
Phosphorus–tungsten dual-doping boosts acidic overall seawater splitting performance over RuOx nanocrystals†
Junyang
Ding‡
ab,
Zimo
Peng‡
b,
Zhiwei
Wang
c,
Chunhui
Zeng
*a,
Yanhong
Feng
c,
Miaosen
Yang
*a,
Guagnzhi
Hu
 d,
Jun
Luo
e and
Xijun
Liu
*c
d,
Jun
Luo
e and
Xijun
Liu
*c
aSchool of Chemical Engineering, Northeast Electric Power University, Jilin 132012, Jilin, China. E-mail: zengchunhuijia@163.com; ymiaosen@163.com
bInstitute for New Energy Materials & Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China
cState Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, Guangxi Key Laboratory of Processing for Non-ferrous Metals and Featured Materials, MOE Key Laboratory of New Processing Technology for Nonferrous Metals and Materials, School of Resources, Environment and Materials, Guangxi University, Nanning, 530004 Guangxi, China. E-mail: xjliu@gxu.edu.cn
dInstitute for Ecological Research and Pollution Control of Plateau Lakes School of Ecology and Environmental Science, Yunnan University, Kunming 650504, China
eShenSi Lab, Shenzhen Institute for Advanced Study, University of Electronic Science and Technology of China, Longhua District, Shenzhen 518110, China
First published on 28th August 2024
Abstract
Although it is more cost-effective, producing hydrogen through acidic seawater splitting systems is still difficult, especially when it comes to creating highly effective, highly resistant chlorine corrosion bifunctional catalysts for both the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). Herein, under the addition of phosphotungstic acid and the etching effect of oxalic acid, we develop bifunctional P/W dual-doped RuOx nanocrystals for acidic overall seawater splitting, showing overpotentials of 45 and 158 mV for the HER and OER at 10 mA cm−2, respectively, and accompanied by a strong corrosion resistance towards seawater. Accordingly, the two-electrode system requires only 1.47 V to reach a current density of 10 mA cm−2 and maintains a high stability over 50 h. The P/W dual doping modulates the electrical environment of Ru sites, and the addition of oxalic acid encourages the exposure of more active sites, both of which contribute to the enhancement of seawater electrolysis. Theoretical calculations further manifest that the energy barriers of the HER and OER can be lowered by the dual-doping. Moreover, we found a positive influence of Cl− on the catalytic activity of acidic seawater splitting. Our work offers a promising strategy to significantly increase the catalytic efficiency of Ru-based catalysts for the generation of green hydrogen from acidic seawater in a sustainable manner.
1 Introduction
Hydrogen (H2), due to its plentiful supply, powerful energy density, and zero emissions of greenhouse gases, is viewed as the most suitable renewable carrier and feedstock for substituting exhausted fossil fuels and addressing environmental concerns.1,2 However, grey hydrogen, which accounts for more than 96% of total hydrogen production, is still largely dependent on steam-methane reforming for commercial production.3 Meanwhile, given the notion of “carbon neutrality” that has been put forth in many nations worldwide as well as the disadvantages of hydrogen in storage and transportation, the convenient and efficient production of green hydrogen through electrochemical water splitting is an ideal strategy to achieve sustainable development.4–6 The total efficiency of water splitting depends heavily on two significant half-reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER).7–9 Large overpotentials and sluggish reaction kinetics at both electrodes are typically improved, especially for the OER with four-electron transfer, via the addition of alkali or acids to the electrolyte, e.g., 1 M KOH and 0.5 M H2SO4, to increase ionic conductivity.10–12On the other hand, using high-activity electrocatalysts is also indispensable to further address these problems. It has been found that in alkaline environments, earth-abundant transition metal-based catalysts, including alloys, oxides, phosphides, hydroxides, chalcogenides, phosphides, nitrides and carbides, can catalyze the HER and OER effectively and steadily.13–15 In contrast, in acidic environments, they are typically unstable and are deactivated for corrosion. Nevertheless, compared to alkaline water electrolysis (AWE), acidic proton exchange membrane water electrolysis (PEMWE) has drawn attention because of its distinct benefits such as strong proton conductivity, large current density, quick reaction time, compact equipment footprint, and high hydrogen purity.16–18 The most advanced PEMWE electrocatalysts available nowadays are centered on commercially accessible noble metals, such as Pt/C for the cathodic HER as well as IrOx/RuOx for the anodic OER, for which considerable effort has been made into developing highly active rare-metal-based electrocatalysts for acidic water electrolysis.19 For instance, Ir single atoms were incorporated into the lattice of Ir0.08Co2.92O4 NW oxides, where the lattice oxygens supplied lone-pair electrons and charge equilibrium to stabilize the neighboring Ir atoms, leading to increased OER activity and durability.20 Advanced physical characterization of the alloyed RuMo composites with air or Ar treatment confirms that Ru oxides contained in MoO3 with Ru in an oxidized state make up the OER-activity, whereas Ru active sites surrounded by Mo2C in a reduced state make up the HER-activity.21 The Ir@SrIrO3 heterojunctions can control interfacial electron redistribution and, in the end, generate low-energy barriers for the OER and HER, according to theoretical simulations.22 It was discovered that the dopant in FeCoNiCrTi–RuO2 materials might modify the electronic structure in such a way that electrons could be grouped around the unsaturated Ru sites, therefore reducing Ru solvation and markedly enhancing catalytic stability/activity.23,24 Among them, two of these elements, phosphorus and tungsten, are frequently used to enhance the HER/OER activity of catalysts.25,26 For phosphides, their special metal/phosphorus coordination structure and ordered surface reconstruction can change the electronic properties of the catalyst surface, enhance electron transfer, and increase the reaction rate.27 For example, in the prepared NiCoPv@NF, Pv lowers the surface electro-oxidative reconstruction energy barrier, which makes it easier to promote the local conversion of crystals to reactive oxy(hydroxide) in the OER, and the Ni site of NiCoPv is the active site of the HER process.28 As for tungsten, its introduction could enhance the electrocatalytic activity of the material via altering its crystal structure, surface morphology, and surface electronic state of the substrate, e.g., tungsten in H–FeCoNiMnW forms a surface protective oxide layer during water splitting, which results in excellent catalytic durability and corrosion resistance in acidic media.29,30 Moreover, just a few of the recently reported ultrafine defective RuO2,31 nanostructured Pt@RuOx,32 “5%Pt-containing” Ru oxide nanosheets,33 highly crystalline Ir–Ni nanocages,34 ultrafine Ru–Ir–Te nanotubes,35etc. have also demonstrated excellent activity for acidic HER/OER; nevertheless there are still limited amounts of relevant research.
Furthermore, challenges such as global freshwater scarcity resulting from commercial water decomposition technologies based on electrolysis, mainly PEMWE and AWE that use high-purity water as a reactant, should not be disregarded, while seawater, which makes up 96.5% of the Earth's surface, can be considered a useful resource option.36,37 The precipitation of Mg2+/Ca2+ cations at the cathode makes it difficult to maintain high current densities due to the poor electrical conductivity. The chlorine oxidation reaction (ClOR) and the generation of chlorine or hypochlorous acid (ClO−) at the anode are just a few of the difficulties presented by the complex seawater environment.38 Pourbaix plots from 0.5 M NaCl aqueous solutions show that the presence of a wider window of overpotentials in acidic media favors the OER over the ClOR, which also has a positive effect on increasing conductivity and avoiding cation precipitation.39 Thus, the development of effective and structurally stable electrocatalysts in acidic seawater settings is imperative, as is the understanding of the mechanism that underlies them.
