
Enhancing cell adhesion in synthetic hydrogels via physical confinement of peptide-functionalized polymer clusters†
Shohei
Ishikawa
 *,
Hiroyuki
Kamata
* and
Takamasa
Sakai
*,
Hiroyuki
Kamata
* and
Takamasa
Sakai
Department of Chemistry & Biotechnology, School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. E-mail: ishikawa@gel.t.u-tokyo.ac.jp; hiroyuki-kamata@g.ecc.u-tokyo.ac.jp
First published on 24th June 2024
Abstract
Artificially synthesized poly(ethylene glycol) (PEG)-based hydrogels are extensively utilized as biomaterials for tissue scaffolds and cell culture matrices due to their non-protein adsorbing properties. Although these hydrogels are inherently non-cell-adhesive, advancements in modifying polymer networks with functional peptides have led to PEG hydrogels with diverse functionalities, such as cell adhesion and angiogenesis. However, traditional methods of incorporating additives into hydrogel networks often result in the capping of crosslinking points with heterogeneous substances, potentially impairing mechanical properties and obscuring the causal relationships of biological functions. This study introduces polymer additives designed to resist prolonged elution from hydrogels, providing a novel approach to facilitate cell culture on non-adhesive surfaces. By clustering tetra-branched PEG to form ultra-high molecular weight hyper-branched structures and functionalizing their termini with cell-adhesive peptides, we successfully entrapped these clusters within the hydrogel matrix without compromising mechanical strength. This method has enabled successful cell culture on inherently non-adhesive PEG hydrogel surfaces at high peptide densities, a feat challenging to achieve with conventional means. The approach proposed in this study not only paves the way for new possibilities with polymer additives but also serves as a new design paradigm for cell culturing on non-cell-adhesive hydrogels.
 Shohei Ishikawa | I am a biomaterial scientist engaged in the research of functional polymer hydrogels in the fields of tissue engineering, regenerative medicine, and medical materials. Currently, I am working as an Assistant Professor at the Department of Chemistry & Biotechnology, School of Engineering, the University of Tokyo. In 2020, I earned PhD under the guidance of Professor Hidenori Otsuka at Tokyo University of Science. From 2020 to 2023, I worked as a postdoctoral researcher under Professor Takamasa Sakai and Professor Ungil Chung at the Department of Bioengineering, the University of Tokyo. In 2023, I transferred to the Department of Chemistry & Biotechnology at the University of Tokyo. Currently, I am focusing on the elucidation of novel gel science using hydrogels with phase separation structures and the development of scaffolds for tissue regeneration. |
Introduction
Hydrogels, composed of three-dimensional polymer networks and water, are highly versatile due to the tunability of their properties such as mesh density, substance permeability, and mechanical characteristics by modulating the polymers' composition and properties.1,2 This controllability has spurred the fabrication of various biomaterials, including tissue scaffolds that mimic the extracellular matrix, cell culture platforms with regulated cell adhesion, and drug delivery carriers with controlled release behaviors, thereby providing detailed and broad insights into cell behavior.3,4 Hydrogels made from natural polymers like collagen are predominantly used for cell cultures due to their superior biochemical signaling properties, including cell adhesion.5 However, the complexity of their signal transduction and batch-to-batch variability makes it challenging to dissect cell behavior in detail. This complexity could complicate the understanding of how material properties, such as mechanical stiffness, affect cells, potentially leading to misleading conclusions. On the other hand, hydrogels made from synthetically derived polymers like poly(ethylene glycol) (PEG) typically lack cell adhesion domains, showing no inherent cell adhesion behavior.6,7 Nevertheless, this limitation can be overcome by incorporating cell-adhesive molecules like arginine–glycine–aspartic acid (RGD) peptides into the hydrogel network, not only enabling cell adhesion but also introducing new properties like angiogenesis and enzymatic degradability.8,9 Studies have demonstrated that modifying PEG hydrogels with RGD peptides to produce hydrogels of various strengths and culturing mesenchymal stem cells on them can evaluate the influence of hydrogel stiffness on the cells’ differentiation abilities, revealing that this mechanical property significantly impacts differentiation.10–12 Thus, adding new additives to inherently non-cell-adhesive hydrogels opens up possibilities for analyzing how the hydrogels' intrinsic properties affect cell behavior, making it an exceptionally attractive strategy.However, this method is limited to cases where additives can be modified on the hydrogel network, and the capping of crosslinking points significantly reduces the hydrogel's mechanical strength, limiting the polymer types and applications. For example, hydrogels with highly uniform tetra-branch PEG networks pioneered by our group can be formed using two types of tetra-branch PEG with terminal reactivity, leading to a variety of applications such as tissue and cell scaffold.13–16 The terminal modification by additives, through reactions with amine or maleimide groups, is chemically fixated to the hydrogel network. Even in a uniform hydrogel network, bonding between PEG molecules is inhibited, leading to an unavoidable decrease in mechanical strength, complicating the biological evaluation of varying modification densities. On the other hand, agarose, a representative polysaccharide polymer featuring alternating galactose bonds and abundant hydroxyl functional groups, forms hydrogels upon cooling through hydrogen bond formation.17 While it is possible to chemically bind various additives to this polysaccharide network, the synthesis process under harsh alkaline conditions can compromise the sugar chain's properties, and strength reduction due to damaged crosslinking points remains a concern. To overcome this issue, the development of polymer additives has been pursued.18–20 Techniques like PEGylation, where peptides or bioactive substances are modified at the polymer ends and then encapsulated within hydrogels, are expected to prevent mechanical strength reduction without compromising the hydrogel network's crosslinking points. This method allows for more precise physiological analysis by distinctly evaluating the impacts of the hydrogel's properties and the additives under identical conditions. However, the challenge remains that additives may leach out of the hydrogel because the mesh size of the hydrogel is larger than the polymer additives.4,21 This characteristic, albeit unavoidable due to osmotic pressures within the hydrogel, can be mitigated by increasing the size of the polymer additives relative to the mesh size.
In this study, we introduced the development of polymer additives capable of being physically entrapped within the 3D network of hydrogels over extended periods and an approach that allows for the evaluation of cell behavior in non-cell-adhesive hydrogels through long-term encapsulation. While typical methods require molecular design each time to covalently attach functional molecules, such as cell-adhesive peptides, to the polymer chain of hydrogels,22 our physical confinement method offers the distinct advantage that no such customization is required. Therefore, we considered that this strategy, which overcomes the challenges of existing methods, to be promising for the functionalization and application of a wide variety of hydrogels. As polymer additives, we designed clustered-PEG (cP) obtained by clustering two types of tetra-branched PEG (tP) with terminal reactivity, resulting in a high molecular weight entity approximately 10 times the diameter of the tP prepolymer (Scheme 1). We hypothesized that the clustering of tP prepolymers would be sufficiently large compared to the tP hydrogel's mesh size, thereby physically trapping them within the hydrogel for an extended period without adversely affecting the hydrogel's mechanical properties, enabling the assessment of cell functionalities such as cell culture. To validate this hypothesis, we fabricated tP hydrogels using tP as the prepolymer, encapsulated cP with sizes larger than the hydrogel's mesh size, and evaluated their release behavior. Due to the reactivity of the cP termini, it is possible to modify them with peptides, thus enabling the development of functionalized cP (fcP). As anticipated, while tP was quickly released from the hydrogel, cP remained relatively longer within the hydrogel matrix, consequently showing cell adhesion in tP hydrogels with RGD-cP encapsulation.
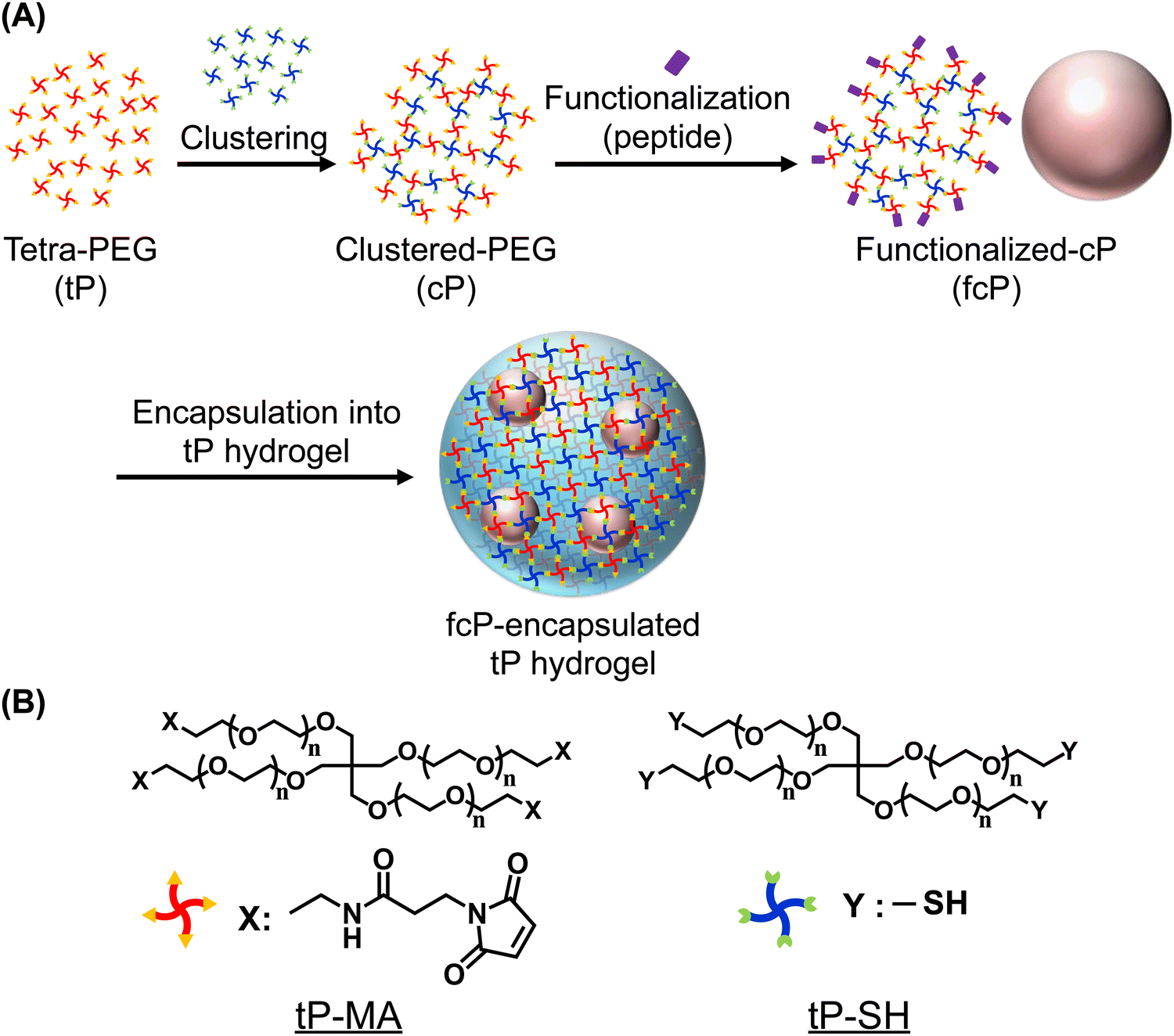 | ||
| Scheme 1 Design and synthesis of clustered-poly(ethylene glycol) within tetra-PEG hydrogels. (A) Schematic representation of the process for fabricating tP hydrogels encapsulating cP functionalized with peptides (fcP). This involves non-equivalent mixing of two end-reactive tP species and peptide functionalization, which is subsequently encapsulated within tP hydrogels. (B) Chemical structures of tP end-functionalized with maleimide (tP-MA) and sulfhydryl (tP-SH) groups. | ||
Result and discussion
Preparation and characterization of clustered-PEG
In the initial phase of synthesizing fcP, we dissolved tP prepolymers end-functionalized with maleimide (tP-MA) and sulfhydryl (tP-SH) in a buffer. These were then mixed under non-equivalent conditions to form a sol rather than a hydrogel. This distinction is crucial for our application since the sol state better facilitates the modification process, which is essential for achieving the desired functionalization. By significantly altering the mixing ratio (r), we induced the formation of ultra-high molecular weight hyper-branched clusters.23 For the modification of additives with maleimide reactivity, we prepared a polymer solution by mixing tP-MA in excess to tP-SH. Subsequently, we then identified the critical mixing ratio for optimal cluster formation using dynamic light scattering (DLS) to analyze the autocorrelation function of a polymer solution prepared at 20 g L−1 (Fig. 1A). The function peaked at 10−3 ms and decays gradually towards 101 ms, indicative of polymer relaxation within this timescale. However, the peak value at r = 0.7, markedly lower than that of other polymer solutions including tP-MA, suggests restricted polymer movement. While other polymer solution showed higher peak due to more vigorous movement. This observation led us to conclude that sols are predominantly formed when the mixing ratio exceeds r = 0.75, highlighting the importance of this threshold for achieving the polymer solution with desired state for clustering.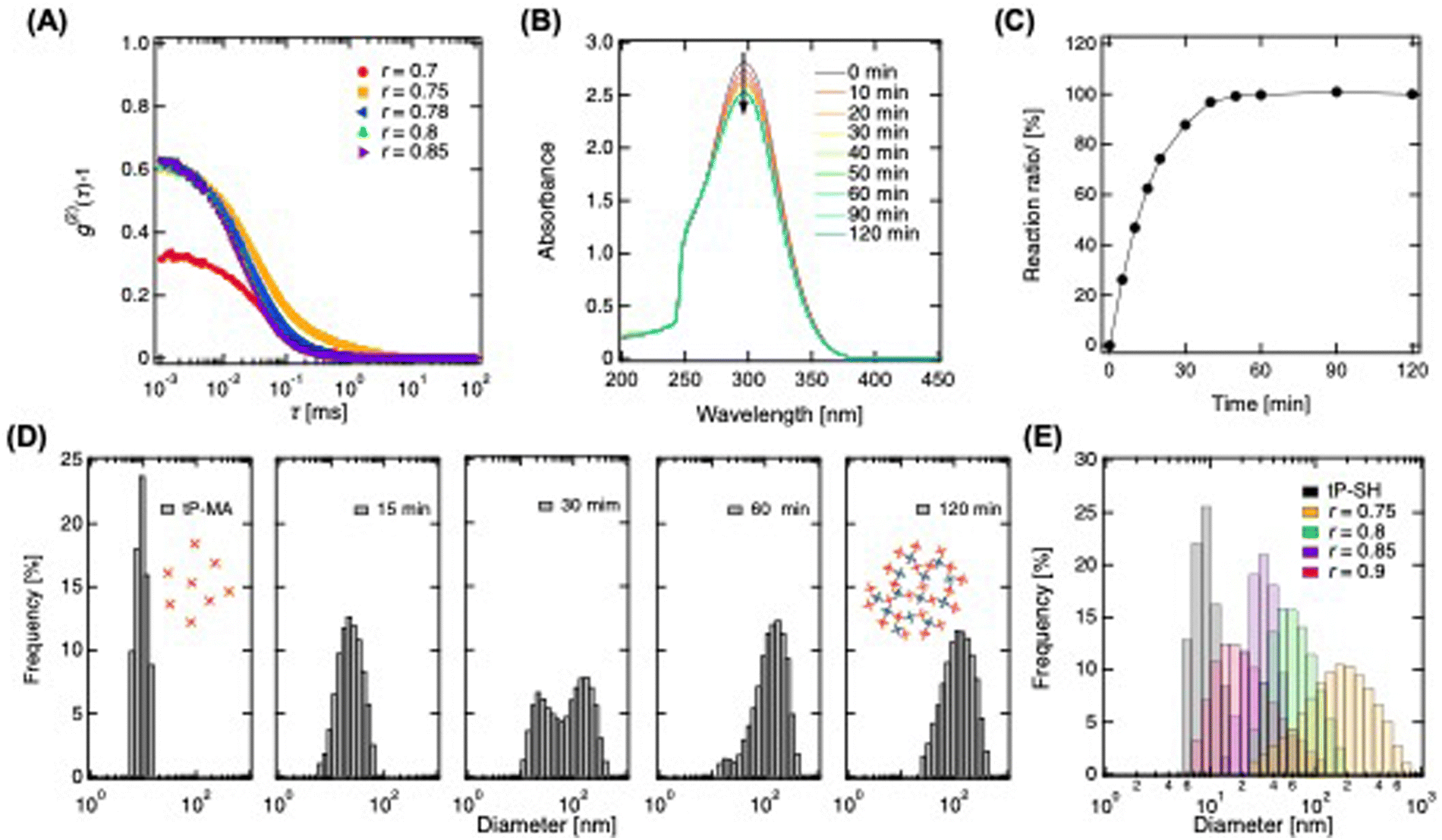 | ||
| Fig. 1 Preparation and characterization of clustered-PEG. (A) Time-correlation function of cP prepared by stoichiometrically imbalanced mixing, where tP-MA was mixed in excess relative to tP-SH (r = 0.7–0.85). (B) UV-Visible spectra of the reaction between tP-MA and tP-SH under stoichiometrically imbalanced conditions (r = 0.78). (C) Reaction ratio of maleimide to sulfhydryl groups at the PEG terminus, calculated from the absorbance at 300 nm. (D) Size distribution under cluster growth process (r = 0.78). (E) Size distribution of cP synthesized under stoichiometrically imbalanced conditions (r = 0.75, 0.8, 0.85, 0.9). | ||
To further evaluate the reaction between tP-MA and tP-SH, we monitored the process in a polymer solution with a mixing ratio of r = 0.78 using UV-vis spectroscopy (Fig. 1B). The spectrum of maleimide groups around 300 nm24 decreases over time, stabilizing after 60 minutes, which allows for a qualitative assessment of the click reaction. Assuming completion after 120 minutes, we observed a rapid reaction progression to approximately 90% within 30 minutes, eventually reaching completion of reaction between tP-SH and tP-MA (Fig. 1C). Moreover, DLS revealed the particle size distribution evolution from monomodal around 10 nm to larger clusters through inter-cluster linkage, eventually forming clusters around 100 nm (Fig. 1D). This tenfold growth under extreme non-equivalent mixing conditions showcases to create hyper-branched, heterogeneously reactive cP. Adjusting the mixing ratio allows for controlled modulation of cluster sizes, from larger clusters near the critical mixing ratio to smaller ones as the ratio increases, with diameters controllably ranging from about 20 nm to 130 nm (Fig. 1E and Fig. S1, ESI†). Given that the mesh size of tP hydrogels using the prepolymers is approximately 5–20 nm,25,26 clusters exceeding this size are expected to be physically trapped within the hydrogel, illustrating our method's potential for creating hydrogels with tunable releasing properties.
Functionalization of cell adhesive peptide
In the subsequent step of our study, we focused on purifying the synthesized cP solution via freeze-drying, a process crucial for its subsequent incorporation into hydrogel matrices (Fig. 2A). The key challenge addressed was ensuring the water solubility of cP post-purification, essential for its seamless integration into hydrogels. Initially, freeze-dried cP formed a white, spongy structure that was water-insoluble. This issue was attributed to the formation of an ultra-concentrated matrix during freeze-drying, which increased the molecular weight due to unreacted maleimide and sulfhydryl groups reacting, similar to creating a structured form by freeze-drying a sponge.27 To overcome the water insolubility of cP, alkaline treatment was employed, effectively inducing the ring-opening of maleimide groups to form carboxylic groups. This transformation eliminated the reactivity between tP-SH and tP-MA, achieving water solubility (Fig. 2A). The success of this treatment was validated by monitoring the UV-vis spectrum of cP suspended in a pH 7.4 buffer (Fig. 2B). A decrease in spectrum at 300 nm, indicative of maleimide groups, along with an increase in spectrum at 255 nm, characteristic of ring-opening reaction's progression.28 This process proved to be pH-dependent, with ring-opening facilitated in neutral and alkaline conditions, a pivotal step enabling the subsequent functionalization with cell-adhesive peptides (Fig. 2C). After this alkali treatment, the polymer formed a structure consisting of numerous tPs, strongly supporting the formation of cP (Fig. S2, ESI†). | ||
| Fig. 2 Functionalization of clustered-PEG. (A) UV-Visible spectra of the hydrolysis process of cP (r = 0.78) in phosphate buffer at pH 7.4. (B) Time-dependent changes in absorbance at 300 nm of cP (r = 0.78) in various buffers at different pH levels (3.8, 5.0, 7.4, 9.8). (C) Water solubility of cP powder (r = 0.78) post-treatment with and without buffer at pH 9.8. (D) UV-Visible spectra of the reaction between GRGDSPC peptide and cP (r = 0.78). (E) 1H-NMR spectrum of tP-MA, base-treated tP-MA, base-treated cP, and fcP. (F) Comparison of the experimentally calculated modification ratio of RGD peptide with the theoretical limit. | ||
For the modification of cP's MA terminals, we employed the cell-adhesive peptide sequence GRGDSPC, observing the reaction via UV-vis spectroscopy (Fig. 2D and E). This combination results in the thiol-maleimide reaction between the cysteine residue and the MA terminal, which forms covalent thioether bonds. The reduction in spectrum at 300 nm over time indicated the successful click reaction between cP and the peptide, eventually reaching completion of reaction after around 200 min. Modification of these functional peptides did not alter the size distribution of the clusters (Fig. S3, ESI†). With tP solution, the reaction between the tetra-branch terminals and the cysteine residue of the peptide increases theoretically as the concentration rises, thus elevating the modification density (Fig. 2F). A similar linear increase in modification rate is observed with cP solution, nearly reaching the theoretical limit. In such instances, the impact on mechanical strength is minimal, and the release from the hydrogel is expected to be inhibited, facilitating not only the assessment of functional peptide effects across a broad concentration but also improving cell culture on traditionally non-adhesive surfaces. Consequently, this significantly broadens the scope and application of hydrogels in biomedical engineering and related fields.
Release profile of clustered-PEG from tetra-PEG hydrogels
Next, we encapsulated the designed cP within tP hydrogels to assess their release behavior (Fig. 3A). The encapsulation process, involving the cP in tP solutions followed by mixing, resulted in tP hydrogels containing uniformly dispersed cP, as evidenced by the absence of phase separation like cloudiness. This uniform dispersion was further confirmed by confocal laser scanning microscopy, which showed no aggregated structures of fluorescence-labeled cP within the hydrogel, indicating a consistent dispersion across all designed clusters (Fig. 3B). This confinement process did not significantly change the mechanical properties of hydrogels (Fig. S4, ESI†). To elucidate the release dynamics of cP from the hydrogel, we immersed the hydrogel in PBS and monitored the attenuation of fluorescence intensity, revealing a clear dependency on the cluster size (Fig. 3C). In all hydrogels, a rapid decrease in fluorescence intensity observed after 2–3 h was attributed to osmotic pressure acting within the hydrogel, leading to the release of polymers. Notably, tP encapsulated within the hydrogel showed almost complete fluorescence attenuation within approximately 6 h, whereas smaller cP clusters (r = 0.9) demonstrated a gradual decrease in intensity, persisting even after 48 h. In contrast, larger cP clusters (r = 0.78) exhibited a more pronounced retention of fluorescence intensity, indicative of their prolonged retention within the hydrogel (Fig. 3D). The quantitative analysis of cP release into PBS further highlighted the release dynamics, with an initial burst release observed, followed by a significant reduction in the release rate for larger clusters, and negligible release detected after 72 h (Fig. 3E). Subsequently, while tP continued to be gradually released, the release of cP effectively ceased. This pattern underscores that tP release aligns with the hydrogel's mesh size and the osmotic pressure within the hydrogel, whereas larger cP clusters remain retained. This observation underscores the potential to enhance the functional efficacy of additives by controlling the size of the clusters, thereby offering a strategy to tailor release profiles and maintain sustained functionality of hydrogel-encapsulated additives. | ||
| Fig. 3 Release profile of clustered-PEG from hydrogels. (A) Photographs of tP hydrogels encapsulating cP (left) and without cP (right). (B) Images of hydrogels encapsulating alexa-labeled cP, shown via bright field (left), fluorescence (middle), and merged (right) observation. Scale bars represent 500 μm. (C) Merged images of various hydrogels encapsulating alexa-labeled cP during PBS immersion for 48 h. Scale bars represent 500 μm. (D) Fluorescence intensity relative to time 0 h. (E) Cumulative release ratio of alexa -labeled cP from hydrogels over time. | ||
Additionally, when encapsulating cP within agarose hydrogels, we observed retention within the hydrogel, albeit to a lesser extent than in tP hydrogels, attributed to agarose's larger mesh size.29 This finding suggests that increasing the cluster size or utilizing longer tP as the prepolymer could significantly augment retention, underscoring the adaptability and controllability of our strategy for broader hydrogel applications. Thus, our study presents a novel approach to optimizing hydrogel functionality through the strategic encapsulation of functional clusters, with implications for their use in various biomedical applications.
Cell culture on hydrogels encapsulating functionalized, clustered-PEG
Finally, our study encapsulated functionalized cP (cP-RGD, r = 0.78) within tP hydrogels to assess its impact on fibroblast adhesion and proliferation on hydrogel surfaces (Fig. 4A). In this study, we used the well-known cell-adhesive RGD peptide, which can confer cell adhesion properties to various hydrogels, to simply evaluate hydrogel functionalization by observing the adhesion of fibroblasts—whose adhesion mechanisms are well understood—to the hydrogel surface.22 On the first day, fibroblasts demonstrated adhesion and slight extension on the hydrogel surface, exhibiting good survival and non-cytotoxicity of cP. Over time, these behaviors became more pronounced, culminating in significant cell proliferation by day 7. This enhancement in cell-adhesive properties, attributable to the RGD modification of cP, marks a notable contrast to the non-adhesive nature of hydrogels with unmodified cP or traditional tP hydrogels (Fig. 4B). When incorporating RGD-cP into agarose hydrogels, we observed similar initial adhesion and survival as in tP hydrogels. However, a distinct shift occurred by day 4, with cells detaching and forming spheroids, likely due to the efficient release of fcP from the agarose matrix. This behavior underscores the role of fcP in imparting cell adhesiveness not just to tP hydrogel but also to agarose hydrogels, despite their inherent differences in mesh size and composition.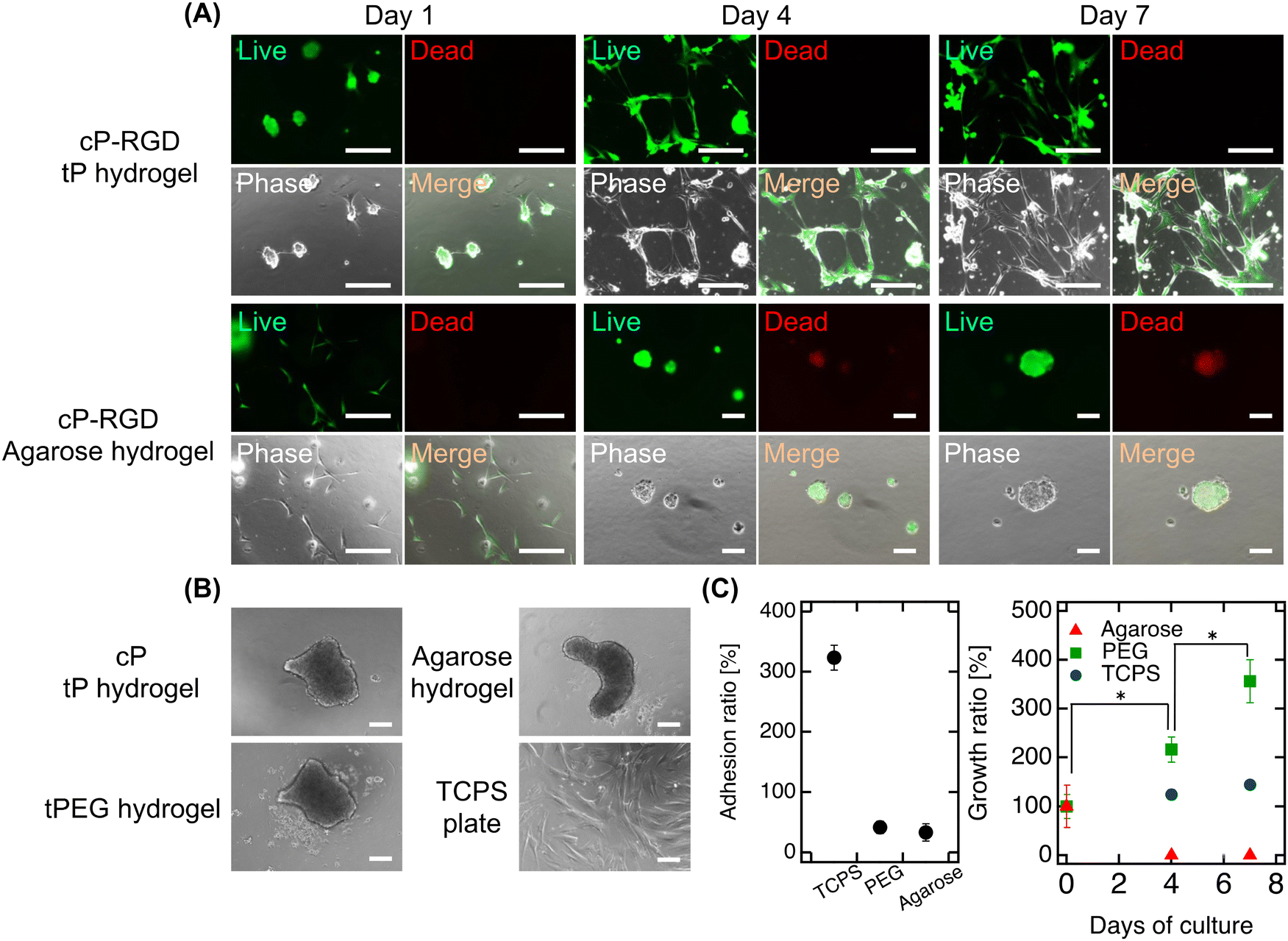 | ||
| Fig. 4 Two-dimensional cell culture on hydrogels. (A) Live/dead staining images of cells cultured for 7 days on tP hydrogels (upper section) and agarose hydrogels (lower section) both encapsulating cP (r = 0.78). Scale bars represent 200 μm. Photos of each block show live (left-upper), dead (right-upper), phase-contrast (left-lower), and merge (right-lower) images. (B) Phase-contrast images of cells on various hydrogels after 1 day of culture. Scale bars represent 200 μm. (C) Quantitative analysis of cell adhesion, expressed as the ratio of adhered cells to the number of cells seeded (left), and cell growth, expressed as the growth ratio after 1 day of culture (right). *P < 0.05 was considered statistically significant. | ||
Although initial adhesion rates were lower compared to fibroblasts cultured on standard TCPS plates, the observed proliferation in tP hydrogels containing cP indicates that the larger cP clusters function effectively without being released (Fig. 4C). This adhesion and proliferation can be further enhanced by optimizing the RGD modification or concentration on cP. Our approach not only simplifies the chemical modification of additives to cP but also opens avenues for integrating peptides that promote angiogenesis or wound healing, paving the way for the development of innovative functional cP variants. Moreover, by employing synthetically derived materials, our strategy presents a reproducible and potentially safer alternative to animal-derived additives, minimizing concerns about cytotoxicity in biomedical applications. It heralds new possibilities for hydrogel-based technologies in hydrogel engineering, drug delivery, and regenerative medicine, suggesting exciting directions for future research and application.
Conclusions
Our study presents a breakthrough in hydrogel technology by encapsulating and functionalizing clustered-PEG within PEG hydrogels, enhancing cell adhesion and proliferation. This advancement not only overcomes the limitations of traditional non-cell-adhesive hydrogels but also introduces a controlled release mechanism based on cluster size, showcasing our method's precision in hydrogel property modulation. The observed uniform dispersion and sustained functionality of cP within the hydrogels demonstrate the potential for creating multifunctional biomaterials applicable to tissue engineering, regenerative medicine, and drug delivery. Furthermore, the successful application of this strategy across different hydrogel matrices, combined with the use of synthetically derived materials, highlights its versatility and safety for biomedical applications. Looking forward, this work paves the way for incorporating a wider range of functional peptides into hydrogels, expanding their biological activities and applications. Our findings set the stage for further investigations into the in vivo performance of these advanced hydrogels and their potential to revolutionize biomaterials science.Experimental section
Materials
4-Armed poly(ethylene glycol) (tetra-PEG (tP), Mw = 10 kg mol−1) end-functionalized with maleimide (tP-MA) and sulfhydryl (tP-SH) was purchased from SINOPEG Biotech Co., Ltd (Fujian, China). GRGDSPC was purchased from GenScript (Tokyo, Japan). Alexa Fluor™ 488 NHS ester (succinimidyl ester) (Alexa488-NHS), Dulbecco's modified Eagle's medium (DMEM), penicillin/streptomycin (PS), fetal bovine serum (FBS), trypsin-EDTA (0.05%), LIVE/DEAD™ viability/cytotoxicity kit, and D-PBS(−) (PBS) were purchased from Thermo Fisher Scientific Inc. (Massachusetts, USA). Fluorescein-4-isothiocyanate (FITC) was purchased from Sigma-Aldrich (Missouri, USA). Dimethyl sulfoxide (DMSO) and methanol were purchased from FUJIFILM Wako Pure Chemical Corporation (Tokyo, Japan). 0.45 μm filter (Minisart, Sartorius AG, Göttingen, Germany), dynamic light scattering (DLS) (Zeta-potential & Particle size Analyzer ELSZneo, Otsuka Electronics Co., Ltd, Osaka, Japan), confocal laser scanning microscope (CLSM, LSM 900, Carl Zeiss AG, Jena, Germany), and ARVO™ X3 microplate reader (PerkinElmer, Inc., Massachusetts, USA) were used in all experiment. In order to adjust the level of pH to control the reaction rate of the thiol-maleimide coupling, citrate phosphate buffer (CPB) was prepared according to manufacture protocol.Preparation of clustered-PEG
tP-SH and tP-MA were dissolved in citrate phosphate buffer (CPB, 20 mM, pH3.8) to achieve a PEG concentration (CPEG) of 20 g L−1. To prepare the pre clustered-PEG (pre cP) solution, tP-MA solution was mixed with the tP-SH solution under stoichiometrically imbalanced conditions, where tP-MA was added in an excess ratio relative to tP-SH, defined by the ratio r = [tP-MA/(tP-SH + tP-MA)]. This pre cP solution was then incubated at 25 °C for 24 h, resulting in the formation of cP.Dynamic light scattering analysis
cP solutions with varying ratios (r = 0.75, 0.78, 0.80, 0.85, 0.90) were diluted in CPB to achieve a final CPEG of 2.0 g L−1. Each solution was transferred into a plastic cuvette (path length: 10 mm) and placed within a dynamic light scattering device. The particle size and distribution of both pre cP and cP solutions were assessed at 25 °C by measuring the time correlation function.Reaction analysis between between PEGs via UV-vis spectroscopy
The pre cP solution with a ratio (r = 0.78) was placed into a plastic cuvette (path length: 10 mm) and incubated at 25 °C for 24 h. Subsequently, the absorbance across the 200–450 nm range was recorded using a UV-vis spectrophotometer (V-670, JASCO Corp., Tokyo, Japan) at 25 °C. To assess the reaction ratio between tP-MA and tPEG-SH, we specifically measured the absorbance at 300 nm. This reaction ratio was calculated as the absorbance ratio of AbsTime (absorbance at a given time) to Abs120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h (absorbance after 120 h), based on the assumption that the reaction reaches completion after 120 h.
h (absorbance after 120 h), based on the assumption that the reaction reaches completion after 120 h.
Mechanical strength of hydrogels
Pre-hydrogel solution was poured in a silicon mold (diameter: 30 mm, thickness: 2 mm) and incubated at 25 °C for 24 h. Subsequently, each hydrogel was removed carefully from the mold and put on the measurement plate. Hydrogels were then compressed with PP25 sensor until detecting the road to 1.5 N, and G′ and G′′ were evaluated at a γ of 1% and ω of 0.6–60 rad s−1 at 25 °C.Hydrolysis process of clustered-PEG solutions
The cP solution with a ratio (r = 0.78) was mixed in a 1:1 volume ratio with CPB at pH 3.8 and 5.8 (200 mM), phosphate buffer at pH 7.4 (200 mM), and carbonate buffer at pH 9.5 (200 mM). The mixtures were incubated at 25 °C. Subsequently, each solution was transferred to a plastic cuvette (path length: 10 mm), and the absorbance within the 200–450 nm range was measured immediately after mixing and then continuously for 48 h using a UV-vis spectrophotometer. Following incubation, the solutions underwent dialysis against water for 24 h using a Spectra/Por® 4 dialysis membrane (MWCO: 12–14 kD, Repligen, Massachusetts, USA), with the water being exchanged three times to ensure thorough removal of small molecules. The dialyzed solutions were then lyophilized for 24 h, resulting in dried cP. To assess water solubility differences, PBS was added to both the alkaline treated (pH 9.5) cP and the non-treated cP to achieve a final CPEG of 10 g L−1. This comparison aimed to elucidate the impact of pH treatment on cP's solubility in aqueous environments.
Observation by transmission electron microscopy
Morphologies of cP (r = 0.78) and tP were observed by transmission electron microscopy (TEM). Only the cP solution was mixed with 2% uranyl acetate solution. The cP and tP solution were dripped onto collodion film on the TEM grid and air-dried. The air-dried samples were measured by TEM (Bio-TEM JEM-1400, JEOL, Japan).Peptide functionalization process
GRGDSPC peptide was dissolved with CPB (pH 3.8, 200 mM) to achieve a peptide concentration of 50 mM. This peptide solution was then added to the cP solution (r = 0.78), adjusting the final peptide concentration to range from 0.5 to 8 mM. The resulting mixture was incubated at 25 °C for 6 h. Subsequently, the absorbance within the 200–450 nm range was measured using a UV-vis spectrophotometer, with particular attention to the absorbance at 300 nm to evaluate the reaction ratio between the maleimide groups of cP and the peptide. Following the completion of the reaction, the mixed solution was combined with carbonate buffer (pH 9.5) at a 1:1 volume ratio and incubated for an additional 6 h. at 25 °C. Finally, the incubated solution underwent dialysis against water and was lyophilized, resulting in the functionalized cP ready for further applications.
Characterization of RGD functionalization
FITC was first dissolved in methanol to achieve a concentration of 10 g L−1. Following the incubation of the RGD-cP solution with carbonate buffer, phosphate buffer (pH 7.4) was added to the cP solution in a 1:1 volume ratio. Subsequently, the FITC solution was introduced to achieve 100 equivalents (eq.) relative to the tP-MA in cP. The mixture was then incubated at 25 °C for 24 h. Post-incubation, the solution underwent dialysis against water and was subsequently lyophilized to obtain the functionalized cP. The lyophilized cP was redissolved in PBS to a final CPEG of 10 g L−1. The fluorescence intensity of this solution was measured using a microplate reader (excitation: 485 nm, Emission: 535 nm). Based on these fluorescence readings, the reaction ratio of the RGD functionalization was evaluated, providing a quantitative measure of the functionalization's efficacy.
Preparation of alexa-labelled clustered-PEG
Alexa488-NHS was first dissolved in DMSO to achieve a concentration of 1 g L−1. Following the incubation of the cP solution with carbonate buffer, phosphate buffer (pH 7.4) was added to the cP solution in a 1:1 volume ratio. Subsequently, the Alexa488-NHS solution was introduced to achieve a molar ratio of 0.1 equivalents (eq.) relative to the tP-MA present in cP. The mixture was incubated at 25 °C for 24 h to facilitate the conjugation of Alexa488 to cP. After the incubation period, the solution underwent dialysis against water to remove unreacted dye and small molecules, followed by lyophilization to obtain the Alexa-labeled cP in dry form.
Observation by confocal laser scanning microscopy
The lyophilized cP was redissolved in CPB at pH 3.8 to a final concentration of 5 g L−1. Separately, solutions of tP-MA and tP-SH were prepared in the cP solution to reach a concentration of 80 g L−1 each. These solutions were then mixed in a 1:1 volume ratio to form the pre-hydrogel solution containing cP. The mixture was transferred into glass capillaries (diameter: 2 mm) for gelation. After a 24 h incubation at 25 °C, the capillary-shaped hydrogels were gently extracted and sectioned into 1 cm lengths. These sections were placed in glass-bottom dishes, submerged in 1 mL of PBS, and covered with a glass slide to minimize water evaporation. The prepared samples were then analyzed in an LSM800 confocal microscope to monitor fluorescence intensity changes over a 48 h period. Observations were conducted at a constant temperature of 25 °C.
Release observation of clustered-PEG from hydrogel
200 μL of the pre-hydrogel solution containing alexa-labeled cP was dispensed into a 1.5 mL sampling tube. Subsequently, 1.2 mL of PBS was added to the tube to initiate the release process. At predetermined time intervals, 1.2 mL of the PBS solution was carefully removed for analysis and replaced with fresh PBS to maintain the volume and conditions consistent throughout the observation period. The fluorescence intensity of the sampled solutions was measured using a plate reader excitation: 485 nm, emission: 535 nm. The release ratio of cP from the hydrogel was determined by accumulating the fluorescence intensities of the sampled solutions, providing a quantitative measure of cP released over time.Culture of human dermal fibroblasts
Live human dermal fibroblasts (HDFs) at passage 14, with a density of 1.0 × 106 cells, were cultured in 10 cm dishes (Corning Inc., New York, USA) using DMEM supplemented with 10% FBS and 1% PS, at 37 °C in an atmosphere of 5% CO2. The culture medium was refreshed every 2–3 days to maintain optimal growth conditions. Separately, solutions of tP-SH and tP-MA were prepared in PBS to a concentration of 80 g L−1 and subsequently filtered through 0.45 μm filters for sterilization. Dried cP with a ratio r of 0.78 was also dissolved in PBS. These PEG solutions and cP solution were then combined in a 1:1 volume ratio to form the pre-hydrogel mixture. This mixture was dispensed into a 24-well plate, using sufficient volume to ensure hydrogel formation upon a 30 min incubation at 37 °C. The final concentrations in the wells were adjusted to 40 g L−1 for the PEG hydrogel and 5 g L−1 for cP. HDFs were seeded onto the prepared hydrogels at a density of 2.0 × 104 cells per well and incubated at 37 °C under 5% CO2. Cell viability and proliferation were assessed on days 1, 4, and 7 using a live/dead staining solution prepared with 1 μM calcein AM and 2 μM ethidium homodimer in DMEM to distinguish live from dead cells effectively. Moreover, cell number was evaluated by trypan blue staining method.
Author contributions
S. I. and H. K. designed the overall approach. S. I. carried out all the experiments. S. I. and H. K. drafted and reviewed manuscript. Administrative, technical, and supervisory tasks were handled by S. I. All authors discussed, commented, and have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that H.K. and T.S. are employees of Gellycle Co., Ltd, Japan.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 23K17208, 23K08327, JST CREST Grant Number JPMJCR1992, Data Creation and Utilization-Type Material Research and Development Project Grant Numbers JPMXP1122714694, and Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP1224UT0182.References
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- H. Cao, L. Duan, Y. Zhang, J. Cao and K. Zhang, Signal Transduction Targeted Ther., 2021, 6, 1–31 CrossRef PubMed.
- J. Nicolas, S. Magli, L. Rabbachin, S. Sampaolesi, F. Nicotra and L. Russo, Biomacromolecules, 2020, 21, 1968–1994 CrossRef CAS PubMed.
- S. Correa, A. K. Grosskopf, H. Lopez Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Chem. Rev., 2021, 121, 11385–11457 CrossRef CAS PubMed.
- Q. Xu, J. E. Torres, M. Hakim, P. M. Babiak, P. Pal, C. M. Battistoni, M. Nguyen, A. Panitch, L. Solorio and J. C. Liu, Mater. Sci. Eng., R, 2021, 146, 1–98 CrossRef PubMed.
- J. Shi, L. Yu and J. Ding, Acta Biomater., 2021, 128, 42–59 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets and T. Hoare, RSC Adv., 2015, 5, 35469–35486 RSC.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- Z. Xiao, S. Zhao, X. Zhang, G. Wei and Z. Su, Macromol. Mater. Eng., 2022, 307, 1–14 Search PubMed.
- S. A. Maynard, A. Gelmi, S. C. Skaalure, I. J. Pence, C. Lee-Reeves, J. E. Sero, T. E. Whittaker and M. M. Stevens, ACS Nano, 2020, 14, 17321–17332 CrossRef CAS PubMed.
- R. Randriantsilefisoa, Y. Hou, Y. Pan, J. L. C. Camacho, M. W. Kulka, J. Zhang and R. Haag, Adv. Funct. Mater., 2020, 30, 1905200 CrossRef CAS.
- K. Ye, L. Cao, S. Li, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2016, 8, 21903–21913 CrossRef CAS PubMed.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Il Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- S. Ishikawa, H. Kamata, U. Il Chung and T. Sakai, RSC Adv., 2021, 11, 23637–23643 RSC.
- S. Ishikawa, Y. Yoshikawa, H. Kamata, U. Chung and T. Sakai, ACS Appl. Mater. Interfaces, 2022, 14, 35444–35453 CrossRef CAS PubMed.
- S. Ishikawa, Y. Iwanaga, T. Uneyama, X. Li, H. Hojo, I. Fujinaga, T. Katashima, T. Saito, Y. Okada, U. il Chung, N. Sakumichi and T. Sakai, Nat. Mater., 2023, 22, 1564–1570 CrossRef CAS PubMed.
- M. Khodadadi Yazdi, A. Taghizadeh, M. Taghizadeh, F. J. Stadler, M. Farokhi, F. Mottaghitalab, P. Zarrintaj, J. D. Ramsey, F. Seidi, M. R. Saeb and M. Mozafari, J. Controlled Release, 2020, 326, 523–543 CrossRef CAS PubMed.
- M. Lunzer, L. Shi, O. G. Andriotis, P. Gruber, M. Markovic, P. J. Thurner, D. Ossipov, R. Liska and A. Ovsianikov, Angew. Chem., Int. Ed., 2018, 57, 15122–15127 CrossRef CAS PubMed.
- H. Otsuka, A. Hirano, Y. Nagasaki, T. Okano, Y. Horiike and K. Kataoka, ChemBioChem, 2004, 5, 850–855 CrossRef CAS PubMed.
- Y. Xu, H. Wang, C. Luan, Y. Liu, B. Chen and Y. Zhao, Biosens. Bioelectron., 2018, 100, 404–410 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- K. Hayashi, F. Okamoto, S. Hoshi, T. Katashima, D. C. Zujur, X. Li, M. Shibayama, E. P. Gilbert, U. Il Chung, S. Ohba, T. Oshika and T. Sakai, Nat. Biomed. Eng., 2017, 1, 1–7 CrossRef.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656–8664 RSC.
- Y. Tsuji, X. Li and M. Shibayama, Gels, 2018, 4, 50 CrossRef PubMed.
- T. Sakai, Physics of Polymer Gels, Wiley-VCH Verlag, 2020 Search PubMed.
- H. Schoof, J. Apel, I. Heschel and G. Rau, J. Biomed. Mater. Res., 2001, 58, 352–357 CrossRef CAS PubMed.
- S. Kirchhof, A. Strasser, H. J. Wittmann, V. Messmann, N. Hammer, A. M. Goepferich and F. P. Brandl, J. Mater. Chem. B, 2015, 3, 449–457 RSC.
- J. Narayanan, J. Y. Xiong and X. Y. Liu, J. Phys. Conf. Ser., 2006, 28, 83–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00761a |
| This journal is © The Royal Society of Chemistry 2024 |