
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparing direct pyrolysis and post-impregnation in the synthesis of atomic Fe active sites for solvent-free aerobic coupling of benzylamine†
Guilong
Lu
a,
Zewen
Shen
b,
Philipp
Schwiderowski
a,
Jannik
Böttger
a,
Tim
Herrendorf
c,
Wolfgang
Kleist
 c,
Xiaoyu
Li
a,
Guixia
Zhao
b,
Baoxiang
Peng
a,
Xiubing
Huang
*d and
Martin
Muhler
*a
c,
Xiaoyu
Li
a,
Guixia
Zhao
b,
Baoxiang
Peng
a,
Xiubing
Huang
*d and
Martin
Muhler
*a
aLaboratory of Industrial Chemistry, Ruhr University Bochum, 44780 Bochum, Germany. E-mail: martin.muhler@ruhr-uni-bochum.de
bNorth China Electric Power University, 102206 Beijing, PR China
cFachbereich Chemie, RPTU Kaiserslautern-Landau, 67663 Kaiserslautern, Germany
dBeijing Key Laboratory of Function Materials for Molecule & Structure Construction, University of Science and Technology Beijing, 100083 Beijing, PR China
First published on 18th October 2024
Abstract
Both direct pyrolysis and post-impregnation are employed for the synthesis of a 3D macroporous carbon-supported Fe/N-C catalyst in this study. By comparing the microstructure, physicochemical properties, and local electronic structure of the products obtained from both preparation methods, the underlying formation mechanisms of atomic Fe sites are proposed based on the degree of carbonization of carbon-based precursors. As embedded Fe ions significantly facilitate the carbonization of carbon-based precursors to form highly stable carbon supports with a certain degree of graphitization, Fe–Nx active sites are the predominant active sites in the catalysts prepared by the direct pyrolysis method, as expected. In contrast, the absence of metal salts in the precursor results in an inadequate carbonization process, leading to pyrolyzed products with limited resistance to concentrated NaOH. This deficiency accounts for the presence of a significant number of oxygen defects in the bare carbon supports. Since metal impregnation and subsequent reduction treatment possess a limited capacity to recover these O-defects, the catalysts obtained via the post-impregnation method exhibit distinct Fe–OyNx configurations. The solvent-free oxidative coupling of benzylamine was used as a probe reaction to evaluate the catalytic activity of these atomic Fe sites. The catalytic results demonstrate the superior catalytic activity and recyclability of Fe–Nx active sites compared with Fe–OyNx moieties, thereby confirming the superiority of the direct pyrolysis approach over the post-impregnation method.
Introduction
Transition metal and nitrogen codoped carbon catalysts (M/N-C), featuring well-defined atomic metal active sites (M–Nx), ultrahigh atom efficiency, and unique structure–activity relationships, have recently attracted significant attention.1–3 However, many reported M/N-C catalysts to date still cannot meet the requirements of commercial applications in the fields of chemical production, environmental catalysis, and energy conversion and storage. Specifically, most of them possess a micropore-dominated porous structure, which severely hinders the accessibility of the internal metal active sites by slow pore diffusion, leading to unsatisfactory catalytic performance.4–7 To tackle this issue, researchers have placed significant emphasis on modifying carbon substrates in terms of their dimensions, geometries, and pore structures. Three-dimensional (3D) macroporous carbon-supported M/N-C catalysts stand out from various candidates because their micropores hosting robust metal active sites are directly fabricated with meso- and macropores for the fast diffusion of reactants and products toward and away from the active sites, respectively. Moreover, the open and interconnected macroporous framework contributes to increasing the specific surface area and enhancing the accessibility of micropores with isolated metal atoms, which is beneficial for obtaining highly efficient M/N-C catalysts.8–10Owing to the rapid advancement in synthetic technology, it has become feasible to integrate atomically dispersed transition metal sites with 3D macroporous N-doped carbon substrates through both the direct pyrolysis approach and the post-impregnation method.11 The former is the direct pyrolysis of hard template-incorporated metal-coordinated polymers or ionic liquids, wherein atomic metal sites are in situ formed during the thermal decomposition of the precursor.12–16 Another strategy is based on wet chemistry. In detail, metal ions are firstly anchored on as-prepared 3D macroporous carbon supports by the impregnation method and subsequently transformed to atomic metal sites after an annealing and/or reduction treatment.17–20 Recently, Hai and coworkers have made a significant breakthrough in this field by successfully fabricating various metal single-atomic sites on different carriers through conventional impregnation and subsequent reduction treatment.21
However, there are several issues regarding these methods. As for the direct pyrolysis approach, the precise role of the metal salt in the thermal decomposition of the C- and N-containing precursors as well as the evolution of the coordination configuration of the atomic metal site during the pyrolysis process still remain elusive.22–24 Although the post-impregnation method ensures a consistent and gentle process for anchoring metal atoms onto various carbon supports, the resulting coordination mode of the atomic metal centre is still influenced by the functional groups present on the carbon surface. This influence could also be ascribed to the pyrolysis of C- and N-containing precursors, as the coordination configuration of the atomic metal site is strongly influenced by specific N or O functional groups on the surface of the final pyrolyzed products. Unfortunately, most studies only focus on assessing the feasibility and effectiveness of the method, while neglecting an in-depth investigation of the interaction between the impregnated metal ions and the surface defects on carbon supports during subsequent annealing and/or reduction processes.25–28 Even worse, hardly any literature exists on the simultaneous synthesis of 3D M/N-C catalysts using both synthetic methods, which hinders our understanding of the impact of different methods on the formation mechanism and catalytic activity of atomic Fe sites.
Thus, a comparative experiment is elaborately designed in this study. To minimize the influence of experimental variables, we employed the same C- and N-containing precursors and identical pyrolysis procedures for the direct synthesis of 3D M/N-C catalysts as well as the preparation of 3D carbon supports for subsequent post-impregnation. The oxidative coupling of primary amines is selected as a probe reaction to evaluate the catalytic performance of the Fe-based catalysts. Notably, most reported mesoporous M/N-C catalysts only exhibited favourable catalytic performance under diluted reaction conditions using solvents due to their limited mass transfer efficiency.12,15 In contrast, our developed 3D Fe/N-C catalysts featuring a unique trimodal pore structure with interconnected micropores, mesopores and macropores can catalyse this reaction under solvent-free conditions, which provides an easier and more sustainable synthetic process for imine production.
Through advanced characterization techniques and rational design of reference samples, a comprehensive investigation is conducted to explore the differences between both series of catalysts in microstructure, physicochemical properties, local electronic structure, as well as catalytic activity and stability. Consequently, a plausible mechanism for the formation of atomic Fe sites is proposed based on the extent of carbonization of carbon-based precursors.
Experimental
Preparation of 3D Fe-X catalysts
Unless otherwise noted, all reagents were obtained from Sigma-Aldrich or Alfa Aesar and used as received without further purification.The 3D Fe-X (X = 700, 800, and 900 °C) catalysts were fabricated through a modified coordinated polymer pyrolysis strategy.13 Typically, 1.5 g SiO2 template and 3 g dicyandiamide (35.68 mmol) were mixed in 20 mL deionized water. After vigorous stirring for 1.5 h, 343.4 mg Fe(NO3)3·9H2O (0.85 mmol) and 3.3 mL 37 wt% formaldehyde solution (46.10 mmol) were successively added, and the mixed solution was heated to 60 °C under stirring and kept for 4 h in order to make dicyandiamide fully polymerize with formaldehyde. Afterwards, the temperature was set to 105 °C for the evaporation of solvent. The as-obtained solids were next placed in a quartz boat and first heated to 600 °C for 2 h under an Ar atmosphere, then pyrolyzed at a higher temperature (700 °C, 800 °C, and 900 °C) for 1 h. Those pyrolysis products SiO2@3D Fe-X were subsequently immersed in a hot 5 M NaOH solution for 15 h for the removal of the SiO2 template. The resulting products were washed thoroughly with deionized water twice and then treated with a 1 M HCl aqueous solution for 30 min. After twice water washing to remove Cl−, the desired catalysts 3D Fe-X were obtained. The preparation procedures for 3D DF-X (X = 700, 800, and 900 °C) carbon supports were similar to those for 3D Fe-X catalysts, except that no metal was involved in the precursors.
Preparation of 3D Fe/DF-X catalysts
The 3D Fe/DF-X (X = 700, 800, and 900 °C) catalysts were prepared based on a reported method with minor modifications.21 To achieve maximum metal loadings, 85.0 mg FeCl2·4H2O (0.43 mmol) and 0.4 g of 3D DF-X carbon supports were dispersed in an ethanol solution (50 mL) and sonicated for 20 min, followed by natural evaporation at room temperature with stirring (400 rpm). After impregnation, the as-obtained black powder was first annealed at 300 °C for 5 h in an Ar atmosphere. After cooling to room temperature, the powder was thoroughly washed with a water–ethanol mixture and then dried at 80 °C for 12 h. Finally, the powders were heated to 550 °C and kept for 5 h under the protection of an Ar flow. The final products were denoted as 3D Fe/DF-X. For comparison, metal-free 3D DF-X-R reference samples were prepared by a similar synthetic procedure used for 3D Fe/DF-X catalysts, with the only difference that metal impregnation was omitted.Material characterisation
X-ray diffraction measurements were carried out with a Bruker D8 Discover X-ray diffractometer, and Kα radiation with a wavelength of 1.5406 Å was used as the X-ray source. The patterns were recorded in a 2θ range from 5 to 80 with a scan speed of 5° per minute. Scanning electron micrograph (SEM) images were recorded using a Quanta 3D FEG scanning electron microscope (FEI) at 30.0 kV in high-vacuum mode. N2 physisorption measurements were performed at 77 K in a BEL-mini apparatus. The as-prepared powders were first degassed at 200 °C under a vacuum for 6 h to remove adsorbed water. The specific surface areas were derived from the adsorption isotherms by using the Brunauer–Emmett–Teller (BET) method. The pore volume and pore size distribution were obtained by the BJH method. Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were carried out in a Thermo Fisher iCAP RQ using a KED cell and a quadrupole mass spectrometer. High-resolution and high-angle annular dark-field scanning transmission electron microscopy (HR-TEM, HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDX) characterisation was performed using a probe-side aberration-corrected JEM-2200FS instrument (JEOL, Akishima, Japan) with an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was performed in an ultrahigh vacuum setup equipped with a Gammadata Scienta SES 2002 analyser. The spectra were obtained at a pass energy of 200 eV at a base pressure of 6 × 10−10 mbar with monochromatic Al Kα radiation (1486.3 eV, 14.5 kV, 45 mA). Synchrotron radiation X-ray absorption fine spectroscopy (XAFS) measurements were performed at the 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF). Gas chromatography (GC) analysis was carried out using an 8860 GC instrument from Agilent Technologies, which was equipped with an Agilent DB-XLB column (30 m × 180 μm × 0.18 μm) and an FID detector.General procedure for the oxidation of amines to imines
In a typical oxidation reaction, 30 mg of catalyst and 2 mL of benzylamine (18.3 mmol) were introduced into a 10 mL two-neck flask with a magnetic bar. The flask was evacuated and purged with O2 twice before it was finally connected to an O2 balloon. Then, the flask was heated at 110 °C for 18 h with stirring (700 rpm). After the reaction had been completed, 20 mL of ethyl acetate was added as a diluent, along with 500 mg of biphenyl as an internal standard. The catalyst was subsequently separated through centrifugation. The quantitative determination of the reactant and products was performed by GC. After completion of the reaction, the reaction mixture was cooled to room temperature, and then the catalyst was filtered, followed by thorough washing with ethyl acetate and ethanol drying at 80 °C overnight for the subsequent recycling reaction.Results and discussion
Materials design and synthesis
As illustrated in Fig. 1, a series of N-doped 3D macroporous carbon substrates, 3D DF-X (X = 700, 800, or 900 °C), were prepared by pyrolyzing a mixture of 300 nm SiO2 spheres and a dicyandiamide–formaldehyde (DF) polymer at different temperatures, followed by NaOH etching. Considering the strongly oxidizing properties of concentrated NaOH, as-prepared carbon substrates possess a certain amount of O functional groups that serve as favourable anchoring sites for binding metal precursors. Afterwards, an elaborately designed reduction strategy was employed to convert adsorbed Fe ions into isolated atomic Fe sites. Specifically, the Fe ion-impregnated carbon supports were initially annealed at a lower temperature of 300 °C, followed by rinsing using H2O and ethanol. This step is designed to preliminarily remove unbound Fe species and nonmetal ligands, ensuring the presence of only chemisorbed metal precursors. The remaining ligands were completely eliminated through a subsequent annealing step conducted at a higher temperature of 550 °C. In this way, the transformation of chemisorbed Fe ions into Fe atomic active sites was accomplished. The final products were designated as the 3D Fe/DF-X series. For comparison purposes, the samples prepared by the direct pyrolysis approach were denoted as the 3D Fe-X series. Notably, to achieve the maximum loading of atomic metal sites, both methods employed a higher amount of metal precursor, and the excess metal was eliminated through washing and acid leaching. The selection of the metal precursors in the different synthesis approaches was guided by their specific characteristics and requirements.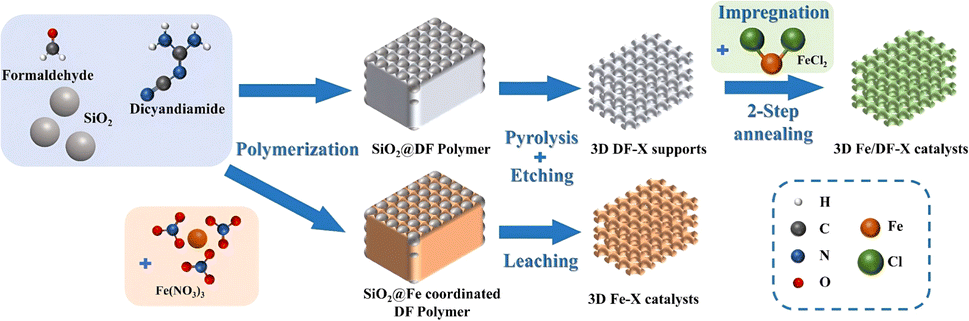 | ||
| Fig. 1 Illustration of the synthetic procedures of 3D Fe/DF-X and 3D Fe-X catalysts. | ||
Effect of the synthesis method on the formation of atomic Fe sites
The X-ray diffraction (XRD) patterns reveal that the diffraction peaks corresponding to the graphite (002) reflection in the 3D Fe-X series are significantly sharper compared with those of the 3D Fe/DF-X catalysts (Fig. 2a) and 3D DF-X supports, indicating a higher degree of crystallinity and graphitization of the carbon matrix in 3D Fe-X catalysts. Notably, the 3D DF-X supports exhibit two additional diffraction peaks at 18° and 32° (Fig. 2b), which can be attributed to structural disorders in the carbon layers induced by concentrated NaOH, such as surface hydroxylation and intercalation of Na ions within conjugated planes.29 These diffraction peaks can be completely eliminated by the following metal impregnation and reduction treatments. In fact, the involvement of metal plays a pivotal role in recovering those defects and enhancing the structural stability of carbon supports. When 3D DF-X supports directly underwent reduction treatment without metal impregnation (3D DF-X-R), a new diffraction peak presumably also corresponding to structural disorder appeared in the XRD patterns of the obtained samples (Fig. 2c). It is also worth noting that the post-impregnation method hardly affects the graphitization degree of carbon substrates according to the sharpness of the graphite (002) reflection. Thus, it is sensible to infer that the presence of Fe ions during the pyrolysis process contributes to the formation of partially graphitized carbon substrates with enhanced stability for the 3D Fe-X series catalyst by significantly enhancing the carbonization of the DF polymer. In addition, although the XRD patterns of 3D Fe/DF-900 and 3D Fe-900 exhibit characteristic diffraction peaks associated with Fe-based NPs, there is a noticeable difference in their precise positions, suggesting a disparity in the method of metal implantation between the two methods. In particular, the characteristic diffraction peaks at 35.5°, 36.1°, and 42.1° originate from γ-Fe2O3, and the peaks at 37.9°, 43.0°, 43.9°, and 44.8° correspond to Fe/Fe3C.30,31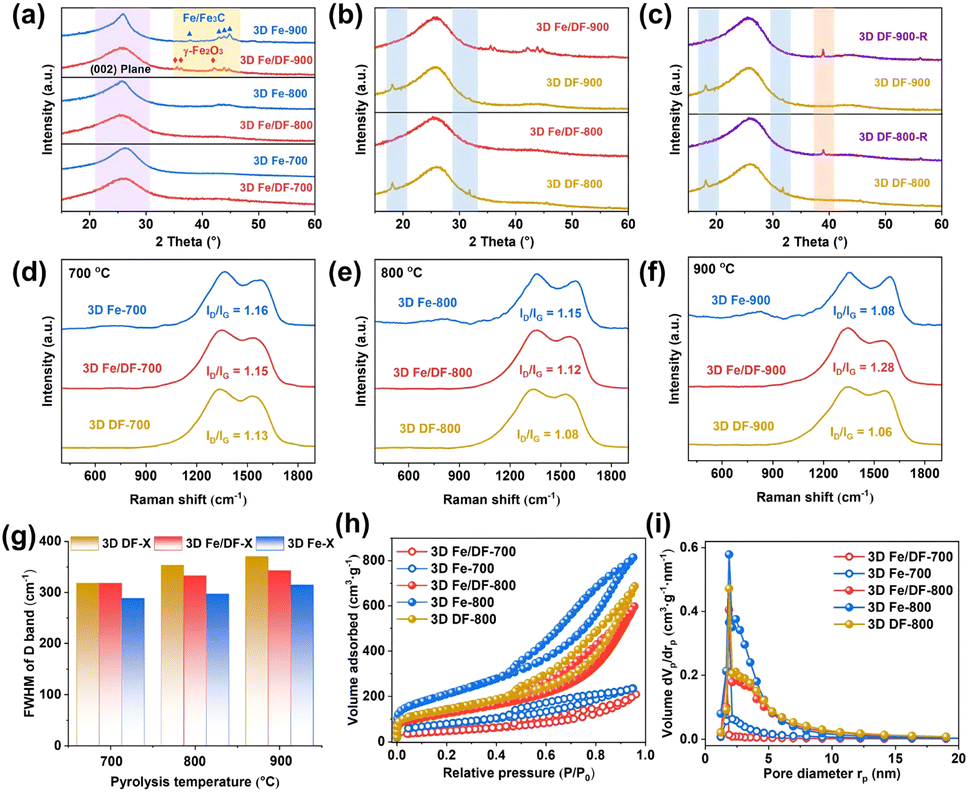 | ||