Considering the advantages of acidic electrolytic water devices, the shortage of freshwater resources, the abundance of seawater reserves, and the unsatisfactory stability and resistance to Cl− corrosion facing current electrocatalysts in the OER process, using wet chemistry and annealing treatment, phosphotungstic acid (PW) as the W and P source, chitosan as the C source, and oxalic acid (OA) which was also added during the synthesis process PW–OA–RuOx@C catalysts were created. RuOx was changed by dual-doping phosphorus and tungsten (P/W) components in response to the experimental findings, which improved its catalytic performance and stability. Furthermore, the increased surface's extra active sites were caused by the OA's etching effect. The P/W dual-doping and OA's etching effect work together to improve PW–OA–RuOx@C's intrinsic HER/OER activities. In acidic seawater electrolytes containing 0.5 M H2SO4, the overpotentials of the HER and OER at 45 and 158 mV, respectively, resulted in current densities up to 10 mA cm−2. In addition, an interesting phenomenon was found that the Ru-based catalysts showed higher OER activity and water splitting performance in acidic seawater than in 0.5 M H2SO4 aqueous solution alone, which could be attributed to the presence of large amounts of Cl− in seawater. Through a series of experimental results and theoretical calculations, the roles of P/W and OA in RuOx were elucidated, laying the foundation for the development of new acidic seawater electrocatalysts with higher activity and stability.
2 Experimental Section
2.1 Chemicals
Ruthenium(IV) oxide (99.9%), chitosan (deacetylation degree ≥95%, viscosity 100–200 mpa s), oxalic acid (99.9%, OA), phosphotungstic acid hydrate (H3O40PW12·xH2O, AR), sodium borohydride (NaBH4, 98%), and ethanol (≥99.5%) were purchased from Aladdin. Reference samples of 20 wt% Pt/C and RuO2 were obtained from Hesen (Shanghai) Electric Co. and Meryer (Shanghai) Biochemical Technology Co., Ltd, respectively. Without additional purification, any of the aforementioned chemicals can be directly utilized in this work. Through purification equipment (Aqua Solutions), distilled water (18 Ω) was prepared.2.2 Synthesis of PW–OA–RuOx@C, PW–RuOx@C, and OA–RuOx@C
Using the wet chemistry method and subsequent annealing treatment, the PW–OA–RuOx@C and comparison samples, i.e., PW–RuOx@C without OA and OA–RuOx@C without PW, were prepared. 100 mg of chitosan and 3 mL RuCl3 solution (10 mg mL−1) were dissolved in 20 mL of deionized water. Then 10 mL of NaBH4 solution (2 mg mL−1) was added and stirred with a magnetic agitator at room temperature. While stirring, 10 mL solution containing 7.5 mg PW and 7.5 mg OA was continued to be injected and stopped after 0.5 h. A 0.1 M NaOH solution was used to bring the composition's pH down to an alkaline level, and then stirring was carried out for 1.0 h at 60 °C. Following centrifugation, the solid product from the solution was collected and vacuum-dried for 12 h at 70 °C. After being dried, the powder is placed in a muffle furnace and heated at a rate of 5 °C per minute for 2 h at 350 °C. At the end of the heat treatment, the PW–OA–RuOx@C catalyst was obtained. As comparative samples, PW–RuOx@C and OA–RuOx@C catalysts were synthesized by adding just PW and OA, respectively, to the construction route utilizing the same preparation method as that of the above-mentioned PW–OA–RuOx@C.3 Results and discussion
3.1 Synthesis and characterization
There are two main components to the preparation route for the PW–OA–RuOx@C hybrid: basic wet chemical treatments and follow-up thermo-chemical treatments. The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 1a and b) of PW–OA–RuOx@C showed that this sample consists of aggregated nanoparticles with an average diameter of about 2.5 nm. The high-resolution TEM (HRTEM) image further exhibits obvious lattice fringes with an interlayer spacing of 0.32 nm, which can be indexed to the (110) plane in bulk RuO2. The elemental mapping, as displayed in Fig. 1d–i, shows that C, N, O, Ru, P, and W are uniformly distributed across the sample region. This suggests that the PW–OA–RuOx@C samples were created effectively and that the nano-scale particles help fully expose the marginal active sites. | ||
| Fig. 1 (a) SEM image, (b) TEM image, (c) HRTEM image, and (d–i) corresponding elemental mapping of C, N, O, Ru, P and W for PW–OA–RuOx@C. | ||
The resultant samples of PW–OA–RuOx@C, PW–RuOx@C, and OA–RuOx@C were characterized utilizing the powder X-ray diffraction (XRD) patterns and X-ray photoelectron spectroscopy (XPS) seen in Fig. 2 in order to gain a deeper knowledge of their crystal structures and chemical states. As shown in Fig. 2a, after PW doping of RuOx or introduction of OA, no new diffraction peaks appeared in the prepared PW–OA–RuOx@C, indicating that the dominant phase is still RuO2 (PDF#88-0286), where these high-intensity peaks located at 28.0°, 35.1°, and 54.3° can be assigned to the (110), (101), and (211) crystalline facets of RuO2, and this result is in accordance with the HRTEM findings (Fig. 1c).
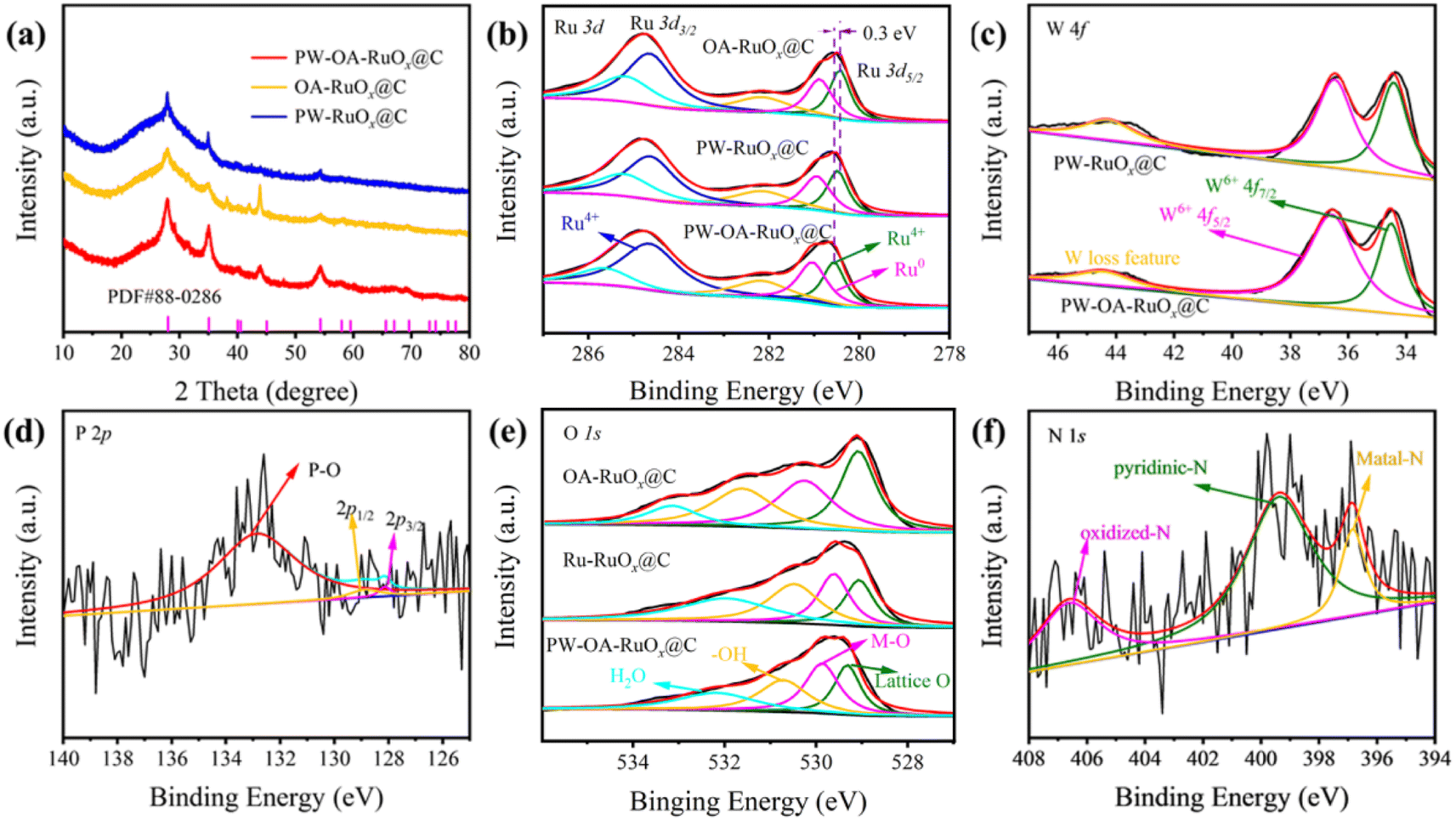 | ||
| Fig. 2 (a) XRD patterns, high-resolution XPS spectra: (b) Ru 3d and (e) O 1s for PW–OA–RuOx@C, PW–RuOx@C, and OA–RuOx@C; (c) W 4f for PW–OA–RuOx@C and PW–RuOx@C; (d) P 2p and (f) N 1s for PW–OA–RuOx@C. | ||
Fig. 2b presents the high-resolution XPS spectra for the Ru 3d; two main peaks attributed to Ru 3d3/2 (284.6 eV) and Ru 3d5/2 (280.7 eV) were observed in PW–OA–RuOx@C, PW–RuOx@C, and OA–RuOx@C. The two main peaks at 281.2 and 280.7 eV in Ru 3d5/2 of PW–OA–RuOx@C, which correspond to Ru0 and Ru4+, respectively, are lower in Ru 3d5/2 than in standard RuO2 (280.9 eV), indicating that Ru species have a lower surface valence state than 4+. This further suggests that P/W doping can effectively inhibit Ru's overoxidation thus contributing to the catalysts' improved stability.40 Meanwhile, the P/W dopants caused a slight shift of the signal peak of Ru 3d5/2 in the positive direction, indicating that the presence of electron transfer from Ru to other components leads to enhanced interaction.
In the case of the W 4f region (Fig. 2c), a pair of 4f7/2 and 4f5/2 peaks belonging to WO3 were noticed at 36.5 and 34.5 eV, which might be the result of W being oxidized during the annealing process.41 Besides, the electron losses of WO3 are reflected by a small peak with a binding energy of 44.3 eV. The P 2p spectra in Fig. 2d show that the P–O bond from air-oxidized phosphorus species is responsible for the peak at around 133.4 eV, while P 2p1/2 and P 2p3/2 binding energies are accountable for the other two centered at 128.9 eV and 128.1 eV, respectively.42 In the deconvoluted O 1s spectra (Fig. 2e), the four peaks at 529.3, 529.9, 530.7, and 532.1 eV correspond to lattice O, metal–oxygen (M–O), −OH, and adsorbed H2O molecules, in that order.43,44 The N 1s XPS spectra (Fig. 2f) give three peaks: a predominant metal–N signal (396.4 eV), pyridinic N (398.2 eV), and oxidized N (406.6 eV). As a consequence, it is evident from these XRD and XPS data that P/W was effectively doped into the RuOx particles to create PW–OA–RuOx@C.
3.2 Hydrogen evolution performance
Using a conventional three-electrode configuration, the HER/OER catalytic capabilities of PW–OA–RuOx@C were investigated in acidic seawater and 0.5 M H2SO4 electrolytes. Also, PW–RuOx@C without OA, OA–RuOx@C without PW, and benchmark Pt/C with the same mass loading were evaluated for their comparative purposes. As seen in the linear sweep voltammetry (LSV) curves in Fig. 3a, PW–OA–RuOx@C shows favorable hydrogen evolution performance compared to others in acidic seawater. With an overpotential of 48 mV at 10 mA cm−2, it even demonstrates a level superior to Pt/C (51 mV@10 mA cm−2), whereas two comparison samples, including PW–RuOx@C and OA–RuOx@C, showed higher overpotentials of 77 and 91 mV, which were obtained at the same current density, respectively. Meanwhile, the dominant position of PW–OA–RuOx@C was not affected by the electrolyte types, as confirmed through the comparative results of these catalysts in 0.5 M H2SO4 (Fig. 3b), with PW–OA–RuOx@C (39 mV@10 mA cm−2) close to Pt/C (23 mV@10 mA cm−2), lower than PW–RuOx@C (50 mV@10 mA cm−2) as well as OA–RuOx@C (87 mV@10 mA cm−2), suggesting that co–modulation of the catalyst via introducing PW and OA during the preparation of Ru-based catalysts is beneficial for the improvement of HER performance. In the meantime, PW–OA–RuOx@C exhibits catalytic activity that is on par with or even higher than that of other proven effective acidic HER electrocatalysts (Table S1†). Furthermore, the corresponding Tafel slopes of the as-prepared Ru-based catalysts in various electrolytes were examined in order to analyze the differences in reaction kinetics between them.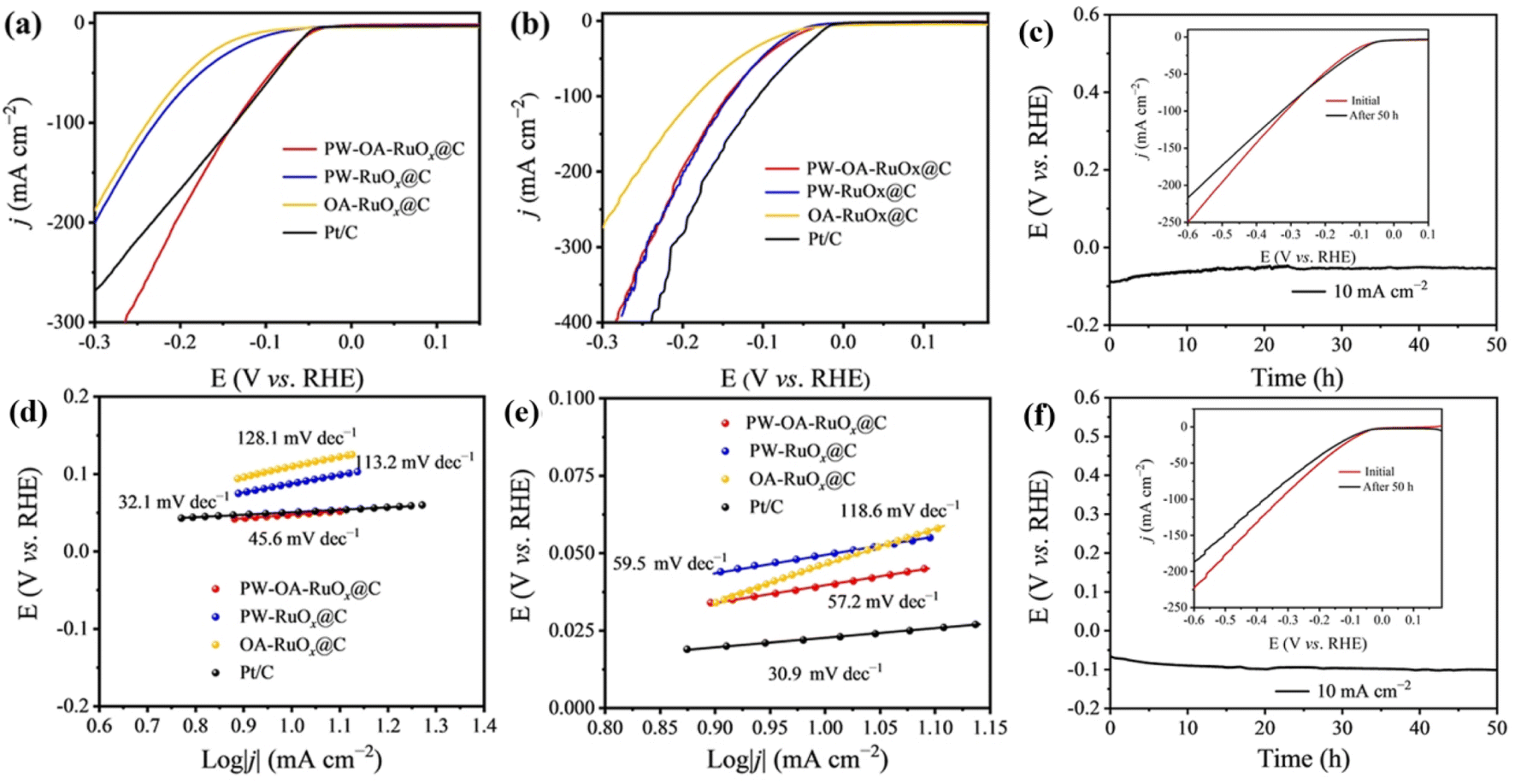 | ||
| Fig. 3 (a) HER LSV curves, (d) Tafel slopes, and (c) stability tests for PW–OA–RuOx@C at a constant current density of 10 mA cm−2 in acidic seawater. (b) HER LSV curves, (e) Tafel slopes, and (f) stability tests for PW–OA–RuOx@C at a constant current density of 10 mA cm−2 in 0.5 M H2SO4. | ||
