
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of TFSI post-treatment on surface doping and passivation of lead halide perovskites†
Konstantina
Gkini
a,
Spyros
Orfanoudakis
ab,
Filippos
Harlaftis
a,
Panagiotis
Dallas
 a,
Christos
Kouzios
a,
Polychronis
Tsipas
a,
Athanassios G.
Kontos
b,
Maria
Konstantakou
a and
Thomas
Stergiopoulos
*a
a,
Christos
Kouzios
a,
Polychronis
Tsipas
a,
Athanassios G.
Kontos
b,
Maria
Konstantakou
a and
Thomas
Stergiopoulos
*a
aInstitute of Nanoscience and Nanotechnology, NCSR Demokritos, 15341, Aghia Paraskevi, Athens, Greece. E-mail: t.stergiopoulos@inn.demokritos.gr
bDepartment of Physics, School of Applied Mathematical and Physical Sciences, National Technical University of Athens, 15780, Zografou, Athens, Greece
First published on 22nd October 2024
Abstract
Bis(trifluoromethane)sulfonimide (TFSI) treatment results in near-unity photoluminescence quantum yields in monolayer transition-metal dichalcogenides, such as MoS2, due to passivation of native defects. Surprisingly, this simple post-treatment process has never been tested in the case of metal halide perovskites which suffer from limited radiative recombination due to charge carrier trapping. Here, we adopt this strategy and treat methylammonium lead iodide perovskite films with TFSI solutions. By employing photoluminescence spectroscopy, the appearance of brighter films proves a net passivation effect, while chemical analysis explains that this is due to strong interactions between S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of TFSI and under-coordinated Pb2+. A simultaneous passivation of iodide vacancies also leads to a reduction of n-doping at the perovskite surface and thus better hole extraction through spiro-MeOTAD which is deposited on top. These two effects combined (chemical passivation and de-doping) result in enhanced stabilized efficiencies for the as-fabricated n–i–p solar cells. The findings pave the way for the use of TFSI-based solutions to improve the performance of perovskite optoelectronic devices.
O groups of TFSI and under-coordinated Pb2+. A simultaneous passivation of iodide vacancies also leads to a reduction of n-doping at the perovskite surface and thus better hole extraction through spiro-MeOTAD which is deposited on top. These two effects combined (chemical passivation and de-doping) result in enhanced stabilized efficiencies for the as-fabricated n–i–p solar cells. The findings pave the way for the use of TFSI-based solutions to improve the performance of perovskite optoelectronic devices.
Introduction
Organic–inorganic halide perovskite solar cells (PSCs) are now a key player in emergent photovoltaics, achieving record power conversion efficiencies (PCE) over 26%.1 Despite defect tolerance of the material itself, accompanied by a range of traps that are relatively benign, several surface (and bulk) defects still exist, providing a pathway for undesirable non-radiative recombination processes for photo-excited charge carriers, thus limiting further improvement of PCE and challenging the long-term performance of photovoltaic devices.2–5The ionic nature of hybrid perovskite materials enables the neutralization of trap states through the passivation of charged defects.6–10 The most prominent strategy to achieve this is the Lewis acid–base approach, where the passivators coordinate with unwanted defects and form Lewis adducts by dative bonds; a Lewis acid contributes to passivation of negatively charged defects (e.g., under-coordinated halides and Pb–I anti-sites) and the Lewis base passivates positively charged defects (e.g., under-coordinated Pb2+ and Pb2+ interstitials).11–17 Towards this approach, pseudo-halide anions (like [PF6]− and [BF4]−) have recently emerged as the next generation of passivation agents in perovskite-based optoelectronics.18–20 One characteristic example of the pseudo-halide family is the [TFSI]− anion.21 Notably, there is limited literature on the surface treatment of perovskite films with TFSI−-based solutions. In these studies, TFSI− anions are utilized in conjunction with their counterparts which are hydrophobic, organic (usually imidazolium-based) or even polymeric, cations. However, even though the passivation is proven, the effect is limited only in regions where large polymer-containing islands are covering the perovskite surface.22 Additionally, the binding energy environment of the TFSI−-based solutions is strongly correlated with the coordination strength between anions and cations; the coordination strength then depends on the size of the cation.23
A solution to this issue could be the incorporation of a TFSI− anion without the presence of any large organic component. For instance, if a proton is used as a counter ion, the simplest and shortest form of the TFSI salt is created, bis(trifluoromethane)sulfonimide (abbreviated as TFSI). Interestingly, TFSI has been already adopted to passivate traps in silicon24 and InAs.25 Even more importantly, TFSI has improved the photoluminescence (PL) quantum yields of monolayers of metal chalcogenides such as MoS2, from <1% up to close to 100%, making them a promising platform for innovative optoelectronic devices.26–29 Since TFSI− is very electronegative and has been frequently adopted for p-doping in typical organic semiconductors,30–32 it has been argued that the substantial increase in the PL intensity is due to electrostatic doping, rather than actual chemical passivation. Additionally, even though the above effects are also related to the presence of labile protons in TFSI solution,33 there are recent studies which suggest that (super)acidity is not a prerequisite for effective passivation.34–36
With these properties in mind, we sought to investigate the effect of TFSI solution treatment on polycrystalline perovskite films. Let us underline that our work is different to what is already reported on the effect of TFSI as an additive on the MAPI precursor solution37 or the treatment of CsPbI3 with a huge excess of TFSI which leads to a net reaction of the perovskite with protons in TFSI, controlling – besides photoluminescence – the crystal shape, size, and structure of the nanocrystals.38 Here, TFSI is incorporated on top of the archetypal methylammonium lead iodide (abbreviated as MAPI). We use PL and Ultraviolet Photoelectron Spectroscopies (UPS) to investigate how TFSI passivates and energetically changes the surface of MAPI. Then we carry out a detailed chemical analysis to identify and explain the exact mechanism behind the TFSI–MAPI interactions. Finally, we fabricate and electrically characterize planar devices based on TFSI-treated MAPI films and examine whether the positive effects of TFSI are translated to improved photovoltaic performance.
Experimental section
Materials
The fluorine-doped tin oxide (FTO) glass substrates were supplied by Ossila Ltd. Lead iodide (PbI2, 99.99%) was supplied by TCI EUROPE N.V., while 2,2′,7,7′-Tetrakis[N,N di(4-methoxyphenyl)amino]-9,9′-spiro-bifluorene (Spiro-OMeTAD, 99.5%) was purchased from Borun New Material Technology LTD. Titanium tetraisopropoxide (97+%) was purchased from Alfa Aesar. Methylammonium iodide (99.99%), was acquired from Great cell Solar Materials Pty Ltd. 4-tert-Butylpyridine (99+%), N,N-dimethylformamide (99.8%), dimethylsulfoxide (99.7%), isopropyl alcohol (99.5%), chlorobenzene (CB, 99.5%) and acetonitrile (99.9%) were supplied from Acros Organics. Lithium bis(trifluoromethylsulfonyl)imide (Li-TFSI, 99.95%) and bis(trifluoromethane)sulfonimide (TFSI, 95%) were purchased from Sigma-Aldrich. All materials were used without further purification.Device fabrication
The FTO substrates were etched by using Zn powder and 6 M HCl solution and successively cleaned by sonication washing with a detergent (Helmanex III 2% in deionized water), deionized water, ethanol and isopropanol for 20 min. The cleaned FTO substrates were then treated with ultraviolet ozone for 20 min.For the compact TiO2 layer, an acidified (HCl) solution of titanium tetraisopropoxide in ethanol was prepared and 60 μl of it was deposited by spin coating at 2000 rpm for 60 s. The obtained films were annealed at 500 °C for 45 min in air. The precursor solution of the perovskite film was prepared by adding PbI2 and CH3NH3I in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio into a 9:1 (v/v) solution of N,N-dimethylformamide and dimethylsulfoxide. 60 μl of the solution were spin coated at 3000 rpm for 25 s. At the 10th second, 150 μl chlorobenzene was dropped on the spinning substrate. The resulting films were immediately annealed at 100 °C for 45 min. After the perovskite annealing process, 60 μl of the TFSI solution (0.018 M in chlorobenzene) were spin-coated on the surface of the film at 4000 rpm for 25 s and subsequently annealed at 100 °C for 2 min. For the HTL, a 70 mM solution of spiro-MeOTAD was prepared in chlorobenzene, along with 36 μl 4-tert-butylpyridine and 20 μl Li-TFSI salt (520 mg in 1 ml acetonitrile). The solution (50 μl) was spin-coated on the perovskite films at 4000 rpm for 10 s. Every step described above was carried out inside a N2-filled glovebox (Jacomex GP Campus T2) with minimum oxygen and water (<1 ppm). Then, the samples were transferred out of the glovebox and stored overnight in a desiccator to oxidize spiro-MeOTAD. Finally, 100 nm of silver was thermally evaporated on top of spiro-MeOTAD, under 10−6 Torr vacuum at a ∼1.2 Å s−1 rate. For stability tests, 100 nm of Au was deposited instead.
:1 molar ratio into a 9:1 (v/v) solution of N,N-dimethylformamide and dimethylsulfoxide. 60 μl of the solution were spin coated at 3000 rpm for 25 s. At the 10th second, 150 μl chlorobenzene was dropped on the spinning substrate. The resulting films were immediately annealed at 100 °C for 45 min. After the perovskite annealing process, 60 μl of the TFSI solution (0.018 M in chlorobenzene) were spin-coated on the surface of the film at 4000 rpm for 25 s and subsequently annealed at 100 °C for 2 min. For the HTL, a 70 mM solution of spiro-MeOTAD was prepared in chlorobenzene, along with 36 μl 4-tert-butylpyridine and 20 μl Li-TFSI salt (520 mg in 1 ml acetonitrile). The solution (50 μl) was spin-coated on the perovskite films at 4000 rpm for 10 s. Every step described above was carried out inside a N2-filled glovebox (Jacomex GP Campus T2) with minimum oxygen and water (<1 ppm). Then, the samples were transferred out of the glovebox and stored overnight in a desiccator to oxidize spiro-MeOTAD. Finally, 100 nm of silver was thermally evaporated on top of spiro-MeOTAD, under 10−6 Torr vacuum at a ∼1.2 Å s−1 rate. For stability tests, 100 nm of Au was deposited instead.
Characterization
The UV-vis transmittance spectra of the perovskite films were recorded with a Carry 60 Agilent UV-Vis spectrometer. Steady-state photoluminescence spectra were recorded in air on a Horiba Fluoromax + spectrofluorometer, equipped with a 150 W Xe lamp as the excitation source and a Hamamatsu R13456 PMT (185–980 nm nominal) as the emission detector. For the time-resolved photoluminescence spectra, a time-correlated-single-photon-counting (TCSPC) method via a Fluorohub single photon counting controller, a Delta-hub laser diode as an excitation source (404 nm, pulse duration <150 ps) and a PMT detector were applied, along with a 450 nm optical filter between the sample and the PMT. The lifetimes were determined by exponential fitting of the as-collected data with the DAS6 software.The surface morphologies of the perovskite films were examined with Scanning Electron Microscopy (SEM) using a JEOL 7401f Field Emission SEM and Atomic Force Microscopy (AFM) using a Solver Nano AFM in contact mode with a CSG30 probe. Kelvin Probe Force Microscopy (KPFM) measurements were also conducted with a Solver Nano AFM using a NSG30/PtIr probe. Fourier-transform Infrared Spectroscopy (FTIR) spectra where recorded using a PerkinElmer Spectrum 100 FT-IR. The crystal structure of the perovskite films was analyzed by using X-ray diffraction (XRD) patterns, which were obtained with a Smart Lab Rigaku diffractometer (Cu Kα radiation). X-ray photoelectron spectroscopy (XPS) measurements were carried out in order to analyze the chemical state and composition of the MAPbI3 films. The XPS data were collected with a PHOIBOS 100 (SPECS) hemispherical analyzer using an Mg Kα X-ray source with a photon energy of 1253.64 eV. Voigt functions were used for the fitting analysis after standard Shirley background subtraction. Work function (WF) and valence band maximum (VBM) values for the perovskite films were extracted by ultraviolet photoelectron spectroscopy (UPS) measurements using a helium excitation source with He I radiation at 21.22 eV.
The J–V curves were recorded with an Ossila Solar Cell I–V Test System while the PSCs were illuminated by using an AAA LED Solar Simulator, emitting AM 1.5 G light of 1000 W m−2 (Wavelabs Sinus-70). Transient Photo-Voltage (TPV) measurements were carried out by keeping the devices at the open-circuit voltage under various white light biases. When Voc is stabilized at the steady-state, an additional light pulse is applied that leads to an increase in voltage; the pulse is small enough to keep the ratio of the perturbation peak versus voltage bias less than 10%. After the light pulse is turned off, the voltage decays back to its previous value. From the resulting voltage decay, the charge carrier recombination lifetime is determined after fitting with a mono-exponential function. Likewise, similar Transient Photo-Current (TPC) measurements were conducted on the devices, but this time, the devices are held under short-circuit conditions, before turning off the light and let the photocurrent decay after a light pulse. Again, charge extraction times are determined through fitting using a mono-exponential function. The Impedance Spectra (IS) were acquired by applying a small sinusoidal voltage signal (±50 mV) over 0 V to the device in the dark. Average values were calculated based on measurements from 4 control devices and 4 TFSI-modified devices. The stability tests of the ISOS-L-1 under specific conditions (un-encapsulated, in the ambient atmosphere, at 25 °C, employing maximum power point-tracking, subjected to the equivalent of 1 sun illumination) were conducted employing a white LED, with adjusted intensity to match the short-circuit current obtained under 1 sun illumination. All the above measurements were conducted using Paios and Litos hardware platforms (provided by Fluxim).
Results and discussion
The brightening of various semiconductors by TFSI post-treatment is an effect highly dependent on the processing conditions.39 Thus, as a first step we optimized the experimental procedure. We performed all the deposition steps inside a glovebox, as TFSI should be handled exclusively under anaerobic conditions due to its hygroscopic and volatile characters. Given the harsh nature of TFSI, we should highlight here that we chose MAPI over other typical perovskites (FAPbI3 or CsPbI3) as, at least under our laboratory conditions, MAPI is a much more humidity-tolerant material. In any case, our idea was that hygroscopic TFSI on top of MAPI could serve as a desiccant and prevent the degradation of the perovskite by moisture.40,41Considering the good compatibility of TFSI with chlorinated organic solvents,42 we identified chlorobenzene (CB) as the optimum solvent, providing the most complete dissolution among several other candidates (such as dichloroethane and dichlorobenzene). We recall that CB is an antisolvent of perovskite, and thus it will not harm the surface of the perovskite. This is an advantage of our approach over other surface treatments with KTFSI and LiTFSI which utilize isopropanol and acetonitrile as solvents.43,44 Then, we prepared solutions of TFSI in CB with varying concentrations (0.036 M, 0.018 M, and 0.009 M) based on reported applications of TFSI.45,46 The concentration of 0.018 M provided the optimal result, based on the intensities of the acquired photoluminescence (PL) spectra, as shown in Fig. S1a.† After spin-coating the solution on top of MAPI, we dried the films at 100 °C to activate the TFSI effect by thermal annealing, as we obtained more reproducible results in PL.47 As a reference test, we poured CB on top of MAPI without any TFSI on top. The PL results underline that chlorobenzene alone has no effect on the perovskite film (Fig. S1b†). Additionally, in order to exclude the case that the processing solvent (CB) of the hole transport material (HTM) which is deposited on top of the perovskite would wash TFSI off the perovskite film's surface, we spin-coated pure CB after TFSI's deposition. Again, the PL spectrum (Fig. S1c†) suggests that TFSI's effect on the perovskite film remains even after treatment with the HTM's solvent. This fact could be also an indication that TFSI does not just exist as an ultrathin layer above the perovskite film, but interacts chemically with it.
We, then, confirmed the successful TFSI deposition on top of the perovskite film (see Fig. S2† and the relevant discussion). Subsequently, we proceeded with recording UV-vis absorption spectra (Fig. 1a) and XRD diffractograms (Fig. 1b). Both indicated virtually no effect of the TFSI treatment on the crystallinity of tetragonal MAPbI3.48 On the other hand, top-view SEM imaging of bare and treated films (Fig. 1c and e) revealed that TFSI-modified films have fewer pinholes, while grain boundaries appear less prominent. Likewise, contact mode AFM imaging (Fig. 1d and f) has shown a slight decrease (of 1.2 nm) in the root mean square roughness of the treated film in comparison with the pristine sample, suggesting that a smoother and more uniform surface is obtained.
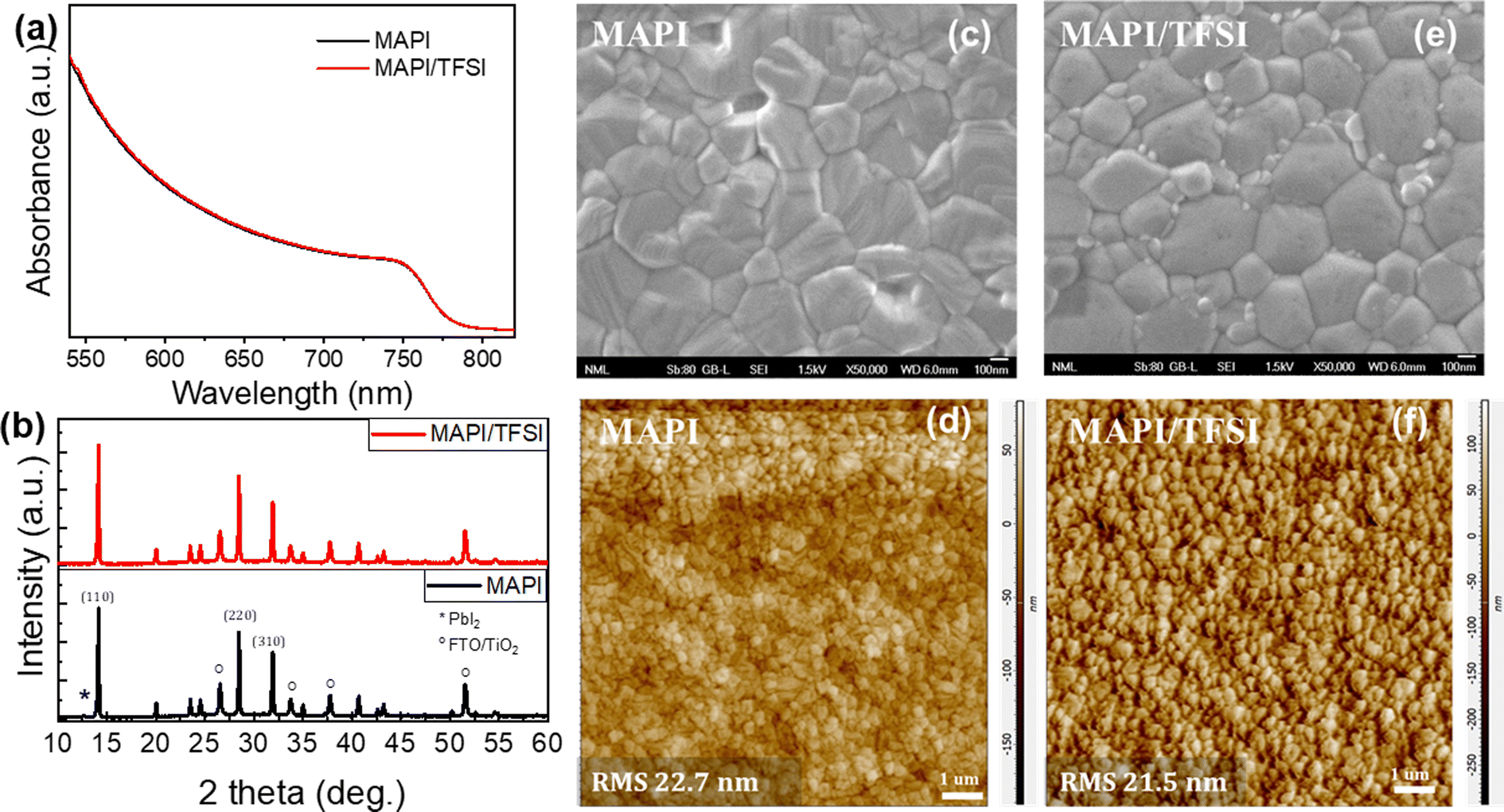 | ||
| Fig. 1 UV-vis absorption spectra (a), XRD diffractograms (b), high magnification top-view SEM images (c and e) and contact mode AFM images (d and f) of MAPI and MAPI/TFSI films. | ||
The next step of our experiments was to investigate the potential passivating character of TFSI through PL spectroscopy. Taking into consideration the strong absorption coefficient of MAPI in short wavelength range spectra, we excited the perovskite/glass films from the perovskite side at 450 nm. PL spectra of 5 representative films were recorded (the average is shown in Fig. 2a), showing an intensity increase (of around 48%) in the case of the treated samples, which translates to enhanced radiative charge carrier recombination and, thus, successful passivation of trap states.49
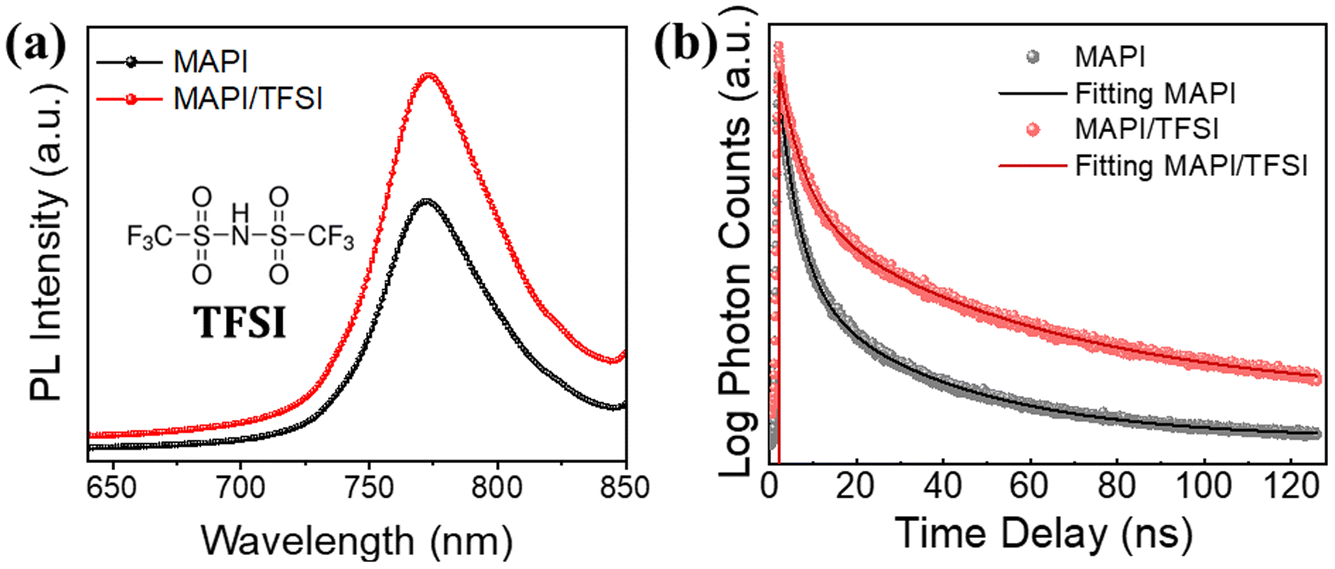 | ||
| Fig. 2 Steady-state PL spectra (a) and time-resolved PL decays (b) of MAPI films, with and without TFSI passivation. The inset of (a) is the structural formula of TFSI. | ||
The results were further confirmed by using time-resolved photoluminescence (TRPL) decay (Fig. 2b). The decay curves were fitted by using a bi-exponential function.50 The first decay (τ1), which corresponds to the fast component, is correlated with the trap filling process of the photogenerated carriers into sub-gap traps, while the second decay (τ2), attributed to the slow component, results from the band-to-band recombination process.51,52 The fittings show longer PL lifetimes for the modified samples in both components, in comparison with those of the pristine perovskite films. The average lifetimes were calculated to be 38.1 ns for the TFSI treated perovskite film and 19.5 ns for the control films (Table S1†).
Trap passivation on MAPI perovskite could arise from various chemical and/or charge transfer mechanisms. To examine such effects, we first conducted XPS measurements. To have a clearer idea of the interactions between TFSI and MAPI, an additional (relatively high) concentration of TFSI (0.12 M) was also examined. The corresponding Pb4f core level spectra are presented in Fig. 3a. The Pb4f scans of the MAPI film show two main peaks at binding energies of 138.7 eV and 143.6 eV related to Pb4f7/2 and Pb4f5/2 signals. The spin–orbit splitting between the Pb 4f7/2 and Pb 4f5/2 levels in all cases remains at 4.9 eV, which is in accordance with the literature values, suggesting that the oxidation state is Pb2+.53 After TFSI treatment, we observe a shift in all core levels of the perovskite elements towards lower binding energies compared to those of the reference sample. The shifts are more obvious (of the order of 0.3 to 0.35 eV) in the case of 0.12 M TFSI, indicating strong interaction between TFSI and the perovskite. The observed shifts imply an increase in the electron density on the Pb atoms (Fig. 3b).54 We argue that this shift is a clear indication of the interaction of TFSI− with Pb2+ (which is left under-coordinated).
 | ||
| Fig. 3 Pb4f XPS spectra of MAPI and MAPI/TFSI films (a), schematic illustration of the correlation between XPS shifts and an atom's electron density (b), I3d XPS spectra (c) and Pb4f XPS spectra focused on 4f7/2 peaks (d) of MAPI and MAPI/TFSI films. | ||
On the other hand, the I3d core level spectra are presented in Fig. 3c. The two main peaks at 619.6 eV and 631.1 eV of the I3d spectrum are ascribed to I3d5/2 and I3d3/2, respectively.55 Also, here, there is a clear shift towards lower binding energies. Surprisingly, this shift is very similar (in eV) to that observed in the case of Pb.56 This phenomenon is more complicated to explain as the electron density of I− also increases. Since this fact excludes that this shift is due to interactions of –CF3 and/or H+ (of TFSI) with I−, we hypothesize that it could be ascribed to a halogen bond between oxygen (of the –SO2 group) and iodide.57 In any case, similar phenomena have been observed in the XPS of perovskite films after passivation with sulfonate anions and were previously attributed to space charge effects associated with the increased work function of the film (which we prove in the UPS discussion later).58
To move a step deeper in our XPS analysis, we acquired high-resolution scans of Pb4f7/2 spectra (Fig. 3d) and we were able to unveil an additional peak at 136.9 ± 0.1 eV. According to the literature, this peak is attributed to metallic lead clusters, which result from the formation of stable Pb–Pb dimers, which have been suggested to be a key defect, increasing the recombination rate in a metal halide perovskite solar cell.59 These dimers form when, upon light illumination, an electron gets trapped in an iodide vacancy, attracting two nearby Pb atoms that detach from I atoms; thus, to interrupt the formation of the Pb dimers, it is imperative to circumvent iodide vacancies.60 Surprisingly, the peak of Pb0 completely vanishes upon TFSI treatment. Given the above mechanism, we argue that this result comes from elimination of iodide vacancies. This assumption agrees well with what we observed in the XPS of Pb; TFSI− anions interact with under-coordinated Pb2+ of the [PbI6]4− octahedron at sites where iodide is missing (thus eliminating iodide vacancies).20
Fourier transform infrared spectroscopy (FTIR) was then employed in order to identify the type of interactions between TFSI and the MAPI perovskite. As a first step, the FTIR spectrum of the pristine TFSI molecule was recorded and can be seen in Fig. 4a (blue line). Strong vibrational modes at 1252, 1233, and 1200 cm−1 are assigned to the –CF3 groups. The asymmetric and symmetric SO stretching vibrations are observed at 1335 and 1118 cm−1 respectively.61 Finally, the secondary amine –N–H modes are observed as single peaks at 3191 cm−1. As a control experiment, TFSI was mixed with lead iodide (PbI2) in DMF at a molar ratio of 1:2. The spectrum is presented in Fig. 4a (orange line). In previous studies, strong interactions between the SO and the lead cations62 as well as hydrogen bonding between the –H of the organic cation and the fluorine of the polar –Cδ+–Fδ− bonds were reported.63 After complexation with lead iodide, the –N–H band at 3191 cm−1 disappears. This is a strong indication that TFSI is completely dissociated. In addition, the asymmetric SO mode splits. This indicates that the molecule inside the matrix is participating in two different environments. On the other hand, the shifts observed in the C–F region between 1200 and 1300 cm−1 are minor. When TFSI reacts with the MAPI perovskite and its strongly basic methylammonium component (pKb = 3.36), the pattern drastically changes, with the SO and C–F bands now appearing as a broad spectrum, while SO also shifts to higher energies (Fig. 4a-red line). In general, the –Cδ+–Fδ- bond appears in a wide range in the FTIR spectrum due to the sensitivity of the stretching frequency to other functional groups and the environment. Taking into consideration the shifts in Pb4f XPS spectra also, we propose a coordination of Pb2+ with SO, which justifies the passivation of under-coordinated Pb2+ defects by TFSI− anions. The abovementioned scenario could be further supported by the O1s XPS spectra (Fig. S3†) where an additional peak at 530.7 eV (indicative of the Pb–O bond) appears for the treated film.64
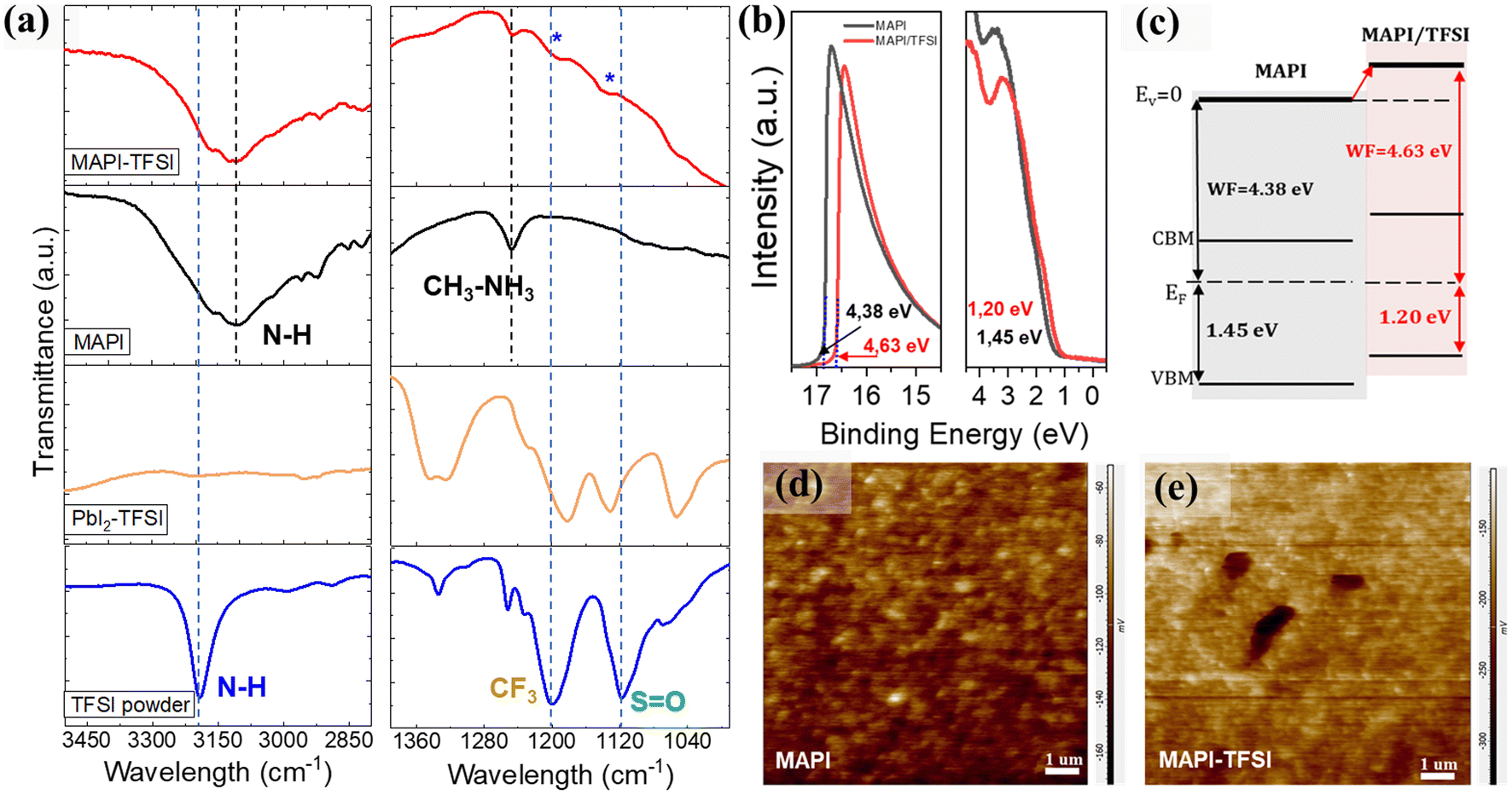 | ||
| Fig. 4 FTIR spectra of the TFSI powder, PbI2–TFSI mixture, MAPI film and MAPI/TFSI film in a comparative presentation (a). UPS spectra (b), schematic diagram of the relative energy level positions (c), and Kelvin probe AFM images (d and e) of FTO/TiO2/MAPI and FTO/TiO2/MAPI/TFSI films of the corresponding devices. | ||
We have detected some particular trends in PL and XPS spectra which we previously attributed to charging of the perovskite surface upon TFSI treatment. To further understand these effects, UPS measurements were performed. From the corresponding spectra (Fig. 4b), the work function (WF) was estimated by applying the relation WF = hv − Ecutoff, where hv is the excitation energy, ∼21.22 eV, and Ecutoff is the low energy cutoff, and found to be ∼4.38 eV and ∼4.63 eV for the bare and the TFSI-treated perovskite films, respectively. Likewise, the distance between the VBM and Fermi level (EF) could be estimated from the lowest binding energy cutoff in UPS spectra, giving the values of ∼1.45 eV and ∼1.20 eV, respectively. Taking these data in hand and also extracting the optical bandgap of the perovskite from the Tauc plots (Fig. S4a†), we sketched a diagram representing the relative positions of the energy levels, after the Fermi level equilibrium, for perovskite films before and after TFSI passivation (Fig. 4c). First, the schematic reveals that the control films show a typical n-type behavior with an EF close to the conduction band maximum (CBM); n-doping in MAPI usually results from the presence of iodide vacancies, which are donor defects.65,66 Compared to the control film, the work function of the TFSI-treated films increased by 0.25 eV and, with this, the EF was shifted downward to the VBM, demonstrating the de-doping of the perovskite surface, that is its transformation from n-type into less n-type (more p-type).67–69 We argue that this is due to the elimination of iodide vacancies which we have previously shown. This effect will provide more favorable energetics at the perovskite/hole transport layer interface for accelerating hole extraction. The UPS results are in agreement with the XPS downshifts of both Pb4f and I3d core levels, confirming the change in surface energy levels.56,70–72 The results are further supported by Kelvin-Probe Force Microscopy (KPFM) measurements (Fig. 4d and e), as the contact potential difference (CPD) reduces from −113 mV for MAPI films to −190 mV for the TFSI treated films. The CPD is related to the sample's surface work function by using the equation CPD = (Φtip − Φsample)/IeI; thus a lower CPD is translated into higher WF (which we also determined from UPS analysis).73–75
Proving both chemical passivation and surface de-doping of MAPI after the TFSI post-treatment, we then turned our attention to check whether these effects translate into performance improvement in operational devices. For this, we fabricated n–i–p devices and their photovoltaic performance was evaluated under 1 sun AM1.5G illumination. Indicative forward and reverse scan J–V curves are shown in Fig. 5a, while the corresponding photovoltaic metrics for the pristine and treated devices are shown in Table 1. A statistical analysis of 30 different devices is also presented in Fig. S5.† For clarity, we present the results of all devices in Tables S3 and S4.† The TFSI-treated devices outperform the control devices, presenting a higher Voc and reaching a mean reverse-scan power conversion efficiency of 17.7%, while the PCE of the control device was 17.3%. To verify the positive effect of TFSI and also examine possible efficiency advancements, we also fabricated two other interesting configurations with TFSI on top and LiTFFSI-free spiro, and TFSI as an additive during crystallization (in the antisolvent) with standard doped spiro-MeOTAD. Even though the results were interesting (Fig. S6 and S7†), the concepts did not deliver any better efficiencies.
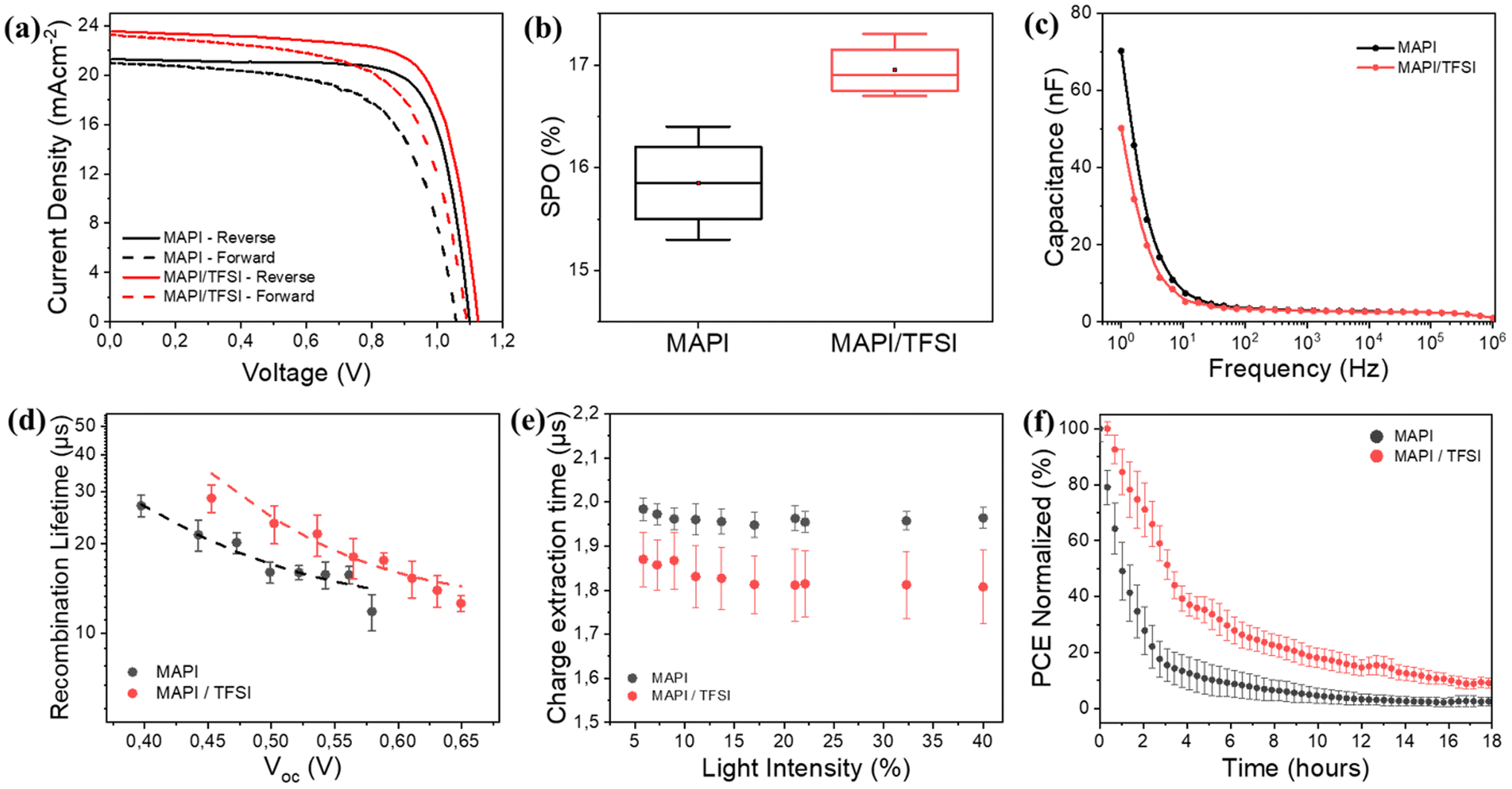 | ||
| Fig. 5 J–V characteristics under forward (dashed lines) and reverse scan (a), statistical analysis of the SPO (b), capacitance–frequency plots (c), recombination lifetime against VOC (d), charge extraction time under different illuminations (e) and PCE stability under light stress (ISOS-L-1) (f), for planar PSCs based on MAPI films, with and without TFSI passivation. | ||
| Sample | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | SPO (%) |
|---|---|---|---|---|---|
| MAPI | 21.7 ± 0.9 (23.4) | 1.08 ± 0.02 (1.11) | 72.8 ± 1.6 (76.4) | 17.3 ± 0.8 (18.8) | 15.8 ± 0.5 (16.4) |
| MAPI/TFSI | 22.5 ± 0.8 (23.9) | 1.10 ± 0.02 (1.13) | 73.5 ± 1.9 (76.9) | 17.7 ± 0.8 (19.4) | 17.0 ± 0.3 (17.3) |
However, due to hysteresis effects observed in the current–voltage characteristics of perovskite solar cells and in order to not report overestimated efficiencies by using a favorable scan, we also recorded the stabilized current density under maximum power point bias for 120 s.76 The measurements resulted in a mean stabilized power output (SPO) of 17% for the passivated devices, superior to a SPO of 15.8% for the control devices (Fig. 5b). Notably, in the case of TFSI-treatment, the current (efficiency) plateaus more quickly and without losing much of the initial value during the initial seconds of stabilization (Fig. S8†). This increase in the SPO agrees well with a concomitant hysteresis index (HI)77 reduction which was found to decrease from 0.18 to 0.12 for representative devices without and with TFSI, respectively (Table S2†).
It is well accepted that ion migration is the dominant contribution to hysteresis.78 To this end, we were able to detect ion effects through proper impedance measurements on our devices. Typical C–f plots are shown in Fig. 5c. The capacitance recorded at low frequencies (1–10 Hz) in the dark is mainly dominated by ions accumulated at the surface.76,79 We observe that this capacitance decreases for TFSI-modified devices, pointing to lower mobile ion concentration. This result is in agreement with what we observe with the J–V analysis. In MAPI, the governing mechanism for ion mobility is vacancy-assisted migration of iodide ions.80 Therefore, the elimination of iodide vacancies by TFSI is responsible for reducing hysteresis effects and increasing the SPO.
To continue with the impedance analysis, Nyquist plots (Fig. S9†) present one visible arc, which is attributed to the coupling of the parallel resistance (which we abbreviate as R1) and the total geometric capacitance (Cg), along with a transmission-line at lower frequencies, which is associated with ionic/charge transport and diffusion in perovskite films.81 A typical equivalent circuit, which also employs a series resistance (Rs), was adopted for fitting,82 from which we extracted (among other parameters) R1 that corresponds to the recombination resistance, by assuming that the shunt resistance is much higher. The larger value of recombination resistance (24.1 kΩ) for the TFSI-modified devices, in comparison with that of control devices (10.6 kΩ), is indicative of reduced recombination due to effective trap passivation.
The above results were further justified by transient photovoltage (TPV) measurements. We plot the calculated recombination lifetimes (τ) against Voc (Fig. 5d), which are dictated by the white light bias, according to the following equation: τ = τ0eβVoc, where β is related to the sensitivity to Voc change. The TPV graphs represent the average values, calculated from 4 different devices. We observe that the recombination lifetimes are longer for TFSI-modified devices (∼25 μs at 0.5 V) in comparison to control devices (∼17 μs at 0.5 V). Likewise, by plotting Voc against the light intensity (Fig. S10a†), we also notice that at the same light intensity, a higher Voc is attained in the case of the TFSI-based solar cells. The ideality factor was calculated by using the graphs' slopes which is equal to nkBT/q, where n is the ideality factor, kB the Boltzmann constant, T the absolute temperature and q the electron's charge. The results demonstrated similar ideality factors for both device systems (1.61 for the modified and 1.60 for the pristine). Similar ideality factors imply that, even though the recombination decreases (verified by TPV), the mechanism of the recombination from which the charges are lost in the device, is not changing with or without the TFSI post-treatment.83
Now, to examine whether the reduction of n-doping contributes to better hole extraction, the charge extraction times were determined through transient photocurrent (TPC) decays (Fig. S10b†).84 Again, we report average times from 8 different devices against the background light intensity (Fig. 5e). We observe that TPC lifetimes of TFSI-modified devices are shorter (∼1.8 μs) than those of the control devices (∼2.0 μs), implying a better charge extraction. We assume that this enhancement is due to increased hole-selectivity and extraction, attributed to better energetics at the perovskite/HTM interface.
Finally, we conducted light stress tests (ISOS-L-1)85 to investigate the passivation effect of TFSI on the working stability of the devices. Fig. 5f (and Fig. S11†) shows the PCE's progress (average values of 4 different devices) after 18 hours of continuous illumination. Control devices demonstrate a decrease in PCE from 100% to 80% within less than an hour. In contrast, the TFSI-modified devices maintain 80% of their initial PCE for up to 2 hours under continuous illumination. This result also complies with the trap passivation/reduction of ions effect of TFSI, since degradation via chemical reactions, driven by ion migration and trapped-charges in the presence of oxygen and water, degrade the photovoltaic performance of a perovskite solar cell under heat or light stress.86
Conclusions
In summary, we have demonstrated that TFSI can be effective, not only for transition metal (Mo2+ and W2+) sulfides, but also for lead halide perovskites. By adopting MAPI as a model perovskite, we provide evidence that the treatment of the perovskite films with TFSI results in a synergistic positive effect. First, TFSI passivates the undercoordinated Pb2+ and iodide vacancies at the perovskite surface through its sulfonyl groups. Second, the elimination of iodide vacancies reduces the n-doping near the perovskite surface. These effects lead to suppressed non-radiative recombination and improved hole extraction at the perovskite/hole transport layer interface. As a result, TFSI-treated n–i–p solar cells present significant SPO efficiencies and an enhanced operational stability under light stress. This study suggests that the field can take advantage of established passivation methods which work efficiently in similar electronic materials.Data availability
Data for this article are either included in the manuscript and the ESI† or are available upon request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the Hellenic Foundation for Research and Innovation (H. F. R. I.) under the “1st Call for H. F. R. I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment” (Project Number: 1027). T. S. also acknowledges the financial support from the European Research Council (ERC) through a Consolidator Grant (818615-MIX2FIX). We also thank Maria-Christina Skoulikidou for taking the SEM images.Notes and references
- J. Park, J. Kim, H.-S. Yun, M. Jae Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 137, 460–466 CrossRef.
- M. Kim, G. H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368, 6497 Search PubMed.
- A. Jamshaid, Z. Guo, J. Hieulle, C. Stecker, R. Ohmann, L. K. Ono, L. Qiu, G. Tong, W. Yin and Y. Qi, Energy Environ. Sci., 2021, 14, 4541–4554 RSC.
- D. Bi, C. Yi, J. Luo, J. D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. En., 2016, 1, 16142 CrossRef CAS.
- T. Wu, Y. Wang, X. Li, Y. Wu, X. Meng, D. Cui, X. Yang and L. Han, Adv. Energy Mater., 2019, 9, 1803766 CrossRef.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J. M. Liu, A. Petrozza, J. Wang, L. M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 136, 418–424 CrossRef.
- R. Wang, J. Xue, L. Meng, J. W. Lee, Z. Zhao, P. Sun, L. Cai, T. Huang, Z. Wang, Z. K. Wang, Y. Duan, J. L. Yang, S. Tan, Y. Yuan, Y. Huang and Y. Yang, Joule, 2019, 3, 1464–1477 CrossRef CAS.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- X. Zheng, Y. Deng, B. Chen, H. Wei, X. Xiao, Y. Fang, Y. Lin, Z. Yu, Y. Liu, Q. Wang and J. Huang, Adv. Mater., 2018, 30, 1803428 CrossRef.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 27, 1–9 Search PubMed.
- F. Wang, W. Geng, Y. Zhou, H. H. Fang, C. J. Tong, M. A. Loi, L. M. Liu and N. Zhao, Adv. Mater., 2016, 28, 9986–9992 CrossRef CAS PubMed.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS PubMed.
- A. Abate, M. Saliba, D. J. Hollman, S. D. Stranks, K. Wojciechowski, R. Avolio, G. Grancini, A. Petrozza and H. J. Snaith, Nano Lett., 2014, 14, 3247–3254 CrossRef CAS PubMed.
- Y. Lin, B. Chen, F. Zhao, X. Zheng, Y. Deng, Y. Shao, Y. Fang, Y. Bai, C. Wang and J. Huang, Adv. Mater., 2017, 29, 1700607 CrossRef PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 51, 1–7 Search PubMed.
- J. Xu, H. Chen, L. Grater, C. Liu, Y. Yang, S. Teale, A. Maxwell, S. Mahesh, H. Wan, Y. Chang, B. Chen, B. Rehl, S. M. Park, M. G. Kanatzidis and E. H. Sargent, Nat. Mater., 2023, 22, 1507–1514 CrossRef CAS PubMed.
- S. Liu, Y. Lu, C. Yu, J. Li, R. Luo, R. Guo, H. Liang, X. Jia, X. Guo, Y.-D. Wang, Q. Zhou, X. Wang, S. Yang, M. Sui, P. Müller-Buschbaum and Y. Hou, Nature, 2024, 628, 306–312 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- J. Foropoulos and D. D. Desmarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef CAS.
- P. Caprioglio, D. S. Cruz, S. C. -Davila, F. Zu, A. A. Sutanto, F. Pena-Camargo, L. Kegelmann, D. Meggiolaro, L. Gregori, C. M. Wolff, B. Stiller, L. Perdigon-Toro, H. Kobler, B. Li, E. Gutierrez-Partida, I. Lauermann, A. Abate, N. Koch, F. De Angelis, B. Rech, G. Grancini, D. Abou-Ras, M. K. Nazeeruddin, M. Stolterfoht, S. Albrecht, M. Antonietti and D. Neher, Energy Environ. Sci., 2021, 14, 4508 RSC.
- L. Du Hill, M. De Keersmaecker, A. E. Colbert, J. W. Hill, D. S. Placencia, J. E. Boercker, N. R. Armstrong and E. L. Ratcliff, Mater. Horiz., 2022, 9, 471 RSC.
- A. I. Pointona, N. E. Granta, E. C. Wheeler-Jonesb, P. P. Altermattc and J. D. Murphya, Sol. Energy Mater. Sol. Cells, 2018, 183, 164–172 CrossRef.
- Y. Zeng, S. Khandelwal, K. F Shariar, Z. Wang, G. Lin, Q. Cheng, P. Cui, R. Opila, G. Balakrishnan, S. Addamane, P. Taheri, D. Kiriya, M. Hettick and A. Javey, IEEE Trans. Electron Devices, 2019, 66, 1856–1861 CAS.
- T. Greg Tucker, Y. Ansari, C. Austen Angell -, Y.-Y. Yue, Z. Wang, L. Wang, J. Liu, N. Liu, D. Liu, D. Kiriya and D.-H. Lien, Nano Express, 2022, 3, 034002 CrossRef.
- H. Lu, A. Kummel and J. Robertson, APL Mater., 2018, 6, 066104 CrossRef.
- M. Amani, D. H. Lien, D. Kiriya, J. Xiao, A. Azcatl, J. Noh, S. R. Madhvapathy, R. Addou, K. C. Santosh, M. Dubey, K. Cho, R. M. Wallace, S. C. Lee, J. H. He, J. W. Ager, X. Zhang, E. Yablonovitch and A. Javey, Science, 2015, 350, 1065–1068 CrossRef CAS PubMed.
- Y. Hou, Z. R. Zhou, T. Y. Wen, H. W. Qiao, Z. Q. Lin, B. Ge and H. G. Yang, Nanoscale Horiz., 2018, 4, 208–213 RSC.
- S. M. Kim, Y. W. Jo, K. K. Kim, D. L. Duong, H.-J. Shin, J. H. Han, J.-Y. Choi, J. Kong and Y. H. Lee, ACS Nano, 2010, 4, 6998–7004 CrossRef CAS.
- Y. Xiao, S. Zhou, Y. Su, L. Ye, S.-W. Tsang, F. Xie and J. Xu, Org. Electron., 2014, 15, 3702–3709 CrossRef CAS.
- A. Abate, D. J. Hollman, J. Teuscher, S. Pathak, R. Avolio, G. D'Errico, G. Vitiello, S. Fantacci and H. J. Snaith, J. Am. Chem. Soc., 2013, 135, 13538–13548 CrossRef CAS PubMed.
- S. N. Suarez, J. R. P. Jayakody, S. G. Greenbaum, T. Zawodzinski Jr and J. J. A Fontanella, J. Phys. Chem. B, 2010, 114, 8941–8947 CrossRef CAS.
- S. L. Pain, N. E. Grant and J. D. Murphy, ACS Nano, 2022, 16, 1260–1270 CrossRef CAS PubMed.
- Z. Li, H. Bretscher, Y. Zhang, G. Delport, J. Xiao, A. Lee, S. D. Stranks and A. Rao, Nat. Commun., 2021, 12, 6044 CrossRef CAS PubMed.
- B. F. M. Healy, S. L. Pain, J. Lloyd-Hughes, N. E. Grant and J. D. Murphy, Adv. Mater. Interfaces, 2024, 2400305 CrossRef CAS.
- C. Tian, Y. Zhao, X. Han, B. Li, Y. Rui, H. Xiong, Y. Qiu, W. An, K. Li, C. Hou, Y. Li, H. Wang and Q. Zhang, J. Chem. Eng., 2023, 452, 139345 CrossRef CAS.
- M. Liu, L. Ma, K. Xie, P. Zeng, S. Wei, F. Zhang, C. Li and F. Wang, J. Phys. Chem. Lett., 2022, 13, 1519–1525 CrossRef CAS.
- D. Kiriya, Y. Hijikata, J. Pirillo, R. Kitaura, A. Murai, A. Ashida, T. Yoshimura and N. Fujimura, Langmuir, 2018, 34, 10243–10249 CrossRef CAS.
- C. Ma and N.-G. Park, ACS Energy Lett., 2020, 5, 3268–3275 CrossRef CAS.
- M. Kim, S. G. Motti, R. Sorrentino and A.-M. Petrozza, Energy Environ. Sci., 2018, 11, 2609 RSC.
- W. Zhao and J. Sun, Chem. Rev., 2018, 118, 10349–10392 CrossRef CAS PubMed.
- Q. Chen, G. Zhai, J. Ren, Y. Huo, Z. Yun, H. Jia, Y. Gao, C. Yu and B. Xu, Org. Electron., 2022, 107, 106544 CrossRef CAS.
- L. Zhao, Q. Li, C.-H. Hou, S. Li, X. Yang, J. Wu, S. Zhang, Q. Hu, Y. Wang, Y. Zhang, Y. Jiang, S. Jia, J. J. Shyue, T. P. Russell, Q. Gong, X. Hu and R. Zhu, J. Am. Chem. Soc., 2022, 144, 1700–1708 CrossRef CAS PubMed.
- M. Amani, P. Taheri, R. Addou, G. H. Ahn, D. Kiriya, D. H. Lien, J. W. Ager, R. M. Wallace and A. Javey, Nano Lett., 2016, 16, 2786–2791 CrossRef CAS.
- H. Bretscher, Z. Li, J. Xiao, D. Y. Qiu, S. Refaely-Abramson, J. A. Alexander-Webber, A. Tanoh, Y. Fan, G. Delport, C. A. Williams, S. D. Stranks, S. Hofmann, J. B. Neaton, S. G. Louie and A. Rao, ACS Nano, 2021, 15, 8780–8789 CrossRef CAS.
- Y. Yamada, K. Shinokita, Y. Okajima, S. N. Takeda, Y. Matsushita, K. Takei, T. Yoshimura, A. Ashida, N. Fujimura, K. Matsuda and D. Kiriya, ACS Appl. Mater. Interfaces, 2020, 12(32), 36496–36504 CrossRef CAS PubMed.
- R. Brenes, C. Eames, V. Bulović, M. Saiful Islam and S. D. Stranks, Adv. Mater., 2018, 30, 1706208 CrossRef PubMed.
- J. Zhu, S. Park, O. Y. Gong, C. H. Sohn, Z. Li, Z. Zhang, B. Jo, W. Kim, G. S. Han, D. H. Kim, T. K. Ahn, J. Lee and H. S. Jung, Energy Environ. Sci., 2021, 14, 4903–4914 RSC.
- A. Solanki, P. Yadav, S. H. Turren-Cruz, S. S. Lim, M. Saliba and T. C. Sum, Nano Energy, 2019, 58, 604–611 CrossRef CAS.
- T. Leijtens, G. E. Eperon, A. J. Barker, G. Grancini, W. Zhang, J. M. Ball, a A. Ram Srimath Kandada, H. J. Snaith and A. Petrozza, Energy Environ. Sci., 2016, 9, 3472–3481 RSC.
- T.-Y. Hsieh, M. Pylnev, E. Palomares and T.-C. Wei, Adv. Funct. Mater., 2020, 30, 1909644 CrossRef CAS.
- R. Lindblad, D. Bi, B.-w. Park, J. Oscarson, M. Gorgoi, H. Siegbahn, M. Odelius, E. M. J. Johansson and H. Rensmo, J. Phys. Chem. Lett., 2014, 5, 648–653 CrossRef CAS PubMed.
- C. Li, N. Zhang and P. Gao, Mater. Chem. Front., 2023, 7, 3797 RSC.
- S. Béchu, M. Ralaiarisoa, A. Etcheberry and P. Schulz, Adv. Energy Mater., 2020, 10, 1904007 CrossRef.
- N. K. Noel, S. N. Habisreutinger, A. Pellaroque, F. Pulvirenti, B. Wenger, F. Zhang, Y.-H. Lin, O. G. Reid, J. Leisen, Y. Zhang, S. Barlow, S. R. Marder, A. Kahn, H. J. Snaith, C. B. Arnold and B. P. Rand, Energy Environ. Sci., 2019, 12, 3063 RSC.
- K.-S. Shin, M. Brezgunova, O. Jeannin, T. Roisnel, F. Camerel, P. Auban-Senzier and M. Fourmigué, Cryst. Growth Des., 2011, 11(12), 5337–5345 CrossRef CAS.
- T. Shao, Y. Zang, Y. Tu, Y. Liu, P. Zhou, G. Li, R. Liu, G. Yang, J. Ye, C. Zhu, H. Chen, Q. Zhou, Y. Wang and W. Yan, Cell Rep. Phys. Sci., 2024, 5(6), 101996 CrossRef CAS.
- R. A. Kerner, T. H. Schloemer, P. Schulz, J. J. Berry, J. Schwartz, A. Sellinger and B. P. Rand, J. Mater. Chem. C, 2019, 7, 5244 RSC.
- X. Feng, B. Liu, Y. Peng, C. Gu, X. Bai, M. Long, M. Cai, C. Tong, L. Han and J. Yang, Small, 2022, 18, 2201831 CrossRef CAS PubMed.
- J. Tan, X. Ao, A. Dai, Y. Yuan, H. Zhuo, H. Lu, L. Zhuang, Y. Ke, C. Su, X. Peng, B. Tian and J. Lu, Energy Storage Mater., 2020, 33, 173–180 CrossRef.
- A. P. Côté and G. K. H. Shimizu, Coord. Chem. Rev., 2003, 245, 49064 CrossRef.
- R. Wang, L. Xie, T. Wu, C. Ge and Y. Hua, Chem. Sci., 2023, 14, 2877 RSC.
- J. S. Stevens, S. Coultas, C. Jaye, D. A. Fischer and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2020, 22, 4916–4923 RSC.
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- L. A. Frolova, N. N. Dremova and P. A. Troshin, Chem. Commun., 2015, 51, 14917 RSC.
- A. Zohar, I. Levine, S. Gupta, O. Davidson, D. Azulay, O. Millo, I. Balberg, G. Hodes and D. Cahen, ACS Energy Lett., 2017, 2, 2408–2414 CrossRef CAS.
- S. Xiong, Y. Dai, J. Yang, W. Xiao, D. Li, X. Liu, L. Ding, P. Gao, M. Fahlman and Q. Bao, Nano Energy, 2021, 79, 105505 CrossRef CAS.
- N. Phung, R. Félix, D. Meggiolaro, A. Al-Ashouri, G. Sousa e Silva, C. Hartmann, J. Hidalgo, H. Köbler, E. Mosconi, B. Lai, R. Gunder, M. Li, K.-L. Wang, Z.-K. Wang, K. Nie, E. Handick, R. G. Wilks, J. A. Marquez, B. Rech, T. Unold, J.-P. Correa-Baena, S. Albrecht, F. De Angelis, M. Bär and A. Abate, J. Am. Chem. Soc., 2020, 142(5), 2364–2374 CrossRef CAS PubMed.
- S. Xiong, Y. Dai, J. Yang, W. Xiao, D. Li, X. Liu, L. Ding, P. Gao, M. Fahlman and Q. Bao, Nano Energy, 2021, 79, 105505 CrossRef CAS.
- X. Guo, J. Su, Z. Lin, X. Wang, Q. Wang, Z. Zeng, J. Chang and Y. Hao, iScience, 2021, 24, 102276 CrossRef CAS PubMed.
- Q. Jiang, Z. Ni, G. Xu, Y. Lin, P. N. Rudd, R. Xue, Y. Li, Y. Li, Y. Gao and J. Huang, Adv. Mater., 2020, 32, 2001581 CrossRef CAS PubMed.
- Y. Lin, Y. Shao, J. Dai, T. Li, Y. Liu, X. Dai, X. Xiao, Y. Deng, A. Gruverman, X. C. Zeng and J. Huang, Nat. Commun., 2021, 12, 7 CrossRef CAS PubMed.
- D. Cao, W. Li, X. Zhang, L. Wan, Z. Guo, X. Wang, D. Eder and S. Wang, J. Mater. Chem. A, 2022, 10, 19278–19303 RSC.
- W.-Q. Wu, Q. Wang, Y. Fang, Y. Shao, S. Tang, Y. Deng, H. Lu, Y. Liu, T. Li, Z. Yang, A. Gruverman and J. Huang, Nat. Commun., 2018, 9, 1625 CrossRef PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T. W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
- Z. L. Zhang, B. Q. Men, Y. F. Liu, H. P. Gao and Y. L. Mao, Nanoscale Res. Lett., 2017, 12, 1–8 CrossRef PubMed.
- V. M. Le Corre, J. Diekmann, F. Peña-Camargo, J. Thiesbrummel, N. Tokmoldin, E. Gutierrez-Partida, K. P. Peters, L. Perdigón-Toro, M. H. Futscher, F. Lang, J. Warby, H. J. Snaith, D. Neher and M. Stolterfoht, Sol. RRL, 2022, 6, 2100772 CrossRef CAS.
- H. Yu, H. Lu, F. Xie, S. Zhou and N. Zhao, Adv. Funct. Mater., 2016, 26, 1411–1419 CrossRef CAS.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- V. Gonzalez-Pedro, E. J. Juarez-Perez, W.-S. Arsyad, E. M. Barea, F. Fabregat-Santiago, I. Mora-Sero and J. Bisquert, Nano Lett., 2014, 14(2), 888–893 CrossRef CAS PubMed.
- H. Wang, A. Guerrero, A. Bou, A. M. Al-Mayouf and J. Bisquert, Energy Environ. Sci., 2019, 12, 2054–2079 RSC.
- T.-Y. Hsieh, M. Pylnev, E. Palomares and T.-C. Wei, Adv. Funct. Mater., 2020, 30, 1909644 CrossRef CAS.
- A. Abudulimu, R. Sandoval-Torrientes, I. Zimmermann, J. Santos, M. K. Nazeeruddin and N. Martín, J. Mater. Chem. A, 2020, 8, 1386–1393 RSC.
- M. V. Khenkin, E. A. Katz and A. Abate, et al. , Nat. Energy, 2020, 5, 35–49 CrossRef.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06018k |
| This journal is © The Royal Society of Chemistry 2024 |