| Fig. 2 (a) The XRD patterns of the 3D Fe/DF-X and 3D Fe-X catalysts; (b) 3D DF-X supports and 3D Fe/DF-X catalysts; (c) 3D DF-800-R and 3D DF-900-R; (d–f) Raman spectra of the 3D DF-X support, 3D Fe/DF-X, and 3D Fe-X catalysts; (g) the full width at half maximum of the carbon D band for all samples; (h) N2 sorption isotherms and (i) pore size distributions of 3D Fe/DF-700, 3D Fe-700, 3D Fe/DF-800, 3D Fe-800, and 3D DF-800. | ||
Raman spectroscopy was employed to analyse the structure of the carbon matrix further. As shown in Fig. 2d–f, two distinct peaks observed at approximately 1380 and 1600 cm−1 are attributed to the D band associated with structural defects and disorder and the G band corresponding to graphitic structure, respectively.32 Compared with bare 3D carbon supports, a slight enhancement is observed in the intensity ratios of the D and G bands (ID/IG) for the 3D Fe/DF-X and 3D Fe-X series, indicating the immobilization of ionic metal impurities onto the carbon substrate.33 Among them, 3D Fe/DF-900 unexpectedly displayed the highest ID/IG value, which was attributed to the severe structural distortion in the carbon skeleton caused by the implantation of irregular Fe-based NPs. Moreover, the full width at half maximum of the D band (FWHM(D)), a more meaningful structural parameter, was also employed to evaluate the structural regularity of carbon substrates (Fig. 2g).34 It is obvious that the FWHM(D) values of the 3D Fe-X catalysts are lower than those of the corresponding 3D Fe/DF-X catalysts and 3D DF-X carbon substrates, indicating a higher graphitization degree of the carbon substrates for 3D Fe-X catalysts. Notably, the validity of those parameters for investigating the chemical structure of M/N-C catalysts remains uncertain due to the interference from defects, excess charge and strain, as well as the statistical uncertainties associated with curve fitting.34–36 It is necessary to employ another deconvolution method to further analyse the Raman spectra. Based on a combined experimental–theoretical study, the Raman spectrum can be deconvoluted into nine peaks.37 Among them, the A band located between the D band and the G band (the valley) refers to the assorted vibrations caused by O and N defects. In our case, the depth of the valley in 3D Fe-X catalysts is greater than that observed in the corresponding 3D Fe/DF-X catalysts and 3D DF-X carbon substrates, indicating that the presence of metal salt during pyrolysis is advantageous for enhancing the carbonization of the DF polymer. Overall, Raman analyses based on three individual parameters consistently support the hypothesis derived from the XRD analysis.
The porous nature was investigated through N2 adsorption/desorption measurements. The 3D Fe-X catalysts exhibit larger Brunauer–Emmett–Teller (BET) specific surface areas (Fig. 2h and S1a†) and pore volumes (Fig. 2i and S1b†) when compared to the 3D Fe/DF-X series (Table S1†). Using 3D DF-800 as a reference, we found that metal implantation through the post-impregnation method resulted in a slight decline in specific surface area, while direct pyrolysis of metal-coordinated DF polymer was beneficial to increase the specific surface area, which can also be ascribed to the metal-induced enhancement in the carbonization of carbon-based precursors during pyrolysis.38
The structural information was further visualized and elucidated by scanning electron microscopy (SEM) (Fig. S2†) and transmission electron microscopy (TEM) studies. As shown in Fig. 3a–c and S3,† there is no obvious change in morphology after metal impregnation onto 3D DF-X series carbon supports. The 3D Fe/DF-X and 3D Fe-X catalysts both exhibit a characteristic macroporous structure with an average pore diameter of 300 nm. Their corresponding energy dispersive X-ray spectroscopy (EDS) elemental mapping images clearly demonstrate the well-dispersed Fe and N atoms within the carbon skeleton (Fig. S4†). The evidence supporting the existence of Fe single atoms was further substantiated through aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) investigations (Fig. S5†). Consistent with the XRD results, Fe-based nanoparticles were observed in 3D Fe/DF-900 and 3D Fe-900 catalysts, as expected. Interestingly, a few small nanoparticles (∼10 nm) were observed on the surface of 3D Fe-800, which are not observed in 3D Fe/DF-800. Considering the relatively higher Fe loading content in 3D Fe/DF-800 catalysts compared with 3D Fe-800 (Table S2†), it is reasonable to assume that the metal-involved pyrolysis of the DF polymer significantly promotes the carbonization degree and simultaneously enhances the graphitization degree of the obtained carbon supports, resulting in limited N sites for anchoring atomic Fe sites. Significantly, the onion-like graphitic carbon layers with a d-spacing of about 0.35 nm were only observed in the HR-TEM image of 3D Fe-800 (Fig. 3d and e), which provides further support for the above conjecture.39
 | ||
| Fig. 3 (a–c) TEM and (d–f) HR-TEM images of the 3D DF-800, 3D Fe/DF-800, and 3D Fe-800. | ||
The identification of the changes in functional groups was initially accomplished using attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy. The IR spectra of most samples exhibit two prominent bands centred at 1290 and 1595 cm−1 (Fig. S6†). The former band (A band) is attributed to the stretching vibrations of C–O and C–N bonds, while the latter band (B band) arises from the vibrations of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds as well as of in-plane deformations of N–H. The distinct disparities in the position of the A band and intensity of the B band observed between 3D Fe/DF-X and 3D Fe-X catalysts indicate significant variations in chemical environments resulting from the diverse preparation methodologies.40
C and CN bonds as well as of in-plane deformations of N–H. The distinct disparities in the position of the A band and intensity of the B band observed between 3D Fe/DF-X and 3D Fe-X catalysts indicate significant variations in chemical environments resulting from the diverse preparation methodologies.40
To further unveil the chemical state of these catalysts, X-ray photoelectron spectroscopy (XPS) was applied. According to published literature, the high-resolution C 1s spectrum (Fig. 4a–c) is fitted with six peaks, namely aromatic CC (284.8 eV), aliphatic sp3 C–C (285.7 eV), N–CN/C–N (286.6 eV), C–NH2 (287.7 eV), C–O/CO (288.7 eV), and π–π* satellite (291.2 eV).41 The presence of C–NH2 is ascribed to insufficient thermal decomposition of the precursor at 700 °C, which aligns with the high nitrogen content derived from the XPS peak areas (Table S3†).42 Additionally, the Fe-based catalysts synthesized through the direct pyrolysis approach exhibit significantly higher carbon contents and a narrower FWHM of the aromatic CC peak compared with the 3D Fe/DF-X series, again demonstrating the crucial role of metal in promoting the carbonization of the DF polymer.41
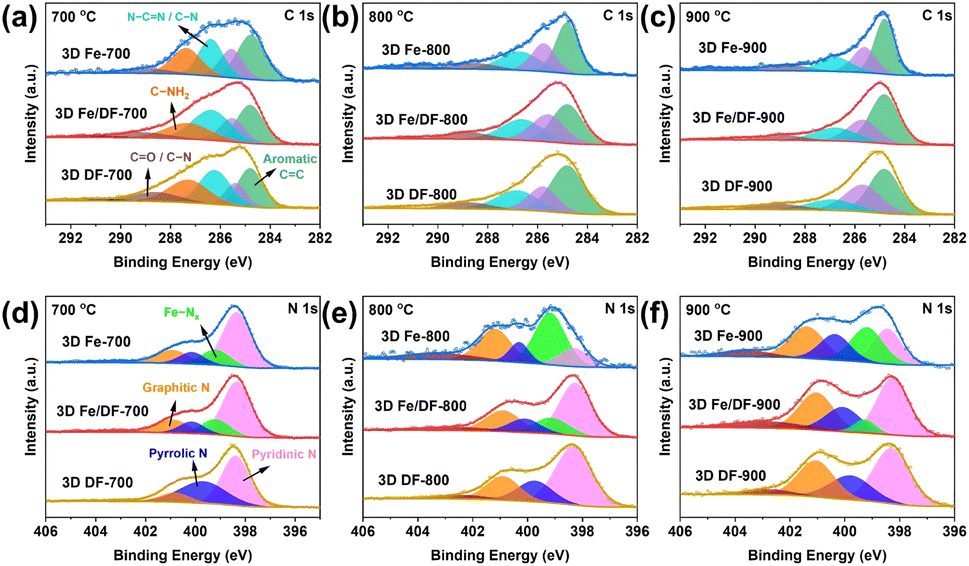 | ||
| Fig. 4 Deconvoluted (a–c) C 1s and (d–f) N 1s XP spectra of the 3D Fe/DF-X and 3D Fe-X series with the comparison of the 3D DF-X supports. | ||
In Fig. 4d–f, the 3D DF-X series is utilized as a reference to identify the positions of pyridinic N, pyrrolic N, graphitic N, and oxidized N, as well as to validate the presence of atomic Fe–Nx sites by comparing them with the N 1s spectra of 3D Fe/DF-X and 3D Fe-X catalysts.15 Generally, 3D Fe-X catalysts exhibit a higher percentage of Fe–Nx than their counterparts prepared by the post-impregnation method, even though the 3D Fe/DF-X catalysts have a higher density of atomic Fe active sites. In this regard, it is reasonable to assume that O atoms are coordinated with the atomic Fe centers in 3D Fe/DF-X. In addition, the ultra-low pyridinic N content observed in 3D Fe-800 suggests that the Fe ions were able to induce the redistribution of N species during pyrolysis at 800 °C, resulting in a distinctive coordination configuration of Fe–Nx that cannot be achieved by the post-impregnation method.