As shown in Fig. 3d and e, the Tafel slope values of PW–OA–RuOx@C are 45.6 mV dec−1 in acidic seawater and 57.2 mV dec−1 in 0.5 M H2SO4, which are much lower than those of PW–RuOx@C (113.2 mV dec−1; 59.5 mV dec−1) and OA–RuOx@C (128.1 mV dec−1; 118.6 mV dec−1), indicating that PW–OA–RuOx@C has the most favorable hydrogen evolution kinetics, which also explains, in one aspect, its origin of high catalytic activity in both electrolytes. Relevant research has demonstrated that W and Ru experience a non-negligible electron donation effect45 and that P may regulate the charge distribution surrounding Ru atoms to co-activate Ru atoms,46,47 thus increasing Ru's reactivity with the HER. A successful acidic water electrolysis catalyst should, in general, have both excellent corrosion resistance and high catalytic activity. In order to prove this concept, a V–t test of PW–OA–RuOx@C was conducted (Fig. 3c and f). The findings demonstrated that the catalyst worked stably at 10 mA cm−2 for 50 h in acidic seawater and 0.5 M H2SO4, and there was only a very slight variation between the LSV curves before and after the tests (8 mV and 12 mV, respectively), which suggests that this catalyst can successfully sustain the HER catalytic activity for a long term.
3.3 Oxygen evolution performance
To determine whether PW–OA–RuOx@C is a bifunctional catalyst, its OER performance was evaluated via using the same configuration as that used for the HER. Fig. 4a and b show the OER LSV curves for each sample with a scan rate of 5.0 mV s−1. To deliver a current density of 10 mA cm−2, PW–OA–RuOx@C demonstrated optimal OER activity, i.e., the lowest overpotentials of 158 and 241 mV in acidic seawater and 0.5 M H2SO4, respectively, surpassing those of PW–RuOx@C (163 mV; 251 mV), OA–RuOx@C (170 mV; 244 mV), commercial RuO2 (187 mV; 256 mV) and most advanced acidic OER electrocatalysts reported in recent years (Table S2†), confirming the superior OER performance of PW–OA–RuOx@C. Likewise, the corresponding Tafel plots in Fig. 4d and e, which were created using the polarization curves, show that PW–OA–RuOx@C promotes the kinetics of the reaction with relatively smaller Tafel slope values: 51.7 mV dec−1 in acidic seawater and 72.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mV dec−1 in 0.5 M H2SO4, lower than those of PW–RuOx@C (53.8 and 75.6 mV dec−1), OA–RuOx@C (55.9 and 73.5 mV dec−1), and standard RuO2 (77.5 and 80.2 mV dec−1). It is noticed that the Tafel slope values of all four catalysts in acidic seawater are less than 0.5 M H2SO4, suggesting that some components of acidic seawater were able to enhance OER performance due to accelerating the catalysts' inherent kinetics, which might be attributed to the fact that appropriate P/W dual-doping can optimize the free energy required for adsorbed key OER intermediates.48 As displayed in Fig. 4c and f, the long-term stability of PW–OA–RuOx@C was further evaluated; even after a long period with a 50 h (acidic seawater) or 20 h (0.5 M H2SO4) OER test, there is only slight degradation of both V–t curves and LSV curves. It is well known that one of the drawbacks of ruthenium-based catalysts is their tendency to dissolve during the electrolysis process.49,50 The above results show that PW–OA–RuOx@C not only achieves high HER/OER catalytic activity but also maintains strong stability in two acidic media, indicating that this material is a promising catalyst for water splitting.
mV dec−1 in 0.5 M H2SO4, lower than those of PW–RuOx@C (53.8 and 75.6 mV dec−1), OA–RuOx@C (55.9 and 73.5 mV dec−1), and standard RuO2 (77.5 and 80.2 mV dec−1). It is noticed that the Tafel slope values of all four catalysts in acidic seawater are less than 0.5 M H2SO4, suggesting that some components of acidic seawater were able to enhance OER performance due to accelerating the catalysts' inherent kinetics, which might be attributed to the fact that appropriate P/W dual-doping can optimize the free energy required for adsorbed key OER intermediates.48 As displayed in Fig. 4c and f, the long-term stability of PW–OA–RuOx@C was further evaluated; even after a long period with a 50 h (acidic seawater) or 20 h (0.5 M H2SO4) OER test, there is only slight degradation of both V–t curves and LSV curves. It is well known that one of the drawbacks of ruthenium-based catalysts is their tendency to dissolve during the electrolysis process.49,50 The above results show that PW–OA–RuOx@C not only achieves high HER/OER catalytic activity but also maintains strong stability in two acidic media, indicating that this material is a promising catalyst for water splitting.
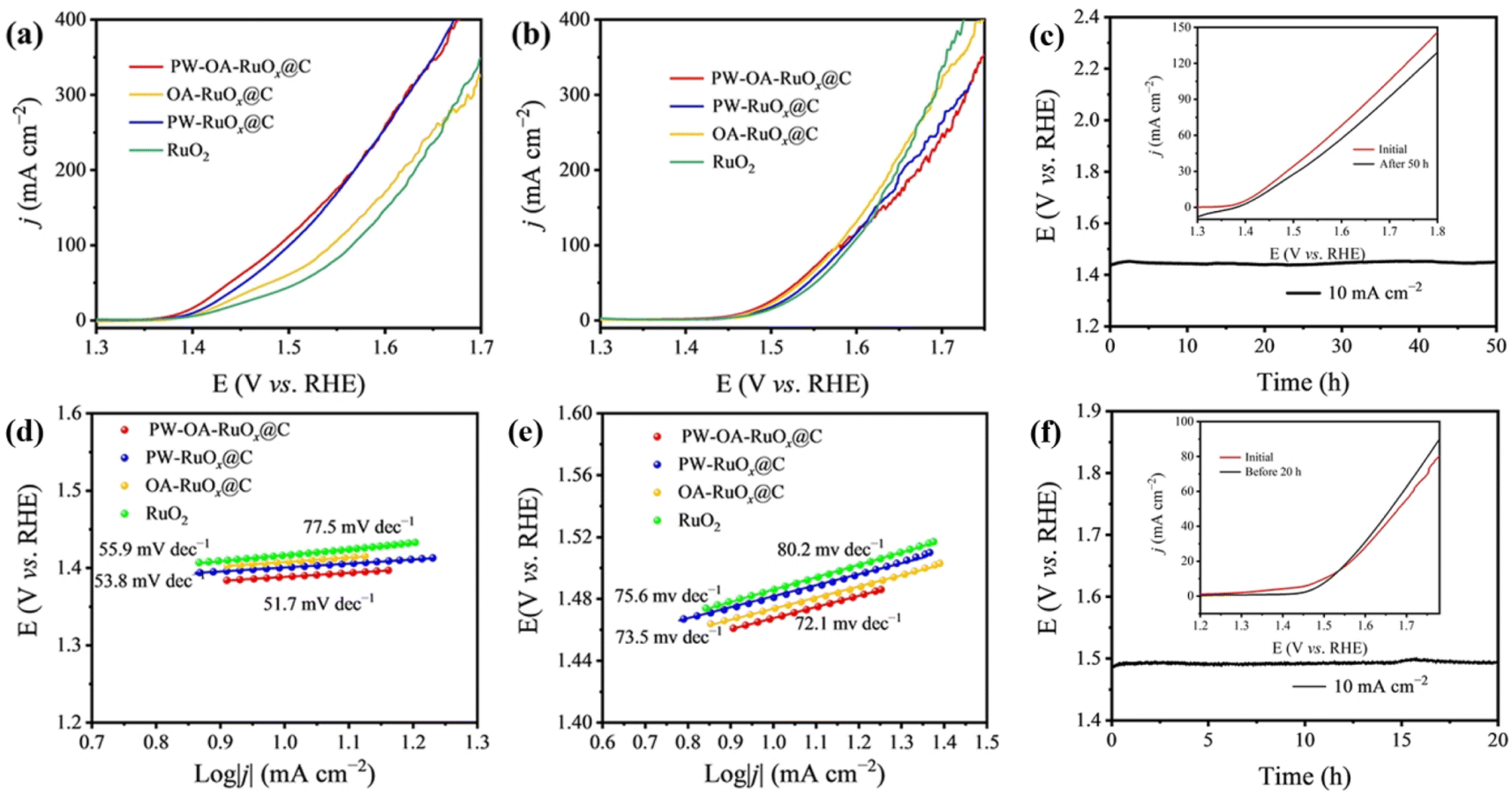 | ||
| Fig. 4 (a) OER LSV curves, (d) Tafel slopes, and (c) stability tests for PW–OA–RuOx@C at a constant current density of 10 mA cm−2 in acidic seawater. (b) OER LSV curves, (e) Tafel slopes, and (f) stability tests for PW–OA–RuOx@C at a constant current density of 10 mA cm−2 in 0.5 M H2SO4. | ||
As exhibited in Fig. S2 and S3,† electrochemical surface area (ECSA) as well as electrochemical impedance spectroscopy (EIS) tests were performed to investigate the origin of the high HER/OER activity. The double-layer capacitance (Cdl) using cyclic voltammetry curves with different rates in the non-faradic area can be used to determine the ECSA.51–53 PW–OA–RuOx@C exhibits an extremely high Cdl value (98.54 mF cm−2), which is about three times that of Pt/C (32.66 mF cm−2), while the Cdl values of PW–RuOx@C and OA–RuOx@C control samples are 48.01 and 74.96 mF cm−2, respectively. It can be seen that the OA-treated catalyst has a larger Cdl compared to PW–RuOx@C without OA, suggesting that the etching action of OA can expose more active sites on the catalyst surface.54 From the semicircle that is attributed to the charge-transfer resistance (Rct) in the high-frequency range of the Nyquist diagram, the resistance of PW–OA–RuOx@C (27.6 Ω) is close to that of Pt/C (16.7 Ω) and only half of that of previously as-prepared pure RuOx NPs (57.9 Ω).32 This result shows that the PW–OA–RuOx@C catalysts exhibit rapid charge transfer, confirming the increase in conductivity that can result from P/W doping and OA etching.
3.4 Overall seawater splitting performance
Given that PW–OA–RuOx@C is a superb bifunctional material, a two-electrode cell was established in order to electrolyze water. The nanostructured PW–OA–RuOx@C powder catalyst loaded on carbon paper was directly employed as an anode and cathode. In acidic seawater and 0.5 M H2SO4 solutions, respectively, this handmade PW–OA–RuOx@C‖PW–OA–RuOx@C remarkably obtained a potential of 10 mA cm−2 at only 1.47 V and 1.49 V, respectively (Fig. 5a and b). This is superior to that of Pt/C‖RuO2 (1.52 V; 1.53 V) and most recently reported electrocatalytic materials for acidic water splitting (Table S3†). Interestingly, we found that PW–OA–RuOx@C exhibited better OER activity and water splitting performance in acidic seawater compared to 0.5 M H2SO4 (Fig. 4 and 5). Based on our previously reported work,32 the enhanced catalytic activity of PW–OA–RuOx@C in acidic seawater electrolyte is mainly attributed to the effect of a large amount of Cl− in seawater. The applied voltage of commercial PtC‖RuO2 was about 1.7 V after the operating current density was set to 10 mA cm−2 for 24 h of continuous operation (Fig. S4†) in acidic seawater, whereas PW–OA–RuOx@C‖PW–OA–RuOx@C did not have any significant voltage drop in the two acidic electrolytes for 50 h in both acidic electrolytes (∼1.5 V in acidic seawater; ∼1.7 V in 0.5 M H2SO4), as shown in Fig. 5c and d, suggesting that P/W elemental doping and interfacial modification play an important role in the stability of nanostructured RuOx composites.55,56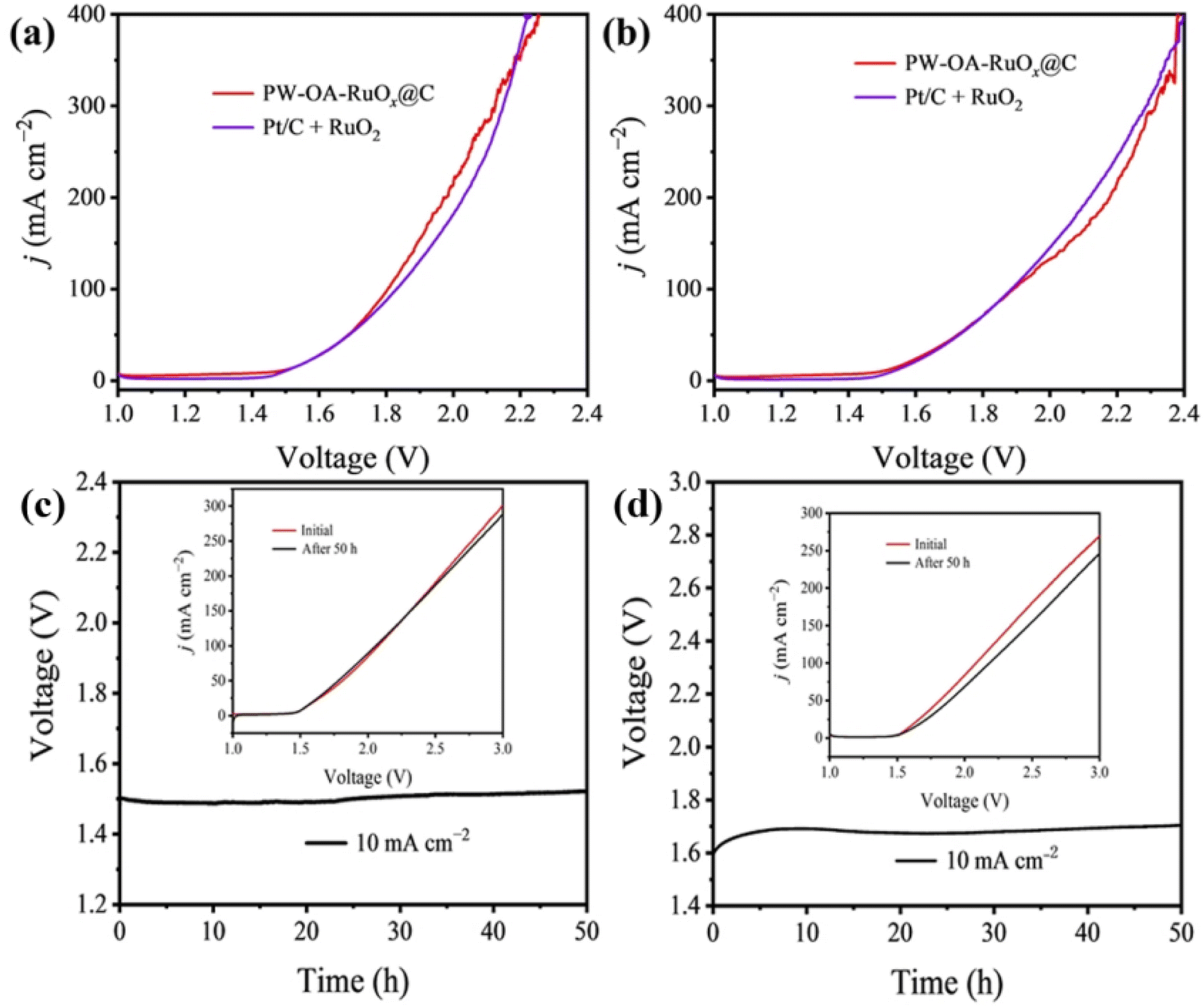 | ||
| Fig. 5 (a) LSV curves of a two-electrode system and (c) long-term durability test of PW–OA–RuOx@C at a constant current density of 10 mA cm−2 for the overall water splitting in acidic seawater. (b) LSV curves of a two-electrode system and (d) long-term durability test of PW–OA–RuOx@C at a constant current density of 10 mA cm−2 for the overall water splitting in 0.5 M H2SO4. | ||
3.5 Theoretical calculations
Using density functional theory (DFT) calculations, an understanding of the source of PW–OA–RuOx@C's intrinsic activity toward the HER/OER in acids was obtained at the atomic level. The Gibbs free energy of *H adsorption (ΔG*H) of P/W co-modified Ru sites on PW–OA–RuOx@C and conventional Ru sites on pure RuO2 are displayed in Fig. 6a. Ru in PW–OA–RuOx@C had the lowest ΔG*H value (0.35 eV) close to 0 eV when compared to pure RuO2 (0.61 eV), suggesting that W/P tailoring could optimize hydrogen adsorption strength and enhance HER kinetics.57,58 The OER, including three key intermediates (*OH, *O, and *OOH) on the catalyst surfaces, is a four-electron process. For the pristine RuO2 system, the production of OOH* from O* is believed to be a rate-determining step (RDS) with an energy barrier of 2.11 eV, while the RDS of PW–OA–RuOx@C exhibits a low energy barrier of 1.97 eV, mirroring the remarkably high active OER electrocatalyst (Fig. 6b).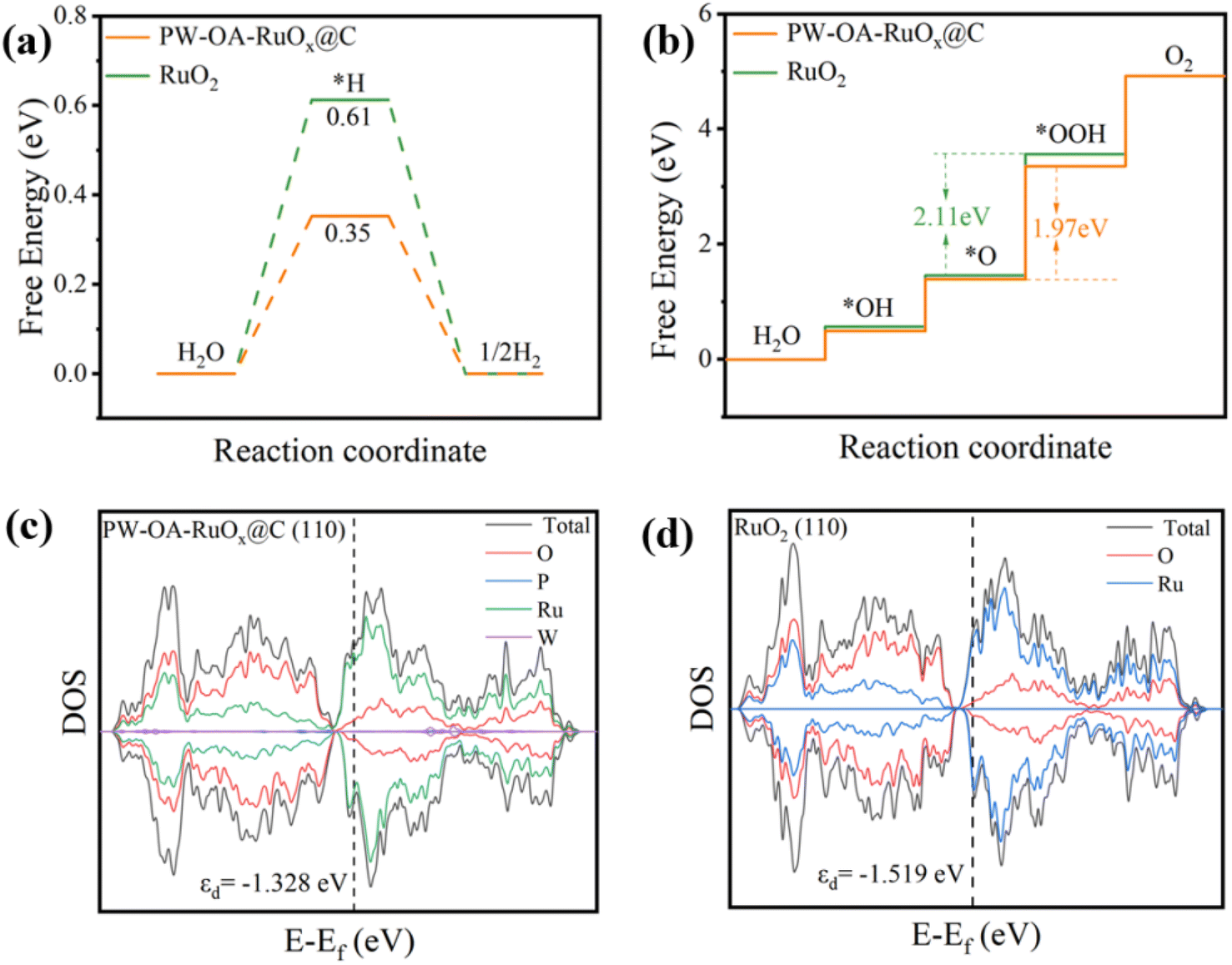 | ||
| Fig. 6 Gibbs free energy diagram of PW–OA–RuOx@C and RuO2 for (a) HER and (b) OER. The projected DOS diagrams of (c) RuO2 and (d) PW–OA–RuOx@C. | ||
Moreover, by analyzing the d-band center (εd) in the d-band from the projected density of states (DOS), the electronic structures of RuO2 and PW–OA–RuOx@C (Fig. 6c and d) were further examined. The εd of PW–OA–RuOx@C (−1.328 eV) are closer to the Fermi energy level (Ef) than that of RuO2 (−1.519 eV), which is advantageous for the electrocatalytic activity since this suggests a larger electron density and easier electron transfer (Fig. S5†).59–62 Consequently, P/W dual-doping can improve the electronic structure, reduce energy barriers, and accelerate reaction kinetics for PW–OA–RuOx@C, hence raising its HER/OER electrocatalytic activity.
4 Conclusion
