
Boosting energy-efficient hydrogen evolution by electronically modulating Ni nodes in a framework for methanol oxidation in fresh and seawater†
Nabeen K.
Shrestha
 *,
Akbar I.
Inamdar
,
Hyunsik
Im
and
Sangeun
Cho
*
*,
Akbar I.
Inamdar
,
Hyunsik
Im
and
Sangeun
Cho
*
Division of System Semiconductor, College of AI Convergence, Dongguk University, Seoul 04620, Republic of Korea. E-mail: Nabeenkshrestha@hotmail.com; sangeun.c@dongguk.edu
First published on 14th October 2024
Abstract
The hydrogen evolution reaction (HER) represents the key step in sustainable energy development. This study presents a novel approach to the HER through value-added formate synthesis utilizing methanol electrolysis in both fresh and seawater environments. This approach leverages electronically modulated nickel nodes of a metal–organic framework (e-Ni MOF) via zinc doping, demonstrating significant enhancement in catalytic performance and energy efficiency owing to the synergistic effects of zinc and nickel, thereby facilitating electron transfer and thus lowering the energy barrier for high-valent Ni-active catalytic phase formation. Specifically, the e-Ni MOF anode-based electrolyzer using a 1.0 M methanol solution in an industrially relevant 30 wt% KOH-seawater electrolyte demonstrated a remarkably lower cell voltage of 1.10 V at 10 mA cm−2. Additionally, it achieved an 11.5-fold higher hydrogen evolution rate by replacing the oxygen evolution reaction (OER) with the methanol oxidation reaction (MeOR). At an industrially relevant current density of 250 mA cm−2, the electrolyzer could lead to an energy saving of 640 W h of electricity compared to a conventional OER system. Moreover, the e-Ni MOF facilitated hydrogen evolution at the cathode and produced formate as a value-added chemical from the MeOR at the anodic side. This dual-benefit approach underscores the potential of e-modulated Ni MOFs in transforming hydrogen evolution processes, offering a sustainable and energy-efficient pathway for hydrogen production from seawater and supporting the goal of carbon-neutral energy solutions.
1. Introduction
The growing global energy demand, climate change, and reliance on fossil fuels necessitate the exploration of sustainable and clean energy sources.1–3 Hydrogen, with its high energy content and clean combustion, is emerging as a promising renewable energy carrier.4,5 However, traditional hydrogen production methods are energy-intensive and emit greenhouse gases.6–8 Water electrolysis powered by renewable energy offers a greener alternative;9–13 however, its efficiency is hampered by the thermodynamically high energy barrier and sluggish kinetics of the anodic process involving the oxygen evolution reaction (OER).14–25 To overcome this thermodynamic limitation,researchers have explored alternative anodic reactions that are thermodynamically favorable and can enhance the overall energy efficiency of hydrogen production at the cathode.26–30 One such promising approach involves the oxidation of small organic molecules, such as alcohols, formic acid, formate, sugars, urea, and hydrazine, as the anodic process of water electrolysis.31–40 Methanol is an abundantly available biomass product and easily oxidizable organic molecule, offering a lower overpotential and faster kinetics compared to the OER, thereby potentially leading to more energy-efficient hydrogen generation.41–43 In addition, the methanol oxidation reaction (MeOR) produces formate as the value-added oxidative product.44–47Studies show that Ni-based catalysts are promising for the MeOR, with electrochemically in situ formed Ni3+–OOH on the surface of the catalyst acting as the active catalytic phase for the MeOR, subsequently producing formate as the value-added oxidative product, as demonstrated by eqn (1) and (2):44–47
| 4Ni2+–(OH)2 + 4OH− → 4Ni3+–OOH + 4H2O + 4e− | (1) |
| CH3OH + 4Ni3+–OOH + OH− → HCOO− + 4Ni2+–(OH)2 | (2) |
Therefore, this work focuses on developing an efficient Ni-based electrocatalyst, particularly Ni-based metal–organic frameworks (Ni MOFs), to facilitate methanol oxidation and hydrogen production because MOFs are known for their highly porous nature with a high surface area, which allow for the efficient dispersion of Ni sites within the frameworks, and hence are emerging as attractive candidates for catalysis.19,36,37,39,41–49 Furthermore, by doping Ni MOFs with zinc (Zn), electronic coupling occurs between the Ni and Zn nodes due to d–d transition and, thus, modulates the electronic structure of the Ni-nodes. This reduces energy barriers for the formation of catalytic active Ni3+–OOH sites. Thus, by leveraging the synergistic effects of Zn and Ni within the MOF structure, we aim to lower the energy barrier for the Ni-high valent active catalytic phase formation to assist the MeOR, thereby promoting more efficient hydrogen production and value-added formate formation, both in freshwater and the challenging seawater environment.50 The e-Ni MOF is expected to offer a stable and efficient matrix for methanol adsorption and oxidation, promoting sustained catalytic performance even under a corrosive environment of seawater.
Thus, using industrially relevant 30 wt% KOH in seawater, the electrolyzer achieved a lower cell voltage of 1.10 V, which is, to our knowledge, the lowest reported for methanol electrolysis. Additionally, the electrolyzer showed a 11.5-fold increase in the hydrogen evolution rate when the OER at the e-Ni MOF/NF anode was replaced with MeOR. At an industrial current density of 250 mA cm−2, the electrolyzer required 4.70 kW h of electricity per cubic meter of hydrogen using OER, but only 4.06 kW h with MeOR, saving 640 W h and substantially reducing hydrogen production costs.
2 Experimental details
2.1. e-Ni-MOF film coating
First, nickel foam (NiF) obtained from MTI Korea (>99.99% purity, 1.0 mm thick, porosity: ≥95% 80–110 pores per inch, average hole diameters: ∼0.25 mm) was cut into 5 cm × 1 cm pieces and washed by ultrasonic agitation sequentially in 3 M HCl (ACS reagent, 37%, Sigma-Aldrich) water, ethanol (≥99.5%, ACS reagent, Sigma-Aldrich) and acetone (≥99.5%, ACS reagent, Sigma-Aldrich). Air-dried NiF at room temperature was used as a current collecting substrate. A thin film of e-modulated Ni-MOF was deposited onto the NiF substrate through a solvothermal route. For this, 1.4375–1.250 millimole of NiCl2·6H2O (≥98%, Sigma-Aldrich), 0.0625–0.250 millimole of Zn(NO3)2·6H2O (98%, Sigma-Aldrich) and 1.50 millimole of H2NC6H3-1,4-(CO2H)2 (NH2-BDC ligand, 99%, Sigma-Aldrich) were dissolved in a 50 mL of anhydrous dimethylformamide (DMF, 99.8%, Sigma-Aldrich) by ultrasonic agitation, followed by immersing two pieces of NiF and conducting solvothermal reaction in a Teflon lined stainless steel autoclave at 150 °C for 20 h. The solvothermally obtained MOF films and the precipitate were washed thoroughly with DMF and methanol (≥99.8%, ACS reagent, Sigma-Aldrich) and dried at 60 °C for 24 h.2.2. Characterization of the e-modulated Ni-MOF film
The morphology of the film was examined using a scanning electron microscope (FE-SEM, Hitachi S-4800, Japan) and a scanning/transmission electron microscope (S/TEM, Talos™ F200X). The crystal structure of the MOFs was investigated by applying an X-ray diffractometer (Malvern PANalytical), high-resolution TEM (HR-TEM) and selected area electron diffraction (SAED). For elemental distribution, an energy dispersive X-ray analyzer coupled with scanning transmission electron microscopy (STEM-EDX) was employed. The chemical composition and elemental binding state of the MOFs were investigated by surface analysis of the samples using an X-ray photoelectron microscope (ESCALAB 250Xi spectrometer, ThermoFisher).| E(RHE) = E(Hg/HgO) + 0.059 pH + 0.098 | (3) |
Electrochemical impedance measurement was conducted at a bias of 1.35 V vs. RHE, and the obtained data were fitted with an equivalent circuit using Z-view software.
