.20,22 Production of H2O2 in proton exchange membrane electrolyzers is preferable over hydroxide conducting membranes, primarily due to their superior energy efficiency, improved stability against H2O2, and greater scalability.23–25 Thus, it is desirable to develop carbon-based catalysts with improved H2O2 selectivity and activity in neutral or weakly acidic pH conditions.12,26,27
The introduction of boron dopants into nitrogen doped carbon frameworks is a broadly reported strategy to enhance the ORR activity vs. pure nitrogen doped carbons.28–36 Boron dopants have been hypothesized to serve as active sites or to impact the electronic properties of surrounding carbon atom active sites upon incorporation into the carbon matrix.29 Several studies have investigated boron and nitrogen co-doped carbon catalysts (BNC), employing a variety of synthetic approaches, including chemical vapor deposition and thermal annealing, in attempts to induce the most active and selective defect-site structures.37,38 However, despite significant progress in the development of BNC catalysts, many of these studies focused on the 4e− ORR pathway to H2O in alkaline media.36,39–41 There are very few reports on high-performance toward the 2e− ORR pathway to H2O2, especially in neutral pH media.42 Moreover, detailed information of the nature of the active B–N–C moieties and understanding their role in the origin of ORR activity and selectivity is lacking.
In the present work, we synthesize a series of BNC catalysts and study their activity and selectivity toward 2e− ORR to H2O2 in neutral pH solutions. We report a facile preparation method for the BNC catalysts and tune the content and nature of B and N dopants via varied pyrolysis temperatures. Spectroscopic characterizations confirm the presence of diverse B–N, B–N–C, and N–C dopant structures in the carbon matrix. The catalyst performance toward H2O2 activity and selectivity are investigated in three different reactor geometries, using a rotating ring disk electrode (RRDE), a gas diffusion electrode (GDE) flow cell, and a dual membrane electrode assembly (MEA) solid electrolyte (SE) flow cell. Benefiting from the high efficiency of active sites and abundant interfacial B–N–C moieties, BNC pyrolyzed at 900 °C (900-BNC) demonstrates the best performance in each of these reactor geometries. The 900-BNC catalyst reaches H2O2 selectivity and Faradaic efficiency up to 80% and 70%, respectively, in an RRDE geometry, and achieves 78% H2O2 Faradaic efficiency at a production rate of 0.9 mmol cm−2 h−1 in a GDE flow cell. While similar production rates have been achieved in strongly alkaline conditions, this production rate is ∼1.75 times greater compared to previously reported non-metal carbon-based electrocatalysts in similar reactors in neutral media.43 This work provides collective insights into the development of heteroatom-doped carbon catalyst materials for the electrosynthesis of H2O2 and enables the design of improved materials toward other applications, such as electrochemical capacitors44 and alkali metal batteries.45,46
2. Experimental section
2.1. Materials and chemicals
Zinc nitrate hexahydrate, 2-methylimidazole, potassium bicarbonate, phenylboronic acid, potassium sulfate, hydrogen peroxide (30 wt%), hydrochloric acid, isopropyl alcohol, and cobalt sulfate heptahydrate were purchased from Sigma Aldrich. All electrolytes and deionized (DI) water were prepared from Millipore-quality water (18.2 MΩ cm at 25 °C). All gases used (O2 and N2) in this work had a purity of >99.995% (Airgas).2.2. Synthesis of catalysts
The zeolitic imidazolate framework 8 (ZIF-8) precursor was prepared according to previous literature.47,48 Briefly, 1.47 g of Zn (NO3)2·6H2O was dissolved in 100 mL methanol; a second solution consisting of 3.24 g of 2-methylimidazole in 100 mL methanol was prepared in parallel. The Zn2+ solution was rapidly poured into the 2-methylimidazole solution, and after 3 hours of stirring at room temperature, ZIF-8 nanocrystals were recovered from the mother liquor by centrifuging at 8000 rpm for 15 minutes and washing three times with fresh methanol. The final product was then dried in an oven overnight at 80 °C in air.To prepare the BNC catalyst materials, 50 mg of the prepared ZIF-8 precursor was suspended in 2 mL of ethanol, followed by the addition of 5.4 mg phenylboronic acid. The resultant mixture was sonicated for 4 hours and subsequently dried in an oven overnight at 80 °C in air. These BNC pre-catalysts were then pyrolyzed at different temperatures ranging from 700 °C to 1100 °C for 3 hours under 100 s.c.c.m N2 with a heating rate of 10 °C min−1. Once cooled to room temperature, the resultant powders were acid treated in 5 mL 1 M HCl for 1 hour with sonication, centrifuged, washed several times with water, and dried overnight at 80 °C. The boron and nitrogen co-doped carbon catalysts are denoted as Y-BNC, where Y represents the pyrolysis temperature. For comparison, nitrogen doped carbon pyrolyzed at 900 °C (900-NC) was synthesized in a similar manner without addition of the boron precursor.
2.3. Material characterization
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) elemental mapping were performed on a Hitachi SU8030 with an Oxford Aztec X-max 80 SDD EDS detector at 10 and 20 keV, respectively. X-ray photoelectron spectroscopy (XPS) was conducted with a Thermo Scientific ESCALAB 250Xi with a monochromatic Al Ka source and 500 μm spot size. All XPS spectra were fitted using CasaXPS software and electron binding energy spectra were calibrated to adventitious carbon C 1s peak set to 284.8 eV. Raman spectra were collected on a laser confocal microscopy Raman with laser excitation at 532 nm.Powder X-ray diffraction (PXRD) was conducted using a Rigaku Ultima IV with Cu Kα radiation source (λ = 0.1540 nm). A scan rate of 5° min−1 was used to record the PXRD patterns for a 2θ range 10–90°. Inductively coupled plasma-mass spectrometry (ICP-MS) was conducted on a Thermo iCapQ ICP-MS (Thermo Fisher Scientific, Waltham, MA, USA) operating in KED mode and equipped with an ESI SC-2DX PrepFAST autosampler (Omaha, NE, USA). N2 adsorption–desorption curves were performed at liquid nitrogen temperature (77 K) on a Micromeritics 3Flex instrument. Brunauer–Emmett–Teller (BET) surface area was calculated based on the adsorption data (p/p0 = 0.05–0.25). Pore size distributions were calculated using non-local density functional theory assuming slit carbon pores. Electrode hydrophobicity was determined by the measurement of water contact angle which was performed using a contact angle goniometer (Ossila). Ex situ soft X-ray absorption spectroscopy (XAS) spectra at the K-edges of carbon, oxygen, nitrogen, and boron were collected at beamline 8–2 at Stanford Synchrotron Radiation Lightsource (SSRL). Total electron yield (TEY) data were acquired for samples in a single load at room temperature and under ultrahigh vacuum (10−9 torr).
2.4. Electrochemical RRDE measurements
The RRDE experiments were performed in a four-electrode configuration using a bipotentiostat (Biologic VSP) in a heart-shaped electrochemical cell equipped with a gas dispersion tube, a silver/silver chloride (Ag/AgCl sat'd KCl, Fisher) reference electrode, a Pt wire counter electrode, and two parallel working electrodes (a glassy carbon (GC) disk with deposited catalyst and a Pt ring, Pine Research Instrumentation, AFE7R9GCPT). The RRDE collection efficiency was determined experimentally to be ∼37% using a ferrocyanide/ferricyanide half reaction system.49 Catalyst ink for the GC disk working electrode was prepared by mixing 3.3 mg of catalyst, 1.00 mL of isopropyl alcohol (ThermoFisher Scientific) and 10 μL of cation-exchanged Nafion solution (5%, Sigma-Aldrich). Then, the resultant ink was sonicated for three hours and 7.6 μL of the catalyst ink was drop-casted onto a clean and polished GC disk substrate (0.246 cm2) to obtain a catalyst loading of 0.1 mg cm−2.Prior to the ORR measurements, the Pt ring electrode was cleaned by successive cyclic voltammetry (CV) in N2-saturated 0.5 M K2SO4 electrolyte within the potential range 0.05 to 1.2 V vs. RHE at 20 mV s−1 until steady CVs were obtained. Baseline electrochemical assessment of the GC disk/catalyst included measuring background (capacitive) current between 0.8 V to 1.0 V vs. RHE, followed by electrochemical impedance spectroscopy (EIS) at open circuit voltage to determine the series resistance (Rs) of the liquid electrolyte for the subsequent iR compensation. All potentials were controlled with respect to a Ag/AgCl reference electrode in saturated KCl and converted to an RHE scale using the expected redox potential and pH shift via E(V vs. RHE) = E(V vs. Ag/AgCl) + 0.197 (V) + 0.059 (V dec−1) × pH.
To assess catalytic activity, electrolyte was purged with O2 for 30 minutes and then linear sweep voltammetry (LSV) measurements were carried out between 1.1 to 0.05 V vs. RHE at 5 mV s−1 with an RRDE rotation speed of 1600 rpm. To obtain Faradaic currents on the disk electrode, the capacitive current was subtracted from the total measured current. The amount of H2O2 produced at the disk was quantified by holding the Pt ring at 1.2 V vs. RHE to oxidize the H2O2 species transported from the disk electrode via rotation. The molar selectivity of H2O2, which represents the ratio between O2 consumption rate toward H2O2 and total O2 consumption rate at the disk electrode, and the Faradaic efficiency of H2O2, which represents the electron efficiency towards driving 2e−vs. 4e− ORR, were calculated according to the following eqn (1) and (2):49
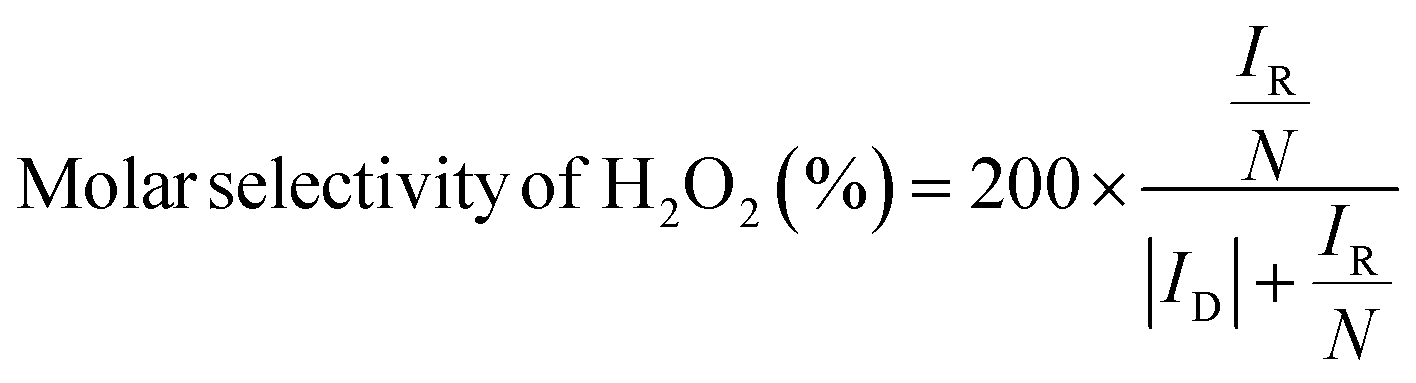 | (1) |
 | (2) |
Tafel slopes (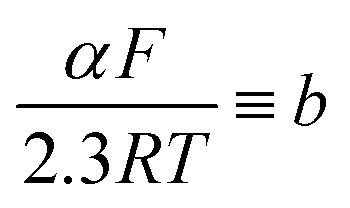 ) can be calculated using the Tafel equation (eqn (3)) obtained from the linear relationship between the overpotential (η) vs. log kinetic disk current density (jdisk,kin).
) can be calculated using the Tafel equation (eqn (3)) obtained from the linear relationship between the overpotential (η) vs. log kinetic disk current density (jdisk,kin).
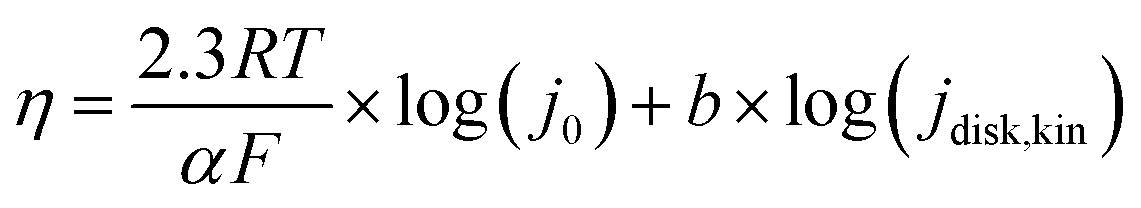 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), R is the ideal gas constant (8.314 J mol−1), T is temperature (298 K), and j0 is the exchange current densities. The kinetic disk current densities (jdisk,kin) can be determined according to the Koutecky–Levich equation (eqn (4)):50
485 C mol−1), R is the ideal gas constant (8.314 J mol−1), T is temperature (298 K), and j0 is the exchange current densities. The kinetic disk current densities (jdisk,kin) can be determined according to the Koutecky–Levich equation (eqn (4)):50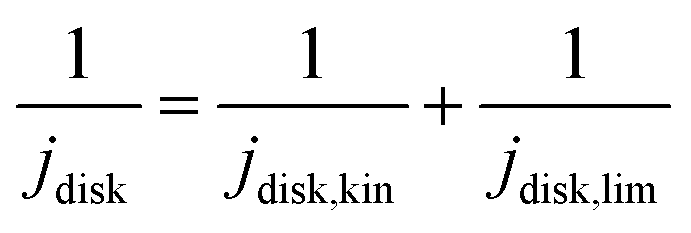 | (4) |
 | (5) |
485 C mol−1), C is the bulk concentration of oxygen (1.2 × 10−6 mol cm−3), is the diffusion coefficient of oxygen (1.9 × 10−5 cm2 s−1), ν is the kinetic viscosity (0.01 cm2 s−1), and ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed).21,52
2.5. Electrochemical GDE flow cell measurements
As shown in Fig. 4A and S1,† a custom-made three-electrode GDE flow cell was constructed to enhance reactant mass transport and mimic geometry of an applied reactor design that could be used for larger-scale electrochemical generation of H2O2. The catalyst ink for the working electrode was prepared by mixing 4.5 mg of catalyst with 900 μL isopropanol and 45 μL Nafion binder. The resulting catalyst ink was ultrasonicated for 1 hour to obtain a homogeneous dispersion of catalyst powder. Afterward, the catalyst ink was spray coated onto a 3 × 3 cm2 hydrophobic carbon cloth (Fuel Cell Store, W1S1011) resulting in a mass loading of ∼0.5 mg cm−2. After drying overnight at room temperature, the spray coated carbon cloth was cut into four 1.5 × 1.5 cm2 electrodes. The anode catalyst for the oxygen evolution reaction was an Ir-based electrocatalyst on Ti felt (platinized, fuel cell store) prepared by a dip coating and decomposition method.53 Ti felt was first etched in 0.5 M oxalic acid (anhydrous 98%, Fisher) at 80 °C for 1 hour. Next, the electrode was dipped into a solution containing 30 mg IrCl3·xH2O (TCI), 1 mL concentrated HCl, and 9 mL isopropyl alcohol. The as-prepared Ti felt was then dried at 100 °C for 5 minutes followed by calcination at 500 °C for 15 minutes in air. This process was repeated 5 times. A fresh piece of IrOx/Ti anode was used for each electrochemical experiment. The GDE flow cell was assembled with catalyst layers facing the flowing liquid electrolyte; the exposed geometric surface area of each catalyst was 1 cm2. The cathode and anode compartments were separated with a Nafion-115 proton exchange membrane (fuel cell store) and accommodated 1 mL electrolyte each. Prior to operation, the Nafion-115 membrane was pre-treated with 5% (v/v) H2O2 for 1 hour at 80 °C, followed by 10% (v/v) H2SO4 for 1 hour at 80 °C, then DI water for 1 hour at 80 °C. Membranes were stored in DI water. Both catholyte and anolyte liquid flow rates were set to 3 mL min−1 using a peristaltic pump (BT100L with dual YZ15 pump heads, Golander) from respective 10 mL reservoirs containing 0.5 M K2SO4. 20 s.c.c.m of O2 was supplied to the cathode side via a mass flow controller (MKS Instruments). ORR was conducted using a potentiostat (Biologic VMP3) with a Ag/AgCl reference electrode. All potentials were corrected to the RHE potential scale and manually compensated for iR to 100%. CVs were performed at a scan rate of 20 mV s−1 with a cell voltage range from 1.1 to 0.05 V vs. RHE. Experiments were carried out using chronoamperometry (CA) at 0.20 V vs. RHE to evaluate the H2O2 production rates by measuring H2O2 concentration after set reaction times (See Section 2.7).2.6. Electrochemical MEA flow cell measurements
H2O2 electrosynthesis was further evaluated using a two-electrode setup in a dual MEA SE flow cell with Ti flow fields (5 cm2 formic acid electrolyzer, Dioxide Materials), comprising a porous SE compartment sandwiched by a cation and an anion exchange membrane (CEM = Nafion-115 and AEM = Sustainion X37-50 (Grade RT), respectively) on the anode and cathode side, respectively. Outside of these membranes, the anode/cathode catalyst layers and flow plates are compressed (with the anode adjacent to the CEM and the cathode adjacent to the AEM). Polytetrafluoroethylene (PTFE) gaskets were used to seal the electrolyzer. The cell configuration is illustrated in Fig. S2.† The middle chamber SE material was chosen as a styrene-divinylbenzene sulfonated copolymer Amberchrom 50WX8 hydrogen form cation conductor (50–100 mesh, Sigma-Aldrich). Prior to use, the SE was washed in a beaker with 0.5 M K2SO4 several times until the pH reached approximately 5. In the SE chamber, Milli-Q water was flowed in a single pass at 0.5 mL min−1 to wash out H2O2 produced, while the anode side was circulated with 1 M H2SO4 at 3 mL min−1. The O2 flow rate to the cathode flow plate was supplied with 20 s.c.c.m. of humidified O2 controlled by a mass flow controller and water/electrolyte flow rates were controlled by a peristatic pump. A Biologic VMP3 potentiostat with 10 A booster was employed to record the electrochemical response. Two pieces of copper tape (3 M) were used to connect the electrolyzer to potentiostat leads. The electrolyzer was first cycled between open circuit potential and 2.5 V, then held at 2.5 V for 10 minutes for activation. Then, 40 minutes chronopotentiometry (CP) holds in increasing current densities from 10 to 200 mA cm−2 were applied to the electrolyzer. The product streams of the first 10 minutes of each CP hold were discarded, and product streams from the next 30 minutes were collected to examine the H2O2 concentration produced.2.7. H2O2 product quantification
The produced H2O2 was quantified using a cobalt-carbonate (Co/CO3) assay coupled with UV-vis spectrophotometry (Agilent Cary 5000).54 The Co/CO3 assay contains a mixture of KHCO3 and cobalt sulfate heptahydrate (CoSO4·7H2O) both of which dissociate in water and exhibit a light pink color solution. H2O2 oxidizes Co2+ in the presence of bicarbonate likely forming a ‘carbonatocobaltate’ complex (Co(CO3)33−) which strongly absorbs at 290 nm and weakly in the visible region (440 and 635 nm), exhibiting a dark green color. The measured absorbance of the Co(CO3)33− complex is linearly proportional to the concentration of H2O2 present; the 290 nm feature was used for quantification unless otherwise specified. The Co/CO3 assay was prepared by first mixing 1.35 mL of 2 M KHCO3 and 35.8 μL of 70 mM CoSO4·7H2O in a cuvette. Next, 35.8 μL of our H2O2 sample (known or unknown concentration) was added to the cuvette and mixed. Immediately afterward, 1.425 mL of 2 M KHCO3 was added to the cuvette and mixed. The absorbance of these samples was measured in the wavelength range 200–800 nm with 1 nm resolution and scanning rate of 2 nm s−1. The UV-vis spectra were background-subtracted using a baseline with 35.8 μL of H2O added instead of H2O2-containing samples. A typical calibration curve was plotted by linear fitting the absorbance values at wavelength of 290 nm for various known concentrations of H2O2 from 20 ppm to 750 ppm (Fig. S3†). The Faradaic efficiency for H2O2 production is calculated using the following equation: | (6) |
3. Results and discussion
3.1. Structural characterization
ZIF-8 was chosen as the precursor for the catalyst preparation due to their superior thermal and chemical stability compared to other metal–organic-frameworks (MOFs). During the carbonization process, ZIF-8 decomposes to form microporous carbon frameworks, which provide abundant active sites and high surface areas for electrocatalytic reactions. Furthermore, the high nitrogen content in the ZIF-8 precursor results in incorporation of nitrogen into the carbon framework during the carbonization process, making ZIF-8 attractive as both the carbon support and nitrogen precursor.55 As illustrated in Fig. 1A, a two-step method to prepare the BNC catalysts was adopted. Briefly, phenylboronic acid, the chosen boron source, was mixed with ZIF-8 in ethanol overnight, followed by pyrolysis under N2 for 3 hours, then acid washed to remove any residual Zn from the ZIF-8 support. Remaining Zn in the structure forms Zn(CN)2 during calcination, which further decomposes and vaporizes at temperatures above 800 °C.56 Further acid treatment would deactivate Zn towards ORR, as Zn has not been shown to be an effective ORR catalyst. Due to the different formation energies of the B–N, B–C and N–C species, the B–N–C configurations can be tuned by adjusting the pyrolysis temperature. To systematically study the effects of pyrolysis temperature and identify the optimal conditions to support H2O2 production, catalyst materials were carbonized at temperatures ranging from 700 °C to 1100 °C. For comparison, nitrogen doped carbon pyrolyzed at 900 °C (900-NC) was prepared in a similar manner without the addition of the boron precursor.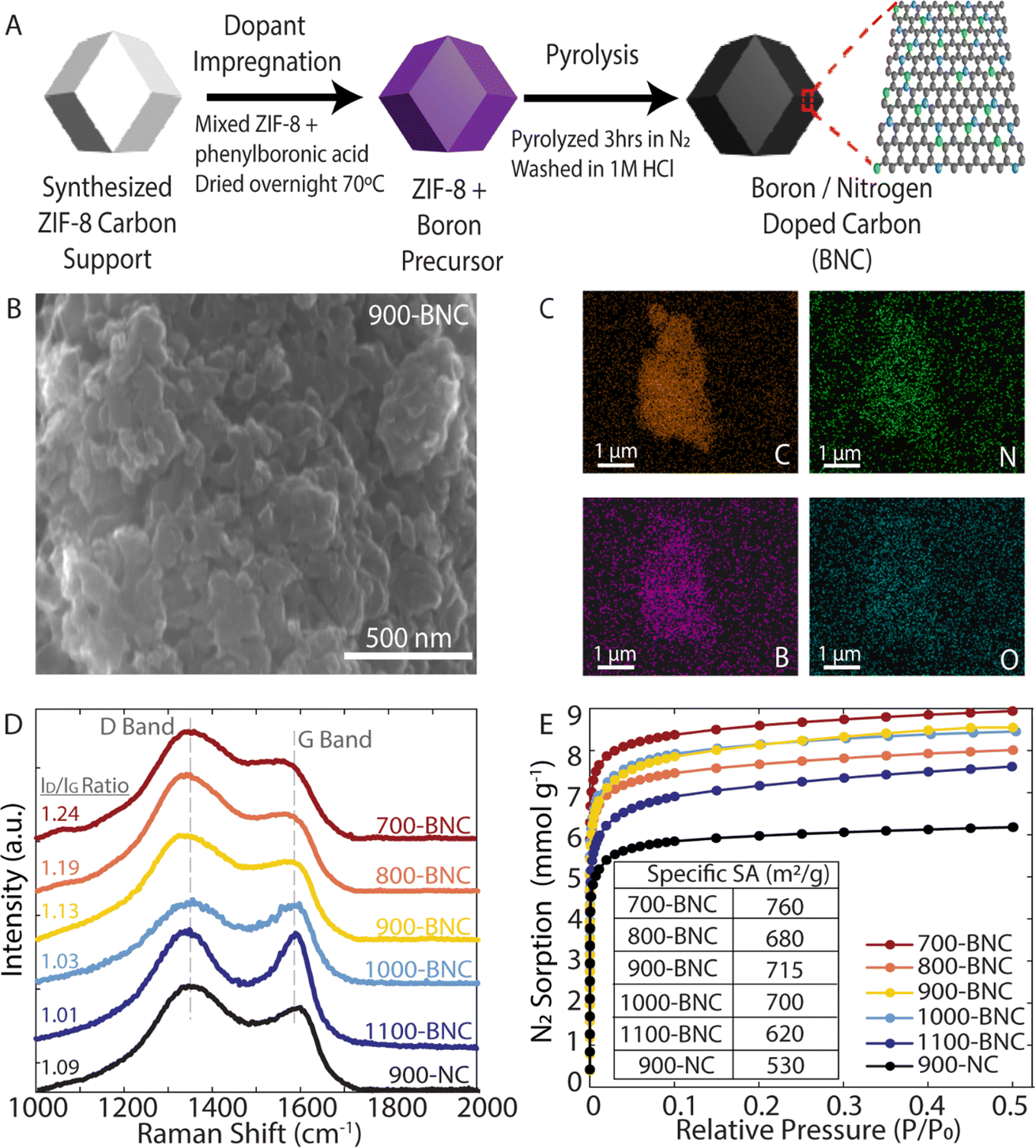 | ||
| Fig. 1 Synthesis and characterization of the catalysts. (A) Schematic illustration of the BNC catalyst synthesis. (B) SEM image of the 900-BNC catalyst and (C) corresponding EDS mapping images. (D) Raman spectra. (E) N2 adsorption/desorption isotherms including the corresponding calculated specific surface areas. | ||
To characterize the morphology of the BNC catalyst materials, SEM was employed, as shown in Fig. 1B for 900-BNC and in Fig. S4† for all other catalysts. SEM images for all the catalysts show similar nanoparticulate morphologies and highly porous structures which are favorable for facilitating mass transport of reactants and products. The EDS elemental mapping of the as-prepared catalyst materials (Fig. 1C and S5†) exhibits uniform distribution of C, N, B, and O elements, validating the successful doping of B and N atoms into the carbon framework.
Raman spectra were also acquired to examine the degree of graphitization, the defect content, and extent of disordered structures. As shown in Fig. 1D, all materials exhibit typical D band (1350 cm−1) and G band (1580 cm−1) peaks; the D band is related to disordered graphitic structures and the G band reflects the degree of graphitization and presence of sp2-hybridized carbon.57 Therefore, the extent of defect formation in the BNC catalysts can be quantified by using the intensity ratio of the D and G bands (ID/IG). The ID/IG ratio (Table S1†) decreases with increasing pyrolysis temperature, indicating an improved degree of graphitization. Comparing 900-NC and 900-BNC catalysts, the intensity ratio or D band increases with the addition of boron, which we attribute to the creation of additional defects in the carbon matrix. The structural properties of the samples were further characterized by PXRD. As displayed in Fig. S6,† all samples show two broad diffraction peaks at 25° and 44°, corresponding to the (002) and (100) lattice planes, respectively.58,59 This indicates that the structures are mainly amorphous, which is typical for nanoporous carbon materials, also supported by the absence of peaks from crystalline hexagonal boron nitride (h-BN) in the as-prepared samples.60 We further observe the (002) and (100) peaks becoming more prominent and less broad with increasing pyrolysis temperatures, indicating a higher degree of graphitization. Prior studies have reported that the graphitic carbon moieties formed during pyrolysis increase the conductivity of carbon materials (i.e., charge transport),61 which may lead to a boost in ORR performance.
Nitrogen adsorption–desorption isotherms were measured to determine specific surface area and pore size characteristics. As displayed in Fig. 1E, all BNC catalysts display higher BET surface areas than 900-NC, indicating that the introduction of boron is beneficial to the enhancement of surface area. However, we find that BET surface area and total pore volume generally decrease with increasing pyrolysis temperature from 700-BNC (760 m2 g−1; 0.31 cm3 g−1) to 1100-BNC (620 m2 g−1; 0.26 cm3 g−1) consistent with the carbon structures becoming more graphitic, and in agreement with our Raman and XRD results (Fig. 1D and Table S2†). All catalysts showed typical type I isotherms indicative of a characteristic micropore morphology, similar to other previously reported ZIF-8 derived carbon materials.62 These results match the patterns of pore size distributions in Fig. S7† where the dominant pore size is between ca. 1–1.5 nm.63,64 Combining this characterization and analysis, it can be concluded that the incorporation of boron is favorable for enhancing the surface area and inducing formation of a highly defective structure, while tuning pyrolysis temperature allows for more fine tuning of the graphitic carbon structure, thereby providing advantages for further catalytic applications.
X-ray photoelectron spectroscopy (XPS) was conducted to examine the electronic structure and chemical composition of the BNC catalysts and 900-NC. As shown in Table 1 and Fig. S8,† all BNC materials primarily consist of C, N, O, and B elements at their surface, with residual amounts of Zn remaining from the ZIF-8 precursor. Although pyrolysis was performed under a N2 gas environment, oxygen content is observed in the BNC materials, likely originating from either oxidation of NC support after being in contact with air or residual phenylboronic acid form the B precursor.65 Continuous loss of N content was observed with increasing pyrolysis temperature from 25.2% for 700-BNC followed by 21.7%, 13.8%, 6.3%, and 4.7% for 800-BNC, 900-BNC, 1000-BNC, and 1100-BNC, respectively. This is due to the nitrogen in the ZIF-8 being removed via the N2 gas flow when the framework is destroyed at elevated temperatures. We note that the N contents for these materials are higher than that of typically reported N doped carbon materials under the same temperature conditions.66 Based on previous reports, this likely arises due to the synergistic effect of co-doping with boron and nitrogen, where boron dopants alter the electronic properties of the carbon structure, thereby promoting additional nitrogen incorporation.39 On the other hand, C concentrations trend closely with increasing pyrolysis temperature from 64.6% to 85.6% for 700-BNC and 1100-BNC, respectively. For boron composition, the total B content generally increases from low to high pyrolysis temperature, although 900-BNC uniquely displays the highest atomic percent (1.78%). We postulate the decrease in B content above 900 °C may be due to structural changes (e.g., fewer defects) or surface chemical changes (e.g., lower N content) at elevated temperatures.
| Sample | C (at%) | O (at%) | Zn (at%) | N (at%) | B (at%) |
|---|---|---|---|---|---|
| 700-BNC | 64.6 | 6.9 | 2.6 | 25.2 | 0.71 |
| 800-BNC | 68.6 | 5.4 | 3.2 | 21.7 | 0.98 |
| 900-BNC | 75.4 | 7.5 | 1.6 | 13.8 | 1.78 |
| 1000-BNC | 85.0 | 7.3 | 0.4 | 6.3 | 1.01 |
| 1100-BNC | 85.6 | 7.9 | 0.2 | 4.7 | 1.63 |
| 900-NC | 80.9 | 5.6 | 1.8 | 11.7 | — |
Different bonding configurations can be formed in the carbon framework by tuning pyrolysis temperature and incorporating boron atoms. As shown in Fig. 2A, the high-resolution XPS B 1s spectra can be deconvoluted into three peaks at 190 eV, 191 eV and 192 eV assigned to B–N–C (e.g., BN2C, BNC2), B–N, and B–O bond structures, respectively.67–70 We note that the FWHM peak widths for the B 1s spectra range between 1.9 and 2.5 eV, which are significantly greater than h-BN FWHM value of 1.5 eV. This broadening may be due to the superposition of multiple peaks in the spectra, in agreement with previous reports.71 No significant features are observed between 187–188.7 eV; previous work has assigned peaks in this region to B–C bonding similar to boron carbide (B4C).72 At lower pyrolysis temperatures, the boron atoms present are predominately bonded with oxygen (B–O), consistent with the O 1s XPS spectra (Fig. S11B†). It is noteworthy that the proportion of B–N–C and B–N configurations, previously correlated with improved 2e− ORR performance,32,42 gradually increases with increasing pyrolysis temperature, while B–O slightly decreases (Table S4†). More specifically, for the 900-BNC catalyst, the B–N, B–N–C, and B–O configurations account for approximately 44%, 29%, and 27%, respectively. Interestingly, the sum of the B–N and B–N–C in 900-BNC accounts for ∼73% of the total boron configurations and is the highest among the catalysts studied.
 | ||
| Fig. 2 Composition investigation of the catalysts. (A) High-resolution B 1s and (B) N 1s XPS spectra. (C) Boron K–edge and (D) nitrogen K-edge X-ray absorption spectroscopy (XAS). | ||
Similarly, the N 1s spectra were deconvoluted and peaks were identified at 397.7 eV, 398.5 eV, 399.5 eV, 400.7 eV, and 403 eV, which are assigned to N–B, pyridinic-N (N6), pyrrolic-N (N5), graphitic-N (NQ), and oxide-N (NO), respectively (Fig. 2B).32,34,73,74 We acknowledge the exact chemical state of the N–B species cannot be clearly defined due to difficulty in deconvoluting the spectra. Based on previous reports, the N–B species likely comprise a B–N–C type structure or a B–N–B moiety similar to h-BN (Fig. S9†).68 While the surface boron atomic percent and the ratio of total B to total N are both small (such that the absolute content of N–B moieties is small), 900-BNC exhibits the highest relative N–B content (7.5%), followed by 1000-BNC (6.1%), 1100-BNC (5.6%), 800-BNC (4.8%), and 700-BNC (4.0%). In addition, we observe in Fig. S10 and Table S5† that, as total nitrogen content decreases with increasing pyrolysis temperature, the relative amount of pyridinic-N decreases whereas the graphitic-N increases, since the stability of graphitic-N is superior to that of pyridinic-N at higher carbonization temperatures.75 Prior studies have shown that a higher ratio of graphitic-N and N–B species is correlated with improved H2O2 selectivity, suggesting these species may act as active sites for the ORR catalysis.21,39
We also deconvolute the high-resolution XPS C 1s spectra, but it is difficult to discern between different types of C in these materials because C species such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–OH, CO, and OC–OH exhibit similar chemical shifts.16,43 For the purpose of simplifying the analysis, the C 1s spectra are divided into four different peaks centered at 283.9 eV, 284.5 eV, 286 eV, and 288 eV, corresponding to C–B, sp2 carbon (CC), C–N, and CO, respectively (Fig. S11A and Table S6†).43,74 Based on this fitting, we observe that the percentage of CC bonds increases from ∼38% to ∼49% with the rise in pyrolysis temperature, which is consistent with the increase in sp2 hybridized carbon observed from Raman. Additionally, the contribution of C–B increases gradually with increasing pyrolysis temperature, while the C–N appears to decrease. On the other hand, CO does not appear to trend, consistent with the O 1s spectra in Fig. S11B.† These XPS results demonstrate that by tuning the pyrolysis temperature, one can control the formation of different N–B, B–N–C, and N–C bonds, and therefore, the nature and content of the B–N–C moieties.76
C, C–C, C–OH, CO, and OC–OH exhibit similar chemical shifts.16,43 For the purpose of simplifying the analysis, the C 1s spectra are divided into four different peaks centered at 283.9 eV, 284.5 eV, 286 eV, and 288 eV, corresponding to C–B, sp2 carbon (CC), C–N, and CO, respectively (Fig. S11A and Table S6†).43,74 Based on this fitting, we observe that the percentage of CC bonds increases from ∼38% to ∼49% with the rise in pyrolysis temperature, which is consistent with the increase in sp2 hybridized carbon observed from Raman. Additionally, the contribution of C–B increases gradually with increasing pyrolysis temperature, while the C–N appears to decrease. On the other hand, CO does not appear to trend, consistent with the O 1s spectra in Fig. S11B.† These XPS results demonstrate that by tuning the pyrolysis temperature, one can control the formation of different N–B, B–N–C, and N–C bonds, and therefore, the nature and content of the B–N–C moieties.76
To provide a more in-depth understanding of the local geometric structure of the B–N–C moieties in the as-prepared carbon catalysts, soft X-ray absorption spectroscopy (XAS) measurements were performed at the respective elemental K edges. For comparison, h-BN was included as a reference material to provide insight on the types of dopant bond motifs. As shown in Fig. 2C, the B K-edge spectra of h-BN exhibits the typical intense peak at 192.3 eV, with two smaller peaks at 193.0 eV and 193.6 eV. The peak at 192.3 eV is a characteristic B 1s → π* transition in h-BN, reflecting sp2 hybridization of boron trigonally coordinated with nitrogen (BN3). The BNC materials pyrolyzed above 700 °C exhibit three peaks at these same energy positions as h-BN; however, the relative intensities are much more even, and the total intensity of these features increases with pyrolysis temperature, coinciding with a decrease in total N content (Table 1). As discussed previously, a more complete carbonization process at higher temperatures generates additional sp2 hybridized carbon and the boron incorporates into the carbon matrix forming a trigonally coordinated boron. Similarly to h-BN, these features in the BNC catalyst spectra are attributed to the π* transitions of B–N bonds as in h-BN (e.g., BN3), but also include signal from trigonal BN2C and BNC2 moieties.69,77,78 At slightly higher energy, the BNC materials display a sharp peak at 194.4 eV that is absent in h-BN, corresponding to B–O bonding consistent with reports of bulk B2O3. However, the absence of the σ* feature at ∼202 eV observed in B2O materials suggests the formation of a mixture of BO3 species that are distinct from bulk B2O3 on the surface of the BNC catalysts.79 At even higher energies, we observe several peaks in the σ* region between 199–202 eV for BNC materials pyrolyzed at lower temperatures (700–900 °C) which are ascribed to the central boron atom binding to species in the carbon matrix forming sp3 hybridization with a tetrahedral structure. Moreover, the broad peak around 203 eV represents contributions from both trigonal and tetrahedral boron, in agreement with previous reports.80 These features suggest that, at lower carbonization temperatures, the BO3 species and its derivatives are highly embedded in the carbon matrix and likely bind with additional species (e.g., N, C) forming a tetrahedral coordination. Furthermore, the peaks located at 199–202 eV and 203 eV in the σ* region decrease in intensity indicating less boron sp3 hybridization with increasing pyrolysis temperature. Consistent with the B 1s XPS results, no features assigned to boron carbide are observed; when present in other reported materials, these boron carbide species are characterized by a 1s → π* transition at lower photon energy (∼190.9 eV).67 The lack of a feature in this energy region indicates a low concentration or simply undetectable distinct B–C bonds in the BNC catalyst materials.
Further insights were obtained from the N K-edge XAS spectra of the BNC catalysts. As shown in Fig. 2D, h-BN displays a strong π* resonance at 400.3 eV and two features at 407 eV and 414.5 eV in the σ* region, respectively, validating typical h-BN domains with sp2 hybridized N–B bonding.32 As noted previously from the XPS results, the B: N ratio is quite small in the BNC materials, such that B–N moieties contribute a significant signal to B K XAS spectra, but do not necessarily contribute a significant signal to N K XAS spectra, relative to intensity of other N-containing species (especially N–C). For 900-NC and all the BNC catalyst materials, we observe three distinct peaks in the π* region between 397–402.5 eV. Compared to the h-BN feature at 400.3 eV, these π* transition features are shifted to lower photon energy indicating formation of an N bonding structure that is weaker than N–B in a h-BN moiety. The two lower energy peaks at 398.4 eV and 399.5 eV are in close agreement with the π* transitions from pyridinic-N and pyrrolic-N, respectively.81,82 The feature at ∼400 eV is present in both the 900-NC catalyst and h-BN; while this feature likely represents another N–C species for the 900-NC sample (such as graphitic-N), this may represent B–N–C type bonding (BN2C or BNC2) for the BNC materials.32,42,83,84 Previous computational simulations suggest that a decreased BN domain size in carbon could also lead π* peaks to shift to a lower energy, which is attributed to the increased ratio of interfacial N–B–C species compared to that of N–B in h-BN.84 Focusing on the higher energy features, we observe shifts in energy position and broadening of the σ* resonances (which becomes even more pronounced at higher pyrolysis temperatures) for the BNC materials vs. h-BN; these differences are representative and have been identified as a “fingerprint” of N–B–C bonding.77,85 Therefore, based on these XAS results, it is evident that the N–B–C coordination structure (e.g. N2BC, NBC2) becomes more dominant with increasing pyrolysis temperature resulting in weaker N–C structure.
In the C K-edge XAS spectra, we observe a characteristic π* resonance from sp2-like carbon (CC) at 285.4 eV and σ* resonance centered around 293 eV (Fig. S12†).69,86,87 The intensity of these features increase with carbonization temperature demonstrating an enhanced graphitic network at elevated temperatures, in agreement with Raman and XPS results. In addition, the features at 287.8 and 288.8 eV are consistent with C atoms that are attached to O containing groups, such as C–O bonds or carbonyls (CO).88 Interestingly, the lack of the C–B feature in the lower photon energy region, as observed in the XPS results, suggests that B is most likely to be in the form of B–N or B–N–C moieties. The feature at 287 eV is consistent with C–N bonds,89,90 which appears to decrease for catalysts pyrolyzed at higher temperatures, as the C is more likely to graphitize and lose N content.
3.2. Electrocatalytic ORR performance
The ORR electrocatalytic activity and selectivity of the BNC catalysts were first evaluated using a three-electrode RRDE in O2-saturated 0.5 M K2SO4 (pH = 7). Fig. 3A shows linear sweep voltammogram (LSV) curves for the disk with thes associated ring currents, corresponding to the total ORR current (i.e., both H2O2 and H2O production) and H2O2 production current, respectively. The disk onset potential (defined in this study as the potential when disk current density reaches −0.20 mA cm−2) shifts to less cathodic potentials (lower overpotentials) with increasing pyrolysis temperature such that 1100-BNC demonstrates the least cathodic onset potential (0.42 V vs. RHE) and 700-BNC demonstrates the most cathodic onset potential (0.17 V vs. RHE). Additionally, 1100-BNC shows the highest total disk current density (−1.55 mA cm−2) at 0.1 V vs. RHE, followed by 1000-BNC (−1.47 mA cm−2), 900-BNC (−1.41 mA cm−2), 800-BNC (−0.96 mA cm−2), and 700-BNC (−0.36 mA cm−2), indicating overall ORR activity improves with increasing pyrolysis temperature. We postulate that this improvement in overall electrocatalytic activity is due to the higher degree of disordered graphitic structure (Fig. S19A†) and an increase in the B:N ratio (Fig. S19B†), which allows for more facile electron transfer and enhances the accessibility of active sites.61
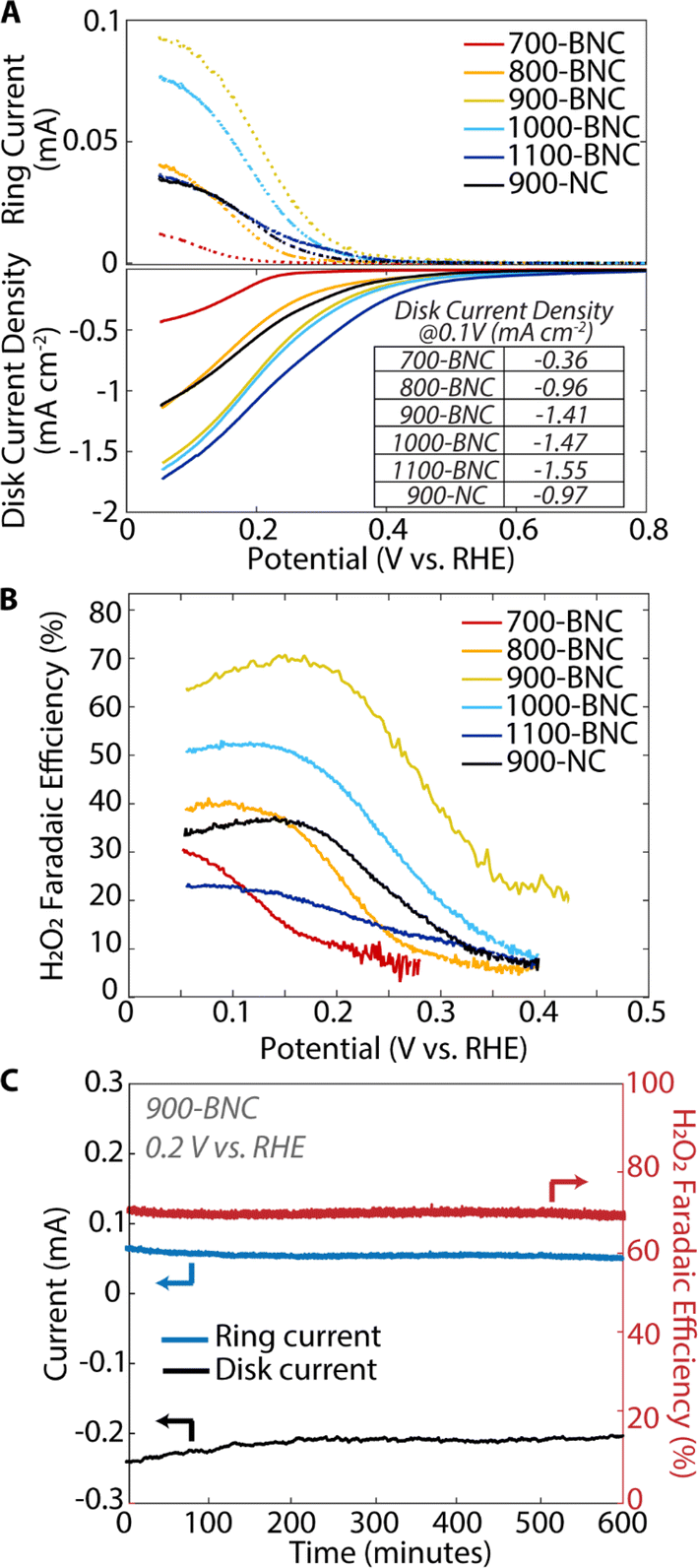 | ||
| Fig. 3 ORR performance using a RRDE of the catalysts in neutral pH solutions. (A) Linear sweep voltammetry (LSV) curves at 1600 rpm for the disk electrode (solid lines) and simultaneous H2O2 detection currents at the ring electrode (dashed lines) in 0.5 M K2SO4. (B) Calculated H2O2 Faradaic efficiency (%) over the applied potential range derived from RRDE data. (C) Chronoamperometric responses of 900-BNC at 0.20 V vs. RHE for 10 hours. | ||
With respect to the selective 2e− ORR activity, or the onset of the ring current density, a different activity trend is observed with 900-BNC demonstrating the least cathodic onset potential, followed by 1000-BNC, 1100-BNC, 900-NC, 800-BNC, and 700-BNC which all demonstrated higher overpotentials for onset. To understand these trends, we first examined the accessibility of active sites for these BNC materials. We note that although BET surface area generally decreased at elevated temperatures, these surface areas may not accurately reflect the accessible active sites in an electrocatalytic reaction. Moreover, the pore size and volumes of microporous and mesoporous structures show no correlation with H2O2 electrosynthesis performance (Fig. S20†). Thus, we also estimated the electrochemically active surface area (ECSA) by measuring the electrochemical double-layer capacitance of all catalysts in N2-saturated electrolyte (Fig. S13–18†). As displayed in Fig. S19C,† the estimated ECSA of BNC catalysts follows the order 900-BNC > 1100-BNC > 1000-BNC > 900-NC > 800-BNC > 700-BNC; this surface area trend is similar to the order of selective 2e− ORR activity, suggesting that a high ECSA plays an important role in improving H2O2 performance. Interestingly, 700-BNC shows a similar BET surface area as the 900-BNC but displays a much lower ECSA. Based on prior studies, we found this poor charge transfer for 700-BNC is related to the high N content and lower degree of graphitization.91 Additionally, despite a higher ECSA value, 1100-BNC shows inferior activity compared with 1000-BNC, suggesting that ECSA is not the only decisive factor accounting for 2e− ORR activity.
The calculated H2O2 Faradaic efficiencies (Fig. 3B) and molar selectivities (Fig. S22†) reveal a slightly different behavior for 2e− ORR performance. Notably, the 900-BNC catalyst delivers the highest H2O2 Faradaic efficiency and molar selectivity of these catalysts, at 70% and 80%, respectively, demonstrated at 0.20 V vs. RHE. The other BNC catalysts demonstrate H2O2 Faradaic efficiency between 20–50% at the same potential with 700-BNC and 1100-BNC exhibiting the overall lowest 2e− ORR performance. Moreover, 900-BNC also demonstrated the highest partial kinetic current density at 0.20 V vs. RHE of 0.82 mA cm−2 (Table S7†). In addition to activity and selectivity, the electrochemical stability of the 900-BNC catalyst was evaluated using chronoamperometry (Fig. 3C). After 10 hours of continuous electabkletrocatalytic H2O2 production (constant voltage of 0.20 V vs. RHE), 900-BNC retained ∼90% of its initial activity demonstrating excellent overall stability under these reaction conditions. In a solid membrane electrode assembly electrolyzer, 900-BNC catalyst maintained its activity over 50 h of operation, producing a pure H2O2 stream with average H2O2 concentration above 300 mM (11000 ppm) (Fig. S28†). Overall, the 900-BNC catalyst demonstrates a superior 2e− ORR performance in neutral pH compared to other reported carbon-based catalysts (Table S9†), exhibiting a relatively high H2O2 activity, high selectivity, and excellent stability.
We postulate that the differences in ORR activity and selectivity observed for the BNC catalyst materials may be attributed to the synergistic effects of the chemical bonding between B and N dopants in the carbon matrix. From combined XAS and XPS analyses, we found that by changing the pyrolysis temperature, the relative formation of B–N, N–C and B–N–C dopant structures can be tuned. With increasing pyrolysis temperature from 700–900 °C, there is an energetic driving force for the formation of local BNC dopant structures, consisting of sp3 hybridized boron and nitrogen atoms in the carbon matrix (e.g., BN2C or BNC2) with tetrahedral coordination. Previous work investigating the ORR energetics using density functional theory for boron-carbon nitride materials demonstrated that the B–N–C dopant structures, namely the BNC2 configuration, exhibits an optimal charge state of B sites and OOH* adsorption energy, thus favoring H2O2 production.92 In contrast, samples obtained at higher pyrolysis temperatures above 1000 °C, form distinct B–N and graphitic-N bonds, similar to h-BN (e.g., BN3) and N-doped carbon, which comprise the dominant dopant bond motifs. The formation of these isolated B and N graphitic dopants likely leads to a reduction in H2O2 performance, thus shifting to the 4e− pathway to H2O. Therefore, our results indicate that the higher content of local B–N–C dopant configurations in the 900-BNC catalyst likely contributes to its enhanced electrocatalytic performance (Fig. S19D†).
Moreover, based on the previous N content analysis, we postulate that the moderate N doping of the 900-BNC catalyst is more favorable to the 2e− ORR (Fig. S19E†). Conversely, excessive N doping and lower degree of carbon graphitization, particularly in the 700-BNC and 800-BNC catalysts, may lead to lower ORR rates due to poor charge transfer and selectivity shift towards the 4e− pathway, as reported for other N-doped carbon materials.91 On the other hand, the very low N content of the 1100-BNC catalyst suggests a deficiency in catalytic ORR active sites, which is unfavorable for achieving a high catalytic current. Moreover, the specific N configurations may contribute to the improved H2O2 performance. Pyrrolic and pyridinic moieties were found to impede H2O2 formation, similar to previous reports (Fig. S19F and G†) while graphitic moieties improve H2O2 production (Fig. S19H†). Graphitic-N has been reported to be favorable for the 2e− pathway due to the lower energy barrier for O2 adsorption compared to pyridinic-N.20 Notably, pyrrolic-N is widely regarded to be less active for the 4e− ORR, and more recently, has been correlated with high activity for the 2e− ORR in alkaline conditions.88 In this work, the 900-BNC catalyst possesses primarily graphitic-N as well as the highest N–B content, further confirming the important role of B–N–C moieties in catalyzing the 2e− ORR to H2O2.
3.3. H2O2 electrosynthesis in flow cell
To investigate the as-prepared BNC catalysts for practical applications, we employed a three-electrode flow cell equipped with a gas diffusion electrode (GDE) in neutral pH electrolyte (Fig. 4A). A GDE cell design alleviates the intrinsic mass transport limitations by utilizing a hydrophobic gas diffusion layer (GDL, carbon cloth) to support formation of a solid–liquid–gas triple phase boundary with the catalyst, electrolyte, and gaseous O2 reactant, enabling improved mass transport.93–95 The bare GDE support (carbon cloth) exhibits a water contact angle of 135 ± 2°) which is critical to ensure efficient gas transport and prevent electrode flooding under reaction conditions.93,96,97 After BNC catalysts are deposited on the carbon paper, the electrodes exhibit water contact angles of 110 ± 2 to 125 ± 2° (Fig. S23†); the 900-BNC electrode presents moderately higher hydrophobicity than the other electrodes, which may be beneficial in enhancing the accessibility of O2 to the catalyst surface.98,99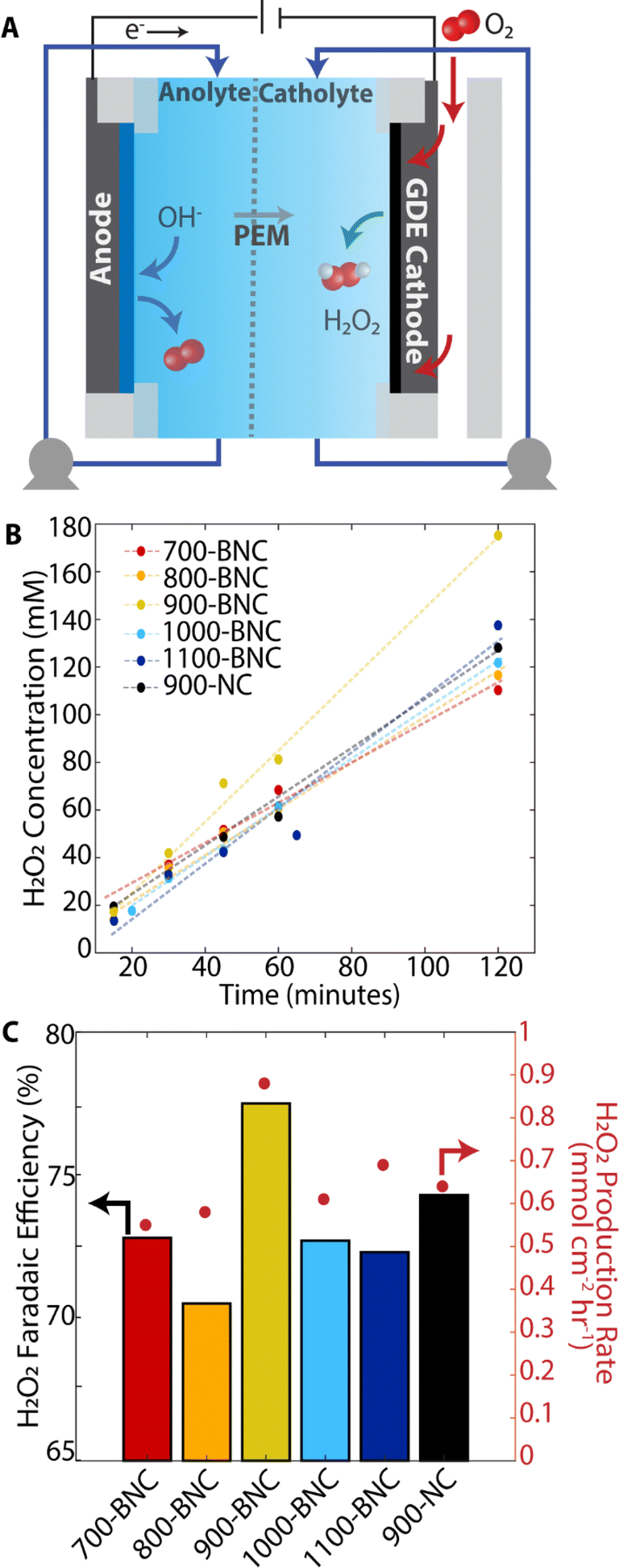 | ||
| Fig. 4 Practical application of H2O2 production in flow cell device. (A) Schematic of electrochemical flow cell set-up. (B) H2O2 concentration measured over time. (C) H2O2 Faradaic efficiencies (right axis) and H2O2 production rate (left axis) for different catalysts. All measurements performed at 0.20 V vs. RHE for 2 hours in 0.5 M K2SO4. | ||
To evaluate H2O2 electrosynthesis performance, we conducted chronoamperometry at 0.20 V vs. RHE for two hours in 0.5 M K2SO4 and measured the corresponding current response (Fig. S24†). In this set-up, the electrolyte and H2O2 product are continuously recycled such that the product concentration builds up over time (Fig. 4B). For all BNC catalyst materials, H2O2 concentration increases linearly with time. Remarkably, 900-BNC delivers superior performance reaching ∼180 mM H2O2 concentration, 78% H2O2 Faradaic efficiency, and 0.9 mmol cm−2 h−1 H2O2 production rate, outperforming most of the previously reported non-metal carbon-based catalysts in neutral pH (Table S10†). Following electrochemical testing, the catholyte composition was sampled via ICP-MS to examine the catalyst stability. As displayed in Table S8,† very little B content dissolved in the catholyte after the two-hour measurement, demonstrating the stability of BNC materials under these ORR conditions. The H2O2 production performance of 900-BNC was further evaluated via chronopotentiometric measurements, by applying constant current densities ranging from −10 to −100 mA cm−2 for 20 minutes (Fig. S25A†). H2O2 production rates (Fig. S25B†) and H2O2 concentrations (Fig. S25C†) increased with increasing applied current densities reaching 40 mM H2O2 and 1.6 mmol cm−2 h−1 production rate at −100 mA cm−2. Notably, this high current ORR process exhibits a H2O2 Faradaic efficiency of 85% (Fig. S23B†), further verifying the excellent performance for practical H2O2 electrosynthesis.
The stability of 900-BNC was further examined under a constant current density of 100 mA cm−2 for 10 hours (Fig. S26†). The measured H2O2 concentrations over this time suggest a slight decrease in H2O2 Faradaic efficiency (Fig. S26A†) and decline in H2O2 production rate (Fig. S26B†) from ∼1.6 to ∼1.3 mmol cm−2 h−1. However, this could result from a reduction in catalytic performance or from self-decomposition of H2O2 into H2O and O2, due to the high H2O2 concentrations. Interestingly, we also find that the final H2O2 solution pH is alkaline (∼pH 10), which may exacerbate the self-decomposition of H2O2 leading to a reduction in measured H2O2 concentrations.100 Despite the possible self-decomposition, the H2O2 produced at this current density reaches a concentration of 1290 mM (4.39 wt%), which is more than sufficient to be used for disinfection or wastewater treatment (<1 wt% typically used for these applications).101
One potential drawback of this three-electrode GDE flow cell is that the H2O2 generated is mixed with an electrolyte, which would require additional separation steps to enable further use. To avoid the extra cost and energy of such separation steps, H2O2 electrosynthesis can be performed in a dual membrane electrode assembly SE electrolyzer where pure H2O2 solutions can be obtained as illustrated in Fig. S3.† CP measurements were performed from 10 mA to 200 mA cm2 for 30 minutes each using the 900-BNC catalyst, as shown in Fig. S27.† At 200 mA cm2, H2O2 concentration reaches 600 mM (∼2 wt%), with a corresponding 72% H2O2 Faradaic efficiency and 2.7 mmol cm−2 h−1 production rate, further demonstrating the practical applicability of this H2O2 electrosynthesis technology.
4. Conclusions
In summary, we successfully synthesized a boron and nitrogen co-doped carbon catalyst that achieves high activity and selectivity for H2O2 production under neutral pH conditions. By tuning the pyrolysis temperature, we controlled the structural and electronic properties, chemical species, and bonding motifs of the BNC materials. Among the catalysts, 900-BNC demonstrated the highest H2O2 performance with 78% Faradaic efficiency and 0.9 mmol cm−2 h−1 production rate in a three electrode GDE flow cell as well as 72% H2O2 Faradaic efficiency and 2.7 mmol cm−2 h−1 production rate in a dual MEA SE flow cell. We attribute the superior performance of 900-BNC in part to its large electrochemically active surface area, higher degree of graphitization, and enhanced hydrophobicity. Moreover, combined XAS and XPS reveals a key factor to the enhanced H2O2 performance for 900-BNC may be the formation of a relatively high content of BNC moieties in the carbon matrix in the form of interfacial B–N–C bonds. An improved understanding of these catalyst material properties in neutral pH electrolyte can provide insights into the design and optimization of electrochemical systems for various applications, including energy conversion, water treatment, and chemical synthesis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
BR contributed to the conceptualization, data curation, formal analysis, investigation, methodology, and writing of the original draft of this work. XL contributed to the collection of XRD experiment, design of electrochemical flow cell experiments, analysis, and review and editing of this work. BL contributed to materials synthesis and review and editing of this work. AD contributed to the design of electrochemical flow cell experiments and review and editing of this work. DN supported soft XAS experiments and contributed to data analysis. JMN contributed valuable insights to project conception and experimental planning. LCS contributed to the conceptualization, methodology, resources, supervision, and review and editing of this work. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by INVISTA. This work made use of the Jerome B. Cohen X-Ray Diffraction Facility supported by the MRSEC program of the National Science Foundation (DMR-1720139) at the Materials Research Center of Northwestern University and the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). REACT acknowledged funding from Northwestern University Office of Research for purchase of the Micromeritics 3Flex instrument. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the International Institute for Nanotechnology (IIN), and Northwestern's MRSEC program (NSF DMR-1720139). Metal analysis was performed at the Northwestern University Quantitative Bioelement Imaging Center (QBIC) generously supported by the NIH under grant S10OD020118. This work made use of the EPIC, Keck-II, and/or SPID facility(ies) of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the IIN; the Keck Foundation; and the State of Illinois, through the IIN. This work made use of beamline 8–2 at the Stanford Synchrotron Radiation Lightsource (SSRL).The authors graciously acknowledge the assistance of Rebecca Sponenburg with metal analysis at QBIC.References
- B. S. B. K. Lane, Metal-Catalyzed Epoxidations of Alkenes with Hydrogen Peroxide, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed.
- R. Hage and A. Lienke, Applications of transition-metal catalysts to textile and wood-pulp bleaching, Angew Chem. Int. Ed. Engl., 2005, 45(2), 206–222 CrossRef PubMed.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Recent Progress of Electrochemical Production of Hydrogen Peroxide by Two-Electron Oxygen Reduction Reaction, Adv. Sci., 2021, 8(15), e2100076 CrossRef PubMed.
- W. Chen, H. Yang, C. Peng and T. Wu, Resolving the "health vs environment" dilemma with sustainable disinfection during the COVID-19 pandemic, Environ. Sci. Pollut. Res. Int., 2023, 30(9), 24737–24741 CrossRef PubMed.
- K. H. Pandey, Hydrogen Peroxide Market Size by End-User (Paper & Pulp, Chemical, Waste Water Treatment, Mining) Report GMI851, Global Market Insights, 2016 Search PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45(42), 6962–6984 CrossRef CAS PubMed.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte, Science, 2019, 366(6462), 226–231 CrossRef CAS PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Enabling direct H2O2 production through rational electrocatalyst design, Nat. Mater., 2013, 12(12), 1137–1143 CrossRef CAS PubMed.
- B.-H. Lee, H. Shin, A. S. Rasouli, H. Choubisa, P. Ou, R. Dorakhan, I. Grigioni, G. Lee, E. Shirzadi, R. K. Miao, J. Wicks, S. Park, H. S. Lee, J. Zhang, Y. Chen, Z. Chen, D. Sinton, T. Hyeon, Y.-E. Sung and E. H. Sargent, Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis, Nat. Catal., 2023, 6, 234–243 CrossRef CAS.
- J. S. Adams, M. L. Kromer, J. Rodriguez-Lopez and D. W. Flaherty, Unifying Concepts in Electro- and Thermocatalysis toward Hydrogen Peroxide Production, J. Am. Chem. Soc., 2021, 143(21), 7940–7957 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Trends in the electrochemical synthesis of H2O2: Enhancing activity and selectivity by electrocatalytic site engineering, Nano Lett., 2014, 14(3), 1603–1608 CrossRef CAS PubMed.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Promoting H2O2 production via 2-electron oxygen reduction by coordinating partially oxidized Pd with defect carbon, Nat. Commun., 2020, 11(1), 1–9 CrossRef PubMed.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H 2O 2 production, J. Am. Chem. Soc., 2011, 133(48), 19432–19441 CrossRef PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Single-atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions, Angew. Chem., Int. Ed., 2016, 55(6), 2058–2062 CrossRef CAS PubMed.
- Y. Bu, Y. Wang, G. F. Han, Y. Zhao, X. Ge, F. Li, Z. Zhang, Q. Zhong and J. B. Baek, Carbon-Based Electrocatalysts for Efficient Hydrogen Peroxide Production, Adv. Mater., 2021, e2103266 CrossRef PubMed.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials, Nat. Catal., 2018, 1(2), 156–162 CrossRef CAS.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts, Nat. Catal., 2018, 1(4), 282–290 CrossRef.
- G.-f. Han, F. Li, S.-j. Kim, Y. Bu, J.-b. Baek, W. Zou, M. Karamad, J.-p. Jeon, S.-w. Kim, Z. Fu, Y. Lu and S. Siahrostami, Building and identifying highly active oxygenated groups in carbon materials for oxygen reduction to H2O2, Nat. Commun., 2020, 11(1), 2209 CrossRef CAS PubMed.
- R. Hu, C. Wu, K. Hou, C. Xia, J. Yang and L. Guan, Tailoring the electrocatalytic oxygen reduction reaction pathway by tuning the electronic states of single-walled carbon nanotubes, Carbon, 2019, 147, 35–42 CrossRef CAS.
- J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji, H. Lan, H. Liu and J. Qu, Graphitic N in nitrogen-Doped carbon promotes hydrogen peroxide synthesis from electrocatalytic oxygen reduction, Carbon, 2020, 163, 154–161 CrossRef CAS.
- Y. Sun, S. Li, Z. P. Jovanov, D. Bernsmeier, H. Wang, B. Paul, X. Wang, S. Kühl and P. Strasser, Structure, Activity, and Faradaic Efficiency of Nitrogen-Doped Porous Carbon Catalysts for Direct Electrochemical Hydrogen Peroxide Production, ChemSusChem, 2018, 11(19), 3388–3395 CrossRef CAS PubMed.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, N-Doped Graphitized Carbon Nanohorns as a Forefront Electrocatalyst in Highly Selective O2 Reduction to H2O2, Chem, 2018, 4(1), 106–123 CAS.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Advanced Electrocatalysis for Energy and Environmental Sustainability via Water and Nitrogen Reactions, Adv. Mater., 2021, 33(6), e2000381 CrossRef PubMed.
- S. Shiva Kumar and V. Himabindu, Hydrogen production by PEM water electrolysis – A review, Mater. Sci. Energy Technol., 2019, 2(3), 442–454 Search PubMed.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Membrane Electrolyzers for Impure-Water Splitting, Joule, 2020, 4(12), 2549–2561 CrossRef CAS.
- J. Zhang, H. Zhang, M.-j. Cheng and Q. Lu, Tailoring the Electrochemical Production of H2O2 : Strategies for the Rational Design of High-Performance Electrocatalysts, Small, 2020, 16(15), e1902845 CrossRef PubMed.
- Y. Pang, K. Wang, H. Xie, Y. Sun, M. M. Titirici and G. L. Chai, Mesoporous Carbon Hollow Spheres as Efficient Electrocatalysts for Oxygen Reduction to Hydrogen Peroxide in Neutral Electrolytes, ACS Catal., 2020, 10(14), 7434–7442 CrossRef CAS.
- Y. Gong, H. Fei, X. Zou, W. Zhou, S. Yang, G. Ye, Z. Liu, Z. Peng, J. Lou, R. Vajtai, B. I. Yakobson, J. M. Tour and P. M. Ajayan, Boron- and Nitrogen-Substituted Graphene Nanoribbons as Efficient Catalysts for Oxygen Reduction Reaction, Chem. Mater., 2015, 27(4), 1181–1186 CrossRef CAS.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Two-step boron and nitrogen doping in graphene for enhanced synergistic catalysis, Angew Chem. Int. Ed. Engl., 2013, 52(11), 3110–3116 CrossRef CAS PubMed.
- Z. Qiang, Y. Xia, X. Xia and B. D. Vogt, Generalized Synthesis of a Family of Highly Heteroatom-Doped Ordered Mesoporous Carbons, Chem. Mater., 2017, 29(23), 10178–10186 CrossRef CAS.
- J. Wang, J. Hao, D. Liu, S. Qin, D. Portehault, Y. Li, Y. Chen and W. Lei, Porous Boron Carbon Nitride Nanosheets as Efficient Metal-Free Catalysts for the Oxygen Reduction Reaction in Both Alkaline and Acidic Solutions, ACS Energy Lett., 2017, 2(2), 306–312 CrossRef CAS.
- Z. Tian, Q. Zhang, L. Thomsen, N. Gao, J. Pan, R. Daiyan, J. Yun, J. Brandt, N. Lopez-Salas, F. Lai, Q. Li, T. Liu, R. Amal, X. Lu and M. Antonietti, Constructing Interfacial Boron-Nitrogen Moieties in Turbostratic Carbon for Electrochemical Hydrogen Peroxide Production, Angew Chem. Int. Ed. Engl., 2022, 61(37), e202206915 CrossRef CAS PubMed.
- X. Li, X. Wang, G. Xiao and Y. Zhu, Identifying active sites of boron, nitrogen co-doped carbon materials for the oxygen reduction reaction to hydrogen peroxide, J. Colloid Interface Sci., 2021, 602, 799–809 CrossRef CAS PubMed.
- T. Sun, J. Wang, C. Qiu, X. Ling, B. Tian, W. Chen and C. Su, B, N Codoped and Defect-Rich Nanocarbon Material as a Metal-Free Bifunctional Electrocatalyst for Oxygen Reduction and Evolution Reactions, Adv. Sci., 2018, 5(7), 1800036 CrossRef PubMed.
- W. S. Li, Z. Y. Sun, J. M. Yang, G. Y. Huang and H. B. Zhu, Efficient Metal-free ZIF-8 Derived B, N-codoped Carbon Electrocatalyst toward Oxygen Reduction, Z. fur Anorg. Allg. Chem., 2021, 647(13), 1326–1333 CrossRef CAS.
- Z. Jiang, X. Zhao, X. Tian, L. Luo, J. Fang, H. Gao and Z. J. Jiang, Hydrothermal Synthesis of Boron and Nitrogen Codoped Hollow Graphene Microspheres with Enhanced Electrocatalytic Activity for Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2015, 7(34), 19398–19407 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan, H. Yu, H. Zhao, Y. Zhang and G. Chen, Boron and Nitrogen Codoped Nanodiamond as an Efficient Metal-Free Catalyst for Oxygen Reduction Reaction, J. Phys. Chem. C, 2013, 117(29), 14992–14998 CrossRef CAS.
- R. Huang, J. Lv, J. Chen, Y. Zhu, J. Zhu, T. Wagberg and G. Hu, Three-dimensional porous high boron-nitrogen-doped carbon for the ultrasensitive electrochemical detection of trace heavy metals in food samples, J. Hazard. Mater., 2023, 442, 130020 CrossRef CAS PubMed.
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, Can boron and nitrogen co-doping improve oxygen reduction reaction activity of carbon nanotubes?, J. Am. Chem. Soc., 2013, 135(4), 1201–1204 CrossRef CAS PubMed.
- M. Fan, Z. Wang, K. Sun, A. Wang, Y. Zhao, Q. Yuan, R. Wang, J. Raj, J. Wu, J. Jiang and L. Wang, N B OH Site-Activated Graphene Quantum Dots for Boosting Electrochemical Hydrogen Peroxide Production, Adv. Mater., 2023, 35(17), e2209086 CrossRef PubMed.
- Y. Xue, D. Yu, L. Dai, R. Wang, D. Li, A. Roy, F. Lu, H. Chen, Y. Liu and J. Qu, Three-dimensional B,N-doped graphene foam as a metal-free catalyst for oxygen reduction reaction, Phys. Chem. Chem. Phys., 2013, 15(29), 12220–12226 RSC.
- S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund, D. Sokaras, T. R. Kim, Y. Liu, X. Yan, E. Nilsson, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, Designing Boron Nitride Islands in Carbon Materials for Efficient Electrochemical Synthesis of Hydrogen Peroxide, J. Am. Chem. Soc., 2018, 140(25), 7851–7859 CrossRef CAS PubMed.
- Z. Bao, J. Zhao, S. Zhang, L. Ding, X. Peng, G. Wang, Z. Zhao, X. Zhong, Z. Yao and J. Wang, Synergistic effect of doped nitrogen and oxygen-containing functional groups on electrochemical synthesis of hydrogen peroxide, J. Mater. Chem. A, 2022, 10(9), 4749–4757 RSC.
- J. Hao, J. Wang, S. Qin, D. Liu, Y. Li and W. Lei, B/N co-doped carbon nanosphere frameworks as high-performance electrodes for supercapacitors, J. Mater. Chem. A, 2018, 6(17), 8053–8058 RSC.
- B. Ni, Y. Li, T. Chen, T. Lu and L. Pan, Covalent organic frameworks converted N, B co-doped carbon spheres with excellent lithium ion storage performance at high current density, J. Colloid Interface Sci., 2019, 542, 213–221 CrossRef CAS PubMed.
- M. Wang, Y. Yang, Z. Yang, L. Gu, Q. Chen and Y. Yu, Sodium-Ion Batteries: Improving the Rate Capability of 3D Interconnected Carbon Nanofibers Thin Film by Boron, Nitrogen Dual-Doping, Adv. Sci., 2017, 4(4), 1600468 CrossRef PubMed.
- M. Erkartal, U. Erkilic, B. Tam, H. Usta, O. Yazaydin, J. T. Hupp, O. K. Farha and U. Sen, From 2-methylimidazole to 1,2,3-triazole: a topological transformation of ZIF-8 and ZIF-67 by post-synthetic modification, Chem. Commun., 2017, 53(12), 2028–2031 RSC.
- J. Cravillon, S. Münzer, S.-j. Lohmeier, A. Feldhoff, K. Huber and M. Wiebcke, Rapid Room-Temperature Synthesis and Characterization of Nanocrystals of a Prototypical Zeolitic Imidazolate Framework, Chem. Mater., 2009, 21(8), 1410–1412 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disk electrode study, J. Electroanal. Chem., 2001, 495(2), 134–145 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical Methods Fundamentals and Applications, 2004, vol. 677, p. 10 Search PubMed.
- S. Xu, Y. Kim, D. Higgins, M. Yusuf, T. F. Jaramillo and F. B. Prinz, Building upon the Koutecky-Levich Equation for Evaluation of Next-Generation Oxygen Reduction Reaction Catalysts, Electrochim. Acta, 2017, 255, 99–108 CrossRef CAS.
- A. Bonakdarpour, D. Esau, H. Cheng, A. Wang, E. Gyenge and D. P. Wilkinson, Preparation and electrochemical studies of metal-carbon composite catalysts for small-scale electrosynthesis of H2O2, Electrochim. Acta, 2011, 56(25), 9074–9081 CrossRef CAS.
- W. Luc, J. Rosen and F. Jiao, An Ir-based anode for a practical CO2 electrolyzer, Catal. Today, 2017, 288, 79–84 CrossRef CAS.
- T. M. Gill and X. Zheng, Comparing Methods for Quantifying Electrochemically Accumulated H2O2, Chem. Mater., 2020, 32(15), 6285–6294 CrossRef CAS.
- T. Palaniselvam, B. P. Biswal, R. Banerjee and S. Kurungot, Zeolitic imidazolate framework (ZIF)-derived, hollow-core, nitrogen-doped carbon nanostructures for oxygen-reduction reactions in PEFCs, Chemistry, 2013, 19(28), 9335–9342 CrossRef CAS PubMed.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- L. Han, Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, In-Plane Carbon Lattice-Defect Regulating Electrochemical Oxygen Reduction to Hydrogen Peroxide Production over Nitrogen-Doped Graphene, ACS Catal., 2019, 9(2), 1283–1288 CrossRef CAS.
- F. M. Hassan, V. Chabot, J. Li, B. K. Kim, L. Ricardez-Sandoval and A. Yu, Pyrrolic-structure enriched nitrogen doped graphene for highly efficient next generation supercapacitors, J. Mater. Chem. A, 2013, 1(8), 2904–2912 RSC.
- G. Srinivas, Y. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Synthesis of graphene-like nanosheets and their hydrogen adsorption capacity, Carbon, 2010, 48(3), 630–635 CrossRef CAS.
- S. Yuan, B. Toury, C. Journet and A. Brioude, Synthesis of hexagonal boron nitride graphene-like few layers, Nanoscale, 2014, 6(14), 7838–7841 RSC.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, Metal-free doped carbon materials as electrocatalysts for the oxygen reduction reaction, J. Mater. Chem. A, 2014, 2(12), 4085–4110 RSC.
- C. Xuan, B. Hou, W. Xia, Z. Peng, T. Shen, H. L. Xin, G. Zhang and D. Wang, From a ZIF-8 polyhedron to three-dimensional nitrogen doped hierarchical porous carbon: an efficient electrocatalyst for the oxygen reduction reaction, J. Mater. Chem. A, 2018, 6(23), 10731–10739 RSC.
- Z. Yang, Y. Gao, L. Zuo, C. Long, C. Yang and X. Zhang, Tailoring Heteroatoms in Conjugated Microporous Polymers for Boosting Oxygen Electrochemical Reduction to Hydrogen Peroxide, ACS Catal., 2023, 13(7), 4790–4798 CrossRef CAS.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, Activity-Selectivity Trends in the Electrochemical Production of Hydrogen Peroxide over Single-Site Metal-Nitrogen-Carbon Catalysts, J. Am. Chem. Soc., 2019, 141(31), 12372–12381 CrossRef CAS PubMed.
- Q. Wang, T. Ina, W. T. Chen, L. Shang, F. Sun, S. Wei, D. Sun-Waterhouse, S. G. Telfer, T. Zhang and G. I. N. Waterhouse, Evolution of Zn(II) single atom catalyst sites during the pyrolysis-induced transformation of ZIF-8 to N-doped carbons, Sci. Bull., 2020, 65(20), 1743–1751 CrossRef CAS PubMed.
- J. W. F. To, J. Wei, D. Ng, S. Siahrostami, A. L. Koh, Y. Lee, Z. Chen, K. D. Fong, S. Chen, J. He, W.-g. Bae, J. Wilcox, H. Y. Jeong, K. Kim, F. Studt, J. K. Nørskov, T. F. Jaramillo and Z. Bao, High-performance oxygen reduction and evolution carbon catalysis : From mechanistic studies to device integration, Nano Res., 2017, 10(4), 1163–1177 CrossRef CAS.
- P. Giusto, H. Arazoe, D. Cruz, P. Lova, T. Heil, T. Aida and M. Antonietti, Boron Carbon Nitride Thin Films: From Disordered to Ordered Conjugated Ternary Materials, J. Am. Chem. Soc., 2020, 142(49), 20883–20891 CrossRef CAS PubMed.
- C. I. Sathish, G. Kothandam, P. Selvarajan, Z. Lei, J. Lee, J. Qu, A. H. Al-Muhtaseb, X. Yu, M. B. H. Breese, R. Zheng, J. Yi and A. Vinu, Ordered Mesoporous Boron Carbon Nitrides with Tunable Mesopore Nanoarchitectonics for Energy Storage and CO(2) Adsorption Properties, Adv. Sci., 2022, 9(16), e2105603 CrossRef CAS PubMed.
- T. Schiros, D. Nordlund, L. Palova, L. Zhao, M. Levendorf, C. Jaye, D. Reichman, J. Park, M. Hybertsen and A. Pasupathy, Atomistic Interrogation of B-N Co-dopant Structures and Their Electronic Effects in Graphene, ACS Nano, 2016, 10(7), 6574–6584 CrossRef CAS PubMed.
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, A. Srivastava, Z. F. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, Atomic layers of hybridized boron nitride and graphene domains, Nat. Mater., 2010, 9(5), 430–435 CrossRef CAS PubMed.
- S. Beniwal, J. Hooper, D. P. Miller, P. S. Costa, G. Chen, S.-Y. Liu, P. A. Dowben, E. C. H. Sykes, E. Zurek and A. Enders, Graphene-like Boron–Carbon–Nitrogen Monolayers, ACS Nano, 2017, 11(3), 2486–2493 CrossRef CAS PubMed.
- I. Jimenez, D. G. J. Sutherland, T. van Buuren, J. A. Carlisle and L. J. Terminello, Photoemission and x-ray-absorption study of boron carbide and its surface thermal stability, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57(20), 13167 CrossRef CAS.
- F. Zheng, Y. Yang and Q. Chen, High lithium anodic performance of highly nitrogen-doped porous carbon prepared from a metal-organic framework, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- Y. Zhao, S. Zhang, C. Han, Q. Lu, Q. Fu, H. Jiang, L. Yang, Y. Xing, Q. Zheng, J. Shen, L. Yan and X. Zhao, Effect of boron and nitrogen modulation in metal atoms anchoring on flower-like carbon superstructure for efficient ammonia electrosynthesis, Chem. Eng. J., 2023, 468, 143517 CrossRef CAS.
- Y.-H. Lee, F. Li, K.-H. Chang, C.-C. Hu and T. Ohsaka, Novel Synthesis of N-Doped Porous Carbons from Collagen for Electrocatalytic Production of H2O2, Appl. Catal., B, 2012, 126, 208–214 CrossRef CAS.
- Q. Shentu, Z. Wu, W. Song, S. Pan, Z. Zhou, W. Lv, C. Song and Y. Yao, Carbon doped boron nitride nanosheet as efficient metal-free catalyst for peroxymonosulfate activation: Important role of B-N-C moieties, Chem. Eng. J., 2022, 446, 137274 CrossRef CAS.
- S. C. Ray, H. M. Tsai, J. W. Chiou, J. C. Jan, K. Kumar, W. F. Pong, F. Z. Chien, M. H. Tsai, S. Chattopadhyay, L. C. Chen, S. C. Chien, M. T. Lee, S. T. Lin and K. H. Chen, X-Ray absorption studies of boron–carbon–nitrogen (BxCyNz) ternary alloys, Diamond Relat. Mater., 2004, 13(4–8), 1553–1557 CrossRef CAS.
- P. S. Hoffmann, O. Baake, M. L. Kosinova, B. Beckhoff, A. Klein, B. Pollakowski, V. A. Trunova, V. S. Sulyaeva, F. A. Kuznetsov and W. Ensinger, Chemical bonds and elemental compositions of BCxNylayers produced by chemical vapor deposition with trimethylamine borane, triethylamine borane, or trimethylborazine, X-Ray Spectrom., 2012, 41(4), 240–246 CrossRef CAS.
- J. Kim, K.-Y. Doh, S. Moon, C.-W. Choi, H. Jeong, J. Kim, W. Yoo, K. Park, K. Chong, C. Chung, H. Choi, S.-Y. Choi, D. Lee and J. K. Kim, Conformal Growth of Hexagonal Boron Nitride on High-Aspect-Ratio Silicon-Based Nanotrenches, Chem. Mater., 2023, 35(6), 2429–2438 CrossRef CAS.
- M. E. Fleet and L. X, Boron K-edge XANES of boron oxides: tetrahedral B-O distances and near-surface alteration, Phys. Chem. Miner., 2001, 28, 421–427 CrossRef CAS.
- T. Schiros, D. Nordlund, L. Palova, D. Prezzi, L. Zhao, K. S. Kim, U. Wurstbauer, C. Gutierrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Connecting dopant bond type with electronic structure in N-doped graphene, Nano Lett., 2012, 12(8), 4025–4031 CrossRef CAS PubMed.
- I. Shimoyama, G. Wu, T. Sekiguchi and Y. Baba, Evidence for the existence of nitrogen-substituted graphite structure by polarization dependence of near-edge x-ray-absorption fine structure, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62(3), R6053 CrossRef CAS.
- X. Chen, P. Ye, H. Wang, H. Huang, Y. Zhong and Y. Hu, Discriminating Active BN Sites in Coralloidal B, N Dual-Doped Carbon Nano-Bundles for Boosted Zn-Ion Storage Capability, Adv. Funct. Mater., 2023, 33, 2212915 CrossRef CAS.
- G. Fronzoni, O. Baseggio, M. Stener, W. Hua, G. Tian, Y. Luo, B. Apicella, M. Alfe, M. de Simone, A. Kivimaki and M. Coreno, Vibrationally resolved high-resolution NEXAFS and XPS spectra of phenanthrene and coronene, J. Chem. Phys., 2014, 141(4), 044313 CrossRef PubMed.
- C. Morant, P. Prieto, J. Bareño, J. M. Sanz and E. Elizalde, Hard BCxNy thin films grown by dual ion beam sputtering, Thin Solid Films, 2006, 515(1), 207–211 CrossRef CAS.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, Defective Carbon-Based Materials for the Electrochemical Synthesis of Hydrogen Peroxide, ACS Sustainable Chem. Eng., 2018, 6(1), 311–317 CrossRef CAS.
- P. A. Bruhwiler, A. Maxwell, C. Puglia, A. Nilsson, S. Andersson and N. Martensson, pi * and sigma * Excitons in C 1s Absorption of Graphite, Phys. Rev. Lett., 1995, 74(4), 614–617 CrossRef CAS PubMed.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S. Z. Qiao, Tailoring Selectivity of Electrochemical Hydrogen Peroxide Generation by Tunable Pyrrolic-Nitrogen-Carbon, Adv. Energy Mater., 2020, 10(21), 2000789 CrossRef CAS.
- J. Ding, H. F. Wang, X. Yang, W. Ju, K. Shen, L. Chen and Y. Li, A Janus heteroatom-doped carbon electrocatalyst for hydrazine oxidation, Natl. Sci. Rev., 2022, 10(3), nwac231 CrossRef PubMed.
- Yu. V. Fedoseeva, M. L. Kosinova, S. A. Prokhorova, I. S. Merenkov, L. G. Bulusheva, A. V. Okotrub and F. A. Kuznetsov, X-ray spectroscopic study of the electronic structure of boron carbonitride films obtained by chemical vapor deposition on Co/Si and CoOx/Si substrates, J. Struct. Chem., 2012, 53, 690–698 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, Efficient Electrochemical Hydrogen Peroxide Production from Molecular Oxygen on Nitrogen-Doped Mesoporous Carbon Catalysts, ACS Catal., 2018, 8(4), 2844–2856 CrossRef CAS.
- T. Zhang, Y. Wang, X. Li, Q. Zhuang, Z. Zhang, H. Zhou, Q. Ding, Y. Wang, Y. Dang, L. Duan and J. Liu, Charge state modulation on boron site by carbon and nitrogen localized bonding microenvironment for two-electron electrocatalytic H2O2 production, Chin. Chem. Lett., 2023, 34(5), 107596 CrossRef CAS.
- P. J. M. Cordeiro-Junior, J. Lobato Bajo, M. R. V. Lanza and M. A. Rodrigo Rodrigo, Highly Efficient Electrochemical Production of Hydrogen Peroxide Using the GDE Technology, Ind. Eng. Chem. Res., 2022, 61(30), 10660–10669 CrossRef CAS PubMed.
- M. Inaba, A. W. Jensen, G. W. Sievers, M. Escudero-Escribano, A. Zana and M. Arenz, Benchmarking high surface area electrocatalysts in a gas diffusion electrode: Measurement of oxygen reduction activities under realistic conditions, Energy Environ. Sci., 2018, 11(4), 988–994 RSC.
- K. Ehelebe, N. Schmitt, G. Sievers, A. W. Jensen, A. Hrnjić, P. Collantes Jiménez, P. Kaiser, M. Geuß, Y.-P. Ku, P. Jovanovič, K. J. J. Mayrhofer, B. Etzold, N. Hodnik, M. Escudero-Escribano, M. Arenz and S. Cherevko, Benchmarking Fuel Cell Electrocatalysts Using Gas Diffusion Electrodes: Inter-lab Comparison and Best Practices, ACS Energy Lett., 2022, 7(2), 816–826 CrossRef CAS.
- Q. Zhang, M. Zhou and X. Du, Highly efficient electrosynthesis of hydrogen peroxide on a superhydrophobic three-phase interface by natural air diffusion, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- J. M. M. Ihonen and G. Lindbergh, Flooding of Gas Diffusion Backing in PTFEs: Physical and Electrochemical Characterization, J. Electrochem. Soc., 2004, 151, A1152 CrossRef CAS.
- Z. Chen, H. Chen, K. Wang, J. Chen, M. Li, Y. Wang, P. Tsiakaras and S. Song, Enhanced TiO2 Photocatalytic 2 e– Oxygen Reduction Reaction via Interfacial Microenvironment Regulation and Mechanism Analysis, ACS Catal., 2023, 6497–6508 CrossRef CAS.
- M. Li, X. Qin, M. Gao, T. Li and Y. Lv, Graphitic carbon nitride and carbon nanotubes modified active carbon fiber cathode with enhanced H2O2 production and recycle of Fe3+/Fe2+ for electro-Fenton treatment of landfill leachate concentrate, Environ. Sci.: Nano, 2022, 9(2), 632–652 RSC.
- W. Zhou, L. Rajic, L. Chen, K. Kou, Y. Ding, X. Meng, Y. Wang, B. Mulaw, J. Gao, Y. Qin and A. N. Alshawabkeh, Activated carbon as effective cathode material in iron-free Electro-Fenton process: Integrated H2O2 electrogeneration, activation, and pollutants adsorption, Electrochim. Acta, 2019, 296, 317–326 CrossRef CAS PubMed.
- M. Umar, H. A. Aziz and M. S. Yusoff, Trends in the use of Fenton, electro-Fenton and photo-Fenton for the treatment of landfill leachate, Waste Manage., 2010, 30(11), 2113–2121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04613g |
| This journal is © The Royal Society of Chemistry 2024 |