
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel multi-functional sites in boron-based bi-atom catalysts synergistically boost C–C coupling for efficient CO electroreduction towards ethanol†
Huong T. D.
Bui
 and
Tore
Brinck
*
and
Tore
Brinck
*
Department of Chemistry, CBH, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: tore@kth.se
First published on 4th November 2024
Abstract
The electrochemical CO reduction reaction (CORR) is faced by challenges in achieving high-value-added C2 products due to inefficient C–C bond formation and low selectivity. Using first-principles calculations, we propose a framework for boron-based bi-atom doping into a silicene monolayer (B–X@Si) to improve CORR catalytic efficiency. Transition metal (TM)-free B–B@Si and TM-containing B–Cu@Si serve as efficient bi-atom catalysts (BACs) with low limiting potentials (−0.28 and −0.63 V) and low activation barriers for C–C coupling (0.54 and 0.53 eV). The CO* binding strength of active sites with co-adsorbed CO* species follows the order TM < B < B–TM. Remarkably, the interplay within the B–TM pair strengthens CO* adsorption, driven by increased TM involvement, as characterized by the upward shift of the d-band center of TM in B–TM@Si relative to Fermi level. The coupling kinetics depend on the reactivity of C(CHO*) and CO* fragments within the decoupled CHO–CO* intermediate. Intriguingly, hetero-B–TM@Si systems display a trade-off between stronger CHO* and weaker CO* binding compared to the moderate binding observed in homo-B–B@Si. Among the TMs, Cu appears the most appropriate partner with B; the moderate synergistic effect of the B–Cu pair resulting in the smallest augmented C-affinity (CHO*) is offset by the weakest CO* binding strength on Cu itself, ensuring rapid C–C coupling similar to that of B–B@Si. Our BACs offer unique multi-functional active sites due to participation of host atoms (Si*) adjacent to the bi-dopants; these Si-atoms stabilize adsorbates, facilitate the subsequent C–C coupling step, and protect the C–O bond for selective ethanol production. This study provides theoretical insights for the development of advanced BACs with novel multi-adsorbing sites and tailored charge redistribution that enhance CO-to-C2 conversion.
1. Introduction
The ever-increasing consumption of carbon-intensive fossil energy sources such as oil, coal and natural gas has led to the continuous increase in carbon dioxide (CO2) in the atmosphere.1,2 Electroreduction of CO2 (CO2RR) and CO (CORR) into fuels and chemical feedstocks has been regarded a promising strategy to reach the carbon neutrality target.2,3 Nevertheless, the diversity of final products formed through complex multi-reaction pathways has unfortunately become a selectivity bottleneck. While numerous studies have successfully demonstrated efficient CO2RR/CORR into low-value C1 products,4–7 progress in enhancing the selectivity towards higher energy density multi-carbon chemicals (C2+), such as ethylene (C2H4) and ethanol (C2H5OH), has been limited. The lack of progress can be traced to a challenging catalytic mechanism with sluggish kinetics/high thermodynamic energy of the C–C coupling step, high complexity of the post C–C coupling stage, and hard-to-control formation of H2/C1 side products.8 Notably, the formation of C2+ compounds, where both CO2RR and CORR share a common CO* intermediate towards same products,9–11 underscores the potential to improve catalytic performance of CORR more effectively than that of CO2RR.12,13 Therefore, it is paramount to rationalize design strategies and elucidate mechanistic insights to improve CORR efficiency towards C2 products.Cu-based materials have generally been considered as the most promising candidates for efficient catalysis of CO2/CO to a wide array of carbon-containing products, but due to their low C2+ selectivity and high overpotential they have so far not been able to meet the commercial standards.14,15 Recently, progress has been made in enhancing the catalytic performance of Cu-containing bimetallic electrocatalysts through the implementation of engineering strategies such as heteroatom doping16–18 and alloying.19,20 These approaches provide dual active sites that can assist the C–C coupling process and optimize surface binding energies of adsorbed species, and thereby control the selectivity. In particular, exceptional binding of CO* has been observed in systems such as PdCu atom pairs in Pd–Cu3N,16 ZnCu binary sites in Zn alloyed with Cu(211)19 and at the PdCu interface of a PdCu bimetal alloy,20 promoting the CO dimerization process and resulting in high C2 product selectivity and activity.
Among the abovementioned dimeric active-site platforms, bi-atom catalysts (BACs) have emerged as cutting-edge hetero-catalysts that maximize atomic utilization efficiency and minimize usage of material, thus accelerating CO2/CO-to-C2 conversion. Recently, Xia et al.21 reported that two adjacent Cu atoms (Cu–N3 moiety) in a graphitic sheet promote C–C coupling for further C2H5OH formation with faradaic efficiency (FE) of ∼81.9%, and Zhao et al.22 demonstrated that MOF homometallic Cu–Cu and heterometallic Cu–Sn with the hexaiminobenzene (HAB) ligand promote the production of C2H4 and C2H5OH, respectively. Theoretical studies have predicted that Cu2 supported on a porous C2N layer (Cu2@C2N)23 and Cu–B atomic pair decorated graphitic carbon nitride (B–Cu@g-C3N4)24 are promising electrocatalysts for C2 production. Interestingly, several recent works have highlighted the superior performance of non-Cu-based BACs over their Cu-based counterparts, contributing to the development of high-performance CO2RR/CORR-to-C2 cathode catalysts.25–27 For example, FeCo/Fe2@C2N prefers to form C2H4 whereas CuCo/Cu2@C2N is selective towards CH4 formation,25 and FeB@C2N is anticipated to be more efficient for CO2RR towards C2 products than B–Cu@C2N.26
Silicene (Si) is a member of 2D semi-metallic xene family that has complementary properties to the well-known graphene, as exemplified by its enhanced chemical reactivity resulting from lowered aromaticity.28–32 Our recent study unveiled that the introduction of a single B into Si monolayer, here termed B@Si, enables CO chemisorption via a unique combination of σ donation (C to B) and π-backdonation (B to C).33,34 Additionally, the initial hydrogenation step of CO* to create CHO* consumes significantly less energy due to the participation of a neighboring host atom (Si*) alongside the B dopant, favorably binding O and C, respectively, thereby stabilizing the CHO* adsorbate on the B–Si* moiety. However, one drawback of the single-atom catalysts, SACs, is their insufficient number of active sites to facilitate the local CO concentration necessary for the vital C–C coupling in the formation of C2 products. To address this, we explore a scenario where one B atom is co-doped with an X atom (X: B or TM (Mn–Cu)) into the Si monolayer to construct BACs, denoted as B–X@Si. Our comprehensive investigation uncovers that both TM-free B–B@Si and TM-based B–Cu@Si exhibit superior catalytic performance with low limiting potentials of −0.28 and −0.63 V, respectively, predominantly generating ethanol. Notably, while B demonstrates moderate CO* binding strength, TM itself exhibits weaker binding. However, the synergy of the B–TM pair enhances CO* adsorption in proportion to the extent of TM involvement (Cu < Ni < Co), which is correlated with the closeness of the d-band center of TM to the Fermi level (EF). Moreover, the stability of the CHO* segment within CHO–CO* on B–X@Si, which is similar to that observed on single B@Si, renders the first reduction step of CO–CO* → CHO–CO* marginally endergonic. The subsequent kinetic favorability of CHO–CO* → OCHCO* coupling on B–B@Si and B–Cu@Si with low activation barriers of 0.54 and 0.53 eV, respectively, stems from the high reactivity of the C(CHO*) and CO* fragments of the decoupled CHO–CO* intermediate. The presence of TM in hetero-B–TM@Si regulates the electronic configuration of the B–TM pair, and fine-tunes the binding strength of adsorbed species involved in the coupling process. Moreover, host substrate atoms (Si*) serve as crucial active sites for stabilizing adsorbates that aid the initial C–C coupling and steer the post C–C coupling towards ethanol rather than ethylene. These findings not only shed light on the mechanism behind the pivotal C–C coupling process but also provide guidance for extending the concept of BACs with unique multi-active sites for the enhancement of CORR towards a single C2 product.
2. Computational methods
Spin-polarized density functional theory (DFT) calculations were performed using VASP35–37 with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional38 and projector augmented wave (PAW) potentials39,40 for the treatment of core electrons. An energy cutoff was set to be 500 eV. DFT-D3 by Grimme was used to describe the long-range van der Waals (vdW) interactions.41 The convergence criteria for force and electronic structure iterations were 0.02 eV Å−1 and 1 × 10−5 eV, respectively. The k-point in Brillouin zone was sampled with a 3 × 3 × 1 Monkhorst–Pack grid. The vacuum space in the z direction was set to 20 Å for all structure models. Solvation effects were included by using the implicit solvation model implemented in VASPsol.42 Bader charge analysis was deployed to compute charge population.43 The projected Crystal Orbital Hamilton Populations (pCOHP) calculations, Löwdin and Mulliken charge analyses were analyzed by the LOBSTER package.44 Vaspkit was used for data post-processing of PDOS.45Ab initio Molecular dynamics (AIMD) simulation was conducted under the NVT ensemble at 500 K with a 2 fs time step to evaluate the thermal stability of the structures.46 Transition states searches and energy barriers were carried out using the climbing image nudged elastic band (CI-NEB) method.47 Heyd–Scuseria–Ernzerhof hybrid functional (HSE06)48 was used to analyze the electronic structures of selected structures, investigating the accuracy of the PBE functional for these types of systems. Further calculational details are given in the ESI.†3. Results and discussion
3.1 Structure and stability
The pristine silicene model is designed and optimized in a stable two-dimensional low-buckled honeycomb configuration, featuring Si atoms in sp2–sp3 hybridization state (Fig. S1†). The calculated lattice constant (3.87 Å), buckling height (0.44 Å), interatomic Si–Si distance (or Si–Si bond length, dSi–Si = 2.28 Å) align well with previous studies.49–51 Two dopants, boron (B) and X, are deliberately placed into the double vacancy 5 × 5 Si supercell lattice, termed as B–X@Si, consisting of 48 Si atoms and 2 dopants, as illustrated in Fig. 1a. A homogeneous B–B@Si structure is built where the second dopant X is also B, while heterogeneous B–TM@Si structures are constructed where X represents one of the five first-row non-precious transition metals (TM: Mn, Fe, Co, Ni and Cu). The stability of the six BACs is initially examined based on their formation energies (Eform) (Fig. 1b). Most structures display negative Eform values, indicative of their thermodynamic favorability, except for B–Fe@Si. We then calculate the dissolution potentials (Udiss) of TM in single B-embedded silicene (B@Si) to assess the electrochemical stability of these metal-based catalysts. The Udiss values tend to become more positive with an increase in the atomic number of TM. As such, both B–Mn@Si and B–Fe@Si with negative Udiss values appear unstable in electrochemical environment. Therefore, three hetero-BACs (B–Co@Si, B–Ni@Si and B–Cu@Si) along with the homo-B–B@Si structure are selected for our further CORR investigation.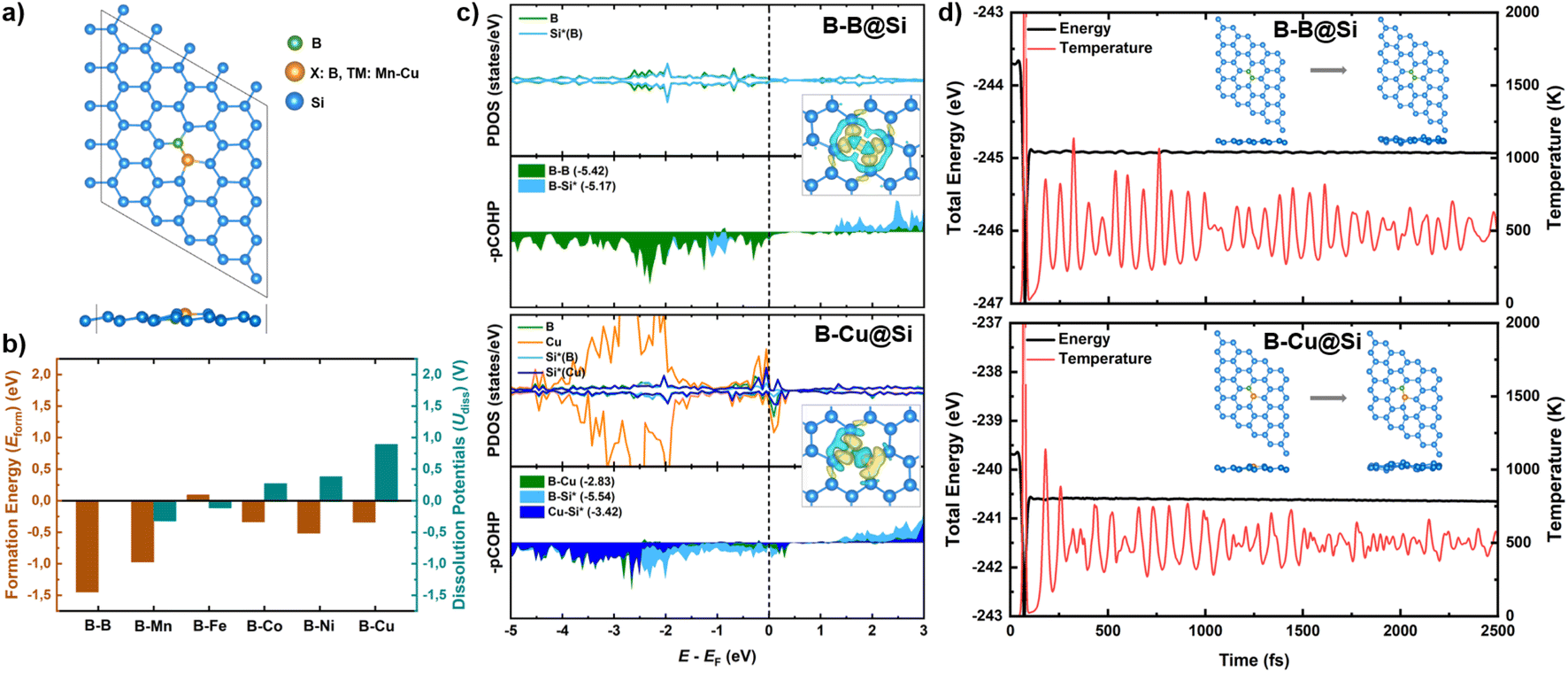 | ||
| Fig. 1 (a) Designed models of B–X bi-atom doped into silicene (B–X@Si) from top and side views. (b) Formation energies (Eform) and dissolution potentials (Udiss) of transition metals (TM) in B-embedded silicene (B@Si). (c) Partial density of states (PDOS) of dopants (B/Cu) and one adjacent substrate atom (Si*); projected crystal orbital Hamilton population (pCOHP) of the dopant–dopant and dopant–host atom bonds, and corresponding integrated COHP (IpCOHP), taking homo-B–B@Si and hetero-B–Cu@Si as examples. The Fermi level displayed in dashed line is set to zero. Their corresponding charge density difference are also displayed. The isosurface value is 0.0025 e Å−3, and the yellow (cyan) regions represent charge accumulation (depletion). (d) Ab initio molecular dynamics (AIMD) simulation of B–B@Si and B–Cu@Si at 500 K with the initial and final structures. | ||
In terms of atomic structure, the Si monolayer maintains its buckled honeycomb configuration after doping. While the introduction of two B substituents creates a nearly planar conformation in the vicinity of dopant sites in homo-B–B@Si, the TM dopants in hetero-B–TM@Si slightly protrudes out of the host substrate. The bond lengths between two dopants (dB–X), or between a dopant and its nearest surrounding host atom (Si*) (dB–Si*/dX–Si*), as detailed in Table S1,† are in good accordance with the sum of their respective covalent radii, indicating strong covalent bonds.
The structural stability of our BACs is further scrutinized through electronic structure calculations and chemical bonding analysis (Fig. 1c and S2†). Differential charge density analysis unveils substantial electron density accumulation at the bonds between X and adjacent substrate Si* atoms, as well as in the proximity of X, consistent with the Löwdin charge analysis (Fig. S3†). To be more specific, a significant amount of electrons is transferred from Si* atoms towards X, with the B dopant accepting a much more negative charge than its TM partners in hetero-B–TM@Si. This charge transfer phenomenon can be attributed to the electronegativity order: B (2.04) > TM (Co – 1.88, Ni – 1.91 and Cu – 1.90) – Si (1.90). Moreover, partial density of state (PDOS) and projected crystal Hamilton population reveal pronounced PDOS overlaps between X and neighbour Si* atoms around the Fermi level (EF), situated within bonding regions, verifying strong X–Si* bonds in the B–X@Si systems. Note that the interaction of B–Si* is stronger than that of TM–Si*, suggesting that the strong covalent bonds between non-metallic B and substrate Si* atoms primarily contribute to the high overall stability of the B–X@Si structure. More importantly, we observe a greater PDOS overlap between the two B atoms within homo-B–B@Si, visually signified by higher electron density concentrated in the B–B bond and quantitatively supported by a more negative integrated pCOHP (IpCOHP) value of the B–B bond, compared to that for B and TM within hetero-B–TM@Si, thus affirming the superior stability of the non-TM B–B@Si material over the B–TM@Si catalysts.
Intriguingly, hetero-B–TM@Si systems have a small portion of PDOS mixing between B and TM in the antibonding region across the EF, with the degree of B–TM antibonding states following the order B–Cu < B–Ni < B–Co. This suggests a higher propensity for initiating and capturing CORR-adsorbed species, following the same trend.
We further perform MD simulations of our materials, taking B–B@Si and B–Cu@Si as examples, to examine their thermal stability. From Fig. 1d, both energy and temperature show slight fluctuations over the simulation period at 500 K. Moreover, two adatoms still prefer to anchor at their favored sites in the defected Si substrate and no significant geometric distortion is observed. This stability originates from the strong covalent bonds between the two dopants and Si host atoms as well as between the two dopants themselves, confirming that the BACs are thermodynamically stable.
3.2 CORR versus HER
Hydrogen Evolution Reaction (HER), a primary side reaction during the CORR process, needs to be suppressed to increase FE of the CORR. Indeed, strong CO adsorption on the catalyst surface can hinder H from blocking active sites for CORR, thus minimizing the unexpected impact of HER on the CORR performance. Therefore, all possible configurations of H and two CO molecules adsorbed on four material surfaces were examined to identify the most stable one based on the lowest total energy (Etotal), taking B–B@Si as an example, see Fig. S4.† For the first step of HER – the Volmer reaction, a proton tends to absorb on the bridge site of two dopants to form H* (Fig. S5†). Meanwhile, these dimers appear to act as favorable active sites for binding two CO molecules via upright chemisorption, which can serve as a key 2CO* intermediate and facilitate the C–C coupling step towards C2 selectivity, as further discussed below. The adsorption energies of H* are less negative than the average ones of 2CO* in the four structures (Eads [H*] > avg_Eads [CO*]), indicating weaker adsorption of H+ compared to CO (Fig. 2). These results suggest that H+ is not likely to poison the catalytic surface; instead, the bi-doped active site thermodynamically prefers to be occupied by CO. Consequently, all four BACs exhibit a high selectivity preference for CORR over HER. | ||
| Fig. 2 Adsorption energies (Eads) of H* and average 2CO* species on B–B@Si, B–Cu@Si, B–Ni@Si and B–Co@Si. Units: eV. | ||
3.3 CORR activity and selectivity
 | ||
| Fig. 3 Free energy profiles of different C–C coupling paths on (a) B–B@Si, (b) B–Cu@Si, (c) B–Ni@Si and (d) B–Co@Si at 0 V (vs. RHE). Inserts display the corresponding optimized structures of reaction intermediates and numbers show the corresponding reaction free energy change. The solid lines indicate the C–C coupling step. Color code: B – green, Cu – orange, Ni – silver, Co – dark blue, Si – light blue, C – brown, O – red and H – light pink. | ||
The kinetic energy barriers of this coupling on the three materials are calculated to be 0.54, 0.53 and 0.94 eV, respectively (refer to Fig. 4), which is in accord with the trend of the above reaction free energy changes. Note that B–B@Si and B–Cu@Si require less activation energy for CHO* coupled to CO* than Cu (211) (0.68 eV).55 A coupling reaction step at room temperature with a kinetic barrier below 0.75 eV (corresponding to a TOF of 1 s−1) is indicative of fast kinetics.56 B–B@Si and B–Cu@Si are found to be promising candidates, thermodynamically and kinetically favoring the CO–CO* → CHO–CO* → OCHCO* coupling route.
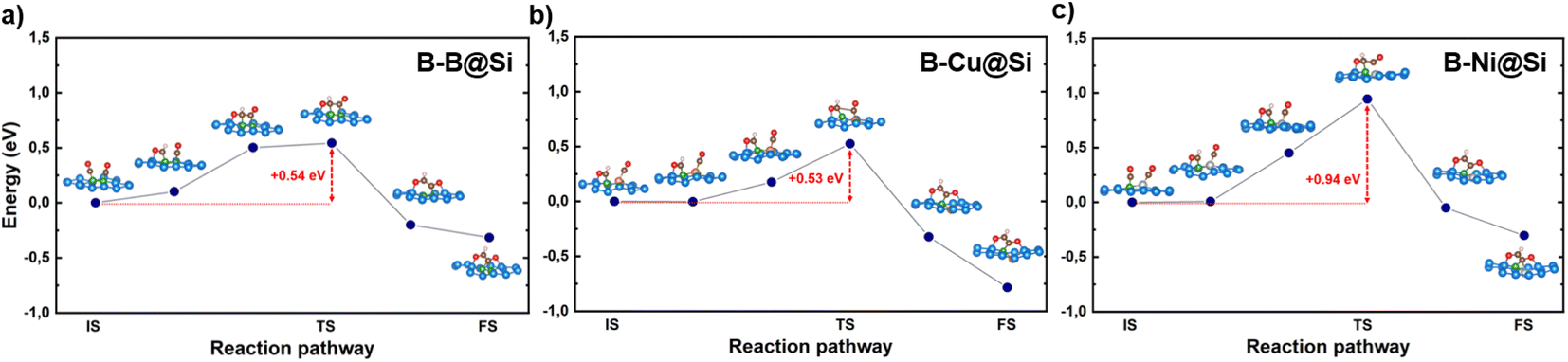 | ||
| Fig. 4 The reaction path energy diagrams for the coupling step CHO–CO* → OCHCO* on (a) B–B@Si, (b) B–Cu@Si and (c) B–Ni@Si. The insets show the atomic structures of initial state (IS), transition state (TS) and final state (FS). Red numbers indicate the activation energy barriers. Color code: B – green, Cu – orange, Ni – silver, Si – light blue, C – brown, O – red and H – light pink. | ||
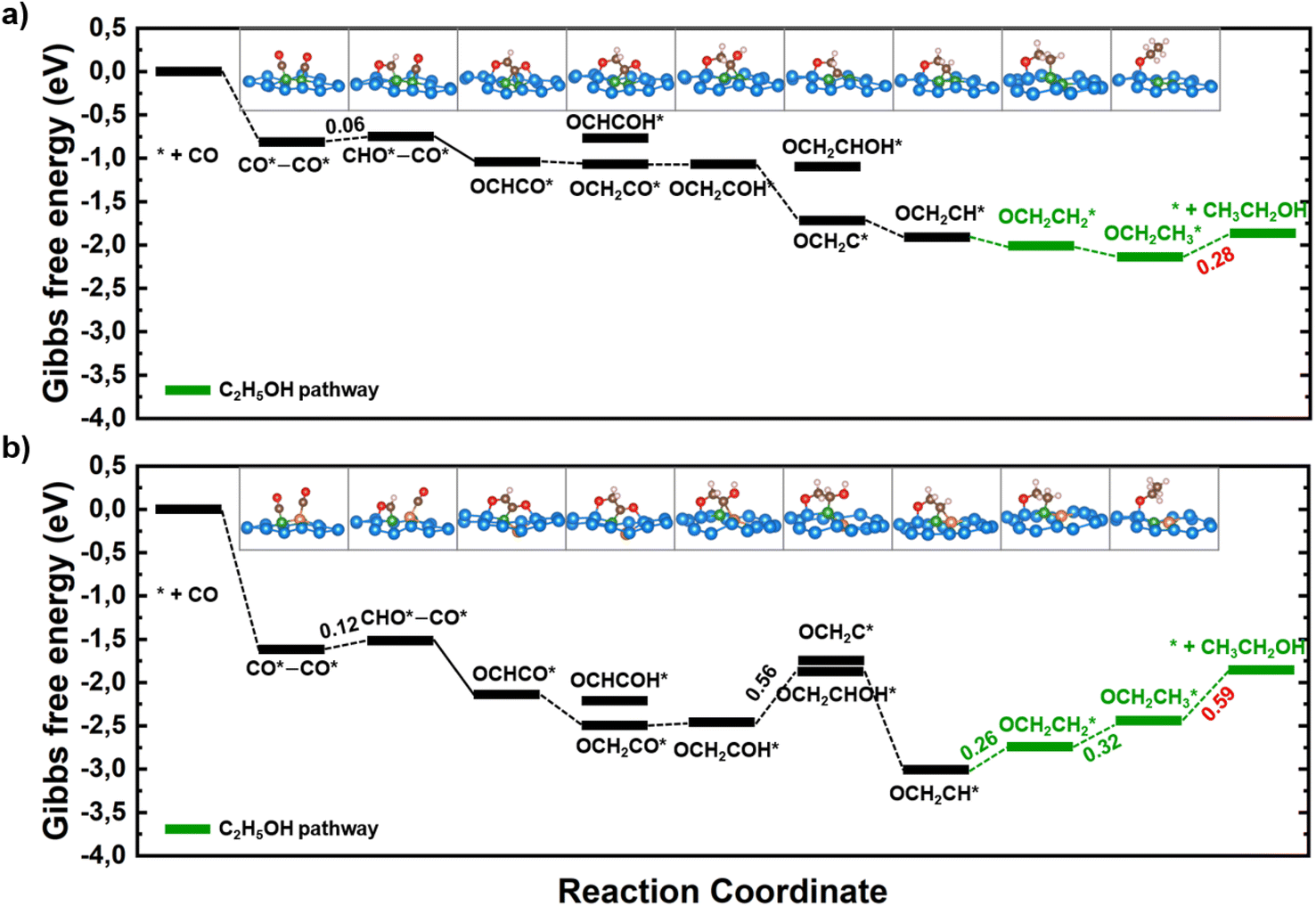 | ||
| Fig. 5 Free-energy profiles for CO reduction on (a) B–B@Si and (b) B–Cu@Si. The potential determining step is labeled with the corresponding free energy change in red color. The green line demonstrates the best overall pathway towards CH3CH2OH. The insets show the lowest-energy optimized intermediate geometries along all the most thermodynamically CORR pathway. The solid lines indicate the C–C coupling step. Color code: B – green, Cu – orange, Si – light blue, C – brown, O – red, H – light pink. | ||
The hydrogenation of the OCHCO* adsorbate on B–B@Si and B–Cu@Si yields OCH2CO* rather than OCHCOH* due to its persistent strong binding frame via B–C and two O–Si* bonds. The OCH2CO* is then protonated at the O* site to generate OCH2COH* with negligible ΔG values. Subsequently, although the free energy level of the created OCH2C* intermediate together with the dehydration on two structures is approximately equal (around −1.80 eV), the formation of OCH2CHOH* on B–B@Si (−1.07 eV) is higher than on B–Cu@Si (−1.87 eV). In this regard, the OCH2COH* is favorably hydrogenated to produce OCH2C* + H2O on B–B@Si and OCH2CHOH* on B–Cu@Si. The subsequent exergonic protonation step leads to the creation of OCH2CH*, a selectivity-determining intermediate, which bifurcates the pathway and determines the main C2 product. That is, the subsequent hydrogenation of OCH2CH* may result in HOCH2CH*, 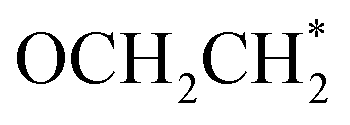 , or O* + C2H4. No C–O bond cleavage yielding C2H4 is observed after the protonation at C(OCH2CH*) to form the stable
, or O* + C2H4. No C–O bond cleavage yielding C2H4 is observed after the protonation at C(OCH2CH*) to form the stable  species (see Fig. S7†). It is understandable that after the formation of the stable key coupled OCHCO* species, the host atom (Si*) with moderate oxygen affinity preserves the C–O bond well, implying a preference to produce CH3CH2OH over C2H4. Furthermore, the
species (see Fig. S7†). It is understandable that after the formation of the stable key coupled OCHCO* species, the host atom (Si*) with moderate oxygen affinity preserves the C–O bond well, implying a preference to produce CH3CH2OH over C2H4. Furthermore, the  part within OCH2CO* retains its binding backbone with Si* over a series of sequential reduction reactions. Consequently, the
part within OCH2CO* retains its binding backbone with Si* over a series of sequential reduction reactions. Consequently, the 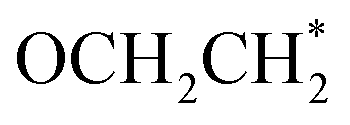 is more likely to be converted into
is more likely to be converted into  than
than  . It is predicted that both B–B@Si and B–Cu@Si undergo an endergonic reaction with a ΔG value of 0.28 and 0.59 eV, respectively; a proton attacks
. It is predicted that both B–B@Si and B–Cu@Si undergo an endergonic reaction with a ΔG value of 0.28 and 0.59 eV, respectively; a proton attacks  to yield HOCH2CH3, which is considered the rate-determining step (RDS) of the entire CORR process. Noteworthily, although the two adsorbed species, OCH2COH* and OCH2CH*, exhibit analogous favorable interaction behaviors with B–B@Si and B–Cu@Si systems via O–Si* and C–(B–B)/C–(B–Cu), they show much stronger adsorption on B–Cu@Si than on B–B@Si, making the subsequent reduction steps on B–Cu@Si endergonic. This can be mainly explained by the fact that, as C in OCH2(C)OH* and OCH2(C)H* forms four bonds, the more weakly bound electrons in B–Cu (B–Cu@Si), as previously mentioned, are more likely to strongly bind with the C to form C–B and C–Cu bonds than in B–B@Si. In other words, the optimal electronic configuration in B–B@Si leads to moderate binding strength with CORR-adsorbed species in general, resulting in slightly more favorable CORR performance on B–B@Si than on B–Cu@Si from a thermodynamic perspective.
to yield HOCH2CH3, which is considered the rate-determining step (RDS) of the entire CORR process. Noteworthily, although the two adsorbed species, OCH2COH* and OCH2CH*, exhibit analogous favorable interaction behaviors with B–B@Si and B–Cu@Si systems via O–Si* and C–(B–B)/C–(B–Cu), they show much stronger adsorption on B–Cu@Si than on B–B@Si, making the subsequent reduction steps on B–Cu@Si endergonic. This can be mainly explained by the fact that, as C in OCH2(C)OH* and OCH2(C)H* forms four bonds, the more weakly bound electrons in B–Cu (B–Cu@Si), as previously mentioned, are more likely to strongly bind with the C to form C–B and C–Cu bonds than in B–B@Si. In other words, the optimal electronic configuration in B–B@Si leads to moderate binding strength with CORR-adsorbed species in general, resulting in slightly more favorable CORR performance on B–B@Si than on B–Cu@Si from a thermodynamic perspective.
The solvation effects on the electrochemical CO reduction of BACs are examined by comparing the Gibbs free energy diagrams generated using VaspSol with those in vacuum. Specifically, as shown in Fig. 3 and S8,† the energy required for the first crucial protonation step, CO–CO* → CHO–CO*, is slightly lower in the aqueous solution compared to the vacuum on B–B@Si (0.06 vs. 0.09 eV), B–Cu@Si (0.12 vs. 0.16 eV), B–Ni@Si (0.11 vs. 0.14 eV) and B–Co@Si (0.10 vs. 0.12 eV). Similarly, from Fig. 5 and S9,† the free energy change for the PDS  is reduced with VaspSol relative to that in vacuum for two potential catalysts, B–B@Si (0.28 vs. 0.36 eV) and B–Cu@Si (0.59 vs. 0.68 eV). These comparisons show that the aqueous environment has a slightly favorable effect on the CORR performance of our BACs.
is reduced with VaspSol relative to that in vacuum for two potential catalysts, B–B@Si (0.28 vs. 0.36 eV) and B–Cu@Si (0.59 vs. 0.68 eV). These comparisons show that the aqueous environment has a slightly favorable effect on the CORR performance of our BACs.
3.4 Origin of the C–C coupling enhancement
 | ||
| Fig. 6 Adsorption energies (Eads) of 2CO* and CHO–CO* species on B–B@Si, B–Cu@Si, B–Ni@Si and B–Co@Si; and d-band centers of TM in bare hetero-B–TM@Si structures. | ||
 | ||
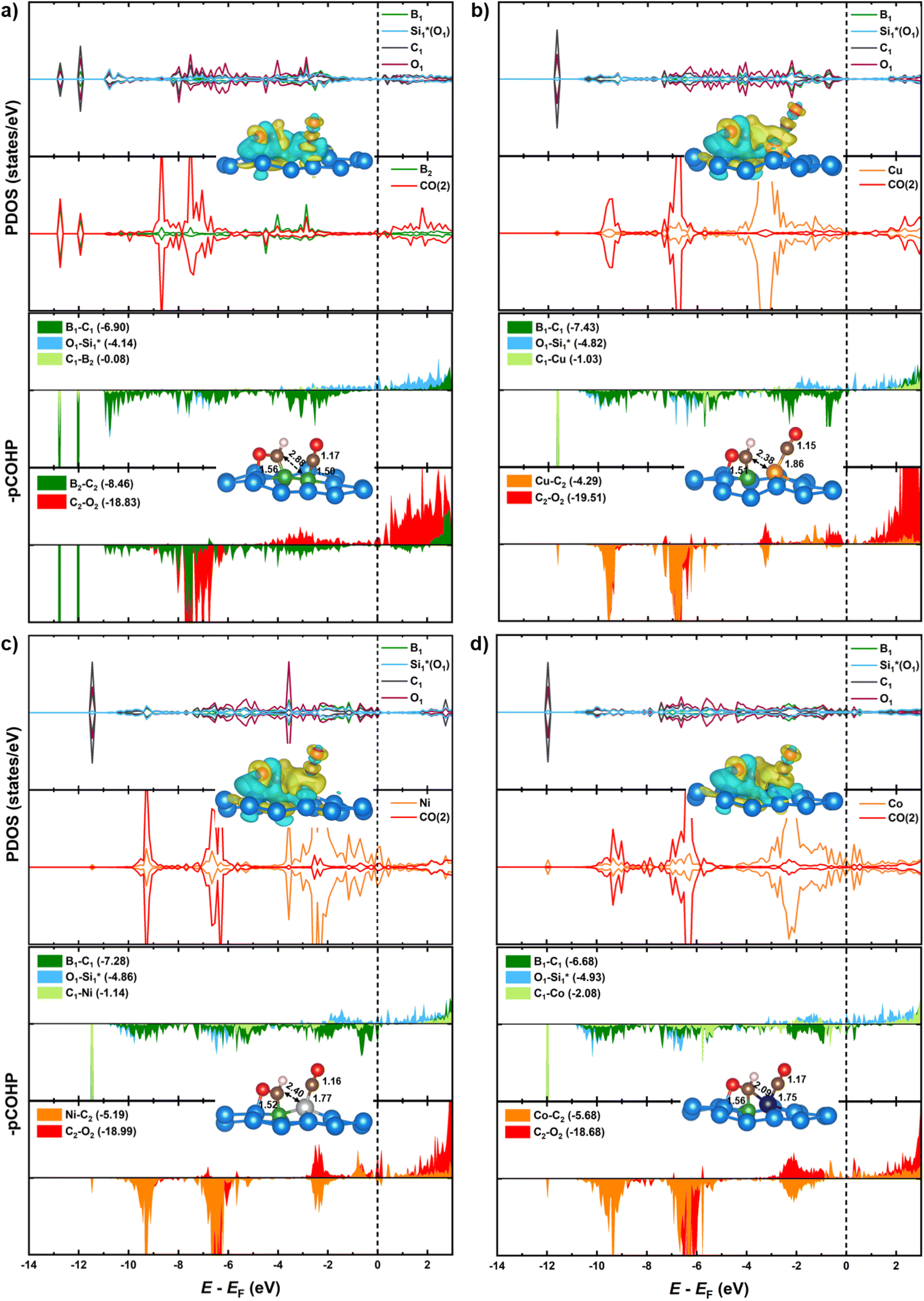
| Fig. 7 Partial density of states (PDOS) of dopants and two adsorbed CO* species (upper panels) on (a) B–B@Si, (b) B–Cu@Si, (c) B–Ni@Si and (d) B–Co@Si. Their corresponding charge density difference are also displayed. The isosurface value is set to 0.0025 e Å−3, and the yellow (cyan) regions represent charge accumulation (depletion). Projected crystal orbital Hamilton population (pCOHP) of the 2CO* intermediate. The Fermi level displayed in dashed line is set to zero. Their geometries with bond lengths are also displayed. | ||
Substituting the second B (B2) with a TM in the homo-B–B@Si system reinforces CO* co-binding with the catalytic surface, with a strengthening trend as B–B@Si < B–Cu@Si < B–Ni@Si < B–Co@Si. This strengthening can be attributed to the greater DOS contribution and denser charge clouds of TM (Cu < Ni < Co) together with the first dopant (B1) for the pronounced interaction between dual active sites (B1–TM) and the first CO molecule (CO(1)), leading to a combination of lengthened dB1–C1 and shortened dC1–TM in 2CO*-adsorbed-B–TM@Si. Illustrated in pCOHP (lower panels), more populated bonding states of the C1–TM bond (highlighted in light green), especially around EF, follow the order of B–Cu@Si < B–Ni@Si < B–Co@Si, and this is quantitively verified by more negative IpCOHP (C1–TM) values of −0.58, −1.19 and −3.12, respectively. This tendency aligns with the higher d-band center relative to the EF of TM in B–TM@Si, i.e. −2.51 (Cu), −1.58 (Ni), and −1.24 eV (Co) (see Fig. 6), resulting in an increased binding affinity of active sites (B–Cu < B–Ni < B–Co) with CO(1) as well as (Cu < Ni < Co) with CO(2). Note that the involvement of B2 (within homo-B–B@Si) in interacting with CO(1) is negligible (IpCOHP = −0.16), emphasizing the nearly independent roles of the two B in moderately binding with each CO molecule, in contrast to the hetero-B–TM@Si. In addition, the binding ability of TM (without B1) with CO(2) is markedly weaker than that of TM–B1 with CO(1) due to the PDOS of TM–C2 interaction lying in the antibonding regions across the EF (indicated by orange pCOHP curve), resulting in less negative IpCOHP values of TM–C2 bond than the IpCOHP sum of B1–C1 and TM1–C1 bonds. Concurrently, the C–O triple bond in CO(2)* is less activated than that of CO(1)*, as reflected by the lesser elongation of dC2–O2 and less negative IpCOHP (C2–O2) values. Clearly, the pCOHP shows more antibonding states of the C1–O1 bond around the EF as the B–TM pair simultaneously interacts with CO(1), suggesting that CO(1) is more likely to be reduced than CO(2) further along the CORR process. The PDOS and pCOHP observations also verify the increasing binding strength of TM with CO(2) in the sequence of Cu < Ni < Co, which originates from a higher degree of DOS mixing in three energy ranges (−10 to −9 eV, −7 to −6 eV, and −3 to −2 eV) for the engagement in TM–C2 bonding. In short, although the binding of CO(2)* on TM in hetero-B–TM@Si is weakened, the synergistic effect of the B–TM pair substantially enhances the binding of CO(1), thereby bolstering the overall stability of the co-adsorbed CO* intermediate, in comparison with the moderate binding of both CO(1) and CO(2) with homogeneous B–B@Si.
We have conducted hybrid functional calculations using the HSE06 functional and compare them with the PBE results to examine the sensitivity of the electronic structures of the 2CO*-adsorbed-B–X@Si system to the level of theory, as illustrated in Fig. S10.† Overall, the PDOS patterns with their corresponding bonding/antibonding states (depicted in pCOHP curves) are similar between two methods across the four systems; however, HSE06 shows a slight downward shift in the PDOS relative to EF compared to PBE. This leads to slightly more negative IpCOHP values of all key bonds of the system when using HSE06, but the relative order of these IpCOHP values from HSE06 is consistent with the trend observed with PBE (see Table S2† for details). For instance, in the interaction with CO(1), while the engagement of B2 in homo-B–B@Si remains minor with less negative IpCOHP values obtained by HSE06 (PBE in parentheses) of −0.20 (−0.16), the interplaying contribution of TM (Cu < Ni < Co) in hetero-B–TM@Si is more pronounced, with more negative IpCOHP values as follows: −0.67 (−0.58), −1.32 (−1.19) and −3.47 (−3.12). These comparisons validate the accuracy of the PBE functional used in our study to elucidate the mechanistic insights of CORR on our materials.
 | ||
| Fig. 8 Partial density of states (PDOS) of dopants, C and O atoms of CHO* part, and CO*(2) part (upper panels) on (a) B–B@Si, (b) B–Cu@Si, (c) B–Ni@Si and (d) B–Co@Si. Their corresponding charge density difference are also displayed. The isosurface value is set to 0.0025 e Å−3, and the yellow (cyan) regions represent charge accumulation (depletion). Projected crystal orbital Hamilton population (pCOHP) of the decoupled CHO–CO* intermediate. The Fermi level displayed in dashed line is set to zero. Their geometries with bond lengths are also displayed. | ||
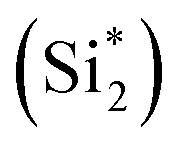 . In this manner, the high reactivity of CO*(2) and C1(CHO*) within the decoupled CHO–CO* intermediate is responsible for the coupling process. Because both CHO* and CO*(2) species are adsorbed on B dopants in the decoupled CHO–CO* intermediate with relatively moderate binding strengths, the homo-B–B@Si system favors the C–C bond formation. In the hetero-B–TM@Si system, the lower energy of the d-band center (Cu < Ni < Co) results in weaker interactions of CO*(2) and TM as well as C1(CHO*) and hybrid bi-dopants (B1–TM), as evidenced by less negative values of IpCOHP for TM–C2 and C1–TM, respectively (see lower panels in Fig. 8). It indicates that the higher reactivity of CO*(2) and C1(CHO*), governed by the more moderate TM engagement, leads to favorable C–C coupling kinetics in the order of B–Cu@Si > B–Ni@Si > B–Co@Si. There is a trade-off between stronger CHO* and weaker CO*(2) adsorption on hetero-B–TM@Si compared to homo-B–B@Si. Notably, the moderate synergistic effect of B–Cu in the B–Cu@Si results in the slightest increase in its binding strength with CHO* and the weakest binding of Cu with CO*(2) among the three hetero-B–TM@Si systems; these features allow the B–Cu@Si to kinetically favor the CHO–CO* → OCHCO* coupling step, achieving similar performance to that of B–B@Si. We also calculated magnetic moments at the individual active sites (B and B/TM) for the CHO–CO* → OCHCO* reaction step to investigate the potential role of spin for the reactivity, see Table S3.† However, the magnetic moments of the two intermediates for the two best performing catalysts (B–B@Si and B–Cu@Si) are consistently zero. Thus, we conclude that magnetization is not a significant activity descriptor for these systems.
. In this manner, the high reactivity of CO*(2) and C1(CHO*) within the decoupled CHO–CO* intermediate is responsible for the coupling process. Because both CHO* and CO*(2) species are adsorbed on B dopants in the decoupled CHO–CO* intermediate with relatively moderate binding strengths, the homo-B–B@Si system favors the C–C bond formation. In the hetero-B–TM@Si system, the lower energy of the d-band center (Cu < Ni < Co) results in weaker interactions of CO*(2) and TM as well as C1(CHO*) and hybrid bi-dopants (B1–TM), as evidenced by less negative values of IpCOHP for TM–C2 and C1–TM, respectively (see lower panels in Fig. 8). It indicates that the higher reactivity of CO*(2) and C1(CHO*), governed by the more moderate TM engagement, leads to favorable C–C coupling kinetics in the order of B–Cu@Si > B–Ni@Si > B–Co@Si. There is a trade-off between stronger CHO* and weaker CO*(2) adsorption on hetero-B–TM@Si compared to homo-B–B@Si. Notably, the moderate synergistic effect of B–Cu in the B–Cu@Si results in the slightest increase in its binding strength with CHO* and the weakest binding of Cu with CO*(2) among the three hetero-B–TM@Si systems; these features allow the B–Cu@Si to kinetically favor the CHO–CO* → OCHCO* coupling step, achieving similar performance to that of B–B@Si. We also calculated magnetic moments at the individual active sites (B and B/TM) for the CHO–CO* → OCHCO* reaction step to investigate the potential role of spin for the reactivity, see Table S3.† However, the magnetic moments of the two intermediates for the two best performing catalysts (B–B@Si and B–Cu@Si) are consistently zero. Thus, we conclude that magnetization is not a significant activity descriptor for these systems.
From Fig. 9, the main binding framework, consisting of one B1–C2 bond and two 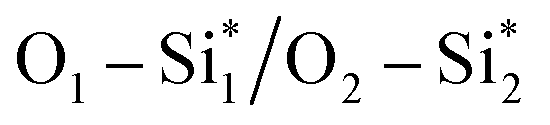 bonds, contributes to the stability of coupled OCHCO*. In fact, there is a substantial DOS overlap between B1 and C2 in the bonding region (green pCOHP) near the EF with more negative IpCOHP (B1–C2) values. Notably, the C–C bond length of the OCHCO* adsorbate on all four structures (dC1–C2 = 1.39 ∼ 1.42 Å) is shorter than the average of double and single C–C bond lengths (dC–C ≈ 1.44 Å), implying that the OCHCO* intermediate should be more stable as C2 binds primarily to B1. Indeed, a higher TM involvement (Cu < Ni < Co) in the TM–B pair within hetero-B–TM@Si for the interaction with C2 weakens the stability of OCHCO*, resulting in significantly increased ΔG values for the coupling. Notably, the charge density difference indicates a low electron density between C2 and Cu, accounting for the high stability of OCHCO*-adsorbed B–Cu@Si. Similarly, homo-B–B@Si, despite two B dopants sharing nearly equal moderate binding strength with C2, exhibits less exergonic coupling than B–Cu@Si. Additionally, two positively charged substrate atoms (Si*) close to the two dopants preferentially bind with the negatively charged O*, as indicated by charge density distribution and relatively negative IpCOHP (
bonds, contributes to the stability of coupled OCHCO*. In fact, there is a substantial DOS overlap between B1 and C2 in the bonding region (green pCOHP) near the EF with more negative IpCOHP (B1–C2) values. Notably, the C–C bond length of the OCHCO* adsorbate on all four structures (dC1–C2 = 1.39 ∼ 1.42 Å) is shorter than the average of double and single C–C bond lengths (dC–C ≈ 1.44 Å), implying that the OCHCO* intermediate should be more stable as C2 binds primarily to B1. Indeed, a higher TM involvement (Cu < Ni < Co) in the TM–B pair within hetero-B–TM@Si for the interaction with C2 weakens the stability of OCHCO*, resulting in significantly increased ΔG values for the coupling. Notably, the charge density difference indicates a low electron density between C2 and Cu, accounting for the high stability of OCHCO*-adsorbed B–Cu@Si. Similarly, homo-B–B@Si, despite two B dopants sharing nearly equal moderate binding strength with C2, exhibits less exergonic coupling than B–Cu@Si. Additionally, two positively charged substrate atoms (Si*) close to the two dopants preferentially bind with the negatively charged O*, as indicated by charge density distribution and relatively negative IpCOHP ( and
and 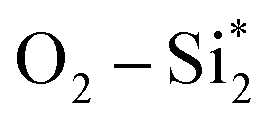 bonds). It is worth noting that the pCOHP curves of the O–Si* bonding fall into antibonding areas around the EF, indicating that the Si* atoms haves moderately strong O affinity, reducing the likelihood of C–O bond scissoring in oxygen-bound intermediates such as OCHCO* or further OCH2CO*. This preservation of the C–O bond may direct the CORR pathway bifurcation on our Si-based BACs towards C2H5OH rather than O* and C2H4, as discussed in Section 3.3.2.
bonds). It is worth noting that the pCOHP curves of the O–Si* bonding fall into antibonding areas around the EF, indicating that the Si* atoms haves moderately strong O affinity, reducing the likelihood of C–O bond scissoring in oxygen-bound intermediates such as OCHCO* or further OCH2CO*. This preservation of the C–O bond may direct the CORR pathway bifurcation on our Si-based BACs towards C2H5OH rather than O* and C2H4, as discussed in Section 3.3.2.
 | ||
| Fig. 9 Partial density of states (PDOS) of dopants, host atoms Si* (bonded to O), O1 atom (OCH*), O2 atom (CO*(2)), and C2 atom (CO*(2)) (upper panels) on (a) B–B@Si, (b) B–Cu@Si, (c) B–Ni@Si and (d) B–Co@Si. Their corresponding charge density difference are also displayed. The isosurface value is set to 0.0025 e Å−3, and the yellow (cyan) regions represent charge accumulation (depletion). Projected crystal orbital Hamilton population (pCOHP) of the coupled OCHCO* intermediate. The Fermi level displayed in dashed line is set to zero. Their geometries with bond lengths are also displayed. | ||
The proposed C–C coupling mechanism of our homo/hetero-B–X@Si is summarized in Scheme 1 to provide an overview. First, two CO molecules vertically chemisorb with C pointing downwards at the dopant-pair sites. Each of the two B dopants in homo-B–B@Si independently binds one CO molecule with moderate binding strength. In hetero-B–TM@Si, the B–TM pair collaboratively strengthens the CO(1) adsorption (highlighted in orange dashed arrow), while the TM itself displays a relatively weak binding with CO(2). Next, CO(1)* is hydrogenated to form the stable adsorbed CHO* part via and B1–C1 in homo-B–B@Si or (B1–TM)–C1 in hetero-B–TM@Si, with similar TM contribution as in CO(1)* of the 2CO* intermediate. Then, within the decoupled CHO–CO*, CO(2)* with its inherent moderate-to-weak adsorption on X is more likely to shift towards B1, concurrent with the movement of C(CHO*) out of the surface, leading to the formation of a C–C bond. Evidently, three main active centers, B1 and two substrate neighbors (Si*), are responsible for stabilizing the adsorbed species involved in the C–C coupling process (highlighted in gray dashed circles). Furthermore, B1 and X play a crucial role in regulating the reactivity of C(CHO*) for the C–C coupling (in red dashed circle). Indeed, when X is B, there seems to be no engagement of X in the moderate adsorption of CHO* on B1, explaining the high mobility of C(CHO*). Conversely, if X is TM, a reduced TM contribution (Cu < Ni < Co) to the favorable C(CHO*) adsorption results in a more active C(CHO*). More importantly, X acts as a temporary active site to retain CO(2) before supplying it as a source for C–C coupling. In this manner, weaker CO(2)* adsorption is advantageous for boosting the migration of CO(2) towards the coupling.
and B1–C1 in homo-B–B@Si or (B1–TM)–C1 in hetero-B–TM@Si, with similar TM contribution as in CO(1)* of the 2CO* intermediate. Then, within the decoupled CHO–CO*, CO(2)* with its inherent moderate-to-weak adsorption on X is more likely to shift towards B1, concurrent with the movement of C(CHO*) out of the surface, leading to the formation of a C–C bond. Evidently, three main active centers, B1 and two substrate neighbors (Si*), are responsible for stabilizing the adsorbed species involved in the C–C coupling process (highlighted in gray dashed circles). Furthermore, B1 and X play a crucial role in regulating the reactivity of C(CHO*) for the C–C coupling (in red dashed circle). Indeed, when X is B, there seems to be no engagement of X in the moderate adsorption of CHO* on B1, explaining the high mobility of C(CHO*). Conversely, if X is TM, a reduced TM contribution (Cu < Ni < Co) to the favorable C(CHO*) adsorption results in a more active C(CHO*). More importantly, X acts as a temporary active site to retain CO(2) before supplying it as a source for C–C coupling. In this manner, weaker CO(2)* adsorption is advantageous for boosting the migration of CO(2) towards the coupling.
 | ||
| Scheme 1 Proposed scenarios of C–C coupling mechanism on B–X@Si (homogeneous B–B@Si and heterogeneous B–TM@Si). Taking the coupling process on B–B@Si and B–Cu@Si for examples with both top and side views of corresponding optimized intermediate geometries. Dark gray and red circles symbolize ‘stabilize’ and ‘mobilize’ functions of active sites. Green narrow displays C–C bond and orange dashed line indicates the TM involvement. | ||
4. Conclusion
In summary, we introduce a theoretical model of boron-based bi-atom doping into a silicene monolayer (B–X@Si) for CO reduction to C2 end-products. Two bi-atom catalysts (BACs), namely TM-free homo-B–B@Si and TM-containing hetero-B–Cu@Si, stand out as promising electrocatalysts with high thermodynamic stability, low limiting potentials of −0.28 and −0.63 V, respectively, and a high selectivity towards CORR over HER.The B dopant serves as the preferred active site for CO capture and activation with its moderate binding strength via a noticeable “σ-donation – π-backdonation” pattern compared to its TM counterparts. The synergy between B and TM (B–TM) increases the adsorbed CO* stability through the greater degree of TM contribution, induced by the upward shift of the d-band center of TM towards EF. The initial reduction of CO*-bound B (or B–TM) motifs within CO* co-adsorption on homo-B–B@Si (or hetero-B–TM@Si), respectively, requires significantly less energy owing to the unique CHO* binding mode with B (or B–TM) together with an adjacent host atom (Si*). Moreover, the combined high reactivity of C(CHO*) and CO* units within the decoupled CHO–CO* drives the CHO–CO* → OCHCO* coupling on B–B@Si and B–Cu@Si with low activation energy barriers of 0.54 and 0.53 eV. The moderately strong CHO* and CO* adsorption on homo-B–B@Si makes the C–C coupling viable at room temperature. Meanwhile, the feasibility trend of hetero-B–TM@Si for this coupling is observed in decreasing order: B–Cu@Si > B–Ni@Si > B–Co@Si due to the higher engagement of TM into the synergistic effect with B for increased C-affinity (CHO*) accompanied by the stronger affinity of TM with CO*. The fine-tuned electronic structure of B–Cu@Si strikes a balance between the least sluggish C(CHO*) and the most mobile CO* and promotes the coupling on B–Cu@Si. The TM dopant thereby acts as an electronic modulator for the hybrid B–TM active sites and serves as a transient site for initial CO* adsorption before participating in the coupling process. Crucially, silicene is an excellent substrate not only for stable accommodation of dopants but also for furnishing binding sites (Si*) that stabilize adsorbates, enhance C–C coupling and protect the C–O bond for the formation of ethanol. Our results pave the way for advancing a class of BACs featuring outstanding synergistic multi-active sites and illuminate mechanistic origins of CO2RR/CORR enhancement towards high C2 selectivity.
Data availability
The data supporting this article have been included as part of the Supplementary Information.Author contributions
Huong T. D. Bui: conceptualization, data curation, formal analysis, methodology, investigation, and writing. Tore Brinck: conceptualization, methodology, investigation, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (VR) grant number 2021-05881. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the National Supercomputer Centre in Linköping University (NSC) and the PDC Centre for High Performance Computing (PDC-HPC) partially funded by the Swedish Research Council through grant agreement no. 2022-06725.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- A. Bagger, L. Arnarson, M. H. Hansen, E. Spohr and J. Rossmeisl, J. Am. Chem. Soc., 2019, 141, 1506–1514 CrossRef CAS PubMed.
- J. Cai, Q. Zhao, W.-Y. Hsu, C. Choi, Y. Liu, J. M. P. Martirez, C. Chen, J. Huang, E. A. Carter and Y. Huang, J. Am. Chem. Soc., 2023, 145, 9136–9143 CrossRef CAS PubMed.
- K. Fernández-Caso, G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, ACS Energy Lett., 2023, 8, 1992–2024 CrossRef.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- H. T. D. Bui, V. Q. Bui, X. Shao, A. Kumar, S.-G. Kim, H. M. Le, Y. Kawazoe and H. Lee, J. Phys. Chem. C, 2021, 125, 13176–13184 CrossRef CAS.
- T. Yan, X. Chen, L. Kumari, J. Lin, M. Li, Q. Fan, H. Chi, T. J. Meyer, S. Zhang and X. Ma, Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS PubMed.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, NY, 2008, pp. 89–189 Search PubMed.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- B. Ruqia, G. M. Tomboc, T. Kwon, J. Kundu, J. Y. Kim, K. Lee and S.-I. Choi, Chem Catal., 2022, 2, 1961–1988 CrossRef CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, J. Zhu, C. Ye, Y. Mao, B. Wang, G. Zhou, L. Mai, Z. Wang, X. Liu and D. Wang, Nano Lett., 2023, 23, 2312–2320 CrossRef CAS PubMed.
- X. Zhi, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Commun., 2021, 57, 9526–9529 RSC.
- Z. Li, P. Wang, X. Lyu, V. K. R. Kondapalli, S. Xiang, J. D. Jimenez, L. Ma, T. Ito, T. Zhang, J. Raj, Y. Fang, Y. Bai, J. Li, A. Serov, V. Shanov, A. I. Frenkel, S. D. Senanayake, S. Yang, T. P. Senftle and J. Wu, Nat. Chem. Eng., 2024, 1, 159–169 CrossRef.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- C. Zhu, A. Chen, J. Mao, G. Wu, S. Li, X. Dong, G. Li, Z. Jiang, Y. Song, W. Chen and W. Wei, Small Struct., 2023, 4, 2200328 CrossRef CAS.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou, X. Dong, J. Zhai, J. Li, J. He, D. Jiang, Y. Yamauchi, M. He, H. Wu and B. Han, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS PubMed.
- Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2023, 145, 26783–26790 CrossRef CAS PubMed.
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS.
- T. He, K. Reuter and A. Du, J. Mater. Chem. A, 2020, 8, 599–606 RSC.
- H. Liu, Q. Huang, W. An, Y. Wang, Y. Men and S. Liu, J. Energy Chem., 2021, 61, 507–516 CrossRef CAS.
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 17, 2104445 CrossRef CAS PubMed.
- H. T. D. Bui, V. Q. Bui, S.-G. Kim, Y. Kawazoe and H. Lee, Phys. Chem. Chem. Phys., 2021, 23, 25143–25151 RSC.
- X. Lin and J. Ni, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075440 CrossRef.
- J. Feng, Y. Liu, H. Wang, J. Zhao, Q. Cai and X. Wang, Comput. Mater. Sci., 2014, 87, 218–226 CrossRef CAS.
- W. Xia, W. Hu, Z. Li and J. Yang, Phys. Chem. Chem. Phys., 2014, 16, 22495–22498 RSC.
- S. S. Raya, A. S. Ansari and B. Shong, Surf. Interfaces, 2021, 24, 101054 CrossRef CAS.
- W. Liao, G. Yu, L. Zhao, H. Zhu and W. Chen, Nanoscale, 2022, 14, 10918–10928 RSC.
- H. T. D. Bui and T. Brinck, J. Mater. Chem. A, 2024, 12, 2110–2120 RSC.
- T. Brinck and S. K. Sahoo, Phys. Chem. Chem. Phys., 2023, 25, 21006–21019 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- D. M. Bylander and L. Kleinman, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 13756–13761 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- S. Lebègue and O. Eriksson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115409 CrossRef.
- J. C. Garcia, D. B. de Lima, L. V. C. Assali and J. F. Justo, J. Phys. Chem. C, 2011, 115, 13242–13246 CrossRef CAS.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Şahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, Fundamental Concepts in Heterogeneous Catalysis, John Wiley & Sons, Ltd, 1st edn, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PDOS, pCOHP and charge density difference of B–Ni@Si and B–Co@Si, Bader/Löwdin/Mulliken charge variations of four bare BACs, optimized geometries of 2CO* and H* intermediates, OCH2CH* analysis, bond lengths of four optimized BACs structures. See DOI: https://doi.org/10.1039/d4ta04897k |
| This journal is © The Royal Society of Chemistry 2024 |