3 Results and discussion
3.1. MOF film fabrication and characterizations
Fig. 1a and b depict the surface view of a nickel foam (NF) through SEM before and after the deposition of MOF. After the solvothermal reaction, the metallic color of the NF turned into a faint greenish color. Microscopically, the smooth surface of the NF backbones looks rough with the deposition of some granules, as shown in Fig. 1a and b. At a higher magnification SEM view, the Ni MOF film reveals that it consists of layered nano-sheets, as depicted in Fig. 1c. However, after adding a small amount of Zn ions (Zn/Ni mole ratio = 0.082), the morphology of the e-modulated Ni MOF film changed slightly. In this case, in addition to the flat-layered sheets, a few vertically oriented sheets in different directions can also be observed, as shown in Fig. 1d. Similar to the surface morphology, the crystal structure of the e-modulated MOF changed very slightly. The XRD examination of the pristine and e-modulated Ni MOF shows similar patterns with the major peaks located approximately at 2θ of 10.9°, 16.4°, 19.7°, 20.3°, 22.7°, and 26.1°, as shown in Fig. 2a. This reveals that both pristine and e-modulated MOFs essentially have a similar crystal structure. However, the close examination reveals that the XRD peaks of the e-Ni MOF have shifted slightly toward a lower 2θ value, indicating the insertion of larger zinc (atomic radii of Zn and Ni are 0.138 and 0.124 nm, respectively) into the nickel lattice of the framework. Employing the widely used Scherrer equation, the average crystallite size of the e-Ni MOF was determined to be 29.23 nm, while that of the pristine Ni MOF was 32.84 nm. Most importantly, the XRD patterns and peak positions of the Ni MOF well matched those of the simulated XRD patterns. In addition, these XRD patterns well matched those of the previously reported Ni MOF.51–53 Moreover, the FTIR spectra of the pristine and e-Ni MOF showed all characteristic N–H, C–H, COOH and C–N signature bands of the NH2-BDC ligand together with the metal-oxo (Zn/Ni–O) moiety (Fig. 2b), suggesting the presence of the metal–ligand coordination bond.51,54 Thus, these findings indicate that the pristine and e-modulated MOFs have been synthesized successfully. | ||
| Fig. 1 SEM top views of (a) blank nickel foam (NF), (b) e-Ni MOF deposited on NF, (c) Ni MOF film deposited on NF, and (d) e-Ni MOF deposited on NF. | ||
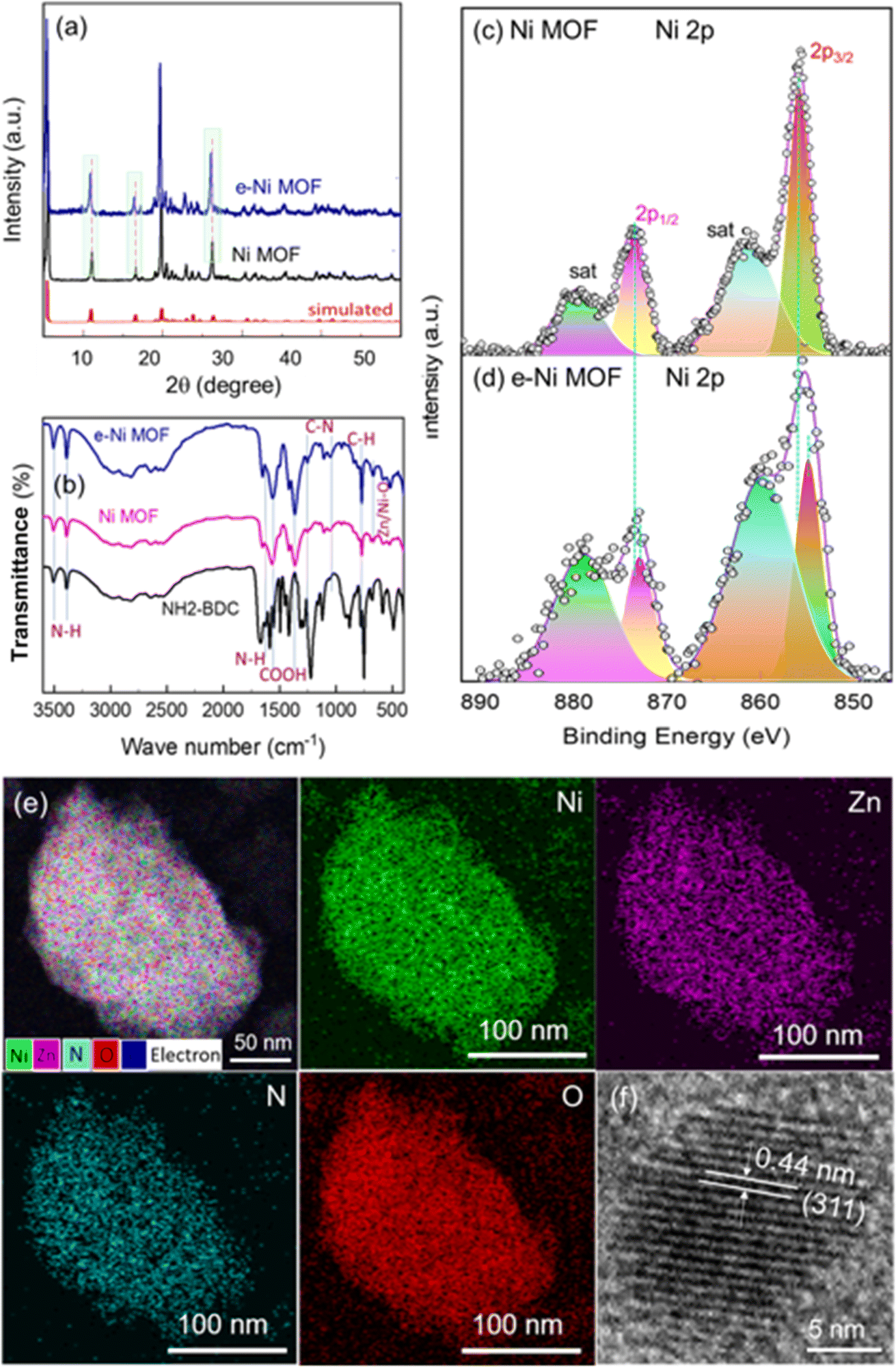 | ||
| Fig. 2 (a) PXRD of Ni MOF and e-Ni MOF bulk powder, (b) FTIR spectra of the MOF and NH2-BDC organic ligand. XPS core level spectra of (c) the Ni 2p of Ni MOF and (d) Ni 2p of e-Ni MOF. (e) STEM-EDX layered image and the corresponding elemental mapping images of Ni, Zn, N and O for the e-Ni MOF film. (f) HR-TEM image of the e-Ni MOF. | ||
Fig. S1† displays a doublet located at about 1021.6 eV and 1044.7 eV, corresponding to the 2p3/2 and 2p1/2 orbitals, respectively, with a spin–orbit splitting energy of 23.1 eV in the Zn 2p XPS spectrum. This reveals the Zn nodes in the Zn2+ oxidation state in the framework. Similarly, Fig. 2c and d depict the XPS spectra of the Ni 2p cores of the pristine and e-Ni MOF. In the former case, a doublet located at about 856.0 eV and 873.6 eV can be observed, corresponding to the 2p3/2 and 2p1/2 orbitals of the Ni2+ oxidation state, respectively. However, as in the case of the XRD, these XPS peaks of the e-modulated framework are slightly shifted toward a lower binding energy region. This indicates that after entering the Ni-lattice, the doped Zn interacts electronically, thereby transferring electrons from a less electronegative Zn2+ node to a higher electronegative Ni2+ node in the frameworks. The detailed electron transfer mechanism and the resulting impact on the electrocatalytic performance are discussed in the subsequent section. The e-modulated MOF film was further investigated with scanning transmission electron microscopy (STEM), and the local distribution of the constitutional elements, particularly Ni, Zn, N and O was studied. Fig. 2e shows the overlay and EDX mapping images of Ni, Zn, N and O, which reveals that the constitutional elements in the e-modulated MOF film are distributed uniformly. Furthermore, the high-resolution transmission electron microscopy (HR-TEM) image of the film showed a clear crystal lattice fringe of the (311) plane with a lattice spacing of 0.44 nm.
