
Cooling rate uncovers epimer-dependent supramolecular organization of carbohydrate amphiphiles†
Vânia I. B.
Castro
ab,
Yuting
Gao
abc,
Alexandra
Brito
ab,
Jie
Chen
 c,
Rui L.
Reis
ab,
Iva
Pashkuleva
*ab and
Ricardo A.
Pires
*ab
c,
Rui L.
Reis
ab,
Iva
Pashkuleva
*ab and
Ricardo A.
Pires
*ab
a3B's Research Group, I3Bs – Research Institute on Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Parque de Ciência e Tecnologia, Zona Industrial da Gandra, 4805-017 Barco, Guimarães, Portugal. E-mail: rpires@i3bs.uminho.pt; pashkuleva@i3bs.uminho.pt
bICVS/3B's–PT Government Associate Laboratory, Braga/Guimarães, Portugal
cDepartment of Chemical Engineering, School of Environmental and Chemical Engineering, Shanghai University, Shangda Road 99, Shanghai 200444, P. R. China
First published on 27th June 2024
Abstract
We show distinct CH–π interactions and assembly pathways for the amphiphile N-(fluorenylmethoxycarbonyl)-galactosamine and its epimer N-(fluorenylmethoxycarbonyl)-glucosamine. These differences result in the formation of supramolecular nanofibrous systems with opposite chirality. Our results showcase the importance of the carbohydrates structural diversity for their specific biointeractions and the opportunity that their ample interactome offers for synthesis of versatile and tunable supramolecular (bio) materials.
Introduction
Molecular self-assembly is a ubiquitous process in Nature used by most living entities for building-up functional structures.1,2 It has been explored in the development of supramolecular biomaterials, in which specifically designed synthetic peptide amphiphiles (PAs) are assembled to generate systems for different applications, as for example: hydrogel-based biomaterials;3–5 vaccine adjuvants;6,7 drug-delivery systems;8,9 among others.10 Recently, carbohydrate amphiphiles (CAs) emerged as alternative, or complementary, building blocks to PAs due to their ample supramolecular biointeractome: because of their intrinsic structure even the simplest carbohydrate units, i.e. monosaccharides, can display a vast variety of ligand structures that render their multivalent interactions specific and selective.11–14 However, our understanding of the mechanism(s) and the effect of distinct structural elements on the CAs assembly is still vague. In particular, to our knowledge, there have been no systematic studies on the effect of carbohydrate stereochemistry on the morphology, stability, and gelation capacity of the CAs.Herein, we investigated two of the simplest CAs described to date, namely N-(fluorenylmethoxycarbonyl)-glucosamine (Fmoc-GlcN, 1) and N-(fluorenylmethoxycarbonyl)-galactosamine (Fmoc-GalN, 2) that assemble into nanofibers and gel upon undergoing a heating–cooling cycle.151 and 2 differ by their stereochemistry at C4 position (Fig. 1). Moreover, we also included N-(fluorenylmethoxycarbonyl)-mannosamine (Fmoc-ManN, 3) in this study to assess the impact of changing the spatial orientation of the fluorenylmethoxycarbonyl (Fmoc) moiety (at the C2 position) in the ability of the CA to assemble and gel.
 | ||
| Fig. 1 Chemical structure of the studied carbohydrate amphiphiles N-(fluorenylmethoxycarbonyl)-glucosamine (Fmoc-GlcN, 1), N-(fluorenylmethoxycarbonyl)-galactosamine (Fmoc-GalN, 2) and N-(fluorenylmethoxycarbonyl)-mannosamine (Fmoc-ManN, 3). The arrows indicate the stereoisomeric position of 2 and 3 in respect to 1. | ||
Results and discussion
Synthesis of the carbohydrate amphiphiles
We synthesized the stereoisomers 1–3 (Fig. 1) by one-step reaction between the respective glycosylamine and N-fluorenylmethoxycarbonyl chloride. The structure and purity of each CA was confirmed by HPLC, FTIR, MS and NMR (Fig. S1–S6, ESI†).Temperature-mediated assembly and gelation
The CAs were dissolved in water upon heating to 363 K and then cooled to room temperature at different rates (slow: 5 K min−1; fast: 40 K min−1) because of the known effect of the cooling rate on the thermodynamics of the self-assembly process.16 Of note, the stereoisomer 3 was soluble at high temperature, but precipitated upon cooling. Previous studies with similar carbohydrate amphiphiles have demonstrated that the structure of the aromatic portion and the link between this portion and the carbohydrate can influence the gelation by altering the hydrophilic/hydrophobic balance.15,17 Herein, we add a stereochemical perspective for tailoring the gelation: the axial position of the hydrophobic Fmoc at C2 in stereoisomer 3 is torsionally strained and crowded, thus, disturbing the mannose hydration and the intermolecular π-interactions, i.e. the assembly of stable long-range ordered supramolecular structures.18–20 The behaviour of 3 is in good agreement with previous data showing a disruptive effect of axial substituents in CH–π stacking.21The other two stereoisomers 1 and 2 formed stable hydrogels at the studied heating–cooling cycles as confirmed by the inverted vial test (Fig. S7, ESI†), with Young's modulus in the range of 7–9 kPa (concentration of 12 mM, Fig. S8, ESI†).
Characterization of the assembly process
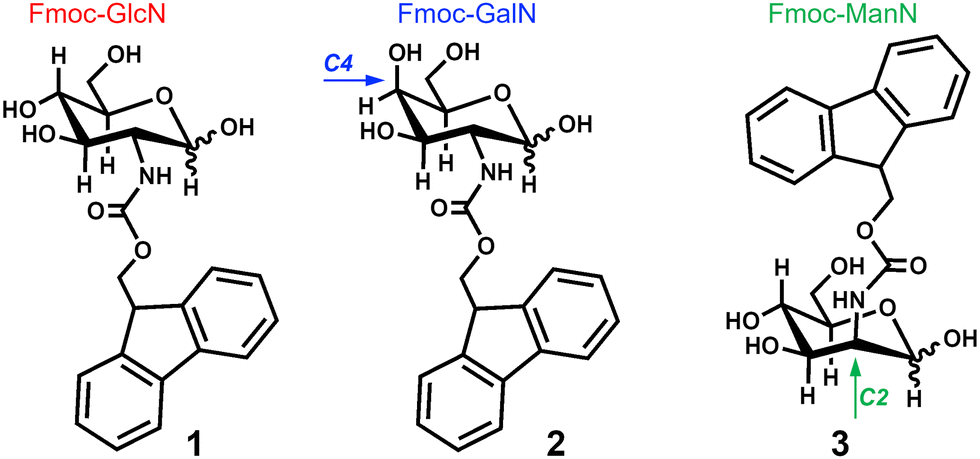
Fluorescence (FL) spectroscopy revealed contribution of the Fmoc moiety to the assembly of both CAs (Fig. 2A and B): a drastic reduction of FL intensity upon cooling is indicative of assembly and gelation, as previously observed for fluorenyl-based PAs and CAs.15,22,23 Circular dichroism (CD) spectroscopy showed an emergence of a characteristic signal at ∼304 nm (π–π* transitions)17,24 upon cooling (Fig. 2C and D), evidencing the formation of chiral structures by 1 and 2.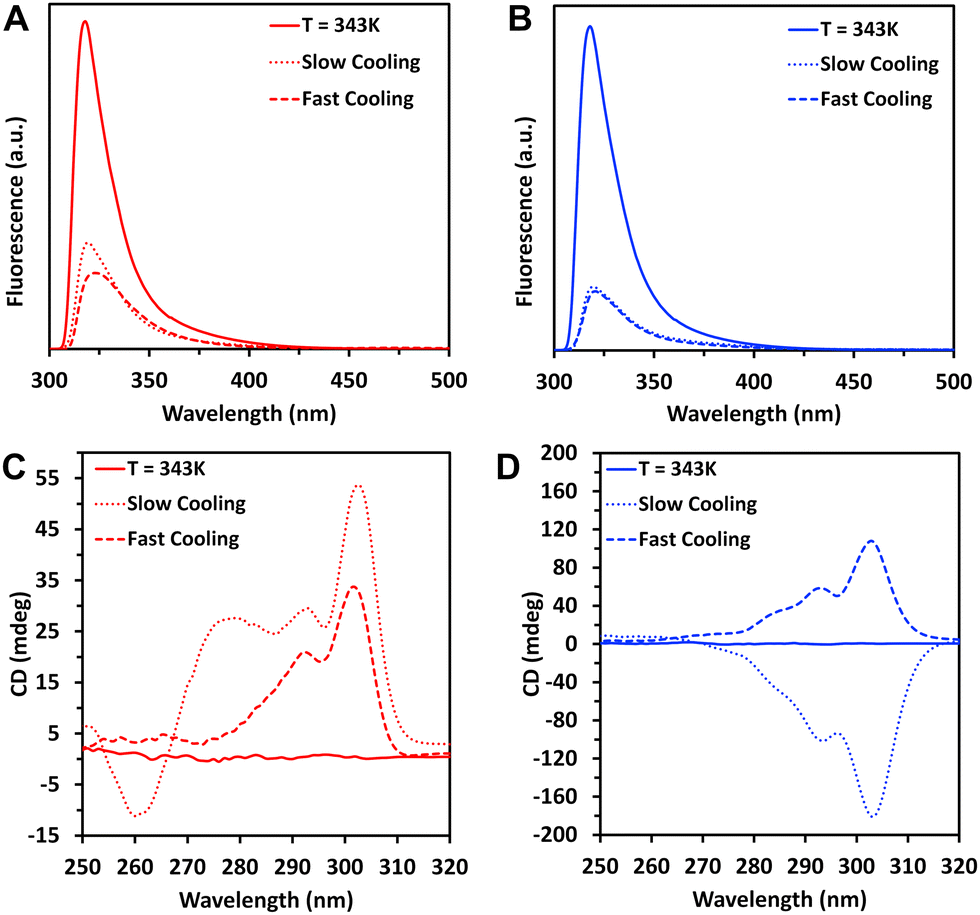 | ||
| Fig. 2 Fluorescence (A) and (B) and CD (C) and (D) spectra of the hydrogels assembled from 1 (A) and (C) and 2 (B) and (D) in water (16 mM) under two cooling rates (slow: ∼5 K min−1 and fast: ∼40 K min−1). | ||
These data agree with a previous studies showing that the assembly of 1 and 2 is driven by different non-covalent interactions, such as: π–π stacking of adjacent fluorenyl groups (T-stacking); H-bonding of the carbohydrate OH groups and the NH group of the carbamate; and CH–π interactions.15,25,26 Hydrophobic CH–π interactions are crucial for carbohydrate recognition in biological systems and occur between CH from the carbohydrate and the aromatic residues of another molecule (e.g. aromatic amino acids from a protein in biosystems or Fmoc moieties in the case of 1 and 2).21,27 These interactions are driven by weak London dispersion forces and thus the cooperative presence of three CH groups oriented towards the planar aromatic regions enhances the binding.28,29 Because 1H NMR spectroscopy showed predominance of the β form (∼80%) with a low contribution of the α anomer for 1 and 2 at high temperature (Fig. 3A, Fig. S6, ESI†), CH–π stacking can occur via only one planar region (C1–C3–C5) in the glucosamine amphiphile 1, while in the case of 2 there are two possibilities (C1–C3–C5 and C3–C4–C5) (Fig. 3B).27,30
 | ||
| Fig. 3 (A) 1H NMR spectra (T = 353 K, 16 mM in D2O) of 1 (red) and 2 (blue) showing the predominance of the β anomers (δ–4.4 ppm). (B) Schematic presentation of the CH planar regions that can participate in CH–π interactions for stereoisomers 1 (red) and 2 (blue). | ||
Upon fast-cooling, similar CD spectra were observed for 1 and 2 (Fig. 2C and D), consistent with an assembly mediated by the C1–C3–C5 planar region for both amphiphiles. At slow cooling rate, an additional peak at ∼280 nm appeared in the CD spectrum of 1 (but not 2) suggesting an increased complexity of the assembly process. At these conditions, an inversion of the signals’ polarity (from positive to negative) was observed in the CD spectrum of 2 (Fig. 2D). Such behaviour has been previously reported for other systems, and is related with helicity changes of the assembled supramolecular structures.31,32 Thus, we used scanning electron microscopy (SEM) to observe the systems generated by the assembly of 1 and 2 and to assess their helicity (Fig. 4).
 | ||
| Fig. 4 SEM images of the nanofibers generated from 12 mM solutions of 1 (A) and (C) or 2 (B) and (D) in water upon a fast (40 K min−1; A and B) or slow (5 K min−1; C and D) cooling rate. Scale bar represents 500 nm. | ||
The SEM images confirmed the unidirectional assembly of both 1 and 2 resulting in the formation of nanofibers that interact with each other laterally and form fibre bundles. The bundling made challenging to visualize the morphology/twisting of the individual nanofibers but it was possible to observe the helicity of the bundles themselves. Upon fast cooling, the assemblies generated by 1 and 2 presented a clockwise helicity (Fig. 4A and B). When the cooling process was slow, we observed bundles with the same helicity for 1 (Fig. 4C) and with opposite, i.e., counterclockwise, helicity for 2 (Fig. 4D). These results are consistent with the CH planar regions in 1 and 2 (Fig. 3B), as well as with the CD data. Of note, the CD signal accounts all contributions, i.e., the individual fibres and bundles, and reflects the predominant type of helicity.
Kinetics of the assembly
Assessing the assembly at different concentrations and temperatures can give information about the mechanism of the process.33 The assembly can follow two main mechanisms that have been proposed for the supramolecular fibre formation: isodesmic and cooperative.34,35 The isodesmic process is characterized by an even growth of the fibre: it is defined by one association constant and a characteristic near-sigmoidal kinetics. The cooperative assembly, also known as nucleation-growth, is an extension of the isodesmic model that involves an additional nucleation step at the beginning of the process. The nucleation step demands higher energy when compared to the growth phase,35i.e., there is a kinetic barrier at the nucleation stage that is described by an additional association constant.33 The two steps, nucleation and fibre growth, are distinct in the experimental aggregation curves and a deviation from the sigmoidal conversion is observed. In all the cases, a parameter that is commonly used to characterize the assembly process is the temperature of elongation (Te), i.e., the temperature at which the elongation of the supramolecular structures starts.We recorded the FL spectra of 1 and 2 at different temperatures using a slow cooling rate (i.e., 5 K min−1) and the normalized intensity of the FL signal at ∼300 nm was plotted as a function of temperature (Fig. 5). The curve obtained for 1 at 8 mM presented a steady FL decrease, suggesting an isodesmic assembly at these conditions. At higher concentrations of 1 (i.e., 12 mM and 16 mM), we observed curves with two regions: a steep decrease between ∼338 K and ∼318 K followed by a slower one between ∼318 K and ∼283 K that is compatible with cooperative assembly. Similar type of nucleation-growth mechanism (i.e., cooperative assembly) was observed for 2 within the whole range of tested concentrations.
 | ||
| Fig. 5 Normalized fluorescence (FL) of the assemblies of 1 (A) and 2 (B) as a function of temperature at different concentrations (8, 12 and 16 mM). FL intensities were recorded at 318 nm. | ||
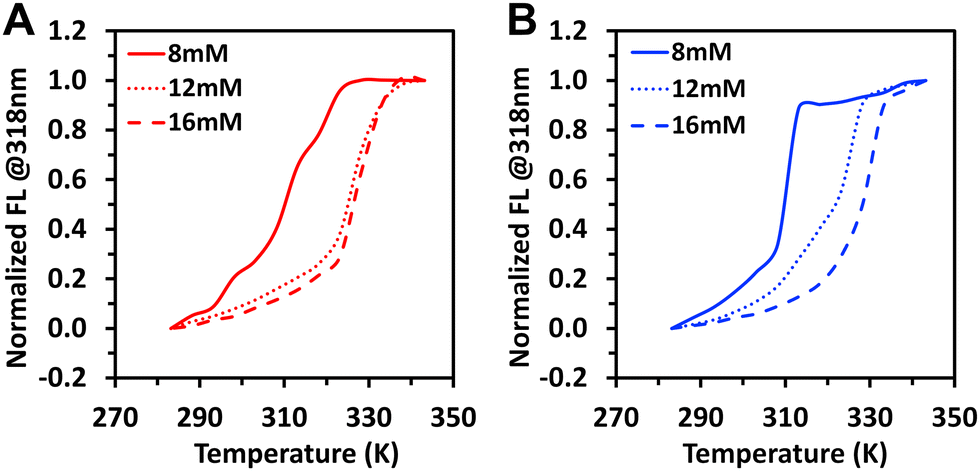
The Te was determined from the CD spectra at different temperatures. Te was assessed upon cooling (slow rate) but also under the heating regime (Te′). In this experimental setup, the Te′ is not influenced by the nucleation as in the case of Te. We used the signal at ∼304 nm to follow the assembly, and Te (or Te′) was defined as the temperature at which the signal deviates from zero in the case of Te, or reaches zero in the case of Te′ (examples shown in Fig. 6B and C). At the studied conditions, the two amphiphiles assembled via a cooperative mechanism as shown by the difference in Te or Te′ associated with the nucleation step (Fig. 6A). While Te′ of 1 and 2 were identical (heating curves, Fig. 6A), the Te were significantly different (cooling curves, Fig. 6A), with higher temperatures observed for 1.
 | ||
| Fig. 6 (A) Elongation temperature of the assemblies at different concentrations of the amphiphiles 1 and 2 during cooling (Te) and heating (Te′) regime determined from the CD spectra (signal at ∼304 nm). (B) and (C) Intensity of the CD signal at ∼304 nm recorded upon heating and cooling (5 K min−1) of 12 mM solutions of 1 (B) and 2 (C) in water. | ||
Of note, the assembly of 2 (cooling curve, Fig. 6C) exhibits a profile characteristic for a cooperative mechanism with a nucleation step, while the observed typical sigmoidal heating curve is characteristic of an isodesmic mechanism, i.e., without contribution from a nucleation stage. This observation was expected as the heating cycle is initiated with the nanofibers already formed, and there is no nucleation under these conditions.
A closer look at the CD data for the assembly of 1 and 2 during cooling at lower temperatures (e.g., below 295 K, Fig. 6B and C) revealed more differences between the assembly of these amphiphiles. In the case of 2 (Fig. 6C), the decrease of the CD signal during assembly stopped at ∼295 K and an increase is observed at the end of the assembly. Such behaviour has been previously reported for other systems and explained by the fibres’ bundling and formation of superhelices with different helicities that revert the overall CD signal.16,31 This behaviour was not observed for 1 (see Fig. 6B). Altogether, our data showed that the assembly of 2 is more complex than 1 and can follow different pathways that depend on the cooling regime, resulting in different supramolecular structures.
Conclusions
We show that carbohydrate stereochemistry can influence the self-assembly process of CAs via CH–π interactions. Among the biomolecules that code and transfer bioinformation (proteins, nucleic acids and glycans), the carbohydrates have the richest stereoisomer diversity that can be used to direct different assembling pathways, affecting the assemblies’ morphological presentation. Thus, this stereochemical diversity of carbohydrates offers possibilities for the development of adaptable and tuneable systems relevant for several (bio) applications.Author contributions
Conceptualization: IP and RAP; investigation: VIBC, YG and AB; writing: VIBC, IP and RAP; supervision: JC, RLR, IP and RAP; funding acquisition: JC, RLR, IP and RAP.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Commission H2020 programme and the Portuguese Foundation for Science and Technology (FCT) for financial support: M-ERA-NET3/0007/2021 (RePark); RAP - CEECIND/05623/2022; VIBC – PD/BD/135256/2017; COVID/BD/152018/2021. We also acknowledge the project “TERM RES Hub – Scientific Infrastructure for Tissue Engineering and Regenerative Medicine” - PINFRA/22190/2016 (Norte-01-0145-FEDER-022190, co-funded by FCT and the Northern Portugal Regional Coordination and Development Commission, CCDR-N), for providing lab facilities, state-of-the art equipment and highly qualified human resources.Notes and references
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wires Nanomed. Nanobitechnol., 2013, 5, 582–612 CrossRef CAS PubMed.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- H. M. Wang, Z. Luo, Y. C. Z. Wang, T. He, C. B. Yang, C. H. Ren, L. S. Ma, C. Y. Gong, X. Y. Li and Z. M. Yang, Adv. Funct. Mater., 2016, 26, 1822–1829 CrossRef CAS.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- D. Kalafatovic, M. Nobis, J. Y. Son, K. I. Anderson and R. V. Ulijn, Biomaterials, 2016, 98, 192–202 CrossRef CAS PubMed.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS PubMed.
- S. S. Lee, T. Fyrner, F. Chen, Z. Alvarez, E. Sleep, D. S. Chun, J. A. Weiner, R. W. Cook, R. D. Freshman, M. S. Schallmo, K. M. Katchko, A. D. Schneider, J. T. Smith, C. W. Yun, G. Singh, S. Z. Hashmi, M. T. McClendon, Z. L. Yu, S. R. Stock, W. K. Hsu, E. L. Hsu and S. I. Stupp, Nat. Nanotechnol., 2017, 12, 821 CrossRef CAS PubMed.
- A. Brito, Y. M. Abul-Haija, D. S. da Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem. Sci., 2019, 10, 2385–2390 RSC.
- J. Zhou, X. W. Du, X. Y. Chen and B. Xu, Biochemistry, 2018, 57, 4867–4879 CrossRef CAS PubMed.
- V. I. B. Castro, A. R. Araujo, F. Duarte, A. Sousa-Franco, R. L. Reis, I. Pashkuleva and R. A. Pires, ACS Appl. Mater. Interfaces, 2023, 15, 29998–30007 CrossRef CAS PubMed.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- A. Osypenko, E. Moulin, O. Gavat, G. Fuks, M. Maaloum, M. A. J. Koenis, W. J. Buma and N. Giuseppone, Chem. – Eur. J., 2019, 25, 13008–13016 CrossRef CAS PubMed.
- Z. M. Yang, G. L. Liang, M. L. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845 RSC.
- L. L. Kiessling and R. C. Diehl, ACS Chem. Biol., 2021, 16, 1884–1893 CrossRef CAS PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- C. He, S. Wu, D. Liu, C. Chi, W. Zhang, M. Ma, L. Lai and S. Dong, J. Am. Chem. Soc., 2020, 142, 17015–17023 CrossRef CAS PubMed.
- E. Jiménez-Moreno, G. Jiménez-Osés, A. M. Gómez, A. G. Santana, F. Corzana, A. Bastida, J. Jiménez-Barberodef and J. L. Asensio, Chem. Sci., 2015, 6, 6076–6085 RSC.
- Z. M. Yang, H. W. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- Z. M. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC.
- W. P. Wang, Z. M. Yang, S. Patanavanich, B. Xu and Y. Chau, Soft Matter, 2008, 4, 1617–1620 RSC.
- S. Gim, G. Fittolani, Y. Nishiyama, P. H. Seeberger, Y. Ogawa and M. Delbianco, Angew. Chem., Int. Ed., 2020, 59, 22577–22583 CrossRef CAS.
- S. Gim, G. Fittolani, Y. Yu, Y. T. Zhu, P. H. Seeberger, Y. Ogawa and M. Delbianco, Chem. – Eur. J., 2021, 27, 13139–13143 CrossRef CAS.
- J. L. Asensio, A. Ardá, F. J. Cañada and J. Jiménez-Barbero, Acc. Chem. Res., 2013, 46, 946–954 CrossRef CAS PubMed.
- K. Ramirez-Gualito, R. Alonso-Rios, B. Quiroz-Garcia, A. Rojas-Aguilar, D. Diaz, J. Jimenez-Barbero and G. Cuevas, J. Am. Chem. Soc., 2009, 131, 18129–18138 CrossRef CAS PubMed.
- A. Brito, S. Kassem, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem, 2021, 7, 2943–2964 CAS.
- V. Spiwok, Molecules, 2017, 22 Search PubMed.
- M. Hifsudheen, R. K. Mishra, B. Vedhanarayanan, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2017, 59, 12634–12638 CrossRef PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–U103 CrossRef CAS PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- Y. T. Sang and M. H. Liu, Chem. Sci., 2022, 13, 633–656 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00728j |
| This journal is © The Royal Society of Chemistry 2024 |