
Antibacterial activity of hydrophobicity modulated cationic polymers with enzyme and pH-responsiveness†
Desoshree
Ghosh‡
 a,
Sandeep
Yadav‡
b,
Sagar
Bag
a,
Amirul Islam
Mallick
*b and
Priyadarsi
De
*a
a,
Sandeep
Yadav‡
b,
Sagar
Bag
a,
Amirul Islam
Mallick
*b and
Priyadarsi
De
*a
aPolymer Research Centre and Centre for Advanced Functional Materials, Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia, West Bengal 741246, India. E-mail: p_de@iiserkol.ac.in
bDepartment of Biological Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia, West Bengal 741246, India. E-mail: amallick@iiserkol.ac.in
First published on 20th February 2024
Abstract
The membrane lipid compositions of prokaryotic and eukaryotic cells are inherently different in many aspects, although some similarities exist in their structure and composition. Therefore, selective targeting of membrane lipids with a compound of therapeutic value, such as an antibacterial copolymer, is often challenging. Hence, developing an ideal copolymer with antibacterial properties demands hydrophobicity/hydrophilicity balance with a high biosafety profile. To integrate hydrophobic/hydrophilic balance and cationic charge in an alternating antibacterial copolymer with enzyme and pH-responsiveness, a lysine appended styrenic monomer was copolymerized with a fatty acid (octanoic acid (OA) or myristic acid (MA)) tethered maleimide monomer via reversible addition–fragmentation chain transfer (RAFT) polymerization. A range of microscopic analyses, including dynamic light scattering (DLS), confirmed the formation of nanoaggregates (size ∼30–40 nm) by these polymers in aqueous solution with positive zeta potential (cationic surface charge). Hydrophobic Nile red (NR) dye was successfully encapsulated in the nanoaggregates, and the in vitro release kinetics of the NR dye were monitored at different pHs and in the presence or absence of esterase/lipase. The in vitro release kinetics of NR revealed ∼85% dye release in the presence of pH 5.5 and lipase, suggesting their suitability for pH/enzyme-triggered therapeutic payload delivery. The standard broth microdilution assay showed significant bactericidal activity against both Gram-positive (Bacillus subtilis) and Gram-negative (Escherichia coli) bacteria with an MIC50 value <30 μg mL−1. The effect of polymeric nanoaggregates on bacterial morphology and in vitro survival was further confirmed by field emission scanning electron microscopy (FESEM), agar gel disk diffusion assay, and bacterial live/dead cell count. The significantly low hemolytic activity against red blood cells (RBCs) (HC50 >103 μg mL−1) and nontoxic effect on human intestinal epithelial cells (INT 407) (EC50 >500 μg mL−1) ensure that the polymer nanoaggregates are safe for in vivo use and can serve as a potent antibacterial polymer.
Introduction
Since 1928, when penicillin, the first antibiotic, was discovered, morbidity and mortality rates of bacterial infections have dramatically decreased. Unfortunately, the surge in antibiotic resistance among the wide range of bacterial pathogens has emerged as a major threat to human and animal health globally. This is primarily because of the indiscriminate and injudicious use of antimicrobials in agriculture, animal husbandry, and human clinical practices.1 According to a current assessment sourced from the World Health Organization (WHO), annually 2 million fatalities could result from multidrug resistance (MDR) infections, and if the issue is not adequately addressed, approximately 10 million people could perish every year by 2050.2,3 In addition, the emergence of MDR infections also incurs higher treatment costs along with a significant effect on the quality of life and, consequently, has placed a tremendous socioeconomic strain on the fast-growing population.2 Despite the tireless efforts of the scientific community around the globe, there have been many challenges in the synthesis and development of novel antibiotics, leading to a “discovery void” in the antibiotic arsenal to fight against MDR pathogens.To this end, antimicrobial peptides (AMPs) have emerged as a potential alternative to antibiotics for fighting against microbial pathogens.4,5 These host defense peptides (HDPs) are normally synthesized locally by specialized cells, contributing to innate host defense against a wide range of pathogens. Armed with intrinsically different “modes of action”, AMPs are considered potential antimicrobials capable of effectively tackling antibiotic resistance.6,7 These peptides are made up of a few amino acid residues comprising cationic, hydrophilic, and hydrophobic groups. The amphiphilic nature of AMPs enables them to bind to a range of precursor molecules essential for bacterial cell walls and membrane formation, disrupting their membrane architecture and subsequent cell death. In addition, AMPs can interact with negatively charged cell walls of bacteria by disrupting their phospholipid membrane.8,9 Despite these advantages, the exogenous application of AMPs as an antibiotic alternative is questionable due to their non-specific targeting and low to moderate degrees of cell cytotoxicity, such as hemolytic activity on red blood cells (RBCs). The other limiting factors for AMPs are cost-intensive production, susceptibility to proteolysis, and their short-term stability in the biological milieu.10
Influenced by the architecture of the peptides and with the recent developments in polymer chemistry, antimicrobial polymers have emerged as prospective substitutes for antibiotics or AMPs.11,12 These antibacterial polymers are less prone to proteolysis, have cost-effective production, and are easy to scale up for large-scale synthesis. Owing to these advantages, several amphiphilic antibacterial polymers were reported in the recent past, namely norbornene,13 maleimide,14 acrylamide,15 meth(acrylate),16 urea,17etc. Although these polymers show significant antibacterial activity, they often exhibit high toxicity to host cells, reducing their potential applications. This has warranted careful tuning of the synthesis process of antimicrobial polymers, ensuring minimal cell cytotoxicity and enhancing biocompatibility without diminishing their antibacterial activity.18 Growing evidence suggests that the molar mass and composition of the polymer, like amphiphilic balance,19 can significantly improve the antibacterial specificity and activity of the polymer.20 Such polymers are normally synthesized by copolymerizing cationic/hydrophilic and hydrophobic monomers to regulate amphiphilicity. Moreover, the nature and quantity of cationic and hydrophobic moieties present in the polymer also determine the target specificity.21,22 In this regard, primary amine-containing amphiphilic copolymers exhibit a strong antibacterial effect toward Gram-negative bacteria, while quaternary ammonium-containing polymers are highly effective toward mycobacteria.15,23 Various hydrophobic monomers have been investigated to enhance selectivity because hydrophobic groups damage both the bacterial and mammalian cell membranes.24 Due to the amphiphilic balance in such polymers, the formation of nanoaggregates in an aqueous medium is easily achievable and a variety of therapeutic cargos can be encapsulated as well. Such cargo-loaded nanoaggregates are one of the most attractive modalities for combating drug-resistant bacteria more efficiently.25,26
Macromolecular features of the polymer, such as folding and self-assembly, can be influenced by controlling the composition, sequence of monomers and architectures.27 The significance of sequence in macromolecular architectures can be easily assessed from natural biopolymers such as deoxyribonucleic acid (DNA), peptides and proteins.28 The diversity, adaptability and complexity of life extensively depend on the organized monomer sequence. In the cellular system, the monomer sequence additionally regulates genetics, self-replication and molecular recognition.29 Therefore, the development of sequence-controlled macromolecular architectures is the most attractive field of current research.30 The performances of sequence-controlled polymers are enhanced by functional group modification at the alternate position compared to less periodic polymers like block, random and gradient polymers.31
Based on these perspectives, styrene and N-substituted maleimide (or maleic anhydride) monomer pairs are utilized to develop sequence-controlled alternating polymers with regioregularity.32,33 Recently, our group synthesized several sequence-controlled alternating copolymers demonstrating their multimodal applications, including macromolecular luminogen,34,35 pH-induced morphological transformation,36,37 drug delivery,38 UV and pH-responsive polymeric nitric oxide donors,39 and lectin recognition ability.40
To integrate the alternating sequence, hydrophobic/hydrophilic balance, and cationic charge in a single macromolecular architecture armed with antibacterial activity and enzyme/pH-responsive drug/dye releasing capability, a lysine appended styrenic monomer was copolymerized with fatty acid (octanoic acid (OA) or myristic acid (MA)) tethered maleimide monomer via reversible addition–fragmentation chain transfer (RAFT) polymerization (Scheme 1). Here, L-lysine, a natural amino acid, was utilized for its higher cationic nature due to the presence of two primary amine moieties, biocompatibility, and most importantly its widespread antibacterial uses.41 Several ester groups, present in the side-chain of the polymer, are prone to biodegradation by two kinds of enzymes: (i) lipase (L) enzyme secreted by bacteria,42 and (ii) lysosomal esterase (E) enzyme of the endothelial cells when they are unwantedly encountered by the mammalian cells of normal healthy tissues.43 In an aqueous medium, the cationic polymer formed self-assembled nanoaggregates, with antibacterial activity against a Gram-positive bacteria, Bacillus subtilis (B. subtilis) and Gram-negative Escherichia coli (E. coli). We demonstrated that the synthesized copolymers are biologically safe for mammalian cells and erythrocytes owing to their excellent therapeutic value for long-term ramifications in treating infectious diseases.
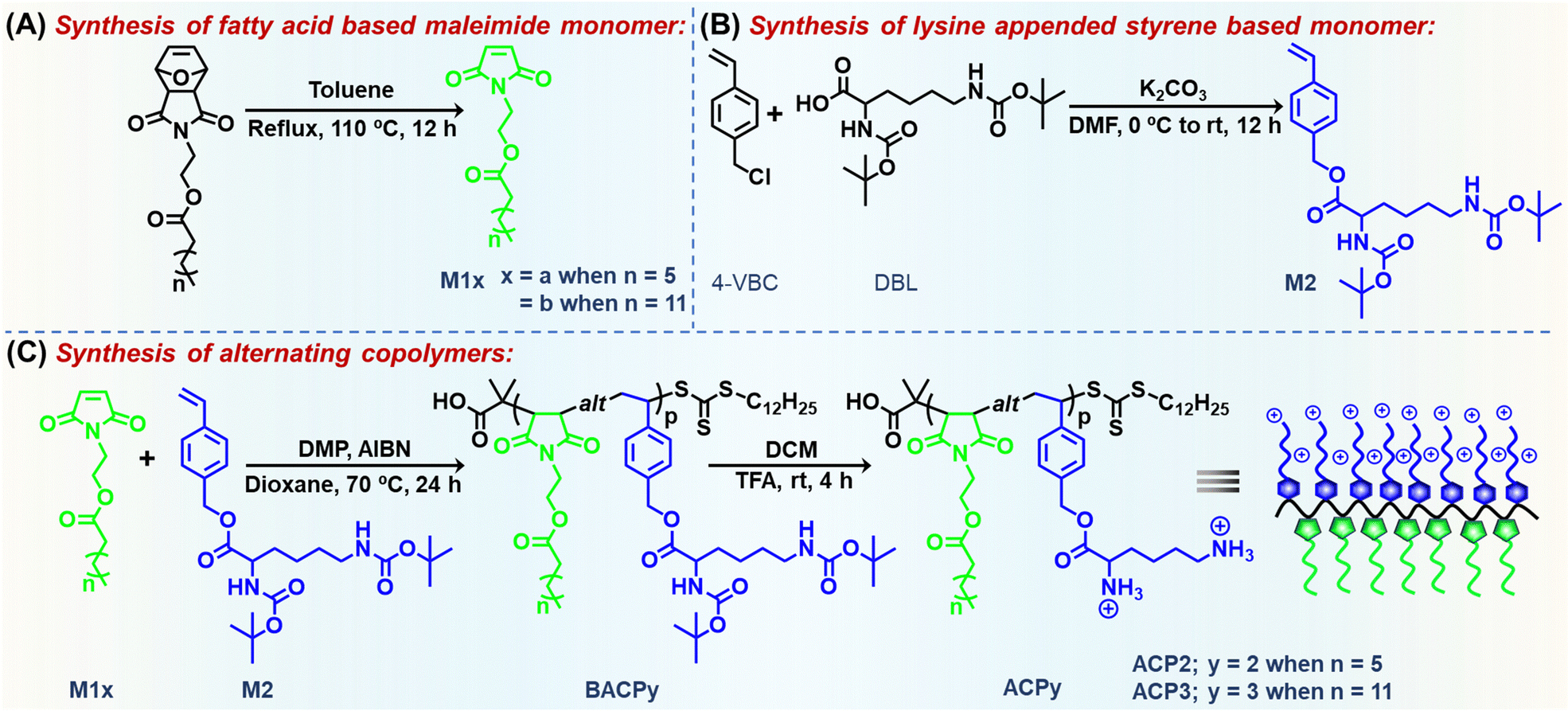 | ||
| Scheme 1 Synthetic scheme of (A) M1x, (B) M2 and (C) amphiphilic alternating copolymers (ACPy). | ||
Experimental section
Materials and methods
Details of chemicals, instruments and characterization methods are given in the ESI.†Synthesis of M1a and M1b
Maleimide monomers with an octanoate moiety (M1a) and myristate moiety (M1b) were prepared by following a previous literature report.44 These monomers were characterized by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS) (Fig. S1–S6, ESI†).Synthesis of M2
M2 monomer was synthesized by a typical nucleophilic substitution reaction. In a 250 mL round-bottom (RB) flask, 4-vinyl benzyl chloride (4-VBC, 1.95 g, 12.8 mmol), N,N′-di-tert-butylcarbamate (Boc)-L-lysine (DBL, 3.70 g, 10.6 mmol) and potassium carbonate (K2CO3, 1.76 g, 12.8 mmol) were dissolved in 150 mL N,N-dimethylformamide (DMF), stirred in an ice bath for 30 min and kept overnight at room temperature (rt). When the reaction was completed, the reaction mixture was diluted with 300 mL brine solution and extracted with 200 mL dichloromethane (DCM) in a portion. The resulting DCM solution was further extracted with brine solution (3 × 100 mL), dried with anhydrous sodium sulfate (Na2SO4) and purified by column chromatography using hexane and ethyl acetate (80/20, v/v) as an eluent to get a white solid M2. Yield = 80%. 1H NMR (CDCl3, δ, ppm, Fig. 1(A)): 7.42 (ArCH, 2H, d), 7.33 (ArCH, 2H, d), 6.73 (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, 1H, dd), 5.78 (–CHCH2, 1H, t), 5.30 (–CHCH2, 1H, d), 5.23–5.06 (–CHNHCOO, –ArCH2COO, 3H, m), 4.55 (–CH2NHCOO, 1H, s), 4.33 (–NHCHCH2, 1H, t), 3.07 (–CH2CH2NH, 2H, t), 1.79 (–CHCH2CH2, 2H, m), 1.65 (–CH2CH2CH2CH2, 4H, m) 1.44 (C(CH3)3, 18 H, s).
CH2, 1H, dd), 5.78 (–CHCH2, 1H, t), 5.30 (–CHCH2, 1H, d), 5.23–5.06 (–CHNHCOO, –ArCH2COO, 3H, m), 4.55 (–CH2NHCOO, 1H, s), 4.33 (–NHCHCH2, 1H, t), 3.07 (–CH2CH2NH, 2H, t), 1.79 (–CHCH2CH2, 2H, m), 1.65 (–CH2CH2CH2CH2, 4H, m) 1.44 (C(CH3)3, 18 H, s).
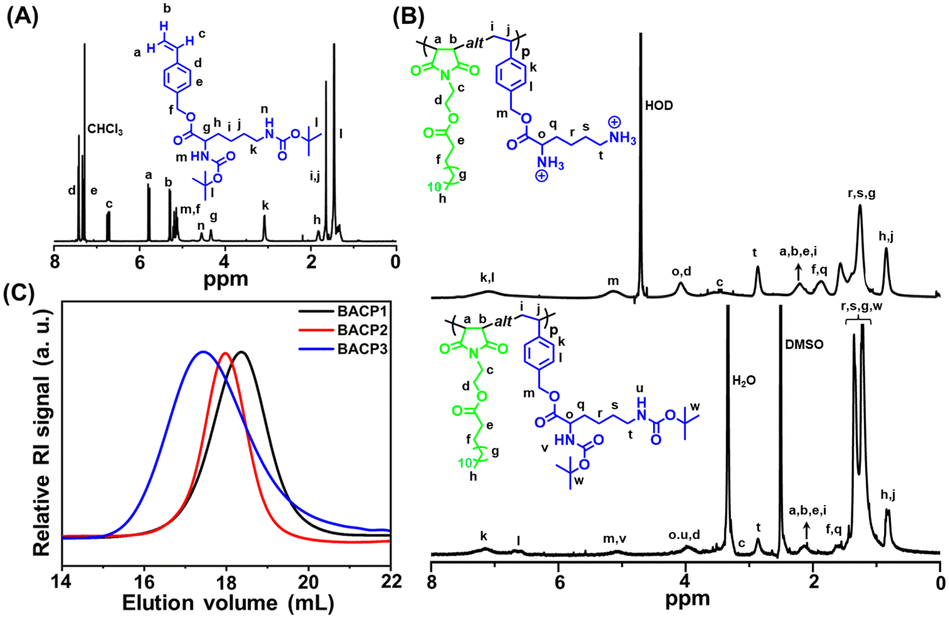 | ||
| Fig. 1 (A) 1H NMR spectrum of M2. (B) 1H NMR spectra of the protected polymer (BACP3) in DMSO-d6 and deprotected polymer (ACP3) in D2O. (C) SEC RI traces of BACP1–BACP3 in DMF. | ||
Synthesis of alternating copolymers, BACP2 and BACP3
For the preparation of BACP2, the monomers M1a (300 mg, 1.122 mmol) and M2 (519 mg, 1.122 mmol), 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid (DMP, 27 mg, 0.074 mmol) and 2,2′-azobis(2-methylpropionitrile) (AIBN, 6.1 mg, 0.037 mmol) were added in a 20 mL glass vial according to a predetermined feed ratio (Table 1) equipped with a magnetic stir bar. Dioxane (3 mL) was added to the vial to dissolve all the components. The vial was sealed tightly, kept under continuous N2 flow for 10 min, and stirred at 70 °C under continuous stirring for 24 h. Then, the vial was kept in ice-cold water briefly to quench the polymerization and the solvent was removed by applying a vacuum. The residue was then dissolved in a minimum amount of DCM and precipitated by excess methanol. The process was repeated four times, and the final polymer was dried in a desiccator under a high vacuum. Similarly, BACP3 was prepared using M1b (300 mg, 0.854 mmol), M2 (395 mg, 0.854 mmol), DMP (20.7 mg, 0.057 mmol) and AIBN (4.6 mg, 0.028 mmol) in dioxane at 70 °C (Table 1).| Polymer | [M]/[M2]/[DMP]/[AIBN] | Conv.d (%) | M n,theo (g mol−1) | M n,UV-Vis (g mol−1) | M n,SEC (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| a [M] = HEMI. b [M] = M1a. c [M] = M1b. All the polymerization reactions were carried out in 1,4-dioxane at 70 °C. d Calculated gravimetrically. e M n,theo = [([M1x + M2]/[DMP] × (molar mass of M1x + molar mass of M2) × conv.) + molar mass of DMP].35 f Determined from the 308 nm absorbance peak of the trithiocarbonate functional group of the CTA chain end. g Determined by SEC analysis in DMF. | ||||||
| BACP1 | 15/15/1/0.5a | 65 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
11000 |
9800 | 1.23 |
| BACP2 | 15/15/1/0.5b | 70 | 15600 |
13500 |
12600 |
1.14 |
| BACP3 | 15/15/1/0.5c | 68 | 16900 |
18300 |
14000 |
1.36 |
Deprotection of the alternating copolymers
The Boc groups in BACP2 and BACP3 were removed by treatment of trifluoroacetic acid (TFA) in DCM at room temperature (Scheme 1) to produce ACP2 and ACP3, respectively. Typically, 200 mg Boc-protected polymer was dissolved in 4 mL of DCM, and 2 mL TFA was added dropwise into it and kept for 4 h at room temperature. After the removal of the solvent under vacuum, the residue was dissolved in a minimum amount of methanol and precipitated from diethyl ether. This process was repeated 3 times, and the final polymer was dried in a desiccator under a high vacuum.Neutralization of the polymers
Prior to any biological experiment, the deprotected polymers were neutralized with aqueous ammonium hydroxide solution and dialyzed against deionized (DI) water (pH ∼ 6.5) for 48 h to remove TFA salts and other impurities generated during Boc group removal.Self-assembled nanoaggregate formation
The formation of self-assembled nanoaggregates from the cationic alternating copolymer was performed using the solvent evaporation method.45 In a typical procedure for dynamic light scattering (DLS) measurement, 2 mg of polymer was first dissolved in 500 μL of acetone, added dropwise to 1× phosphate buffer saline (PBS, pH 7.4), acidic buffer (pH 5.0) and alkaline buffer (pH 9.0) under stirring, keeping the concentration of polymer at 1 mg mL−1 and was left open overnight. Similarly, samples were prepared for field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM), where the concentration of the polymer solution was maintained at 0.1 mg mL−1 and DI water was used instead of PBS buffer.For the preparation of Nile red (NR) encapsulated nanoaggregates, 1 mg polymer in 200 μL acetone and 50 μL NR solution in acetone (8 × 10−8 M) were added subsequently to 10 mL 1× PBS buffer (pH 7.4). After stirring the solution for 12 h in open conditions, the resulting solution was passed through a 0.45 μm nylon filter to remove unencapsulated NR.
Determination of dye loading content (DLC) and dye loading efficiency (DLE)
Both the DLC and DLE were determined from the below mentioned equations,46| DLC (%) = [weight of loaded NR/weight of NR loaded NPs] × 100% |
| DLE (%) = [weight of loaded NR/weight of NR in feed] × 100% |
Zeta potential (ξ) measurement
The surface charge of all polymers (ACP1–ACP3) was measured by dissolving the polymers in three different pH solutions (pH 5.0, 7.4 and 9.0).Critical aggregation concentration (CAC) determination
CAC of the amphiphilic cationic copolymers was determined using fluorescence spectroscopy and pyrene as a fluorescent probe.47 A known concentration of pyrene in acetone (10−3 M), and polymer aqueous solutions ranging in concentrations from 10−1 mg mL−1 to 10−4 mg mL−1 each with 2 mL volume were prepared. A constant amount of pyrene-containing acetone solution was added to each polymer solution, keeping the final pyrene concentration constant at 10−7 M. Then, all the polymer solutions were left open for 6 h to encapsulate pyrene in the hydrophobic core of the polymeric nanoaggregates and evaporation of acetone. Subsequently, the fluorescence emission intensity was measured for each polymer solution at an excitation wavelength of 337 nm. The emission intensity ratio I393/I373 was plotted against the logarithm of polymer concentration to determine the CAC value from the intersection point of the plot.pH and enzyme-triggered degradation of NR encapsulated nanoaggregates
pH and enzyme-triggered degradation of NR encapsulated nanoaggregates was performed using the dialysis method. In a typical procedure, NR encapsulated 4 mL of 0.1 mg mL−1 polymer solution was kept inside a semipermeable dialysis membrane (MWCO = 2000 g mol−1) along with enzymes i.e. esterase (E) and lipase (L) at a concentration of 10 units per mL (U mL−1). Then, the sealed dialysis membrane was immersed in 400 mL buffer solution with different pH (7.4 and 5.0). The NR release kinetics were recorded by the fluorescence emission intensity measurement of aliquots (1 mL) withdrawn from the dialysis membrane at regular time intervals.Results and discussion
Synthesis of polymers
The M2 was synthesized by a typical nucleophilic substitution on 4-VBC with DBL using K2CO3 as a base and DMF as a solvent at room temperature. It was successfully characterized by 1H NMR (Fig. 1(A)), 13C NMR spectroscopy (Fig. S7, ESI†), and HRMS (Fig. S8, ESI†). Next, fatty acid appended maleimide monomers M1a and M1b were copolymerized with M2 to form alternating copolymers BACP2 and BACP3, respectively, by RAFT polymerization in 1,4-dioxane at 70 °C using DMP as a chain transfer agent (CTA) and AIBN as an initiator (Scheme 1) at [M1x]/[M2]/[DMP]/[AIBN] = 15/15/1/0.5. The control polymer (BACP1) without a fatty acid moiety in the maleimide monomer was also prepared from the RAFT polymerization of 2-hydroxyethyl maleimide (HEMI) with M2 (Scheme S1, ESI†).Protected polymers (BACPy) were successfully analyzed by 1H NMR spectroscopy and a representative 1H NMR spectrum of BACP3 is depicted in Fig. 1(B) (lower curve). The disappearance of maleimide (from M1b) and vinyl (from M2) protons confirms the purity of the copolymers with the successful removal of unreacted monomers. All the peaks from the two repeating units were identified in the 1H NMR spectrum of BACP3 in DMSO-d6 (Fig. 1(B), lower curve). We could not determine the molar mass of alternating copolymers from 1H NMR spectra because there was no distinguishable peak observed for chain-end protons due to the overlap of peaks from copolymer repeating unit protons. Thus, the molar mass of the copolymers (Mn,UV-Vis) was determined by UV-Vis spectroscopy (Table 1), following the literature procedure.44 The number average molar mass (Mn,SEC) and dispersity (Đ) of the copolymers were also measured by size exclusion chromatography (SEC) in DMF solvent, summarised in Table 1. The SEC curves were monomodal, as shown in Fig. 1(C). The theoretical number-average molar mass (Mn,theo) values were determined according to monomer conversion (Conv.) (Table 1). Thus, reasonable agreement between the Mn,theo, Mn,UV-Vis and Mn,SEC values in Table 1 was observed. Previously, several literature reports have supported the formation of alternating copolymers by an equimolar mixture of maleimide/maleic anhydride and styrene-based monomer.33,48 For the BACP3 polymer (Fig. 1(B), lower curve), the integration ratio of the ‘’t” proton peak for 2 protons at 2.87 ppm and “h,j” proton peak for 4 protons at 0.82 ppm is almost close to 1:2, suggesting 1:1 monomer incorporation in the copolymer. Similar observations were also noticed from the 1H NMR spectra of BACP1 (Fig. S9, ESI†) and BACP2 (Fig. S10, ESI†). The alternating sequence of BACP3 was further assessed by 13C NMR spectroscopy (Fig. S11, ESI†).35 The NMR peaks at 33.7 ppm and 135.4 ppm correspond to C3 and C5 carbon, respectively, supporting the alternating position of the two monomers in the polymer.
The BACPy copolymers were successfully deprotected by TFA in DCM at room temperature, producing water-soluble cationic copolymers ACPy. The disappearance of the proton peak of the tert-butyl moiety at 1.34 ppm in the 1H NMR spectrum confirmed the successful deprotection of the Boc groups in ACP1, ACP2 and ACP3 (Fig. S9, S10, ESI† and Fig. 1(B); upper curve). Upon deprotection, the copolymers exhibited high positive charge due to the presence of free amine moieties, as confirmed by zeta potential measurements using DLS (Table 2).
| Polymer | CAC (mg L−1)a | D h (nm)b | PDIb | Zeta potential (ξ) (mV)b | FE-SEM size (nm) | TEM size (nm) |
|---|---|---|---|---|---|---|
| a CAC values were determined by fluorescence spectroscopy using pyrene dye. b D h, PDI and surface charges were measured by a DLS instrument at pH 7.4 and 25 °C. | ||||||
| ACP1 | — | 3 ± 1 | 0.320 | +26.7 | — | — |
| ACP2 | 6 | 33 ± 8 | 0.218 | +40.0 | 20 ± 2 | 20 ± 4 |
| ACP3 | 10 | 41 ± 10 | 0.303 | +37.9 | 30 ± 3 | 30 ± 4 |
Self-assembly of polymers
The deprotected copolymers comprise hydrophilic lysine moieties and hydrophobic fatty acid units, providing the necessary amphiphilicity to form self-assembled nanoaggregates in an aqueous medium.47 At first, self-assembled nanoaggregate formation from ACP2–ACP3 was confirmed by CAC measurement using fluorescence spectroscopy by encapsulating pyrene as a hydrophobic dye (Fig. 2(A) and (B)). CAC values of all polymers are summarized in Table 2, where the CAC value of ACP1 could not be determined as it was freely soluble in water due to the absence of hydrophobic fatty acid pendants. The sizes of the ACP1–ACP3 copolymers in PBS buffer (pH 7.4) were investigated by dynamic light scattering (DLS), which are depicted in Fig. S12 (ESI†) and Fig. 2(A) and (B), respectively and summarized in Table 2. Furthermore, it was also noticed that the sizes of the ACP2 and ACP3 nanoaggregates were almost the same. But, in the case of ACP1, a very small size confirms that this polymer did not form self-assembled nanoaggregates due to the absence of the hydrophobic/hydrophilic balance. Then, the hydrodynamic diameter (Dh) of ACP2 and ACP3 was measured in acidic buffer (pH 5.0) and alkaline buffer (pH 9.0). Fig. S13 (ESI†) depicts that in acidic buffer (pH 5.0) and alkaline buffer (pH 9.0), both the polymers (ACP2 and ACP3) showed higher Dh. This is because of hydrolysis of the ester bond at acidic pH,38 and at alkaline pH deprotonation of the primary amine moieties resulted in an insoluble agglomerated structure. The stability of the ACP2 and ACP3 polymer nanoaggregates was also confirmed by DLS measurement of the polymer solution for 5 days (Fig. S14, ESI†). It was observed that there is no significant change in Dh for both polymer nanoaggregates, which suggests the formation of a stable nanoassembly. To investigate the surface charge of the ACP1–ACP3 polymers, zeta potential (ξ) measurement was carried out in three different pH solutions (pH 5.0, pH 7.4 and pH 9.0). At pH 5.0 and pH 7.4, positive ξ values were noticed for the ACP1–ACP3 polymers (Table S2, ESI†). However, ξ values almost close to zero were obtained at pH 9.0 (Table S2, ESI†), because of the deprotonation of the primary amine moieties present in the polymers at pH 9.0.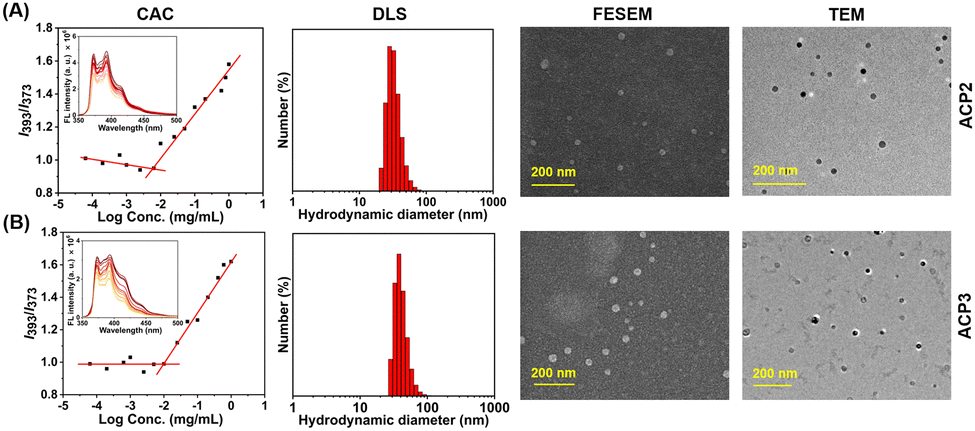 | ||
| Fig. 2 CAC determination, DLS curve, FE-SEM and TEM images of (A) ACP2 and (B) ACP3. | ||
To gain more insights into the morphology of the nanoparticles in an aqueous medium, FE-SEM and TEM images of ACP2 and ACP3 were investigated. In Fig. 2(A) and (B), FE-SEM images confirmed that both the two copolymers, ACP2 and ACP3 form spherical nanoaggregates with an approximate size of 20–30 nm. TEM images of both polymers also were consistent with the FE-SEM data (Fig. 2(A), (B) and Table 2). In Table 2, the sizes obtained from the DLS study are to some extent greater than the sizes determined from microscopy images of the polymers, because the microscopy images were recorded for vacuum-dried samples, whereas the DLS instrument measures the hydrodynamic diameter of solvated polymer nanoaggregates.38 Nevertheless, the spherical nanoparticles could be used to fabricate intelligent drug delivery vehicles to utilize the enhanced permeability and retention (EPR) effect because of their perfect size.49 The high positive zeta potentials (Table 2) of the polymeric nanoparticles were also expected to be a potential candidate for antibacterial activity.16
Enzymatic degradation of polymers
The ACP2 and ACP3 cationic polymers carry lysine and octanoate/myristate pendants, which were attached to the polymers by ester linkages. Aliphatic esters were reported to be degraded by intracellular enzymes, i.e., mammalian cell esterase and bacterial lipase.43 Depending on whether the aliphatic chains are short or long, esterase and lipase enzymes can easily hydrolyze the ester bonds in lipid molecules.50 Recent investigations have also confirmed that E. coli contains considerable levels of the esterase and lipase enzymes necessary for biocatalytic ester hydrolysis in lipids.51 Inspired by the studies on biodegradation of polymers by esterase and lipase enzymes,42,52 we have selected these enzymes for the biodegradation of ACP2 and ACP3. The bacterial extracellular lipase enzymes are expected to degrade the ester linkages of the polymers, causing disassembly of the self-assembled nanostructure (Fig. 3(A)). Therefore, hydrophobic antibacterial drugs can be loaded for on-demand delivery at the specific bacterial microenvironment.53,54 Secondly, the undesired cytotoxic effect caused by cationic polymers traveling through the cell membranes of mammalian cells via the endocytic pathway would be attenuated by biodegradation of the polymers intracellularly by lysosomal esterase enzymes.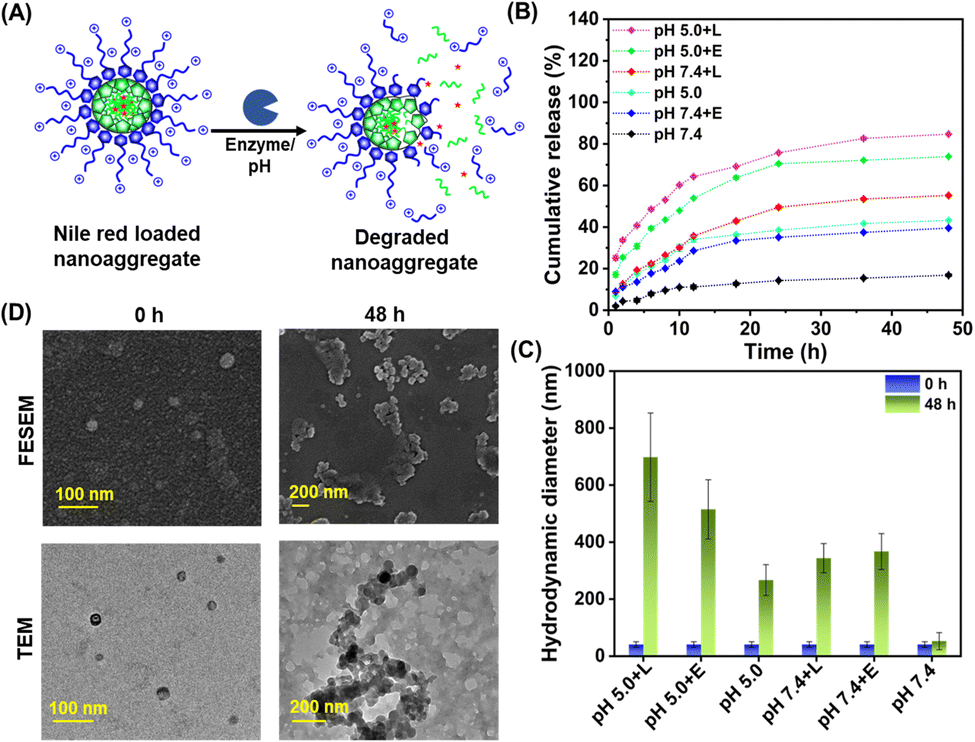 | ||
| Fig. 3 (A) Schematic illustration of the degradation of NR-loaded nanoaggregates in the presence of enzyme and altered pH. (B) Time-dependent cumulative release plot of NR from the interior of ACP3 nanoaggregates in the absence/presence of enzyme and altered pH. (C) Change in hydrodynamic diameter of ACP3 before and after treatment of pH/enzyme. (D) FE-SEM and TEM images of ACP3 nanoaggregates before and after treatment of pH 5.0 and lipase. In (B) and (C), lipase and esterase are represented as L and E, respectively. | ||
To analyze the enzymatic biodegradation of ACP2 and ACP3, NR release kinetics from their nanoaggregates were assessed by the dialysis method.36 During the time of incubation, the side chain appended ester linkages of the polymer were cleaved by the enzyme, resulting in the disassembly of the nanoaggregates and the release of NR from its cavity. Fig. 3(B) depicts the cumulative release of NR from NR-loaded ACP3 polymer nanoaggregates by lipase and esterase enzymes at two different pHs (pH 5.0 and 7.4) for different incubation times. Data from Fig. 3(B) suggest that the percentage of NR release from the polymeric nanoaggregates increased with incubation time in the presence of enzymes and changes in pH. At pH 5.0, the action of lipase enzyme achieved almost 80% dye release. Also, at pH 5.0, a slightly higher percentage of dye release happened than at pH 7.4. This phenomenon can be attributed to the aliphatic ester linkage in the copolymer's side chain being prone to hydrolysis at endo-lysosomal pH (pH 4.5 to 6.5).38 Furthermore, the DLC and DLE were determined to be 1.8% and 15.6%, respectively. Similar observations were also noticed for the NR-loaded ACP2 polymer nanoaggregates (Fig. S15, ESI†), with DLC and DLE values of 1.2% and 12.8%, respectively. This enzymatic biodegradation of ACP3 was also monitored by DLS (Fig. 3(C)). Due to the hydrolysis of the ester bonds, the hydrophobic octanoate/myristate pendants and hydrophilic lysine units were separated from the polymeric backbone. This phenomenon caused the formation of larger agglomerates in the solution, thus increasing the hydrodynamic size of the particle.43 Furthermore, it was confirmed by NMR spectroscopy (Fig. S16, ESI†), FE-SEM and TEM images (Fig. 3(D)) that, after 48 h of incubation, the self-assembled nanoaggregates of the ACP3 polymer degraded with the lipase enzyme at pH 5.0. After treatment with lipase enzyme at pH 5.0, the newly generated peaks at 5.3 ppm and 3.5–4 ppm, suggest the successful degradation of the ACP3 polymer (Fig. S16, ESI†). Control experiments were carried out without enzymes to analyze the leakage of NR from the ACP3 nanoaggregates under identical incubation conditions in PBS at pH 7.4. In 48 h, 40% and 16% NR release were observed at pH 5.0 and 7.4, respectively (Fig. 3(B)). There was a marginal change in Dh at pH 7.4, but ∼260 nm large aggregated particles were observed at pH 5.0 (Fig. 3(C)), possibly due to the pH-induced degradation of ester linkages.38
Cell cytotoxicity assessment by standard MTT and hemolysis assay
Under physiological conditions, the cationic polymers can directly (a) adhere to healthy cells and tissues, (b) hemolyze erythrocytes, and (c) attach with negatively charged bacterial membranes via electrostatic interaction.43 Regardless of the difference in magnitude of the negative potential of erythrocytes, eukaryotic and bacterial cell membranes, all three types of biological membranes have a strong affinity towards cationic polymers due to the electrostatic interactions.43 Thus, it is important to investigate the specific affinity of ACP1–ACP3 cationic polymers towards biological membranes of differential origin. For this, we assessed the degree of cell cytotoxicity of three cationic polymers in human intestinal epithelial cells (INT 407), using varied concentrations of the test polymers (Fig. 4(A)). For all polymers (ACP1–ACP3), the EC50 values (polymer concentration at which 50% cells are viable) are >500 μg mL−1; however, at 1000 μg mL−1 concentration, the cell viability dropped significantly up to 30% for ACP1 and ACP2. The contrast in cell viability exhibited by the three copolymers is presumably due to preferential interaction between ACP1 and ACP2 with the eukaryotic cell membrane than that of ACP3.55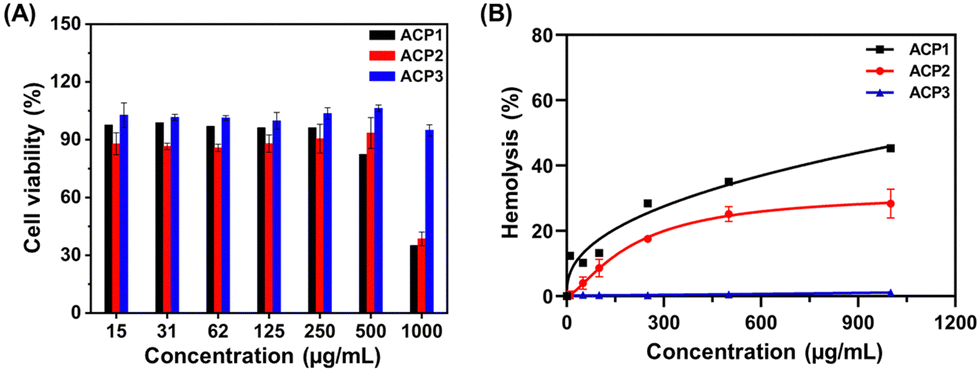 | ||
| Fig. 4 (A) Cytotoxicity analysis of ACP1–ACP3 to the INT 407 cell line. (B) Hemolysis assay of ACP1–ACP3. | ||
The extent of hemolysis delivers key information on the relative rupture of the erythrocyte membrane upon contact with a cationic polymer. Hence, we investigated how ACPy interacts with RBCs using the standard hemolysis assay.43 10% triton X-100 was chosen as the positive control, causing 100% RBC deterioration due to osmosis, whereas PBS was used as the negative control. All the test polymers (ACP1–ACP3) were subjected to a hemolysis assay using increasing polymer concentrations ranging from 10 to 1000 μg mL−1 (Fig. 4(B)). An overall finding revealed that the hydrophobicity variation in the side chains significantly impacted hemolytic activity.55,56 According to ISO/TR 7405-1984(f), if the %hemolysis is less than 5%, the samples are considered nonhemolytic in nature. Samples with %hemolysis values in the 5 to 10% range are categorized as slightly hemolytic, but above 10% are considered extremely hemolytic.47 With increasing hydrophobicity from octanoate (ACP2) to myristate (ACP3), the hemolytic property of the polymer decreased (Fig. 4(B)). ACP2 caused significant hemolysis above 50 μg mL−1 and ACP3 was nonhemolytic in nature even at 1000 μg mL−1. But, ACP1 showed high hemolytic activity at a very low concentration, 10 μg mL−1. Thus, hydrophobic balance is also needed to minimize the cytotoxicity of the polymers to RBCs caused by higher cationic charge. Therefore, ACP1 was excluded from further study because of its high hemolytic behavior.
To evaluate the relative selectivity of the cationic polymers to bacterial cells in comparison with RBCs, the HC50 (polymer concentration at which 50% treated erythrocytes are viable) and MIC50 (polymer concentration that inhibits 50% growth of bacteria) values of ACP2 and ACP3 were determined (Table S1, ESI†). The selectivity index is defined by the HC50/MIC50 factor.43 The maximum concentrations tested for hemolysis activity (1000 μg mL−1) of the polymers were almost 40–90 times greater than the MIC50 value of the polymers. This demonstrates that the polymers' activity was highly selective toward bacterial cell membrane disruption and it is nontoxic to RBCs. This can be explained by the fact that bacterial membranes contain more phospholipid content than RBCs, which contributes to larger anionic potential with better selectivity.57ACP2 and ACP3 exhibit strong electrostatic interactions with the bacterial cell membrane over INT 407 cell lines and RBCs, which results in a high selectivity index (Table S1, ESI†). It should be noted that ACP2 and ACP3 are absolutely nontoxic to both RBCs (HC50 >103 μg mL−1) and INT 407 (EC50 >500 μg mL−1) cells while highly active against bacterial membranes.
Antibacterial activity and mechanism
Polymers with cationic tethered primary ammonium groups on their side chains have been reported to have strong antibacterial activity,58 and these polymers can greatly emulate the amphiphilic characteristics and cationic properties of AMPs.59 The ACP2 and ACP3 cationic polymers with lysine and fatty acid pendants were examined for assessing antibacterial activity against B. subtilis and E. coli using the broth microdilution method (Fig. 5(A) and (B)) and zone of inhibition (ZOI) (Fig. 6 and Table 3). A marked decline in bacterial growth, as evident from the reduction in optical density of the bacterial culture medium (OD600), suggests that ACP2 and ACP3 polymer treatment can effectively restrict bacterial growth. In contrast, a steady and consistent increase in the growth profile of bacteria (grown in the planktonic phase) without the test polymers (control) suggests healthy cells in the stationary phase under experimental circumstances. The MIC50 values of ACP2 and ACP3 for B. subtilis and E. coli are summarized in Table S1 (ESI†). It is evident from Fig. 5(A) that the myristate appended polymer (ACP3) achieved a greater reduction of B. subtilis growth than the shorter chain fatty acid tethered polymer, ACP2. This may be because of differences in hydrophobicity amongst copolymer systems. Along with electrostatic interactions, it is well-established that the hydrophobicity of a polymer has an effect on its antibacterial activity.60,61 Many studies have supported that the insertion of hydrophobic substituents into the polymer would lead to penetrating the cell membrane of the bacteria which causes cytoplasmic leakage and ultimate cell death. But in the case of E. coli, the MIC50 values of both the polymers (ACP2 and ACP3) are almost similar, suggesting that there is no additional role of hydrophobicity of these polymers in exerting an antibacterial effect towards E. coli (Fig. 5(B) and Table S1, ESI†).55 For further assessment of the bactericidal effect, FE-SEM was used to analyze the morphology of the bacterial cells before and after treatment with polymers to understand better how polymeric nanoparticles affect bacterial survival (Fig. 5(C)). FE-SEM images of control B. subtilis and E. coli cells, confirming their rod-like morphology and unaffected membranes. However, upon treatment with polymers both the bacterial cell membranes were found to be disrupted. This is presumably due to the strong interaction between the polymer and the bacterial cell membrane, which disrupts membrane integrity and releases intracellular contents, resulting in cell death. Therefore, ACP2 and ACP3 cationic polymers efficiently inhibit the growth of B. subtilis as well as E. coli via membrane-disrupting pathways. Additional evidence of the bactericidal effect of the positively charged copolymers was further analyzed by the ZOI experiment. Different concentrations of ACP2 and ACP3 polymer (20, 50, 100 and 200 μg mL−1) were cast on designated wells created on sterile Mueller Hinton (MH) agar plates. With the increase of the concentration of the polymer, the ZOI for both the bacterial cultures was also increased, which is depicted in Fig. 6 and summarized in Table 3.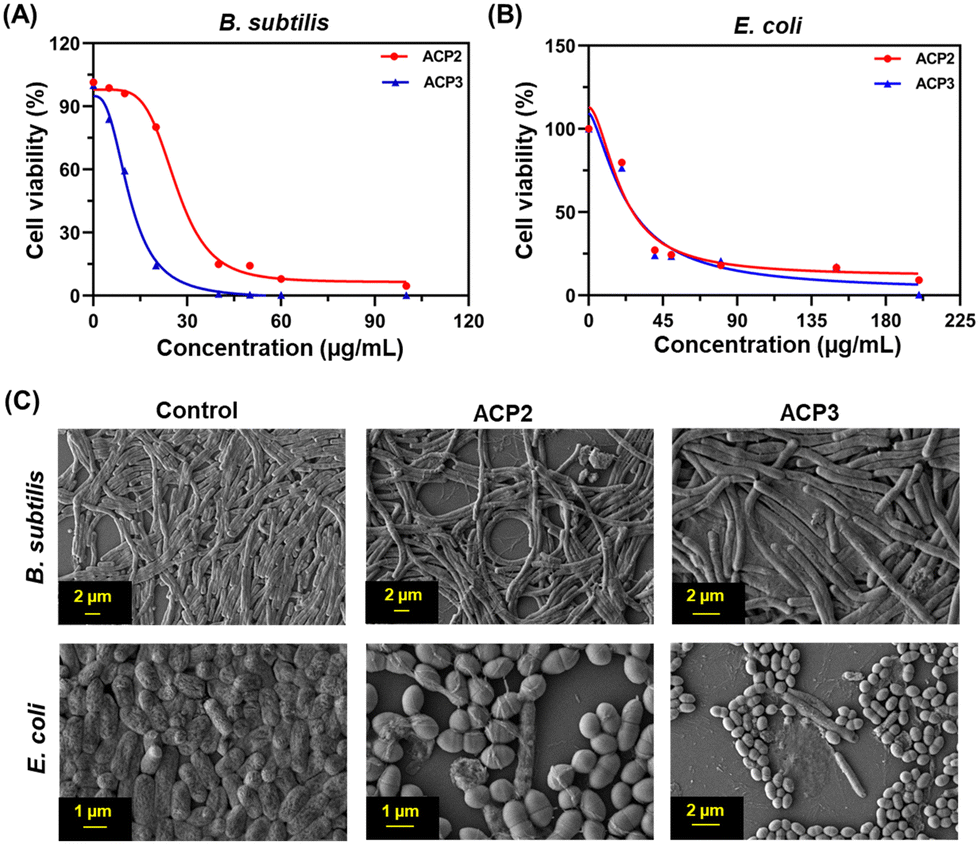 | ||
| Fig. 5 Bactericidal activity of ACP2 and ACP3 for (A) B. subtilis and (B) E. coli. (C) FE-SEM images of the B. subtilis and E. coli controls and those treated with ACP2 and ACP3. | ||
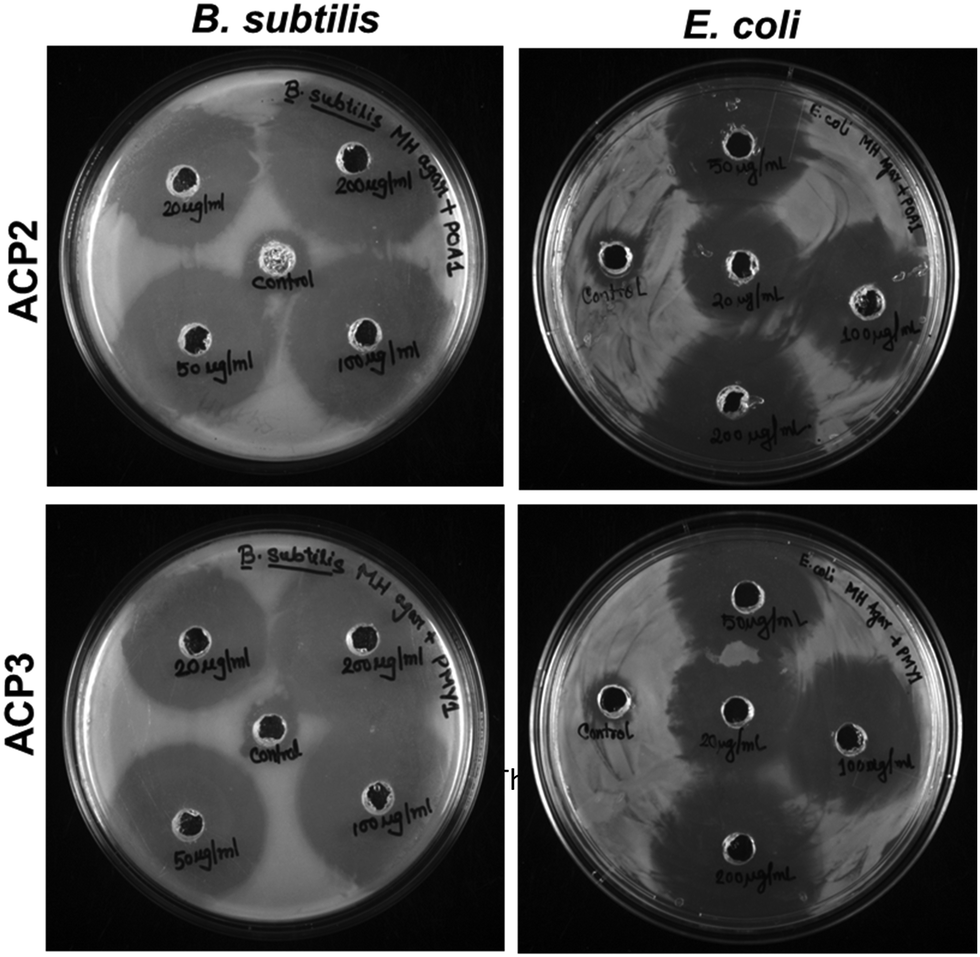 | ||
| Fig. 6 ZOI against B. subtilis and E. coli exhibited by ACP2 and ACP3 using increasing concentrations of both the polymers (20, 50, 100 and 200 μg mL−1). | ||
| Polymer | Conc. (μg mL−1) | Radius of ZOI (cm) (R) | ZOI (cm2)a | ||
|---|---|---|---|---|---|
| B. subtilis | E. coli | B. subtilis | E. coli | ||
| a ZOI = π(R2 − r2), r = radius of well = 0.35 cm. | |||||
| ACP2 | Control | 0.57 | 0.55 | 0.63 | 0.56 |
| 20 | 1.50 | 1.25 | 6.68 | 4.52 | |
| 50 | 1.60 | 1.50 | 7.65 | 6.68 | |
| 100 | 1.75 | 1.70 | 9.23 | 8.68 | |
| 200 | 2.00 | 1.75 | 12.18 | 9.23 | |
| ACP3 | Control | 0.55 | 0.56 | 0.56 | 0.60 |
| 20 | 1.55 | 1.20 | 7.16 | 4.13 | |
| 50 | 1.75 | 1.50 | 9.23 | 6.68 | |
| 100 | 1.80 | 1.65 | 9.79 | 8.16 | |
| 200 | 2.00 | 1.75 | 12.18 | 9.23 | |
In vitro live/dead cell assay by confocal microscopy
The live vs. dead cell population was determined using propidium iodide (PI), which stains dead bacteria cells, while acridine orange (AO) was used to stain both living and dead bacteria cells.62 Based on this principle, the confocal images of bacterial cells showed direct evidence of antibacterial actions of cationic polymers. To demonstrate the bactericidal effect of the polymers, both bacteria (B. subtilis and E. coli) were cultured in MH media, then exposed to the cationic polymers (ACP2 and ACP3) and the images were taken 4 h after staining with AO and PI. In Fig. 7(A) and (B), the first panel (control) shows emission in the AO channel but no emission in the PI channel, which indicates the existence of live bacterial cells. The second and third panels correspond to ACP2 and ACP3 treated cells, suggesting two important observations: (a) significant green emission observed due to the AO intercalation to the double-stranded DNA in bacteria, and (b) the disruption of the bacterial cell membranes by both polymers makes it easier for PI, a membrane-impermeable dye, to enter into the cell and give a bright red fluorescence. The colocalization of red and green fluorescent regions in merged images indicates the dead bacteria as yellowish. Additionally, a few cells in the merged images were only marked in green fluorescence of AO. These observations suggest that both the polymers (ACP2 and ACP3) significantly affected a majority of B. subtilis and E. coli bacteria, attaining their antibacterial activity. | ||
| Fig. 7 Confocal microscopy images of (A) B. subtilis and (B) E. coli cells. Control (first row), incubated with ACP2 (middle row) and ACP3 (bottom row). Cells were incubated for 4 h at 37 °C and stained by AO and PI. | ||
Conclusions
The present investigation of the self-assembled amphiphilic copolymers with lysine and octanoate/myristate pendants has demonstrated antibacterial properties without significant host cell cytotoxicity. Due to the perfect hydrophobic and hydrophilic balance, the deprotected cationic polymers formed self-assembled nanoaggregates in an aqueous solution. The lysine and octanoate/myristate moieties were tethered to the polymer side chain via the ester group of these polymers, enabling side-chain biodegradation in the presence of bacterial lipase and lysosomal esterase enzyme. After biodegradation, hydrophobic/hydrophilic imbalance resulted in the formation of larger agglomerates in solution, facilitating effective payload release.It was also observed that the presence of hydrophobic/hydrophilic balance in these polymers makes them a superior antibacterial material while maintaining nontoxicity towards erythrocytes and human intestinal epithelial cells. The positive surface charge due to lysine units and hydrophobic fatty acid chains is found to be critical for exhibiting antibacterial activity against B. subtilis and E. coli. FE-SEM images of the polymer-treated bacterial cells confirmed irreversible changes in the bacterial surface architecture. The comparative analysis of the live/dead cells by confocal microscopy further suggests the superior antibacterial efficacy of ACP2 and ACP3 polymers. Altogether, the synthesized self-assembled amphiphilic cationic copolymers demonstrated an antibacterial effect and sustained hydrophobic dye release in the presence of enzyme/pH. We envision that our present research on fabricating enzyme/pH-responsive antibacterial polymers will play a pivotal role in developing next-generation antibacterial agents to combat MDR bacteria-mediated disease.
Author contributions
P. D. has designed and supervised the work. D. G. and S. B. have synthesized and characterized the monomers, polymers and further carried out all the biophysical experiments. S. Y. and A. I. M. performed the biological studies. The manuscript was written through the contributions of all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
D. G. and S. B. acknowledge the University Grants Commission (UGC) and Council of Scientific and Industrial Research (CSIR), respectively, for their senior research fellowship (SRF). S. Y. thanks IISER Kolkata for his fellowship (JRF).References
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS.
- J. O’Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance, Welcome Trust, London, UK, 2016 Search PubMed.
- L. J. V. Piddock, Lancet Infect. Dis., 2016, 16, 767–768 CrossRef.
- L. Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS.
- M. Riool, A. de Breij, J. W. Drijfhout, P. H. Nibbering and S. A. J. Zaat, Front. Chem., 2017, 5, 1–13 Search PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 CrossRef CAS.
- H. Moravej, Z. Moravej, M. Yazdanparast, M. Heiat, A. Mirhosseini, M. Moosazadeh Moghaddam and R. Mirnejad, Microb. Drug Resist., 2018, 24, 747–767 CrossRef CAS.
- T. N. Gevrek, K. Yu, J. N. Kizhakkedathu and A. Sanyal, ACS Appl. Polym. Mater., 2019, 1, 1308–1316 CrossRef CAS.
- P. Pham, S. Oliver and C. Boyer, Macromol. Chem. Phys., 2022, 224, 2200226–2200254 CrossRef.
- A. Kumar, J. Sharma, P. Srivastava and L. Nebhani, J. Mater. Chem. B, 2023, 11, 2234–2248 RSC.
- X. Shen, Y. Rao, D. Liu, J. Wang, X. Niu, Y. Wang, W. Chen, F. Liu, L. Guo and H. Chen, J. Mater. Chem. B, 2023, 11, 5786–5793 RSC.
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nüsslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS.
- D. S. S. M. Uppu, S. Samaddar, J. Hoque, M. M. Konai, P. Krishnamoorthy, B. R. Shome and J. Haldar, Biomacromolecules, 2016, 17, 3094–3102 CrossRef CAS.
- P. R. Judzewitsch, L. Zhao, E. H. H. Wong and C. Boyer, Macromolecules, 2019, 52, 3975–3986 CrossRef CAS.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2017, 2, 1633–1644 CrossRef CAS PubMed.
- H. Tang, R. J. Doerksen and G. N. Tew, Chem. Commun., 2005, 1537–1539 RSC.
- A. C. Engler, J. P. K. Tan, Z. Y. Ong, D. J. Coady, V. W. L. Ng, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2013, 14, 4331–4339 CrossRef CAS PubMed.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS.
- R. Liu, X. Chen, S. Chakraborty, J. J. Lemke, Z. Hayouka, C. Chow, R. A. Welch, B. Weisblum, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4410–4418 CrossRef CAS.
- E. F. Palermo, D.-K. Lee, A. Ramamoorthy and K. Kuroda, J. Phys. Chem. B, 2011, 115, 366–375 CrossRef CAS.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2018, 3, 769–780 CrossRef CAS.
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS.
- X. Hou, L. Yang, J. Liu, Y. Zhang, L. Chu, C. Ren, F. Huang and J. Liu, Biomater. Sci., 2020, 8, 6350–6361 RSC.
- İ. S. Goregen and O. Ozay, J. Macromol. Sci., Part A: Pure Appl. Chem., 2023, 1–12 Search PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, 4th edn, Garland Science, New York, 2002, pp. 401–405 Search PubMed.
- J. M. Heather and B. Chain, Genomics, 2016, 107, 1–8 CrossRef CAS.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149–1238158 CrossRef PubMed.
- K. Nishimori and M. Ouchi, Chem. Communλ., 2020, 56, 3473–3483 RSC.
- G. Q. Chen, Z. Q. Wu, J. R. Wu, Z. C. Li and F. M. Li, Macromolecules, 2000, 33, 232–234 CrossRef CAS.
- S. Bag, S. Ghosh, S. Paul, M. E. H. Khan and P. De, Macromol. Rapid Commun., 2021, 42, 2100501 CrossRef CAS PubMed.
- K. Bauri, B. Saha, J. Mahanti and P. De, Polym. Chem., 2017, 8, 7180–7187 RSC.
- B. Saha, N. Choudhury, A. Bhadran, K. Bauri and P. De, Polym. Chem., 2019, 10, 3306–3317 RSC.
- P. Sar, S. Ghosh, Y. D. Gordievskaya, K. G. Goswami, E. Y. Kramarenko and P. De, Macromolecules, 2019, 52, 8346–8358 CrossRef CAS.
- A. Dey, U. Haldar, R. Tota, R. Faust and P. De, J. Macromol. Sci., Part A: Pure Appl. Chem., 2023, 60, 161–170 CrossRef CAS.
- B. Saha, N. Choudhury, S. Seal, B. Ruidas and P. De, Biomacromolecules, 2018, 20, 546–557 CrossRef PubMed.
- S. Paul, S. Pan, A. Chakraborty, P. De and A. Mukherjee, ACS Appl. Polym. Mater., 2021, 3, 2310–2315 CrossRef CAS.
- S. Sahoo, S. Paul, S. Pan, D. Pal, S. Das, S. Maiti, B. Mukhopadhyay, P. Tecilla and P. De, ACS Appl. Polym. Mater., 2023, 5, 1474–1486 CrossRef CAS.
- M. Zheng, M. Pan, W. Zhang, H. Lin, S. Wu, C. Lu, S. Tang, D. Liu and J. Cai, Bioact. Mater., 2021, 6, 1878–1909 CAS.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-C. Wang, B. Sun and J. Wang, J. Am. Chem. Soc., 2012, 134, 4355–4362 CrossRef CAS PubMed.
- R. Ghosh, M. Malhotra, R. R. M. Sathe and M. Jayakannan, Biomacromolecules, 2020, 21, 2896–2912 CrossRef CAS PubMed.
- S. Mete, K. G. Goswami, E. Ksendzov, S. V. Kostjuk and P. De, Polym. Chem., 2019, 10, 6588–6599 RSC.
- S. Desgouilles, C. Vauthier, D. Bazile, J. Vacus, J.-L. Grossiord, M. Veillard and P. Couvreur, Langmuir, 2003, 19, 9504–9510 CrossRef CAS.
- N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2018, 19, 3572–3585 CrossRef CAS PubMed.
- A. Bera, D. Mukhopadhyay, K. Goswami, P. Ghosh, R. De and P. De, Biomater. Sci., 2022, 10, 3466–3479 RSC.
- M. Yu, Y. Gao, A. Ying, L. Li, G. Xie, S. Gong, X. Gao, T. Wang and C. Yang, Macromolecules, 2023, 56, 5381–5389 CrossRef CAS.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- D. Romano, F. Bonomi, M. C. de Mattos, T. de Sousa Fonseca, M. da, C. F. de Oliveira and F. Molinari, Biotechnol. Adv., 2015, 33, 547–565 CrossRef CAS.
- J.-M. Ahn, P. Wentworth, Jr. and K. D. Janda, Chem. Commun., 2004, 364–365 RSC.
- D.-Z. Xu, X.-Y. Sun, Y.-X. Liang, H.-W. Huang, R. Liu, Z.-L. Lu and L. He, Bioconjugate Chem., 2023, 34, 248–256 CrossRef CAS.
- M. Chen, S. Xie, J. Wei, X. Song, Z. Ding and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36814–36823 CrossRef CAS.
- T. Wang, Y. Li, E. J. Cornel, C. Li and J. Du, ACS Nano, 2021, 15, 9027–9038 CrossRef CAS.
- M. M. Konai, S. Barman, R. Issa, S. MacNeil, U. Adhikary, K. De, P. N. Monk and J. Haldar, ACS Infect. Dis., 2020, 6, 703–714 CrossRef CAS.
- P. T. Phuong, S. Oliver, J. He, E. H. H. Wong, R. T. Mathers and C. Boyer, Biomacromolecules, 2020, 21, 5241–5255 CrossRef CAS.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS.
- A. Punia, A. Mancuso, P. Banerjee and N. Yang, ACS Macro Lett., 2015, 4, 426–430 CrossRef CAS.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS.
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., Int. Ed., 2008, 47, 1250–1254 CrossRef CAS.
- G. J. Gabriel, J. A. Maegerlein, C. F. Nelson, J. M. Dabkowski, T. Eren, K. Nüsslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 433–439 CrossRef CAS.
- C. Vipin, K. Saptami, F. Fida, M. Mujeeburahiman, S. S. Rao, Athmika, A. B. Arun and P. D. Rekha, PLoS One, 2020, 15, e0241304 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C and HRMS of the monomers; 1H NMR spectra of protected and deprotected polymers; 13C NMR of BACP3; 1H NMR spectra of the degradation of ACP3 polymer by lipase enzyme and at pH 5.0; DLS of ACP1; stability of the polymer nanoaggregates (ACP2 and ACP3) by DLS, cumulative release of NR from ACP2 in the presence of enzyme and pH, and table containing HC50, EC50, MIC50 and selectivity index. See DOI: https://doi.org/10.1039/d3tb02801a |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |