
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing a biocatalyzed reaction cycle for transient and pH-dependent host–guest supramolecular hydrogels†
Bo
Su
,
Teng
Chi
 ,
Weike
Chen
,
Sijie
Xian
,
Dongping
Liu
,
Christopher J.
Addonizio
,
Yuanhui
Xiang
and
Matthew J.
Webber
*
,
Weike
Chen
,
Sijie
Xian
,
Dongping
Liu
,
Christopher J.
Addonizio
,
Yuanhui
Xiang
and
Matthew J.
Webber
*
Department of Chemcial & Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana, USA. E-mail: mwebber@nd.edu
First published on 22nd April 2024
Abstract
The formation of transient structures plays important roles in biological processes, capturing temporary states of matter through influx of energy or biological reaction networks catalyzed by enzymes. These natural transient structures inspire efforts to mimic this elegant mechanism of structural control in synthetic analogues. Specifically, though traditional supramolecular materials are designed on the basis of equilibrium formation, recent efforts have explored out-of-equilibrium control of these materials using both direct and indirect mechanisms; the preponderance of such works has been in the area of low molecular weight gelators. Here, a transient supramolecular hydrogel is realized through cucurbit[7]uril host–guest physical crosslinking under indirect control from a biocatalyzed network that regulates and oscillates pH. The duration of transient hydrogel formation, and resulting mechanical properties, are tunable according to the dose of enzyme, substrate, or pH stimulus. This tunability enables control over emergent functions, such as the programmable burst release of encapsulated model macromolecular payloads.
Introduction
Many structures in nature, from microtubule self-assembly to motion of protein motors, form and function through a constant supply of energy.1,2 Powered by continuous consumption of high energy molecules, these processes exist in high-energy states away from local thermodynamic equilibrium and dissipate when deprived of their energy source.3 In spite of frequent natural inspiration, synthetic supramolecular materials typically reside in equilibrium states, where the features of a dynamic system are determined by the relative thermodynamic stabilities of association.4,5 A new frontier in supramolecular design has instead envisioned soft materials that mimic the transience of natural dissipative and non-equilibrium states of matter.6 This approach could unlock new function and application through active materials, spatiotemporal patterns, autonomous machines, and self-replicators.7–13Both natural and synthetic out-of-equilibrium materials exist from dynamic assemblies with a net exchange of matter and/or energy with their environment.14 The energy source can be chemicals or light, with these inputs then coupled to chemical reaction networks directly or indirectly.15–23 A pioneering synthetic example of dynamic self-assembly was reported using N,N′-dibenzoyl-L-cystine (DBC) as a building block, with this compound activated for self-assembly by direct reaction with methyl iodide.24 The resulting DBC dimethyl ester self-assembled into transient one-dimensional fibers, with DBC restored via spontaneous ester hydrolysis leading to dissipation and fiber disassembly. Subsequent work used dimethyl sulfate (DMS) as the reactive agent, demonstrating out-of-equilibrium material formation with tunable lifetime, stiffness, and self-regeneration capabilities.7 This work also inspired a directly regulated dynamic supramolecular hydrogel based on molecular recognition of cucurbit[7]uril (CB[7]) and bicycle[2.2.2]octane (BO) host–guest pair, which formed transient hydrogels upon conversion of the BO guest to its methyl ester form through reaction with DMS, altering the affinity and dynamics of CB[7]–BO recognition.25 In this way, the lifetime of transient hydrogels and the corresponding dynamic modulus were regulated by tuning rates of methyl ester formation (via DMS) and hydrolysis (via pH),25 rather than by equilibrium host–guest complex formation.26 However, DMS is very reactive to other constituents of a functional system, and is also highly toxic and readily absorbed through the skin, mucous membranes, and gastrointestinal tract.27 The lifetime of this DMS-based dissipative supramolecular hydrogel was also quite long and difficult to control, especially as the hydrolysis rate for ester-containing guest species is dramatically reduced upon CB[7] binding.25,28 Accordingly, these two features rendered the approach unsuitable for further application.
Instead of using chemical reactions to directly modify supramolecular building blocks to drive dissipative assemblies, indirect control mechanisms can be used to drive transient material formation. Enzymes are a particularly appealing approach for use as actuators in the design of transient materials due to their specificity, biocompatibility, high turnover rates, and tunable reaction kinetics. Some examples of this approach include building blocks that bind adenosine triphosphate (ATP) to achieve assembled structures, subsequently disassembling when ATP is consumed by enzymes.16,29–32 Pulsating polymeric micelles and transient de-gelation of host–guest microgels have also been achieved from polymers modified with β-cyclodextrin by pairing supramolecular recognition to the enzymatic consumption of ATP.33,34 Transient host–guest supramolecular materials have been likewise reported that were indirectly regulated by enzymatic consumption of the macrocycle or control of the guest redox state.35,36 Feedback systems governed by enzymatic regulation of pH have also demonstrated promise for indirect coupling with transient self-regulating supramolecular assembly.37–43
Increasingly, host–guest chemistry has been explored for out-of-equilibrium complex formation.44 Among different synthetic host macrocycles, CB[7] has received interest for its ability to bind to diverse guests molecules with high binding affinity;45–47 tuning affinity offers a useful means to control hydrogel dynamics.48 CB[7]–guest affinity is likewise pH-dependent in cases where guest designs include sites for protonation;49 the CB[7]–BO interaction indeed transitions from no detectable binding at pH 11 to a stable complex at pH 7.25 This result arises from binding between CB[7] and negatively charged guests that is highly unfavorable due to electrostatic repulsion between the electronegative carbonyl portal and a negatively charged guest.50 From the pH- and charge-dependent principles governing CB[7]–guest recognition affinity, CB[7]–BO hydrogels are thus demonstrated here that form transiently upon addition of a citric acid/sodium citrate buffer (CA) and urea under indirect control from the presence of the urease enzyme (Fig. 1). This previously reported pH feedback loop enables an immediate and transient reduction in pH upon CA addition that is corrected as urease consumes urea to produce ammonia, thereby enabling reversible pH oscillation.37,51–53 As such, the transient reduction in pH upgrades CB[7]–BO binding affinity to promote physical crosslinking and hydrogelation, with subsequent and spontaneous pH correction destabilizing the complex and leading to hydrogel dissolution. This work contributes a route for transient host–guest hydrogels to complement many other works in a space predominated by low molecular weight gelators. This biocatalytic approach to supramolecular hydrogel formation also improves on shortcomings of the prior report using DMS,25 offering a milder and more biologically relevant control mechanism. Moreover, this approach enables interesting functionality to be explored in the context of programmed burst release of encapsulated macromolecules. As such, the work presented here effectively translates fundamental concepts derived from foundational out-of-equilibrium materials design to realize a more biocompatible approach with practical application. Furthermore, this general strategy offers a vision of more highly functional biomaterials derived by accessing transient and externally controlled states to achieve emergent function.
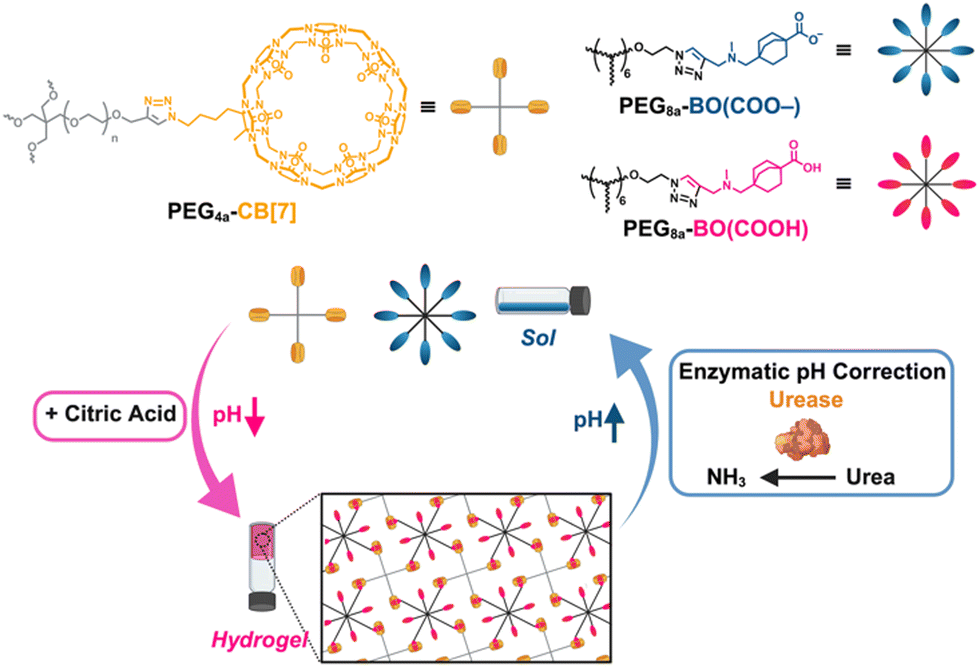 | ||
| Fig. 1 Molecular structures used here (top): a CB[7]-modified 4-arm PEG macromer, PEG4a–CB[7]; a guest-bearing PEG macromer in its negative form, PEG8a–BO(COO–) and uncharged form, PEG8a–BO(COOH). Schematic for transient non-equilibrium host–guest hydrogel formation coupled to an enzymatic network to regulate pH (bottom). The addition of urea and citric acid buffer mixture drives hydrogel formation upon initial acidification from citric acid, with the solution phase restored upon urease-catalyzed conversion of urea to ammonia raising the pH. | ||
Results and discussion
A 10 kDa 4-arm PEG macromer modified with CB[7] (PEG4a–CB[7]) and 20 kDa 8-arm PEG macromer modified with BO guest (PEG8a–BO(COO–)) were synthesized as previously reported.25 The ability of the CA/urea/urease pH feedback mechanism to induce gelation was first explored using samples prepared at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (by mole) of these two macromers at a total concentration of 4% w/v at an initial pH of 8.5. Urease concentration was held constant at 1.0 g L−1. A mixture of CA at 12 mM and urea at either 24, 30, or 36 mM resulted in hydrogel formation immediately following one minute of vortex mixing in all cases (Fig. 2). When urea was added at 36 mM, the hydrogel was stable until beginning to liquify at around 30 min and was fully restored to a solution after approximately 40 min (Fig. 2A). Samples prepared with 30 mM (Fig. 2B) and 24 mM (Fig. 2C) of urea began to liquify in approximately 60 min and 130 min, respectively. This urea-dependent lifetime was further evident in time-lapse videos of these inverted hydrogels (Movies S1–S3, ESI†). The increased hydrogel lifetime as urea was reduced matched expectations for this enzymatic reaction cycle, as less urea should reduce the rate of ammonia production and slow pH reversal following initial acidification by CA.
:1 ratio (by mole) of these two macromers at a total concentration of 4% w/v at an initial pH of 8.5. Urease concentration was held constant at 1.0 g L−1. A mixture of CA at 12 mM and urea at either 24, 30, or 36 mM resulted in hydrogel formation immediately following one minute of vortex mixing in all cases (Fig. 2). When urea was added at 36 mM, the hydrogel was stable until beginning to liquify at around 30 min and was fully restored to a solution after approximately 40 min (Fig. 2A). Samples prepared with 30 mM (Fig. 2B) and 24 mM (Fig. 2C) of urea began to liquify in approximately 60 min and 130 min, respectively. This urea-dependent lifetime was further evident in time-lapse videos of these inverted hydrogels (Movies S1–S3, ESI†). The increased hydrogel lifetime as urea was reduced matched expectations for this enzymatic reaction cycle, as less urea should reduce the rate of ammonia production and slow pH reversal following initial acidification by CA.
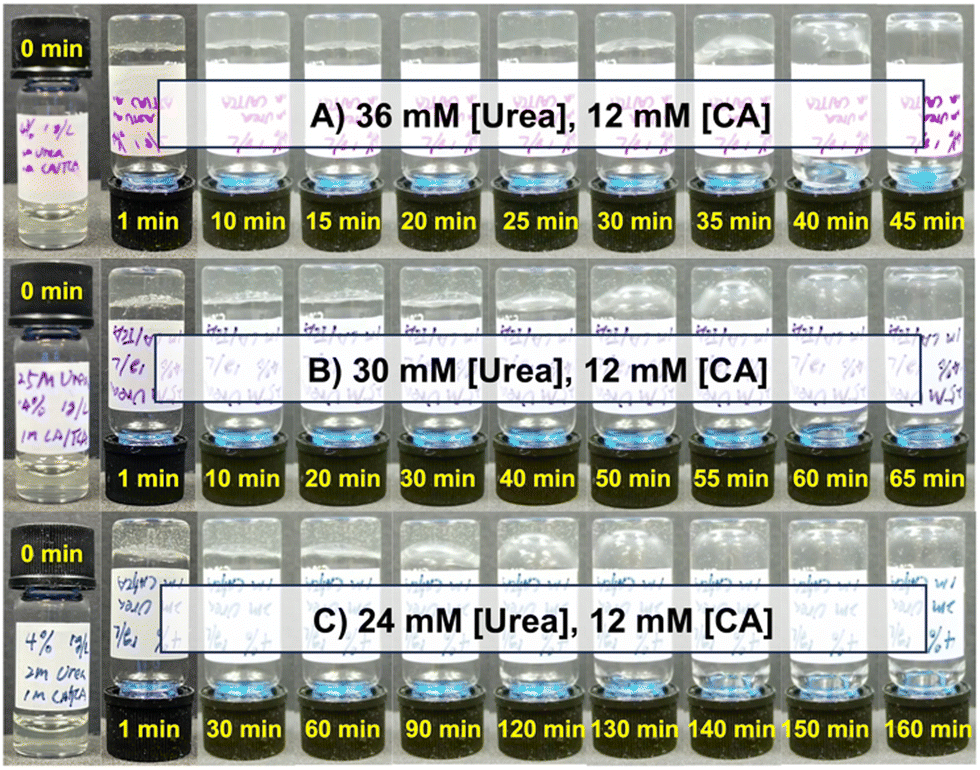 | ||
| Fig. 2 Hydrogel dissolution using a pH-regulatory enzymatic network. PEG8a–BO and PEG4a–CB[7] are mixed 1:1 by mole at 4% w/v at pH 8.5 with [urease] of 1.0 g L−1, then adding (A) 36 mM [urea] and 12 mM [CA], (B) 30 mM [urea] and 12 mM [CA], or (C) 24 mM [urea], 12 mM [CA] with vials inverted and monitored over time for the gel to begin to flow. Times for each condition are shown in each panel. | ||
To better visualize the transient pH reduction, a pH probe was submerged in a sample macromer solution containing 1 g L−1 urease; as 36 mM urea and 12 mM CA were added, the pH transitioned from about 8.5 to about 5 before recovering back to about 8.5 after roughly 40 min (Fig. S1, ESI†). The pH achieved upon CA addition (∼5) and the corrective buffering by urea/urease (∼8.5) are relevant in the context of pKa values for sites on the BO guest. CB[7]–BO complexation is strongly preferred when its tertiary amine and carboxylate sites are both protonated, and prior work showed that hydrogelation occurred for this material when pH was reduced from 11 to 7.25 Alone, the pKa values for the BO guest molecule were measured as 4.25 for the carboxylate and 8.31 for the tertiary amine (Fig. S2, ESI†). However, the actual functional estimation of these pKa values in situ is challenging given that likely complexation-induced pKa shifting upon CB[7] binding favors the preferred binding form.54 It is thus reasonable to expect in the presence CB[7] that the pKa for the tertiary amine group would exist on the higher end of the pH range for the CA and urea/urease system (∼8–10), while the carboxylic acid would have a pKa value closer to the lower end of this range (∼5–6). The triazole is not expected to protonate under pH conditions relevant for the cycling here, as estimates for the pKa of a triazole is ∼2.55
These initial studies suggested a need to better understand the specific parameters of the urease-catalyzed pH feedback loop in order to control hydrogel lifetime. As such, the impact of altering urease concentration ([urease]), urea concentration ([urea]), and citric acid buffer concentration ([CA]) was probed systematically using oscillatory rheology (Fig. 3). By first maintaining [urea] and [CA] constant at 60 mM and 12 mM, respectively, [urease] was varied from 0.4 g L−1 to 1.0 g L−1 (Fig. 3A). Upon addition of CA/urea and brief vortexing, hydrogels were formed by the time samples were loaded onto the rheometer. In all cases, G′ continued to increase in the first ∼10 min after loading, reaching a maximum 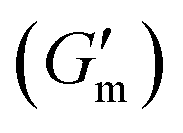 after which point G′ began to spontaneously decrease. The ultimate value of
after which point G′ began to spontaneously decrease. The ultimate value of 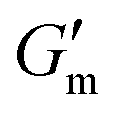 was observed to decrease as [urease] increased (Fig. 3B). Increasing [urease] likewise resulted in a reduction in gel lifetime (tgel) which for the purposes of the studies performed here was defined as the time between
was observed to decrease as [urease] increased (Fig. 3B). Increasing [urease] likewise resulted in a reduction in gel lifetime (tgel) which for the purposes of the studies performed here was defined as the time between 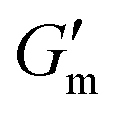 to the point where G′ < 1 Pa. It is noted that while the rheological threshold for a gel is defined as G′ > G′′, methods for tgel estimation here deviated from this since the solution-to-gel G′/G′′ crossover could not be captured on the rheometer due to the time required for sample loading and data collection, while the gel-to-solution G′/G′′ crossover was confounded by a very long tail in the data and occurred at the lower limits of sensitivity for the instrument. These findings for the dependence of hydrogel stiffness and lifetime on [urease] are attributed to more rapid enzymatic conversion of urea to ammonia as [urease] is increased, more quickly correcting the initial acidification caused by CA addition. Accordingly, modulating the concentration of the enzyme, which catalyzes the pH buffering, enables one route to tune hydrogel properties.
to the point where G′ < 1 Pa. It is noted that while the rheological threshold for a gel is defined as G′ > G′′, methods for tgel estimation here deviated from this since the solution-to-gel G′/G′′ crossover could not be captured on the rheometer due to the time required for sample loading and data collection, while the gel-to-solution G′/G′′ crossover was confounded by a very long tail in the data and occurred at the lower limits of sensitivity for the instrument. These findings for the dependence of hydrogel stiffness and lifetime on [urease] are attributed to more rapid enzymatic conversion of urea to ammonia as [urease] is increased, more quickly correcting the initial acidification caused by CA addition. Accordingly, modulating the concentration of the enzyme, which catalyzes the pH buffering, enables one route to tune hydrogel properties.
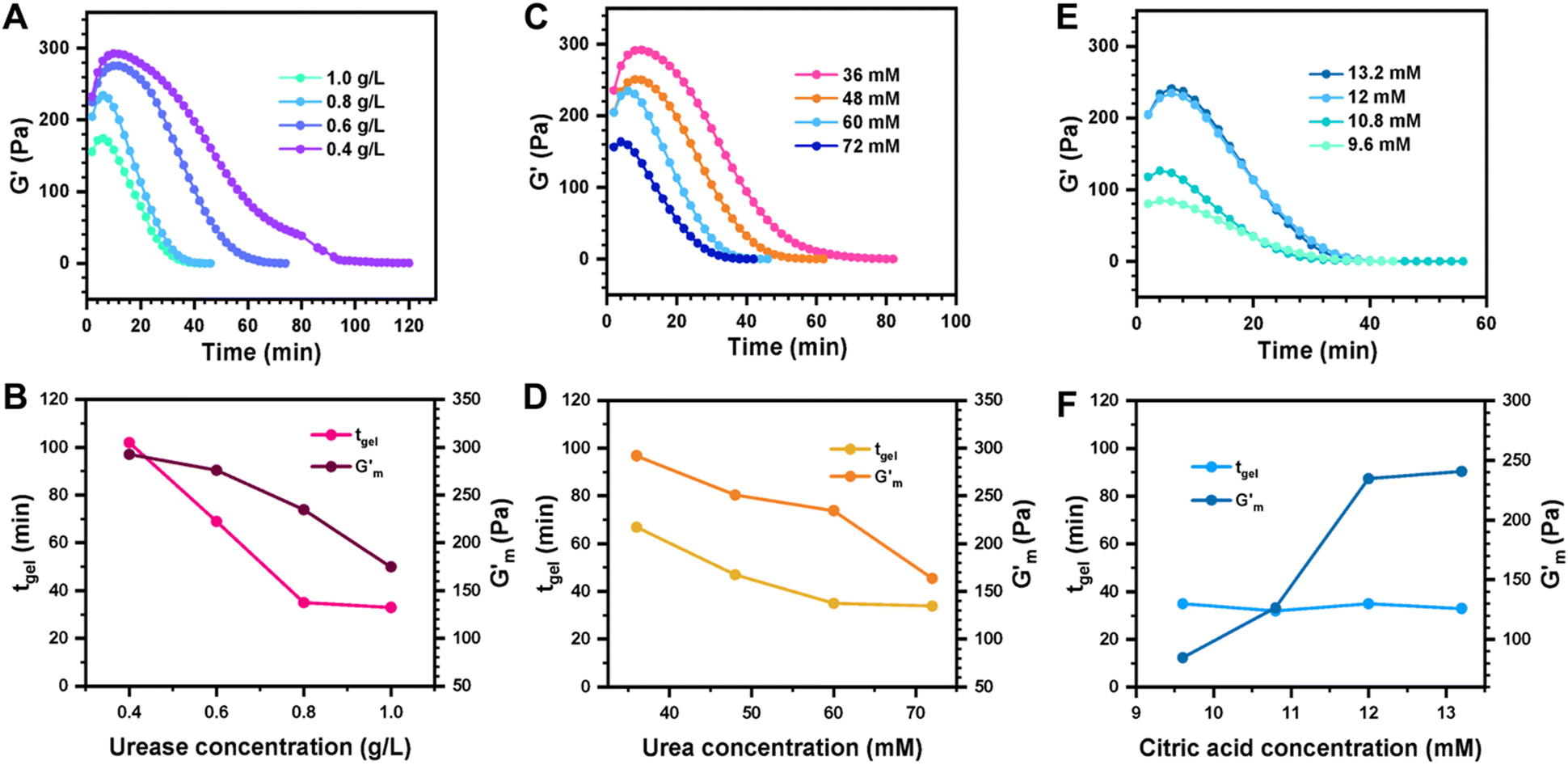 | ||
Fig. 3 Transient hydrogels from mixtures of PEG8a–BO and PEG4a–CB[7] prepared at 1:1 by mole and 4% w/v at pH 8.5. (A) Rheology studies for G′ over time when varying [urease] at constant [urea] of 60 mM and [CA] of 12 mM. (B) Gel lifetime (tgel) and maximum G′ 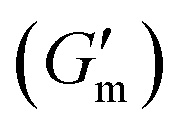 with varying [urease]. (C) Rheology studies for G′ over time when varying [urea] at constant [urease] of 0.8 g L−1 and [CA] of 12 mM. (D) tgel and with varying [urease]. (C) Rheology studies for G′ over time when varying [urea] at constant [urease] of 0.8 g L−1 and [CA] of 12 mM. (D) tgel and 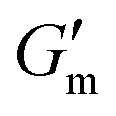 with varying [urea]. (E) Rheology studies for G′ over time when varying [CA] at constant [urease] of 0.8 g L−1 and [urea] of 60 mM. (F) tgel and with varying [urea]. (E) Rheology studies for G′ over time when varying [CA] at constant [urease] of 0.8 g L−1 and [urea] of 60 mM. (F) tgel and 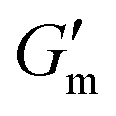 with varying [urea]. Rheology time sweeps in a reaction cycle at varying [CA/Na3C] ([urea] = 60 mM, [urease] = 0.8 g L−1). (F) tgel and with varying [urea]. Rheology time sweeps in a reaction cycle at varying [CA/Na3C] ([urea] = 60 mM, [urease] = 0.8 g L−1). (F) tgel and 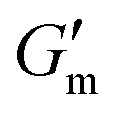 at varying [CA]. For all studies, tgel was defined as the time from at varying [CA]. For all studies, tgel was defined as the time from 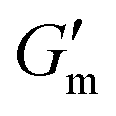 to where G′ is <1.0 Pa. to where G′ is <1.0 Pa. | ||
Next, [urea] was varied from 36 mM to 72 mM, adding this together with a constant [CA] of 12 mM and constant [urease] of 0.8 g L−1 (Fig. 3C). Increased [urea] was expected to increase the biocatalytic production of ammonia underlying pH reversal. Indeed, increased [urea] resulted in decreased values of 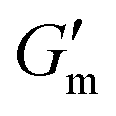 and tgel (Fig. 3D). Finally [urease] and [urea] were kept constant at 0.8 g L−1 and 60 mM, respectively, while [CA] was increased from 9.6 mM to 13.2 mM (Fig. 3E). The resulting hydrogels showed increased
and tgel (Fig. 3D). Finally [urease] and [urea] were kept constant at 0.8 g L−1 and 60 mM, respectively, while [CA] was increased from 9.6 mM to 13.2 mM (Fig. 3E). The resulting hydrogels showed increased 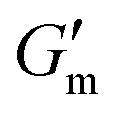 as [CA] was increased, to be expected given the pH-induced gelation mechanism of CB[7]–BO crosslinking is expected to be more responsive to a more significant acid stimulus (Fig. 3F). Interestingly, tgel was not observed to change with the varying [CA], a finding that likely results from the fixed rate of the pH-restoring feedback due to constant [urea] and [urease] coupled with the large excess of [urea] compared to the narrow range of [CA] added in each condition. As such, both hydrogel stiffness and lifetime were directly dependent on [urease] and [urea], driving the forward catalyzed urea conversion reaction that governs hydrogel dissolution through pH reversal. Meanwhile [CA] only served to regulate the stiffness of the hydrogels, arising from differential application of the pH stimulus, but offered limited control over gel lifetime in the concentration ranges evaluated. In all cases, gel lifetimes ranged from 30–100 min. In terms of practical use, these lifetimes were importantly much shorter than the lifetime obtained in the previous report of DMS-driven transient hydrogels, which were on the order of 60+ hours at their shortest.25
as [CA] was increased, to be expected given the pH-induced gelation mechanism of CB[7]–BO crosslinking is expected to be more responsive to a more significant acid stimulus (Fig. 3F). Interestingly, tgel was not observed to change with the varying [CA], a finding that likely results from the fixed rate of the pH-restoring feedback due to constant [urea] and [urease] coupled with the large excess of [urea] compared to the narrow range of [CA] added in each condition. As such, both hydrogel stiffness and lifetime were directly dependent on [urease] and [urea], driving the forward catalyzed urea conversion reaction that governs hydrogel dissolution through pH reversal. Meanwhile [CA] only served to regulate the stiffness of the hydrogels, arising from differential application of the pH stimulus, but offered limited control over gel lifetime in the concentration ranges evaluated. In all cases, gel lifetimes ranged from 30–100 min. In terms of practical use, these lifetimes were importantly much shorter than the lifetime obtained in the previous report of DMS-driven transient hydrogels, which were on the order of 60+ hours at their shortest.25
Repeated gelation cycles were also assessed on 4% w/v samples prepared at pH 8.5 as a solution with 1.2 g L−1 [urease] and serially supplemented with 24 mM [urea] and 12 mM [CA] during rheological assessment (Fig. S3, ESI†). The storage modulus (G′) was tracked to monitor the process of gelation/degelation, and the loss tangent (tanδ = G′′/G′) was also calculated to observe the solution-to-gel transition (tanδ > 1). Over the course of 10 min after loading the sample, G′ increased to reach 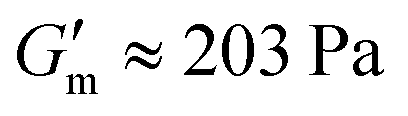 with tan δ ≈ 0.01. After reaching this maximum, the gel began to liquify and the solution was restored at ≈ 110 min after reaching
with tan δ ≈ 0.01. After reaching this maximum, the gel began to liquify and the solution was restored at ≈ 110 min after reaching 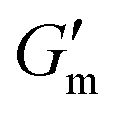 , with G′ ≈ 0.6 Pa and tanδ ≈ 1.1. The process was repeated two additional times, with a general trend for increasing gel lifetimes upon repeat cycling, with times of ≈ 220 min and ≈ 570 min observed for the second and third addition of 24 mM [urea] and 12 mM [CA], respectively. This result may be attributable to some loss in enzymatic function over time or to greater resistance to pH correction with continued accumulation of the CA buffer resulting from successive additions.
, with G′ ≈ 0.6 Pa and tanδ ≈ 1.1. The process was repeated two additional times, with a general trend for increasing gel lifetimes upon repeat cycling, with times of ≈ 220 min and ≈ 570 min observed for the second and third addition of 24 mM [urea] and 12 mM [CA], respectively. This result may be attributable to some loss in enzymatic function over time or to greater resistance to pH correction with continued accumulation of the CA buffer resulting from successive additions.
To explore a possible application for transient hydrogels exhibiting rapid and programmable dissolution through indirect enzymatic feedback control, the burst release of a model macromolecule (70 kDa FITC-labeled dextran) was explored from 4% w/v hydrogel samples (Fig. 4). FITC-labeled dextran has been used as a model macromolecular agent to study controlled release from supramolecular hydrogels.48 The size of this probe is also on the order of the expected pore dimensions of these multi-arm PEG macromer hydrogels,56 thereby limiting the extent of release through passive Fickian diffusion over short times so as to study rapid burst release associated with hydrogel dissolution. As [CA] had limited impact on hydrogel lifetime, [urease] and [urea] were instead altered to realize fast, moderate, and slow burst release profiles with [CA] kept constant at 12 mM. Hydrogels were prepared at a volume of 300 μL and bathed in a bulk phase of 1 mL water and the fluorescence signal of this bulk phase was sampled and monitored over time. At a high level of 1.2 g L−1 [urease] and 72 mM [urea], only 6% of the encapsulated FITC-dextran was released over the first 30 min. However, after an additional 20 min, 100% of the encapsulated FITC-dextran was released into the bulk. Comparatively, when a low level of 0.4 g L−1 [urease] and 32 mM [urea] were used, only 14% had released up to 170 min, with a rapid burst release of the entire remaining dextran then occurring in the ensuing 30 min period. Accordingly, by regulating [urease] and [urea], these transient hydrogels could achieve programmable burst release, offering possibilities for practical biological applications that would not be feasible for harsher methods of creating transiently formed and spontaneously liquifying hydrogels, such as DMS-driven reactions.25 Burst release may confound certain efforts in the field of controlled drug delivery that seek to extend the duration of release with predictable kinetics. However, for certain applications such as oral delivery or vaccination, the stable encapsulation of a payload followed by rapid and timed burst release is desirable.57,58 Indeed, a recent report tuned the molecular weight and monomer ratios of poly(lactic-co-glycolic acid) in order to vary the timing for a rapid burst release from transdermal microneedle devices used for vaccination.59
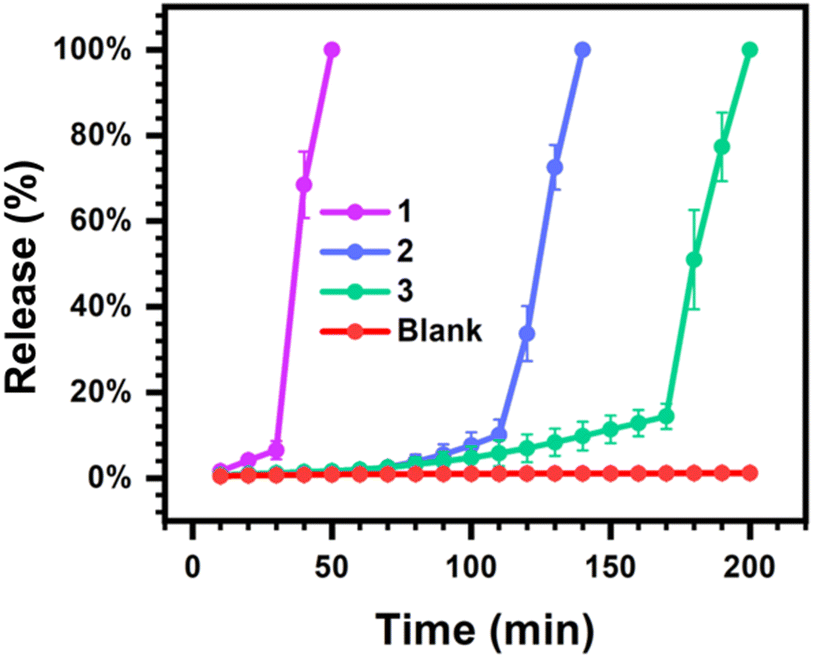 | ||
| Fig. 4 Programmed burst release from mixtures of PEG8a–BO and PEG4a–CB[7] prepared at 1:1 by mole and 4% w/v at pH 8.5 and containing 70 kDa FITC-dextran under the following conditions: (1) fast release: 1.2 g L−1 [urease]; 72 mM [urea]; 12 mM [CA]. (2) Moderate release: 0.6 g L−1 [urease]; 48 mM [urea]; 12 mM [CA]. (3) Slow release: 0.4 g L−1 [urease]; 32 mM [urea]; 12 mM [CA]. (Blank) No release: 0.0 g L−1 [urease]; 32 mM [urea]; 12 mM [CA]. | ||
Conclusions
A new approach is demonstrated here to achieve transient supramolecular host–guest hydrogels through indirect coupling with a biocatalytic network for pH regulation. While numerous examples have demonstrated transient self-assembly and gelation of low molecular weight compounds under both direct and indirect control, the use of related approaches for transient gelation in host–guest supramolecular hydrogels is less common. Building from a prior report of DMS-driven CB[7]–guest hydrogelation,25 the current method achieves a similar outcome of a transient host–guest hydrogels from a biocatalyzed pH-oscillatory network that is more biocompatible than the reactive DMS fueling and affords more rapid gel-to-solution transitions with lifetimes on the order of hours instead of days. Both hydrogel stiffness and lifetime can be tuned and controlled by manipulating the concentration of the urease enzyme or its urea substrate, while the initial citric acid stimulus for gelation only governs hydrogel stiffness. Excitingly, this transient host–guest hydrogel enables programmable burst release of a model macromolecular payload through tuning the concentration of the participants in the biocatalytic control network. Accordingly, beyond mimicking the interesting property of structural transience seen in biological matter, this approach offers opportunities for functional applications towards a new frontier of active and programmable biomaterials.Experimental mthods
Modified PEG synthesis
Synthesis of PEG8a–BO(COO–) and PEG4a–CB[7] was performed as previously reported.25 Starting materials were purchased from commercial suppliers and used without further purification.Preparation of hydrogel samples
A solution of 4% w/v PEG8a–BO(COO–) and PEG4a–CB[7] were prepared at a ratio of 1:1 by mole in DI water at pH 8.5. Urease from Canavalia ensiformis (Type III, powder, 15000–50000 Units per g, solid) purchased from Sigma was added at the designated concentration. A mixture of citric acid buffer and urea was added at the designated concentrations using a pipette, and the samples were mixed by vortexing. The citric acid buffer was prepared as a mixture of citric acid and sodium citrate (9:1 by mole). The initial concentration of citric acid buffer added was 12 mM for all cases except the citric acid screening experiment, where initial concentrations of 9.8, 10.8, 12, and 13.2 mM were explored. The initial urea concentration was 60 mM, except in the urea screening experiment where the initial concentration varied from 36 mM to 72 mM. For cycling experiments, multiple samples were prepared and processed under identical conditions. The initial and reloading concentrations of the citric acid buffer was 12 mM and urea was 24 mM.
Rheology measurements
All rheological measurements were carried out using TA Instruments Discovery HR-2 rheometer fitted with a Peltier stage set to 25 °C. All the samples were prepared as described above. Time sweep measurements from at 10.0 rad s−1 were performed at 2% strain. Data was collected every 2 min. All measurements were carried out using a 25 mm parallel plate and mineral oil was used to minimize sample drying during testing. Fresh samples were reloaded at each cycling point using a transfer pipette, with multiple samples prepared identically and in parallel to afford sufficient sample volumes for each timepoint.pH measurements
To monitor hydrogel pH over time, PEG8a–BO(COO–) and PEG4a–CB[7] hydrogels were prepared at 4% w/v in DI water at pH 8.5 as described above. Urease was premixed in the macromer solution at 1.0 g L−1. A mixture of citric acid buffer and urea at concentrations of 12 mM and 36 mM, respectively, was added using a pipette. The pH was monitored using a pH meter (Mettler Toledo) every five minutes (Fig. S1, ESI†).Release experiments
Sample hydrogels of 300 μL total volume were prepared as described above in microcentrifuge tubes, with a model macromolecule (70 kDa FITC-labeled dextran) included at a concentration of 1 mg mL−1; urease, urea, and citric acid buffer were modified in order to tune the expected lifetime of the hydrogels. The gel samples were then layered with a bulk phase of 1 mL of neutral DI water. Data were collected every 10 min. At each time point, tubes were inverted one time and then 100 μL of solution was taken from the liquid layer above the gels. Fluorescence intensity was measured using a Tecan infinite M200 plate reader (excitation 482 nm, emission 522 nm). Samples were returned to their original tubes after measurement. Samples were tested at n = 3 and the total release percentage was determined as the fluorescence intensity for each point relative to the final fluorescence, in conjunction with a FITC-dextran standard curve (Fig. S4, ESI†).Author contributions
B. S. and M. J. W. conceived of ideas, designed experiments, and analyzed data. M. J. W. provided supervision and oversight to the work. B. S., T. C., W. C., S. X., D. L., C. J. A., and Y. X. contributed to synthesis, experiments, and data collection. B. S. and M. J. W. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. W. gratefully acknowledges funding support from the National Science Foundation (BMAT, 1944875), the National Institutes of Health (NIGMS, R35GM137987), and a 3M Non-Tenured Faculty Award (3M Company). Rheology was performed in the ND Energy Materials Characterization Facility. Schematics were created using https://BioRender.com.Notes and references
- A. Desai and T. J. Mitchison, Annu. Rev. Cell Dev. Biol., 1997, 13, 83–117 CrossRef CAS PubMed.
- M. G. L. van den Heuvel and C. Dekker, Science, 2007, 317, 333–336 CrossRef CAS PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- I. Hwang, R. D. Mukhopadhyay, P. Dhasaiyan, S. Choi, S.-Y. Kim, Y. H. Ko, K. Baek and K. Kim, Nat. Chem., 2020, 12, 808–813 CrossRef CAS PubMed.
- M. Kathan, S. Crespi, N. O. Thiel, D. L. Stares, D. Morsa, J. de Boer, G. Pacella, T. van den Enk, P. Kobauri, G. Portale, C. A. Schalley and B. L. Feringa, Nat. Nanotechnol., 2022, 17, 159–165 CrossRef CAS PubMed.
- M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed.
- M. G. Howlett, A. H. J. Engwerda, R. J. H. Scanes and S. P. Fletcher, Nat. Chem., 2022, 14, 805–810 CrossRef CAS PubMed.
- K. Liu, A. Blokhuis, C. van Ewijk, A. Kiani, J. Wu, W. H. Roos and S. Otto, Nat. Chem., 2023, 1–10 Search PubMed.
- K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS PubMed.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- J. Leira-Iglesias, A. Sorrenti, A. Sato, P. A. Dunne and T. M. Hermans, Chem. Commun., 2016, 52, 9009–9012 RSC.
- A. K. Dambenieks, P. H. Q. Vu and T. M. Fyles, Chem. Sci., 2014, 5, 3396–3403 RSC.
- F. Rakotondradany, M. A. Whitehead, A.-M. Lebuis and H. F. Sleiman, Chemistry, 2003, 9, 4771–4780 CrossRef CAS PubMed.
- J. J. D. de Jong, P. Ralph Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS PubMed.
- D. Samanta and R. Klajn, Adv. Opt. Mater., 2016, 4, 1373–1377 CrossRef CAS.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed.
- Z. Yin, G. Song, Y. Jiao, P. Zheng, J.-F. Xu and X. Zhang, CCS Chem., 2019, 1, 335–342 CrossRef CAS.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- B. Su, T. Chi, Z. Ye, Y. Xiang, P. Dong, D. Liu, C. J. Addonizio and M. J. Webber, Angew. Chem., Int. Ed., 2023, 62, e202216537 CrossRef CAS PubMed.
- S. M. Mantooth, B. G. Munoz-Robles and M. J. Webber, Macromol. Biosci., 2019, 19, e1800281 CrossRef PubMed.
- J. C. R. Rippey and M. I. Stallwood, Emerg. Med. J., 2005, 22, 878–879 CrossRef CAS PubMed.
- M. P. van der Helm, G. Li, M. Hartono and R. Eelkema, J. Am. Chem. Soc., 2022, 144, 9465–9471 CrossRef CAS PubMed.
- J. L.-Y. Chen, S. Maiti, I. Fortunati, C. Ferrante and L. J. Prins, Chemistry, 2017, 23, 11549–11559 CrossRef CAS PubMed.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef PubMed.
- X. Hao, W. Sang, J. Hu and Q. Yan, ACS Macro Lett., 2017, 6, 1151–1155 CrossRef CAS PubMed.
- X. Hao, H. Wang, W. Zhao, L. Wang, F. Peng and Q. Yan, CCS Chem., 2022, 4, 838–846 CrossRef CAS.
- M. Jain and B. J. Ravoo, Angew. Chem., Int. Ed., 2021, 60, 21062–21068 CrossRef CAS PubMed.
- H. Lu, J. Hao and X. Wang, ChemSystemsChem, 2022, 4, e202100050 CrossRef CAS.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS PubMed.
- X. Fan and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 11398–11405 CrossRef CAS PubMed.
- I. Maity, C. Sharma, F. Lossada and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 22537–22546 CrossRef CAS PubMed.
- C. Sharma, I. Maity and A. Walther, Chem. Commun., 2023, 59, 1125–1144 RSC.
- H. Che, S. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS PubMed.
- C. Sharma and A. Walther, Angew. Chem., Int. Ed., 2022, 61, e202201573 CrossRef CAS PubMed.
- G. Li, R. W. Lewis and R. Eelkema, CCS Chem., 2023, 6, 27–40 CrossRef.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- L. Zou, A. S. Braegelman and M. J. Webber, ACS Appl. Mater. Interfaces, 2019, 11, 5695–5700 CrossRef CAS PubMed.
- A. S. Braegelman and M. J. Webber, Theranostics, 2019, 9, 3017–3040 CrossRef CAS PubMed.
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984–12989 CrossRef CAS PubMed.
- A. J. Russell, M. Erbeldinger, J. J. DeFrank, J. Kaar and G. Drevon, Biotechnol. Bioeng., 2002, 77, 352–357 CrossRef CAS PubMed.
- G. Hu, J. A. Pojman, S. K. Scott, M. M. Wrobel and A. F. Taylor, J. Phys. Chem. B, 2010, 114, 14059–14063 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chemistry, 2021, 27, 8928–8939 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- W. Zhai, B. M. Chapin, A. Yoshizawa, H.-C. Wang, S. A. Hodge, T. D. James, E. V. Anslyn and J. S. Fossey, Org. Chem. Front., 2016, 3, 918–928 RSC.
- A. S. Braegelman, R. C. Ollier, B. Su, C. J. Addonizio, L. Zou, S. L. Cole and M. J. Webber, ACS Appl. Bio Mater., 2022, 5(10), 4589–4598 CrossRef CAS PubMed.
- A. Sanchez, R. K. Gupta, M. J. Alonso, G. R. Siber and R. Langer, J. Pharm. Sci., 1996, 85, 547–552 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- K. T. M. Tran, T. D. Gavitt, N. J. Farrell, E. J. Curry, A. B. Mara, A. Patel, L. Brown, S. Kilpatrick, R. Piotrowska, N. Mishra, S. M. Szczepanek and T. D. Nguyen, Nat. Biomed. Eng., 2021, 5, 998–1007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00545g |
| This journal is © The Royal Society of Chemistry 2024 |