3.2. Electrocatalytic performance toward OER
To demonstrate the influence of the Zn incorporation into the Ni lattice toward electrocatalysis, the OER performance of the Ni MOF was evaluated through linear sweep voltammetry (LSV) in a 1.0 M KOH solution. To achieve optimal electrocatalytic performance, we first investigated the influence of the doping level of Zn in the Ni MOF. For this, the Zn2+ ion in the reaction precursor was varied, and the OER activity was measured. As shown in Fig. S2a,† the MOF characterized by a 0.082 mole ratio of Zn/Ni exhibited the best OER activity. This optimized Ni MOF was named an e-modulated framework (e-Ni MOF) and was selected for further studies. Fig. 3a depicts the LSV curves, which demonstrate an apparent difference in the OER activity between the pristine and e-modulated frameworks. Before the commencing of the onset of the OER, the LSV curves show an oxidation peak approximately at 1.30 V and 1.37 V vs. RHE for the e-Ni MOF and pristine MOFs, respectively. This oxidation peak is due to the oxidation of Ni2+ into Ni3+–OOH, which is the active phase for the OER.55 It should be noted that the onset for the peak in the former case appeared toward a lower potential value (1.30 V vs. 1.35 V) and the peak height was also higher in comparison to those of the pristine MOF. This finding implies that the incorporation of Zn into the Ni-lattice assists in the formation of OER active Ni3+–OOH sites. Consequently, the OER onset and sudden rise in current due to the OER in the case of the e-modulated MOF can be observed earlier in the LSV curves. Similarly, the OER overpotentials at various current densities of the e-modulated MOF are relatively lower, as revealed in the bar graph of Fig. 3b. To further understand the influence of doping levels on catalytic activity, MOFs synthesized with various concentrations of the Zn precursor were characterized using XPS, SEM, and EDX. Regardless of the Zn doping level, the Ni 2p and Zn 2p XPS spectra show similar peak positions, as shown in Fig. S3.† These spectra are very similar to those previously studied in detail for the e-Ni MOF (Fig. 2c, d and S1†). As evident from the SEM (Fig. S4†) and EDX (Fig. S5†) results, increasing the Zn-doping concentration led to the formation of layered and curled nanosheets, with dimensions of approximately 2 to 3 μm. The cross-section of the MOF film demonstrated that the deposited MOF had a film thickness of approximately 26 μm (inset image of Fig. S4c†). However, when the Zn/Ni mole ratio exceeded 0.082, the nanosheets were found to agglomerate into bulky masses, as shown in Fig. S4(d) and (e).† This could explain the decrease in OER activity when the Zn/Ni mole ratio was increased beyond 0.082, as shown in Fig. S2a.†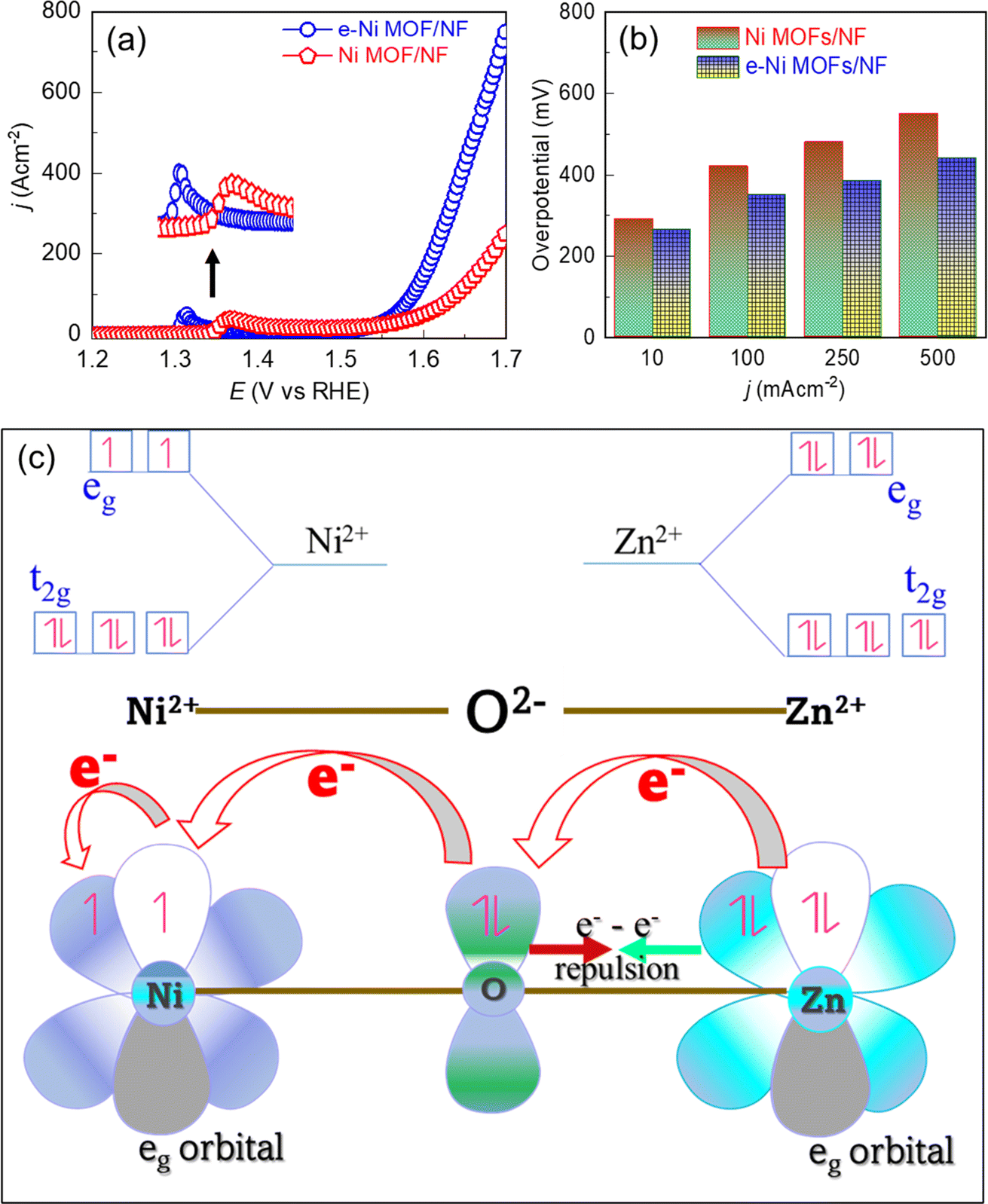 | ||
| Fig. 3 (a) LSV curves of the MOF films on the NF substrate measured in a 1.0 M KOH solution at a scanning rate of 5 mV s−1. Inset shows the enlarged view of the redox peaks. (b) OER overpotential-current density profile executed from the corresponding LSV curves. (c) Schematic showing electronic coupling between Zn2+ and Ni2+ in the Zn-doped Ni MOF (e-Ni MOF). | ||
The observed OER activity of the frameworks shown in Fig. S2a† and 3a can be directly related to the electrochemically active surface area (ECSA), which was determined by measuring CV at various scan rates (Fig. S6a–e†) and estimating the double-layer capacitance (Cdl), as shown in Fig. S6f.† The ECSA was then calculated using the following relation:
| ECSA = (geometrical surface area) × Cdl/Cs, | (4) |
3.3. Electronic modulation of Ni-nodes via Zn doping and electron transfer mechanism
The key mechanism behind the superior electrocatalytic activity of the Zn-doped framework can be attributed to the modulation of the electronic structure of the Ni nodes by the incorporated Zn2+ in their crystal lattice. Based on the crystal field theory, the electron distribution in the d-orbitals and their interaction to stabilize the Ni MOF after doping with Zn2+ can be explained as follows. The carboxylic ligand employed in this work exerts a strong crystal field, thereby forming a low-spin MOF. Thus, based on Hund's rule for filling t2g and eg sets of the d-orbital of a low-spin Zn-doped Ni MOF, the Ni2+ has fully occupied t2g orbitals but has occupied 2 unpaired electrons in the eg set. However, Zn2+ has fully filled d-orbitals. Hence, doping Ni MOF with Zn2+, electronic coupling between Ni2+ and Zn2+ nodes occurred in the framework, which enhanced the electron donation through the e−–e− π-electron repulsion between the bridging O2− and Ni2+ nodes, leading to a partial electron transfer from Zn2+ to Ni2+ nodes,56,57 as illustrated in Fig. 3c. This argument is also supported by a shift of the Ni 2p XPS peaks in the Zn-doped Ni MOF toward a lower binding energy region, as shown in Fig. 2d. This phenomenon leads to the modulation of the local coordination environment of the Ni-nodes, rearranging the electronic cloud of the Ni-nodes in the framework. Consequently, some coordination bonds between the Ni-nodes and catalytically hindered organic ligand counterparts may partially loosen, thereby directly exposing the Ni-nodes and facilitating the redox reaction.58 This improved redox ability decreases the energy barrier of the e-modulated Ni-nodes to promote the oxidation of Ni2+ into the catalytic active Ni3+–OOH phase, subsequently enhancing electrocatalysis.3.4. Electrocatalytic performance toward MeOR
Based on the OER performance of the pristine and e-Ni MOF, their activity toward oxidation of methanol (MeOR) was studied systemically in detail. To understand the influence of Zn-dopant, cyclic voltammograms were measured in a 1.0 M KOH solution, and 1.0 M methanol was added to the 1.0 M KOH solution. In this strong alkaline solution, the Ni-based compound easily oxidizes to hydroxides while polarizing in the anodic direction. Fig. 4a shows a pair of peaks corresponding to the redox reaction of Ni+2–(OH)2 ↔ Ni3+–OOH. This high-valent nickel (i.e., Ni3+–OOH), in addition to the OER, is also the catalytic active phase for the MeOR. It should be noted that after doping the framework with Zn, the onset of the oxidation peak appeared 40 mV earlier, implying that the e-modulation of the Ni-nodes decreased the activation energy barrier for the oxidation of Ni2+–(OH)2 to Ni3+–OOH. In addition, the area under the oxidation peak of the e-modulated framework is relatively larger, signifying that the number of active sites in this case is highly populated (Fig. 4a). In addition, based on similar anodic/cathodic peak heights and area under the peaks, the Ni2+–(OH)2 to Ni3+–OOH oxidation in the pristine Ni MOF is reversible, while that of e-Ni MOF is quasi-irreversible. This finding suggests that in addition to facilitating the formation of the Ni3+–OOH phase, the e-modulation also assists in stabilizing the in situ formed Ni3+–OOH phase. Consequently, the OER activity of the e-Ni MOF is superior to that of the pristine Ni MOF, as shown in Fig. 3a and b. Unlike in the case of OER, when 1.0 M of methanol was added to the KOH solution, no oxidation peaks were obtained (Fig. 4b). The disappearance of the oxidation peak is accompanied by the occurrence of MeOR, which can be noticed clearly by a sudden increase in the anodic current density much earlier than that in the case when methanol was not added to the KOH solution (Fig. 4a). The MeOR on Ni-based catalysts follows a combined electrochemical and chemical reaction-based mechanism, as shown in eqn (2) and (3).46,47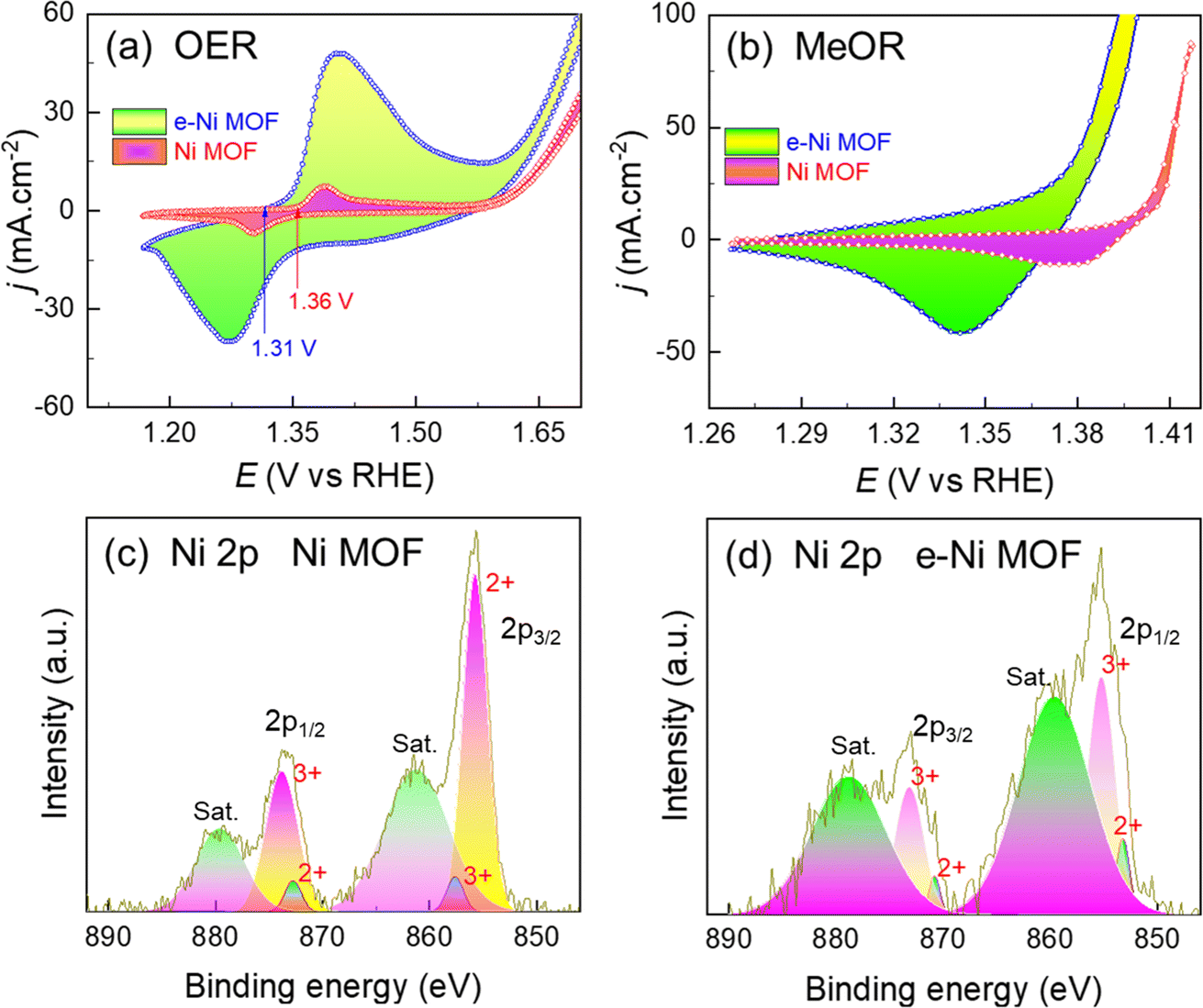 | ||
| Fig. 4 Cyclic voltammograms of the MOF films on the NF substrate in (a) 1.0 M KOH solution and (b) 1.0 M methanol added 1.0 M KOH solution. XPS spectra of Ni 2p cores in (c) pristine and (d) e-modulated Ni MOFs (i.e., Zn doped). | ||
Reaction (2) reveals that the Ni3+–OOH phase participated in the oxidation of methanol to formate. This could explain why the oxidation peak of Ni2+–(OH)2 to Ni3+–OOH is not observed. Most importantly, in addition to the formation of value-added formate, the initial Ni2+–(OH)2 phase is regenerated, which helps to maintain the integrity of the catalyst during the long-term electrocatalysis process. Wang et al. showed that the oxidation peak of Ni2+–(OH)2 to Ni3+–OOH during electrolysis of ethanol is an endothermic reaction, while the reduction of Ni3+–OOH to Ni2+–(OH)2 along with the dehydrogenation of ethanol is Ni2+–(OH)2 is an exothermic or spontaneous reaction.59 Recently, a similar result was revealed by Feng and co-workers, wherein they demonstrated that the catalytic MeOR to formate on the in situ formed Ni3+–OOH is fully exothermic or spontaneous.60 This implies that methanol spontaneously reduces Ni3+–OOH to Ni2+–(OH)2, forming format, as shown in reaction (2). The structural phase change between Ni+2 and Ni3+–OOH during the catalytic progress in an alkaline medium was also confirmed by XPS analysis of the MOF/NF-based anode before (Fig. 2c and d) and after polarizing at 1.42 V vs. RHE in 1.0 M methanol added 1.0 M KOH solution (Fig. 4c and d). In addition, it should be noted that the area under the Ni3+ peak of the e-Ni MOF/NF anode is larger than that of the Ni MOF/NF anode. This implies that the e-modulation potentially decreases the reaction energy barrier, thereby assisting the formation of Ni3+–OOH and promoting the number of MeOR active sites in the framework. This can also be achieved by the larger reduction peak area of the e-Ni MOF (Fig. 4b), which is an indirect measure of the Ni3+–OOH phase density. This is because, for a given redox reaction, the succeeding reduction peak area of a voltammogram is directly proportional to the preceding oxidation peak area. The surface reconstruction and formation of active Ni3+–OOH catalytic sites during MeOR were also confirmed by analyzing the MOF/NF via XPS after the MeOR. Fig. 2c and d shows that the as-synthesized MOF/NF films exhibited peaks for only the Ni2+ oxidation state, while those after MeOR additionally exhibited peaks for the Ni3+ oxidation state, as shown in Fig. 4c and d. Moreover, the peak height and area under the Ni3+ peaks of the e-modulated MOF film are higher compared to that of the pristine MOF film. This finding also supports the earlier argument that e-modulation assists the formation of Ni3+–OOH catalytic sites. Furthermore, the MOF/NF films were polarized at a potential laying before and after the MeOR at 1.20 V and 1.42 V vs. RHE in 1.0 M methanol added 1.0 M KOH solution and were analyzed via Raman shift measurement. In line with the XPS analysis, the Raman shift measurement also reveals that the MOF films do not form the Ni3+–OOH phase when they are polarized at 1.20 V vs. RHE. In contrast, they clearly show the characteristic Raman bands from 400 to 600 cm−1 for the formation of Ni3+–OOH phase45,54 when the MOF films were polarized at 1.42 V vs. RHE (Fig. S7†).
Based on the above facts, compared to the pristine MOF, the LSV curves of the e-modulated framework in Fig. 5a demonstrate superior MeOR activity. Specifically, the e-Ni MOF film achieved MeOR current densities of 10, 100, 250 and 500 mA cm−2 at anodic potentials of 1.31, 1.37, 1.39, and 1.41 V vs. RHE, respectively. This MeOR activity is among the highest previously reported for high-performance electrocatalysts (Table S1†). In contrast, the pristine framework film could drive the MeOR at the same current densities only at higher anodic potentials of 1.37, 1.41, 1.43 and 1.45 V vs. RHE, respectively (Fig. 5b). Most importantly, it should be noted in Fig. 5a that the MeOR occurred much earlier than the OER, which can be attributed to the relatively lower oxidation potential of methanol compared to that of water, thereby lowering the reaction energy barrier and assisting the MeOR. Consequently, the e-Ni MOF at electrolysis current densities of 10, 100, 250 and 500 mA cm−2 exhibited anodic potentials lower by 175, 210, 225 and 260 mV for the MeOR compared to OER. It can be observed that the slope of the LSV curves decreased after 1.45 V vs. RHE, as depicted in Fig. 5a. This can be attributed to the detrimental competition between the MeOR and OER.61,62 As with the OER, the observed MeOR activity of the frameworks is also directly related to the ECSA. Consequently, a similar trend in the MeOR activity of the frameworks with different Zn-doping levels was observed, as displayed in Fig. S8.† To validate the reproductivity of the catalyst synthesis and MeOR activity, five replicas of the e-Ni MOF and pristine Ni MOF samples were synthesized, and their MeOR activity was evaluated using linear sweep voltammetry. As evident in Fig. S9a and b,† the variations in MeOR activity among the samples are minimal and within the margin of measurement error.
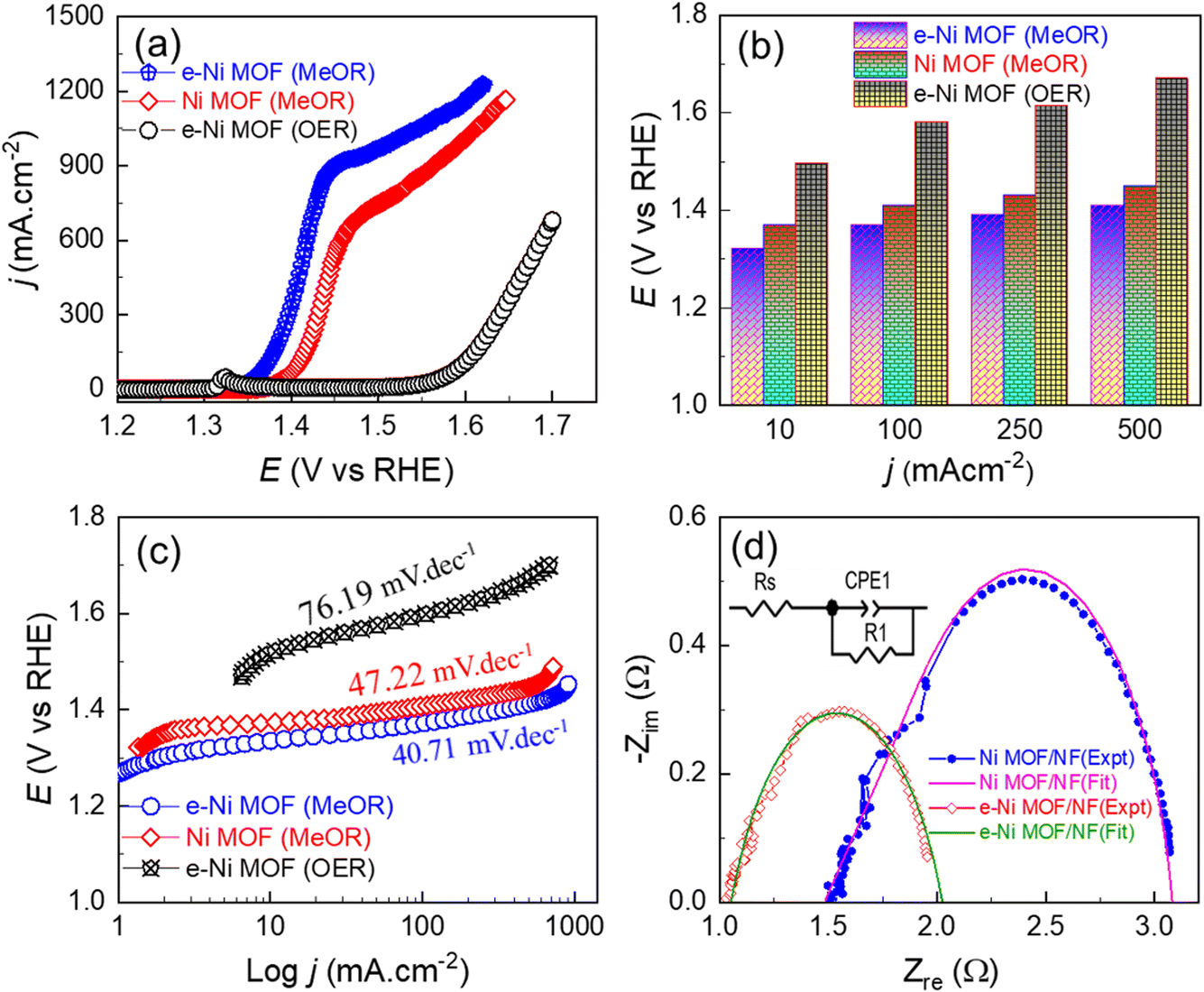 | ||
| Fig. 5 (a) LSV curves of the MOF films on the NF substrate measured with (MeOR) and without (OER) 1.0 M methanol added 1.0 M KOH solution at a scanning rate of 5 mV s−1. (b) Anodic potential (E) vs. current density (j) profile and (c) Tafel slopes derived from the corresponding LSV curves. (d) Nyquist plots of the corresponding MOF films measured at 1.40 V vs. RHE. Inset shows an equivalent circuit employed to fit the experimental data. | ||
In addition to the enhanced electrocatalytic activity, the e-Ni MOF also demonstrated faster kinetics for the MeOR, which can be achieved by the comparatively lower Tafel slope, as illustrated in Fig. 5c. The observed faster MeOR kinetics of the e-Ni MOF electrode are also supported by the lower charge transfer resistance (Rct), which is represented by the smallest semicircle shown in the Nyquist plot in Fig. 5d. Furthermore, to assess the intrinsic activity of the MOFs, the MeOR current density was normalized based on the ECSA. Fig. S10† illustrates that, in addition to the case of geometrical area-based MeOR activity, the e-Ni MOF electrode exhibits the highest activity, implying that doping Zn into the Ni-lattice of the framework efficiently regulates the electronic structure, thereby modulating the charge distribution of the Ni-nodes, and eventually improving the intrinsic activity for the MeOR. However, both the pristine and e-modulated frameworks maintained their MeOR activity for long-term electrolysis studied for 48 h, as displayed in Fig. S11.†
Based on the notable activity and long-term performance of the e-Ni MOF for catalyzing the MeOR in a three-electrode system, a full electrolyzer was devised in a two-electrode configuration (MOF-MeOR‖Pt(20 wt%)/C-HER) for practical MeOR-aided water splitting in comparison to conventional water splitting. Fig. 6a and b shows that the e-Ni MOF anode-based electrolyzer exhibited lower cell voltages of 1.33, 1.64, 1.80, and 1.88 V for electrolyzing methanol in a 1.0 M KOH solution at current densities of 10, 100, 400, and 800 mA cm2, respectively. In contrast, the pristine Ni MOF anode-based electrolyzer exhibited relatively higher cell voltages of 1.47, 1.76, 1.99, and 2.37 V under the same conditions. To understand the anodic reaction and current efficiency of the MeOR product, an NMR analysis of the electrolytic solution was conducted. Fig. S12a† displays 1H NMR spectra before and after the electrolysis of methanol at 50 mAcm−2 for 5 hours. After electrolysis, the NMR spectrum shows that formate is the key product of the MeOR. Using potassium hydrogen phthalate (KHP) as an internal standard, the amount of formate produced was estimated, and the Faraday current efficiency for the formation of formate from the MeOR was determined to be 95.6%. When the methanol electrolysis was performed at a higher current density of 200 mA cm−2, the current efficiency slightly decreased to 93.23%. However, no additional product other than the format was detected through 13C NMR analysis (Fig. S12b†). This indicates high selectivity for formate formation on the e-Ni MOF catalyst.
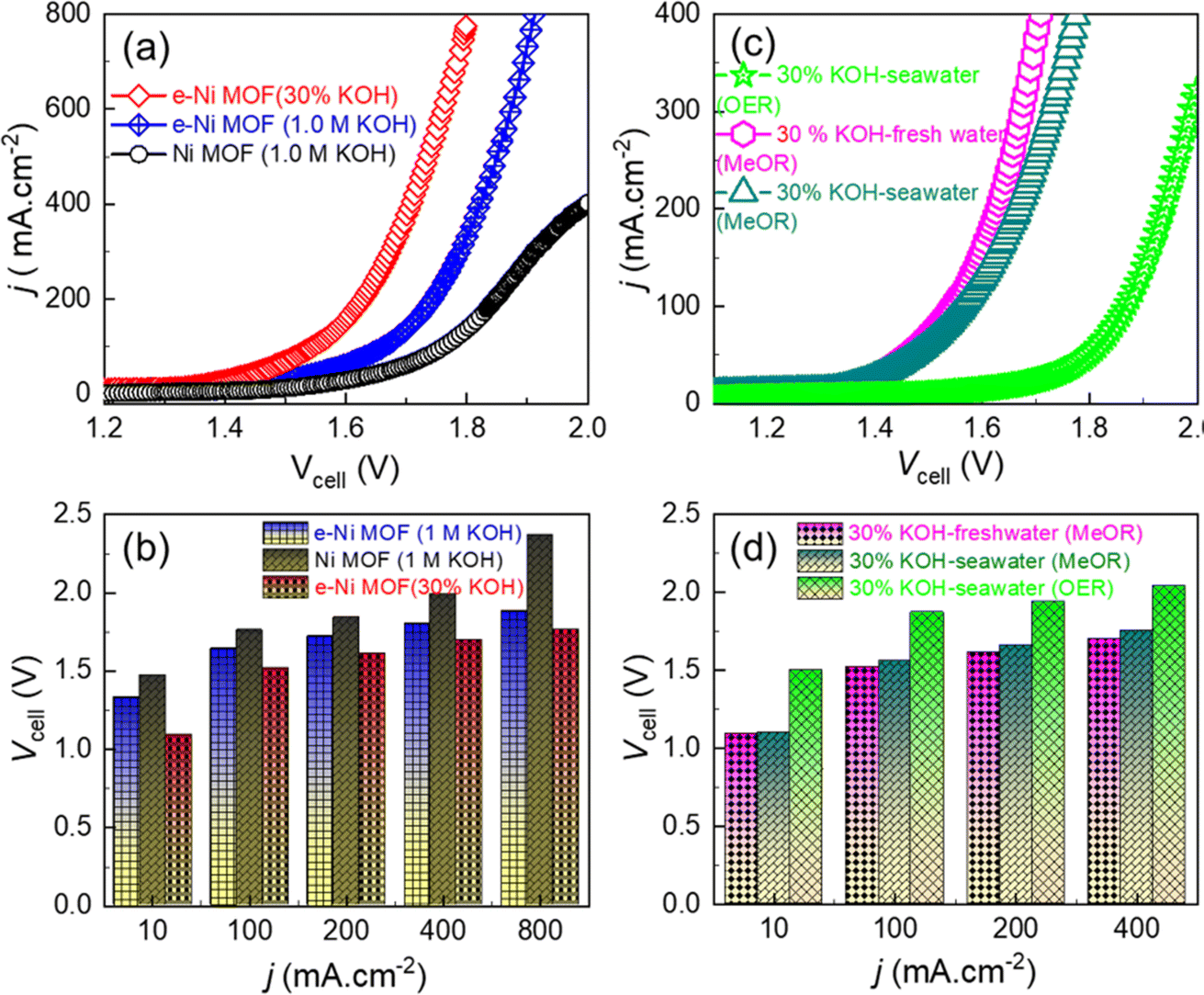 | ||
| Fig. 6 (a) LSV curves of the MOF/NF‖Pt(20 wt%)/C/NF electrolyzers measured in 1.0 M and 30 wt% KOH solution in fresh water, and (b) cell current density-cell voltage profile derived from the corresponding LSV curves. (c) LSV curves of the e-Ni MOF/NF‖Pt(20 wt%)/C/NF electrolyzer measured in 30 wt% KOH prepared in fresh and seawater with and without the addition of 1.0 M methanol, and (d) cell current density–cell voltage profile derived from the corresponding LSV curves. | ||
Interestingly, when an industrially relevant 30 wt% KOH electrolyte was employed, the above e-Ni MOF anode-based electrolyzer demonstrated enhanced MeOR activity with lower cell voltages of 1.09, 1.52, 1.70, and 1.76 V for electrolyzing 1.0 M methanol at current densities of 10, 100, 400, and 800 mA cm−2 (Fig. 6a and b). It is noteworthy that these cell voltages are relatively lower than those reported previously for the Ni(OH)2/NF-based high-performance methanol electrolyzer (Fig. S13 and Table S2†). Based on this notable result, the e-Ni MOF-(MeOR)‖Pt(20 wt%)/C-(HER) electrolyzer was further studied to electrolyze 1.0 M methanol in 30 wt% KOH in both fresh and seawater. The LSV curves (Fig. 6c) and cell voltage–current density profile (Fig. 6d) reveal that the MeOR activity of the e-Ni MOF anode in fresh and seawater-based electrolytes is closer. However, a significant difference in cell voltage was observed between methanol-added (MeOR) and non-methanol-added (OER) 30 wt% KOH electrolytes in seawater. In the former case, the MeOR occurred exclusively, while the OER occurred in the latter case. This was also ascertained visually by the evolution of gas at the electrodes. When the electrolysis was performed with methanol containing seawater electrolyte; no gas bubbles were observed at the anode, while a vigorous evolution of hydrogen gas was observed at the cathode (Fig. S14†). For the MeOR at current densities of 10, 100, and 400 mA cm−2, the electrolyzer attained cell voltages of 1.10, 1.56, and 1.75 V, respectively, while for the OER, the cell voltages were 1.50, 1.87, and 2.04 V, respectively. Most importantly, for comparison when the electrolyzer was operated at 1.70 V to electrolyze 30 wt% KOH in seawater, it generated approximately 2 × 10−4 moles of hydrogen gas at the cathode (Fig. 7a). In contrast, with 1.0 M methanol added to the electrolyte, the same electrolyzer generated approximately 23 × 10−4 moles of hydrogen, thus achieving an 11.5-fold higher rate of hydrogen evolution by replacing the OER at the e-Ni MOF/NF anode with the MeOR. Furthermore, when the electrolysis was performed at an industrial relevant electrolysis current density of 250 mA cm−2, the electrolyzer with the OER as anodic reaction consumed 4.70 kW h of electricity to generate 1 m3 of H2, while the same electrolyzer with MeOR as the proxy anodic reaction consumed only 4.06 kW h. This reduction leads to a cost saving equivalent to 640 W h of electricity, thereby significantly lowering the cost of hydrogen production. This finding highlights the importance of replacing the OER with MeOR at the anode to accelerate hydrogen evolution at the cathode of the electrolyzer.
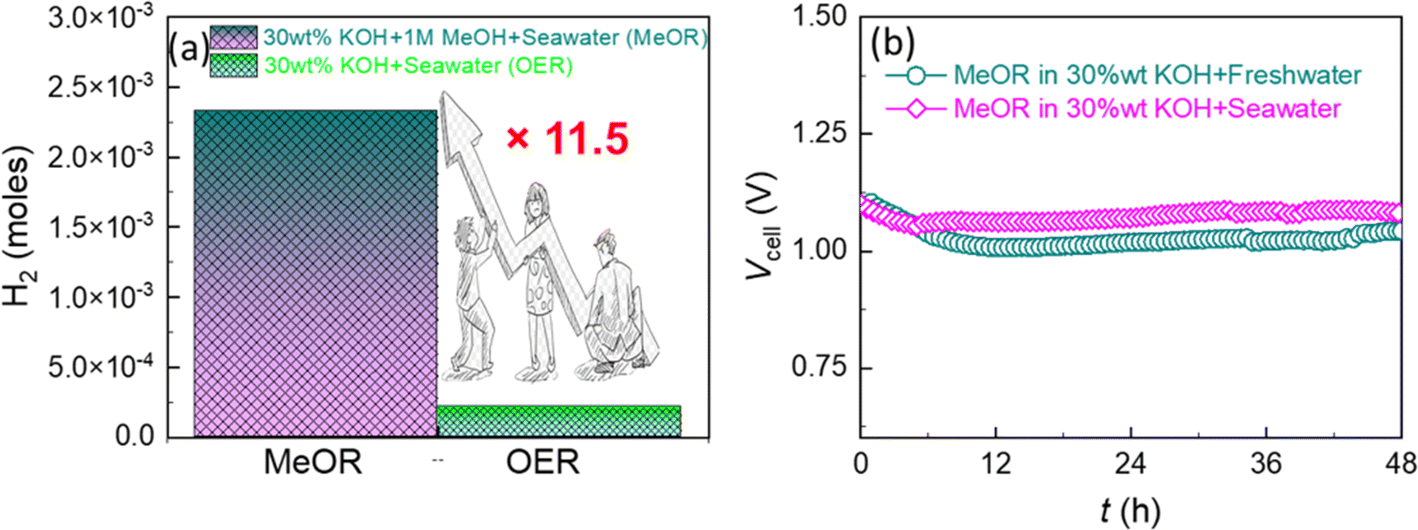 | ||
| Fig. 7 (a) Amount of hydrogen produced by the e-Ni MOF/NF‖Pt(20 wt%)/C/NF electrolyzer measured at 1.70 V for 20 minutes in 30 wt% KOH solution prepared in fresh and seawater with (MeOR) and without (OER) the addition of 1.0 M methanol. The graph shows that the hydrogen production rate can be accelerated 11.5-fold by replacing the OER at the e-Ni MOF/NF anode with the MeOR. (b) Chronopotentiometric curves showing the long-term durability of the e-Ni MOF/NF anode during the MeOR at 10 mA cm−2. | ||
Finally, the long-term stability of the e-Ni MOF electrocatalyst was evaluated using a chronopotentiometry test. For this, chronopotentiometry was performed for over 48 hours by employing an e-Ni MOF-(MeOR)‖Pt(20 wt%)/C-(HER) electrolyzer that maintained a constant current density of 10 mA cm−2 in 1.0 M methanol with 30 wt% KOH electrolyte in fresh and seawater. Fig. 7b displays the resulting chronopotentiometric curves, indicating the excellent stability of the e-Ni MOF on the electrocatalytic performance for MeOR over a prolonged operating time. This approach thus promises to lower the energy requirements for hydrogen production and address the challenges posed by seawater electrolysis, paving the way for more practical and scalable hydrogen generation solutions. To clarify the excellent stability of the e-modulated Ni MOF catalyst in a seawater-based electrolyte, the Tafel corrosion polarization curves of both the pristine and modulated Ni MOF were measured. The corrosion potential and corrosion current density were calculated through extrapolation, as shown in Fig. S15.† Compared to the pristine Ni MOF, the modulated Ni MOF exhibited a significantly lower corrosion current density of 4.43 μA cm−2 (vs. 21.49 μA cm−2) and a higher corrosion potential of 0.75 V vs. RHE (vs. 0.62 V). This result indicates that the modulated Ni MOF has a greater degree of corrosion resistance during the methanol electrolysis process in alkaline seawater. Additionally, the electrolyte solution from the anodic side of the electrolyzer, following the long-term stability test, was analyzed using an N,N-diethyl-1,4-phenylenediamine (DPD) indicator to examine the formation of hypochlorite ions due to the chlorine evolution reaction.63 In principle, the DPD indicator works by being oxidized by hypochlorite, resulting in a magenta color. However, in the present work, no color change was observed upon the addition of the indicator, ruling out the occurrence of the chlorine evolution reaction during the MeOR in alkaline seawater.
Furthermore, PXRD analysis of the e-Ni MOF before and after long-term stability testing was performed. As evident in Fig. S16a,† even after the stability testing for over 48 hours, the e-Ni MOF shows that the majority of the characteristic XRD peaks appeared initially before the stability testing, thus evidencing the crystal structural integrity. However, after the stability testing, the XRD peaks became less intense. In addition, a small broad hump was observed centering approximately at 2θ of 9°, and some of the fine XRD peaks initially appearing at 2θ positions beyond 30° also disappeared. This may be due to the localized amorphization in the framework structure. In line with this finding, the SEM images displayed in Fig. S16b and c† also indicate some changes in the morphology of the e-Ni MOF film. Particularly, the initially observed 3-dimensionally oriented sheets of the film (Fig. S16b†) after the stability testing turned into more flattened morphology (Fig. S16c†). EDX analysis reveals that their composition before and after the stability testing remained similar with a slight increase in Zn/Ni mole ratio from 0.0823 to 0.0833 (Fig. S16d and e†). Additionally, XPS analysis was performed to determine whether surface oxidation of the MOF film occurred during the MeOR process. The Ni 2p XPS spectra in Fig. S17a† indicate that a significant portion of the Ni2+–(OH)2 surface catalyst was oxidized to the Ni3+–OOH phase, while the Zn2+ nodes were also found to be slightly oxidized to Zn2+δ.
4 Conclusions
We successfully demonstrated that zinc doping led to the modulation of the electronic structure of the Ni-nodes in the framework. This strategy improves the catalytic efficiency of methanol oxidation and reduces the energy input required for hydrogen production. Using a combination of electrochemical and surface analysis techniques, the mechanisms by which the electronically modulated Ni-nodes in the framework enhance methanol oxidation are elucidated. The study reveals that the e-modulated Ni-nodes modify the local coordination between the Ni-nodes and the organic linkers, which leads to a partial exposure of the Ni-nodes, thereby facilitating the oxidation of Ni2+–(OH)2 to an active Ni3+–OOH phase to catalyze methanol oxidation reaction. Thus, the e-Ni MOF possesses a higher density of high valent Ni3+–OOH species, which is favorable for boosting the MeOR catalytic sites, thereby lowering thermodynamic barriers and advancing effectively the kinetics of methanol oxidation reaction. However, NMR analysis revealed that the key oxidation product was formate, a value-added chemical. Therefore, environmentally friendly production of valuable products can be achieved. Additionally, the cost of hydrogen generation can be considerably reduced as a value-added organic compound is obtained instead of oxygen at the anode. The implications of this study extend towards the broader field of renewable energy technologies by offering insights into the design and application of advanced materials for more efficient hydrogen generation processes.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
Nabeen K. Shrestha: conceptualization, investigation, experimental analysis, writing original draft & editing, supervision; Akbar I. Inamdar: S/TEM analysis; Sangeun Cho: FE-SEM and X-ray analysis, funding acquisition; Hyunsik Im: project administration and funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from the National Research Foundation of Korea (NRF) through grants funded by the Korea Government Ministry of Science (Grant No. 2022R1F1A070724).References
- A. M. Omer, Renewable Sustainable Energy Rev., 2008, 12, 2265–2300 CrossRef.
- J. L. Holechek, H. M. E. Geli, M. N. Sawalhah and R. Valdez, Sustainability, 2022, 14, 4792 CrossRef.
- E. S. Hanley, J. P. Deane and B. P. Ó. Gallachóir, Renewable Sustainable Energy Rev., 2018, 82, 3027–3045 CrossRef.
- T. da Silva Veras, T. S. Mozer, D. da Costa Rubim Messeder dos Santos and A. da Silva C'esar, Int. J. Hydrogen Energy, 2017, 42, 2018–2033 CrossRef.
- A. Midilli, M. Ay, I. Dincer and M. A. Rosen, Renewable Sustainable Energy Rev., 2005, 9, 255–271 CrossRef.
- F. Suleman, I. Dincer and M. Agelin-Chaab, Int. J. Hydrogen Energy, 2015, 40, 6976–6987 CrossRef.
- I. Dincer and C. Zamfirescu, Int. J. Hydrogen Energy, 2012, 37, 16266–16286 CrossRef CAS.
- M. Granovskii, I. Dincer and M. A. Rosen, Atmos. Environ., 2007, 41, 1777–1783 CrossRef.
- M. Granovskii, I. Dincer and M. A. Rosen, J. Power Sources, 2006, 157, 411–421 CrossRef.
- K. Christopher and R. Dimitrios, Energy Environ. Sci., 2012, 5, 6640–6651 RSC.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 11954–11971 Search PubMed.
- M. Granovskii, I. Dincer and M. A. Rosen, Int. J. Hydrogen Energy, 2007, 32, 927–931 CrossRef.
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2014, 39, 1–12 CrossRef.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef.
- Z.-Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef PubMed.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146 CrossRef.
- N. K. Shrestha, S. A. Patil, J. Han, S. Cho, A. I. Inamdar, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 8989 RSC.
- N. K. Shrestha, S. A. Patil, S. Cho, Y. Jo, H. Kim and H. Im, J. Mater. Chem. A, 2020, 8, 24408–24418 RSC.
- A. I. Inamdar, H. S. Chavan, J. H. Seok, C. H. Lee, G. Shin, S. Park, S. Yeon, S. Cho, Y. Park, N. K. Shrestha, S. U. Lee, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 20497–20508 RSC.
- H. S. Chavan, C. H. Lee, A. I. Inamdar, J. Han, S. Park, S. Cho, N. K. Shreshta, S. U. Lee, B. Hou, H. Im and H. Kim, ACS Catal., 2022, 12, 3821–3831 CrossRef.
- Y. Jo, S. Cho, J. Seo, A. T. A. Ahmed, C. H. Lee, J. H. Seok, B. Hou, S. A. Patil, Y. Park, N. K. Shrestha, S. U. Lee, H. Kim and H. Im, ACS Appl. Mater. Interfaces, 2021, 13, 53725–53735 CrossRef.
- L. Du, Y. Sun and B. You, Mater. Rep.: Energy, 2021, 1, 100004 Search PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 507, 230072 CrossRef.
- Z. Li, L. Sun, Y. Zhang, Y. Han, W. Zhuang, L. Tian and W. Tan, Coord. Chem. Rev., 2024, 510, 215837 CrossRef.
- W. Zhou, S. Chen, X. Meng, J. Li and J. Gao, Int. J. Hydrogen Energy, 2023, 48, 15748–15770 CrossRef.
- C. Chen, Q. Bai, J. Liu, Z. Wang and K. Cen, Int. J. Hydrogen Energy, 2020, 45, 20894–20903 CrossRef.
- W. Liu, X. Niu, J. Tang, Q. Liu, J. Luo, X. Liu and Y. Zhou, Chem. Synth., 2023, 3, 44 CAS.
- E. A. Moges, C. Y. Chang, M. C. Tsai, W. N. Su and B. J. Hwang, EES Catal., 2023, 1, 413–433 RSC.
- D. Li, J. Jiang, H. Dong, H. Li, X. Peng and P. K. Chu, Mater. Sci. Eng., R, 2024, 160, 100829 CrossRef.
- Z. Li, Y. Yan, S. M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS.
- Z.-. Ge, Y. Ding, T.-J. Wang, F. Shi, P.-J. Jin, P. Chen, B. He, S.-B. Yin and Y. Chen, J. Energy Chem., 2023, 77, 209–216 CrossRef CAS.
- X. -L. Liu, Y. -C. Jiang, J. -T. Huang, W. Zhong, B. He, P. -J. Jin and Y. Chen, Carbon Energy, 2023, 5, e367 CrossRef CAS.
- S. A. Patil, A. C. Khot, K. D. Kadam, H. T. Bui, H. Im and N. K. Shrestha, Inorg. Chem. Front., 2023, 10, 7204–7211 RSC.
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar and H. Im, Dalton Trans., 2023, 52, 10933–10941 RSC.
- S. A. Patil, S. Cho, Y. Jo, N. K. Shrestha, H. Kim and H. Im, Chem. Eng. J., 2021, 426, 130773 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, A. I. Inamdar, S. Park, S. Yeon, G. Shin, S. Cho, H. Kim and H. Im, Dalton Trans., 2022, 51, 8994–9006 RSC.
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar and H. Im, J. Mater. Chem. A, 2023, 11, 14870–14877 RSC.
- Z.-M. Wang, Q.-L. Hong, X.-H. Wang, H. Huang, Y. Chen and S.-N. Li, Acta Phys.-Chim. Sin., 2023, 39, 2303028 Search PubMed.
- L. Li, L. Zhang, L. Gou, S. Wei, X. Hou and L. Wu, Chem. Eng. J., 2023, 454, 140292 CrossRef CAS.
- N. Kuramochi, M. Yoshida-Hirahara, H. Ogihara and H. Kurokawa, Sustainable Energy Fuels, 2023, 7, 778–785 RSC.
- E. Ruiz-López, A. Caravaca, P. Vernoux, F. Dorado and A. de Lucas-Consuegra, Chem. Eng. J., 2020, 396, 125217 CrossRef.
- C. Lu, F. Yang, A. Schechter and L. Feng, Advanced Sensor and Energy Materials, 2023, 2, 100055 CrossRef.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS.
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X. Z. Fu and J. L. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS.
- J. Li, C. Xing, Y. Zhang, T. Zhang, M. C. Spadaro, Q. Wu, Y. Yi, S. He, J. Llorca, J. Arbiol, A. Cabot and C. Cui, Small, 2021, 17, 2006623 CrossRef CAS PubMed.
- N. Ullah, S. Ullah, S. Khan, D. Guziejewski and V. Mirceski, Int. J. Hydrogen Energy, 2023, 48, 3340–3354 CrossRef CAS.
- L. Yaqoob, T. Noor, N. Iqbal, H. Nasir and A. Mumtaz, Sci. Rep., 2021, 11, 13402 CrossRef CAS PubMed.
- S. A. Patil, A. C. Khot, V. D. Chavan, I. Rabani, D. Kim, J. Jung, H. Im and N. K. Shrestha, Chem. Eng. J., 2024, 480, 146545 CrossRef CAS.
- S. Chen, G. Hai, H. Gao, X. Chen, A. Li, X. Zhang and W. Dong, Chem. Eng. J., 2021, 406, 126886 CrossRef CAS.
- H. Guo, Z. Zheng, Y. Zhang, H. Lin and Q. Xu, Sens. Actuators, B, 2017, 248, 430–436 CrossRef CAS.
- K. Huang and X. Zhang, Aust. J. Chem., 2021, 74, 282–289 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, J. H. Seok, A. S. Salunke, S. Cho, A. I. Inamdar, Y. Park, S. U. Lee, H. Kim and H. Im, Mater. Today Phys., 2023, 38, 101252 CrossRef CAS.
- J. Zhao, N. Liao and J. Luo, J. Mater. Chem. A, 2023, 11, 9682 RSC.
- S. Zhao, Y. Wang, J. Dong, C. T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- C. Wu, X. Zhang, H. Li, Z. Xia, S. Yu, S. Wang and G. Sun, Chin. J. Catal., 2021, 42, 637–647 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar, H. Kim and H. Im, Dalton Trans., 2023, 52, 13852–13857 RSC.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS PubMed.
- R. Lin, L. Kang, T. Zhao, J. Feng, V. Celorrio, G. Zhang, G. Cibin, A. Kucernak, D. J. L. Brett, F. Corà, I. P. Parkin and G. He, Energy Environ. Sci., 2022, 15, 2386 RSC.
- Y. Yang, W. H. Lie, R. R. Unocic, J. A. Yuwono, M. Klingenhof, T. Merzdorf, P. W. Buchheister, M. Kroschel, A. Walker, L. C. Gallington, L. Thomsen, P. V. Kumar, P. Strasser, J. A. Scott and N. M. Bedford, Adv. Mater., 2023, 35, 2305573 CrossRef CAS.
- C. Feng, Y. Zhou, Z. Xie, Z. Yang, L. Zou, P. Wang, W. Lian, P. Xiaokaiti, Y. Kansha, A. Abudula and G. Guan, Chem. Eng. J., 2024, 495, 153408 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05688d |
| This journal is © The Royal Society of Chemistry 2024 |