In this work, a simple process (wet chemistry and subsequent annealing) was used to introduce P/W components into RuOx nanoparticles and simultaneously utilize the etching effect of OA on them to enhance HER/OER activity. The resulting PW–OA–RuOx@C catalyst delivered a current density of 10 mA cm−2 at a lower overpotential of 158 mV for the OER, a HER overpotential of 45 mV, along with 50 h of stable splitting at 1.47 V and 10 mA cm−2 in acidic seawater. Theoretical calculations prove that this outperforming electrocatalytic performance could be attributed to the improved electronic structure and accelerated reaction kinetics by P/W dual-doping. In addition, we found the promoting effect of Cl− on the catalytic activity of acidic seawater splitting. These findings open new avenues for the construction of highly efficient bifunctional catalysts for acidic seawater-based electrolytes with high resistance to Cl− corrosion.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Guangxi Natural Science Fund for Distinguished Young Scholars (2024GXNSFFA010008), the National Natural Science Foundation of China (22075211) and the Natural Science Foundation of Jilin Province of China (20240101098JC).References
- A. H. Schrotenboer, A. A. T. Veenstra, M. A. J. uit het Broek and E. Ursavas, Renew. Sustain. Energy Rev., 2022, 168, 112744 CrossRef CAS.
- Q. Zhang, K. Lian, G. Qi, S. Zhang, Q. Liu, Y. Luo, J. Luo and X. Liu, Sci. China Mater., 2023, 66, 1681–1701 CrossRef CAS.
- M. Hermesmann and T. E. Müller, Prog. Energy Combust. Sci., 2022, 90, 100996 CrossRef.
- L. Wang, D. Wang and Y. Li, Carbon Energy, 2022, 4, 1021–1079 CrossRef CAS.
- H. Kong, Y. Sun, Z. Li, H. Zheng, J. Wang and H. Wang, Appl. Energy, 2023, 347, 121451 CrossRef CAS.
- J. Li, S. Yan, G. Li, Y. Wang, H. Xu and G. Duan, China Powder Sci. Technol., 2023, 29, 101–109 Search PubMed.
- J. T. Kleinhaus, J. Wolf, K. Pellumbi, L. Wickert, S. C. Viswanathan, K. junge Puring, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2023, 52, 7305–7332 RSC.
- Q. Zhang, K. Lian, Q. Liu, G. Qi, S. Zhang, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 646, 844–854 CrossRef CAS PubMed.
- J. Mei, Y. Deng, X. Cheng, X. Wang and Q. Wu, Chin. Chem. Lett., 2024, 35, 108900 CrossRef CAS.
- G. Venugopalan, D. Bhattacharya, E. Andrews, L. Briceno–Mena, J. Romagnoli, J. Flake and C. G. Arges, ACS Energy Lett., 2022, 7, 1322–1329 CrossRef CAS.
- H. Zhang, G. Meng, T. Wei, J. Ding, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 12144–12147 RSC.
- X. Chen, K. Xu, J. Li, X. Wang, T. Zhao, F. Liu, M. Yu and F. Cheng, Chin. Chem. Lett., 2023, 34, 108713 CrossRef CAS.
- Y. Li, L. Zhou and S. Guo, EnergyChem, 2021, 3, 100053 CrossRef CAS.
- J. Li, C. Zhang, C. Wu, Y. Liu, X. Zhang, X. Li, Y. Li, J. Sun and Z. Su, Chin. Chem. Lett., 2024, 35, 108782 CrossRef CAS.
- C. Wan, X. Liu, J. Wang, F. Chen and D.-G. Cheng, Nano Res., 2023, 16, 6260–6269 CrossRef CAS.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 CrossRef CAS PubMed.
- L. Wenxian, N. Xinxin, T. Jiawei, L. Qian, L. Jun, L. Xijun and Z. Yingtang, China Powder Sci. Technol., 2023, 3, 44 Search PubMed.
- C. Wan, R. Li, J. Wang, D.-g. Cheng, F. Chen, L. Xu, M. Gao, Y. Kang, M. Eguchi and Y. Yamauchi, Angew. Chem., Int. Ed., 2024, 63, e202404505 CrossRef CAS PubMed.
- J. Cai, R. Javed, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2020, 8, 22467–22487 RSC.
- W. Zhao, F. Xu, Z. Wang, Z. Pan, Y. Ye, S. Hu, B. Weng and R. Zhu, Small, 2022, 18, 2205495 CrossRef CAS PubMed.
- C. Li, J. Chen, K. C. Chong, L. Wang and B. Liu, Small Struct., 2024, 5, 2300394 CrossRef CAS.
- L. Zhao, Z. Tao, M. You, H. Xiao, S. Wang, W. Ma, Y. Huang, B. He and Q. Chen, Adv. Sci., 2024, 11, 2309750 CrossRef CAS PubMed.
- X. Wu, J. Wu, Y. Hu, L. Zhu, B. Cao, K. M. Reddy, Z. Wang and H.-J. Qiu, Small, 2024, 2404019 CrossRef PubMed.
- T. Wang, Y. Shi, J. Fei, J. Zhu, L. Song, C. Li, T. Zhan, J. Lai and L. Wang, Appl. Catal., B, 2024, 358, 124367 CrossRef CAS.
- C. Wan, G. Li, J. Wang, L. Xu, D.-g. Cheng, F. Chen, Y. Asakura, Y. Kang and Y. Yamauchi, Angew. Chem., Int. Ed., 2023, 62, e202305371 CrossRef CAS PubMed.
- W. Zhong, Q.-L. Hong, X. Ai, C. Zhang, F.-M. Li, X.-F. Li and Y. Chen, Adv. Mater., 2024, 36, 2314351 CrossRef CAS PubMed.
- C. Wan, Y. Liang, L. Zhou, J. Huang, J. Wang, F. Chen, X. Zhan and D.-g. Cheng, Green Energy Environ., 2024, 9, 333–343 CrossRef CAS.
- L. Guo, J. Chi, T. Cui, J. Zhu, Y. Xia, H. Guo, J. Lai and L. Wang, Adv. Energy Mater., 2024, 14, 2400975 CrossRef CAS.
- S.-Q. Chang, C.-C. Cheng, P.-Y. Cheng, C.-L. Huang and S.-Y. Lu, Chem. Eng. J., 2022, 446, 137452 CrossRef CAS.
- T.-J. Wang, L.-B. Sun, X. Ai, P. Chen, Y. Chen and X. Wang, Adv. Mater., 2024, 36, 2403664 CrossRef CAS PubMed.
- R. Ge, L. Li, J. Su, Y. Lin, Z. Tian and L. Chen, Adv. Energy Mater., 2019, 9, 1901313 CrossRef.
- Z. Peng, Q. Zhang, G. Qi, H. Zhang, Q. Liu, G. Hu, J. Luo and X. Liu, Chin. J. Struct. Chem., 2024, 43, 100191 CrossRef CAS.
- Q. Yao, J. Le, S. Yang, J. Cheng, Q. Shao and X. Huang, Chin. J. Catal., 2022, 43, 1493–1501 CrossRef CAS.
- H. Ding, C. Su, J. Wu, H. Lv, Y. Tan, X. Tai, W. Wang, T. Zhou, Y. Lin, W. Chu, X. Wu, Y. Xie and C. Wu, J. Am. Chem. Soc., 2024, 146, 7858–7867 CrossRef CAS PubMed.
- M. Liu, S. Liu, Q. Mao, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 2021–2026 RSC.
- X. Wang, M. Geng, S. Sun, Q. Xiang, S. Dong, K. Dong, Y. Yao, Y. Wang, Y. Yang, Y. Luo, D. Zheng, Q. Liu, J. Hu, Q. Wu, X. Sun and B. Tang, J. Mater. Chem. A, 2024, 12, 634–656 RSC.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, eScience, 2023, 3, 100111 CrossRef.
- Y. Zhao, Z. Yu, A. Ge, L. Liu, J. L. Faria, G. Xu and M. Zhu, Green Energy Environ., 2024 DOI:10.1016/j.gee.2024.02.001.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- Y. Li, W. Wang, M. Cheng, Y. Feng, X. Han, Q. Qian, Y. Zhu and G. Zhang, Adv. Mater., 2023, 35, 2206351 CrossRef CAS PubMed.
- W. Wang, Z. Yang, F. Jiao and Y. Gong, Appl. Surf. Sci., 2020, 529, 146987 CrossRef CAS.
- H. Wang, M. Ma, J. Li, Z. Zhang, W. Zhou and H. Liu, J. Mater. Chem. A, 2021, 9, 25539–25546 RSC.
- Z. Huang, Z. Liu, M. Liao, L. Wang, Z. Luo, T. T. Isimjan and X. Yang, Chem. Eng. J., 2023, 462, 142281 CrossRef CAS.
- J.-Y. Jeong, J. M. Lee, Y. S. Park, S. Jin, S.-W. Myeong, S. Heo, H. Lee, J. G. Albers, Y.-W. Choi, M. H. Seo, S. M. Choi and J. Lee, Appl. Catal., B, 2024, 356, 124220 CrossRef CAS.
- G. Meng, H. Tian, L. Peng, Z. Ma, Y. Chen, C. Chen, Z. Chang, X. Cui and J. Shi, Nano Energy, 2021, 80, 105531 CrossRef CAS.
- X. Liu, F. Liu, J. Yu, G. Xiong, L. Zhao, Y. Sang, S. Zuo, J. Zhang, H. Liu and W. Zhou, Adv. Sci., 2020, 7, 2001526 CrossRef CAS PubMed.
- W. Liu, W. Liu, T. Hou, J. Ding, Z. Wang, R. Yin, X. San, L. Feng, J. Luo and X. Liu, Nano Res., 2024, 17, 4797–4806 CrossRef CAS.
- Z. Chen, R. Zheng, M. Graś, W. Wei, G. Lota, H. Chen and B.-J. Ni, Appl. Catal., B, 2021, 288, 120037 CrossRef CAS.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. Norooz Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- J. Ding, H. Yang, H. Zhang, Z. Wang, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, Int. J. Hydrogen Energy, 2024, 53, 318–324 CrossRef CAS.
- Z. Tongxue, L. Shuai, W. Fumin, L. Wenxian, H. Xinyuan, L. Qian, Z. Xubin and L. Xijun, Microstructures, 2024, 4, 2024043 Search PubMed.
- Y. Ji, Z. Yu, L. Yan and W. Song, China Powder Sci. Technol., 2023, 29, 100–107 Search PubMed.
- J. Yang, F. Zhang and J. Chen, China Powder Sci. Technol., 2024, 30, 161–170 Search PubMed.
- T. Wei, G. Meng, Y. Zhou, Z. Wang, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 9992–9995 RSC.
- Z. Lu, H. Yang, G. Qi, Q. Liu, L. Feng, H. Zhang, J. Luo and X. Liu, Small, 2024, 20, 2308841 CrossRef CAS PubMed.
- Y. Zhu, H. A. Tahini, Z. Hu, Z.-G. Chen, W. Zhou, A. C. Komarek, Q. Lin, H.-J. Lin, C.-T. Chen, Y. Zhong, M. T. Fernández–Díaz, S. C. Smith, H. Wang, M. Liu and Z. Shao, Adv. Mater., 2020, 32, 1905025 CrossRef CAS PubMed.
- X. Liu, M. Chen, J. Ma, J. Liang, C. Li, C. Chen and H. He, China Powder Sci. Technol., 2024, 30, 35–45 Search PubMed.
- K. Chen, D. Ma, Y. Zhang, F. Wang, X. Yang, X. Wang, H. Zhang, X. Liu, R. Bao and K. Chu, Adv. Mater., 2024, 36, 2402160 CrossRef CAS PubMed.
- H. Wu, G. Fei, X. Gao, X. Guo, X. Gong, X. Ma, Q. Wang and S. Xu, China Powder Sci. Technol., 2023, 29, 70–80 Search PubMed.
- X. Zou, J. Xie, C. Wang, G. Jiang, K. Tang and C. Chen, Chin. Chem. Lett., 2023, 34, 107908 CrossRef CAS.
- Q. Pan, J. Li and L. Yan, China Powder Sci. Technol., 2024, 30, 90–102 Search PubMed.
- Y. Zhang, Z. Li, K. Chen, X. Yang, H. Zhang, X. Liu and K. Chu, Adv. Energy Mater., 2024, 2402309 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05277c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |