
Advances in drug delivery-based therapeutic strategies for renal fibrosis treatment
Sida
Huang†
b,
Hanqi
Lu†
 a,
Jin
Chen†
a,
Chengyi
Jiang
b,
Guanmin
Jiang
*c,
Govindhan
Maduraiveeran
*d,
Ying
Pan
b,
Jianqiang
Liu
*b and
Li-Er
Deng
*a
a,
Jin
Chen†
a,
Chengyi
Jiang
b,
Guanmin
Jiang
*c,
Govindhan
Maduraiveeran
*d,
Ying
Pan
b,
Jianqiang
Liu
*b and
Li-Er
Deng
*a
aDepartment of Nephrology, Dongguan Hospital of Guangzhou University of Traditional Chinese Medicine, Dongguan, Guangdong 523000, China. E-mail: dengruiqiao@126.com
bDongguan Key Laboratory of Drug Design and Formulation Technology, Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Guangdong Medical University, Dongguan, 523808, China. E-mail: jianqiangliu2010@126.com
cDepartment of Oncology, Affiliated Dongguan Hospital, Southern Medical University (Dongguan people's hospital), 78 Wandao Road South, Dongguan, 523059 Guangdong, China. E-mail: terryjzm@gmail.com
dMaterials Electrochemistry Laboratory, Department of Chemistry, SRM Institute of Science and Technology, Kattankulathur – 603 203, Chengalpattu, Tamil Nadu, India. E-mail: maduraig@srmist.edu.in
First published on 7th June 2024
Abstract
Renal fibrosis is the result of all chronic kidney diseases and is becoming a major global health hazard. Currently, traditional treatments for renal fibrosis are difficult to meet clinical needs due to shortcomings such as poor efficacy or highly toxic side effects. Therefore, therapeutic strategies that target the kidneys are needed to overcome these shortcomings. Drug delivery can be attained by improving drug stability and addressing controlled release and targeted delivery of drugs in the delivery category. By combining drug delivery technology with nanosystems, controlled drug release and biodistribution can be achieved, enhancing therapeutic efficacy and reducing toxic cross-wise effects. This review discusses nanomaterial drug delivery strategies reported in recent years. Firstly, the present review describes the mechanisms of renal fibrosis and anti-renal fibrosis drug delivery. Secondly, different nanomaterial drug delivery strategies for the treatment of renal injury and fibrosis are highlighted. Finally, the limitations of these strategies are also discussed. Investigating various anti-renal fibrosis drug delivery strategies reveals the characteristics and therapeutic effects of various novel nanosystem-derived drug delivery approaches. This will serve as a reference for future research on drug delivery strategies for renal fibrosis treatment.
1 Introduction
Acute kidney injury (AKI) and chronic kidney disease (CKD) are the two major categories of kidney disease, with CKD accounting for far more patients than AKI. According to the Global Burden of Disease (GBD) and World Health Organization (WHO) estimates, CKD caused 1.2 million deaths in 2015.1,2 Based on WHO forecasts, the death rate from CKD is expected to increase by 14 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by 2030.3 The quantity of people with kidney disease is amplified as a result of the New Crown epidemic, and the mortality rate for CKD is likely to be higher than the prediction of WHO.4 A study of global chronic kidney disease from 1990–2017 showed that the majority of CKD patients worldwide were concentrated in stages 1–3, accounting for 97.8 percent of all patients. The CKD stage 4 and CKD stage 5 accounted for 1.76 percent and 0.77 percent of the total patients, respectively. Many CKD patients were concentrated in stages 1–3, which accounted for 97.8 percent of all patients, while CKD stage 4 and CKD stage 5 accounted for 1.76 percent and 0.77 percent of the total proportion of patients, respectively. As a result, renal fibrosis is increasingly becoming a global health problem.5,6
000 by 2030.3 The quantity of people with kidney disease is amplified as a result of the New Crown epidemic, and the mortality rate for CKD is likely to be higher than the prediction of WHO.4 A study of global chronic kidney disease from 1990–2017 showed that the majority of CKD patients worldwide were concentrated in stages 1–3, accounting for 97.8 percent of all patients. The CKD stage 4 and CKD stage 5 accounted for 1.76 percent and 0.77 percent of the total patients, respectively. Many CKD patients were concentrated in stages 1–3, which accounted for 97.8 percent of all patients, while CKD stage 4 and CKD stage 5 accounted for 1.76 percent and 0.77 percent of the total proportion of patients, respectively. As a result, renal fibrosis is increasingly becoming a global health problem.5,6
Renal fibrosis is a pathological change that progresses over time, causing the functionality of the kidneys to alter from healthy to damaged or injured. This change can be majorly categorized into three stages: the inflammatory response phase, the fibrosis formation phase, and the scar formation phase.7,8 High levels of matrix deposition and interstitial fibroblast activation and proliferation are hallmarks of renal fibrosis. Additionally, damaged renal tubular epithelial cells and inflammatory cells secrete several growth factors and mediators that support angiogenesis and lymphangiogenesis. These observations are typical of a range of renal fibrosis illnesses. Conventional treatments for renal fibrosis mainly include drug therapy, lifestyle modification, and renal replacement therapy. However, the shortcomings of these treatment modalities gradually make it difficult to meet clinical needs. Pharmacological treatment of renal fibrosis usually slows down the process of renal fibrosis by using immunosuppressants, hormone-regulating drugs, and blood pressure-controlling drugs. However, the ability of these drugs to control the progression of renal fibrosis is limited. The adverse effects of drug therapy are hard to ignore, such as those due to using immunosuppressants, which may lead to an increased risk of infection, and hormone therapy, which may lead to osteoporosis. Clinical investigations on other proposed options, such as connective tissue growth factor inhibitors, bone morphogenetic protein (BMP)-7 agonists, and transforming growth factor (TGF)-β antagonists, have yielded substandard results.9 Renal replacement therapies such as haemodialysis and peritoneal dialysis can only partially replace the functioning of the kidneys. They do not fully mimic normal physiological processes, and long-term reliance on these therapies may have a serious impact on the quality of life of patients. In addition, renal replacement therapy carries the risk of complications, such as cardiovascular problems and infections. Drug delivery challenges are a major contributor to the struggle to meet expectations for renal fiber therapy.
A set of technologies known as “drug delivery technology” controls all aspects of the distribution of medication in an organism, including its location, timing, and dosage. Drug delivery can be achieved by improving drug stability, addressing controlled release, and battered drug delivery in the delivery category. Incorporating drug delivery into renal antifibrotic therapy enhances drug targeting to the kidneys, improves bioavailability, and reduces damage to other organs.10 More importantly, the use of drug delivery technology holds the promise of developing a novel clinical treatment strategy for renal fibrosis (Fig. 1). This review focuses on research advances in drug delivery-based therapeutic strategies for renal fibrosis. It focuses on the drug delivery strategies for renal antifibrosis, as well as the methods of administrating medications to the kidneys and renal fibrosis. We also present these summaries to guide research on drug delivery strategies for renal fibrosis treatment.
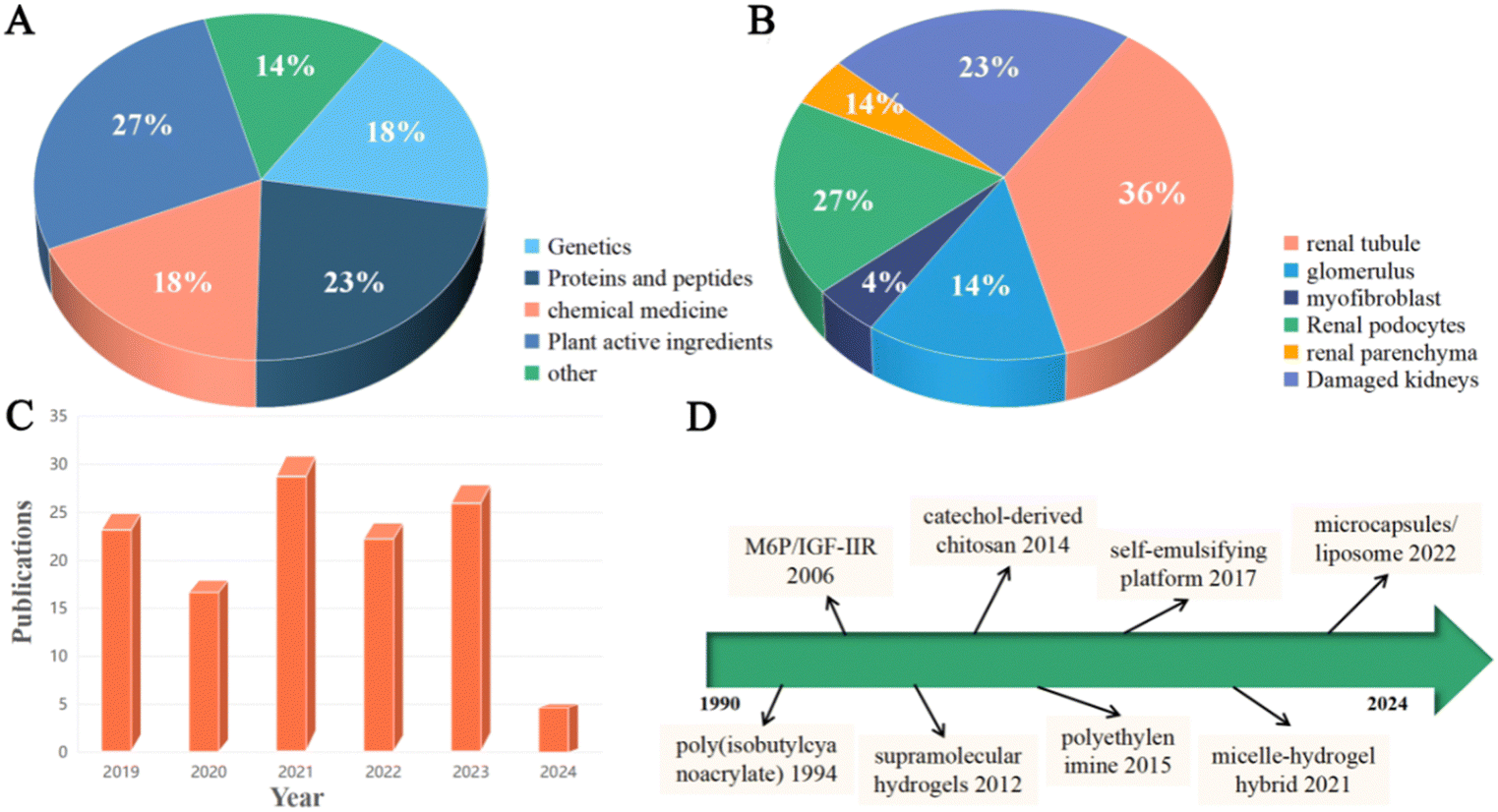 | ||
| Fig. 1 (A) Percentage of therapeutic agents used for renal fibrosis drug delivery. (B) Percentage of target sites for renal fibrosis drug delivery. (C) Research advances in renal fibrosis drug delivery therapy in the last 5 years (source: web of Science, 25 March 2024). (D) Key developments in drug delivery system-based therapy for renal fibrosis. | ||
2 Mechanisms of renal fibrosis
Fibrosis is a pathological extension of the normal healing method for wounds, marked by damage, inflammation, myofibroblast activation and migration, deposition of the matrix, and remodeling. Much of the pathophysiology of renal fibrosis is the same as that of other fibrotic diseases, such as cirrhosis,11 cardiomyopathy,12 and idiopathic pulmonary fibrosis.13 However, there are currently no clinically approved targeted antifibrotic therapies for the kidneys, possibly since there is still much to learn about the intricate processes that create and propel fibrosis. Among the key mechanisms of renal fibrosis that have increased more agreements are the heterogeneousness and procedures involved in myofibroblast formation, and the role of immune cells and injured epithelial cells and the mechanisms of their interaction (Fig. 2).8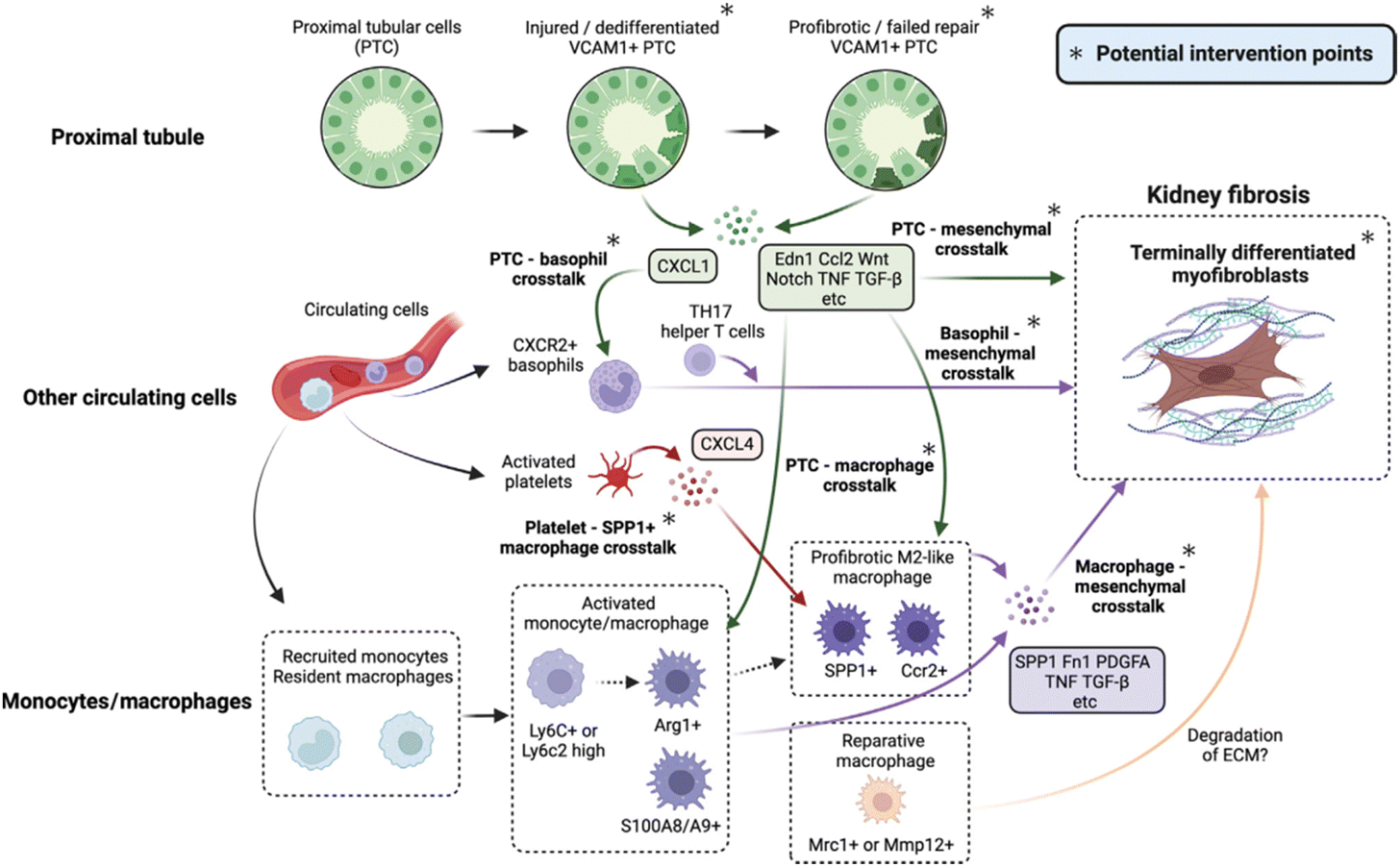 | ||
| Fig. 2 Potential intervention targets and the cellular crosstalk that drives kidney fibrosis.8 Reproduced with permission. Copyright 2023 Elsevier Ltd. | ||
The main source of resident fibroblasts, pericytes, and their subpopulations is myofibroblast-derived tissue. Smooth muscle cells-mainly reactive cells found in pathological circumstances like injury or cancer-combine characteristics of fibroblasts and myofibroblasts. Alpha-smooth muscle actin (α-SMA), which generates stress fibers – bunches of myofilaments, is the greatest indicator of the myofibroblasts. These fibers under stress are essential for attaching myofibroblasts to the extracellular matrix (ECM), and excessive deposition of the ECM is characteristic of renal fibrosis. The extracellular matrix (ECM) is a complex reticular dynamic structure composed of macromolecules secreted by cells into the extracellular mesenchyme; under normal physiological conditions, ECM catabolism and synthesis are in dynamic equilibrium; under pathological conditions, there is an increase in synthesis and a decrease in catabolism, with an eventual overdeposition of the ECM and an accompanying up-regulation of myofibroblast activity, resulting in a chronic macrophage- and immune-cell infiltrating inflammatory environment with infiltration of macrophages and immune cells. As the structure of the matrix changes and hardens with excessive ECM deposition, mechanical forces are exerted on the matrix as the cells contract, causing reorganisation and wound closure during the healing process. PDGFR-β, the receptor for the platelet-derived growth factor, is expressed by all myofibroblasts. Furthermore, one important regulator of myofibroblast development during fibrosis is transforming growth factor-beta 1 (TGF-β1). Researchers have demonstrated the major role of myofibroblasts in renal fibrosis by deleting αV integrins and cutting off signaling that transmits pro-fibrotic TGF-β using the PDGFR-β-Cre driver, which led to the observation of a significant reduction in renal fibrosis.14
In addition to myofibroblasts, renal tubulointerstitial fibrosis in the kidneys is caused by a variety of cells, including tubular epithelial cells, mesenchymal stromal cells, endothelial cells, and inflammatory cells. The formation and progression of renal fibrosis is coordinated by these cells’ intimate communication.15 One of the main processes causing renal fibrosis in these many cellular components is believed to be crosstalk between the injured renal tubular epithelial cells and mesenchyme. The researchers observed renal fibrosis driven by specific damage to the renal tubules of model mice, suggesting that damaged tubules are a critical component in the progress of renal fibrosis. The fibrosis is greatly influenced by the potential interaction between the injured tubules and the immune cells or mesenchyme.16,17 Vascular damage and capillary thinning are also major contributors to renal fibrosis. Renal damage in humans due to chronic kidney disease is characterized by peritubular capillary thinning. Tissue oxygenation measurements in several CKD experimental models validate hypoxia.18,19 Hypoxia-inducible factor-1α promotes pro-fibrotic signaling pathways and is up-regulated in hypoxic renal tubular epithelial cells, especially those in patients with chronic kidney disease.20,21 Loss of peritubular capillaries from acute or chronic injury results in local hypoxia, further damage to nearby tubules, up-regulation of pro-fibrotic pathways, and a self-reinforcing vicious cycle.
3 Drug delivery in renal fibrosis
Among the most significant treatment-related obstacles of kidney disease is the selective delivery of drugs to achieve therapeutically relevant concentrations in the target organ to reduce the amount of drugs required and minimize side effects. In 1994, a targeted drug delivery method was reported for glomerular mesangial cells using actinomycin D-loaded poly (isobutylcyanoacrylate) nanoparticles that are concentrated in rat mesangial cells. Since the beginning of the 1990s, research has been conducted on kidney-targeted drug delivery systems.22 Currently, renal drug delivery mainly targets tubules and glomeruli, in addition to targeting giant proteins, tubules, etc.23 However, glomerular filtration makes drug delivery to renal cells difficult. Podocytes, glomerular endothelial cells, and glomerular basement membranes make up the glomerular filtration barrier. Nanoparticles with a hydrodynamic diameter of less than 10 nm can pass via the glomerulus and be removed, however particles larger than 150 nm cannot cross the glomerular filtration barrier and collect in diseased areas.24 More importantly, podocytes in the glomeruli were the target of nanoparticles that could pass through the glomerular filtration barrier at a certain size of 5–30 nm.25 Studies have shown that nanoparticle accumulation is limited to the glomerular tunica and the extracellular matrix of the kidneys, and that maximum glomerular deposition can be achieved with about 80 nm size of the nanoparticles.26,27 Furthermore, one crucial factor for renal targeting is charge selectivity. Long circulation durations and little macrophage uptake are observed in nanoparticles with a surface charge of 15 mV.28 Cells within the mononuclear phagocyte system quickly eliminate highly positively charged nanoparticles from the bloodstream, whereas anionic nanoparticles are also eliminated by these cells.Renal therapy has demonstrated encouraging results using nanosystems that have diverse physicochemical qualities (dimension, form, surface, and charge) and biological attributes (high cellular internalization, low cytotoxicity, regulated pharmacokinetics, and biodistribution). Nanomaterial-derived drug delivery systems based on anti-renal fibrosis trigger extensive research discussions among researchers (Fig. 3).29 Among many nanomaterials, metal organic frameworks (MOFs) have attracted wide attention due to their porosity, large specific surface area, stable results and modifiability. MOFs are a class of porous materials formed by metal ions or metal clusters connected with organic ligands through strong coordination bonds. They are widely used in gas storage, separation, catalysis, drug release, sensing and other fields. In biological applications, MOFs are mainly used in drug carriers, bioimaging technology, and biosensing technology and as antimicrobial materials. Among them, the most promising is the drug carrier technology of MOFs, which often outperforms the therapeutic effect of drugs alone by carrying anti-tumour drugs and so on. In addition to the field of drug delivery, MOFs have been extensively studied in other fields, especially in battery storage and sewage treatment, where they have been practically applied and have high economic potential.
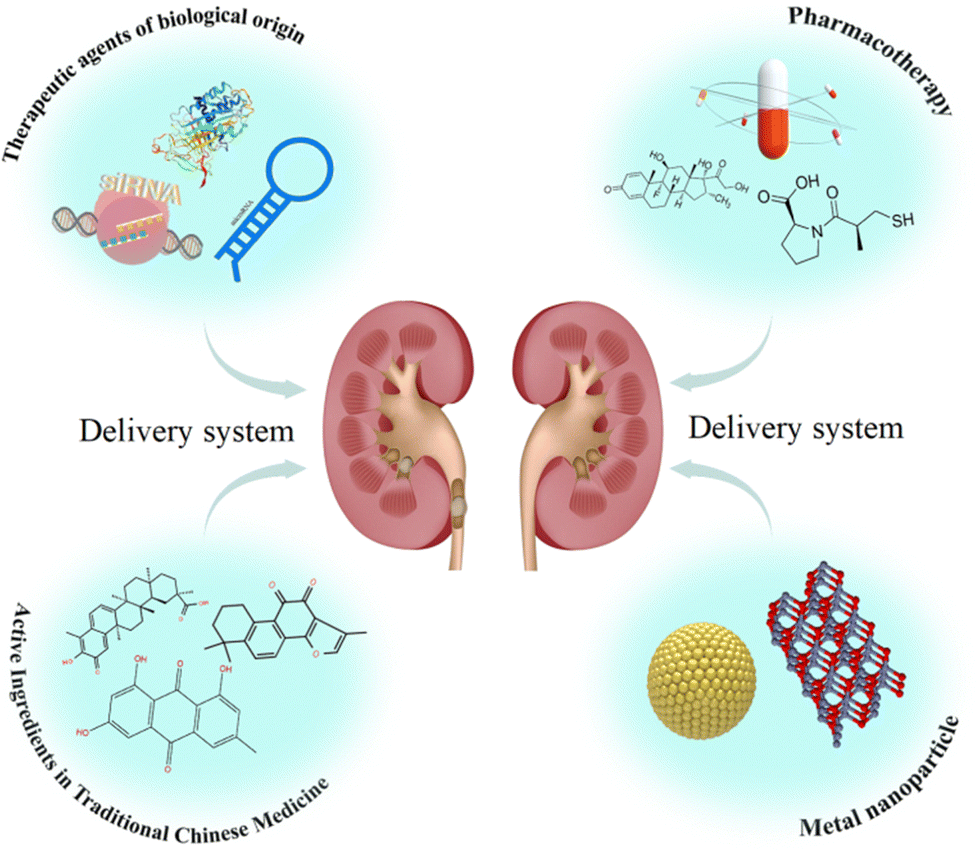 | ||
| Fig. 3 Renal drug delivery system. | ||
4 Drug delivery strategies for renal antifibrosis
Due to the special physiological structure and physiological function of the kidneys, drug delivery systems for delivery to the kidneys are often on the nanoscale. Different nanomaterials, such as microcapsules, lipid bilayers, nanocopolymers, etc., are modified and adapted to improve their biocompatibility and renal targeting. Thus, anti-renal fibrosis therapeutic agents, including peptides, medicines, active ingredients of traditional Chinese medicines, etc. can be efficiently delivered to the damaged kidneys (Table 1).| Drug delivery strategy | Therapeutic agent | Delivery vehicle | Particle size (nm) | Route of drug delivery | Target site | Ref. |
|---|---|---|---|---|---|---|
| Cyclo(RGDfC) | siRNA | cyclo (RGDfC)-polymeric | 65.31 ± 1.83 | Peritoneal injection | Renal podocytes | 30 |
| SAMiRNA-AREG | siRNA | Self-assembling micelles | — | Intravenous injection | Renal tubules | 31 |
| miR-146a-PEI-NPs | miRNA | Polyethyleneimine nanoparticles | — | Caudal vein injection | UUO Kidneys | 32 |
| DTmiAnp-cRGD-30 a mm | miRNA-30a | Cyclo (RGDfC)-gated polymeric-nanoplexes | 73.51 ± 1.43 | Peritoneal injection | Renal podocytes | 33 |
| CTLA-4-lg | CTLA-4-Ig | Microbubbles | — | Peritoneal injection | Renal podocytes | 34 |
| TKI-LZM | TGF-β type-I receptor kinase inhibitor | Protein lysozyme | — | Intravenous injection | Renal tubules | 35 |
| PTRAs EPLP-VEGF | Vascular endothelial growth factor | Elastin-like polypeptide | — | Renal Endovascular Implant | Renal parenchyma | 36 |
| SAP-TNF-α/HGF | Anti-TNF-α, hepatocyte growth factor | Self-assembling peptides/heparin hydrogel | 10–20 | Intrarenal injection | Renal tubules | 37 |
| IL-10@PLT | Interleukin-10 | Platelets | 965 | Caudal vein injection | Inflammatory renal cells | 38 |
| MV-DEX | Dexamethasone | RAW 264.7 macrophage cell-derived MVs | 140.7 ± 4.8 | Intravenous injection | Inflammatory renal cells | 39 |
| RDYH58-NP | Sorafenib | Lactic-co-glycolic acid | 76.0 ± 13.7 | Intravenous injection | Myofibroblasts | 40 |
| DXMS/CAP@PLGA-ILs | Dexamethasone, captopril | Liposome–nanoparticle hybrid | 119.1 ± 2.31 | Caudal vein injection | Glomerulus | 41 |
| KGM/PBA-PGA hydrogel | Insulin, liraglutide | Glucose-responsive hydrogel | — | Hypodermic injection | Renal tubules | 42 |
| SMEDDS | Emodin | Self-microemulsifying drug delivery systems | 18.31 ± 0.12 | Oral administration | Renal tubules | 43 |
| CPP/PEG-NPs | Emodin, tanshinone IIA | Microcapsules | 156.3 ± 2.4 | Oral administration | UUO Kidneys | 44 |
| PPP-RH-NPs | Rhein | Polyethyleneglycol-co-polycaprolactone-co-polyethylenimine triblock | 75 ± 25 | Intravenous injection | Diabetic kidneys | 45 |
| Plum pudding | Celastrol | Self-assembled triblock polymeric micelles and injectable hydrogels | 35.6 | Intravenous injection | Glomerulus | 46 |
| PLGA-Gyp XLIX | Gypenoside | Polylactic acid-co-glycoside (PLGA) | 35.6 | Intravenous injection | Renal tubules | 47 |
| BBR-BC 12-NPs | Berberine | Brij-grafted-chitosan | 102.0 ± 0.7 | Oral administration | Glomerulus | 48 |
| NTCC | CoCl2 | 50% N-acetylation-thiolated chitosan | 50–100 | Peritoneal injection | Renal tubules | 49 |
| GLAuNPs-Co | Co, AuNPs | GLAuNPs-Co | 22.97 ± 7.43 | Peritoneal injection | Renal tubules | 50 |
| ZnONPs | Zinc oxides | Nanoparticles | — | Peritoneal injection | Renal podocytes | 51 |
4.1 Gene therapy
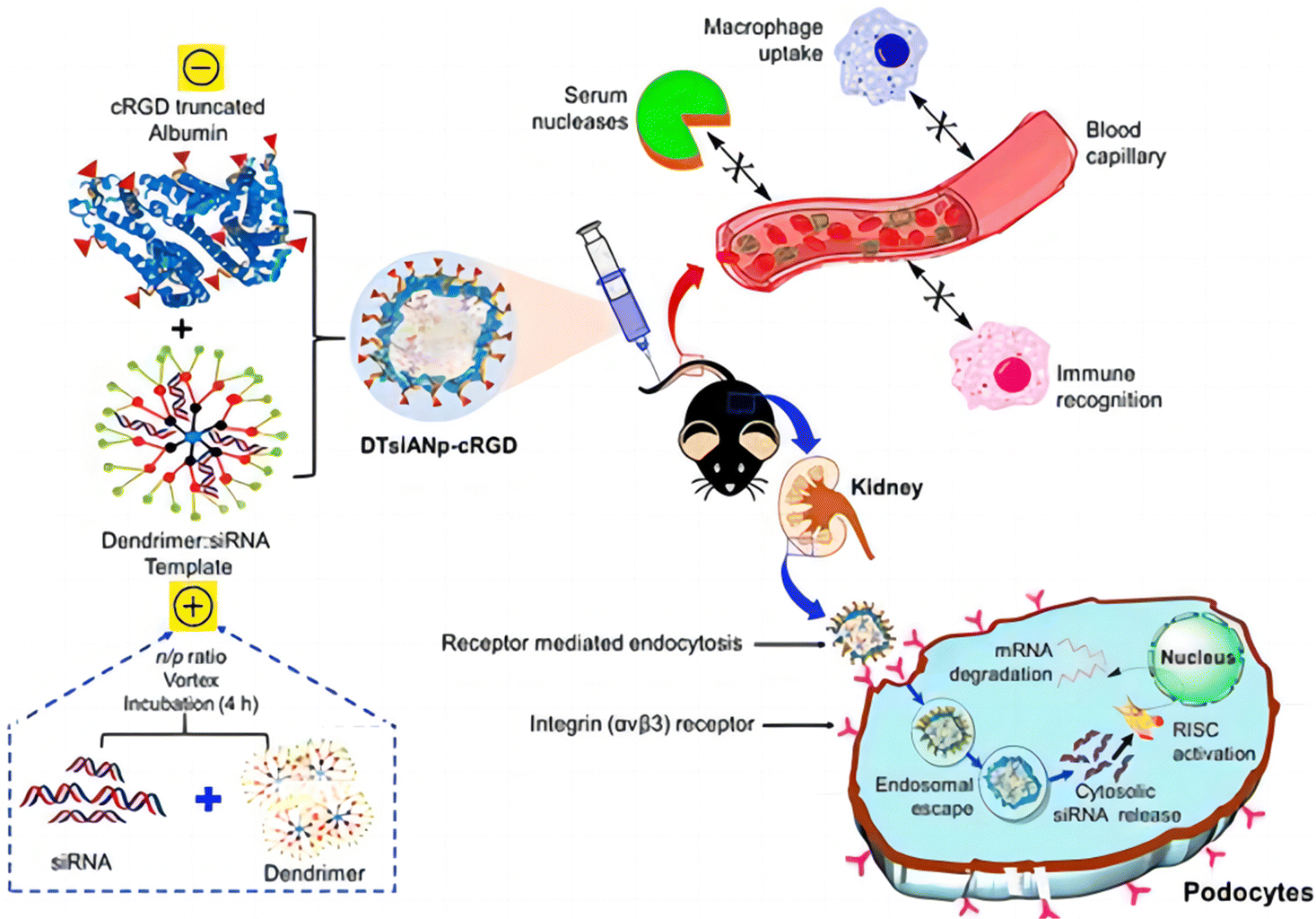 | ||
| Fig. 4 Schematic illustration of the nanoconstruction of cyclo-RGD truncated polymers for the HDAC4 gene silencing target in a mouse with diabetic nephropathy.30 Reproduced with permission. Copyright 2021 American Chemical Society. | ||
Amphiregulin (AREG) is a transmembrane glycoprotein recently found to be associated with renal fibrosis. Son et al.31 demonstrated in vivo suppression of amphiregulin using a new and efficient renal fibrosis through self-assembled micelle (SAM) inhibitory RNA. This self-assembled micelle inhibitory RNA can be blocked with epidermal growth factor receptor (EGFR) signaling. After injection of SAMiRNA-AREG into two types of renal fibrosis mice produced by unilateral ureteral obstruction (UUO) and adenosine diet (AD), due to the small nanoparticle size, it is easier for it to enter into tumours, inflamed or fibrotic tissues. The SAMiRNA-AREG accumulates in damaged kidneys and exhibits excellent renal therapeutic and fibrotic effects through passive targeting by enhanced permeability and retention effects. Finally, it was confirmed that the upregulation of AREG in UUO or AD replicas was largely localized to the distal tubules.
The miRNA-30a is mainly accountable for podocyte homeostasis. In diabetic nephropathy (DN), miRNA-30a is mainly directly inhibited by the hyperglycaemic kidney-induced notch signaling trail, leading to podocyte injury and apoptosis. Thus, delivery of exogenous miRNA-30a to podocytes is expected to ameliorate proteinuria as well as podocyte damage, thus acting as an antifibrotic agent. Raval et al. reported the targeted and effective administration of miRNA-30a mimic by dendrimer-templated cyclo (RGDfC)-gated polymeric nanoplexes to improve podocyte conditions.33 DTmiAnp-cRGD-30a mm had a fluid dynamics dimension of particles of about 73.51 ± 1.43 nm, and a zeta potential of 20.32 ± 1.03 mV. DTmiAnp-cRGD-30a mm was efficiently delivered to podocytes and up-regulated miRNA 30 compared to controls and protected the miRNA-loaded therapeutic agent from the deprivation by RNA enzymes in the process (Fig. 5). The kidney's glomerular region had less fibrosis and glomerular dilatation after its delivery.
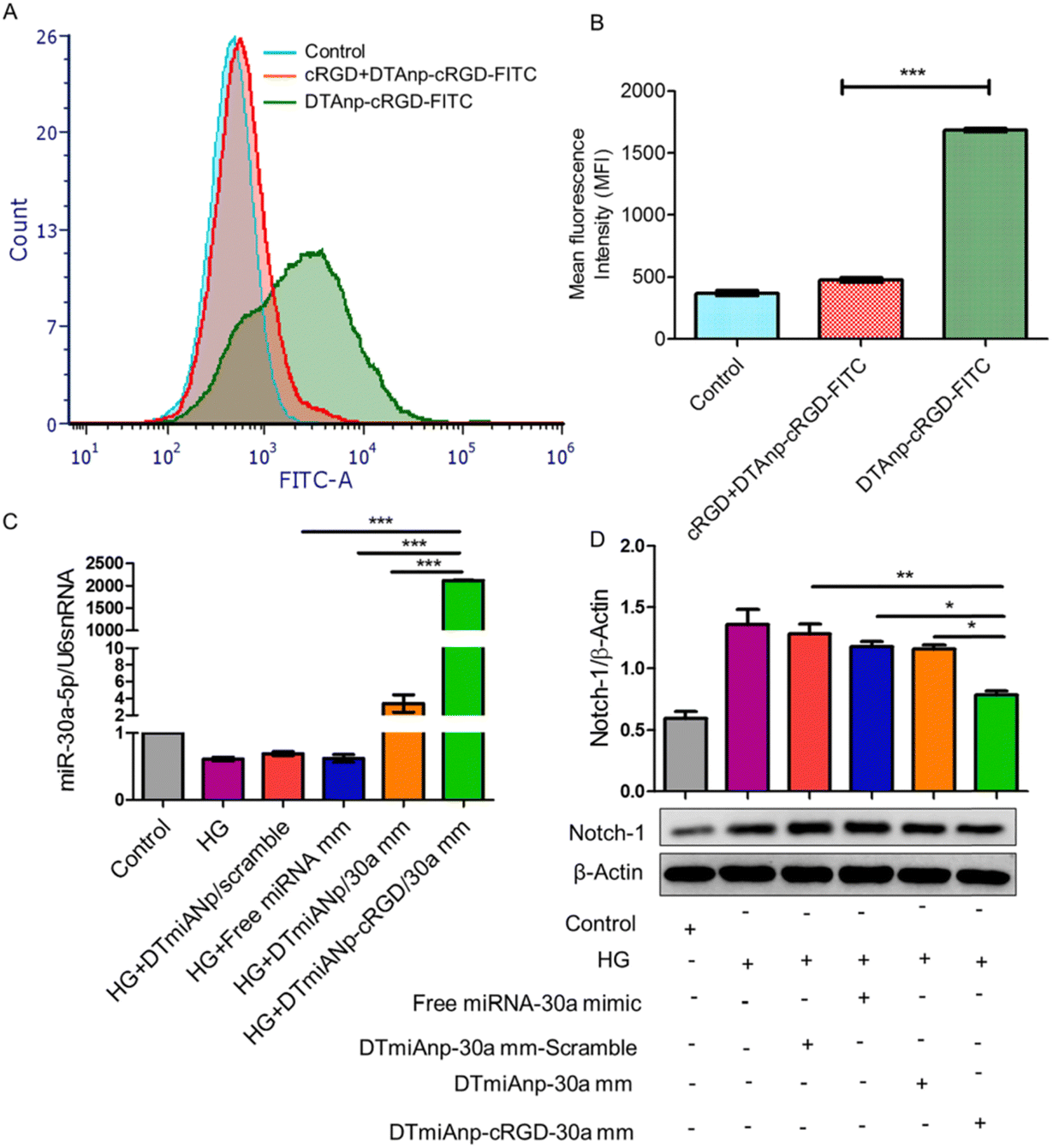 | ||
| Fig. 5 (A) Analysis of HG-treated podocytes using flow cytometry following treatment with free cRGD, DTAnp-cRGD-FITC (+) and DTAnp-cRGD-FITC (−). (B) Quantitative assessment of cell uptake using MFI p < 0.001 in comparison to cRGD + DTAnp-cRGD-FITC. (C) When miRNA-30a mimic loaded nanoplexes were treated, the qRT-PCR data show a relative upregulation of miRNA-30a; in this case, U6snRNA was taken into account as an endogenous control (healthy podocytes). The results show that ***p < 0.001 for both DTmiAnp-cRGD-30a mm and DTmiAnp-cRGD-30a mm vs. free miRNA-30a mimic. (D) Following the incubation of free miRNA-30a mimic, DTmiAnp-30a mm-scramble, DTmiAnp-30a mm, and DTmiAnp-cRGD-30a mm, Notch-1 expression was measured by western blot analysis. The endogenous control used was β-actin. Both **p < 0.01 and ***p < 0.001, respectively, compare DTmiAnp-cRGD-30a mm to DTmiAnp-30a mm and free miRNA-30a mimic.33 Reproduced with permission. Copyright 2021 Elsevier B.V. | ||
4.2 Protein and peptide therapy
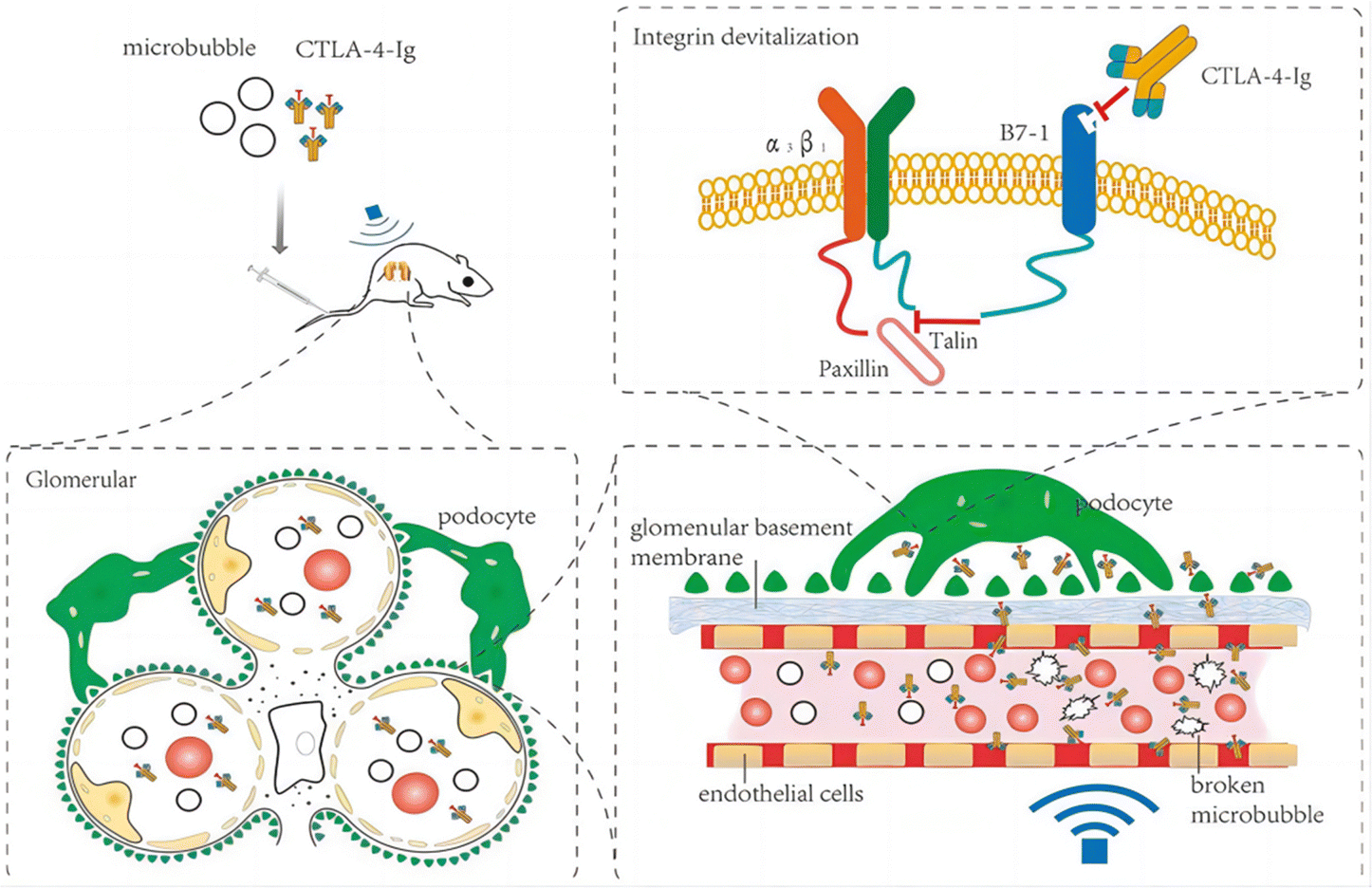 | ||
| Fig. 6 Therapeutic mechanism of CTLA-4-Ig in DN model rats.34 Reproduced with permission. Copyright 2021 Wang et al. | ||
Myofibroblasts are one of the key factors in the progression of renal fibrosis, and myofibroblasts are the foremost source of ECM in the process of renal fibrosis. Thus, the inhibition of myofibroblast production is expected to inhibit the progression of renal fibrosis. Albumin is an abundant multifunctional plasma protein synthesized mainly by hepatocytes.64 The activated stellate cells (SCs) did not naturally express albumin. However, the phenotype of myofibroblasts reverted to that of early activated cells, along with the reappearance of cytoplasmic lipid droplets and decreased expression of collagen type I and α-SMA when forced to express it.65 Cha et al.66 reported an anti-fibrotic recombinant fusion protein (called R-III) that was created by fusing the C-terminus of retinol-binding protein (RBP) with the albumin domain III. RBP can be used to target SCs for delivery, based on which R-III can be targeted to renal stellate cells (Fig. 7). R-III action induced phenotypic reversal of activated/myofibroblast to a fat storage phenotype, but had no substantial effect on renal fibroblasts. Chronic tubular damage has been demonstrated to be a symptom of mitochondrial diseases, including structural and functional deficits, which progresses to fibrosis.67 The regulatory element of the mitochondrial permeability transition pore (MPTP) that controls MPTP opening and MPTP-dependent necrotic cell death is identified as the mitochondrial matrix protein cyclophilin D (CypD). This exhibits cis–trans peptidyl prolyl isomerase activity. Jang et al. showed that CypD in renal tubular injury, which is a key aspect contributing to renal fibrosis in UUO,68 and the results of this study may provide a new paradigm for targeted treatment of fibrotic diseases.
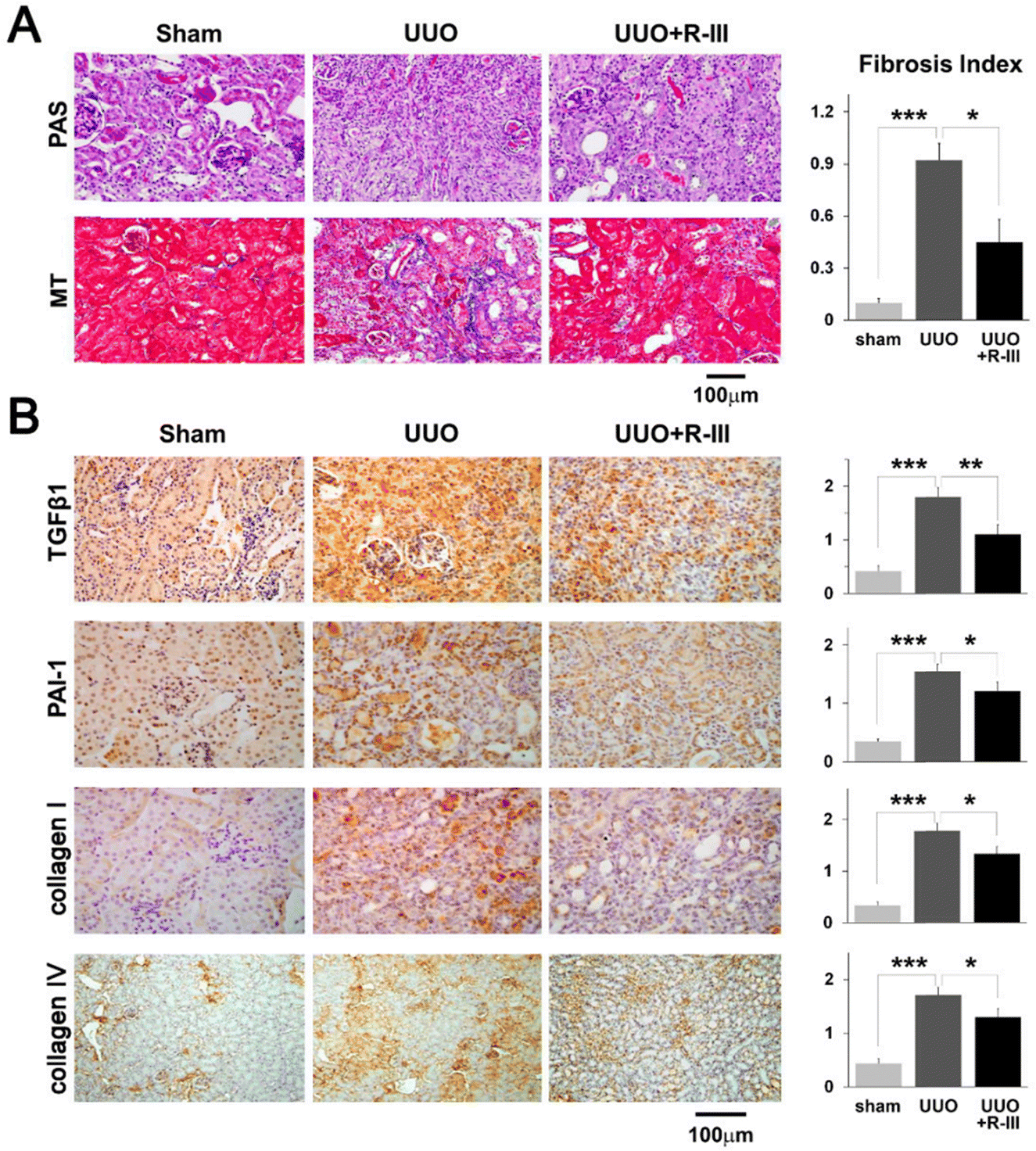 | ||
| Fig. 7 Renal fibrosis caused by unilateral ureteral obstruction (UUO) is lessened by R-III. (A) PAS and MT were used to stain kidney sections from sham, UUO, and UUO + R-III-treated animals. The semiquantitative score for tubulointerstitial fibrosis is displayed in the right panel. (B) TGF-β1, PAI-1, collagen type I and type IV, and kidney sections from sham, UUO, and UUO + R-III-treated mice were immunohistochemically examined. Each research group's representative photographs are displayed. The staining intensity semiquantitative analysis for each group is displayed (right). The data are shown as means ± SD (p-value; Kruskal–Wallis test, then DSCF multiple comparison test). *p < 0.05, **p < 0.01, and ***p < 0.001.66 Reproduced with permission. Copyright 2020, Cha et al. | ||
TGF-β is a multifunctional cytokine that serves an important part in the pathogenesis of several diseases related to renal, leading to renal fibrosis.70 TGF-β activates renal tubular epithelial cells, leading to their alteration into fibroblasts via epithelial–mesenchymal transdifferentiation,71 and ultimately the conversion of fibroblasts into myofibroblasts and construction of extracellular matrix (ECM) proteins.72 Prakash et al.35 developed a renal drug delivery system that inhibits TGF-β receptors (TKI-LZM). The system consists of an inhibitor integrated into renal carrier protein lysozyme (LZM), which targets the TGF-β kinase inhibitor to renal tubular cells and provides sustained delivery of the active substance in the target cells over several days. TKI-LZM accumulated rapidly in renal tubular cells within 1 h after intravitreal injection and no accumulation was observed in the heart, spleen, and lungs. The degradation half-life of TKI–LZM conjugate in the kidney was about 37.7 h. The degradation of TKI–LZM was also observed in the kidneys, and the degradation of the TKI–LZM conjugate in the kidneys was also observed in the heart and spleen. Following binding to its type-II receptor, TGF-β interacts to another type-I receptor called activin receptor-like kinase-5 (ALK5).73 By controlling the deposition of the extracellular matrix, ALK5 triggers downstream signaling cascades, and therefore contributes to the establishment of renal fibrosis.74
Vascular endothelial growth factor (VEGF) is a pro-angiogenic cytokine that plays a crucial role in neovascularisation, healing, and conservation of the MV matrix throughout the body. The bioavailability of the VEGF is reduced in stenotic kidneys with renal vascular disease.75 In a model of progressive renal failure, VEGF treatment improves kidney injury and protects renal microcirculation.76 Guise et al.36 developed a bioengineered fusion of drug delivery carriers with renal targeting. Adjuvant targeted administration of ELP-VEGF enhances the effectiveness of plastic and stenting in renovascular disease (RVD), and has a better therapeutic effect on renal injury such as renal fibrosis.
Self-assembling peptides (SAPs) are designer biomaterials readily available from naturally occurring amino acids that instinctively form cross-linked nanofibers, and are transformed into nanoscale hydrogels.77,78 Liu et al.37 reported the hepatocyte growth factor (HGF) and the TNF-α neutralizing antibody (anti-TNF-α) co-delivered using an injectable, self-assembling peptide/heparin (SAP/Hep) hydrogel (Fig. 8). KLD 2 R/Hep hydrogels are only 10–20 nm in diameter and sequentially release the drug and accumulate in the kidneys after injection into the body. Animal studies have shown that SAP-drug hydrogels are more effective in lowering apoptosis and inflammation, enhancing tubular regeneration and renal function, and thus reducing chronic fibrosis after ischemia-reperfusion I/R hurt, compared to SAPs or free drugs.
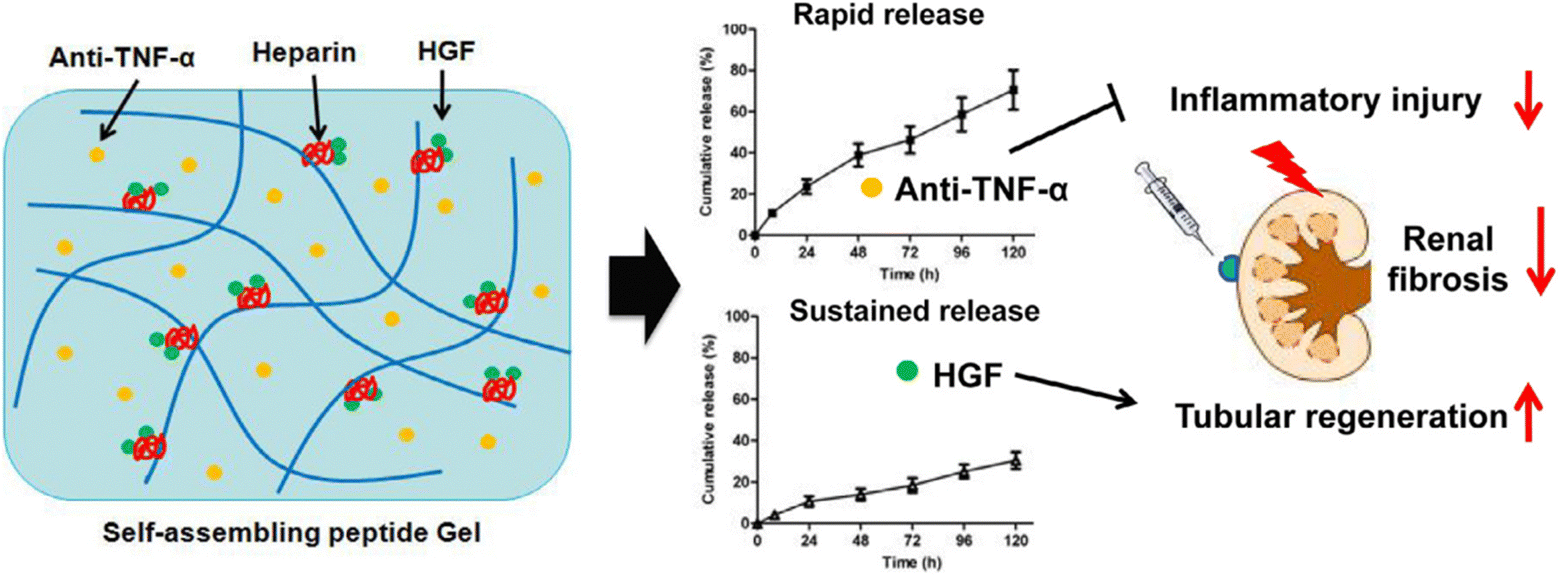 | ||
| Fig. 8 Scheme of the dual-drug delivery KLD2R/Hep hydrogel for improving of renal repair. The embedded anti-TNF-α and HGF were discharged from the hydrogel in a sequential manner, and thus endorsed tissue repair.37 Reproduced with permission. Copyright 2019 Acta Materialia Inc. Published by Elsevier Ltd. | ||
The development of both acute and chronic renal illness has been linked to inflammatory agents, which are now recognized as important pathogenic molecules, yet interleukin-10 (IL-10) deficiency in damaged kidneys affects its anti-inflammatory effects. To address this issue, Gong et al.38 developed a platelet (PLT)-activated IL-10 (IL-10@PLT), which was experimentally demonstrated to target IL-10@PLT to concentrate in the renal tubules of damaged kidneys, reduce inflammatory response, improve renal function, and ameliorate renal fibrosis during the recovery phase of UUO treatment.
4.3 Pharmacotherapy
Current drugs for the treatment of renal fibrosis mainly alleviate renal fibrosis with multi-targets and numerous pathway trails. Among them, angiotensin-converting enzyme inhibitors and glucocorticoids are the main drugs for treating renal diseases, but these drugs generally lack targeting and adverse effects that vary with dosage limiting their clinical application to a large extent. Enhancing the renal targeting of chemical drugs through drug delivery techniques is expected to reduce toxic side effects and improve the therapeutic effect on renal fibrosis.Exosomes and microvesicles are examples of extracellular vesicles (EVs), which are membrane particles generated from host cells and have an inherent means of intercellular communication.79–81 EVs can serve as effective carriers for drugs, RNA drugs and anti-inflammatory agents.82–84 Tang et al.39 used RAW 264.7 macrophage cell-derived MVs as a vehicle for targeted delivery of dexamethasone (DEX) to inflamed kidneys (Fig. 9). The emulsive dosages of MV-DEX considerably reduced renal injury in murine models of LPS- or ADR-induced nephropathy, with improved therapeutic efficacy against renal inflammation and fibrosis, when compared to free DEX treatment. More importantly, the delivery of DEX in MV expressively abridged the consequences of long-term glucocorticoid treatment.
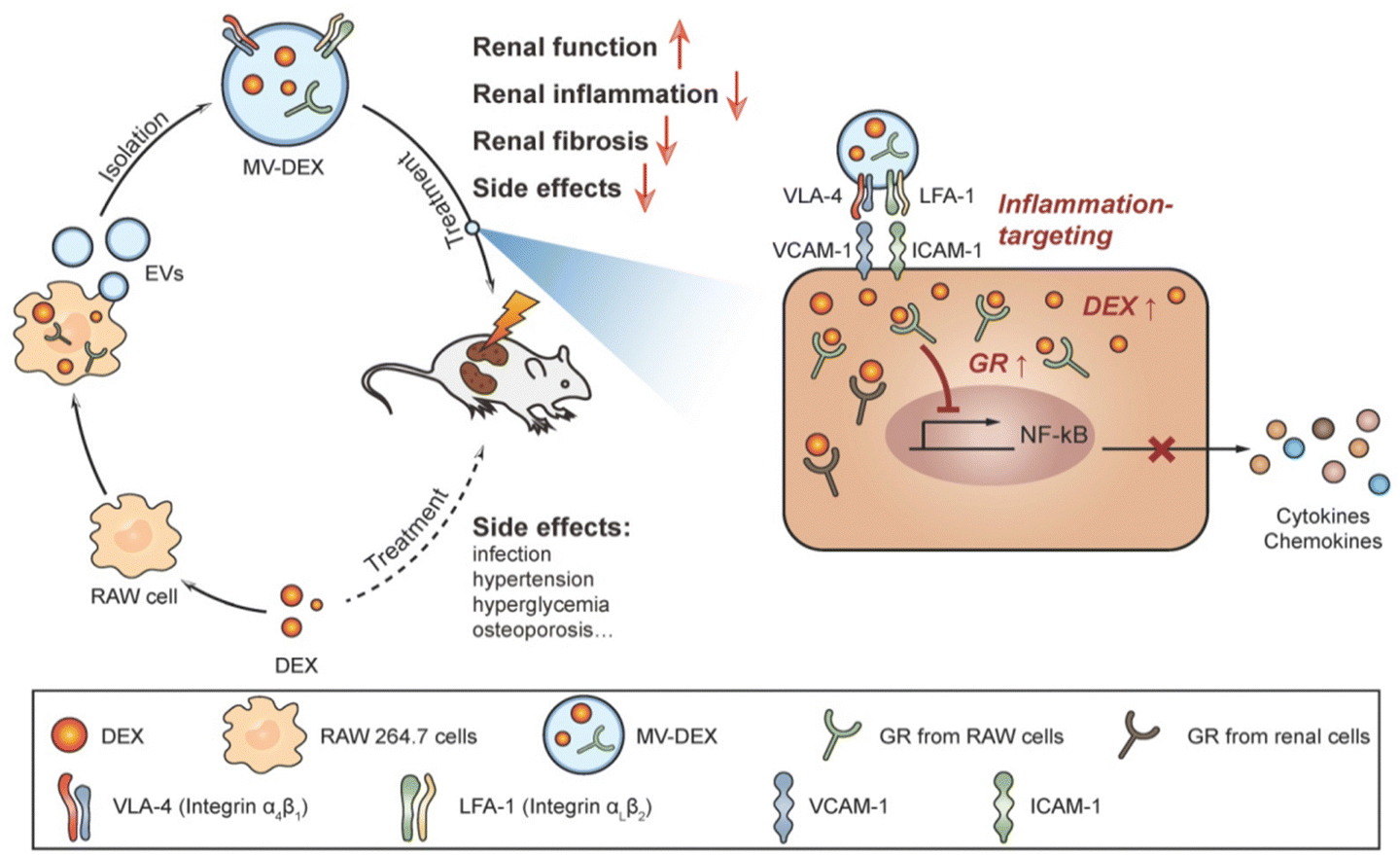 | ||
| Fig. 9 A pictorial representation of the MV-DEX production process and its therapeutic effects against renal disorders.39 Reproduced with permission. Copyright 2020 Tang et al. | ||
Liposomes are tiny lipid vesicles, usually encapsulated by one or more lipid layers, employed to deliver drugs or other active ingredients. Liposomes can improve drug solubility, stability, and to some extent targeting. Cheng et al.40 created lipid-coated poly(lactic-co-glycolic acid) nanoparticles with sorafenib, a powerful anti-tumor multikinase inhibitor, encapsulated in the core and fibrotic kidney homing peptides on the surface. The nanoparticles (NPs) had an average particle size of ∼70 nm, and nanoparticle-loaded sorafenib allowed targeted delivery to myofibroblasts compared to free sorafenib. Peptide-modified NPs were ∼2.6 times more effective than non-targeted NPs in reaching UUO-induced fibrotic kidneys, and significantly ameliorated the progression of renal fibrosis when given intravenously to mice suffering from UUO-induced renal fibrosis. In addition, Zhou et al.41 coated a phospholipid bilayer on the surface of PLGA-NPs to create a liposome-nanoparticle hybrid (PLGA-LNHy). Afterwards, PLGA-LNHy was co-modified with PEG and α8 integrin antibodies to create PLGA immunoliposomes (PLGA-ILs). The loading of dexamethasone (DXMS) and captopril (CAP) resulted in the DXMS/CAP@ PLGA-IL nanoparticles. The prepared nanoparticles contained a core–shell design with a uniform and suitable size (119.1 ± 2.31 nm), 10.22 ± 1.00% for DXMS and 6.37 ± 0.25% for CAP. The DXMS/CAP@ PLGA-IL possesses low cytotoxicity and good tethered cell entry capacity for successful aggregation in the glomerular MC zone. In vivo pharmacodynamic studies showed that DXMS/CAP@PLGA-ILs were effective in ameliorating pathological changes in the glomerular tunica albuginea region and positive expression of glomerular proliferating cell nuclear antigen (PCNA), decreasing the manifestation of factors that cause inflammation, fibrosis, and reactive oxygen species (ROS). Other researchers have used hydrogels, virus-mimetic nanoparticles, etc. loaded with chemical drugs to obtain better renal delivery capacity and renal fibrosis treatment.42,43
4.4 Herbally active compound therapy
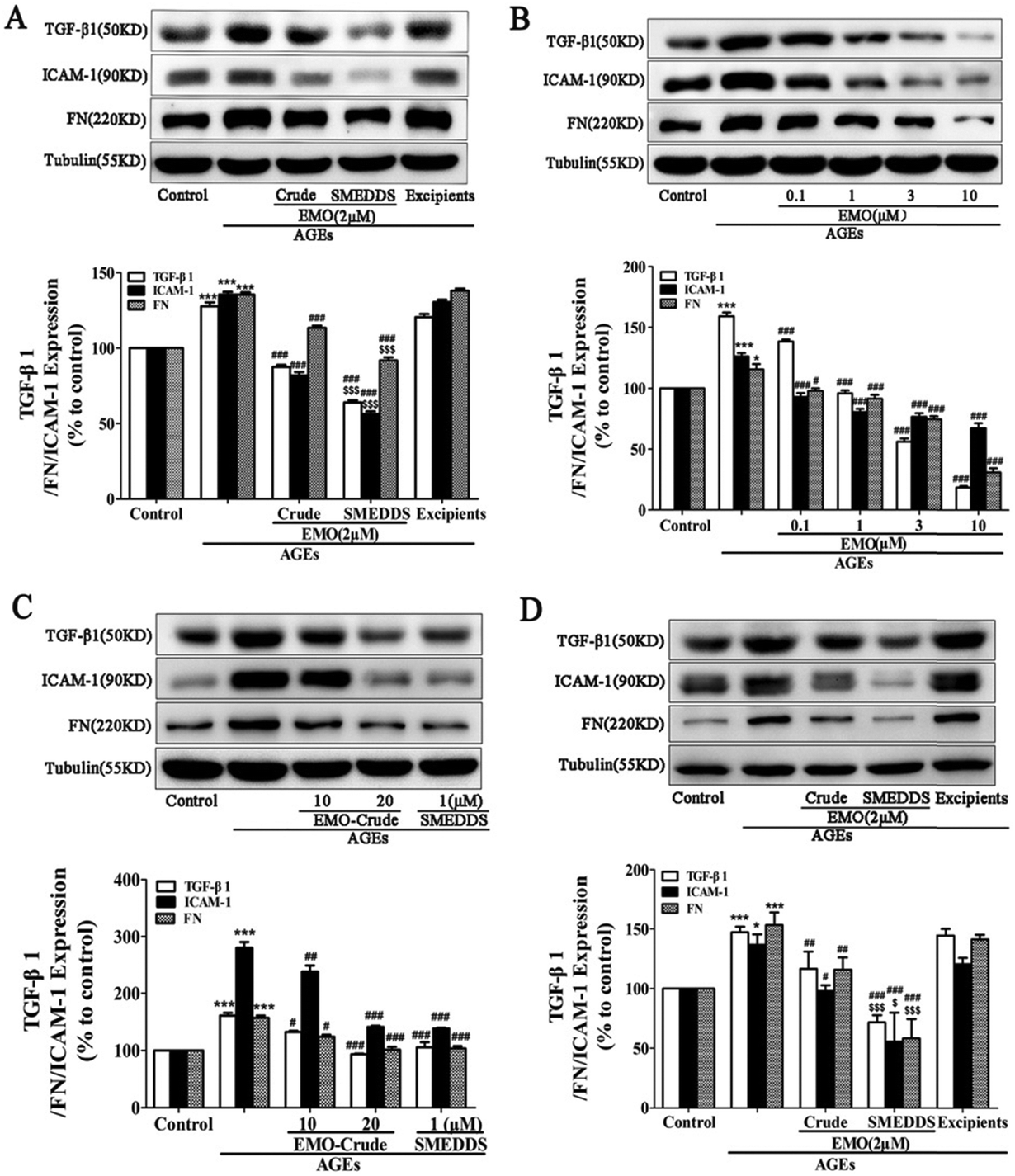 | ||
| Fig. 10 SMEDDS loaded with emodin reduced the expression of FN, TGF-β1, and ICAM-1 in GMCs and NRK-52E cells treated with AGEs. For twenty-four hours, AGEs were used to activate GMCs and NRK-52E cells in the presence or absence of crude emodin (Crude) or SMEDDS. (A) Impact of crude emodin (Crude) and SMEDDS at the same 2 μMEMO (emodin) concentration on FN, TGF-β1, and ICAM-1 expression in GMCs; excipients served as the solvent control. (B) How SMEDDS containing 0.1, 1, 3, and 10 μMEMO affects the expression of TGF-β1, ICAM-1, and FN in GMCs. (C) Impacts of crude emodin (EMO-Crude) with 10, 20 μMEMO and SMEDDS with 1 μMEMO on GMC expression of FN, TGF-β1, and ICAM-1. (D) Impact of crude emodin (Crude) and SMEDDS at the same 2 μMEMO(emodin) concentration on FN, TGF-β1, and ICAM-1 expression in NRK-52E cells; excipients served as the solvent control. *P 0.05, ***P b 0.001 versus control, #P 0.05, ##P b 0.01, ###P b 0.001 versus AGEs, Compared to crude emodin, $P b 0.05 and $$$P b 0.001.87 Reproduced with permission. Copyright 2016 Elsevier B.V. | ||
Tanshinone IIA (Tan IIA) is a component of Danshen. By blocking the activity of glycogen synthase kinase (GSK) 3β, fibroblast recruitment, and tubular epithelial cell fibrosis, Tan IIA reduces renal fibrosis.88 Tan IIA may act as an antirenal fibrosis agent along with emodin; nevertheless, inadequate oral absorption, unforeseen drug–drug interactions, and their potential to modify individual pharmacokinetic profiles when combined significantly restrict their utilization. To alleviate these limitations, Sun et al.44 devised a novel nanomicrosystem-based co-delivery method by embedding Tan IIA-loaded nanoparticles (Tan IIA-NPs) into EMO-containing microcapsules with cell-penetrating peptides (CPPs) and polyethylene glycol (PEG) modifications to obtain CPP/PEG-NPs (Fig. 11). This nano-microsystem exhibited significant sequential drug release. In addition, in vivo tests showed that Tan IIA and EMO had improved oral bioavailability and possessed noteworthy therapeutic effects in UUO rats.
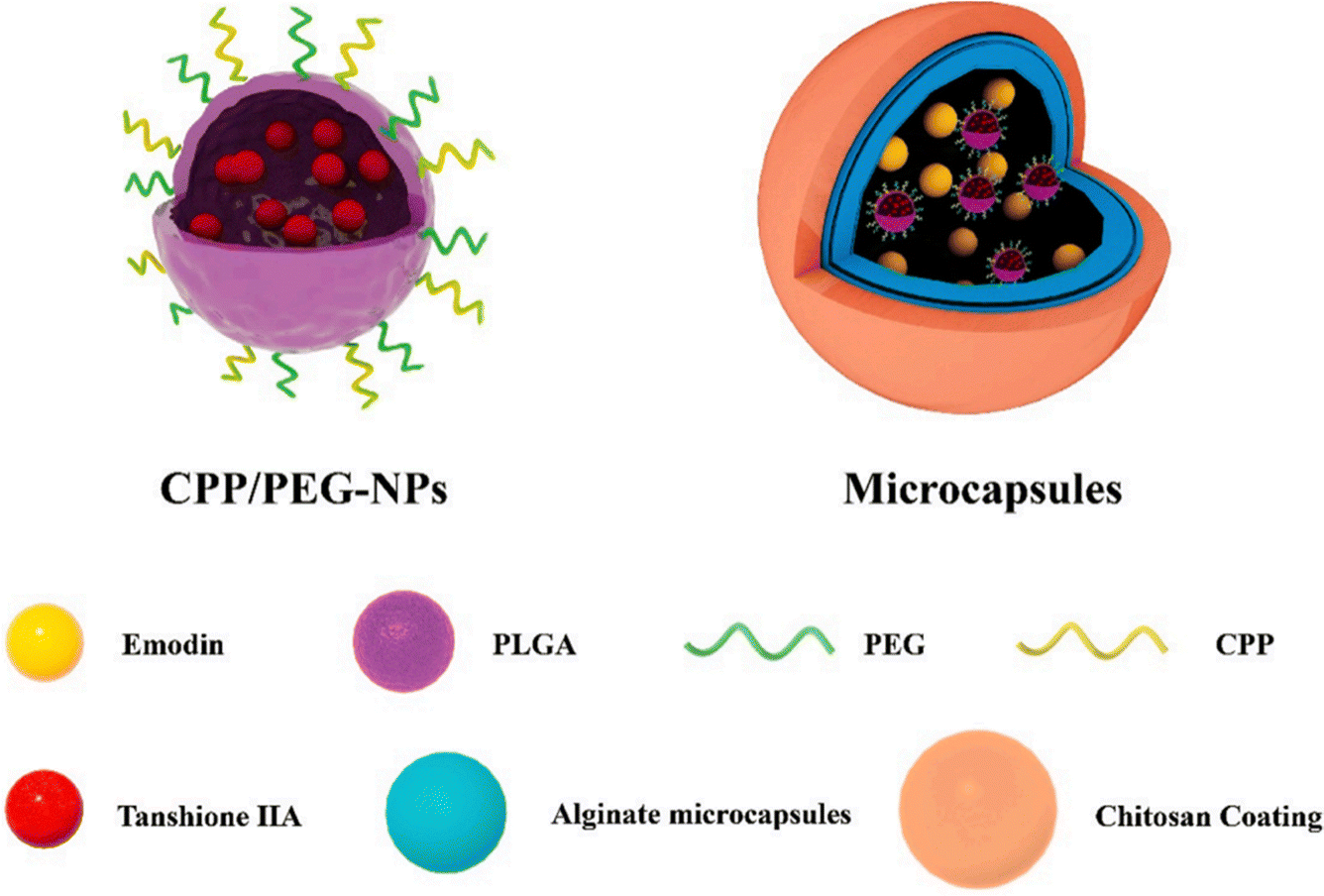 | ||
| Fig. 11 Schematic illustration of hierarchically organized microcapsules.44 Reproduced with permission. Copyright 2022 Elsevier B.V. | ||
All traditional Chinese medicine uses rhine, a lipophilic anthraquinone molecule, as the primary absorbable anthraquinone derivative that enters the bloodstream following the oral treatment. Through a mechanism that inhibits the toll-like receptor 4 (TLR 4) and TGF-β1 release in the kidneys, rhine dramatically improves glomerular disease and tubule interstitial fibrosis. However, clinical administration of Rhine is restricted by adverse effects, low bioavailability, poor solubility, and decreased transport into the kidneys. Chen et al.45 produced polyethyleneglycol-co-polycapro lactone-co-polyethylenimine triblock amphiphilic polymers to create RH-loaded PPP-RH-NPs. PPP-RH-NPs were about 80 nm in size with a zeta potential of 10 mV. The most popular and well-researched gene transfer carrier is polyethylenimine (PEI). It demonstrates renal dispersion for gene delivery to some extent and includes 15–20% protonatable amine groups under physiological conditions. In the streptozotocin (STZ)-induced DN model, PPP-RH-NPs displayed upright renal dispersal and exhibited improved renal function, and antirenal fibrosis effects.
Celastrol (CLT) is a naturally occurring chemical that was isolated from Tripterygium wilfordii. Because of its potent anti-inflammatory and antiproliferation properties, Celastrol was chosen as a model therapy for patients with RIF. Qin et al.46 developed a topical drug delivery platform with controlled-release and long-acting characteristics via injectable hydrogels crosslinked by self-assembled triblock polymer micelles and piggybacked with CLT, which enables long-term release of CLT at the renal site. Gypenosides (Gyp) are a class of dammarane type saponins extracted from five-leafed gynostemma. Among them, Gyp XLIX is one of the most active components, with anti-inflammatory,93 anticancer,94 and anti-hepatic fibrosis effects.95 Studies related to the use of Gyp in the treatment of renal diseases have been continuously reported,96,97 but direct administration of Gyp XLIX is not ideal due to its low solubility and bioavailability, high molecular weight, and significant barriers to absorption in the body. To solve the problems related to limiting the clinical application of Gyp XLIX, Liu et al.47 obtained PLGA-Gyp XLIX nanoparticles using a poly(lactic acid)-glucoside copolymer (PLGA) loaded with Gyp XLIX. The PLGA-Gyp XLIX nanoparticles possessed a particle size of ∼120 nm and could be targeted to UUO kidneys. In contrast to Gyp XLIX, the PLGA-Gyp XLIX nanoparticles can lessen tubular necrosis, decrease collagen deposition, and limit renal fibrosis (Fig. 12).
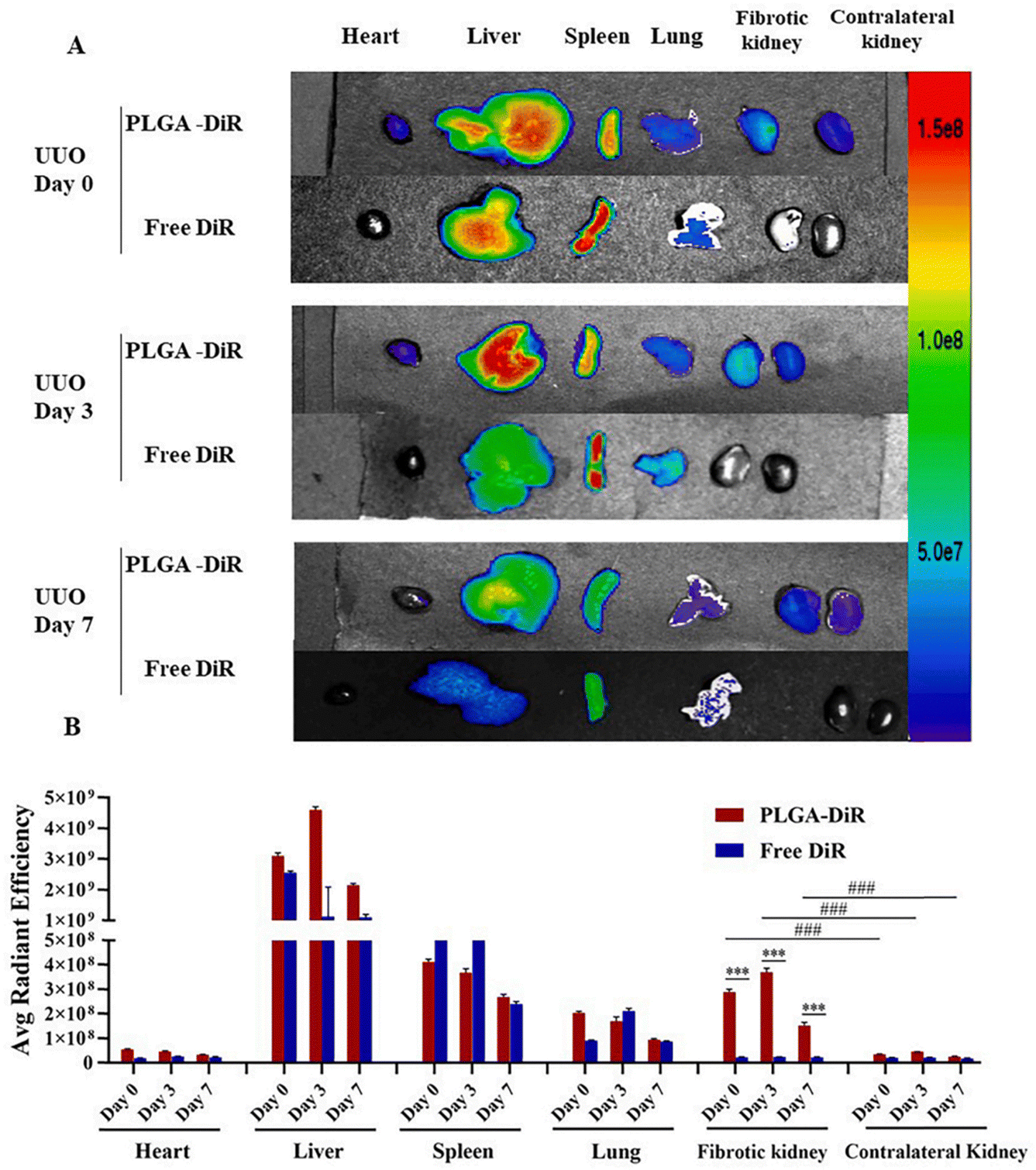 | ||
| Fig. 12 Mouse UUO kidney is the target of PLGA-DiR NPs. (A) The fluorescence signal image of the primary organs removed from UUO mice (heart, liver, spleen, lung, and kidneys) at 0, 3, and 7 days following surgery when PLGA-DiR and Free DiR were administered. (B) The average intensity of fluorescence for every organ in (A). For six to eight separate mouse experiments, the data show the mean ± S.D. Comparing the fibrotic kidney to the contralateral kidney, ***P < 0.001 was found. ###P is less than 0.001. Comparison between PLGA-DiR and Free DiR. PLGA-DiR refers to PLGA nanoparticles loaded with DiR.47 Reproduced with permission. Copyright 2021 Elsevier B.V. | ||
By inhibiting NF-jB activation, dampening the Notch/Snail pathway, upregulating Nrf2 signaling, downregulating the TGF-b/Smad/EMT pathway, reducing a-SMA levels, boosting E-cadherin levels, and inhibiting RhoA/ROCK, berberine prevents ECM buildup and lowers RF.100–103 Because of P-glycoprotein-mediated drug efflux and first-pass effects, BBR has a very limited oral bioavailability. To address this issue, Xiong et al.48 developed a Brij-grafted-chitosan (BC) nanocarrier technology to improve oral bioavailability of berberine, and therapeutic efficacy in diabetic nephropathy. The BBR-BC 12-NP was spherical in shape and had a size of approximately 102.0 ± 0.7 nm. The comparative oral bioavailability (FRel) of the BBR-BC 12-NP was demonstrated by in vivo experiments. The relative oral bioavailability (FRel) of BBR was determined to be 440.8% (Fig. 13). In a STZ-induced diabetic rat model, BBR-BC 12-NP was efficiently distributed in the renal fraction and exerted therapeutic effects.
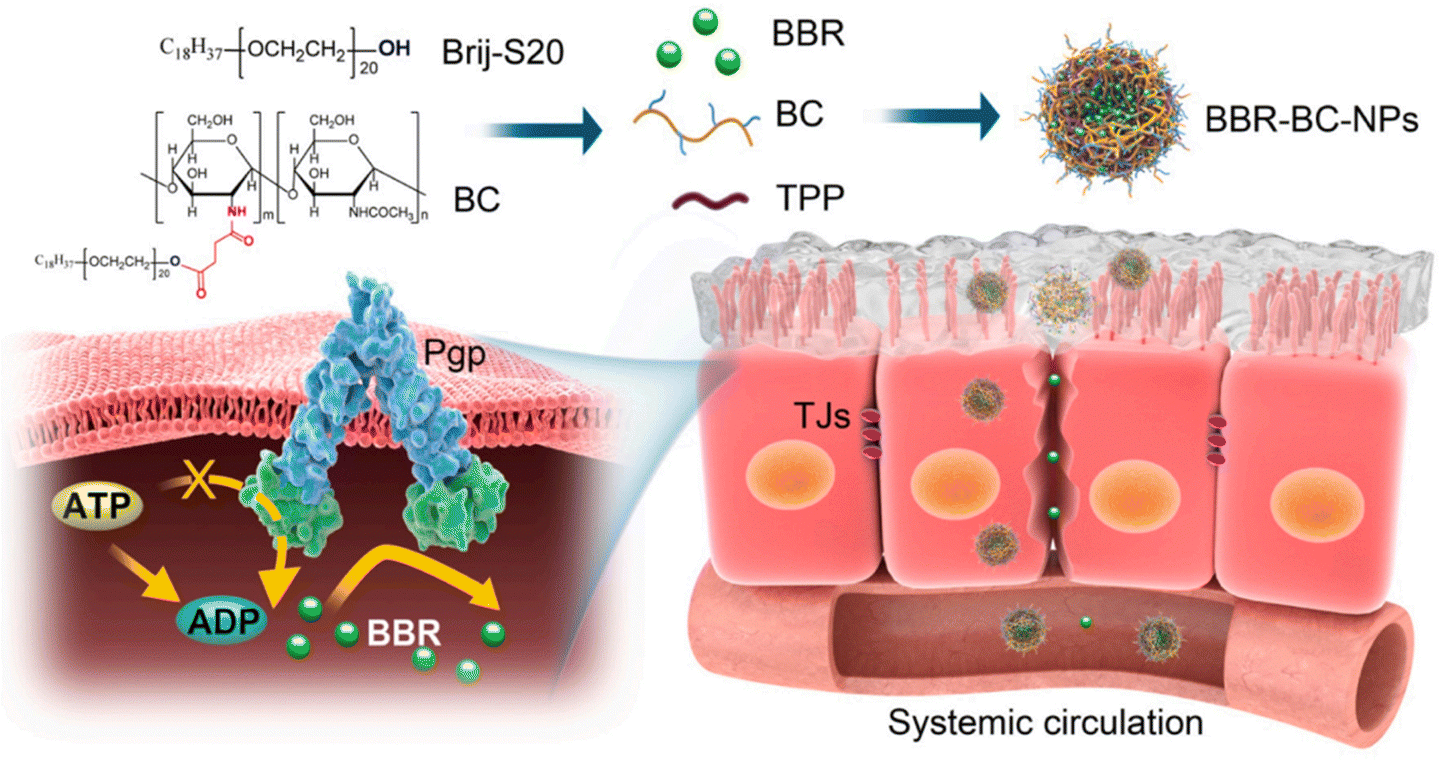 | ||
| Fig. 13 Pictorial representation of BBR-BC-NPs improving the intestinal absorption of berberine (BBR) via transiently and reversibly adjusting the intercellular tight junctions and Pgp-mediated drug efflux.48 Reproduced with permission. Copyright 2021 Elsevier Ltd. | ||
4.5 Other antirenal fibrosis drug delivery systems
Metal ions provide important functions in biological systems such as regulation of electrolyte balance, oxygen transport, electron transfer, catalysis, etc. Many transition metal ions (Mn, Fe, Cu, Co, etc.) are involved in many redox reactions in the body due to their variable and physiologically accessible oxidation states. Metal nanoparticles are often made to give full play to the therapeutic effect of metal ions in vivo and are widely used in anticancer, anti-microbial, imaging, diagnostic and other applications. Therapies based on metal nanoparticles have also been proposed such as photothermal therapy,104 chemodynamic therapy,105 photodynamic therapy,106 and ultrasonodynamic therapy.107 In recent years, the strategy of using metal nanoparticles against renal fibrosis has been continuously proposed by numerous researchers.Cobalt chloride is one of the utmost extensively employed hypoxia inducible-factor activators in biomedicine, and HIF activators protect the kidneys by ameliorating renal hypoxia in chronic kidney disease. Unfortunately, unfavorable side effects and limited renal targeting of cobalt chloride severely restrict its therapeutic uses. Cobalt nanomaterials possessed numerous advantages of low preparation cost, low toxicity, high photothermal conversion ability, and high drug-carrying capacity. They have been proved to have a promising application in anticancer and anti-infection therapy. Li et al.49 designed N-acetylated-thiolated chitosan cobalt (NTCC) nanocompatibles. Cobalt will be liberated from biomaterials in acidic environments, whereas NTCC has exceptional stability in typical physiological circumstances. The NTCC showed a particle size of about 50–100 nm. Upon injection of NTCC into the UUO mouse model, the NTCC targets and accumulates in the kidneys, where it is released within 8 h. Compared with cobalt chloride particles, NTCC had a significant antirenal fibrosis effect.
Excellent biocompatibility, simplicity of surface modification, tunable features, and several uses of Au nanoparticles (AuNPs) in drug delivery, animal and cell imaging, and disease treatment have generated a lot of interest. However, Au nanoparticles have low drug-carrying ability, and poor capability to embed drugs. Providentially, the Au nano-assemblies with greater surface areas and self-assembly possessions were able to address the challenges of Au nanoparticles. Tan et al.50 demonstrated novel stimuli-responsive drug release nanoplatforms. The nanoplatforms involved the self-assembly of GLAuNPs (glutathione-modified Au nanoparticles) and Co2+ into nano-assemblies (GLAuNPs-Co) through coordinated interactions among empty orbitals and lone pair glutathione. GLAuNPs exhibited high drug loading capacity upon assembly with Co2+ and triggered drug release in renal fibrotic tissues. Furthermore, the cytotoxicity of Co2+ in renal tubular cells was significantly decreased by Co2+ ions enclosed in GLAuNPs. Histofluorescence imaging revealed that the kidneys, and particularly the proximal tubules, were the site of a specific accumulation of GLAuNPs-Co.
Elemental Zn plays important roles in biological processes, such as a catalytic cofactor, in immune responses, and as a structural functionality of many proteins. Studies have shown that a high prevalence of diabetes and progressive DN have been linked to systemic zinc deficiency.108–110 In addition, the TGF-β/Smad 2/3 pathway activates renal interstitial fibroblasts.111 Alomari et al.51 studied the therapeutic consequence of zinc oxide nanostructures on STZ-induced DN rats. The results showed that ZnONPs ameliorated renal injury by ameliorating podocyte damage, oxidative stress, inflammation, and aberrant angiogenesis, as well as by enhancing renal function. The experimental DN rat model is more effectively utilized (Fig. 14). This demonstrated that ZnONP nanoparticles were operative in ameliorating the development of renal fibrosis.
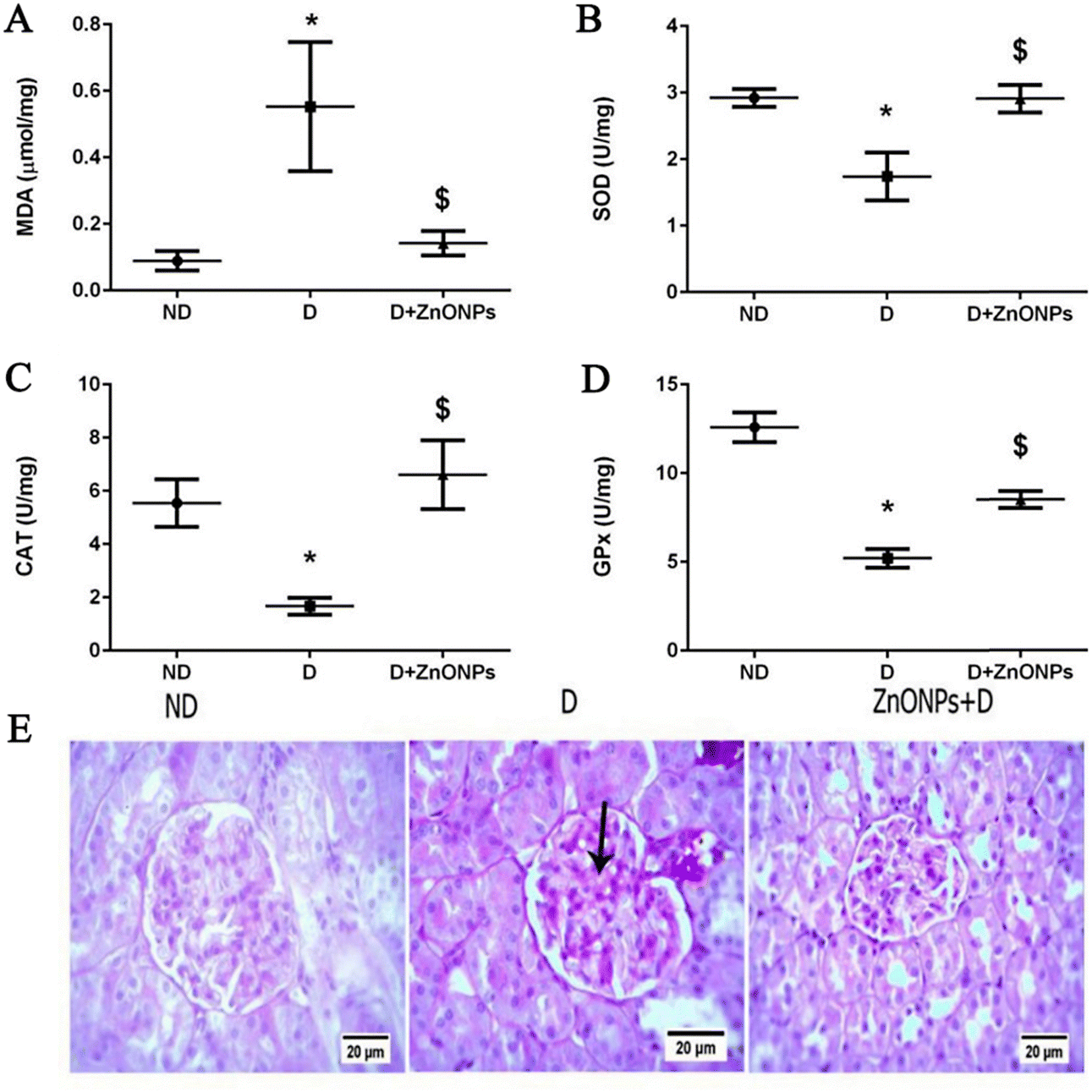 | ||
| Fig. 14 Zinc oxide nitrides and their effects on various oxidative stress markers in renal tissue in various levels (A)–(D); (E) exemplary renal tissue cortex stained with PAS (magnification ×400).51 Reproduced with permission. Copyright 2021 The Authors. | ||
5 Summary and outlook
Drug delivery technologies can effectively deliver antifibrotic therapeutics to the kidneys, which not only enhances therapeutic efficacy, but more importantly reduces the total dose and mitigates toxic side effects. Currently, NP-based delivery systems that overcome barriers to enter into the kidneys have been extensively studied, such as self-assembling micelles, polyethyleneimine nanoparticles, liposome–nanoparticle hybrids, etc. Numerous studies have shown that nanoparticles can effectively piggyback on antifibrotic therapeutics and target the injured kidneys. Through nanomaterial drug delivery strategies, it is possible to concentrate RNA, TGF-β, etc. in the kidneys. Thus, they can act on organ-specific targets. For chemical drugs, drug delivery technology can not only target them to damaged kidneys but also reduce toxic side effects and provide therapeutic effects. In addition, drug delivery technology has even enhanced the use of herbal extracts for the treatment of renal fiber. Due to poor bioavailability, toxic side effects, and other disadvantages of some Chinese medicine extracts, it is difficult to achieve anti-renal fibrosis effects by direct use. However, through drug delivery technology, they can efficiently and safely reach the target site and exert their effect. These studies bring new hope to overcome the challenges of renal fibrosis treatment, but more relevant studies for overcoming some challenges are still needed.The first is that the safety of drug delivery vehicles cannot be fully convincing. In many experiments, the biotoxicity of nanomaterials was observed only for a short period after they were injected or orally administered into a mouse model. There were fewer studies on whether prolonged use triggered the accumulation of toxicity. Secondly, the degradation and metabolism of many nanotherapeutic agents after entering the body have not been described. Moreover, now that the study population is almost exclusively mice with damaged kidneys, elucidating the distinctions between the experimental models of animals and humans with healthy and diseased kidney states may result in a more reliable transition from preclinical to clinical research. Understanding pharmacokinetic characteristics, biodistribution, and potential immunogenicity has become crucial, as has the biosafety of various nanomaterials for clinical usage. Currently, there are still drug delivery strategies whose mechanisms have not been clarified, especially nano-formulations based on active ingredients of herbal medicines. Even though it is well acknowledged that botanical substances are effective in treating RF, there are no standard diagnostic or therapeutic criteria, and the mechanisms of action of several natural compounds are unclear or inconclusive. Natural remedies do not work as quickly as western medications, and their clinical applications. The safety of nano formulations based on metal ions is difficult to be extensively recognized due to the possibility of triggering problems such as metal accumulation.
In conclusion, using a nanoscale therapeutic delivery system is a new approach to renal fibrosis treatment that effectively controls drug release, enhances targeting and improves therapeutic efficacy. Future research on drug delivery for the treatment of renal fibrosis should focus on the drug loading capacity, targeting function, biocompatibility, and pharmacokinetics of drug delivery systems. When the shortcomings and challenges of nanoscale therapeutic delivery systems are overcome one by one, it is believed that they can shine in the clinical renal fibrosis treatment.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank for the Key Scientific Research Project of Colleges and Universities of Education Department of Guangdong Province (20202ZDZX2046 and 2021ZDZX2052, 2022ZDZX2022), Dongguan Social Development Science and Technology Project (20231800936222), Guangdong Medical University Research Project (4SG23285G), Dongguan Social Development Science and Technology Project (20221800906342), the open research fund of Songshan Lake Materials Laboratory (2022SLABFN12), and Guangdong Basic and Applied Basic Research Foundation (2021A1515011616 and 2020A1515110137), Featured Innovation Project of Guangdong Province (2022KTSCX045), Dongguan Science and Technology Bureau, Dongguan Social Science and Technology Development Key Project (grant number 20221800906132 and 20211800904412) and the Natural Science Foundation of Guangdong Province (2023A1515011536).References
- N. R. Hill, S. T. Fatoba, J. L. Oke, J. A. Hirst, C. A. O. Callaghan, D. S. Lasserson and F. D. R. Hobbs, PLoS One, 2016, 11, e158765 Search PubMed.
- H. Wang, M. Naghavi, C. Allen, R. M. Barber and Z. A. Bhutta, Lancet, 2016, 388, 1459–1544 CrossRef PubMed.
- O. Swartling, H. Rydell, M. Stendahl, M. Segelmark, L. Y. Trolle and M. Evans, Am. J. Kidney Dis., 2021, 78, 190–199 CrossRef PubMed.
- V. Mahalingasivam, G. Su, M. Iwagami, M. R. Davids, J. B. Wetmore and D. Nitsch, Nat. Rev. Nephrol., 2022, 18, 485–498 CrossRef CAS PubMed.
- A. S. Levey, R. Atkins, J. Coresh, E. P. Cohen, A. J. Collins, K. U. Eckardt, M. E. Nahas, B. L. Jaber, M. Jadoul, A. Levin, N. R. Powe, J. Rossert, D. C. Wheeler, N. Lameire and G. Eknoyan, Kidney Int., 2007, 72, 247–259 CrossRef CAS PubMed.
- S. L. Friedman, D. Sheppard, J. S. Duffield and S. Violette, Sci. Transl. Med., 2013, 5, 167sr1 Search PubMed.
- B. D. Humphreys, Annu. Rev. Physiol., 2018, 80, 309–326 CrossRef CAS PubMed.
- N. Yamashita and R. Kramann, Trends Endocrinol. Metab., 2024, 35, 31–48 CrossRef CAS PubMed.
- F. Liu and S. Zhuang, Adv. Exp. Med. Biol., 2019, 1165, 625–659 CrossRef CAS PubMed.
- H. Cabral, J. Li, K. Miyata and K. Kataoka, Nat. Rev. Bioeng., 2024, 2, 214–232 CrossRef.
- P. Y. Ke and S. S. Chen, Viruses, 2012, 4, 2251–2290 CrossRef CAS PubMed.
- K. T. Weber, Y. Sun, S. K. Bhattacharya, R. A. Ahokas and I. C. Gerling, Nat. Rev. Cardiol., 2013, 10, 15–26 CrossRef CAS PubMed.
- T. A. Wynn, J. Exp. Med., 2011, 208, 1339–1350 CrossRef CAS PubMed.
- N. C. Henderson, T. D. Arnold, Y. Katamura, M. M. Giacomini, J. D. Rodriguez, J. H. Mccarty, A. Pellicoro, E. Raschperger, C. Betsholtz, P. G. Ruminski, D. W. Griggs, M. J. Prinsen, J. J. Maher, J. P. Iredale, A. Lacy-Hulbert, R. H. Adams and D. Sheppard, Nat. Med., 2013, 19, 1617–1624 CrossRef CAS PubMed.
- L. Gewin, R. Zent and A. Pozzi, Kidney Int., 2017, 91, 552–560 CrossRef CAS PubMed.
- I. Grgic, G. Campanholle, V. Bijol, C. Wang, V. S. Sabbisetti, T. Ichimura, B. D. Humphreys and J. V. Bonventre, Kidney Int., 2012, 82, 172–183 CrossRef CAS PubMed.
- K. Takaori, J. Nakamura, S. Yamamoto, H. Nakata, Y. Sato, M. Takase, M. Nameta, T. Yamamoto, A. N. Economides, K. Kohno, H. Haga, K. Sharma and M. Yanagita, J. Am. Soc. Nephrol., 2016, 27, 2393–2406 CrossRef PubMed.
- D. P. Basile, D. L. Donohoe, K. Roethe and D. L. Mattson, Am. J. Physiol.: Renal Physiol., 2003, 284, F338–F348 CrossRef CAS PubMed.
- M. Goldfarb, C. Rosenberger, Z. Abassi, A. Shina, F. Zilbersat, K. U. Eckardt, S. Rosen and S. N. Heyman, Am. J. Nephrol., 2006, 26, 22–33 CrossRef PubMed.
- D. F. Higgins, K. Kimura, W. M. Bernhardt, N. Shrimanker, Y. Akai, B. Hohenstein, Y. Saito, R. S. Johnson, M. Kretzler, C. D. Cohen, K. U. Eckardt, M. Iwano and V. H. Haase, J. Clin. Invest., 2007, 117, 3810–3820 CAS.
- T. Tanaka and M. Nangaku, Curr. Opin. Nephrol. Hypertens., 2010, 19, 43–50 CrossRef CAS PubMed.
- C. P. Liu, Y. Hu, J. C. Lin, H. L. Fu, L. Y. Lim and Z. X. Yuan, Med. Res. Rev., 2019, 39, 561–578 CrossRef PubMed.
- F. Oroojalian, F. Charbgoo, M. Hashemi, A. Amani, R. Yazdian-Robati, A. Mokhtarzadeh, M. Ramezani and M. R. Hamblin, J. Controlled Release, 2020, 321, 442–462 CrossRef CAS PubMed.
- N. Kamaly, J. C. He, D. A. Ausiello and O. C. Farokhzad, Nat. Rev. Nephrol., 2016, 12, 738–753 CrossRef CAS PubMed.
- R. Bruni, P. Possenti, C. Bordignon, M. Li, S. Ordanini, P. Messa, M. P. Rastaldi and F. Cellesi, J. Controlled Release, 2017, 255, 94–107 CrossRef CAS PubMed.
- C. H. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- L. Guo, S. Luo, Z. Du, M. Zhou, P. Li, Y. Fu, X. Sun, Y. Huang and Z. Zhang, Nat. Commun., 2017, 8, 878 CrossRef PubMed.
- J. Wang, J. J. Masehi-Lano and E. J. Chung, Biomater. Sci., 2017, 5, 1450–1459 RSC.
- H. Miyazawa, K. Hirai, S. Ookawara, K. Ishibashi and Y. Morishita, Nano Rev. Exp., 2017, 8, 1331099 CrossRef PubMed.
- N. Raval, H. Jogi, P. Gondaliya, K. Kalia and R. K. Tekade, Mol. Pharmaceutics, 2021, 18, 641–666 CrossRef CAS PubMed.
- S. S. Son, S. Hwang, J. H. Park, Y. Ko, S. I. Yun, J. H. Lee, B. Son, T. R. Kim, H. O. Park and E. Y. Lee, Sci. Rep., 2021, 11, 2191 CrossRef CAS PubMed.
- Y. Morishita, T. Imai, H. Yoshizawa, M. Watanabe, K. Ishibashi, S. Muto and D. Nagata, Int. J. Nanomed., 2015, 10, 3475–3488 CrossRef CAS PubMed.
- N. Raval, P. Gondaliya, V. Tambe, K. Kalia and R. K. Tekade, Int. J. Pharm., 2021, 605, 120842 CrossRef CAS PubMed.
- L. Wang, P. Wang, X. Li, Y. Dong, S. Wu, M. Xu, X. Chen, S. Wang, C. Zheng and C. Zou, Aging, 2021, 13, 8524–8540 CrossRef CAS PubMed.
- J. Prakash, M. H. de Borst, A. M. van Loenen-Weemaes, M. Lacombe, F. Opdam, H. van Goor, D. K. Meijer, F. Moolenaar, K. Poelstra and R. J. Kok, Pharm. Res., 2008, 25, 2427–2439 CrossRef CAS PubMed.
- E. Guise, J. E. Engel, M. L. Williams, F. Mahdi, G. R. Bidwell and A. R. Chade, Am. J. Physiol.: Renal Physiol., 2019, 316, F1016–F1025 CrossRef CAS PubMed.
- S. Liu, M. Zhao, Y. Zhou, L. Li, C. Wang, Y. Yuan, L. Li, G. Liao, W. Bresette, Y. Chen, J. Cheng, Y. Lu and J. Liu, Acta Biomater., 2020, 103, 102–114 CrossRef CAS PubMed.
- H. Gong, L. Zhang, Y. Ma, Y. Gui, T. Xiang, J. Liu, S. Fei, K. Yue, Q. Li, H. Liu, D. Xia and X. Huang, Chem. Eng. J., 2023, 462, 142258 CrossRef CAS.
- T. T. Tang, L. L. Lv, B. Wang, J. Y. Cao, Y. Feng, Z. L. Li, M. Wu, F. M. Wang, Y. Wen, L. T. Zhou, H. F. Ni, P. S. Chen, N. Gu, S. D. Crowley and B. C. Liu, Theranostics, 2019, 9, 4740–4755 CrossRef CAS PubMed.
- H. T. Cheng, H. C. Huang, T. Y. Lee, Y. H. Liao, Y. H. Sheng, P. R. Jin, K. W. Huang, L. H. Chen, Y. T. Chen, Z. Y. Liu, T. C. Lin, H. C. Wang, C. H. Chao, I. P. Juang, C. T. Su, K. H. Huang, S. L. Lin, J. Wang, Y. C. Sung and Y. Chen, J. Controlled Release, 2022, 346, 169–179 CrossRef CAS PubMed.
- L. Zhou, Z. Ye, E. Zhang, L. Chen, Y. Hou, J. Lin, F. Huang and Z. Yuan, Int. J. Nanomed., 2022, 17, 1531–1547 CrossRef CAS PubMed.
- M. Q. Tong, L. Z. Luo, P. P. Xue, Y. H. Han, L. F. Wang, D. L. Zhuge, Q. Yao, B. Chen, Y. Z. Zhao and H. L. Xu, Acta Biomater., 2021, 122, 111–132 CrossRef CAS PubMed.
- H. Xu, T. Wu and L. Huang, Adv. Drug Delivery Rev., 2021, 177, 113911 CrossRef CAS PubMed.
- J. Sun, Z. Xu, Y. Hou, W. Yao, X. Fan, H. Zheng, J. Piao, F. Li and Y. Wei, Int. J. Pharm., 2022, 616, 121490 CrossRef CAS PubMed.
- D. Chen, S. Han, Y. Zhu, F. Hu, Y. Wei and G. Wang, Int. J. Nanomed., 2018, 13, 3507–3527 CrossRef CAS PubMed.
- X. Qin, Y. Xu, X. Zhou, T. Gong, Z. R. Zhang and Y. Fu, Acta Pharm. Sin. B, 2021, 11, 835–847 CrossRef CAS PubMed.
- Q. Liu, X. Chen, M. Kan, J. Yang, Q. Gong, R. Jin, Y. Dai, J. Jin and H. Zang, Eur. J. Pharmacol., 2021, 910, 174501 CrossRef CAS PubMed.
- W. Xiong, S. H. Xiong, Q. L. Chen, K. G. Linghu, G. D. Zhao, J. Chu, G. Wong, J. Li, Y. J. Hu, Y. T. Wang and H. Yu, Carbohydr. Polym., 2021, 266, 118112 CrossRef CAS PubMed.
- M. Li, L. Tan, L. Tang, A. Li and J. Hu, J. Biomater. Sci., Polym. Ed., 2016, 27, 972–985 CrossRef CAS PubMed.
- L. Tan, X. Lai, M. Zhang, T. Zeng, Y. Liu, X. Deng, M. Qiu, J. Li, G. Zhou, M. Yu, X. Geng, J. Hu and A. Li, Biomater. Sci., 2019, 7, 1554–1564 RSC.
- G. Alomari, B. Al-Trad, S. Hamdan, A. Aljabali, Z. M. Al, K. Al-Batanyeh, J. Qar, G. J. Eaton, A. K. Alkaraki, W. Alshaer, S. Haifawi, K. Jemon, D. K. Chellappan, K. Dua and M. M. Tambuwala, IET Nanobiotechnol., 2021, 15, 473–483 CrossRef PubMed.
- T. C. Roberts, R. Langer and M. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- B. Hu, L. Zhong, Y. Weng, L. Peng, Y. Huang, Y. Zhao and X. J. Liang, Signal Transduction Targeted Ther., 2020, 5, 101 CrossRef CAS PubMed.
- Z. Meng and M. Lu, Front. Immunol., 2017, 8, 331 Search PubMed.
- T. Treiber, N. Treiber and G. Meister, Nat. Rev. Mol. Cell Biol., 2019, 20, 5–20 CrossRef CAS PubMed.
- L. Gebert and I. J. Macrae, Nat. Rev. Mol. Cell Biol., 2019, 20, 21–37 CrossRef CAS PubMed.
- P. Sun, J. Wang, T. Ilyasova, A. Shumadalova, M. Agaverdiev and C. Wang, Non-coding RNA Res., 2023, 8, 593–601 CrossRef CAS PubMed.
- L. L. Lv, Y. Feng, M. Wu, B. Wang, Z. L. Li, X. Zhong, W. J. Wu, J. Chen, H. F. Ni, T. T. Tang, R. N. Tang, H. Y. Lan and B. C. Liu, Cell Death Differ., 2020, 27, 210–226 CrossRef CAS PubMed.
- F. Liu, Z. P. Zhang, G. D. Xin, L. H. Guo, Q. Jiang and Z. X. Wang, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 4252–4260 CAS.
- A. R. Sharma, S. K. Kundu, J. S. Nam, G. Sharma, D. C. Priya, S. S. Lee and C. Chakraborty, Biomed Res. Int., 2014, 2014, 327950 Search PubMed.
- S. Y. Li and S. L. Guo, Colloids Surf., B, 2019, 178, 269–275 CrossRef CAS PubMed.
- R. Karshafian, P. D. Bevan, R. Williams, S. Samac and P. N. Burns, Ultrasound Med. Biol., 2009, 35, 847–860 CrossRef PubMed.
- N. A. Geis, H. A. Katus and R. Bekeredjian, Curr. Pharm. Des., 2012, 18, 2166–2183 CrossRef CAS PubMed.
- T. W. Evans, Aliment. Pharmacol. Ther., 2002, 16(Suppl 5), 6–11 CrossRef CAS PubMed.
- N. Kim, W. Yoo, J. Lee, H. Kim, H. Lee, Y. S. Kim, D. U. Kim and J. Oh, Gut, 2009, 58, 1382–1390 CrossRef CAS PubMed.
- J. J. Cha, C. Mandal, J. Y. Ghee, J. A. Yoo, M. J. Lee, Y. S. Kang, Y. Y. Hyun, J. E. Lee, H. W. Kim, S. Y. Han, J. Y. Han, A. Y. Chung, D. W. Yoon, I. J. Rhyu, J. Oh and D. R. Cha, Biomedicines, 2020, 8, 8–15 CrossRef PubMed.
- H. S. Jang, M. R. Noh, J. Kim and B. J. Padanilam, Front. Med., 2020, 7, 65 CrossRef PubMed.
- T. M. Bauer and E. Murphy, Circ. Res., 2020, 126, 280–293 CrossRef CAS PubMed.
- M. V. Nastase, J. Zeng-Brouwers, M. Wygrecka and L. Schaefer, Adv. Drug Delivery Rev., 2018, 129, 295–307 CrossRef CAS PubMed.
- E. P. Bottinger and M. Bitzer, J. Am. Soc. Nephrol., 2002, 13, 2600–2610 CrossRef PubMed.
- M. Zeisberg and R. Kalluri, J. Mol. Med., 2004, 82, 175–181 CrossRef PubMed.
- A. A. Eddy, J. Am. Soc. Nephrol., 1996, 7, 2495–2508 CrossRef CAS PubMed.
- H. W. Schnaper, T. Hayashida and A. C. Poncelet, J. Am. Soc. Nephrol., 2002, 13, 1126–1128 CrossRef CAS PubMed.
- N. J. Laping, Curr. Opin. Pharmacol., 2003, 3, 204–208 CrossRef CAS PubMed.
- A. R. Chade and S. Kelsen, Am. J. Physiol.: Renal Physiol., 2012, 302, F1342–F1350 CrossRef CAS PubMed.
- R. Iliescu, S. R. Fernandez, S. Kelsen, C. Maric and A. R. Chade, Nephrol., Dial., Transplant., 2010, 25, 1079–1087 CrossRef CAS PubMed.
- A. Hernandez, J. D. Hartgerink and S. Young, Front. Bioeng. Biotechnol., 2023, 11, 1139782 CrossRef PubMed.
- S. Lee, T. Trinh, M. Yoo, J. Shin, H. Lee, J. Kim, E. Hwang, Y. B. Lim and C. Ryou, Int. J. Mol. Sci., 2019, 20, 20–29 Search PubMed.
- S. Stremersch, S. C. De Smedt and K. Raemdonck, J. Controlled Release, 2016, 244, 167–183 CrossRef CAS PubMed.
- M. Tkach and C. Thery, Cell, 2016, 164, 1226–1232 CrossRef CAS PubMed.
- B. Wang, A. Zhang, H. Wang, J. D. Klein, L. Tan, Z. M. Wang, J. Du, N. Naqvi, B. C. Liu and X. H. Wang, Theranostics, 2019, 9, 1864–1877 CrossRef CAS PubMed.
- Y. Wan, L. Wang, C. Zhu, Q. Zheng, G. Wang, J. Tong, Y. Fang, Y. Xia, G. Cheng, X. He and S. Y. Zheng, Cancer Res., 2018, 78, 798–808 CrossRef CAS PubMed.
- S. Kamerkar, V. S. Lebleu, H. Sugimoto, S. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498–503 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, X. Xiang, Y. Liu, S. Zhang, C. Liu, S. Barnes, W. Grizzle, D. Miller and H. G. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- L. Ma, H. Li, S. Zhang, X. Xiong, K. Chen, P. Jiang, K. Jiang and G. Deng, Int. Urol. Nephrol., 2018, 50, 373–382 CrossRef CAS PubMed.
- F. Yang, L. Deng, J. Li, M. Chen, Y. Liu, Y. Hu and W. Zhong, Drug Des., Dev. Ther., 2020, 14, 3567–3575 CrossRef CAS PubMed.
- J. Huang, W. Gong, Z. Chen, J. Huang, Q. Chen, H. Huang and C. Zhao, Eur. J. Pharm. Sci., 2017, 99, 128–136 CrossRef CAS PubMed.
- Y. T. Wu, Y. M. Bi, Z. B. Tan, L. P. Xie, H. L. Xu, H. J. Fan, H. M. Chen, J. Li, B. Liu and Y. C. Zhou, Eur. J. Pharmacol., 2019, 853, 93–102 CrossRef CAS PubMed.
- B. Jian, H. Zhang, C. Han and J. Liu, Molecules, 2018, 23, 387 CrossRef PubMed.
- T. Liu, X. Chen, Y. Hu, M. Li, Y. Wu, M. Dai, Z. Huang, P. Sun, J. Zheng, Z. Ren and Y. Wang, Phytochemistry, 2022, 204, 113428 CrossRef CAS PubMed.
- A. D. Voloshina, A. S. Sapunova, N. V. Kulik, M. G. Belenok, I. Y. Strobykina, A. P. Lyubina, S. K. Gumerova and V. E. Kataev, Bioorg. Med. Chem., 2021, 32, 115974 CrossRef CAS PubMed.
- A. A. Kulkarni, T. H. Thatcher, H. M. Hsiao, K. C. Olsen, R. M. Kottmann, J. Morrissette, T. W. Wright, R. P. Phipps and P. J. Sime, PLoS One, 2013, 8, e63798 CrossRef CAS PubMed.
- H. Cai, Q. Liang and G. Ge, Neural Plast., 2016, 2016, 6362707 CrossRef PubMed.
- J. Ma, X. Hu, C. Liao, H. Xiao, Q. Zhu, Y. Li, Z. Liu, A. Tao, Z. He, C. Xu and K. Zheng, Molecules, 2019, 24, 1054 CrossRef CAS PubMed.
- J. Chen, X. Li, Y. Hu, W. Liu, Q. Zhou, H. Zhang, Y. Mu and P. Liu, Am. J. Chin. Med., 2017, 45, 1061–1074 CrossRef CAS PubMed.
- Q. Yang, H. M. Zang, T. Xing, S. F. Zhang, C. Li, Y. Zhang, Y. H. Dong, X. W. Hu, J. T. Yu, J. G. Wen, J. Jin, J. Li, R. Zhao, T. T. Ma and X. M. Meng, Phytomedicine, 2021, 85, 153541 CrossRef CAS PubMed.
- L. Zhang, X. Wang, S. He, F. Zhang and Y. Li, J. Ethnopharmacol., 2023, 311, 116466 CrossRef CAS PubMed.
- Y. Xiao, C. Peng, Y. Xiao, D. Liang, Z. Yuan, Z. Li, M. Shi, Y. Wang, F. Zhang and B. Guo, Front. Physiol., 2020, 11, 599 CrossRef PubMed.
- E. Tan, Z. Gao, Q. Wang, B. Han, H. Shi, L. Wang, G. Zhu and Y. Hou, Basic Clin. Pharmacol. Toxicol., 2023, 133, 757–769 CrossRef CAS PubMed.
- X. Zhang, H. He, D. Liang, Y. Jiang, W. Liang, Z. H. Chi and J. Ma, Int. J. Mol. Sci., 2016, 17, 1327 CrossRef PubMed.
- G. Yang, Z. Zhao, X. Zhang, A. Wu, Y. Huang, Y. Miao and M. Yang, Drug Des., Dev. Ther., 2017, 11, 1065–1079 CrossRef CAS PubMed.
- K. Huang, W. Liu, T. Lan, X. Xie, J. Peng, J. Huang, S. Wang, X. Shen, P. Liu and H. Huang, PLoS One, 2012, 7, e43874 CrossRef CAS PubMed.
- X. Xie, X. Chang, L. Chen, K. Huang, J. Huang, S. Wang, X. Shen, P. Liu and H. Huang, Mol. Cell. Endocrinol., 2013, 381, 56–65 CrossRef CAS PubMed.
- J. H. Park, E. Jung, H. Lim, J. R. Lee, Y. K. Joung, T. Yu and S. H. Bhang, Tissue Eng. Regener. Med., 2022, 19, 289–299 CrossRef CAS PubMed.
- X. Liu, Y. Jin, T. Liu, S. Yang, M. Zhou, W. Wang and H. Yu, Acs Biomater. Sci. Eng., 2020, 6, 4834–4845 CrossRef CAS PubMed.
- J. Ye, K. Zhang, X. Yang, M. Liu, Y. Cui, Y. Li, C. Li, S. Liu, Y. Lu, Z. Zhang, N. Niu, L. Chen, Y. Fu and J. Xu, Adv. Sci., 2024, 11, e2307424 CrossRef PubMed.
- M. Pourhajibagher and A. Bahador, Photodiagn. Photodyn. Ther., 2021, 35, 102432 CrossRef CAS PubMed.
- D. J. Al-Timimi, D. M. Sulieman and K. R. Hussen, J. Clin. Diagn. Res., 2014, 8, CC4–CC8 Search PubMed.
- S. Barman and K. Srinivasan, Can. J. Physiol. Pharmacol., 2016, 94, 1356–1365 CrossRef CAS PubMed.
- P. Ranasinghe, S. Pigera, P. Galappatthy, P. Katulanda and G. R. Constantine, Daru, 2015, 23, 44 CrossRef PubMed.
- X. Zhang, D. Liang, X. Lian, Z. H. Chi, X. Wang, Y. Zhao and Z. Ping, Mol. Med. Rep., 2016, 14, 5245–5252 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |