
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-crosslinkable poly(urethane)-based hydrogels relying on supramolecular interactions and light-initiated polymerization: promising tools for advanced applications in drug delivery†
Alessandro
Torchio‡
 a,
Monica
Boffito‡
*ab,
Rossella
Laurano
a,
Claudio
Cassino
c,
Mario
Lavella
d and
Gianluca
Ciardelli
*ae
a,
Monica
Boffito‡
*ab,
Rossella
Laurano
a,
Claudio
Cassino
c,
Mario
Lavella
d and
Gianluca
Ciardelli
*ae
aDepartment of Mechanical and Aerospace Engineering, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy. E-mail: monica.boffito@polito.it; gianluca.ciardelli@polito.it
bInstitute for Chemical-Physical Processes, National Research Council (CNR-IPCF), Via G. Moruzzi 1, 56124, Pisa, Italy
cDepartment of Science and Technological Innovation, Università del Piemonte Orientale “A. Avogadro”, Viale Teresa Michel 11, 15121 Alessandria, Italy
dDepartment of Management, Information and Production Engineering, Università degli Studi di Bergamo, Viale G. Marconi, 5, 24044 Dalmine, BG, Italy
eDepartment of Life Sciences, University of Modena and Reggio Emilia, Via Campi 287, 41125 Modena, Italy
First published on 2nd July 2024
Abstract
Physical and chemical hydrogels are promising platforms for tissue engineering/regenerative medicine (TERM). In particular, physical hydrogels are suitable for use in the design of drug delivery systems owing to their reversibility and responsiveness to applied stimuli and external environment. Alternatively, the use of chemical hydrogels represents a better strategy to produce stable 3D constructs in the TERM field. In this work, these two strategies were combined to develop multi-functional formulations integrating both drug delivery potential and TERM approaches in a single device. Specifically, a novel photo-sensitive poly(ether urethane) (PEU) was developed to form supramolecular networks with α-cyclodextrins (α-CDs). The PEU was successfully synthesized using Poloxamer® 407, 1,6-diisocyanatohexane and 2-hydroxyethyl methacrylate, as assessed by infrared spectroscopy, size exclusion chromatography and proton nuclear magnetic resonance (1H NMR) spectroscopy. Subsequently, PEU thermo-responsiveness was characterized through critical micelle temperature evaluation and dynamic light scattering analyses, which suggested the achievement of a good balance between molecular mass and overall hydrophobicity. Consequently, the formation of supramolecular domains with α-CDs was demonstrated through X-ray diffraction and 1H NMR spectroscopy. Supramolecular hydrogels with remarkably fast gelation kinetics (i.e., few minutes) were designed using a low PEU concentration (≤5% w/v). All formulations were found to be cytocompatible according to the ISO 10993-5 regulation. Notably, the hydrogels were observed to possess mechanical properties and self-healing ability, according to rheological tests, and their fast photo-crosslinking was evidenced (<60 s) by photo-rheology. A high curcumin payload (570 μg mL−1) was encapsulated in the hydrogels, which was released with highly tunable and progressive kinetics in a physiological-simulated environment for up to 5 weeks. Finally, a preliminary evaluation of hydrogel extrudability was performed using an extrusion-based bioprinter, obtaining 3D-printed structures showing good morphological fidelity to the original design. Overall, the developed hydrogel platform showed promising properties for application in the emerging field of regenerative pharmacology as (i) easily injectable drug-loaded formulations suitable for post-application stabilization through light irradiation, and (ii) biomaterial inks for the fabrication of patient-specific drug-loaded patches.
Introduction
Physical hydrogels are extremely interesting systems owing to their significant reversibility and processability. Indeed, the main advantage of physically crosslinked networks is represented by the possibility to easily perform injections and adapt them to external morphologies. Another advantage of these systems is their high tunability. Nonetheless, they are associated with some limitations. Most importantly, the development of stable and durable hydrogel networks based on physical interactions is challenging. Indeed, a relevant number of weak crosslinks is necessary to properly stabilize the polymeric network in an aqueous environment. Furthermore, the indispensable hydrophilicity that is characteristic of these systems induces the absorption of fluids from the external environment. This natural process can lead to mass exchange with the external environment, and complete dissolution of the system may occur within few days. Although this behavior is ideal for most drug delivery applications since several therapeutic drugs are characterized by high hydrophobicity and low bioavailability and hence need specific carriers for their delivery, it represents a strong limitation in the prolonged administration of therapies. This issue is well-known and has been widely discussed by various authors.1,2Accordingly, a possible solution to overcome this drawback is the development of chemically crosslinked hydrogel networks. However, this strategy is generally associated with low responsiveness and limited handling.3 Moreover, chemical additives are often necessary to carry out the crosslinking process, which may induce toxic effects.4
In this case, a relatively broader approach involves the integration of both the physical and chemical crosslinking methods in a single hydrogel formulation with the ultimate goal of combining the advantages of both strategies, while reducing the related deficiencies. In this regard, the appropriate selection of the chemical composition and the resulting physical properties are essential factors to successfully develop hybrid strategies based on both chemical and physical crosslinking. Indeed, the tuning of the physical interactions originating from the constituent polymeric units in combination with additional chemical crosslinking can result in highly stiff and stable hydrogels, as reported, for example, in the work by Li et al., in which a hydrogel system characterized by an elastic modulus around 5 MPa and a strength equal to 2.5 MPa was described.5 In detail, the authors discussed how hydrogel network organization and the nature of the occurring interactions account for the development of formulations with a remarkable mechanical response. In this work, they developed a hydrogel network stabilized through crystalline (physical) and covalent (chemical) domains using poly(vinyl alcohol) (PVA) to form crystallites and poly(acrylamide) (PAA) to generate chemical bonds. Consequently, they obtained a hydrogel system with mechanical properties very similar to that of natural tissues, such as cartilage and skin. In this case, chemical crosslinking was pursued through the overnight in situ polymerization of acrylamide in the presence of N,N′-methylenebisacrylamide (MBAA) and tetramethyl-ethylenediamine (TEMED) at room temperature. This approach can be considered a “macromolecular” strategy, in which the organization of the resulting network is based on standard intermolecular interactions.
Nonetheless, a more sophisticated and versatile approach can also be pursued relying on supramolecular (SM) self-assembly. In this case, the initial processability of physical hydrogels is preserved due to the adaptivity of the SM networks and subsequent in situ stabilization can be performed through chemical crosslinking. Hence, highly organized SM networks can be ad hoc engineered to exhibit appropriate functionalities for further and fast chemical stabilization, which induces mechanical strengthening and improves their resistance to solubilization. In this regard, the chemical composition of all the components of the derived hydrogel systems is even more important in terms of tuning and engineering. An interesting example describing this approach can be found in the work conducted by Zhao and co-authors.6 In this case, a modified di-acryloyl block co-polymer was synthesized using Pluronic® F68 (also commercially available as Poloxamer® 188, P188) and poly(ε-caprolactone) (PCL) with the aim to exploit its photo-sensitivity to ensure fast polymerization after complete self-assembly into poly(pseudo)rotaxanes (PPRs) with α-cyclodextrins (α-CDs).7–12 The resulting hydrogel systems were characterized by notably high mechanical properties (i.e., storage modulus (G′) greater than 100 kPa) and good responsiveness in aqueous environments (i.e., the equilibrium swelling ratio of the developed formulations was dependent on the molar feed ratio of α-CDs to the macromer). Another similar work was conducted by the same authors utilizing the same macromer in the presence of β-CDs, thus further proving the versatility of appropriately synthesized amphiphilic polymers in enabling the ad hoc engineering of hydrogels best matching the specific requisites of each envisaged application.13 In contrast, Wei and co-workers developed an interesting system composed of star-shaped photo-curable macromers and α-CDs.14 In detail, they synthesized a methacryloyl-terminated poly(L-lactide)-b-poly(ethylene glycol) (PLA-b-PEG) copolymer consisting in three or four arms and formulated SM hydrogels by adding α-CDs in aqueous solution. The presence of photocurable domains was demonstrated to improve mechanical strength and stability of the derived SM networks in an aqueous environment. Moreover, these systems showed a highly tunable responsiveness in aqueous environment, reaching a swelling ratio of up to 500% within 7 days of incubation in an aqueous medium. The same research group developed other similar systems based on the same strategy.15,16
In the last decade, although few research works focused on the specific design of photo-crosslinkable PPR-based networks, the general importance of developing shear-thinning and self-healing hydrogels retaining potential for secondary crosslinking has attracted continuously increasing interest. Indeed, the combined possibility to easily process a hydrogel matrix that is intrinsically self-sustaining, and then further stabilize it through fast photo-crosslinking also represents an extremely important feature for the development of bioinks and biomaterial inks for extrusion-based 3D printing. The most recent and relevant strategies underpinning this idea have been reviewed and critically analyzed by many researchers.17–19 Specifically, in these publications, the authors debated the key role played by mechanical features in the development of highly effective and functional hydrogel bioinks and biomaterial inks. In this regard, supramolecular-based matrices represent one of the most promising strategies to ensure self-healing and shear-thinning responses, which are elemental features for injection or printing of even cell-laden hydrogels.
Accordingly, based on the promising results reported in our previous publications,20,21 this work was conceived with the aim of designing a new platform of double-crosslinkable hydrogels relying of physical SM interactions and chemical bonds resulting from light-initiated radical polymerization. Specifically, herein, novel SM and photo-curable systems based on a new synthesized poly(ether urethane) (PEU) were developed. In fact, ad hoc designed PEUs retain huge potential as materials for the fabrication of medical devices due to their LEGO-like chemical structure. This peculiarity has been widely exploited in previous studies aiming to properly fulfil various technical and clinical needs.22–28 In detail, our previous findings indicated that Poloxamer® 407 (P407)-based PEUs are the best materials for the formation of PPR-based networks with α-CDs due to their balanced match among molecular mass, hydrophobicity and overall macromolecular functionality induced by chemical domains and structures. Based on these results, a new photo-sensitive PEU based on P407 and 1,6-hexamethylene diisocyanate (HDI) was synthesized by end-capping the pre-polymer chains produced at the end of the first step of the reaction through the addition of 2-hydroxyethyl methacrylate (HEMA). Consequently, a further improvement in the gelling ability of the resulting formulations was expected due to the significant reduction in polymers molar mass and concomitant preservation of the hydrophobic character derived from the partial polymerization reaction. The proposed approach has the advantage of combining both the potential to form physical hydrogels with α-CDs and the exposure of pendant photo-curable moieties in a single polymeric backbone. Subsequently, complete physico-chemical characterization of the synthesized PEU was performed through size exclusion chromatography (SEC) and attenuated total reflectance–Fourier transformed infrared (ATR-FTIR) spectroscopy, and proton nuclear magnetic resonance (1H NMR) spectroscopy. The assessment of PEU thermo-responsiveness was conducted through dynamic light scattering (DLS) analyses and evaluation of the critical micelle temperature (CMT) of its aqueous solutions. Then, a new platform of photo-curable SM hydrogels was designed by mixing PEU and α-CD solutions and the most promising formulations were selected for rheological and photo-rheological characterization, as well as cytotoxicity evaluation according to the ISO 10993-5 regulation. Subsequently, curcumin was encapsulated within these novel PEU-based hydrogels as a notable example of highly hydrophobic (water solubility of approx. 3 × 10−8 M)29 and environment-sensitive (poor chemical stability under physiological conditions) drugs.30–32 The release profiles of curcumin from the physical and photo-cured hydrogels were obtained through incubation in an aqueous medium in highly destabilizing and physiological-like conditions (i.e., in phosphate buffered saline solution at pH 7.4 and 37 °C). Finally, the suitability of the developed formulations for rapid prototyping purposes was preliminary evaluated using a commercial extrusion-based 3D-printing set-up. In summary, this work was conceived to prove the versatility of the PEU synthesis process as an effective tool to produce highly functional and tunable hydrogel systems for potential application as carriers of therapeutic agents in drug delivery and the emerging field of regenerative pharmacology.
Experimental
Materials
Poloxamer® 407 (P407, 12
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 Da, 70% w/w poly(ethylene oxide)), 1,6-hexamethylene diisocyanate (HDI), 2-hydroxyethyl methacrylate (HEMA), dibutyltin dilaurate (DBTDL) and 3-(trimethylsilyl)propionic-2,2,3,3-d 4 acid sodium salt (TSP) were obtained from Merck/Sigma-Aldrich (Milan, Italy). α-CDs (herein, simply indicated with the acronym “CDs”) and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, a photo-initiator) were purchased from TCI Chemicals Europe (Zwijndrecht, Belgium). All solvents were purchased from Carlo Erba Reagents (Milan, Italy) in analytical grade and used as received unless otherwise stated. Before the synthesis, P407 was dried under mild vacuum (ca. 150 mbar) at 100 °C for 8 h, and then kept at 40 °C under vacuum until use. HEMA was dried and stored in a desiccator at low pressure (approx. 1–5 mbar) and room temperature, while 1,2-dichloroethane (DCE) was dehydrated over activated molecular sieves (3 Å, Sigma Aldrich, Milan, Italy) under an N2 flow. HDI was distilled under reduced pressure to completely remove moisture and stabilizers.
600 Da, 70% w/w poly(ethylene oxide)), 1,6-hexamethylene diisocyanate (HDI), 2-hydroxyethyl methacrylate (HEMA), dibutyltin dilaurate (DBTDL) and 3-(trimethylsilyl)propionic-2,2,3,3-d 4 acid sodium salt (TSP) were obtained from Merck/Sigma-Aldrich (Milan, Italy). α-CDs (herein, simply indicated with the acronym “CDs”) and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, a photo-initiator) were purchased from TCI Chemicals Europe (Zwijndrecht, Belgium). All solvents were purchased from Carlo Erba Reagents (Milan, Italy) in analytical grade and used as received unless otherwise stated. Before the synthesis, P407 was dried under mild vacuum (ca. 150 mbar) at 100 °C for 8 h, and then kept at 40 °C under vacuum until use. HEMA was dried and stored in a desiccator at low pressure (approx. 1–5 mbar) and room temperature, while 1,2-dichloroethane (DCE) was dehydrated over activated molecular sieves (3 Å, Sigma Aldrich, Milan, Italy) under an N2 flow. HDI was distilled under reduced pressure to completely remove moisture and stabilizers.
Synthesis of the photo-sensitive poly(ether urethane)
The photo-sensitive PEU used in this work was synthesized in two steps according to the protocol reported in our previous works with slight modifications.21,23,25 In detail, in the first step of the synthesis, a prepolymer was produced by reacting P407 (20% w/v) with HDI (2:1 molar ratio with respect to P407) for 2.5 h at 80 °C in anhydrous DCE under a nitrogen atmosphere. Differently, the second step of the reaction involved the end-capping of the isocyanate-terminated prepolymer chains through the addition of HEMA (3% w/v in anhydrous DCE, 2:1 molar ratio with respect to P407). The reaction was conducted for 3 h at room temperature under stirring and protected from any source of light to avoid the occurrence of side reactions. Then, the reaction was stopped through the addition of methanol to passivate any exposed and unreacted isocyanate groups. The solution containing the end-capped PEU was precipitated in petroleum ether (4:1 volume ratio with respect to total DCE volume) at room temperature under vigorous agitation and in the dark. The supernatant was separated from the precipitated PEU, which was kept under a fume hood overnight to allow drying. Subsequently, the dried PEU was solubilized again in DCE at 20% w/v concentration and purified through precipitation in a mixture of diethyl ether and methanol (98:2 v/v, 5:1 volume ratio with respect to DCE volume) in the dark. The purified PEU was collected through centrifugation (Hettich, MIKRO 220R, Tuttlingen, Germany) at 0 °C and 6000 rpm for 20 min and dried under a fume hood overnight. The resulting dried polymer was collected and stored under vacuum at 3 °C in the dark until use. The obtained PEU was denoted as HHP407, where the first H refers to the chain extender (HEMA), the second H identifies the diisocyanate (HDI) and P407 indicates the macrodiol.
Physico-chemical characterization of HHP407
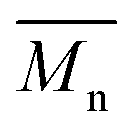 ), weight average molar mass (
), weight average molar mass (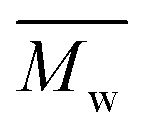 ) and dispersity index (
) and dispersity index (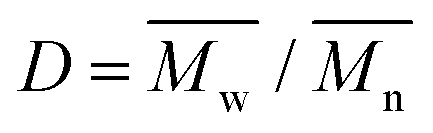 ) were calculated using Microsoft Excel (Office 365, Microsoft Corporation, USA) and a specific calibration curve obtained from poly(ethylene oxide) (PEO) standards with peak molecular weight (Mp) ranging from 982 to 205500 Da. For sample preparation, 2 mg of PEU were dissolved in 1 mL of DMF with LiBr added and filtered using poly(tetrafluoroethylene) (PTFE) syringe filters (0.45 μm, LLG International, Meckenheim, Germany).
) were calculated using Microsoft Excel (Office 365, Microsoft Corporation, USA) and a specific calibration curve obtained from poly(ethylene oxide) (PEO) standards with peak molecular weight (Mp) ranging from 982 to 205500 Da. For sample preparation, 2 mg of PEU were dissolved in 1 mL of DMF with LiBr added and filtered using poly(tetrafluoroethylene) (PTFE) syringe filters (0.45 μm, LLG International, Meckenheim, Germany).
Investigation of the thermo-responsiveness of HHP407 aqueous solutions
Preparation and characterization of HHP407- and CD-based SM complexes
| Recovery (%) = 100 − (winitial − wfinal)/winitial × 100 | (1) |
Preparation and characterization of HHP407- and CD-based SM hydrogels
Self-healing tests were also executed to evaluate the physical reversibility of the hydrogels when a cyclic strain was applied over time at 37 °C, according to the protocol described by Wu and co-workers with few modifications.35 In detail, a constant strain (0.1%, 1 Hz, recovery phase) was initially applied to the SM hydrogels for 120 s; subsequently, complete rupture of the hydrogel network was caused by application of a remarkably higher deformation (100%, 1 Hz, rupture phase) for 60 s. This cyclic strain was applied on each hydrogel system trice, and finally the starting strain (0.1%, 1 Hz, recovery phase) was again applied to estimate the residual mechanical properties of each formulation.
Moreover, due to the photo-responsive nature of HHP407, a photo-rheological characterization was also performed on the same samples. In detail, the storage and loss modulus (G′ and G′′, respectively) trends of the SM hydrogels was registered as a function of time in oscillation (1 Hz, 0.1% strain, 0 N normal force) for 1 min before UV irradiation. Then, UV light exposure was applied for 1 min at 365 nm and 10 mW cm−2 and the parameters were registered for additional 2 min to assess the overall stability of the photo-crosslinked networks.
Preparation of hydrogel extracts and cyclodextrin solutions. Samples for cytotoxicity tests were prepared by weighing 250 mg of self-assembling HHP407-based hydrogels in Bijou sample containers. Three different conditions were tested to isolate the contribution of the main features of the hydrogels (i.e., solvent composition and strategy for hydrogel crosslinking), as summarized in Table 1. The samples were acclimatized at 37 °C for 15 min, 2.5 mL (i.e., 1 mL every 100 mg of hydrogel) of complete medium (37 °C) were gently added and the systems were incubated for 24 h at 37 °C. Then, the eluates were withdrawn and collected for the evaluation of cell cytotoxicity and viability. Extract sterility was ensured through filtration with 0.22 μm poly(ether sulfone) filters (Carlo Erba Reagents, Milan, Italy).
Evaluation of cell cytotoxicity and cell viability. Before cell seeding, 96-well plates were treated to improve cell adhesion. Specifically, gelatin type A (Sigma Aldrich/Merck, Milan, Italy) was first dissolved at a concentration of 1 mg mL−1 in sterile ddH2O; then, the solution was filtered through 0.22 μm poly(ether sulfone) filters (Carlo Erba Reagents, Milan, Italy), and lastly, it was added to a well plate at 50 μL well−1. Subsequently, the plates were incubated at 37 °C for 30 min, UV-sterilized, and finally, they were kept in a sterile cabinet overnight to allow complete water evaporation. Afterwards, NIH-3T3 murine fibroblasts were first collected through trypsinization, re-suspended in complete medium and seeded at 10
000 cells well−1, as recently reported by Laurano et al.36 Then, the plates were incubated under normal culture conditions for 24 h to allow adherence. Subsequently, the medium was removed and replaced with 100 μL of hydrogel extract or CD solution and incubated for 24 h. Cell viability/cytotoxicity was first qualitatively assessed through the Live/Dead assay performed following the procedure suggested by the supplier. Briefly, the supernatant was removed and the cells were washed with Dulbecco's Phosphate-Buffered Saline (DPBS). Subsequently, 100 μL of Live/Dead working solution (i.e., 2 μM Calcein AM and 4 μM ethidium homodimer-1 in DPBS) were added to each well and the plate was incubated for 20 min at 37 °C. Lastly, live (green) and dead (red) cells were imaged under a fluorescence microscope (Leica DM IL LED Fluo, Leica Mycrosystems, Varese, Italy). Cell cytotoxicity was quantitatively investigated by measuring the amount of lactate dehydrogenase released in the medium through the CytoTox-ONE™ homogeneous membrane integrity assay (Promega) according to the manufacturer's instructions. Briefly, the 96-well plate was first equilibrated at room temperature for 20 min, and then 100 μL of CytoTox-ONE reagent were added to each well. After 10 min of reaction, 50 μL of stop solution were added to each well, and lastly the fluorescence was quantified through a multimode plate reader (VICTOR X3, PerkinElmer) at Ex/Em 535/590 nm. Cell viability was quantitatively evaluated by measuring the cell metabolic activity through the CellTiter-Blue® cell viability assay (Promega) following the protocol provided by the manufacturer. Briefly, 20 μL of CellTiter-Blue® solution were added to each well and the 96-well plate was incubated at 37 °C, 5% CO2 for 3 h. Then, the fluorescence was measured through a multimode plate reader (VICTOR X3, PerkinElmer) at Ex/Em 535/590.
For all the experiments, cells cultured in complete medium and cells treated with a lysing solution were used as the negative and positive controls, respectively. Cytotoxicity and cell viability percentages were calculated with respect to the positive and negative control, respectively. Tests were performed in triplicate and results reported as the average value ± standard deviation.
Curcumin encapsulation within SM gels, characterization and release studies. A curcumin (Cur) suspension with a concentration of 1 mg mL−1 was prepared in a CD solution at 14% w/v in PBS/LAP. Ultrasonication (52 W, 20 kHz, Vibracell VCX130, Sonics, USA) was applied for 3 min using a probe and a water-ice bath to avoid evaporation phenomena. The stability of the obtained suspension was evaluated by visual inspection at room temperature (25 °C) to define the temporal window for practical use. Then, this suspension was added to HHP407-based solutions in PBS/LAP, thus obtaining SM hydrogels at specific HHP407, CD and Cur contents. Incubation at 25 °C was carried overnight out to ensure complete sample gelation.
The entire set of rheological and photo-rheological characterizations previously described was also conducted on Cur-loaded SM-based hydrogels to evaluate the effects of drug loading on the development of SM networks and UV light responsiveness.
SM hydrogels (0.5 mL) for release studies of Cur were prepared in glass vials (10 mm inner diameter, 4 mL volume capacity, Sigma Aldrich, Milan, Italy) and denoted as HHP407 X%–CD Y% Cur, where X and Y indicate the PEU and CD concentrations (% w/v), respectively. For all the selected formulations, two sets of 5 samples each were prepared to evaluate the behavior of the purely physical hydrogel networks and the additional contribution of photo-crosslinking. Upon UV light exposure (1 min at 365 nm and 10 mW cm−2), the samples were identified with the acronym HHP407 X%–CD Y% Cur UV. Release studies were conducted on SM hydrogels at 37 °C after acclimatization for 15 min. Then, 1 mL of PBS (37 °C) was gently added on top of each hydrogel sample. At precise time steps (2, 4, 6, 24 h and 3, 4, 7, 14, 21, 28 and 35 days), eluates were withdrawn and completely refreshed with PBS. Cur quantification was performed using a UV/Vis spectrometer (PerkinElmer Lambda 25 UV/VIS spectrometer, Waltham, MA, USA) by evaluating the absorbance peak at around 430 nm. Reference curves were obtained by specific standard solutions produced through hydrogel dissolution and subsequent dilution (Cur concentration range of 1 to 20 μg mL−1).
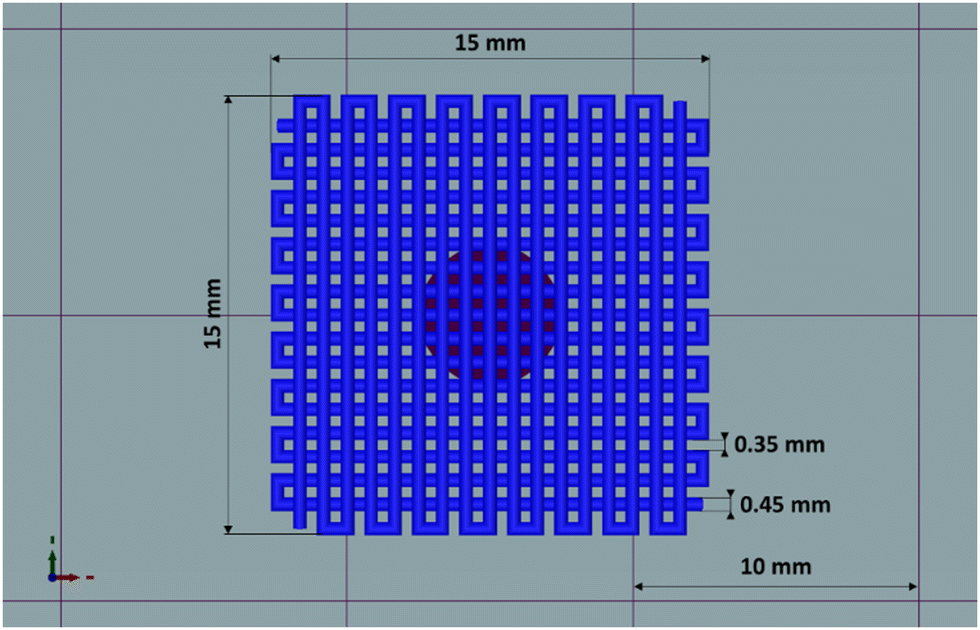 | ||
| Fig. 1 Top view of the CAD model used to evaluate the 3D-extrudability of the HHP407-based hydrogel. | ||
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 8 for Windows 10 (GraphPad Software, La Jolla, CA, USA; https://www.graphpad.com). Two-way ANOVA analysis coupled with the Bonferroni multiple comparison test was performed to compare results, assessing the statistical significance according to Boffito et al.23 The significance level was assigned depending on the p-values, as follows: p < 0.0001 extremely significant (****), 0.0001 < p < 0.001 extremely significant (***), 0.001 < p < 0.01 very significant (**), 0.01 < p < 0.05 significant (*), and p ≥ 0.05 not significant (ns).Results and discussion
Physico-chemical characterization of HHP407
The ATR-FTIR spectra of HHP407 and P407 are presented in Fig. S1 (ESI†). The appearance of the typical vibrational bands of the urethane domains in the HHP407 spectrum proved its successful synthesis. In detail, the peak at around 1720 cm−1 is ascribed to the stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O domains, while the peak at 1530 cm−1 is attributed to the simultaneous bending and stretching of the N–H and C–N chemical regions, respectively. Moreover, the typical broad vibrational band at around 3350 cm−1 corresponds to the N–H stretching. The absence of a signal at ca. 2200 cm−1 suggests the complete conversion of the isocyanate groups of HDI. In addition, the characteristic vibrational bands of P407 were detected at 1250 cm−1 (CH2 stretching), 2880 cm−1 (CH2 rocking) and 1100 cm−1 (C–O–C stretching). Nonetheless, no peaks due to the end-capping molecule (i.e., HEMA) were detectable, probably because they overlapped with the peaks related to the macrodiol and in-chain urethane domains. The SEC results provided
O domains, while the peak at 1530 cm−1 is attributed to the simultaneous bending and stretching of the N–H and C–N chemical regions, respectively. Moreover, the typical broad vibrational band at around 3350 cm−1 corresponds to the N–H stretching. The absence of a signal at ca. 2200 cm−1 suggests the complete conversion of the isocyanate groups of HDI. In addition, the characteristic vibrational bands of P407 were detected at 1250 cm−1 (CH2 stretching), 2880 cm−1 (CH2 rocking) and 1100 cm−1 (C–O–C stretching). Nonetheless, no peaks due to the end-capping molecule (i.e., HEMA) were detectable, probably because they overlapped with the peaks related to the macrodiol and in-chain urethane domains. The SEC results provided  equal to 22 kDa and a dispersity index of 1.7. The low D value proved the synthesis of a PEU with a narrow molecular weight distribution as a result of the good control of the polymerization procedure. The 1H NMR characterization of HHP407 in D2O was performed to detect the presence of HEMA domains, thus proving the effectiveness of the end-capping procedure. As shown in Fig. 2, the typical peaks of HEMA were detected at 6.15, 5.85 and 1.95 ppm. Moreover, the end-capping procedure produced polymeric chains containing approximately 1.25–1.5 HEMA units per chain (i.e., up to 75% end-capping yield), where the maximum theoretical value is 2 (i.e., 100% end-capping yield). This result indicates a reliable end-capping reaction during the second step of PEU synthesis. The thermo-sensitive behavior of HHP407-based solutions (1% w/v concentration) was evaluated in PBS and ddH2O. Fig. S2 and S3 (ESI†) present the CMT curves (i.e., absorbance at 356 nm vs. temperature) and the DLS volume patterns, respectively, for HHP407 solutions with a concentration of 1% w/v. Interestingly, HHP407 solubilized in pure water showed a CMT value of 22.2 °C, while the same formulation prepared in PBS was characterized by a CMT equal to 19.6 °C. This relevant difference can be ascribed to the salting-out effect generated by the presence of dissolved PBS ions, similar to that reported for previously studied P407-based PEUs.21 Interestingly, HHP407 was characterized by comparable CMT values to that of chain-extended P407-based PEUs at an equal concentration (i.e., 1% w/v), although its molecular mass was significantly lower (i.e., around 30%).33 The correlation between these results suggests an enhancement in the thermo-induced self-assembly of the HHP407 chains into micelles probably originated from a better balance between the overall hydrophobicity and length of the polymer chains. It is also plausible that the presence of end-methacrylate groups could induce a significant contribution in this regard. The DLS measurements showed that both analyzed samples underwent polymer chain organization into micellar structures, with some differences ascribed to the solubilization medium, and in particular the absence of dissolved ions in the sample prepared in ddH2O. Indeed, at 25 °C the micelle hydrodynamic diameter was measured to be 18.5 ± 5.2 nm and 34.5 ± 3.5 nm for HHP407 solubilized in ddH2O and PBS, respectively. Irrespective of the solubilization medium, the micelle hydrodynamic diameter increased with an increase in temperature to 37 °C, reaching 41.2 ± 7.3 nm and 44.6 ± 5.8 nm for the samples prepared in ddH2O and PBS, respectively (statistically significant increase for the sample prepared in ddH2O, p = 0.0118). Interestingly, polymer solubilization in PBS produced significantly large micelles at 25 °C (statistically significant difference, p = 0.0115), in accordance with the lower CMT value estimated for this sample, which could initiate the micellization process at lower temperature. Conversely, when HHP407 was solubilized in ddH2O, the same micelle dimensions were achieved with an increase in temperature to 37 °C (34.5 ± 3.5 nm for HHP407 1% w/v in PBS at 25 °C vs. 41.2 ± 7.3 nm for HHP407 1% w/v in ddH2O at 37 °C, not statistically significant difference), in accordance with the higher temperature needed for this sample to initiate the polymeric chain organization into micellar structures.
equal to 22 kDa and a dispersity index of 1.7. The low D value proved the synthesis of a PEU with a narrow molecular weight distribution as a result of the good control of the polymerization procedure. The 1H NMR characterization of HHP407 in D2O was performed to detect the presence of HEMA domains, thus proving the effectiveness of the end-capping procedure. As shown in Fig. 2, the typical peaks of HEMA were detected at 6.15, 5.85 and 1.95 ppm. Moreover, the end-capping procedure produced polymeric chains containing approximately 1.25–1.5 HEMA units per chain (i.e., up to 75% end-capping yield), where the maximum theoretical value is 2 (i.e., 100% end-capping yield). This result indicates a reliable end-capping reaction during the second step of PEU synthesis. The thermo-sensitive behavior of HHP407-based solutions (1% w/v concentration) was evaluated in PBS and ddH2O. Fig. S2 and S3 (ESI†) present the CMT curves (i.e., absorbance at 356 nm vs. temperature) and the DLS volume patterns, respectively, for HHP407 solutions with a concentration of 1% w/v. Interestingly, HHP407 solubilized in pure water showed a CMT value of 22.2 °C, while the same formulation prepared in PBS was characterized by a CMT equal to 19.6 °C. This relevant difference can be ascribed to the salting-out effect generated by the presence of dissolved PBS ions, similar to that reported for previously studied P407-based PEUs.21 Interestingly, HHP407 was characterized by comparable CMT values to that of chain-extended P407-based PEUs at an equal concentration (i.e., 1% w/v), although its molecular mass was significantly lower (i.e., around 30%).33 The correlation between these results suggests an enhancement in the thermo-induced self-assembly of the HHP407 chains into micelles probably originated from a better balance between the overall hydrophobicity and length of the polymer chains. It is also plausible that the presence of end-methacrylate groups could induce a significant contribution in this regard. The DLS measurements showed that both analyzed samples underwent polymer chain organization into micellar structures, with some differences ascribed to the solubilization medium, and in particular the absence of dissolved ions in the sample prepared in ddH2O. Indeed, at 25 °C the micelle hydrodynamic diameter was measured to be 18.5 ± 5.2 nm and 34.5 ± 3.5 nm for HHP407 solubilized in ddH2O and PBS, respectively. Irrespective of the solubilization medium, the micelle hydrodynamic diameter increased with an increase in temperature to 37 °C, reaching 41.2 ± 7.3 nm and 44.6 ± 5.8 nm for the samples prepared in ddH2O and PBS, respectively (statistically significant increase for the sample prepared in ddH2O, p = 0.0118). Interestingly, polymer solubilization in PBS produced significantly large micelles at 25 °C (statistically significant difference, p = 0.0115), in accordance with the lower CMT value estimated for this sample, which could initiate the micellization process at lower temperature. Conversely, when HHP407 was solubilized in ddH2O, the same micelle dimensions were achieved with an increase in temperature to 37 °C (34.5 ± 3.5 nm for HHP407 1% w/v in PBS at 25 °C vs. 41.2 ± 7.3 nm for HHP407 1% w/v in ddH2O at 37 °C, not statistically significant difference), in accordance with the higher temperature needed for this sample to initiate the polymeric chain organization into micellar structures.
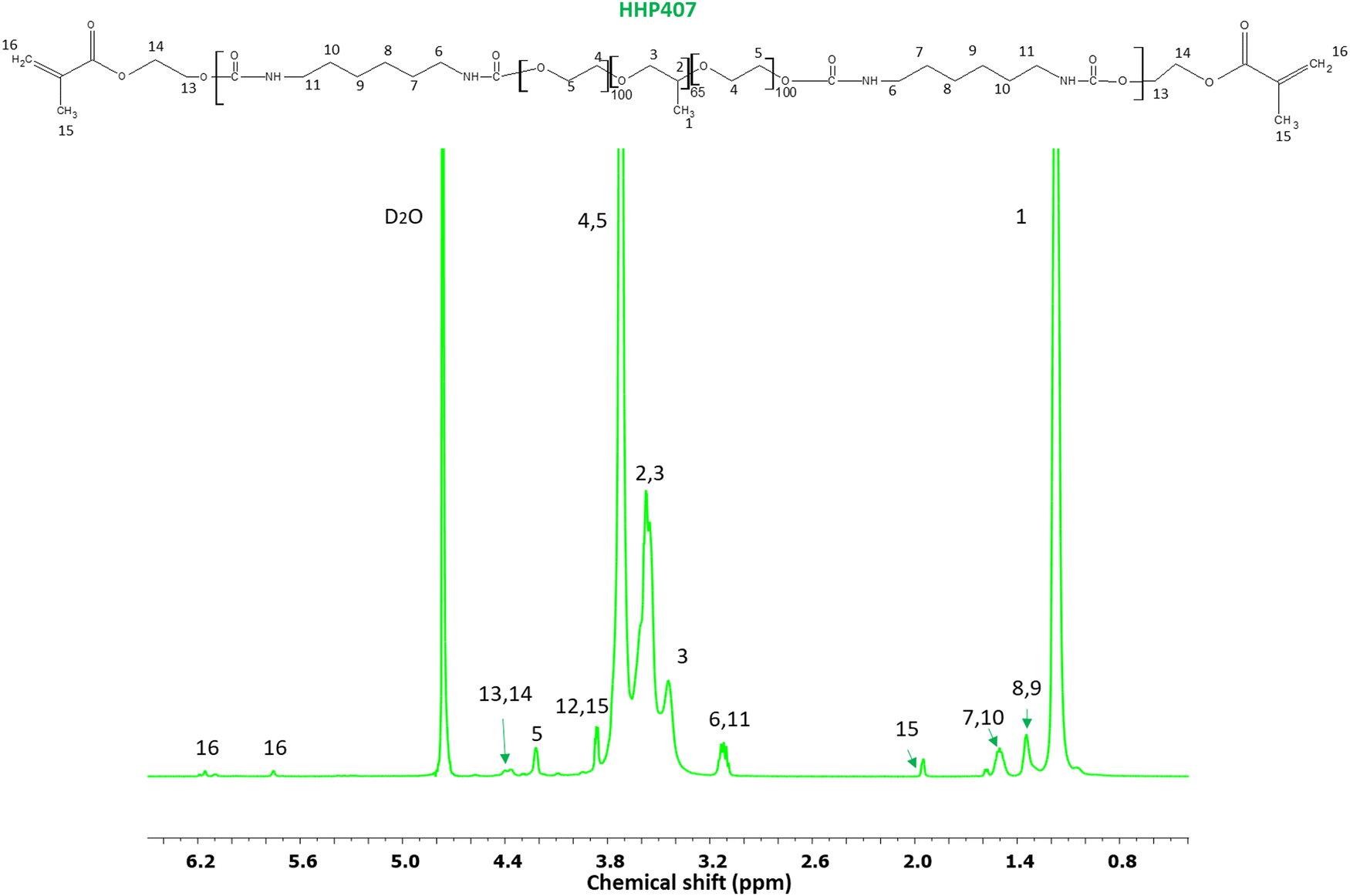 | ||
| Fig. 2 1H NMR spectrum of HHP407. All PEU-related proton peaks are indicated by specific numbers and arrows. The presence of typical HEMA domains is indicated by protons 13 and 14 (about 4.2–4.4 ppm), 15 (1.95 ppm) and 16 (5.85 and 6.1 ppm). | ||
Characterization of HHP407–CD SM complexes
The mixture containing HHP407 at 1% w/v and CDs at 7.6% w/v (i.e., 100% theoretical PEO domain coverage, HHP407 1%–SM 100%) in ddH2O resulted in a 100% recovery (calculated according to eqn (1)). This result indicates that HHP407 was a more effective polymer in terms of interaction with CDs in aqueous environments compared to previously characterized chain-extended P407-based PEUs, which were characterized by a maximum recovery of 60%.20,21 Indeed, HHP407 was probably characterized by the best match between molecular mass and resulting hydrophobicity, which could enhance its self-assembling ability in forming micelles, and thence PPR-based networks in solution. 1H NMR characterization was performed on the resolubilized HHP407 1%–SM 100% crystalline powder to clearly confirm the co-presence of HHP407 and CDs within the collected sample, as reported in Fig. S4 (ESI†). In fact, the presence of the characteristic peaks of CDs was found within the chemical shift range of 4.05 to 3.55 ppm and at 5.08 ppm, while the PEU resonance domains were detected within the chemical shift region between 1.60 and 0.95 and at 3.75 ppm. The XRD patterns demonstrated the formation of PPR-based channel-like crystalline structures within the HHP407 1%–SM 100% sample, as reported in Fig. 3. Indeed, three main peaks at 2θ values of 7.4°, 12.8° and 19.6° were detected and clearly represented the formation of SM crystals within the HHP407-based network, as notably indicated in the literature.37 Instead, the pattern of native HHP407 powders was characterized by the presence of the same peaks that were found in chain-extended P407-based PEUs (i.e., at 2θ values of 19° and 23.3°),20,21 which were comparable to that of P407, thus suggesting that the molecular mass distribution profile and the introduced hydrophobic domains in HHP407 PEU did not influence the crystallization process of the P407 block significantly.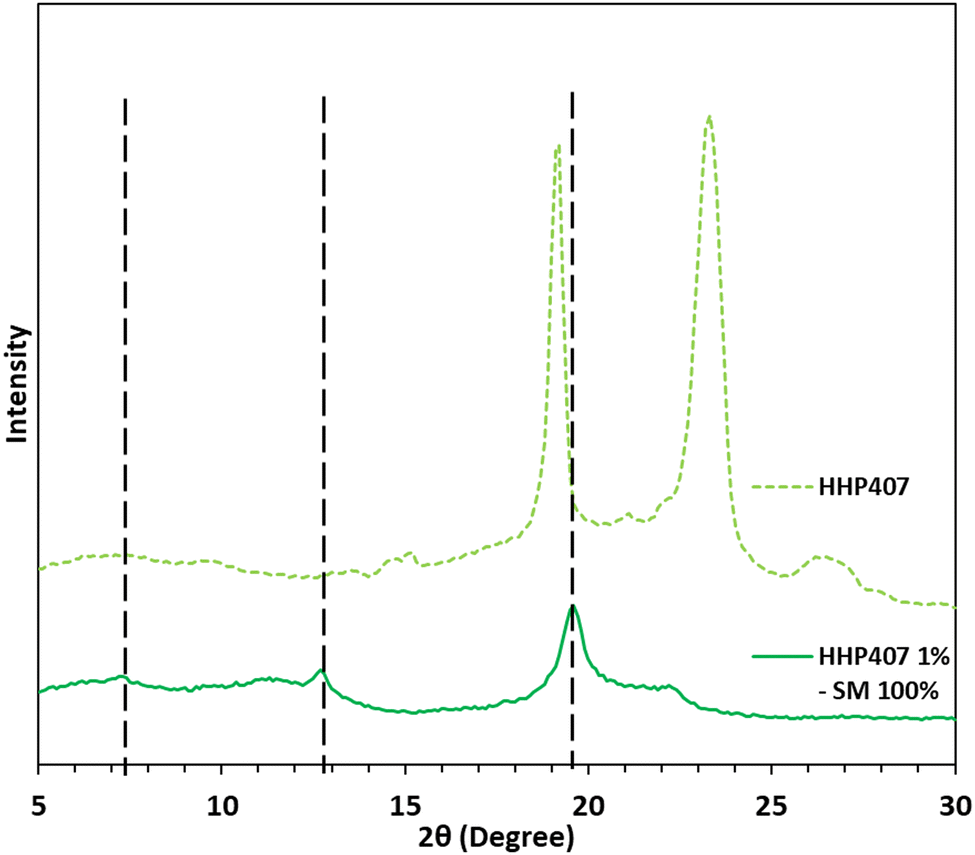 | ||
| Fig. 3 XRD patterns of HHP407 (light green dashed line) and HHP407 1%–SM 100% (green continuous line). Typical peaks of channel-like PPR-based crystals are indicated by black vertical dashed lines at 2θ values of 7.4°, 12.8° and 19.6°. The pattern of pure HHP407 is also reported for comparative purposes. | ||
Design of HHP407-based SM hydrogels
| CDs (% w/v) | |||||
|---|---|---|---|---|---|
| 7 | 8 | 9 | 10 | ||
| HHP407 (% w/v) | 1 | O.N. | 4 h 20′ | 2 h 15′ | 1 h 5′ |
| 5 | O.N. | 3 h | 50′ | 20′ | |
Thus, considering all the above-mentioned reasons, among the HHP407-based hydrogel formulations, the hydrogels containing CDs at 8% w/v concentration were selected for further investigations. This selection was driven by the concurrent possibility to significantly decrease the CD content with respect to the previously developed formulations based on chain-extended PEUs (i.e., CD content 20% lower),21 while keeping the gelation relatively fast. Additionally, this novel design would provide clear advantages in terms of hydrogel production-related costs.
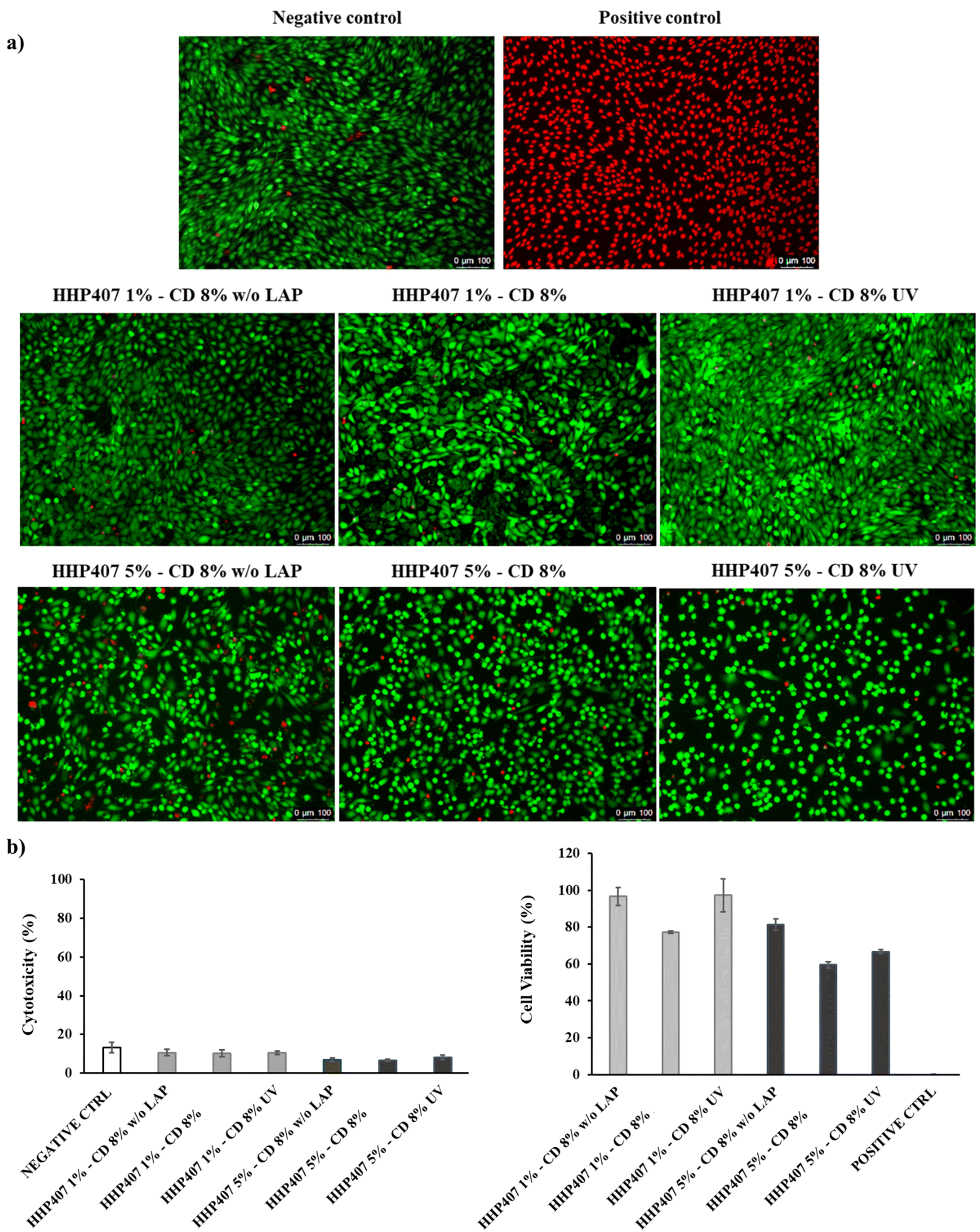 | ||
| Fig. 4 (a) Live and dead staining images of NIH-3T3 murine fibroblasts treated with HHP407 X%–CD 8%-based extracts after 24 h of incubation under normal culture conditions. Live and dead cells are stained in green and red, respectively. (b) Evaluation of NIH-3T3 cytotoxicity and cell viability induced by HHP407 X%–CD 8%-based extracts prepared according to ISO 10993-5 rules and tested through CytoTox-ONE™ and CellTiter-Blue assays, respectively. | ||
Characterization of curcumin-loaded SM hydrogels based on HHP407 and CDs
 | ||
| Fig. 5 G′ (continuous lines) and G′′ (long dashed lines) as a function of applied strain at 37 °C for (a) HHP407 1%–CD 8% (green) and HHP407 1%–CD 8% Cur (orange), and (b) HHP407 5%–CD 8% (green) and HHP407 5%–CD 8% Cur (orange). | ||
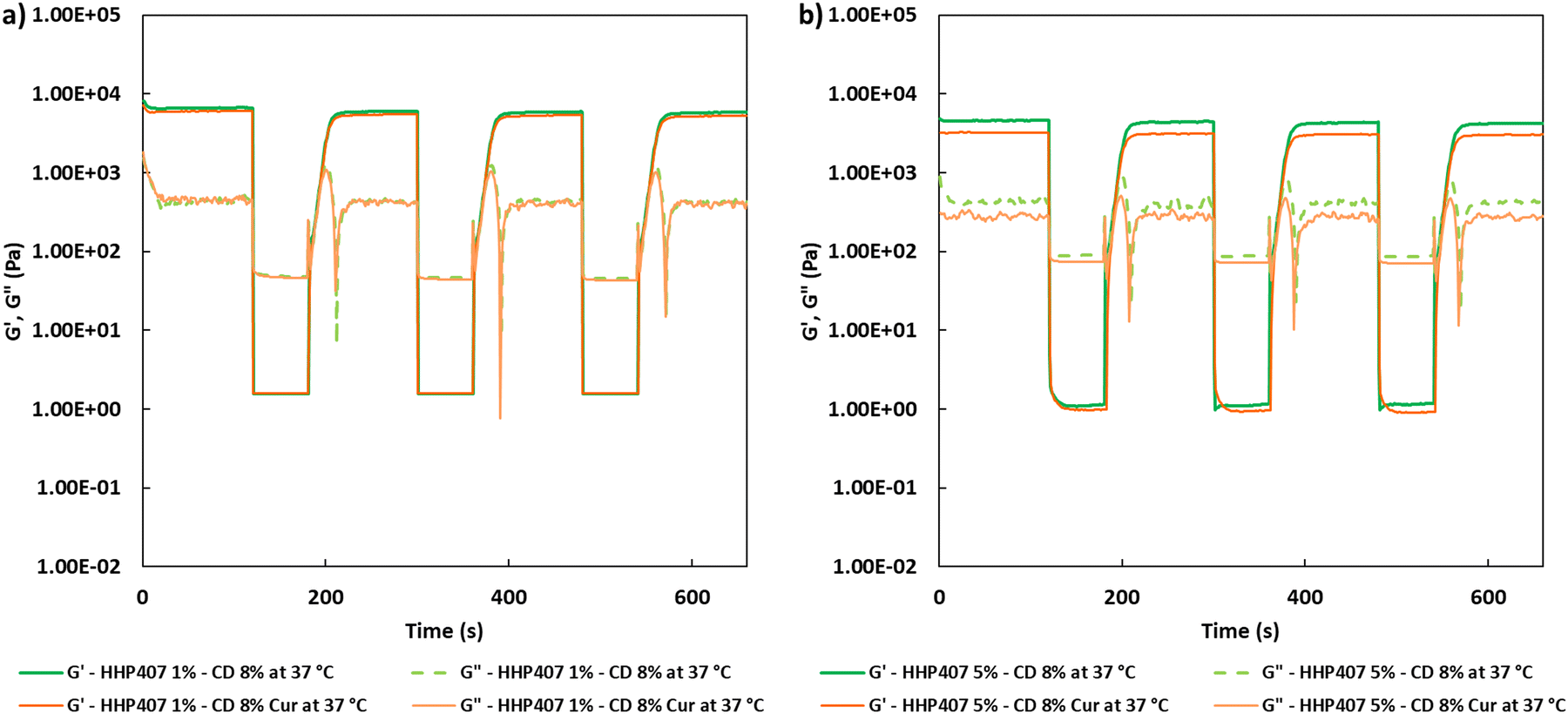
Frequency sweep tests were conducted at 25 °C, 30 °C and 37 °C with the aim to evaluate the behavior of HHP407-based hydrogels in terms of network development and thermo-responsiveness. All the investigated formulations showed a fully developed gel state at each tested temperature and no cross-over points between G′ and G′′ were identified, as reported in Fig. S9 (ESI†). A slight dependence in the G′ and G′′ trends on temperature was observed for all the hydrogel systems. For example, even the formulation containing the lowest amount of thermo-sensitive PEU (i.e., HHP407 1%–CD 8%) showed a G′ equal to 5700 Pa at 25 °C (at 100 rad s−1 applied angular frequency), while that at 37 °C was 7400 Pa. Interestingly, the formulation containing HHP407 at 1% w/v showed a better developed hydrogel network compared to that containing PEU at a concentration of 5% w/v. Indeed, the former was characterized by a less marked dependency of G′ and G′′ on the angular frequency compared to the latter. Even the frequency sweep tests evidenced the better mechanical properties of the hydrogel composed of HHP407 at a concentration of 1% w/v with respect to that containing PEU at 5% w/v. For example, G′ at 37 °C (100 rad s−1) was 7400 Pa for HHP407 1%–CD 8% and 5000 Pa for HHP407 5%–CD 8%. Cur loading induced a slight decrease in the characteristic mechanical parameters, as previously observed and discussed. The contribution of Cur was more evident in the samples containing HHP407 at 5% w/v compared to that at 1% w/v PEU concentration, probably because of the presence of a less developed SM hydrogel network at a higher PEU/CD mass ratio (i.e., HHP407 at 5% w/v). However, no significant effects of Cur on hydrogel development were generally observed, given that G′ and G′′ of the Cur-loaded hydrogels showed consistent trends over the applied angular frequency at all the investigated temperatures with respect to the control samples. The self-healing tests evidenced a remarkable recovery ability of all the investigated HHP407-based hydrogels, as reported in Fig. 6. In fact, a G′ recovery greater than 85% after 3 rupture cycles was observed for all the investigated systems. Higher recovery values were measured for the samples with an HHP407 concentration of 5% w/v (i.e., around 95%) compared to those at 1% w/v (i.e., around 88%). This difference can be ascribed to a better fatigue resistance of the hydrogel systems containing PEU at a higher concentration (i.e., 5% w/v) due to the relatively higher damping effect originating from the more diffused and interacting amorphous domains39 (i.e., PEU chains not involved in the formation of SM crystals) with respect to the hydrogel networks with a lower PEU/CD ratio (i.e., 1% w/v). Indeed, this condition generally favored the organization of the available PEU chains into stiffer PPR-based networks. Finally, the presence of Cur did not significantly affect the mechanical response of the resulting hydrogels to applied rupture cycles and recovery phases. Lastly, photo-rheological tests were performed with the aim to assess the photo-responsiveness of the developed SM hydrogels. In this regard, the typical turbidity of these systems can represent an important impediment to light transmission within the entire hydrogel volume. Nonetheless, all the gels showed good responsiveness to UV light exposure, as highlighted in Fig. 7, showing the trend of complex viscosity (η*) as a function of time during light irradiation. Indeed, the complex viscosity (η*) increased during light irradiation for all the tested formulations, where an η* increase of ca. 16%, 8%, 17% and 12% was measured for HHP407 1%–CD 8%, HHP407 1%–CD 8% Cur, HHP407 5%–CD 8% and HHP407 5%–CD 8%, respectively. A similar trend was also observed for the storage modulus, where G′ increased after UV light exposure to around 19% and 17% for the HHP407 1%–CD 8% and HHP407 5%–CD 8% hydrogels, respectively.
 | ||
| Fig. 6 Strain test curves showing G′ (continuous lines) and G′′ (dashed lines) as a function of time during cyclic rupture (100% strain, 60 s) and recovery (0.1% strain, 120 s) of hydrogel networks at (a) 1% and (b) 5% w/v HHP407 concentration at 37 °C. Control hydrogels (green lines) are compared to the hydrogels containing Cur at 570 μg mL−1 (orange lines). | ||
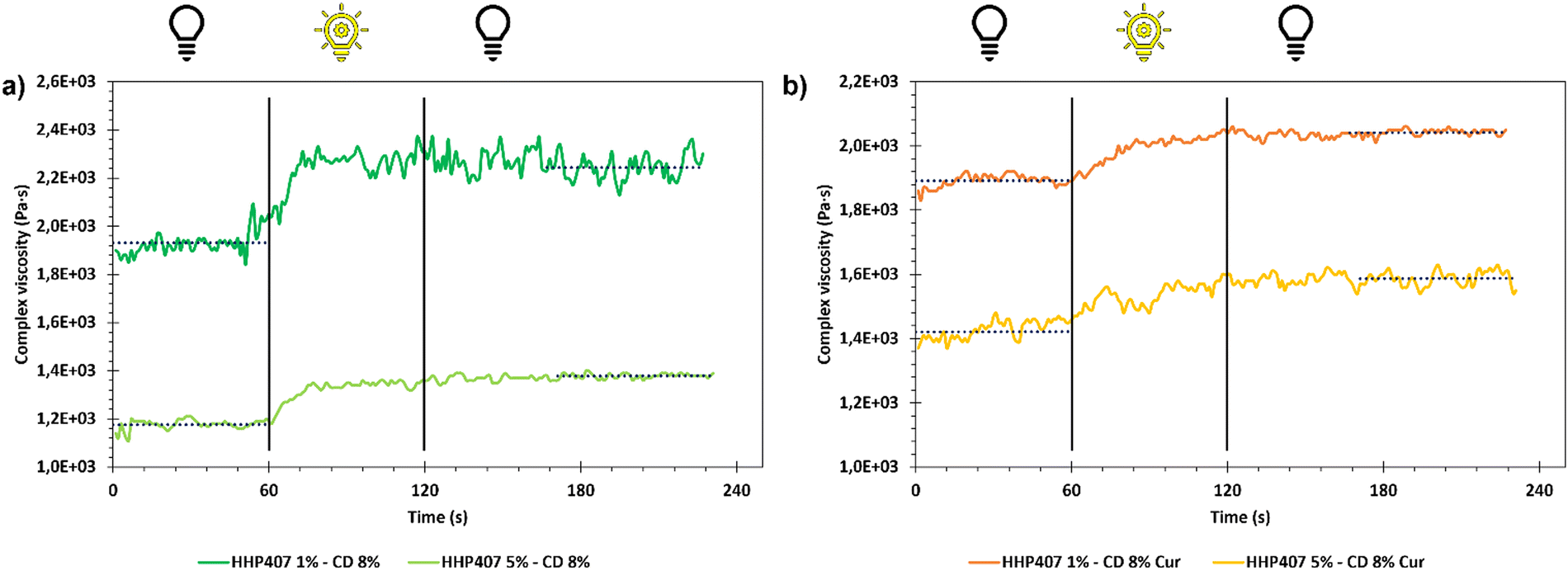 | ||
| Fig. 7 Complex viscosity as a function of time for hydrogels (a) at 1% and 5% w/v HHP407 concentration and (b) containing Cur at 570 μg mL−1. Hydrogels were characterized before photo-crosslinking for 60 s. Then, exposure to UV light (365 nm, 10 mW cm−2) was conducted for 60 s and the resulting mechanical properties were registered for additional 120 s. Horizontal dotted blue lines identify the mean complex viscosity values before (in the first 60 s of observation) and after photo-curing (in the last 60 s of observation). | ||
Differently, an 8% and 11% increase in G′ was observed on average for the hydrogel systems containing Cur at 570 μg mL−1 and PEU at 1% and 5% w/v concentration, respectively. These results suggest a lower photo-curing potential for the Cur-loaded systems due to the typical Cur photo-sensitivity, which provided a contribution in terms of UV light dissipation. Interestingly, the visual inspection of the sample containing HHP407 at 1% w/v and Cur indicated a significant reduction in its original intense orange color. This observation indicated the indisputable degradation of a relevant amount of Cur payload within the hydrogel network containing PEU at a low concentration. Differently, no significant color changes were observed for the HHP407 5%–CD 8% samples, thus suggesting that a higher content of photo-sensitive PEU helped to preserve the Cur payload. This behavior can be ascribed to two main aspects. Firstly, a significantly higher number of photo-sensitive groups was intrinsically present in the formulation containing HHP407 at 5% w/v compared to 1% w/v, thus competing with the Cur degradation process in terms of UV light absorbance through the formation of chemical crosslinks. Secondly, it is likely that a higher content of PEU is beneficial in terms of the stabilization and protection of Cur from degradation through its encapsulation in more available hydrophobic domains. In conclusion, the performed photo-rheological characterization suggested that a shorter UV light exposure was sufficient to achieve complete crosslinking and this aspect was particularly relevant for the samples containing HHP407 at a low concentration (i.e., 1% w/v). Specifically, the results suggested that almost complete crosslinking was achieved within 20–30 s of UV light irradiation for all the investigated hydrogels, as can be observed in the complex viscosity curves reported in Fig. 7.
The release profiles of Cur from all the hydrogels (i.e., HHP407 1%–CD 8% Cur, HHP407 1%–CD 8% Cur UV, HHP407 5%–CD 8% Cur, HHP407 5%–CD 8% Cur UV) showed significantly different trends, which were correlated with the hydrogel formulation and crosslinking method, as shown in Fig. 8. Generally, the systems exposed to UV light showed a delayed delivery of Cur and could sustain this process for up to 35 days of incubation under physiological-like conditions (i.e., in contact with PBS at 37 °C). Surprisingly, the entire payload of Cur (i.e., 285 μg per sample) was also released from the UV-cured systems, thus indicating the optimal preservation of its chemical stability within the SM networks, which indeed acted as protective element for this highly sensitive drug. Interestingly, the SM hydrogels based on HHP407 at a concentration of 1% w/v exhibited slower release kinetics compared to those containing PEU at a concentration of 5% w/v. In particular, at each time point, a significantly higher release rate was observed for HHP407 5%–CD 8% Cur UV with respect to HHP407 1%–CD 8% Cur UV (data not shown, p < 0.0012). This observation can be correlated with the previous rheological and photo-rheological characterizations, in which the hydrogels composed of HHP407 at a concentration of 1% w/v showed the highest G′ values, thus indicating the formation of highly rigid networks, due to the enhanced SM self-assembly into PPR-based crystals.42,43
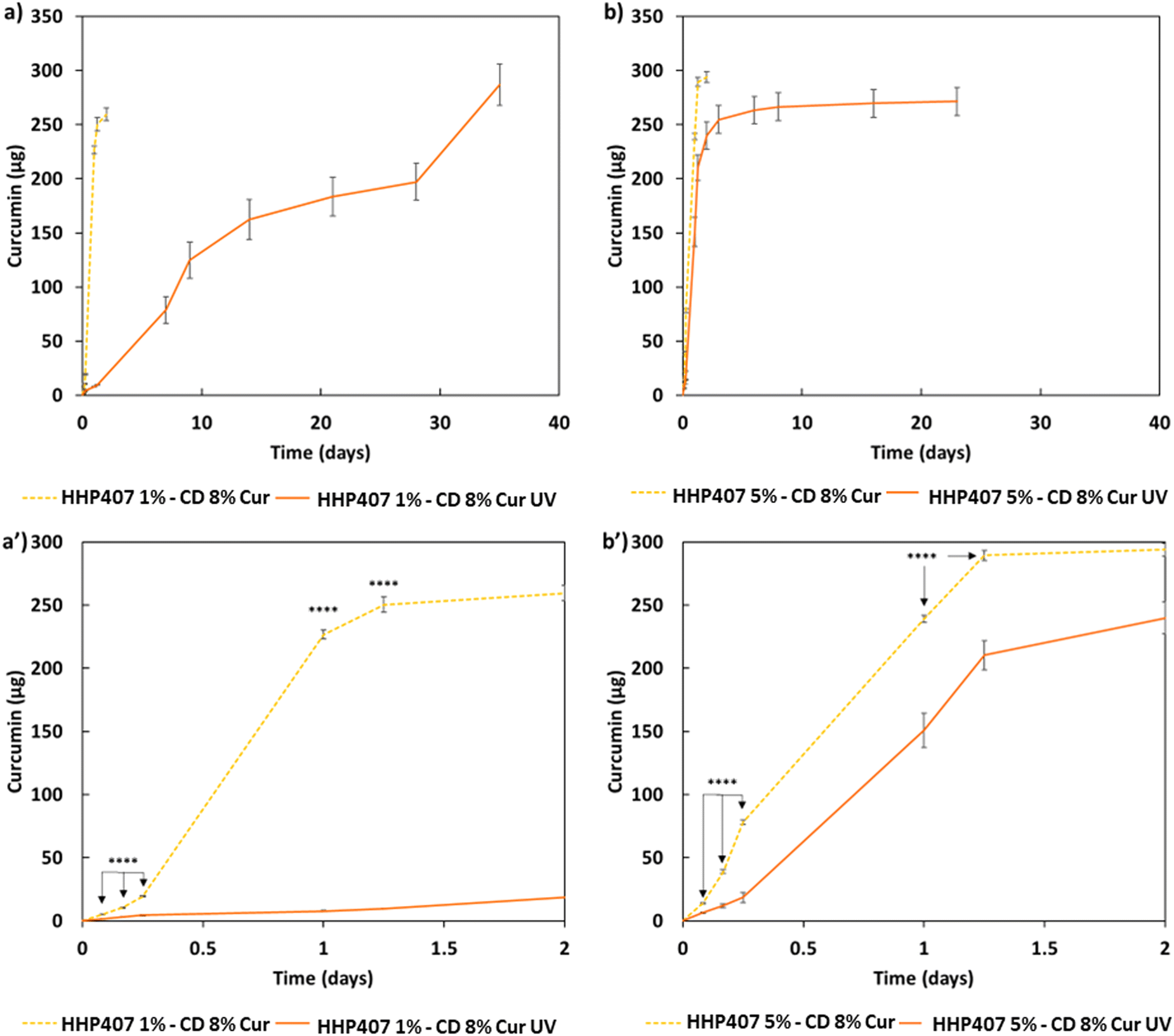 | ||
| Fig. 8 Cur release profiles from hydrogel systems in contact with PBS at 37 °C and characterized by different HHP407 contents: (a) and (a′) 1% w/v (HHP407 1%–CD 8% Cur and HHP407 1%–CD 8% Cur UV) and (b) and (b′) 5% w/v (HHP407 5%–CD 8% Cur and HHP407 5%–CD 8% Cur UV). Physical hydrogels (light orange dashed lines) are compared with photo-cured systems (orange continuous lines). (a′) and (b′) graphs represent the release profile in detail measured within the first 2 days of observation. | ||
Even in this case, the PEU/CD ratio suggested the formation of differently organized networks that could also undergo photo-induced crosslinking and open a way towards the possibility of finely tuning the resulting release kinetics.
Preliminary studies on HHP407-based SM hydrogel extrudability and printing
In this study, a preliminary evaluation of the general extrudability of the HHP407-based hydrogel systems was conducted. The HHP407 5%–CD 8% formulation was selected for this purpose due to its lower rigidity and generally higher damping/self-healing ability, which were observed through rheological characterizations. These parameters are fundamental aspects to consider for the selection of a potential bio-ink or biomaterial ink.44 The printing process was conducted by controlling different parameters including the tip extrusion diameter (DE), extrusion pressure (PE), printhead velocity (νP), syringe temperature (Ts), and bed temperature (TB). The printing process was conducted at 37 °C for both the syringe and bed to favor the stabilization of the hydrogel network through the major contribution of hydrophobic interactions. A preliminary set of optimized parameters was obtained and reported in Table 3.| D E | P E | ν P | T s | T B |
|---|---|---|---|---|
| 250 μm | 35–40 kPa | 10 mm s−1 | 37 °C | 37 °C |
Fig. 9 shows the produced 3D-printed constructs and a qualitative comparison between the resulting geometry and the theoretical CAD design. The printing process produced constructs with good definition and acceptable printing fidelity. In fact, the filament diameter was determined to be 0.46 ± 0.08 mm (average of 5 measurements obtained from 4 different filaments), while the theoretical value was 0.45 mm, thus proving both extrudability and filament formation. The selection of a smaller tip diameter (0.25 mm) with respect to the theoretical value (0.45 mm) was driven by the need to reach the best compromise between printing velocity and required extrusion pressure. Indeed, under the selected conditions, the production of two-layer grids with a 0/90° pattern required an overall printing time of around 1 min. The pore dimensions were larger than the theoretical design, where the pore area of the printed constructs was calculated to be 0.17 ± 0.04 mm2, which is around 40% higher than the theoretical value. This difference can be ascribed to the presence of defects in proximity to the intersections between the two layers. Nonetheless, good printability could be deduced from these preliminary results. Further optimization can be done based on geometry investigation through more precise methodologies (e.g., computed tomography or optical coherence tomography) to better find the correlation between morphological features and printing parameters. Another qualitative characterization was conducted in terms of stability in contact with a large volume of aqueous medium (PBS, 5 mL, 25 °C) (Fig. 10). The UV cured systems (HHP407 5%–CD 8% Cur UV) showed a significantly improved residence time in aqueous medium with respect to the physically-crosslinked sample (HHP407 5%–CD 8% Cur). In fact, as shown in Fig. 10, the UV light-irradiated samples preserved their geometrical integrity for up to 3 days of observation in a highly destabilizing aqueous environment, while physically-crosslinked samples were completely solubilized within an incubation time of 3 h.
 | ||
| Fig. 9 (a) Photo of a 3D-printed construct based on HHP407 5%–CD 8% Cur. (b) UV curing of HHP407 5%–CD 8% Cur structures. (c) Qualitative geometrical match between the 3D-printed HHP407 5%–CD 8% Cur and the starting theoretical CAD design. | ||
 | ||
| Fig. 10 Qualitative evaluation of the stability of the 3D-printed HHP407 5%–CD 8% Cur and HHP407 5%–CD 8% Cur UV structures in contact with 5 mL of PBS at 25 °C (within a Petri support – 50 mm diameter). | ||
Moreover, an evident color change was observed after UV curing for 1 min, thus indicating that the resulting relevant interfacial surface and the limited thickness of the construct probably enhanced the exposure of Cur to photo-degradation. Nonetheless, an evident yellowish color was maintained, indicating the protective effect of HHP407 and CDs on Cur even under these particularly degradative conditions. Indeed, when the same UV curing protocol was applied to the samples utilized for Cur release studies, it did not induce any evident Cur degradation phenomena due to the concurrent protective effect provided by HHP407/CDs and the selected sample geometry (i.e., thickness and overall volume). Lastly, the HHP407 5%–CD 8% hydrogel formulation was successfully used to print 25-layer (300 μm layer thickness) cylinder constructs with a 0/90° pattern. The layer-by-layer photo-curing process effectively stabilized each 3D printed layer, resulting in a self-standing construct with a final height of ca. 0.75 mm, which is in agreement with the expected theoretical value. Fig. 11 presents the macroscopic appearance of the 3D printed structures during fabrication (Fig. 11a) and at the end of the printing process, highlighting the absence of evident structure collapse and consequent pore closing phenomena (Fig. 11b).
 | ||
| Fig. 11 (a) Photograph of HHP407 5%–CD 8%-based 25-layer cylinder construct with a 0/90° pattern taken during the 3D printing process and (b) macroscopic appearance of 3D printed 25-layer cylinder constructs with a 0/90° pattern at the end of the printing process. | ||
Conclusions
In this work, a novel photosensitive PEU was synthesized by modifying a well-known and optimized synthesis process.21,23,25 Indeed, HHP407 PEU was synthesized using HEMA as the end-capping alcohol of the isocyanate-terminated prepolymer chains resulting from the prepolymerization reaction between P407 and HDI.The performed physico-chemical characterization indicated a good functionalization of the resulting PEU chains through the exposure of methacrylate end-groups. Moreover, excellent thermo-sensitivity was assessed by CMT evaluation and DLS analyses, although HHP407 possessed a lower molecular mass compared to the chain-extended 407-based PEUs previously developed by our group.21 This behavior suggested the achievement of a well-balanced polymer structure and hydrophobicity in terms of length and distribution of the available domains (i.e., PPO, HDI and probably also HEMA). Because of the resulting physico-chemical properties, HHP407 was demonstrated to be suitable for the formation of PPR-based crystals in an aqueous environment. Moreover, it was found to be a particularly suitable polymer for the formation of SM networks among the plethora of materials investigated in the literature.21,43 Hence, it is likely that its structural features represent an optimized set of properties for constructive interaction with CDs, resulting in highly stable and simultaneously responsive networks. This particular behavior allowed the design of SM hydrogels with good mechanical properties at a low HHP407 content (i.e., ≤5% w/v) and a relatively low CD concentration (i.e., 8% w/v). In this regard, the formulation showing the lowest PEU/CD mass ratio (i.e., 1% and 8% w/v HHP407 and CD concentration, respectively) was characterized by better mechanical properties with respect to the system containing HHP407 at 5% w/v, as assessed through rheological characterizations. This observation was in agreement with previous results obtained for similar systems, thus further reinforcing the hypothesis of the high functionality and tunability of PEU macromolecules at low concentrations in combination with CDs.20,21 Although HHP407 produced highly crystalline hydrogel networks with CDs, with the consequent possibility of obtaining brittle hydrogels, obvious self-healing ability was observed. Photosensitivity was assessed by means of photo-rheological characterizations. Irrespective of the HHP407 and CD content, all the tested formulations (i.e., purely physical hydrogels and both physically and chemically crosslinked hydrogels) showed high cytocompatibility according to the ISO 10993-5 regulation. The Cur delivery studies evidenced that the hydrogel composition and consecutive photo-crosslinking play a key role in tuning the Cur release kinetics from the HHP407-based networks. Furthermore, the in vitro Cur release tests demonstrated the protective ability of HHP407 and CDs on this photo-sensitive drug, opening the way for the possibility to sustain the release of Cur in aqueous environments for up to 5 weeks. Interestingly, these samples did not show any color change upon light irradiation, resulting in the possibility to completely release the encapsulated Cur payload. Conversely, under different experimental set-ups (i.e., lower sample thickness and higher exposed surface area), we observed a slight color change in the samples (higher for samples with lower PEU content, and thus lower polymer-induced Cur protection), suggesting the occurrence of Cur photo-induced degradation. Overall, our data suggest that depending on the geometrical features of the developed construct (e.g., 3D bio-printed structure or injected deposit filling a cavity), a fine optimization of the polymerization process should be performed (e.g., by minimizing the irradiation time) to protect the loaded Cur molecules from photo-induced degradation. Finally, one formulation among the developed hydrogels was preliminary evaluated for the production of 3D-printed structures with good resolution. These results further indicated that HHP407 is a highly versatile polymer for the development of engineered devices in regenerative medicine. Hence, this study confirmed the great potential of appropriately synthesized PEUs as raw materials for the design of SM hydrogels with a low synthetic polymer content and showing good mechanical performances, progressive release profiles of encapsulated drugs, and easy processability through extrusion-based 3D bioprinting. The observed properties present insights into the future use of the herein-described hydrogel platform for 3D cell culture,45 inject-based bioprinting46 and vat-polymerization 3D bioprinting,47 among others. Overall, we can conclude that the developed hydrogels can find widespread application in the emerging field of regenerative pharmacology, as (i) easily injectable drug-loaded systems suitable for the perfect filling of body defects through a mini-invasive procedure, and (ii) drug-loaded biomaterial inks in the fabrication of patient-personalized patches. Hence, the designed hydrogels hold great promise, warranting their further development and characterization in future investigations targeting specific applications in the biomedical field (e.g., wound healing, endodontics, and cartilage repair).
Author contributions
Alessandro Torchio: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, visualization. Monica Boffito: supervision, resources, conceptualization, methodology, validation, investigation, writing – review & editing, funding acquisition, visualization. Rossella Laurano: methodology, investigation, data curation, writing – original draft. Claudio Cassino: investigation, data curation, writing – review & editing. Mario Lavella: investigation, data curation, writing – review & editing. Gianluca Ciardelli: supervision, resources, writing – review & editing, funding acquisition.Data availability
All relevant data are within the manuscript and ESI.† The data are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The research leading to these results has received funding from the European Union-NextGenerationEU through the Italian Ministry of University and Research under PNRR-M4C2-I1.3 Project PE_00000019 “HEAL ITALIA” to Gianluca Ciardelli CUP E93C22001860006 of University of Modena and Reggio Emilia. Part of this study was carried out within the Ministerial Decree no. 1062/2021 and received funding from the FSE REACT-EU-PON Ricerca e Innovazione 2014–2020. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.References
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS PubMed.
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Biomater. Sci., 2019, 7, 843–855 RSC.
- B. Tian, S. Hua, Y. Tian and J. Liu, J. Mater. Chem. B, 2020, 8, 10050–10064 RSC.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- J. Li, Z. Suo and J. J. Vlassak, J. Mater. Chem. B, 2014, 2, 6708–6713 RSC.
- S. P. Zhao, L. M. Zhang, D. Ma, C. Yang and L. Yan, J. Phys. Chem. B, 2006, 110, 16503–16507 CrossRef CAS PubMed.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821–2823 CrossRef CAS.
- J. Li, A. Harada and M. Kamachi, Polym. J., 1994, 26, 1019–1026 CrossRef CAS.
- X. Li and J. Li, J. Biomed. Mater. Res. A, 2008, 86, 1055–1061 CrossRef PubMed.
- E. Larrañeta and J. R. Isasi, Langmuir, 2012, 28, 12457–12462 CrossRef PubMed.
- J. Li, X. Ni, Z. Zhou and K. W. Leong, J. Am. Chem. Soc., 2003, 125, 1788–1795 CrossRef CAS PubMed.
- X. Ni, A. Cheng and J. Li, J. Biomed. Mater. Res. A, 2009, 88, 1031–1036 CrossRef PubMed.
- S. P. Zhao and W. L. Xu, J. Polym. Res., 2010, 17, 503–510 CrossRef CAS.
- H. Wei, A. Y. Zhang, L. Qian, H. Yu, D. Hou, R. Qiu and Z. G. Feng, J. Polym. Sci. A Polym. Chem., 2005, 43, 2941–2949 CrossRef CAS.
- H. Wei, H. Yu, A. Zhang, L. Sun, D. Hou and Z. Feng, Macromolecules, 2005, 38, 8833–8839 CrossRef CAS.
- H. Wei, J. He, L. Sun, K. Zhu and Z. Feng, Eur. Polym. J., 2005, 41, 948–957 CrossRef CAS.
- D. Chimene, R. Kaunas and A. K. Gaharwar, Adv. Mater., 2020, 32, 1902026 CrossRef CAS PubMed.
- L. Xiaorui, Z. Fuyin, W. Xudong, G. Xuezheng, Z. Shudong, L. Hui, D. Dandan, L. Yubing, W. Lizhen and F. Yubo, Int. J. Bioprint., 2022, 9, 649 CrossRef PubMed.
- S. Boularaoui, G. Al Hussein, K. A. Khan, N. Christoforou and C. Stefanini, Bioprinting, 2020, 20, e00093 CrossRef.
- A. Torchio, M. Boffito, A. Gallina, M. Lavella, C. Cassino and G. Ciardelli, J. Mater. Chem. B, 2020, 8, 7696–7712 RSC.
- A. Torchio, C. Cassino, M. Lavella, A. Gallina, A. Stefani, M. Boffito and G. Ciardelli, Mater. Sci. Eng., C, 2021, 127, 112194 CrossRef CAS PubMed.
- M. Boffito, F. Di Meglio, P. Mozetic, S. M. Giannitelli, I. Carmagnola, C. Castaldo, D. Nurzynska, A. M. Sacco, R. Miraglia, S. Montagnani, N. Vitale, M. Brancaccio, G. Tarone, F. Basoli, A. Rainer, M. Trombetta, G. Ciardelli and V. Chiono, PLoS One, 2018, 13, e0199896 CrossRef PubMed.
- M. Boffito, E. Gioffredi, V. Chiono, S. Calzone, E. Ranzato, S. Martinotti and G. Ciardelli, Polym. Int., 2016, 65, 756–769 CrossRef CAS.
- M. Boffito, A. Torchio, C. Tonda-Turo, R. Laurano, M. Gisbert-Garzarán, J. C. Berkmann, C. Cassino, M. Manzano, G. N. Duda, M. Vallet-Regí, K. Schmidt-Bleek and G. Ciardelli, Front. Bioeng. Biotechnol., 2020, 8, 384 CrossRef PubMed.
- C. Pontremoli, M. Boffito, S. Fiorilli, R. Laurano, A. Torchio, A. Bari, C. Tonda-Turo, G. Ciardelli and C. Vitale-Brovarone, Chem. Eng. J., 2018, 340, 103–113 CrossRef CAS.
- M. Ding, J. Li, H. Tan and Q. Fu, Soft Matter, 2012, 8, 5414 RSC.
- J. Hu and L. Tan, in Polyurethane composites and nanocomposites for biomedical applications, Polyurethane Polymers, Elsevier, 2017, pp. 477–498 Search PubMed.
- R. Laurano, M. Boffito, M. Abrami, M. Grassi, A. Zoso, V. Chiono and G. Ciardelli, Bioact. Mater., 2021, 6, 3013–3024 CAS.
- H. H. Tønnesen, M. Másson and T. Loftsson, Int. J. Pharm., 2002, 244, 127–135 CrossRef PubMed.
- H. H. Tønnesen and J. Karlsen, Z. Lebensm.-Unters. Forsch., 1985, 180, 402–404 CrossRef PubMed.
- Y. Nimiya, W. Wang, Z. Du, E. Sukamtoh, J. Zhu, E. Decker and G. Zhang, Mol. Nutr. Food Res., 2016, 60, 487–494 CrossRef CAS PubMed.
- M. Kharat, Z. Du, G. Zhang and D. J. McClements, J. Agric. Food Chem., 2017, 65, 1525–1532 CrossRef CAS PubMed.
- M. Wright, K. Kurumada and B. Robinson, in Rates of incorporation of small molecules into pluronic micelles, Trends in Colloid and Interface Science XVI, ed. Miguel, M., Burrows, H. D., Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 8–11 Search PubMed.
- X. J. Loh, L. W. I. Cheng and J. Li, Macromol. Symp., 2010, 296, 161–169 CrossRef CAS.
- L. Wu, L. Yu, X. Fu and Z. Li, Chin. J. Polym. Sci., 2015, 33, 1140–1149 CrossRef CAS.
- R. Laurano, V. Chiono, C. Ceresa, L. Fracchia, A. Zoso, G. Ciardelli and M. Boffito, Eng. Reg., 2021, 2, 263–278 Search PubMed.
- J. W. Chung, T. J. Kang and S. Y. Kwak, Macromolecules, 2007, 40, 4225–4234 CrossRef CAS.
- C. Pradal, K. S. Jack, L. Grøndahl and J. J. Cooper-White, Biomacromolecules, 2013, 14, 3780–3792 CrossRef CAS PubMed.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- V. Kant, A. Gopal, D. Kumar, N. N. Pathak, M. Ram, B. L. Jangir, S. K. Tandan and D. Kumar, J. Surg. Res., 2015, 193, 978–988 CrossRef CAS PubMed.
- S. Khan, M. U. Minhas, M. Ahmad and M. Sohail, J. Biomater. Sci., Polym. Ed., 2018, 29, 1–34 CrossRef CAS PubMed.
- R. S. Stowers, S. C. Allen and L. J. Suggs, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1953–1958 CrossRef CAS PubMed.
- A. Domiński, T. Konieczny and P. Kurcok, Materials, 2019, 13, 133 CrossRef PubMed.
- A. Schwab, R. Levato, M. D’Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS PubMed.
- H. Cao, L. Duan, Y. Zhang, J. Cao and K. Zhang, Signal Transduction Targeted Ther., 2021, 6, 426 CrossRef CAS PubMed.
- W. L. Ng and V. Shkolnikov, Int. J. Bioprint., 2024, 2135 CrossRef CAS.
- W. L. Ng, J. M. Lee, M. Zhou, Y. W. Chen, K. A. Lee, W. Y. Yeong and Y. F. Shen, Biofabrication, 2020, 12, 022001 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ATR-FTIR spectra of P407 and HHP407; CMT curves for HHP407 solutions; DLS volume patterns for HHP407 solutions; 1H NMR spectra of CD-HHP407 complexes; appearance of samples after incubation with DMEM for 24 hours; cytocompatility of CDs at different concentrations; appearance of Cur solution in EtOH vs. Cur dispersion within CD solution; strain sweep tests to study the self-healing properties of unloaded- and Cur-loaded formulations; rheological frequency sweep tests of Cur-loaded hydrogels; calibration curves used for Cur quantification. See DOI: https://doi.org/10.1039/d4tb00092g |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |