
Pre-lithiation carbon anodes mitigating potassium loss for high-performance potassium-ion energy storage devices†
Danni
Du
a,
Qingyuan
Liu
*a,
Jing
Gao
a,
Yuying
Qin
a,
Xiaobo
Jiang
a,
Yuanchang
Shi
*a,
Minghao
Hua
*a,
Xiaohang
Lin
 *a,
Zhiwei
Zhang
a,
Chengxiang
Wang
a,
Longwei
Yin
a and
Rutao
Wang
*ab
*a,
Zhiwei
Zhang
a,
Chengxiang
Wang
a,
Longwei
Yin
a and
Rutao
Wang
*ab
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, School of Materials Science and Engineering, Shandong University, Ji'nan 250061, China. E-mail: rtwang@sdu.edu.cn; yuanchangshi@sdu.edu.cn; huaminghao@mail.sdu.edu.cn; 202014124@mail.sdu.edu.cn; lxh12345@sdu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 12th November 2024
Abstract
Pre-potassiation technologies, which serve to provide additional potassium sources and/or mitigate potassium loss during cycling, are capable of enhancing the energy density and cycling life of potassium-ion capacitors (PICs) and potassium-ion batteries (PIBs). However, many reported pre-potassiation strategies involve using highly chemically reactive potassium sources, such as metallic potassium or K-containing additives, which increase both the cost and production risks. Herein, we propose a novel potassium-ion compensation strategy to meet the demand for high-performance potassium-ion full cells without using any high chemical reactivity potassium sources. This strategy is based on the foundation that a pre-lithiation carbon anode with a preformed solid-electrolyte-interphase (SEI) layer can effectively mitigate potassium loss without hindering the K+ diffusion from the electrolyte to the electrode during cell operation. PICs based on pre-lithiation carbon anodes, including soft carbon, hard carbon, and graphite, show better capacitive performance than those based on pre-potassiation carbon counterparts. This versatile strategy is also applicable to high-performance PIBs. We believe that this design principle, which incorporates mature pre-lithiation technologies into potassium-ion energy storage systems, has the potential to resolve some of the challenges with immature pre-potassium technologies.
Introduction
Recently, electrochemical energy storage (EES) technology has attracted increased interest, driven by the surge in large-scale renewable energy storage markets, such as portable electronics, electric vehicles and smart grids.1,2 To satisfy the requirements of these applications in the renewable energy storage markets, an efficient method is to develop low-cost EES devices that offer high energy density, high rate capability and long-term cycling life.3,4 To this end, potassium-ion batteries (PIBs) and capacitors (PICs) have emerged as competitive candidates, benefiting from the more abundant reserves of K in the Earth's crust (1.5% for K versus 0.065% for Li) and a closer standard redox potential (−2.93 V vs. a standard hydrogen electrode (SHE) for K/K+) compared to Li (−3.04 V vs. SHE).5 These advantages enable PIBs and PICs to achieve low cost, high efficiency and large-scale production.6 Thus far, many novel efforts have focused on exploring desired electrode materials and suitable electrolytes to fabricate high-performance PIBs and PICs.7–13 However, the further development of PIBs and PICs for wide-scale use is largely hampered by a so-called “pre-potassiation” problem.14 For example, in PIBs, the low stoichiometric ratio of K in most reported layered potassium transition-metal oxides (such as K0.5MnO2,15 K0.5V2O5,16 K0.6CoO2,17 K0.69CrO2,18 and K0.75[Ni1/3Mn2/3]O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19) cannot provide enough K-ions to mitigate the K loss on the negative electrode due to the formation of the solid-electrolyte-interphase (SEI) and other side reactions,20,21 let alone PICs in which both battery-like anode and capacitive carbon cathode lack K-ion sources.22,23 Therefore, pre-potassiation technologies are becoming increasingly significant for fabricating high-performance PIBs and PICs.24
19) cannot provide enough K-ions to mitigate the K loss on the negative electrode due to the formation of the solid-electrolyte-interphase (SEI) and other side reactions,20,21 let alone PICs in which both battery-like anode and capacitive carbon cathode lack K-ion sources.22,23 Therefore, pre-potassiation technologies are becoming increasingly significant for fabricating high-performance PIBs and PICs.24
Inspired by the mature pre-lithiation technology, several parallel methods of pre-potassiation have been developed. The most intuitive and commonly used strategy is the electrochemical method. In this method, the battery-like electrodes were pre-assembled in a half-cell, in which a sacrificial potassium metal electrode was used as counter and reference electrode.11,22,25–28 After several cycles (e.g., cyclic voltammetry or galvanostatic charge/discharge), a homogenous and stable SEI would be formed on the electrode.29 This method can control the degree of pre-potassiation easily and precisely by adjusting the cut-off voltage and cycling numbers. However, this method is limited by the use of high-risk and extremely reactive potassium metal and the additional battery disassembly process.30,31 To solve these problems, self-sacrificial additives including potassium metal organics/salts were introduced into the activated carbon electrode.5 These additives would be decomposed to release the potassium ions, which then compensated for the consumption of potassium ions. However, this method is built at the expense of the energy density of PICs and the complication of the subsequent production process due to the dead-mass of additives, occupancy, and gas production. Recently, the chemical pre-potassiation treatment has been proposed as an efficient and controllable approach, which is realized by immersing the electrodes into chemically reductive solutions, such as potassium-naphthalene-tetrahydrofuran solution and potassium-biphenyl in dimethoxyethane.23,24 However, this method cannot expand to large-scale industrial production because of its association with expensive relevant regents and stringent experimental requirements. Therefore, it remains a challenge to explore a low-cost, high efficiency and reliable pre-potassiation method for large-scale industrial production.
Herein, we proposed a design principle of implanting mature pre-lithiation technology into PICs for mitigating potassium loss during cell operation. PICs based on pre-lithiated carbon electrodes, including soft carbon (SC), hard carbon (HC), and graphite, exhibited better capacitive performance than their counterparts based on pre-potassiation carbon electrodes. The experimental study and theoretical calculations were carried out to prove that the preformed Li-based SEI layer on the carbon electrode played a role in mitigating potassium loss during cycling and did not hinder the K+ diffusion from the electrolyte to the electrode. In comparison to the traditional electrochemical pre-potassium method, this new design principle avoids the direct use of highly reactive and unsafe metallic potassium, thereby accelerating the commercialization of PICs and PIBs.
Results and discussion
Fig. 1 schematically illustrates the cell configuration of traditional PICs (denoted as K-PIC) fabricated by using a pre-potassiation carbon anode, a home-made activated carbon (AC) cathode and a KPF6-based organic electrolyte. Here, we propose a new cell configuration of PIC (Fig. 1, right; denoted as Li-PIC) which is nearly identical to K-PIC, apart from the use of a pre-lithiation carbon anode in place of the pre-potassiation carbon. The pre-lithiation and pre-potassiation carbon anodes were achieved by electrochemical lithiation and potassiation processes, respectively (see more details in the Experimental section). Meanwhile, the pre-potassiation step leads to the formation of a solid-electrolyte-interphase (SEI) layer with a mosaic structure on the surface of the anode materials, mitigating the irreversible consumption of K+ from the electrolyte. The successful Li-PIC is achieved on the assumption that the pre-lithiation carbon anode plays a similar role with the pre-potassiation carbon anode in mitigating the initial K+ loss during cycling. To demonstrate this design principle of implanting pre-lithiation technology into PICs, a pre-lithiation soft carbon (pre-Li-SC) is used as an empirical example.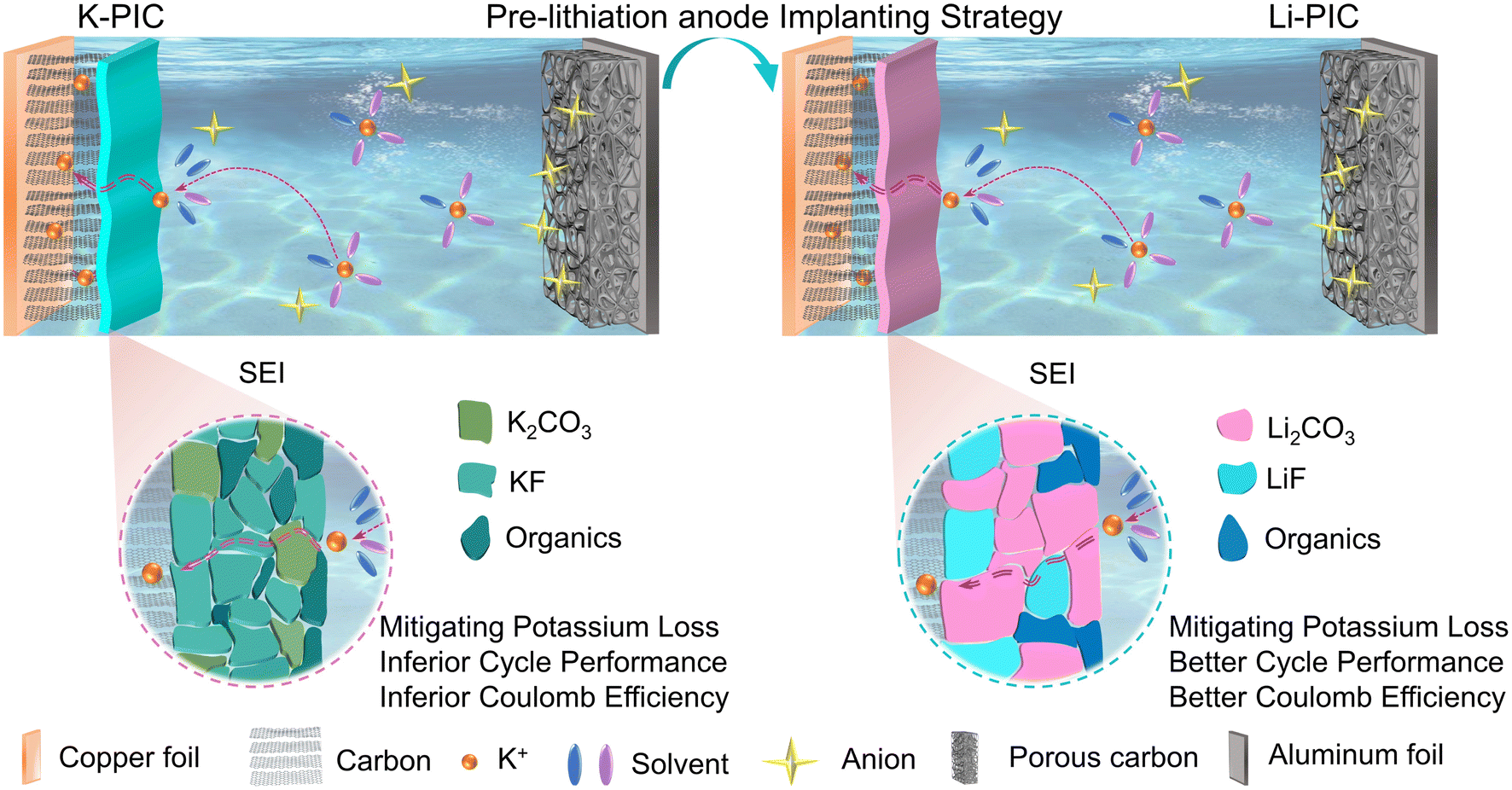 | ||
| Fig. 1 Schematic illustration of the PICs design with the pre-potassiation SC anode and PC cathode (left), and the pre-lithiation SC anode and PC cathode (right). | ||
Fig. 2a shows the initial charge/discharge curves of K-PIC and Li-PIC with a working potential range from 1.0 to 4.0 V at a rate of 0.1 A g−1. Both K-PIC and Li-PIC show the linear charging/discharging profile, indicating good capacitive behaviors. Such good capacitive behaviors of K-PIC and Li-PIC are further demonstrated by the nearly rectangular CV curves shown in Fig. S1.† A closer examination of these linear charge/discharge curves found that the charge/discharge curves of Li-PIC tend to quickly overlap after several cycles, suggesting the improved electrochemical reversibility and decreased parasitic reactions of the as-assembled Li-PIC. On the contrary, the linear charge/discharge curves of K-PIC are unable to overlap well even after ten cycles. The corresponding coulombic efficiencies (CE) during the initial 10 cycles of Li-PIC (shown in Fig. 2b) are slightly higher than that of K-PIC, suggesting fewer side reactions of Li-PIC. We noted that the open circuit voltage (OCV) of Li-PIC is about 3.20 V, which is higher than that of K-PIC. This is mainly because the initial potential of the Li-pre-SC anode versus K/K+ is lower than that of the K-pre-SC anode. It is possible that pre-lithiation is more efficient in lowering the potential of the SC electrode than pre-potassiation.31–34
 | ||
| Fig. 2 (a) Charge/discharge curves for the initial ten cycles at 0.1 A g−1 of Li-PIC (bottom) and of K-PIC (top). (b) Rate capability achieved at the various current densities from 0.1 to 1 A g−1 for Li-PIC and K-PIC. (c) The comparison of the long-term cycling performance and corresponding coulombic efficiencies for 500 cycles at 0.1 A g−1 for Li-PIC and K-PIC. | ||
Fig. 2b comparatively shows the rate performance of Li-PIC and K-PIC under the same rates. At 0.1 A g−1, Li-PIC delivers a specific capacity of 50.1 mA h g−1, which is slightly higher than that of K-PIC (44.3 mA h g−1). As the current density increased to 1 A g−1, Li-PIC delivers a specific capacity of 25.4 mA h g−1, which is still higher than that of K-PIC (17.6 mA h g−1). In addition, Li-PIC exhibits better cycling performance than K-PIC. Li-PIC shows negligible capacity decay at a low current density of 0.1 A g−1 after 500 cycles (Fig. 2c). As the cycling current density increased to 0.5 A g−1 (Fig. S2†), Li-PIC preserves 80% of its initial capacity after 3000 cycles. In contrast, K-PIC achieves a capacity retention value of 76% after 500 cycles at a rate of 0.1 A g−1, and shows a rapid decay at a high rate of 0.5 A g−1. During the whole cycling test, the CE values for both Li-PIC and K-PIC are almost close to 100%. So, considering the excellent electrochemical performance in the rate capability and cycling stability achieved by Li-PIC, the pre-Li-SC anode used in PICs provides nearly identical functionality to the pre-K-SC anode, especially in mitigating the initial potassium loss. However, the underlying reason for the successful anode substitution with pre-Li-SC is still unclear, in addition to how it affects the electrochemical performance of PICs.
To probe the underlying reasons, a series of control experiments were carried out. Three-electrode Swagelok cells with metal K as the reference electrode were first assembled to monitor the cell voltage, and record the potentials of the positive and negative electrodes. Fig. 3a, b and S4† show the typical galvanostatic charge/discharge profiles for Li-PIC and K-PIC for different current densities (0.1 A g−1 and 1 A g−1) within the voltage window between 1.0 and 4.0 V. The working potential of the positive AC electrode from Li-PIC is always lower than 4.0 V versus K/K+ and higher than 1.5 V versus K/K+, which avoids electrolyte oxidation and prevents SEI formation on the surface of the AC electrode.35 The lower potential of the positive AC electrode from K-PIC is always higher than 2.0 V versus K/K+. However, the upper potential of the positive AC electrode from K-PIC is higher than 4.0 V versus K/K+, possibly suffering the risk of electrolyte oxidation. We noted that the working potential range achieved by the positive AC electrode from Li-PIC is wider than that from K-PIC, suggesting that more capacity is stored and released on the AC electrode from Li-PIC. This then provides a plausible explanation for the higher capacity performance achieved by Li-PIC.
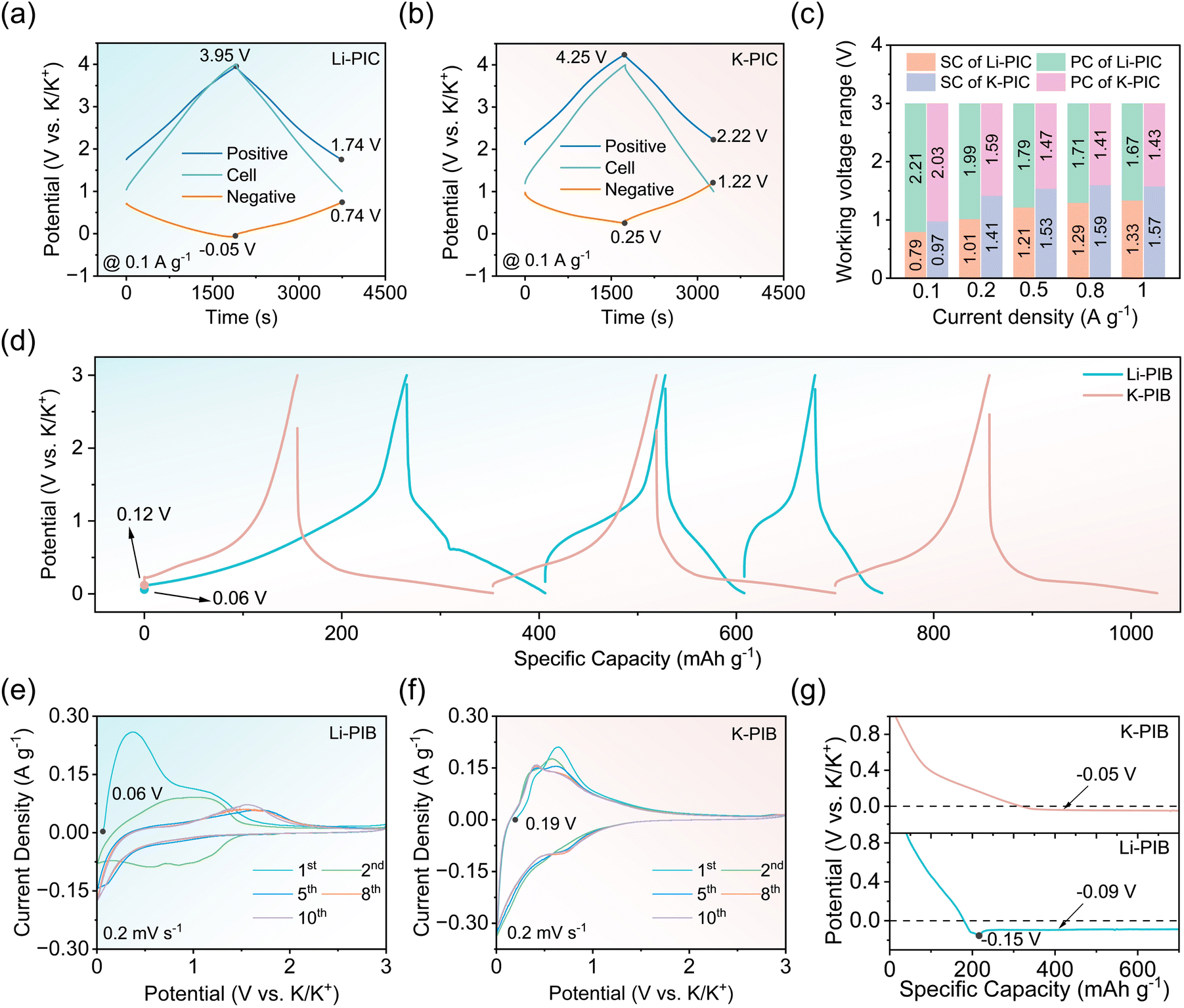 | ||
| Fig. 3 In situ monitoring of the Li-PIC (a) and K-PIC (b) during the galvanostatic charge/discharge (GCD) test using the Swagelok three-electrode system with the reference electrode of K metal at 0.1 A g−1. (c) The working voltage ranges of the negative and positive electrodes of Li-PIC and K-PIC under the different rates. (d) The charge/discharge profiles of the first three cycles at 0.1 A g−1 of the pre-lithiation anode electrodes (denoted as the Li-PIB) or pre-potassiation anode electrodes (denoted as the K-PIB) assembled in a half-cell. A comparison of the CV curves at a scan rate of 0.2 mV s−1 of (e) Li-PIB and (f) K-PIB. (g) Deposition curves of metal K at 0.05 A g−1 in a half-cell for Li-PIB (bottom) and K-PIB (top). | ||
Concerning the pre-Li-SC negative electrode, the potential swing range is located between −0.05 V and 0.74 V versus K/K+ when the Li-KIC is discharged at 0.1 A g−1. As the discharging current density increases to 1 A g−1 (shown in Fig. S3a†), the potential swing range becomes wider where the lower potential limit is −0.11 V (vs. K/K+) and the upper potential limit is 1.22 V (vs. K/K+). The lower potential limit of pre-Li-SC is always lower than 0 V, possibly suffering the risk of K deposition. For the pre-K-SC negative electrode, the potential swing range is located between 0.25 V and 1.22 V versus K/K+ when the K-PIC is discharged at the current density of 0.1 A g−1, which is slightly higher than that of pre-Li-SC. As the discharging current density increases to 1 A g−1, the potential swing range of pre-K-SC becomes wider where the lower potential limit is 0.12 V (vs. K/K+) and the upper potential limit is 1.69 V (vs. K/K+). The lower potential limit of pre-K-SC is always higher than 0 V, which prevents the K deposition. Upon closer examination of the individual charge/discharge curves of the negative SC electrode, positive AC electrode, and these two PICs, K-PIC exhibits a larger IR drop than Li-PIC. Nevertheless, the larger IR drop of K-PIC mainly stems from the negative side, which largely contributes to the inferior rate capability of K-PIC. The voltage windows of the positive and negative electrodes of Li-PIC and K-PIC are summarized in Fig. 3c. The positive electrode swing ranges of Li-PIC are always wider than that of K-PIC at the same rate level, which provide a plausible explanation for the higher capacity achieved by Li-PIC. These results from the three-electrode Swagelok cells, combined with different capacitive performances of Li-PIC and K-PIC, suggest that the K-ion intercalated behavior in SC is affected more by the pre-lithiation process, in contrast to the K-ion intercalated behavior of SC undergoing the pre-potassium process.
We then examined the respective K+ charge-storage behaviors of the pre-Li-SC anode and pre-K-SC anode in the half cells. Typically, the pre-Li-SC or pre-K-SC electrode was detached from the half cells at a pre-lithiation/potassiation state (a cut-off potential of 0.01 V), and then re-assembled in a two-electrode cell using metal K as the counter and reference electrode (or called half cells). Fig. 3d shows a side-by-side comparison of the initial galvanostatic charge/discharge curves of the pre-Li-SC anode and pre-K-SC anode in the half cells achieved at a rate of 100 mA g−1. A close examination shows that the pre-Li-SC anode has a lower OCV of 0.06 V vs. K/K+ as compared to 0.12 V vs. K/K+ of the pre-K-SC anode, which provides a plausible explanation for Li-PIC showing a higher OCV than K-PIC, as observed in Fig. 2a. The lower OCV for the pre-Li-SC anode is largely related to the full pre-lithiation degree of SC detached at a potential of 0.01 V vs. Li/Li+ and the lower standard hydrogen potential of Li+/Li (−3.035 V vs. SHE), as compared with that of K/K+ (−2.930 V vs. SHE). By contrast, the OCV of the pre-K-SC anode is 0.12 V (vs. K/K+), which is close to the full pre-potassiation degree (detached at a potential of 0.01 V vs. K/K+).
The charge/discharge curves are quite different between the pre-Li-SC and pre-K-SC anodes in the re-assembled cells. As shown in Fig. 3d and S4b,† the charge/discharge profiles of the pre-K-SC electrode during the initial and following two cycles show no difference with the SC electrode in the pre-potassium process. By contrast, the pre-Li-SC anode shows a slope voltage profile with a charging capacity of 266 mA h g−1 during the first charge process, which is close to the Li+ storage capacity during the prior pre-lithiation process. In view of there being no K+ intercalated into the pre-Li-SC electrode, the initial charge capacity of the pre-Li-SC anode is mainly caused by the deintercalation of the pre-intercalated Li+ in the SC electrode. During the following charge/discharge process, the voltage profile of the pre-Li-SC changes with the increased polarization occurring in the initial charge stage. The increased polarization suggests that some storage sites in SC are not accessed by K+ in the re-assembled SC/K half cells. The charge/discharge capacities of pre-Li-SC are gradually decreased and eventually holding at 66 mA h g−1 after three cycles. It should be mentioned that the lower IR drop is achieved by the pre-K-SC anode in comparison to the pre-Li-SC anode during the charge process. On the contrary, the pre-K-SC anode exhibits a larger IR drop than the pre-Li-SC anode during the discharge process, which will be amplified at the high rate (Fig. S5†). The large IR drop for the pre-K-SC anode is responsible for the poor rate performance observed for K-PIC. It should be emphasized that the large IR drop observed for the pre-K-SC electrode during the discharge process is largely related to the ohmic polarization from the electrode and electrolytes rather than the electrochemical/concentration polarization. At this stage, K+ storage mainly relies on the double-layer adsorption/desorption process on the surface of the electrode rather than the faradaic intercalation reaction. On the contrary, the large IR drop observed for the pre-Li-SC electrode during the charge process is largely related to the combined electrochemical polarization and ohmic polarization, particular in view of the dominated K+ intercalation behavior being postponed at this stage.
In good agreement with the above observation from the galvanostatic charge/discharge curves, the CV profile for the pre-Li-SC/K half-cell is apparently different from that for the pre-K-SC/K half-cell. The initial CV curve of the pre-Li-SC anode exhibits several oxidation peaks, corresponding to the Li+ deintercalation. These oxidation peaks shift toward high potential, become weaker, and even disappear in the following cycles. Meanwhile, only one weak anodic peak around 1.66 V is preserved after 5 cycles, corresponding to the gentle slope region observed in the charge curves in Fig. 3d. After two cycles, no cathodic peak appears at the high potential region (high than 0.3 V vs. K/K+), whereas a sudden increase in the current density appears at the low potential region (less than 0.3 V vs. K/K+), which can be assigned to K+ insertion into the pre-Li-SC electrode. Meanwhile, there is no sign of peaks assigned to K+ deintercalation during the following anodic process, especially at the low potential region (less than 1.0 V vs. K/K+). Instead, there is an approximately smooth CV curve, which coincided with the broad polarization region in the charge process shown in Fig. 3d. By contrast, the CV profile of the pre-K-SC anode shows no apparent difference with that before the anode transfer shown in Fig. S6b.† It should be noted that during the initial cathodic scan on both pre-Li-SC and pre-K-SC anodes, no sign of new SEI formation is observed, as suggested by the lack of irreversible reduction peaks emerging between 0.5 and 2.0 V vs. K/K+. These results, combined with the CVs and charge/discharge curves from the half cells, suggest that the thermodynamics/kinetics of K-ion intercalation in SC are affected by the pre-lithiation process. However, these results conflict with the above-stated findings from PICs and the three-electrode Swagelok cell, in which the thermodynamics/kinetics of K-ion intercalation in SC are not affected much by the pre-lithiation process and even better than pre-K-SC, as suggested by the better capacitive performance of PICs and low ohmic and concentration polarizations achieved by the pre-Li-SC anode.
In view of the fact that the actual cut-off voltage ranges of the pre-Li-SC and pre-K-SC anodes in PICs are apparently shorter than the testing potential regions (from 0.01–3.00 V vs. K/K+) in the half cells, it is speculated that the wider potential range used in the half-cell may run unparallel to the real K+ intercalation behavior on these two SC anodes. Then, we reset the working potential range from 0.01 to 0.70 V of the pre-Li-SC/K cell to carry out the galvanotactic charge/discharge test at a rate of 0.05 A g−1, with reference to the results from the three-electrode Swagelok cell shown in Fig. 3a and b. As shown at the bottom of Fig. S7,† except for the initial charge process, the specific capacities achieved in the following charge/discharge processes rapidly decay. Accompanied by increasing polarization at the initial charge stage, the gradually elevated charge plateau potential suggests the postponed K+ deintercalation behavior, which is consistent with the above results without resetting the potential range in Fig. 3d. By contrast, the charge/discharge curves of pre-K-SC show no apparent difference before and after resetting the working potential range (at the middle of Fig. S7†). After we detached the cycled pre-Li-SC/K half-cell and then transferred the pre-Li-SC anode into a half-cell with metal Li as the reference (RE)/counter (CE) electrode (denoted as the Li-PIB-LIB), pre-Li-SC recovers the original capacity of Li+ storage, which rules out the hypothesis that pre-Li-SC is out of order during the prior K+ intercalation process.
Given that the lower potential limit of pre-Li-SC in PIC is down to −0.05 V vs. K/K+ at a rate of 0.1 A g−1 and even decreases to −0.11 V vs. K/K+ at a rate of 1 A g−1 (Fig. 3a and S3a†), a question arises that whether the metal K deposition occurs on pre-Li-SC electrode after the potential is down to 0 V. Our previous study on pre-lithiation hard carbon (pre-Li-HC) had a low plating polarization potential of −0.15 V (vs. Na/Na+), which can avoid Na deposition and provide additional capacity to enable a sodium-ion capacitor based on a pre-Li-HC anode with high energy output and safety.31 If so, the additional capacity contributed by the extended potential range would be provided by pre-Li-SC, which may be one of the reasons for the improved electrochemical performance of PICs. We then measured the overall voltage trace in a pre-Li-SC/K half-cell during the first potassium plating process under a current density of 0.05 A g−1. Fig. 3g shows that a slope discharge curve coupled with a significant voltage dip (around −0.15 V vs. K/K+) appears at the beginning of the K deposition, followed by a flat voltage plateau around −0.09 V vs. K/K+. This voltage dip is attributed to the metal deposition at a “critical” concentration, above which the metal starts to agglomerate to form metallic clusters.36–39 By contrast, the case for potassium plating on the pre-K-SC electrode is slightly different with pre-Li-SC, showing no obvious voltage dip. The K plating polarization potential of the pre-Li-SC electrode is evaluated to −0.09 V (vs. K/K+). The decreasing K plating polarization potential is primarily due to the surface chemistry, e.g., SEI.39 Although the lower work potential of the pre-Li-SC electrode monitored from the three-electrode Swagelok cell is −0.05 V (vs. K/K+), the pre-Li-SC electrode is still in the safe region without K deposition. With regard to the practical working potential of pre-Li-SC in Li-PIC being down to −0.05 V (vs. K/K+) and additional capacity being contributed from the extended working potential range, we reevaluated the K-ion storage of the pre-Li-SC anode within an extended potential range from −0.08 to 3.00 V (vs. K/K+). Fig. S8† shows that the voltage profile of the charge/discharge curves shows no difference before and after extending the working potential range. In this case, the calculated capacity of the pre-Li-SC electrode increases 22.4% to 173 mA h g−1. However, the limited additional capacity provided by the extended potential range and the preserved large polarization during the initial charge stage fail to explain the improved capacitive performance of PICs based on the pre-Li-SC anode.
To further probe the underlying reasons for the controversial results between PICs and half cells, we turn to the electrolyte with a consideration of the contaminated K-ion based electrolyte used in the half-cell by the Li+ deintercalated from the pre-Li-SC anode. The above charge/discharge curves in Fig. 3d show that the pre-lithiation lithium-ions of the SC anode would be detached during the first charge process, which cannot be reversibly intercalated into the SC anode. We employed a plasma atomic emission spectrometer (ICP-AES) to study the Li trace in the electrolyte from the pre-Li-SC/K half cells after the initial charge process. It was found that the K/Li molar ratio is ∼2.05 in the electrolyte from the pre-Li-SC/K half-cell (Table S1†). In addition, we found the Li trace in the electrolyte from the PC//pre-Li-SC PICs with a K/Li molar ratio of ∼2.16. Recent studies demonstrated that the introduction of a guest alkaline salt into the host alkaline salt-based organic electrolyte induced the change of the SEI composition and electrostatic shielding effect, thus improving the reversibility of the metal deposition/striping and the electrochemical performance of the cells.40–42 It is speculated that the extra Li+ in the electrolyte has a positive effect on the electrochemical performance of PICs. To confirm this assumption, a fresh SC electrode was transferred into the cycled pre-Li-SC/K half-cell with a cut-off potential of 3.00 V to replace the pre-Li-SC (under the de-potassiation state for providing the extra Li+ in the electrolyte, denoted as the pre-Li-SC/K → fresh SC/K). For comparison, a fresh SC electrode was transferred into the cycled de-Li-SC/K half-cell (denoted as the de-Li-SC/K → fresh SC/K). Electrochemical results in Fig. S9† show that the former SC electrode delivers inferior capacity performance compared to the latter SC, which is still higher than the pre-Li-SC/K half-cell. The capacity values achieved by the latter SC electrode are close to that from the fresh SC/K or pre-K-SC/K half cells. These results show that the extra Li+ in the electrolyte has a negative effect on the electrochemical performance of various SC/K half cells, which still fails to explain the mismatch between the improved capacitive performance of PICs and the reduced capacity performance of the pre-Li-SC/K half cells.
In general, these proposed methods rely on traditional two-electrode half cells, in which various SCs are used as the working electrode (cathode) and metal K is used as the reference and counter electrode (anode). The electrochemical results from full-cell PICs and three-electrode Swagelok cells confirm the excellent K+ storage in the pre-Li-SC electrode. Hence, it is speculated that the main problem here lies within the metal K reference/counter electrode. Recent studies showed that the metal K used in the half-cell configuration has large and unstable and/or irreproducible overpotentials.43 The overpotentials arise from the mass transport of metal cations in the electrolyte, cation desolvation energy and transport through SEI, and the metal nucleation barrier, which lead to the apparent mismatch between the poor K-ion storage in the half-cell and the improved capacitive performance in the full cell.43–45 In our case, the overpotential for metal K in pre-Li-SC/K half cells may be associated with the changed composition of the electrolyte. ICP results in Table S1† show that the partial Li+ detached from the pre-Li-SC will be diffused into the prior K-ion based electrolyte to form a K+/Li+ double-cation electrolyte. Recent studies showed that if the additive cations exhibit an effective reduction potential below the standard reduction potential of the subjective cations, these additive cations will form a positively charged electrostatic shield on the surface of the electrode without reduction and deposition of the additives.42,46 Then, K+ needs to migrate the electrostatic shielding layer formed by Li+ to realize metal K deposition, leading to the large overpotential. To confirm this electrostatic shield, we detached the cycled pre-Li-SC/K and pre-K-SC/K half cells, and found that the surface of the K metal from the pre-Li-SC/K cell is much smoother than that from the pre-K-SC/K cell (Fig. 4a and b). The smoother surface of the K metal from the pre-Li-SC/K cell is consistent with previous studies in that the electrostatic shield prevents metal dendrite growth, transforms the surface uniformity of coatings, and increases the overpotential.42,47,48 In addition, no Li signal was detected in the XPS spectra of the metal K electrode from the cycled pre-Li-SC/K cell, confirming that Li+ would not be reduced into metal Li. As a result, it is therefore urgent to explore alternative testing methods to minimize the impact of the K counter electrodes and to evaluate the electrochemical behaviors accurately for high-performance pre-Li-SC electrodes.
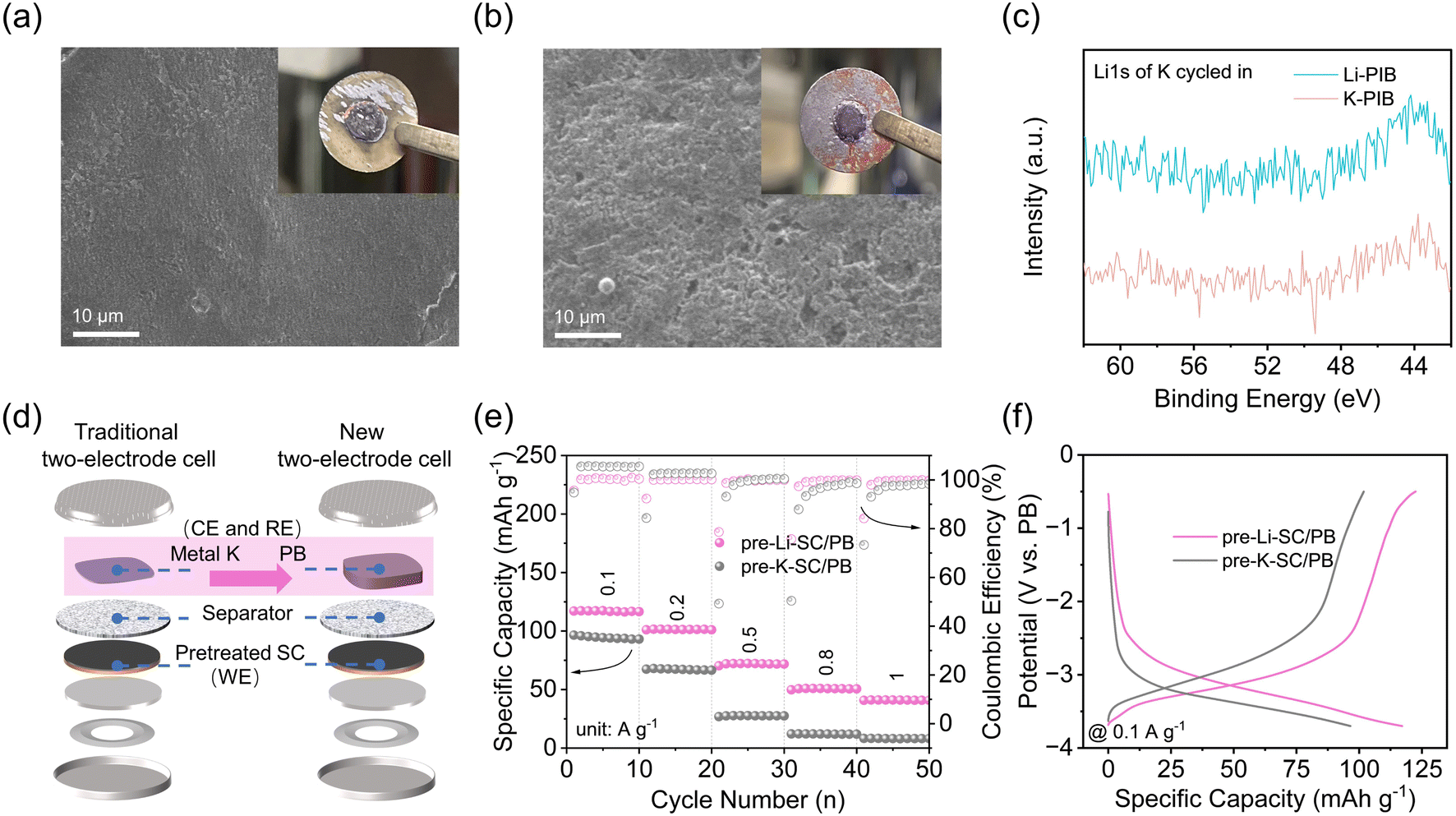 | ||
| Fig. 4 SEM images of metallic potassium disassembled in the de-lithiation/de-potassiation state of the negative electrode from Li-PIB (a) and K-PIB (b), respectively. Inset images are optical images of the corresponding metal K electrodes. (c) High XPS Li 1s partial enlargement spectra at 43–61 eV of metallic potassium disassembled from Li-PIB and K-PIB. (d) Schematic illustration of the new two-electrode cell with PB as the reference electrode compared with a traditional two-electrode cell. (e) Rate capability achieved at the various current densities from 0.1 to 1 A g−1 for pre-Li-SC/PB and pre-K-SC/PB. (f) The typical charge/discharge profiles at 0.1 A g−1 of pre-Li-SC/PB and pre-K-SC/PB. | ||
To avoid the polarizability of the K counter electrodes, we used Fe-substituted Mn-rich Prussian blue (K1.5Mn0.61Fe0.39[Fe(CN)6]0.77·H2O, denoted as PB) as the RE/CE electrode to replace metal Li. As shown in Fig. S10,† the PB electrode delivers a high specific capacity of 107.9 mA h g−1 at 0.05 A g−1, and exhibits a stable discharge plateau around 3.9 V vs. K/K+ and charge plateau around 4.1 V vs. K/K+. As suggested by the preparation process of the Li4Ti4O12 and LiFePO4 RE electrodes proposed by Cui et al.,49 the PB electrode was cycled three times at 3.0–4.2 V and stopped at half charge during the K extraction to obtain a RE potential of 3.7 V vs. K/K+ as much as reproducibly possible. Moreover, in view of the specific capacity delivered by the SC electrode being apparently higher than that of the PB electrode at the same rate level, the mass loading of PB RE/CE was controlled to 10 mg, which is 10 times higher than the SC electrode (N/P capacity ratio close to 1:5). After the preparation, the pre-Li-SC/PB two-electrode configuration (schematically shown in Fig. 4d) was assembled, in which the pre-Li-SC electrode was used as the working electrode (WE) and PB was used as the RE/CE. For comparison, the pre-K-SC/PB two-electrode configuration was assembled according to the same procedure. Compared with the traditional half-cell, the PB films are used instead of metallic potassium as the RE/CE electrode. As shown in Fig. 4e, the pre-Li-SC/PB has good rate performance and stable coulombic efficiency, which is also consistent with the PICs' performance shown in Fig. 2b. Fig. 4f shows the typical charge/discharge curves of the pre-Li-SC/PB and pre-K-SC/PB half cells at a current density of 0.1 A g−1 within a working potential range from −3.7 to −0.5 V (vs. PB) (equal to 0.0–3.0 V vs. K/K+). Pre-Li-SC/PB shows the slope profile and lower electrochemical/concentration polarization, which is similar to the voltage profile of the SC electrode in the SC/K half-cell. However, as the current density increases, the large ohmic polarization and electrochemical concentration polarization cause the rate performance of the pre-K-SC/PB half-cell to drop rapidly, as shown in Fig. S11.†
On the basis of the above discussion, it can be concluded that the thermodynamics/kinetics of the K-ion intercalation in SC are not affected much by the pre-lithiation process, and are even better than pre-K-SC undergoing the pre-potassiation process. Moreover, the pre-lithiation process gives rise to a Li-based SEI layer on the SC negative electrode, which plays similar roles in compensating the large irreversible capacity loss and greatly improving the initial coulombic efficiency. To further understand the electrolyte decomposition mechanisms and the formation and transformation law of the SEI layer, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) analyses were performed on SC electrodes pretreated in the half-cell and then cycled in PICs, respectively. Fig. 5a and b show that the pre-lithiation process gives rise to a rough SEI film with a thinness of ∼8 nm on the pre-Li-SC electrode. After the pre-Li-SC electrode was transferred into PICs and cycled over 10 cycles at a rate of 0.1 A g−1, the prior rough SEI film became smooth and the thickness of SEI was slightly decreased to ∼7.4 nm. This morphological change of the SEI film on the pre-Li-SC anode is mainly associated with the partial dissolution of the SEI film depending on the changed electrolyte compositions. Moreover, the actual working potential range of the SC electrode becomes narrow in PICs, which is responsible for the changed SEI morphology.38 By contrast, a smooth SEI film with a thickness of ∼9.8 nm is coated on the SC anode after the pre-potassiation process shown in Fig. 5e and f, which becomes smoother and thicker (∼10.8 nm) after the pre-K-SC electrode is transferred into the PICs and cycled over 10 cycles (Fig. 5g and h).
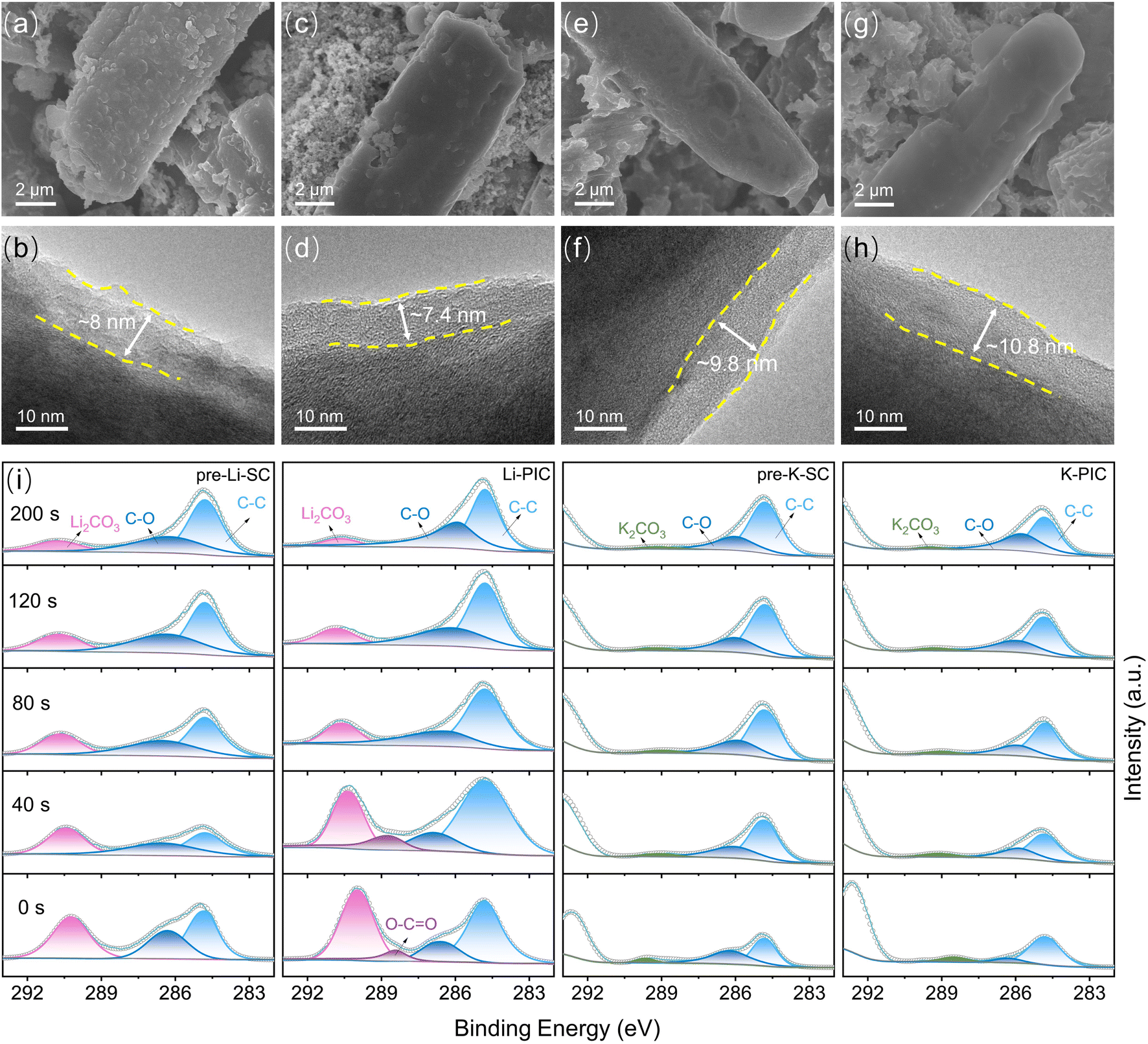 | ||
| Fig. 5 Characterizations of the SEM and HRTEM images on the surface morphology and SEI structure of pre-treated negative electrodes: (a and b) pre-Li-SC electrodes in the half-cell, (c and d) after ten cycles in PIC at 0.1 A g−1, (e and f) pre-K-SC electrodes in the half-cell, (g and h) after ten cycles in PIC at 0.1 A g−1. (i) X-ray photoelectron spectroscopy (XPS) spectra with O 1s sputtered at 0 s, 40 s, 80 s, 120 s and 200 s for the pre-lithiation SC electrodes in the half-cell (denoted as the pre-Li-SC), after ten cycles in PIC (denoted as the Li-PIC), the pre-potassiation SC electrodes in the half-cell (denoted as the pre-K-SC), and after ten cycles in PIC (denoted as the K-PIC). | ||
To study the SEI chemical compositions of the pre-Li-SC and pre-K-SC electrodes, XPS coupled with Ar+ sputtering depth profiling were carried out. The deconvoluted high-resolution C 1s, O 1s, F 1s and Li 1s XPS spectra of the pre-Li-SC electrode suggest that the outer surface of the SEI film is composed of inorganic (LiF and Li2CO3) and organic (Li alkyl carbonates, as suggested by the C–O and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O functional groups) species. With the depth profiling (sputtering time from 0 s to 40 s) of the SEI film of pre-Li-SC, the content of the organic species in the outer layer of SEI is significantly reduced as confirmed by the largely reduced C–O and C–F species (from the C 1s and F 1s spectra). Meanwhile, the Li2CO3 content from the C 1s spectra is reduced, while the LiF content from the F 1s spectra is increased (F as an indicator). XPS elemental analysis in Fig. S12b† shows that the C atomic ratio is reduced from 27.85% to 16.30% after the initial depth profiling. These results suggest that SEI on the pre-Li-SC electrode has a two-sublayer structure, where the outer layer is rich in organic species and the inner layer is rich in inorganic species, which is similar to the Li-rich SEI reported in the other literature.50,51 With the extended Ar+ sputtering time over 40 s, the signals related to the sp2 carbon, C–O functional groups and other inorganic species (Li2CO3 and LiF) on the pre-Li-SC show no apparent change. However, the C atomic ratio begins to increase, which is assumed to be due to the SEI-SC interface. In view of the Ar+ sputtering etching rate of 0.29 nm s−1, the thickness of SEI is calculated to be ∼11.6 nm, which is close to the HRTEM observation in Fig. 5h.
C–O functional groups) species. With the depth profiling (sputtering time from 0 s to 40 s) of the SEI film of pre-Li-SC, the content of the organic species in the outer layer of SEI is significantly reduced as confirmed by the largely reduced C–O and C–F species (from the C 1s and F 1s spectra). Meanwhile, the Li2CO3 content from the C 1s spectra is reduced, while the LiF content from the F 1s spectra is increased (F as an indicator). XPS elemental analysis in Fig. S12b† shows that the C atomic ratio is reduced from 27.85% to 16.30% after the initial depth profiling. These results suggest that SEI on the pre-Li-SC electrode has a two-sublayer structure, where the outer layer is rich in organic species and the inner layer is rich in inorganic species, which is similar to the Li-rich SEI reported in the other literature.50,51 With the extended Ar+ sputtering time over 40 s, the signals related to the sp2 carbon, C–O functional groups and other inorganic species (Li2CO3 and LiF) on the pre-Li-SC show no apparent change. However, the C atomic ratio begins to increase, which is assumed to be due to the SEI-SC interface. In view of the Ar+ sputtering etching rate of 0.29 nm s−1, the thickness of SEI is calculated to be ∼11.6 nm, which is close to the HRTEM observation in Fig. 5h.
After the pre-Li-SC electrode was transferred into a PIC as the negative electrode and cycled for more than 10 cycles, the two-sublayer SEI structure was still preserved (Fig. S12a†). However, the content of the organic species in the outer layer of the SEI was slightly reduced, as confirmed by the reduced C–O and C–F functionalities (Fig. S13 and S14†). This may be the reason why the SEI film becomes smoother and thinner after the transfer (Fig. 5d). The inner layer of SEI predominantly consisted of inorganic (LiF and Li2CO3) components, showing no apparent change in comparison to the pre-Li-SC electrode before transfer. Hence, the preformed Li-based SEI layer is chemically and structural stable, except for the top surface of SEI. As shown in Fig. S12b and c,† the Li atomic ratio decreased by 4.45% after pre-Li-SC cycled in PICs, which is associated with the pre-insertion Li being removed from SC. Interestingly, from the surface to the inner part of SEI, no K 2p peaks appear during the sputtering, accompanied by low atomic ratios (less than 0.1 wt%). This suggests that the K-ions, transferring from the outer electrolyte to the inner SC, do not participate in ion exchange with the Li-ion species in SEI and/or the formation of K-ion based inorganic or organic species filled within the intervals of the primary SEI. These observations are apparently different from a previous report in which the progressive substitution of Li+ with K+ in the preformed Li-based SEI would occur during K+ diffusion through the SEI.52
After the pre-K-SC electrode was transferred into a PIC as the negative electrode and cycled for more than 10 cycles, the two-sublayer SEI structure is still preserved but is very unstable (Fig. S12d–f†). Fig. S15† shows that the surface of the SEI formed in the pre-potassiation process consists of both inorganic (KF and K2CO3) and organic (K alkyl carbonates, as suggested by C–O and O–CO functional groups) species. During the sputtering, the organic species are reduced and the inorganic species are increased, which indicates that the SEI on pre-K-SC is made up of mixed inorganic and organic compounds from the surface to the inner part. Except for the identical two-layer structure of SEI, the chemical compositions of the SEI layers from pre-Li-SC and pre-K-SC are apparently different. Specifically, compared to SEI formed on the pre-Li-SC electrode, the C and F atomic ratios in SEI formed on the pre-K-SC electrode greatly increase. However, the O atomic proportion in SEI deceases. This is because more KF and less C–O species are formed on the pre-K-SC electrode, as indicated by the C 1s and F 1s spectra. We also noted that the content of the organic species in the outer layer of the SEI was slightly reduced. This occurred after the pre-K-SC electrode was transferred into a PIC as the negative electrode and cycled for more than 10 cycles, and was responsible for the smoother SEI film observed in Fig. 5g. Moreover, the greater amount of KF contributes to a thicker SEI on the pre-K-SC electrode after cycling in PIC, which suggests that continuous side reactions occur on the pre-K-SC electrode when it is transferred into PICs (Fig. S16†).
Previous literature results suggest that the critical rate-determining step is largely related to ionic transfer through the inner inorganic layer of SEI.53–55 To understand how these two different SEI layers affect the K-ion transfer, DFT calculations were conducted to evaluate the mechanisms of ionic transfer in the individual SEI components, including LiF, Li2CO3, KF, and K2CO3. Although several migrating mechanisms have been reported,53,54 such as hopping, vacancies, exchange, and knock-off, only those yielding minimum values of migration barriers and most likely to exist are summarized in the Fig. 6 inset. As a result, the hopping mechanism is preferred. Fig. 6 shows the activation energy barriers for Li-ions migrating in single crystal models of LiF, Li2CO3, KF, and K2CO3, emulating the individual SEI components. The DFT results show that for K-ion diffusion in LiF and Li2CO3, the migration barriers are 0.55 and 0.60 eV, respectively, which is higher than that of K-ion diffusion in KF (0.47 eV) and K2CO3 (0.21 eV). With a consideration of KF being the predominant component in the SEI on the pre-K-SC electrode, K-ion diffusion through the K-based SEI is just a little bit faster than that through the Li-ion based SEI. In addition, the SEI layer on the pre-K-SC electrode is thicker than that on the pre-Li-SC electrode, indicating a longer K-ion diffusion path. In fact, the SEI layer always has a mosaic structure composed of various inorganic species and organic species. Hence, many factors (including the organic species, surface morphology, and porosity) also affect the diffusion of K+. However, the relationship between these factors and the K+ diffusion is complex, and requires further investigation. Combining the theoretical and experimental results, it is inferred that the K-ions have faster kinetics in the K-based SEI than that in Li-based SEI. However, the K-ion diffusion path in the K-ion based SEI is longer than that in the Li-ion based SEI.
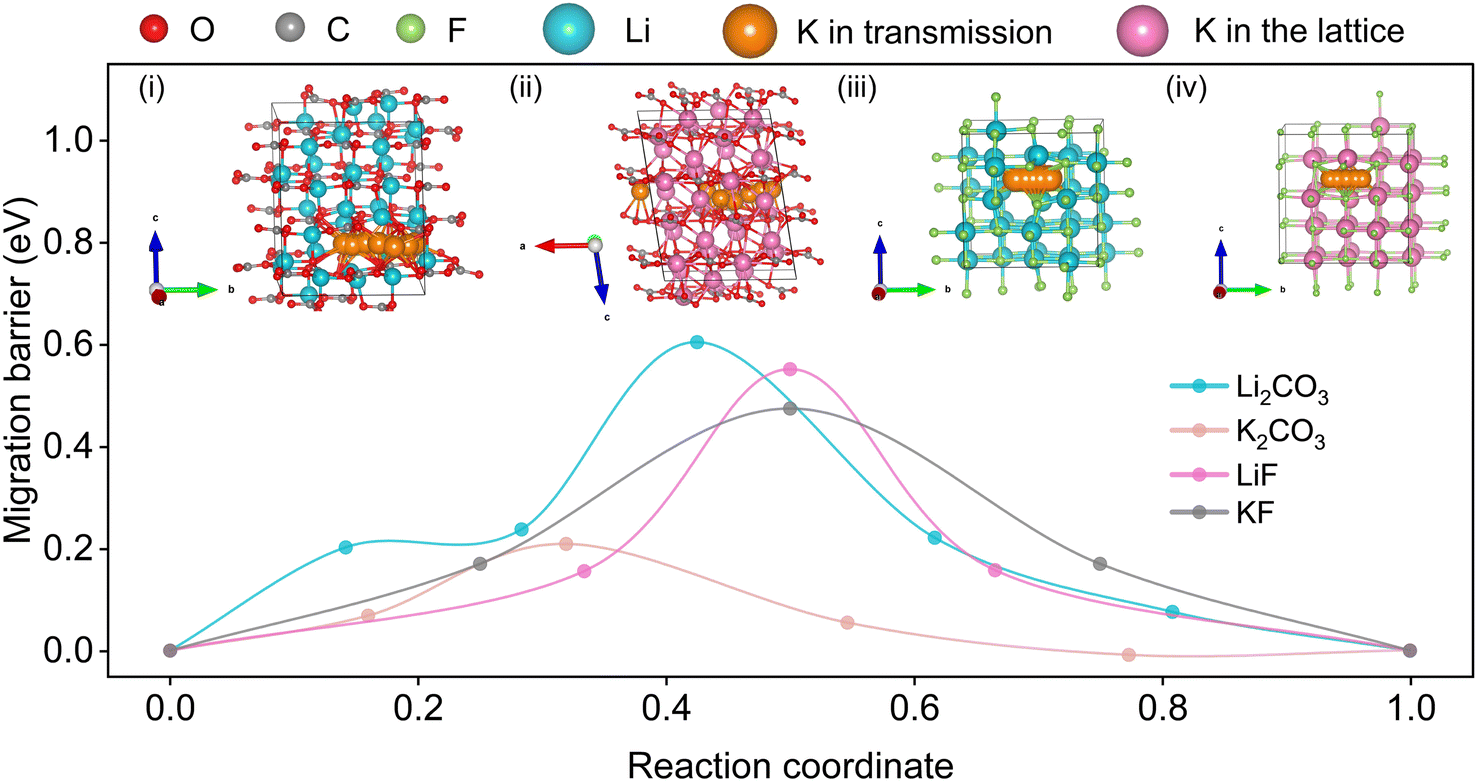 | ||
| Fig. 6 Theoretical calculation of the K migration paths and corresponding migration energy in (i) Li2CO3, (ii) K2CO3, (iii) LiF and (iv) KF. | ||
The results discussed so far suggest that this Li-based SEI layer is stable, and can be transferred and reused in PICs with K-ion based organic electrolytes. More importantly, this Li-based SEI layer is demonstrated to have a function in mitigating potassium loss during cycling, which is nearly identical to a K+ based SEI layer, thus greatly improving the capacitive performance of PICs. We also found that a Li-based SEI enabled more efficient K+ intercalation than a K-based SEI. These new observations on implanting pre-lithiation technology into PICs may apply to other carbonaceous electrodes, and even to PIBs.
To further demonstrate the advantages of the design principle of implanting pre-lithiation technology into PICs, a pre-lithiation hard carbon (pre-Li-HC) was used as the anode to assemble Li-PIC. As a comparison, K-PIC was fabricated using the pre-potassiation hard carbon (pre-K-HC). Fig. 7a shows that both Li-PIC and K-PIC exhibit the triangularly shaped galvanostatic charge/discharge curves at different current densities, confirming the good capacitive properties. Li-PIC based on the pre-Li-HC anode delivers large specific capacity values of 59.2 and 20.1 mA h g−1 at the current densities of 0.1 and 1 A g−1 (Fig. 7a, left), respectively, which are apparently higher than that of K-PIC based on the pre-Li-HC anode. Li-PIC based on the pre-Li-HC anode also exhibits better cycling performance than K-PIC based on the pre-K-HC anode. Specifically, Li-PIC based on the pre-Li-HC anode worked stably for more than 1000 cycles without any apparent decay at a current density of 0.5 A g−1, while K-PIC based on the pre-Li-HC anode preserved only 54% of the initial capacity after 1000 cycles (Fig. S17†). Moreover, as shown in Fig. 7c and d and S18,† a Li-PIC based on a pre-lithiation graphite (pre-Li-graphite) anode also exhibits better capacitive performance than a K-PIC based on a pre-potassiation graphite (pre-K-graphite) anode.
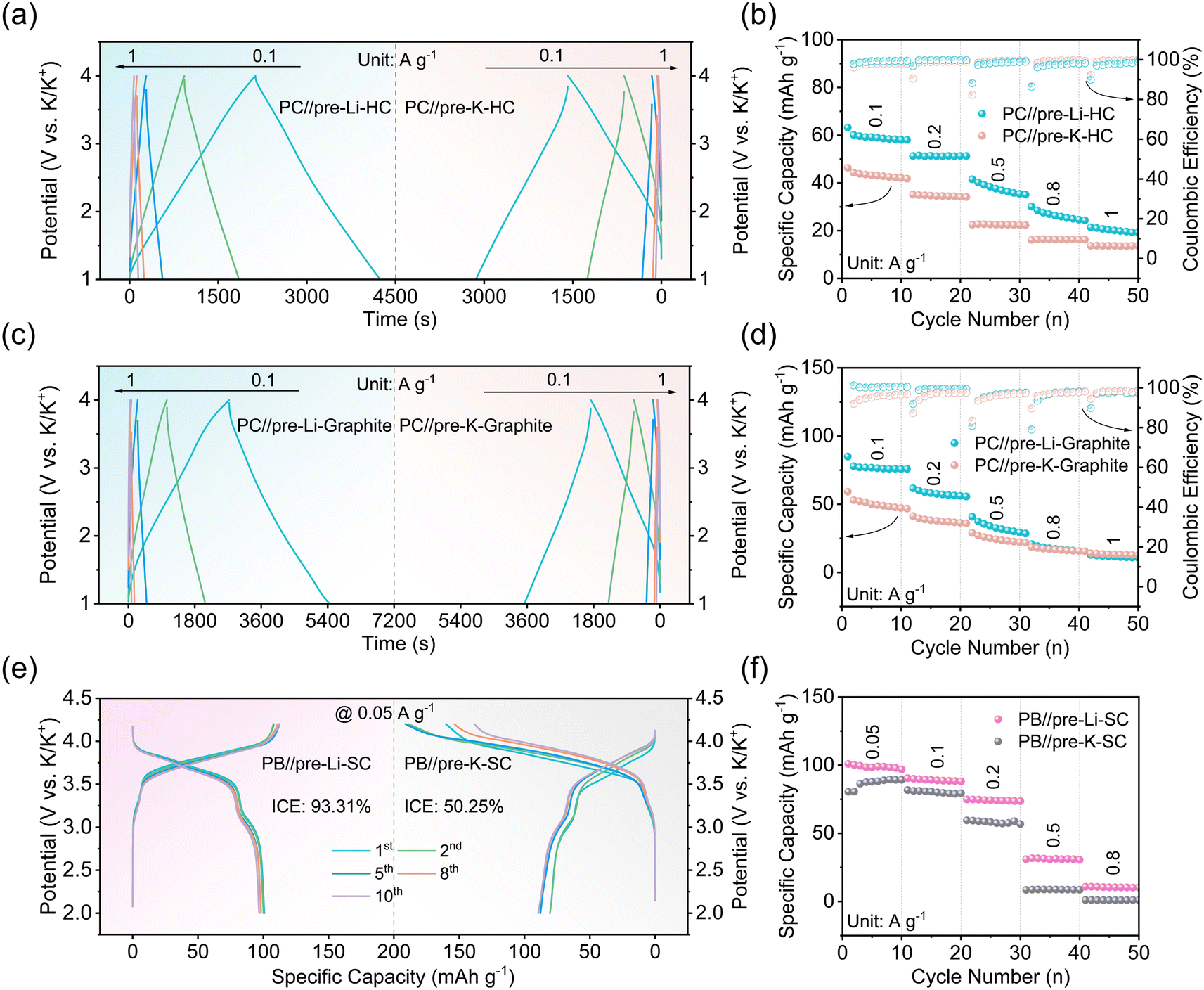 | ||
| Fig. 7 (a) Charge/discharge curves at 0.1–1 A g−1 of PC//pre-Li-HC (left) and PC//pre-K-HC (right). (b) Rate capability achieved at the various current densities from 0.1 to 1 A g−1 for pre-Li-HC and PC//pre-K-HC. (c) Charge/discharge curves at 0.1–1 A g−1 of PC//pre-Li-graphite (left) and PC//pre-K-graphite (right). (d) Rate capability achieved at the various current densities from 0.1 to 1 A g−1 for PICs based on the pre-Li-graphite and PC//pre-K-graphite electrodes. (e) Charge/discharge curves in the initial ten cycles at 0.05 A g−1 of PB//pre-Li-SC (left) and PB//pre-K-SC PIBs (right). (f) Rate capability at 0.05–0.8 A g−1 for PB//pre-Li-SC and PB//pre-K-SC PIBs. | ||
In addition to implanting pre-lithiation technology into PICs, we assembled a PIB by coupling the aforementioned PB as the positive electrode and the pre-Li-SC as the negative electrode with a N/P ratio of 1.25. Fig. 7e and f shows the typical charge–discharge profiles for PB//pre-Li-SC PIBs at a current density of 0.05 A g−1 in the voltage range of 2.0–4.2 V. This PIB exhibits an average discharge voltage of 3.64 V and delivers a high discharge capacity of 98.3 mA h g−1 (based on the positive electrode) with the initial coulombic efficiency of 93.31%. By contrast, the assembled PB//pre-K-SC PIBs delivers a high discharge capacity of 87.7 mA h g−1 with a low initial coulombic efficiency of 50.25%. The high coulombic efficiency and good rate performance are largely associated with the lithium-based SEI layer on the SC surface.56,57 The PB//pre-Li-SC PIBs also exhibit good rate performance with a high capacity of 30.9 mA h g−1 even at a high current density of 0.5 A g−1, which is slightly higher than that of PB//pre-K-SC PIBs. In addition, the PB//pre-Li-SC full cell shows good cycling stability with a 91.9% capacity retention after 30 cycles at a current density of 0.1 A g−1 (Fig. S19†). In addition to the advantage of pre-Li-SC, the enhanced electrochemical performance of the PB//pre-Li-SC PIBs is related because Li+ may be inserted into and leave the PB cathode during cell operation, as confirmed by the Li signal detected in the XPS spectra of the cycled PB cathode after the Ar+ sputtering (Fig. S20†). Ex situ XRD patterns of the PB electrode before and after cycling (Fig. S21†) show that the pristine PB structure in the PB//pre-Li-SC PIBs is still preserved after multiple cycles. These excellent electrochemical properties of the PB//pre-Li-SC PIBs further confirm that the advantages of this design principle reside in using the mature pre-lithiation technology.
Conclusions
In conclusion, we propose an innovative concept of anode pre-lithiation to replace anode pre-potassiation for mitigating potassium loss during the PICs operation. Through a combined theoretical and experimental approach, we demonstrate that the Li-ion based SEI layer preformed on the SC electrode is stable, and can be transferred and reused in PICs. In addition, this Li+-based SEI layer has a function in mitigating potassium loss during cycling, and does not hinder the K+ diffusion from the electrolyte to electrode. PICs based on pre-lithiation carbon electrodes exhibit better capacitive performance than their counterparts based on pre-potassiation carbon electrodes. Compared with the conventional pre-potassiation process for PICs, the advantages of this design principle reside in using the mature pre-lithiation technology. Furthermore, it avoids the use of highly reactive and unsafe metallic K and potassium-ion compounds for pre-potassiation, thus enabling us to fabricate much safer and more economical PICs, and even PIBs.Experimental section
Materials synthesis
Soft carbon (SC) was synthesized by thermal decomposition of red PTCDA (AR, 98%, Aladdin) in a tube furnace under an inert atmosphere at 900 °C for 4 h with a heating rate of 10 °C min−1. Commercial hard carbon (HC) and graphite were purchased from Kureha Battery Materials Japan Co., Ltd, and Btr New Material Group Co., Ltd, respectively. Activated carbon (polyaniline derived porous carbon, denoted as PC) used in this work was prepared by chemical activation on carbonated polyaniline, in which KOH was used as the activating agent in accordance with our previous work.28 The Fe-substituted Mn-rich Prussian Blue (PB, K1.5Mn0.61Fe0.39[Fe(CN)6]0.77·H2O) samples were obtained by a simple modified coprecipitation method, in accordance with previous work.7,12Materials preparation
The anode electrode was composed of 80 wt% SC, 10 wt% acetylene black, and 10 wt% poly(vinylidene fluoride) (PVDF). The evenly mixed power, dissolved into N-methyl-2-pyrrolidone (NMP), was magnetically stirred overnight to form a homogeneous slurry. Then, the slurry was coated on the Cu current collector by squeegee, and dried under a vacuum oven at 80 °C for 8 h for further use. The foil was punched into 14 mm diameter round electrodes with a mass loading of ∼1.00 mg. The PC positive electrode was composed of 80 wt% PC powder, 10 wt% polytetrafluoroethylene (PTFE), and 10 wt% conductive carbon black (super P) to form a paste with the assistance of ethanol, which was rolled into a film pack and dried under vacuum at 120 °C for 6 h. The PC film was cut into 1 cm2 square electrodes with a mass loading of ∼1 mg. The PB cathode electrode was composed of 70 wt% PB, 20 wt% acetylene black, and 10 wt% polytetrafluoroethylene (PTFE), which was rolled into a film and dried under a vacuum oven at 80 °C for 12 h. The film was cut into square electrodes with a mass loading of ∼1 mg. The PB film with a high mass loading of 10 mg was folded into a 1 cm2 square electrode and then was pressed on Al foil current collector, which was used as the reference and counter electrode.Pre-potassiation and pre-lithiation processes
For the pre-potassiation process, the half-cell (CR2032 coin cells) assembled with these carbon electrodes as the working electrode and potassium metal as the counter electrode and reference electrode was cycled for 3 cycles at a current density of 0.1 A g−1 at 0.01–3.00 V, and finally ended in a pre-potassiation state. Then, the working electrode was detached in an argon gas-filled glove box with moisture and oxygen contents of (<0.1 ppm). About 80 μl of 0.8 M KPF6 in EC:DEC (volume ratio = 1:1) electrolyte was added into the half-cell. The pre-lithiation process is similar to the pre-potassiation process, except Li metal and 1 M LiPF6 in EC:DMC:EMC (volume ratio = 1:1:1) were employed as the reference electrode and electrolyte, respectively.
Fabrication of half cells and full cells
For half cells, both the anode and cathode were tested by using the CR2032 coin cells, where a glass fiber membrane (Whatman, GF/D, USA) was used as the separator and K metal foil was used as the reference and counter electrode. For the fabrication of the full cells, the pre-metalation anode coupled with PC or PB cathodes was assembled into CR2032 coin cells, in which the glass fiber membrane (Whatman, GF/D, USA) was used as the separator. For the PIC full cell, the mass loading ratio between the positive electrode and negative electrode is 1, while for the PIB full cell, the N/P ratio (capacity) was set to 1.25. Before assembling PIBs, the PB cathode was pre-cycled in half cells at 0.05 A g−1 for about 3 cycles and finally charged to 4.2 V. For both half-cell and full cell, 0.8 M KPF6 in EC:DEC (volume ratio = 1:1) was employed as the electrolyte. To monitor the potential swings of the respective electrodes in PICs and PIBs, three-electrode Swagelok cells (E200, Tianjin Aida Ltd, China) were assembled, which are identical to the full cell configuration except that metal K was used as the auxiliary electrode. All the above-stated cells were assembled into an argon gas-filled glove box with moisture and oxygen contents of (<0.1 ppm).
Characterization
The morphologies of the samples were characterized by field emission scanning electron microscopy (FESEM, SU-70, Hitachi, Japan) and transmission electron microscopy (TEM, Tecnai F20, FEI company, Hillsboro, OR, USA). The chemical composition of the carbon electrode samples was analyzed by X-ray photoelectron spectroscopy (XPS, PerkinElmer PHI-5702 Spectrometer, PerkinElmer, Waltham, MA, USA). In order to quantify the composition changes of C, O, F, P, Li and K in the depth of the anode material structures, the sputtering time increments were 0, 40, 80, 120, and 200 s with an etch rate of 0.29 nm s−1. An inductively coupled plasma emission spectrometer (ICP-AES, Agilent 730, USA) was employed to measure the contents of the elements. For pre-Li-SC/K, the half-cell assembled with pre-lithiation SC as the working electrode and potassium metal as the counter electrode and reference electrode was cycled for 1 cycle at a current density of 0.1 A g−1 at 0.01–3.00 V, and finally ended in a de-lithiation state. For PC//pre-Li-SC, the full-cell assembled with pre-lithiation SC as the working electrode and PC as the counter electrode and reference electrode was cycled for 1 cycle at a current density of 0.1 A g−1 at 1–4 V, and finally ended in a de-lithiation state. Then, the separated glass fiber was immersed in 3 mL of dimethyl carbonate (DMC) in an argon gas-filled glove box with moisture and oxygen contents of (<0.1 ppm).Theoretical calculation
All the first-principle calculations were performed with the projector augmented wave (PAW) method in the Vienna Ab initio Simulation Package (VASP)x.58,59 The generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) formulation was adopted for structural relaxations and energy calculations.60 The kinetic cut-off energy was set to 520 eV. The convergence criteria for the self-consistent field (SCF) step were 1 × 10−5 eV. Structure optimizations were executed until the force was less than 0.02 eV Å−1. The corresponding k-points sets in our calculations were 3 × 3 × 3 for the (2 × 2 × 2) supercell of LiF, 3 × 3 × 3 for the (2 × 2 × 2) supercell of KF, 4 × 3 × 3 for the (1 × 2 × 2) supercell of Li2CO3, and 4 × 5 × 4 for the (2 × 1 × 2) supercell of K2CO3, respectively. The convergence of the results with respect to these parameters has been carefully checked. The migration barriers of the Li/K-ion in these different crystal structures were calculated using the climbing image nudged elastic band (CI-NEB) method.61 To determine the initial structure, one additional Li/K atom was inserted into different sites and fully relaxed. The lowest energy structure was then used for further analysis. For different NEB calculations, three to five intermediate images were used to find the minimum energy path between the start and the end positions. The energies and forces were converged to 1 × 10−5 eV and 0.05 eV Å−1, respectively.Electrochemical measurements
The cyclic voltammetry (CV) test was performed on a CHI760E Electrochemical Workstation (Shanghai, China). The charge/discharge and cycle-life tests were performed on the LAND Battery Test System (Land CT2001A model, Wuhan, China).Data availability
Data are available on request from the authors.Author contributions
The manuscript was written with contributions from all of the authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272224, 51902188).Notes and references
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef PubMed.
- S. N. Lauro, J. N. Burrow and C. B. Mullins, E-Science., 2023, 3, 100152 Search PubMed.
- F. Degen, M. Winter, D. Bendig and J. Tübke, Nat. Energy, 2023, 8, 1284–1295 CrossRef CAS.
- Y. Xu, Y. Du, H. Chen, J. Chen, T. Ding, D. Sun, D. H. Kim, Z. Lin and X. Zhou, Chem. Soc. Rev., 2024, 53, 7202–7298 RSC.
- S. Zhao, Z. Liu, G. Xie, Z. Guo, S. Wang, J. Zhou, X. Xie, B. Sun, S. Guo and G. Wang, Energy Environ. Sci., 2022, 15, 3015–3023 RSC.
- T. Bashir, S. Zhou, S. Yang, S. A. Ismail, T. Ali, H. Wang, J. Zhao and L. Gao, Electrochem. Energy Rev., 2023, 6, 5 CrossRef CAS.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- J. Liao, X. Zhang, Q. Zhang, Q. Hu, Y. Li, Y. Du, J. Xu, L. Gu and X. Zhou, Nano Lett., 2022, 22, 4933–4940 CrossRef CAS PubMed.
- L. Yu, X. He, B. Peng, W. Wang, G. Wan, X. Ma, S. Zeng and G. Zhang, Matter, 2023, 6, 1604–1621 CrossRef CAS.
- F. Zhang, X. Wang, M. Wu, A. Yang, Y. Li, M. Man, Y. Li and J. Guo, ACS Energy Lett., 2023, 8, 4895–4902 CrossRef CAS.
- Y. Qin, Y. Xie, H. Zhao, C. Zhu, T. Li, S. Zhang, R. Wang, Y. Shi and L. Yin, Chin. Chem. Lett., 2022, 33, 1463–1467 CrossRef CAS.
- J. Chen, S. Lei, S. Zhang, C. Zhu, Q. Liu, C. Wang, Z. Zhang, S. Wang, Y. Shi, L. Yin and R. Wang, Adv. Funct. Mater., 2023, 2215027 CrossRef CAS.
- M. Gu, A. M. Rao, J. Zhou and B. Lu, Energy Environ. Sci., 2023, 16, 1166–1175 RSC.
- L. Liang, M. Tang, Q. Zhu, W. Wei, S. Wang, J. Wang, J. Chen, D. Yu and H. Wang, Adv. Energy Mater., 2023, 13, 2300453 CrossRef CAS.
- H. Kim, D. Seo, J. C. Kim, S. Bo, L. Liu, T. Shi and G. Ceder, Adv. Mater., 2017, 29, 1702480 CrossRef PubMed.
- L. Deng, X. Niu, G. Ma, Z. Yang, L. Zeng, Y. Zhu and L. Guo, Adv. Funct. Mater., 2018, 28, 1800670 CrossRef.
- T. Deng, X. Fan, C. Luo, J. Chen, L. Chen, S. Hou, N. Eidson, X. Zhou and C. Wang, Nano Lett., 2018, 18, 1522–1529 CrossRef CAS PubMed.
- J.-Y. Hwang, J. Kim, T.-Y. Yu, S.-T. Myung and Y.-K. Sun, Energy Environ. Sci., 2018, 11, 2821–2827 RSC.
- J. H. Jo, J. U. Choi, Y. J. Park, Y. H. Jung, D. Ahn, T. Jeon, H. Kim, J. Kim and S. Myung, Adv. Energy Mater., 2020, 10, 1903605 CrossRef CAS.
- W. Xiao, P. Shi, Z. Li, C. Xie, J. Qin, H. Yang, J. Wang, W. Li, J. Zhang and X. Li, J. Energy Chem., 2023, 78, 589–605 CrossRef CAS.
- H. Qian, X. Li, Q. Chen, W. Liu, Z. Zhao, Z. Ma, Y. Cao, J. Wang, W. Li, K. Xu, K. Zhang, W. Yan, J. Zhang and X. Li, Adv. Funct. Mater., 2024, 34, 2310143 CrossRef CAS.
- H. Li, Y. Gong, H. Zhou, J. Li, K. Yang, B. Mao, J. Zhang, Y. Shi, J. Deng, M. Mao, Z. Huang, S. Jiao, Y. Kuang, Y. Zhao and S. Luo, Nat. Commun., 2023, 14, 6407 CrossRef CAS PubMed.
- T. Li, H. Zhao, C. Li, W. Yu, Y. Shi and R. Wang, New Carbon Mater., 2021, 36, 253–277 CrossRef CAS.
- X. Shi, H. Wang, Z. Xie, Z. Mao, T. Zhang, J. Jin, B. He, R. Wang, Y. Gong and H. J. Fan, Adv. Mater., 2024, 2406794 CrossRef CAS PubMed.
- C. Xu, J. Mu, T. Zhou, S. Tian, P. Gao, G. Yin, J. Zhou and F. Li, Adv. Funct. Mater., 2022, 32, 2206501 CrossRef CAS.
- S. Zhao, Z. Liu, G. Xie, X. Guo, Z. Guo, F. Song, G. Li, C. Chen and G. Wang, Angew. Chem., 2021, 60, 26246–26253 CrossRef CAS PubMed.
- T. Li, X. Huang, S. Lei, J. Zhang, X. Li, C. Wang, Z. Zhang, S. Wang, L. Yin and R. Wang, Energy Mater., 2023, 3, 300018 CAS.
- M. Shao, C. Li, T. Li, H. Zhao, W. Yu, R. Wang, J. Zhang and L. Yin, Adv. Funct. Mater., 2020, 30, 2006561 CrossRef CAS.
- X. Liang, Z. Mao, X. Shi, T. Zhang, Z. Zheng, J. Jin, B. He, R. Wang, Y. Gong and H. Wang, J. Mater. Chem. A, 2023, 11, 18097–18105 RSC.
- S. Zhao, G. Li, B. Zhang, T. Li, M. Luo, B. Sun, G. Wang and S. Guo, Joule, 2024, 8, 922–943 CrossRef CAS.
- Q. Liu, J. Chen, D. Du, S. Zhang, C. Zhu, Z. Zhang, C. Wang, L. Yin and R. Wang, J. Mater. Chem. A, 2023, 11, 17491–17502 RSC.
- Y. Qian, B. Wu, Y. Li, Z. Pan, S. Feng, N. Lin and Y. Qian, Angew. Chem., Int. Ed., 2023, 62, e202217514 CrossRef CAS PubMed.
- X. Xiang, D. Liu, J. Song, X. Zhu, Z. Xie, H. Tang, H. Zheng and D. Qu, J. Alloys Compd., 2023, 960, 170864 CrossRef CAS.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- P. Jeżowski, O. Crosnier, E. Deunf, P. Poizot, F. Béguin and T. Brousse, Nat. Mater., 2018, 17, 167–173 CrossRef PubMed.
- Y. Sun, H.-W. Lee, Z. W. Seh, N. Liu, J. Sun, Y. Li and Y. Cui, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718–725 CrossRef CAS.
- N. Qin, L. Jin, Y. Lu, Q. Wu, J. Zheng, C. Zhang, Z. Chen and J. P. Zheng, Adv. Energy Mater., 2022, 12, 2103402 CrossRef CAS.
- R. Guo, K.-H. Kim and B. M. Gallant, J. Electrochem. Soc., 2022, 169, 100523 CrossRef CAS.
- S. Wan, K. Song, J. Chen, S. Zhao, W. Ma, W. Chen and S. Chen, J. Am. Chem. Soc., 2023, 145, 21661–21671 CrossRef CAS PubMed.
- C. Zheng, D. Ji, Q. Yao, Z. Bai, Y. Zhu, C. Nie, D. Liu, N. Wang, J. Yang and S. Dou, Angew. Chem., Int. Ed., 2023, 62, e202214258 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- E. C. Cengiz, J. Rizell, M. Sadd, A. Matic and N. Mozhzhukhina, J. Electrochem. Soc., 2021, 168, 120539 CrossRef CAS.
- S. S. Zhang, J. Electrochem. Soc., 2020, 167, 100510 CrossRef CAS.
- N. Qin, L. Jin, G. Xing, Q. Wu, J. Zheng, C. Zhang, Z. Chen and J. P. Zheng, Adv. Energy Mater., 2023, 13, 2204077 CrossRef CAS.
- C. Zheng, D. Ji, Q. Yao, Z. Bai, Y. Zhu, C. Nie, D. Liu, N. Wang, J. Yang and S. Dou, Angew. Chem., Int. Ed., 2023, 62, e202214258 CrossRef CAS PubMed.
- Y. Yuan, S. D. Pu, M. A. Pérez-Osorio, Z. Li, S. Zhang, S. Yang, B. Liu, C. Gong, A. S. Menon, L. F. J. Piper, X. Gao, P. G. Bruce and A. W. Robertson, Adv. Mater., 2024, 36, 2307708 CrossRef CAS PubMed.
- J. Zheng, Z. Huang, Y. Zeng, W. Liu, B. Wei, Z. Qi, Z. Wang, C. Xia and H. Liang, Nano Lett., 2022, 22, 1017–1023 CrossRef CAS PubMed.
- F. La Mantia, C. D. Wessells, H. D. Deshazer and Y. Cui, Electrochem. Commun., 2013, 31, 141–144 CrossRef CAS.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- T. Liu, L. Lin, X. Bi, L. Tian, K. Yang, J. Liu, M. Li, Z. Chen, J. Lu, K. Amine, K. Xu and F. Pan, Nat. Nanotechnol., 2019, 14, 50–56 CrossRef CAS PubMed.
- J. Hui, N. B. Schorr, S. Pakhira, Z. Qu, J. L. Mendoza-Cortes and J. Rodríguez-López, J. Am. Chem. Soc., 2018, 140, 13599–13603 CrossRef CAS PubMed.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector, H. Li and S. J. Harris, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- S. Shi, Y. Qi, H. Li and L. G. Hector, J. Phys. Chem. C, 2013, 117, 8579–8593 CrossRef CAS.
- Z. Tang, S. Zhou, Y. Huang, H. Wang, R. Zhang, Q. Wang, D. Sun, Y. Tang and H. Wang, Electrochem. Energy Rev., 2023, 6, 8 CrossRef CAS.
- T. Zhang, Z. Mao, X. Shi, J. Jin, B. He, R. Wang, Y. Gong and H. Wang, Energy Environ. Sci., 2022, 15, 158–168 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06451h |
| This journal is © The Royal Society of Chemistry 2024 |