As for the O 1s XP spectra (Fig. 5a–c), the peaks centred at around 530.9 eV, 532.4 eV, 533.4 eV, 534.4 eV, and 535.8 eV are assigned to N–CO, O–(CO*)–C, O*–(CO)–C, O–C–O, and adsorbed H2O, respectively. Notably, the peak at 529.5 eV corresponding to Fe–O only appears in 3D Fe/DF-X catalysts, indicating that the coordination configuration of atomic Fe sites in 3D Fe/DF-700 and 3D Fe/DF-800 is Fe–OyNx, as well as that Fe2O3 nanoparticles are present in 3D Fe/DF-900.43 It is also noted that the 3D DF-X supports possess a much higher O content than the 3D Fe-X catalysts. Based on previous XRD analysis, excessive oxygen atoms in 3D DF-X are attributed to their poor resistance to concentrated NaOH.
 | ||
| Fig. 5 (a–c) Deconvoluted O 1s XP spectra of the 3D Fe/DF-X and 3D Fe-X series with the comparison of the 3D DF-X supports; (d–f) deconvoluted Fe 2p XP spectra of the 3D Fe/DF-X and 3D Fe-X series. | ||
In the high-resolution XP Fe 2p spectrum (Fig. 5d–f), the deconvoluted peaks centred at 709.4 eV, 710.5 eV, and 714.9 eV are attributed to the Fe 2p3/2 peaks of Fe2+, Fe3+, and a satellite peak, respectively. Obviously, the Fe3+ components of 3D Fe/DF-X catalysts exhibit a slight increase compared with those of the 3D Fe-X catalysts, which is evidence for the existence of a Fe–OyNx structure. Additionally, a distinct contrast in the Fe3+ content between 3D Fe/DF-700 and 3D Fe-700 implies a difference in the formation mechanisms of atomic Fe sites between the direct pyrolysis approach and the post-impregnation method. Last but not least, zero-valent Fe, characterized by the peaks located at 707.1 eV and 720.2 eV, is exclusively detected in 3D Fe-900, indicating the presence of metallic Fe or Fe3C, which is consistent with the previous analysis.44
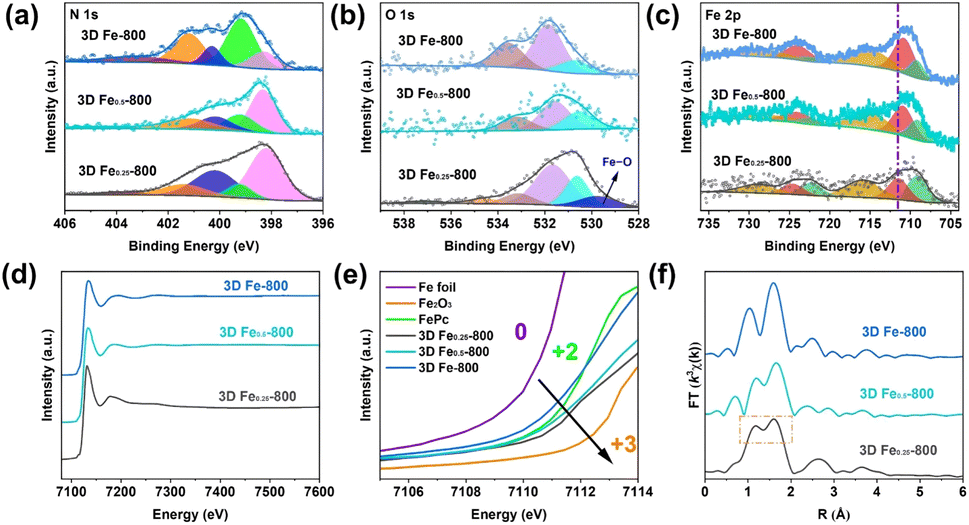
To acquire more precise insights into the electronic structure and coordination environment of the embedded Fe atoms in the carbon substrates, synchrotron radiation X-ray absorption fine structure spectroscopy (XAFS) measurements were conducted using Fe foil, Fe2O3, and FePc as reference materials. The Fe K-edge X-ray absorption near-edge structure spectra (XANES) reveal that the absorption edges of 3D Fe/DF-X catalysts are situated at higher energies compared to those of 3D Fe-X catalysts (Fig. 6a and b), indicating a relatively higher oxidation state of Fe for 3D Fe/DF-X catalysts.45 Notably, the unusually low energy of the absorption edge in 3D Fe-800 is inconsistent with the XPS analysis. Given that XPS is a surface-sensitive technique, it may not effectively detect Fe/Fe3C nanoparticles embedded within the interior of the 3D carbon matrix. This is supported by the absence of metallic Fe peaks in the Fe 2p XP spectrum of 3D Fe-800. Conversely, XANES provides bulk-sensitive information, enabling a more accurate determination of the average oxidation state of Fe throughout the entire sample.
 | ||
| Fig. 6 Fe K-edge (a and b) XANES spectra and (c) FT-EXAFS spectra of 3D Fe/DF-700, 3D Fe-700, 3D Fe/DF-800, and 3D Fe-800, with Fe foil, Fe2O3, and FePc as reference samples. | ||
The Fe K-edge spectra were further processed by Fourier transformation to obtain extended X-ray absorption fine structure spectra (FT-EXAFS) for detailed information about the coordination environment of the Fe atoms. In Fig. 6c, no discernible peaks at ∼2.2 Å corresponding to the Fe–Fe bond are observed in the k3-weighted FT-EXAFS spectra of 3D Fe/DF-700, 3D Fe/DF-800, 3D Fe-700, and 3D Fe-800. Instead, all of them exhibited a prominent peak at ∼1.6 Å, which can be ascribed to the Fe–N or Fe–O bond according to the backscattering paths of Fe2O3 and FePc in their first coordination shell. These results confirm that the Fe atoms in those catalysts predominantly exist as isolated single-atom sites, which is further supported by corresponding wavelet transform (WT) contour plots (Fig. S7†). Notably, only the FT-EXAFS spectrum of 3D Fe/DF-700 exhibits a shoulder peak at around 1.2 Å with noticeable intensity, implying the significant impact of coordinated oxygen atoms on the electronic structure of the central Fe atom. Moreover, 3D Fe/DF-700 and 3D Fe/DF-800 exhibit distinct characteristics in the second coordination shell compared with their counterparts with a typical Fe(–N–)C structure, indicating the presence of Fe–OyNx in 3D Fe/DF-X catalysts. Afterwards, the coordination configuration of the atomic Fe sites was quantitatively analysed through a least-squares EXAFS fitting process. The fitting results (Table S4 and Fig. S8, S9†) indicate that the average coordination number of Fe in 3D Fe/DF-700 is simulated to be 4.2. The central Fe atom coordinates with three O atoms and one N atom, forming a special Fe–O3N structure. Significantly, the isolated Fe atom in 3D Fe/DF-800 is anchored by more N atoms, resulting in Fe–ON3 structure, which could be attributed to the improved structural properties of the 3D DF-800. It is worth noting that these EXAFS fitting results are inconsistent with previous Fe oxidation state analyses. The relatively lower valence state of Fe in 3D Fe/DF-700 can be attributed to the predominance of coordination bond characteristics in most Fe–O bonds, which do not directly affect the oxidation state of metal centres. In contrast, 3D Fe/DF-800 exhibits enhanced covalent coordination interactions that directly alter the electronic structure of the metal centres. Specifically, the high electronegativity of nitrogen and oxygen atoms facilitates electron withdrawal from metal atoms, resulting in an increased valence state of metals.46–48 For 3D Fe-X catalysts, the Fe–O bond is nearly absent due to sufficient carbonization of the DF polymer facilitated by metal salts. Based on the fitting results, the predominant Fe active sites in 3D Fe-700 and 3D Fe-800 are identified as Fe–N4 and Fe–N3 moieties, respectively. These observations indicate that the coordination configuration of atomic Fe sites is strongly determined by the properties of carbon supports.
Since the pyrolysis of a precursor involves the carbonization of DF polymers into carbon substrates and the potential graphitization of formed carbon supports, it is necessary to further investigate the impact of metal salts on the properties of carbon substrates and the coordination configuration of single-atom Fe sites.49,50 In this context, two reference samples were prepared by reducing the amount of Fe(NO3)3 by 50% and 75% in the precursor. The obtained catalysts were denoted as 3D Fe0.5-800 and 3D Fe0.25-800, respectively. No Fe/Fe3C nanoparticles are detected in the TEM images of 3D Fe0.5-800 and 3D Fe0.25-800 (Fig. S10†), and their EDS mapping images (Fig. S11†) demonstrate that Fe and N were homogeneously dispersed onto the carbon skeleton. As the Fe content in the precursor is raised, an increase in the sharpness of the (002) diffraction peak and the specific surface area are observed in the XRD patterns (Fig. S12a†) and N2 adsorption isotherms (Fig. S12b†), respectively, indicating the crucial role of the metal salt in facilitating the carbonization of DF polymers to form carbon supports with a certain graphitization degree. More importantly, a remarkable increase in the percentage of pyridinic N is observed in the N 1s XP spectrum of 3D Fe0.5-800 (Fig. 7a). This observation not only indicates a change in the coordination mode of Fe–Nx compared with 3D Fe-800, but also verifies the pivotal role of Fe ions in regulating the distribution of N species. Interestingly, with a further decrease in the Fe content, the signal of the Fe–O bond appears in the O 1s XP spectrum of 3D Fe0.25-800 (Fig. 7b), and the Fe 2p3/2 peak of Fe3+ shifts to a higher binding energy (Fig. 7c). In this regard, the atomic Fe sites in 3D Fe0.25-800 are presumed to be anchored by both O and N atoms.
 | ||
| Fig. 7 Deconvoluted (a) N 1s, (b) O 1s, and (c) Fe 2p XP spectra of 3D Fe0.25-800, 3D Fe0.5-800, and 3D Fe-800; Fe K-edge (d and e) XANES spectra and (f) FT-EXAFS spectra of 3D Fe0.25-800, 3D Fe0.5-800, and 3D Fe-800. | ||
The Fe K edge XAFS spectra of these samples were further utilized to identify the local electronic structure of each catalyst. As depicted in Fig. 7d and e, the absorption edge of the Fe K-edge XANES spectrum shifts to higher energies as the Fe content decreases, indicating an increase in the valence state of Fe. Interestingly, this observation is inconsistent with XPS results that reveal an increase in the intensity of the Fe 2p2/3 peak of Fe2+ from 3D Fe0.5-800 to 3D Fe0.25-800. In this regard, the valence state of Fe is assumed to be significantly influenced by the state of Fe3+. Additionally, the k3-weighted FT-EXAFS spectrum of 3D Fe0.25-800 also exhibits a distinctive shoulder peak similar to that of 3D Fe/DF-700, suggesting the formation of Fe–O bonds (Fig. 7f). Based on the quantitative least squares EXAFS curve-fitting analysis, the coordination configurations of Fe atoms in 3D Fe0.25-800 and 3D Fe0.5-800 are determined as Fe–ON3 and Fe–N4, respectively.
In general, the above investigation reveals a positive correlation between the metal content in the precursor and the extent of carbonization of the DF polymer. The precursor of 3D Fe0.25-800 contains such a low concentration of iron nitrate that insufficient carbonization occurs, resulting in the formation of a Fe–ON3 moiety instead of the typical Fe–Nx structure. The coordinated O atom should originate from the formaldehyde and/or SiO2 template. Notably, the unsuccessful conversion of the DF polymer to the desired carbon substrate also occurs when the pyrolysis temperature fails to reach a certain threshold, despite an adequate Fe content in the precursor. According to relevant publications, Fe-induced catalytic graphitization does not occur at temperatures below 715 °C.51,52 Thus, the pyrolysis of the precursor at 700 °C cannot achieve sufficient carbonization of the DF polymer, resulting in the presence of abundant O-defects in the carbon substrate. Due to the interference of O atoms, most individual Fe atoms tend to coordinate with both O and N atoms, forming Fe–OyNx moieties. Unfortunately, these O-coordinated atomic Fe sites can be damaged during the NaOH etching treatment due to their limited structural stability. 3D Fe-700 consequently suffers from ultra-low Fe loadings, as only a few stable Fe–N4 sites are formed and well maintained in the end. It is also noteworthy that an excessive Fe content in the precursor is conducive to obtaining a carbon substrate with fewer O-defects, whereas it also results in a significant decrease in the number of available N sites for anchoring isolated Fe atoms. Thus, the coordination number of Fe–N for 3D Fe-800 decreases to 3.1 compared with 3D Fe0.5-800, and Fe-based NPs are also observed in the 3D Fe-800 catalyst.
In addition, the formation mechanism of Fe–OxNy configurations for the post-impregnation method is also proposed. Since previous analyses demonstrate that the isolated Fe atoms prefer bonding with O atoms in carbon substrates during an insufficient carbonization process, the Fe ions should be initially captured by oxygen functional groups via a coordination bond on the surface of 3D DF-X supports during the impregnation process, and then immobilized into the carbon skeleton after the first reduction treatment at 300 °C. During the second reduction treatment at a higher temperature, the transformation of the coordination configuration from Fe–O bonds to Fe–N bonds is achieved by an in situ anion exchange reaction, wherein specific nitrogen atoms could replace the coordinating oxygen atoms and the covalent coordination interactions is enhanced simultaneously.43 In fact, this process is indeed a complex one that is influenced by various factors, such as the initial coordination mode of Fe–O and the availability of suitable N sites. Considering that a higher pyrolysis temperature is also beneficial for promoting the carbonization of the DF polymer, it is reasonable that the coordination number of O in the Fe–OxNy moiety for 3D Fe/DF-700 is higher than 3D Fe/DF-800. In parallel, the increased oxidation state of Fe in 3D Fe/DF-800 indicates enhanced covalent coordination interactions between the metal center and the N and O ligands. In the end, the relationship between the coordination configuration of the atomic Fe site and the extent of carbonization of the DF polymer is illustrated in Fig. 8.
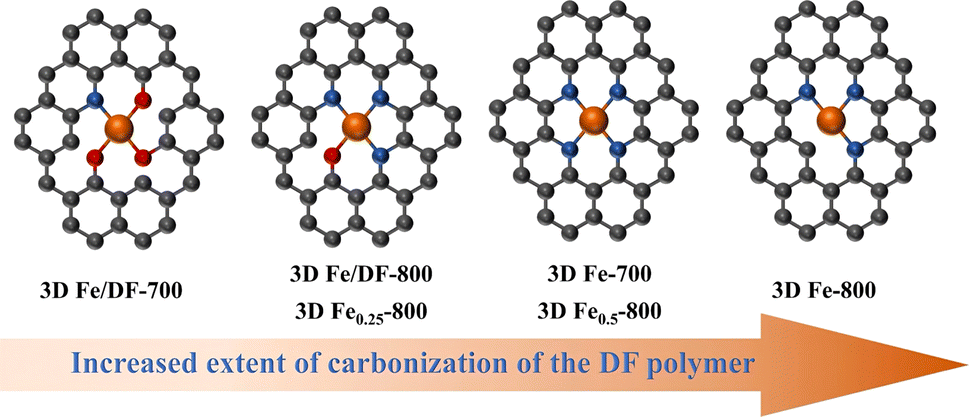 | ||
| Fig. 8 The relationship between the coordination configuration of the atomic Fe site and the extent of carbonization of the DF polymer. | ||
Effect of the synthesis method on the catalytic performance
The catalytic activity was evaluated by catalysing the oxidative coupling of benzylamine under solvent-free conditions (Table S5†). In general, except for 3D Fe-700, 3D Fe-X catalysts exhibit higher imine yields compared with their counterparts obtained through post-impregnation methods, as depicted in Fig. 9a. Among them, 3D Fe-800 displays the highest benzylamine conversion (99.3%) and selectivity (99.1%) toward the desired imine product. Despite the 3D Fe/DF-800 catalysts having approximately twice the Fe content compared with the 3D Fe-800 catalysts, the turnover frequency (TOF) value of 3D Fe-800 (47 h−1) is around twice as high as that of 3D Fe/DF-800 (24 h−1). Similarly, 3D Fe/DF-700 has an approximately three-fold higher Fe content compared with 3D Fe-700, but its imine yield shows only a marginal increase of 6% over that of the latter. These observations indicate the superior catalytic activity of Fe–Nx over Fe–OxNy. Besides, the catalytic efficiency of both series of catalysts exhibits a consistent trend with increasing pyrolysis temperature, and 800 °C is considered the most suitable pyrolysis temperature for obtaining the optimal Fe–Nx coordination configuration. As the pyrolysis temperature increases from 800 °C to 900 °C, there is a certain decrease in catalytic efficiency of 3D Fe/DF-900 and 3D Fe-900, which is ascribed to the limited number of atomic Fe actives and abundant inactive Fe-based nanoparticles caused by the excessive carbonization process. The optimal catalysts selected from the 3D Fe-X and 3D Fe/DF-X catalysts were further assessed using various amine derivatives under the same reaction conditions. As shown in Table S7,† both 3D Fe/DF-800 and 3D Fe-800 exhibited good applicability towards a wide range of aromatic and aliphatic amines, with 3D Fe-800 possessing a higher catalytic activity than 3D Fe/DF-800. | ||
| Fig. 9 (a) Catalytic performances of the 3D DF-X supports, 3D Fe/DF-X, and 3D Fe-X catalysts; (b) the color of the solutions extracted from the specific reaction system (from left to right: 3D Fe/DF-700, 3D Fe-700, 3D Fe/DF-800, 3D Fe-800, and N-benzylidene benzylamine reference); comparison of the recyclability of (c) 3D Fe/DF-700 vs. 3D Fe-700 and (d) 3D Fe/DF-800 vs. 3D Fe-800; (e) XRD patterns and (f and g) TEM images of used 3D Fe/DF-800 and 3D Fe-800. | ||
In addition to catalytic activity, an assessment of the stability of the as-prepared catalysts was also conducted. Upon the separation of the 3D Fe/DF-700 and 3D Fe/DF-800 from their individual reaction systems after the initial reaction, the remaining reaction solutions exhibited a distinct blood-red colour (Fig. 9b), implying the leaching of Fe atoms. This hypothesis was subsequently validated by ICP-OES measurements (Table S6†). It is not surprising that the catalytic activity of these catalysts exhibited a significant decrease during subsequent recycling tests. Specifically, after five reuse cycles, the imine yields of 3D Fe/DF-700 (Fig. 9c) and 3D Fe/DF-800 (Fig. 9d) were reduced to 43.8% and 61.0%, respectively. To some extent, the more pronounced decline in catalytic performance observed in 3D Fe/DF-700 during recyclability experiments also demonstrates a higher fraction of metal–ligand coordination bonds in 3D Fe/DF-700 compared with 3D Fe/DF-800. On the contrary, the colour of the extracted reaction solutions from the 3D Fe-X catalysts initially catalysed reaction systems was pale yellow, corresponding to the desired imine product N-benzylidene benzylamine. No obvious Fe leaching was detected in the used 3D Fe-X catalysts by ICP-OES. Moreover, the 3D Fe-800 catalyst maintained its catalytic performance after 5 reuse circles with a negligible activity loss of approximately 5%. It is noted that the structure and morphology of both 3D Fe/DF-800 and 3D Fe-800 were hardly affected by the stability test (Fig. 9e–g), indicating that metal leaching has very little effect on the carbon skeleton of the 3D Fe/DF-X catalysts. Overall, the direct pyrolysis approach exhibits superior advantages over the post-impregnation method in synthesizing highly active and stable atomic Fe sites.
The catalytic behaviour of different active sites
Notably, the bare 3D DF-700 exhibited an unexpectedly high imine yield of 61.9%, surpassing that achieved by both 3D Fe/DF-700 and 3D Fe-700. According to relevant literature, the functional group (N–CO) possesses favourable catalytic activity toward aerobic oxidation reactions, because the adjacent N atom could modulate the local electronic structure of the carbonyl group, resulting in enhanced activation capacity for C–H bonds and oxygen molecules.53 Considering that the incorporation of Fe atoms into 3D DF-700 resulted in the consumption of a certain amount of O functional groups and a consequent decrease in imine yield for 3D Fe/DF-700, the activity of Fe–O3N active sites should be inferior to N–CO. In other words, the presence of Fe impurities with inappropriate electronic structures can have detrimental effects on the practical application of carbon-based materials.54 Furthermore, the O 1s XP spectra analysis provides convincing evidence to support the above hypothesis. In detail, the content of surface N–CO decreases from 9.39% for 3D DF-700 to 4.72% for 3D Fe/DF-700. It is worth noting that all 3D Fe-X catalysts possess a very low content of N–CO functional groups (less than 0.96%) due to a higher degree of carbonization. Therefore, the catalytic contributions from N–CO can be roughly ruled out in 3D Fe-X catalysts.
Due to the complex distribution of different active sites in as-prepared 3D Fe/N-C catalysts, it is better to identify the role of each active site in the oxidation reaction. Considering the pivotal role of reactive oxygen species (ROS) in the solvent-free aerobic coupling of benzylamine, the electron paramagnetic resonance (EPR) technique was used to elucidate the correlation between active sites and ROS. At first, the activity of atomic Fe active sites was assessed by employing 2,2,6,6-tetramethylpiperidine (TEMP) as a singlet oxygen (1O2) trapping agent, since only Fe single atoms possess the capability to activate molecular oxygen (O2) into 1O2 thermally.12,15,55 As shown in Fig. 10a and S13a,† the characteristic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet EPR signal of TEMP–1O2 is only observed for the catalysts 3D Fe/DF-700, 3D Fe/DF-800, 3D Fe-700, and 3D Fe-800. The close-up observation shows that the signal intensity of the 3D Fe-X catalysts is slightly higher than that of the 3D Fe/DF-X catalysts, suggesting the superior capability of Fe–Nx moieties over Fe–OyNx moieties in generating singlet oxygen. Taking the Fe loadings into account, the activity ranking of these atomic Fe sites should be Fe–N4 > Fe–N3 > Fe–ON3 > Fe–O3N. Notably, while 3D Fe-700 exhibits comparable efficiency in generating 1O2 compared with 3D Fe/DF-800, its catalytic performance is significantly inferior to that of 3D Fe/DF-800. This observation implies that 1O2 is not the sole reactive oxygen species involved in the oxidation of benzylamine.
:1:1 triplet EPR signal of TEMP–1O2 is only observed for the catalysts 3D Fe/DF-700, 3D Fe/DF-800, 3D Fe-700, and 3D Fe-800. The close-up observation shows that the signal intensity of the 3D Fe-X catalysts is slightly higher than that of the 3D Fe/DF-X catalysts, suggesting the superior capability of Fe–Nx moieties over Fe–OyNx moieties in generating singlet oxygen. Taking the Fe loadings into account, the activity ranking of these atomic Fe sites should be Fe–N4 > Fe–N3 > Fe–ON3 > Fe–O3N. Notably, while 3D Fe-700 exhibits comparable efficiency in generating 1O2 compared with 3D Fe/DF-800, its catalytic performance is significantly inferior to that of 3D Fe/DF-800. This observation implies that 1O2 is not the sole reactive oxygen species involved in the oxidation of benzylamine.
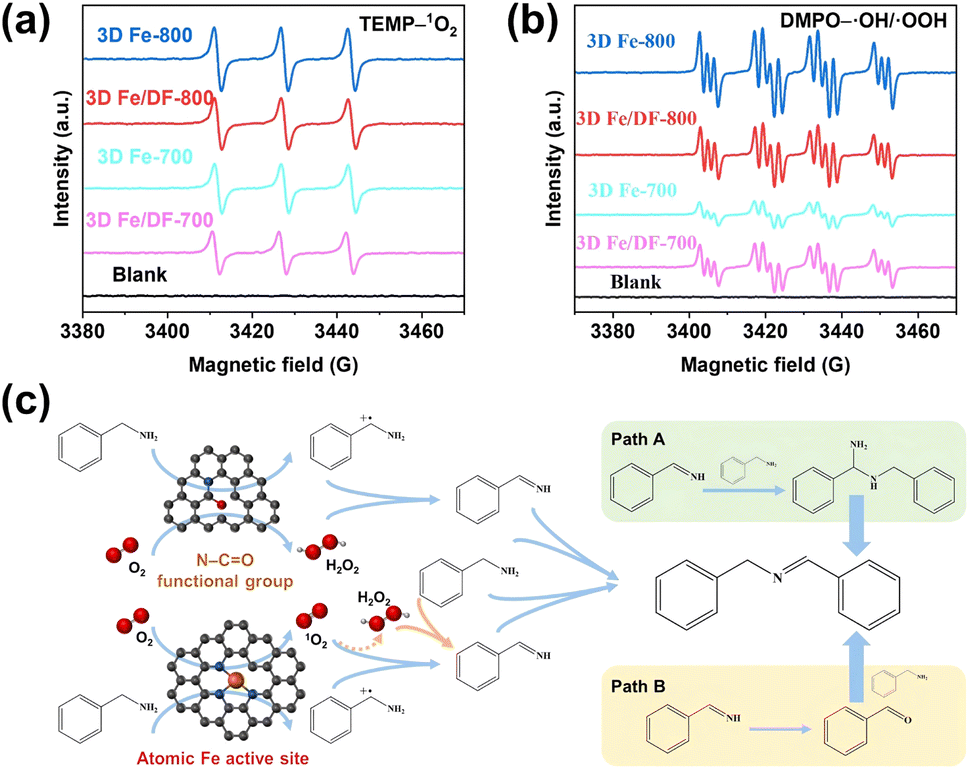 | ||
| Fig. 10 EPR signals of (a) TEMP–1O2 adduct, and (b) DMPO–˙OH/˙OOH adducts in the presence of 3D Fe/DF-700, 3D Fe-700, 3D Fe/DF-800, and 3D Fe-800 at 110 °C; (c) proposed reaction mechanism for the oxidative coupling of benzylamine. | ||
As anticipated, in the presence of 5,5-dimethyl-2-pyrrolidone-N-oxyl (DMPO) as a trapping agent, a combined signal of DMPO–˙OH and DMPO–˙OOH is observed for these catalysts (Fig. 10b). Significantly, the order of their individual intensity is consistent with the ranking of their catalytic performance, highlighting the importance of the hydroxyl radicals (˙OH) and H2O2 toward an efficient oxidative coupling of benzylamine. Besides, a reference experiment confirms the capacity of N–CO functional groups toward efficient activation of O2 (Fig. S13b†). Based on the previously reported reaction pathway (Fig. 10c), we assume that the poor catalytic performance of 3D Fe-700 is attributed to the insufficiency of both Fe–N4 active sites and N–CO functional groups to activate benzylamine and molecular oxygen to form the corresponding cationic amine radical and H2O2.56 Specifically, in the absence of sufficient cationic amine radicals, the generated 1O2 fails to undergo the conversion into H2O2 and to produce abundant key imine intermediates (PhCHNH), thereby impeding the progress of the catalytic process. In contrast to 3D Fe-700, 3D Fe0.5-800 possesses a higher number of Fe–N4 moieties and consequently exhibits a significantly enhanced imine yield (90.8%), demonstrating the robust catalytic activity of the Fe–N4 moiety. In addition, the 3D Fe/DF-800 exhibits a noticeably enhanced ability to generate ROS compared with the 3D Fe/DF-700, suggesting the significance of the lower O coordination number in atomic Fe active sites toward favourable catalytic performance. Thus, its catalytic efficiency in imine production cannot reach the level achieved by 3D Fe0.5-800 and Fe-800. Particularly, the Fe–N3 moiety in 3D Fe-800 exhibits the highest activity among all active sites, with a remarkable ability to simultaneously activate benzylamine and molecular oxygen with exceptional efficiency. Finally, the activity order of these atomic Fe sites can be inferred as follows: Fe–N3 > Fe–N4 > Fe–ON3 > Fe–O3N.
Conclusions
In this study, we aimed to bridge the understanding gap regarding the influence of the synthesis method on the coordination configuration of atomic Fe active sites and their catalytic performance. The experimental results demonstrate that the primary distinction between the direct pyrolysis approach and the post-impregnation method lies in the catalytically induced graphitization of the DF polymer at 800 and 900 °C, which plays a critical role in regulating the coordination mode of atomic Fe sites. Considering that the incorporation of the metal salt in the precursor significantly facilitates the carbonization of the DF polymer and even induces partial graphitization of the carbon matrix, the 3D Fe-X catalysts prepared by the direct pyrolysis approach exhibit typical Fe–Nx structures with favourable reactivity and stability. In contrast, the 3D Fe/DF-X catalysts present a special Fe–OyNx configuration with unsatisfactory structural stability, which is mainly attributed to the absence of metal salts in the preparation of 3D DF-X supports and thus the lack of iron-assisted catalytic graphitization. The carbon substrates derived from an insufficient carbonization process possess poor resistance to concentrated NaOH, resulting in the formation of abundant O-defects on their surface during the etching of the hard template. Although the presence of a certain amount of surface O functional groups benefits the metal impregnation, the subsequent reduction treatment cannot recover residual O-defects in 3D DF-X carbon supports and achieve the transformation of whole Fe–O bonds to Fe–N bonds. The catalytic results and EPR analysis subsequently demonstrate that Fe–Nx moieties exhibit superior performance compared with Fe–OyNx moieties in the solvent-free aerobic coupling of benzylamine. The activity ranking of those atomic Fe sites is inferred as follows: Fe–N3 > Fe–N4 > Fe–ON3 > Fe–O3N, which is consistent with the order of the degree of carbonization of the DF polymer. Hence, the key point underlying the different synthetic strategies for obtaining the desired 3D Fe/N-C catalysts with excellent catalytic activity and favourable stability is the ability to optimize the properties of the carbon matrix. Compared with the post-impregnation method, the direct pyrolysis approach enables the in situ formation of atomic metal sites within the growing carbon frameworks, favouring the precise regulation of the properties of carbon supports. Therefore, the direct pyrolysis approach is expected to gain increasing popularity in future research.Data availability
The data supporting this article have been included as part of the ESI† and are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the “Center for Solvation Science ZEMOS” funded by the German Federal Ministry of Education and Research (BMBF) and by the Ministry of Culture and Science of North Rhine-Westphalia. G. L. gratefully acknowledges the financial support for his PhD studies from the Chinese Scholarship Council (CSC). The authors are grateful to Noushin Arshadi for N2 adsorption measurements, Jessica Aouag for ICP-OES measurements, Dr Jian Zhang for SEM measurements, and Dr Yenting Chen and Dr Markus Heidelmann for TEM measurements.Notes and references
- J. Tian, Y. Zhu, X. Yao, L. Yang, C. Du, Z. Lv, M. Hou, S. Zhang, X. Ma and C. Ca, J. Mater. Chem. A, 2023, 11, 5288 RSC
.
- R. Xu, B. Xu, X. You, D. Shao, G. Gao, F. Li, X. Wang and Y. Yao, J. Mater. Chem. A, 2023, 11, 11202 RSC
- J. Zhao, X. Ren, X. Liu, X. Kuang, H. Wang, C. Zhang, Q. Wei and D. Wu, Chem. Eng. J., 2023, 452, 139533 CrossRef CAS
- M. Shen, J. Qi, K. Gao, C. Duan, J. Liu, Q. Liu, H. Yang and Y. Ni, Chem. Eng. J., 2023, 464, 142719 CrossRef CAS
- H. Li, K. Du, C. Xiang, P. An, X. Shu, Y. Dang, C. Wu, J. Wang, W. Du, J. Zhang, S. Li, H. Tian, S. Wang and H. Xia, J. Mater. Chem. A, 2020, 8, 17136 RSC
- M. Huang, B. Deng, X. Zhao, Z. Zhang, F. Li, K. Li, Z. Cui, L. Kong, J. Lu, F. Dong, L. Zhang and P. Chen, ACS Nano, 2022, 16, 2110–2119 CrossRef CAS PubMed
- Y. Wang, F. Chu, J. Zeng, Q. Wang, T. Naren, Y. Li, Y. Cheng, Y. Lei and F. Wu, ACS Nano, 2021, 15(1), 210–239 CrossRef CAS PubMed
- X. Guo, Q. Zhang, H. He, A. Cai, S. Xi, J. Du, F. Zhang, X. Fan, W. Peng and Y. Li, Appl. Catal., B, 2023, 335, 122886 CrossRef CAS
- Y. Zhao, Y. Guo, X. F. Lu, D. Luan, X. Gu and X. W. Lou, Adv. Mater., 2022, 34, 2203442 CrossRef CAS PubMed
- Y. R. Lv, X. J. Zhai, S. Wang, H. Xu, R. Wang and S. Q. Zang, Chin. J. Catal., 2021, 42, 490–500 CrossRef CAS
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS
- H. He, K. Ma, H. Liu, J. Li, L. Zheng, F. Zhang, X. Fan, W. Peng, J. Ji and Y. Li, J. Mater. Chem. A, 2022, 10, 24831–24838 RSC
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2000896 CrossRef CAS PubMed
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, B. Song, H. Yi, S. Ye and X. Ren, Small, 2020, 16, 2001634 CrossRef CAS PubMed
- Z. Ma, S. Liu, N. Tang, T. Song, K. Motokura, Z. Shen and Y. Yang, ACS Catal., 2022, 12, 5595–5604 CrossRef CAS
- G. Lu, P. Schwiderowski, Z. Shen, X. Li, J. Schulwitz, B. Peng, G. Zhao, X. Huang and M. Muhler, Chem. Mater., 2024, 36, 2049–2060 CrossRef CAS
- Y. Zhou, C. Ma, X. Wang, Z. Xiang, C. Yin, W. Yan, W. He, Y. Liu, C. Lu and X. Li, Mater. Today Chem., 2023, 28, 101340 CrossRef CAS
- Z. Zhang, Y. Chen, L. Zhou, C. Chen, Z. Han, B. Zhang, Q. Wu, L. Yang, L. Du, Y. Bu, P. Wang, X. Wang, H. Yang and Z. Hu, Nat. Commun., 2019, 10, 1657 CrossRef PubMed
- Y. Wang, R. Shi, L. Shang, G. I. N. Waterhouse, J. Zhao, Q. Zhang, L. Gu and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13057–13062 CrossRef CAS PubMed
- J. Büker, X. Huang, J. Bitzer, W. Kleist, M. Muhler and B. Peng, ACS Catal., 2021, 11, 7863–7875 CrossRef
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed
- Z. Ren, X. Wang, S. Wang, H. Zhang, B. Huang, Y. Dai and W. Wei, J. Mater. Chem. A, 2023, 11, 11507 RSC
- W. Chen, X. Luo, T. J. A. Slater, Y. Zhou, S. Ling, R. Bao, J. A. Fernandes, J. Wang and Y. Shen, J. Mater. Chem. A, 2020, 8, 25959 RSC
- Q. Miao, C. Lu, Q. Xu, S. Yang, M. Liu, S. Liu, C. Yu, X. Zhuang, Z. Jiang and G. Zeng, Chem. Eng. J., 2022, 450, 138427 CrossRef CAS
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H. T. Wang, C.-W. Pao, A. R. Head, C. H. Wang, X. Tong, C. J. Sun, W. F. Pong, J. Luo, J. C. Zheng and H. L. Xin, Nat. Mater., 2022, 21, 681–688 CrossRef CAS PubMed
- L. Wu, S. Hu, W. Yu, S. Shen and T. Li, npj Comput. Mater., 2020, 6, 23 CrossRef CAS
- Y. Tang, W. Chen, B. Wu, G. Zhao, Z. Liu, Y. Li and X. Dai, ChemPhysChem, 2019, 20, 2506–2517 CrossRef CAS PubMed
- J. Shan, J. Liao, C. Ye, J. Dong, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202213412 CrossRef CAS PubMed
- C. Hu, W. F. Tsai, W. H. Wei, K. Y. Andrew Lin, M. T. Liu and K. Nakagawa, Carbon, 2021, 175, 467–477 CrossRef CAS
- I. Dirba, C. A. Schwöbel, A. Zintler, P. Komissinskiy, L. Molina-Luna and O. Gutfleisch, Nanoscale Adv., 2020, 2, 4777–4784 RSC
- Y. Tan, K. Zhu, D. Li, F. Bai, Y. Wei and P. Zhang, Chem. Eng. J., 2014, 258, 93–100 CrossRef CAS
- Y. Li, Y. Li, X. Huang, H. Zheng, G. Lu, Z. Xi and G. Wang, Compos. Sci. Technol., 2020, 195, 108197 CrossRef CAS
- X. Li, C. S. Cao, S. F. Hung, Y. R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T. S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS
- B. Apicella, C. Russo, A. Ciajolo, L. Cortese, F. Cerciello, F. Stanzione, A. Wuetscher, M. Muhler and O. Senneca, Fuel, 2019, 241, 264–272 CrossRef CAS
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS
- X. C. Wang, Z. Q. Li, P. Wu, E. Y. Jiang and H. L. Bai, Appl. Surf. Sci., 2006, 253, 2087–2092 CrossRef CAS
- M. Ayiania, E. Weiss-Hortala, M. Smith, J. S. McEwen and M. Garcia-Perez, Carbon, 2020, 167, 559–574 CrossRef CAS
- Y. Liu, S. Bai, Q. Li, Z. Wu, T. Shen, J. Chu and Y. F. Song, Chem. Eng. J., 2023, 477, 146610 CrossRef CAS
- L. Li, Y. Wen, G. Han, F. Kong, L. Du, Y. Ma, P. Zuo, C. Du and G. Yin, Small, 2023, 19, 2300758 CrossRef CAS PubMed
- X. Xu, T. Yuan, Y. Zhou, Y. Li, J. Lu, X. Tian, D. Wang and J. Wang, Int. J. Hydrogen Energy, 2014, 39, 16043–16052 CrossRef CAS
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J. O. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Adv. Mater., 2022, 34, 2110653 CrossRef CAS PubMed
- B. Lin, G. Yang, B. Yang and Y. Zhao, Appl. Catal., B, 2016, 198, 276–285 CrossRef CAS
- L. Zong, K. Fan, L. Cui, F. Lu, P. Liu, B. Li, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309784 CrossRef CAS PubMed
- Z. Zhang, T. Wang, W. Wang, X. Wang, X. Luo, C. Cheng and X. Liu, Adv. Energy Mater., 2023, 13, 2300325 CrossRef CAS
- E. Zhu, C. Sun, C. Shi, J. Yu, X. Yang and M. Xu, Chem. Eng. J., 2023, 463, 142468 CrossRef CAS
- M. Zhang, C. Lai, F. Xu, D. Huang, T. Hu, B. Li, D. Ma, S. Liu, Y. Fu, L. Li, L. Tang and L. Chen, Small, 2023, 19, 2301817 CrossRef CAS PubMed
- W. Song, C. Xiao, J. Ding, Z. Huang, X. Yang, T. Zhang, D. Mitlin and W. Hu, Adv. Mater., 2024, 36, 2301477 CrossRef CAS PubMed
- V. Giulimondi, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2023, 13, 2981–2997 CrossRef CAS PubMed
- H. Zhao, J. Fang, D. Xu, J. Li, B. Li, H. Zhao and Z. Dong, Catal. Sci. Technol., 2022, 12, 641 RSC
- Y. Pan, M. Wang and C. Feng, RSC Adv., 2022, 12, 20578 RSC
- J. He, D. Zhang, Y. Wang, J. Zhang, B. Yang, H. Shi, K. Wang and Y. Wang, Appl. Surf. Sci., 2020, 515, 146020 CrossRef CAS
- A. Gomez-Martin, Z. Schnepp and J. Ramirez-Rico, Chem. Mater., 2021, 33, 3087–3097 CrossRef CAS
- H. Wei, Y. Ma, J. Luo, K. H. Wu, W. Xie, G. Wen, C. L. Chiang, W. Yan, S. Perathoner, G. Centi and Y. Liu, Carbon, 2020, 170, 338–346 CrossRef CAS
- W. Kiciński and S. Dyjak, Carbon, 2020, 168, 748–845 CrossRef
- W. Ma, J. Mao, C. He, L. Shao, J. Liu, M. Wang, P. Yu and L. Mao, Chem. Sci., 2022, 13, 5606–5615 RSC
- B. Chen, L. Wang, W. Dai, S. Shang, Y. Lv and S. Gao, ACS Catal., 2015, 5, 2788–2794 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04731a |
| This journal is © The Royal Society of Chemistry 2024 |