
Soft nanoparticles as antimicrobial agents and carriers of microbiocides for enhanced inhibition activity†
Hui Wen
Yong
 a,
Seyed Mohammad Amin
Ojagh
a,
Gabriel
Théberge-Julien
b,
Laura Sofia Reyes
Castellanos
a,
Faiza
Tebbji
b,
Theo G. M.
van de Ven
a,
Adnane
Sellam
bd,
Éric
Rhéaume
bc,
Jean-Claude
Tardif
bc and
Ashok
Kakkar
*a
a,
Seyed Mohammad Amin
Ojagh
a,
Gabriel
Théberge-Julien
b,
Laura Sofia Reyes
Castellanos
a,
Faiza
Tebbji
b,
Theo G. M.
van de Ven
a,
Adnane
Sellam
bd,
Éric
Rhéaume
bc,
Jean-Claude
Tardif
bc and
Ashok
Kakkar
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, Québec H3A 0B8, Canada. E-mail: ashok.kakkar@mcgill.ca
bResearch Centre, Montréal Heart Institute, 5000 Belanger Street, Montréal, Québec H1T 1C8, Canada. E-mail: jean-claude.tardif@icm-mhi.org
cDepartment of Medicine, Université de Montréal, Montréal, Québec H3T 1J4, Canada
dDépartement de Microbiologie, Infectiologie et Immunologie, Université de Montréal, Montréal, Québec H3T 1J4, Canada
First published on 6th August 2024
Abstract
Antibiotic resistance continues to pose significant health challenges. Considering severe limitations in the discovery and supply of new antibiotics, there is an unmet need to design alternative and more effective strategies for addressing this global issue. Use of polymeric nanoparticles with cationic shell surfaces offers a highly promising approach to coupling their inherent bactericidal action with sustained delivery of small lipophilic microbicides. We have utilized this platform for assembling multi-tasking soft core–shell nanoparticles from star polymers with the desired asymmetric arm composition. These stable nanoparticles with low critical micelle concentration imparted intrinsic antimicrobial potency due to high positive charge density in the corona, as well as the loading of active biocidal agents (such as curcumin and terbinafine) for potential dual and coadjuvant inhibition. This strategic combination allows for both immediate (direct contact) and extended (drug delivery) antibacterial activities for better therapeutic efficacy. Micellar nanoparticles with and without therapeutic cargo were highly efficient against both Escherichia coli (E. coli) and Bacillus subtilis (B. subtilis), representative Gram-negative and Gram-positive bacteria, respectively. Interestingly, we observed bacteria- and concentration-dependent effects, in which higher concentrations of charged nanoparticles were more effective against E. coli, whereas B. subtilis was inhibited only at lower concentrations. This work highlights a valuable platform to achieve combination therapy through nanoparticles with charged coronas and delivery of potent therapeutics to overcome antimicrobial resistance.
Introduction
Bacterial infection is a surging health concern affecting millions of people worldwide. It can lead to life-threatening conditions including sepsis, bacteraemia, tuberculosis, and endocarditis if left untreated.1,2 Patients who are immunocompromised for a variety of reasons such as cancer, organ transplant, or HIV, are especially vulnerable to bacterial infections.3–5 Antibiotics have traditionally played a crucial role; however, the emergence of drug-resistant pathogens is alarming and severely threatens our ability to intervene. The latest World Health Organization (WHO) global priority list of pathogens has highlighted the need for new antibiotics to address this urgency, which further stresses the gravity and research efforts necessary to curb this problem.6Current therapeutic agents used to combat bacterial infections can be classified as either bacteriostatic or bactericidal.7 They work by interfering, for example, with folic acid synthesis to inhibit bacterial DNA synthesis or by disrupting the cell wall structure to induce block in DNA replication or loss of cell integrity, which eventually leads to cell death.8 Some bacteriostatic agents include sulphonamides like sulfamethoxazole or macrolides such as clarithromycin,9,10 whereas fluoroquinolones namely ciprofloxacin or glycopeptides including vancomycin possess bactericidal activity.11 With the increasing instances of drug-resistant strains and deficiency in the discovery and supply of new antibiotics, there is a pressing need to develop alternative strategies for designing effective microbial agents.
Cationic polymers which can disrupt bacterial cell walls have been explored for their activity in preventing cell growth.12–22 These materials disrupt the negatively charged bacterial cell wall through electrostatic interactions with the polymeric cationic moieties. This interaction impairs cell wall integrity, leading to leakage of cytoplasmic contents and eventual cell lysis.23 For example, polyethyleneimine (PEI) and integrated PEI-based hybrid polymers have been extensively explored for antibacterial activity.24–28 Recently, branched PEI-crosslinked cellulose (BC-PEI) was used to prepare antibacterial face masks,29 which were effective in inhibiting the growth of Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa). To work around the high toxicity of such polymers towards mammalian cells,30,31 mannose-modified PEIs were prepared which showed low cytotoxicity against HeLa cells but excellent effectiveness against E. coli.32 Several studies on PEI-grafted polyurethane ureteral stent surfaces have also been shown to reduce the growth of Klebsiella pneumonia (K. pneumonia), Proteus mirabilis (P. mirabilis), and E. coli without significant cytotoxicity against L929 cells.33,34
To enhance our efficacy in addressing escalating antibiotic resistance, we were interested in engineering soft nanoparticles (NPs) with cationic shell surfaces, which could also efficiently encapsulate biocidal agents. These nanoscale alternatives offer opportunities for disrupting cell growth through multiple actions, including electrostatic interactions with the cell membrane; entering cells and interfering with their components; and sustained delivery of active bactericides (Scheme 1). Towards this goal, we report herein an AB2 miktoarm (mikto, Greek meaning different; A = polycaprolactone (PCL), B = polyethylene glycol (PEG) with terminal end amine functionality) polymer-based micelles with a positively charged PEG shell and a hydrophobic PCL core. Branched star polymers offer opportunities for designing NPs of small sizes, low critical micelle concentration (CMCs), high encapsulation efficiencies, and rendering balanced high charged density to hydrophilic shell surfaces.35,36 Such a multifunctional approach presents an efficient platform to address the current challenges of bacterial infections using combination therapy.
 | ||
| Scheme 1 Schematic overview of the dual-purpose polycationic micelles NP for targeting bacteria and drug delivery. | ||
Nanoscale micellar formulations were evaluated for their ability to inhibit microbial growth of E. coli and Bacillus subtilis (B. subtilis). We explored the potential to combine the intrinsic-antimicrobial activity of NPs with physically encapsulated drugs for additive effects. Curcumin (CUR) and terbinafine (TBF) were selected as the antimicrobial agents for this study. CUR is a naturally occurring polyphenol extracted from the rhizome of Curcuma longa.37 It has been widely shown to possess anti-inflammatory, antioxidant, and anticancer properties, as well as antibacterial efficacy on several persistent Gram-negative and Gram-positive bacteria, including P. aeruginosa, S. aureus, B. subtilis, Bacillus cereus (B. cereus), and E. coli.38–42 However, poor pharmacokinetics of CUR, including poor aqueous solubility and stability, have restricted its versatile usage.43
TBF is a lipophilic allylamine that has a wide spectrum of fungicidal activity.44 As it is one of the drugs listed in the WHO's model list of essential medicines, the potency of TBF and its efficiency over several other antifungal drugs have been demonstrated in clinical studies.45 Nevertheless, low penetration rates and poor aqueous solubility of TBF limit its application and require the need for repeated treatments.46 In addition, there has been an increase in clinical failures as certain pathogenic fungi develop resistance to TBF,47 which governs the need for a solution to overcome this hurdle. Although TBF is best known as an antifungal agent, some studies have shown an inhibitory effect against Gram-positive bacterial strains such as S. aureus.48 We examined if TBF alone and loaded into NPs was effective as an antibacterial agent for E. coli and B. subtilis. Our studies present a multifaceted approach in designing alternative antibacterial agents, and we demonstrate that NPs (i) inhibit the growth of both E. coli and B. subtilis and (ii) can be further utilized as drug nanocarriers to achieve sustained inhibition of bacteria.
Experimental
Reagents
Poly(ethylene glycol) (2050 g mol−1, Sigma Aldrich), silver nitrate (AgNO3, ≥99.0%, Sigma-Aldrich), sodium hydroxide (NaOH, ≥97%, ACP chemicals), sodium azide (NaN3, ≥99.5%, Sigma-Aldrich), N-(tert-butoxycarbonyl)-4-aminobutyric acid (Boc-GABA-OH, ≥98%, TCI America), N,N′-dicyclohexylcarbodiimide (DCC, 99%, Sigma-Aldrich), 4-(dimethylamino)pyridine (DMAP, ReagentPlus®, ≥99%, Sigma-Aldrich), propargyl bromide (∼80% in toluene, Sigma-Aldrich), 3,5-dihydroxybenzyl alcohol (99%, Sigma-Aldrich), potassium carbonate (K2CO3, ≥99.0%, Sigma-Aldrich), 18-crown-6 (99%, Sigma-Aldrich), tin(II) 2-ethylhexanoate (Sn(Oct)2, 92.5–100.0%, Sigma-Aldrich), copper(I) bromide (CuBr, 98%, Sigma-Aldrich), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%, Sigma-Aldrich), ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA, 99.0–101.0%, Sigma-Aldrich), trifluoroacetic acid (TFA, ReagentPlus®, 99%, Sigma-Aldrich), curcumin (CUR, >95%, Oakwood Products), and terbinafine HCl (TBF) were used as received. p-Toluenesulfonyl chloride (≥98%, Sigma-Aldrich) was recrystallized from petroleum ether before use. ε-Caprolactone monomer (99%, ACROS Organics) was distilled over CaH2 before use. 13 mm PVDF filters (0.22 μm, non-sterile) were purchased from SyringeFilter.com.Synthesis of (Boc-GABA-PEG2050)2-PCL3900 (P6)
Synthetic details for the hydrophilic PEG (P3) arm and hydrophobic PCL (P5) can be found in the ESI.† P5 (0.186 g, 0.0439 mmol, 1 eq.), P3 (0.200 g, 0.0894 mmol, 2 eq.), and CuBr (0.0162 g, 0.113 mmol, 2.5 eq.) were dissolved in dry toluene under nitrogen with magnetic stirring. A solution of PMDETA (0.0194 g, 0.112 mmol, 2.5 eq.) in dry toluene was then added, and the solution was allowed to react at room temperature for 48 h. The mixture was then concentrated and dialysed once with EDTA and twice with deionized water for 24 h each. The solution was then concentrated and precipitated in cold diethyl ether. The product was collected with vacuum filtration and dried in the desiccator. Yield: 87% (0.32 g). 1H NMR (500 MHz, CDCl3): δ (ppm) 1.34–1.41 (m, 71H, (–OCCH2CH2CH2CH2CH2O)35), 1.43 (s, 18H, –C(CH3)3), 1.60–1.67 (m, 157H, (–OCCH2CH2CH2CH2CH2O)35), 1.81 (m, 4H, –NHCH2CH2CH2CO), 2.29 (t, 69H, (–OCCH2CH2CH2CH2CH2O)35), 2.38 (t, 4H, –NHCH2CH2CH2CO), 3.15 (t, 4H, –NHCH2CH2CH2CO), 3.49 (t, 4H, –OCCH2CH2(CH2CH2O)46), 3.63 (s, 370H, –(CH2CH2O)46), 3.88 (t, 4H, –OCCH2CH2(CH2CH2O)46), 4.05 (t, 67H, (–OCCH2CH2CH2CH2CH2O)33), 4.22 (t, 4H, –(CH2CH2O)46CH2CH2OOC), 4.58 (t, 4H, –(CH2CH2O)46CH2CH2OOC), 5.03 (s, 2H, –ArCH2O), 5.16 (s, 4H, –ArOCH2), 6.59 (s, 3H, –ArH), 7.83 (s, 2H, triazole). 13C NMR (125 MHz, CDCl3): δ (ppm) 24.7, 25.7, 28.5, 34.2, 63.7, 64.3, 69.2, 70.7, 173.7.Synthesis of (+H3N-BA-PEG2050)2-PCL3900 (P7)
P6 (0.136 g, 0.017 mmol, 1 eq.) was dissolved in dichloromethane (0.5 mL) with magnetic stirring. Trifluoroacetic acid (0.5 mL) was added into the solution and allowed to react for 5 min. The solutions were then removed under vacuum and washed extensively with methanol. The product was then precipitated in cold diethyl ether, collected, and dried in the desiccator. Yield: 80% (0.11 g). 1H NMR (500 MHz, CDCl3): δ (ppm) 1.34–1.41 (m, 70H, (–OCCH2CH2CH2CH2CH2O)35), 1.60–1.67 (m, 147H, (–OCCH2CH2CH2CH2CH2O)35), 1.81 (m, 4H, –CH2CH2CH2NH3+), 2.29 (t, 72H, (–OCCH2CH2CH2CH2CH2O)35), 2.38 (t, 4H, –CH2CH2CH2NH3+), 3.06 (t, 4H, –CH2CH2CH2NH3+), 3.49 (t, 4H, –OCCH2CH2(CH2CH2O)45), 3.63 (s, 364H, –(CH2CH2O)45), 3.88 (t, 4H, –OCCH2CH2(CH2CH2O)45), 4.05 (t, 67H, (–OCCH2CH2CH2CH2CH2O)33), 4.27 (t, 4H, –(CH2CH2O)45CH2CH2OOC), 4.55 (t, 4H, –(CH2CH2O)45CH2CH2OOC), 5.03 (s, 2H, –ArCH2O), 5.16 (s, 4H, –ArOCH2), 6.59 (s, 3H, –ArH), 7.83 (s, 2H, triazole). 13C NMR (125 MHz, CDCl3): δ (ppm) 24.7, 25.7, 28.5, 34.3, 64.3, 70.7, 173.7.Preparation of blank and drug-loaded polymeric micelles
To prepare blank NPs, the desired polymer (P6 or P7, 5 mg) was dissolved in 2 mL of HPLC-grade acetone in a vial. The polymer solution was then dropped into Milli-Q water (2 mL) at a rate of 1 drop s−1 while stirring. The acetone was allowed to evaporate overnight in the dark with continuous stirring. The solution was then passed through a 0.22 μm PVDF filter. Milli-Q water was added (if needed) to obtain a final volume of 2 mL.To prepare the CUR-loaded NPs, the desired polymer (P6 or P7, 5 mg) was dissolved in 2 mL of HPLC-grade acetone in a vial. In another vial, CUR (0.5 mg) was dissolved in HPLC-grade acetone (1 mL). The CUR solution was then mixed with the polymer solution and dropped into Milli-Q water (2 mL) at a rate of 1 drop s−1 while stirring. The acetone was allowed to evaporate overnight in the dark with continuous stirring. The solution was then centrifuged for 10 min at 1000 rpm and passed through a 0.22 μm PVDF filter. Milli-Q water was added (if needed) to obtain a final volume of 2 mL. TBF-loaded NPs were also prepared using the same protocol but with HPLC-grade dichloromethane instead of acetone.
CMC determination
A series of polymer concentrations ranging from 0.000122 mg mL−1 to 1.0 mg mL−1 in HPLC-grade acetone were prepared. A 6 μM pyrene in HPLC-grade acetone solution was also prepared separately. Polymer and pyrene solutions were dropped concurrently into Milli-Q water (1 mL) at a rate of 1 drop s−1 while stirring. The acetone was allowed to evaporate overnight in the dark with continuous stirring. Milli-Q water was added to obtain a final volume of 1 mL. Fluorescence spectra were recorded, and the CMC was examined by using a plot of λ338/λ333 intensity against polymer concentration to determine the concentration at which the intensity ratios increased.Drug loading and release
The drug-loaded NP solution was transferred to a spectra/Por 3 dialysis membrane (standard RC, 3.5 kDa MWCO). The solution was dialyzed against 140 mL of phosphate-buffered saline (PBS, 0.01 M, pH 7.4) containing 1% v/v Tween 80 at 37 °C. At desired intervals, 20 μL aliquots were collected from the dialysis membrane and diluted 100 folds with methanol (for CUR) or DMSO (for TBF) for analysis. The absorption intensities (425 nm for CUR and 285 nm for TBF) were measured at room temperature. The results were referred to a standard drug curve to obtain the mass of the drug remaining at each interval. Drug loading (DL%) and encapsulation efficiencies (EE%) were then calculated using the following equations: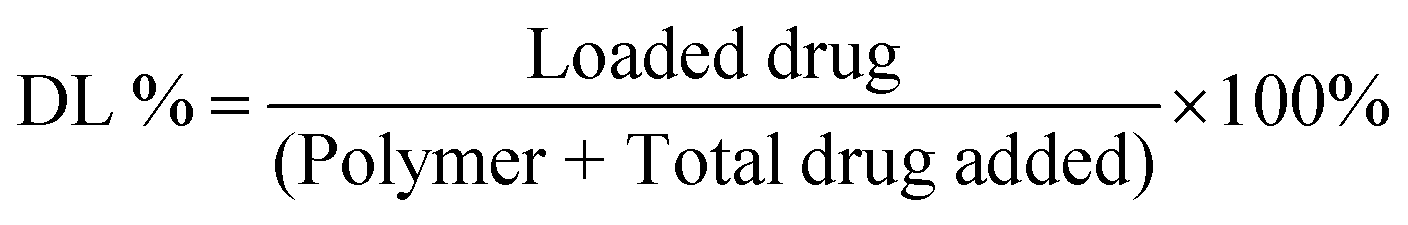
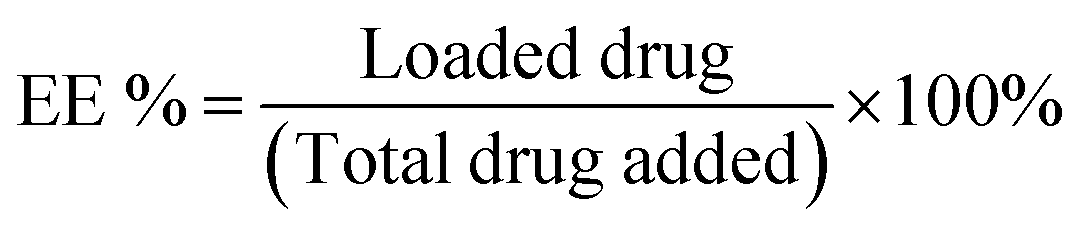
Drug release kinetic studies
The drug release kinetic studies were examined by applying model equations as follows:Zero-order model:
| Ct = C0 + k0t |
First-order model:

Higuchi model:
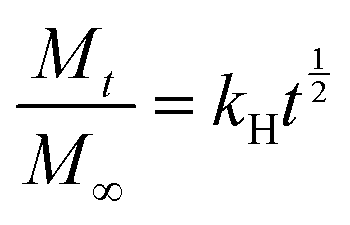
Korsmeyer–Peppas model:

Assessing the antimicrobial activity
To determine the antimicrobial efficiency of P6- and P7-micelles on E. coli ATCC 25922 and B. subtilis ATCC 6633, two-fold serial dilutions were conducted. The bacterial cultures were diluted with LB broth to achieve an optical density of 0.05 at 600 nm (OD600). In parallel, blank P6- and P7-micelles were prepared according to the procedure mentioned previously. The stock micellar solution was then diluted with Milli-Q water to obtain six different concentrations ranging from 78–2500 μg mL−1. Next, 160 μL of bacterial suspension was added to the wells of a 96-well microplate followed by 40 μL of micellar solutions. The positive control was a bacteria suspension treated with PBS and the negative control was a mixture of LB broth and PBS that did not contain any bacteria. The OD600 of each well was assessed every hour up to 24 h at 37 °C using a SynergyH4 multimode plate reader (BioTek, USA). Three replicates were performed for each concentration. The OD600 obtained at each interval was subtracted from the OD of LB media. Thereafter, a graph of corrected OD over time was plotted to observe the change in bacterial growth over 24 h. The effectiveness of P6-CUR, P7-CUR, P6-TBF, and P7-TBF on both E. coli and B. subtilis was also analysed with the same protocol but with drug-loaded micelles. To examine the inhibition of CUR and TBF on both bacteria strains, a similar procedure was applied but the drugs were dissolved in 0.75% DMSO due to their poor aqueous solubility. The drug concentrations prepared ranged from 25–300 μg mL−1.Cell viability assay
5000 HUVEC cells per well were seeded in 96-well plates in EGM-2 medium and incubated at 37 °C and 5% CO2. On the following day, cells were exposed to different concentrations of nanoparticles using two-fold dilutions of P7, P7-CUR, and P7-TBF ranging from 625 to 39.1 μg mL−1 or their loading concentrations of CUR and TBF. CUR and TBF concentrations were equivalent to those loaded into the P7-CUR and P7-TBF NPs (37.5 and 50 μg mL−1, respectively, for the 625 μg mL−1 NP concentration). After 24 h of exposure, cell proliferation was assessed using an MTT assay (Millipore Sigma, #M2128). 0.5 mg mL−1 of 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide was added in EGM-2 and incubated at 37 °C in the dark for 4 h and then the medium was removed, and mitochondrial metabolic activity was measured by dissolving the formazan formed inside the cells in 100 μL of DMSO. Formazan absorbance was measured using a Synergy H1 plate-reader at 575 nm. Survival rates were normalized against unexposed cells.Results and discussion
Synthesis and characterization of cationic miktoarm polymers
Cationic star polymers containing PEG and PCL were synthesized as shown in Scheme 2. PEG offers several unique properties that make it suitable for biomedical applications, including excellent biocompatibility, low toxicity and stealth that minimizes protein adsorption for prolonged circulation half-life.49 PCL is a hydrophobic polyester that confers biodegradability and biocompatibility to NPs.50 The branched polymer was prepared via a combination of arm-first and core-first methods. First, an asymmetric functionalization of PEG2050via the Ag2O-mediated monotosylation was carried out to obtain α-tosyl-ω-hydroxyl PEG2050 (P1). Monotosylation is essential in achieving a positively charged micelle as the free –OH group is then used for esterification with Boc-GABA-OH. The 1H NMR spectrum of the heterobifunctional polymer contained characteristic tosyl peaks (Fig. S1, ESI†). The tosyl group was then displaced using NaN3 to yield α-azido-ω-hydroxyl PEG2050 (P2). Introduction of the azide group to PEG would allow us to attach the hydrophilic arm onto the hydrophobic core through robust click chemistry. 1H NMR spectrum of P2 showed the disappearance of distinct peaks for the tosyl group seen in P1, while the remaining OH group shifted slightly upfield (Fig. S3, ESI†). The presence of the azide group was further confirmed by the band at 2102 cm−1 in the IR spectrum (Fig. S15, ESI†). Subsequently, a Steglich esterification was conducted to link P2 to commercially available Boc-GABA-OH. 1H NMR spectrum showed peaks distinct for Boc groups and the adjacent amide group (Fig. S5, ESI†), while the IR spectrum showed retention of the azide peak at 2102 cm−1, confirming successful heterofunctionalization of PEG. The dipropargylated cores without (P4) and with PCL (P5) were synthesized according to a previously published procedure.51 The degree of polymerization (DP) was calculated by comparing the integral of the benzyl protons between 6.57–6.60 ppm to that of the methylene protons in PCL at 4.06 ppm (Fig. S7, ESI†), and the hydrophobic arm was determined to have a DP of 34, with a number-average molecular weight of 4100 Da. | ||
| Scheme 2 Synthesis details utilizing a mix of arm- and core-first methods to obtain cationic amphiphilic copolymer P7. | ||
We subsequently carried out copper-catalysed azide–alkyne cycloaddition to obtain the miktoarm polymer P6. 1H NMR spectrum of P6 showed the successful addition of characteristic peaks from PEG and Boc onto P5 (Fig. S10, ESI†), while GPC traces confirmed the synthesis without any residual hydrophilic or hydrophobic arms (Fig. S14, ESI†). It is worth noting that the number-average molecular weight of polymers obtained from GPC in THF differs significantly from those obtained from 1H NMR spectra, primarily due to differences in hydrodynamic volumes compared to the PMMA standards used for calibration. Lastly, Boc groups in P6 were removed by addition of trifluoroacetic acid to confer hydrophilicity and positive charges to the miktoarm polymer P7. Complete deprotection of P6 was confirmed by 1H NMR spectra that showed the absence of Boc groups, confirming the successful formation of the P7 copolymer (Fig. S12, ESI†).
Preparation and characterization of micelles
The size and CMC of NPs are important parameters that can significantly influence their performance in biological applications.52 In the NP assemblies, PCL is expected to aggregate to form the particle core and be shielded from the aqueous medium by the hydrophilic PEG. A hydrodynamic diameter of 20–200 nm has been consistently highlighted to be the ideal window in minimizing nonspecific uptake by the reticuloendothelial system while maximizing delivery through the enhanced permeation and retention effect.53 NPs from AB2 polymers (P6 and P7) were prepared with continuous stirring to facilitate solvent/polymer dispersion and solvent evaporation. Both P6 and P7 self-assembled into spherical NPs, subsequently known as P6-micelles and P7-micelles, which were analysed using DLS and TEM (Table 1 and Fig. 1, Fig. S16, ESI†). DLS showed a monomodal size distribution of particles for both, in which hydrodynamic diameters were approximately 44 and 92 nm, respectively. Since both polymers possess hydrophobic PCL with similar molecular weight and composition, the difference in size suggests that it is related to the hydrophilic shell which is positively charged at the termini in P7-micelles, compared to the one without (P6-micelles). The added electrostatic surface repulsion in P7-micelles led to enlargement and an increase in micelle size. Such an expansion has also been observed in other lysine/polyphosphoester/primary amine systems.54–56 TEM analyses showed uniform micelles with an average size smaller than those determined using DLS, approximately 42 nm for P6-micelles and 87 nm for P7-micelles. Zeta (ζ)-potential values suggest that P7-micelles have a positive charge of about 15 mV, while P6-micelles were slightly negative.| Sample | M n,NMR (Da) | M n,GPC (Da) | Đ | Blank-micelles | CMCd (μg mL−1) | |
|---|---|---|---|---|---|---|
| D H (nm) | ζ (mV) | |||||
| a Number-average molecular weight of polymers estimated from 1H NMR. b Number-average molecular weight and dispersity of polymers obtained from GPC in THF. c Hydrodynamic diameter (DH) and zeta potential (ζ) obtained by DLS. d Critical micelle concentration (CMC) determined by pyrene fluorescence studies. | ||||||
| P6 | 8200 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.38 | 44 ± 8 | −5.69 ± 2.07 | 4.66 |
| P7 | 8000 | 5900 | 1.54 | 92 ± 10 | 15.66 ± 3.07 | 7.75 |
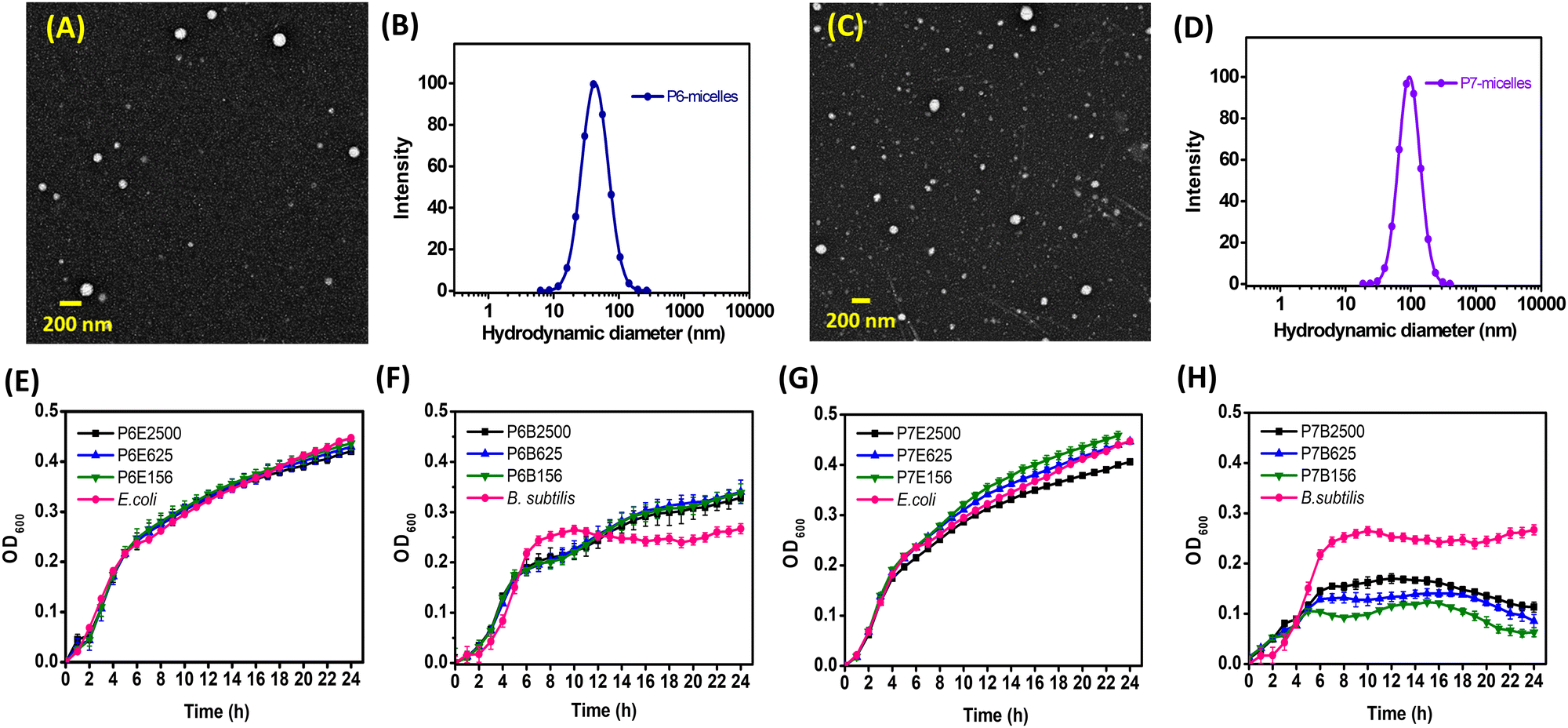 | ||
| Fig. 1 (A) TEM image and (B) DLS analyses of P6-micelles; (C) TEM image and (D) DLS analyses of P7-micelles. Growth curves of E. coli (E) and B. subtilis (F) incubated with P6-micelles, growth curves of E. coli (G) and B. subtilis (H) treated with P7-micelles. Graph legends: P6 represents P6-micelles whereas P7 represents P7-micelles. Letter following sample represents bacterial strains, while the numbers that follow suit represent concentration of micellar solutions (e.g., P6E2500 contains 2500 μg mL−1 of P6-micelle added to E. coli, P7B2500 contains 2500 ug mL−1 of P6-micelle added to B. subtilis). For additional concentrations tested, see ESI† (Fig. S18–S21). | ||
CMC reflects the stability of a nanocarrier when introduced into blood circulation. Low CMC is advantageous to ensure retention of micellar structure and drug solubility in biological media even under extensive dilutions.57 To evaluate the CMC of our system, we encapsulated the fluorescent probe, pyrene, into our polymers as detailed previously (Fig. S17, ESI†). The transition from unimers to well-defined polymeric micelles created different microenvironments for the partition of pyrene that can be easily detected by fluorescence. CMC of P6 was determined to be 4.66 μg mL−1, whereas that for P7 was 7.75 μg mL−1, approximately 1.7-fold higher than P6. This increase is consistent with the increase in size observed in DLS.
Evaluation of antibacterial properties of P6- and P7-micelles
The positively charged shell on NPs facilitates their adhesion to the negatively charged bacteria surface through electrostatic interactions.58 Gram-negative bacteria exhibit a negative charge due to the presence of a lipopolysaccharide layer on the outer leaflet of their membrane, while the negative charge on Gram-positive bacteria stems from the teichoic acid embedded within the peptidoglycan layer.59 Despite Gram-negative bacteria having a thinner peptidoglycan layer, the additional outer membrane serves as an additional diffusion barrier, rendering them more resistant against antibiotic treatments.60Gram-positive B. subtilis and Gram-negative E. coli were chosen to investigate the antibacterial activity of our NPs. E. coli are renowned as a prevalent bacteria in wound infections and hold clinical significance. Meanwhile, the inclusion of B. subtilis stemmed from its genetic affinity with diverse pathogens, notably B. cereus, a bacterium commonly associated with wound infections.61 We performed a 96-well plate method to evaluate the effects of blank P6- and P7-micelles on bacterial growth. Optical density measurements at 600 nm were taken hourly over a 24 h period. In all cases, constant or minimal/decreasing optical densities over time indicated limited bacterial growth or no bacterial growth. Conversely, an increase in optical density suggested enhanced bacterial growth. Given the positive charge of P7-micelles, which enables interaction with the negatively charged bacterial cell membrane, we anticipated these NPs to possess intrinsic antibacterial properties. Consequently, a minimal inhibitory effect from P6-micelles that lack specific interactions with the bacterial cell membrane is expected. The growth curves for both bacterial strains are summarised in Fig. 1E–H, and additional concentrations tested are presented in Fig. S18–S21 (ESI†). As shown in Fig. 1E and F and as expected, P6-micelles demonstrate no inhibitory effect on the growth of E. coli and B. subtilis, even at concentrations up to 2500 μg mL−1, aligning with our hypothesis regarding their lack of specific bacterial cell membrane interactions.
Subsequently, we examined the inhibitory efficacy of P7-micelles on E. coli and B. subtilis bacteria, which was found to be both bacteria- and NP concentration-dependent. Notably, while E. coli inhibition was only evident at higher concentrations of P7-micelles (2500 μg mL−1, Fig. 1G), inhibition of B. subtilis was pronounced even at lower concentrations (156 μg mL−1, Fig. 1H). These results are in agreement with previous reports on cationic polymers, which have shown greater effectiveness against Gram-positive bacteria.62 The growth rates of B. subtilis also displayed several distinct logarithmic, stationary, and death phases. In the absence of P7-micelles, B. subtilis followed a logarithmic phase between 0–7 h, and a relatively stationary phase until 24 h. Treatment with P7-micelles altered this growth curve, with variations depending on the NP concentration. At lower concentrations of 156 mg mL−1, B. subtilis exhibited a logarithmic phase between 0–5 h, a stationary phase between up to 10 h, and a steady increase in cell density until 15 h, before entering the death phase. Conversely, concentrations of 625 and 2500 μg mL−1 demonstrated a very similar but more stable stationary phase before entering the death phase at about 14 h, indicating that higher concentrations are more efficient in consistently inhibiting bacterial growth compared to lower concentrations. These results clearly suggest that P7-micelles exhibit intrinsic antimicrobial activity, with the inhibitory activity dependent on NP concentration. The concentration-dependent biocidal activity has also been shown earlier for cationic polymers. For example, Lim and Hudson examined the antimicrobial activity of S. aureus (Gram-positive bacteria) relative to the concentration of O-acrylamidomethyl-N-[(2-hydroxy-3-trimethylammonium)propyl]chitosan chloride (NMA–HTCC).63 Their study revealed that cationic chitosan effectively inhibited the growth of E. coli and S. aureus, with the peak antimicrobial activity observed at the lowest tested concentration of 10 ppm. The inverse relationship observed, wherein higher concentrations of chitosan (200 ppm) exhibited diminished antimicrobial activity, was attributed to the coating of the bacterial cell membranes which prevents the leakage of intracellular components. Similarly, the concentration-dependent observations with P7-micelles suggest a similar coating effect that influences their efficacy in inhibiting B. subtilis growth. This implies the existence of a potential saturation point, beyond which the inhibitory effect does not increase with concentration, as indicated by the higher OD observed at higher NP concentrations after 24 h. Further investigations will be necessary to elucidate the specific mechanisms by which P7-micelles inhibit B. subtilis growth. Nevertheless, our research highlights the efficacy of cationic P7-micelles against various bacterial strains, demonstrating its versatility in addressing diverse microbial threats.
SEM analyses of bacteria
Inhibition of bacterial growth by cationic NPs occurs typically through electrostatic attractions with the negatively charged bacterial membrane.64 Morphological changes of E. coli and B. subtilis in response to NP solutions were analysed using SEM (Fig. 2). The bacterial cells incubated without any NP treatment were used as blank controls. Fig. 2A shows intact and rod-shaped E. coli, with lengths ranging from 1.5–3.0 μm. Following incubation with P6-micelles for 24 h, the bacterial cells remained intact and were approximately 2.0 μm long (Fig. 2C). In contrast, treatment with P7-micelles induced deformations in E. coli, resulting in hollow and ruptured structures (Fig. 2E). These observations are indicative of E. coli inhibition upon treatment with P7-micelles, which may induce cell leakage by disrupting the bacterial membrane. Chakraborti et al. have reported an analogous disintegration of E. coli structures achieved through treatment with PEI-functionalized ZnO.65 This treatment disrupted bacterial membranes, leading to the leakage of cellular proteins.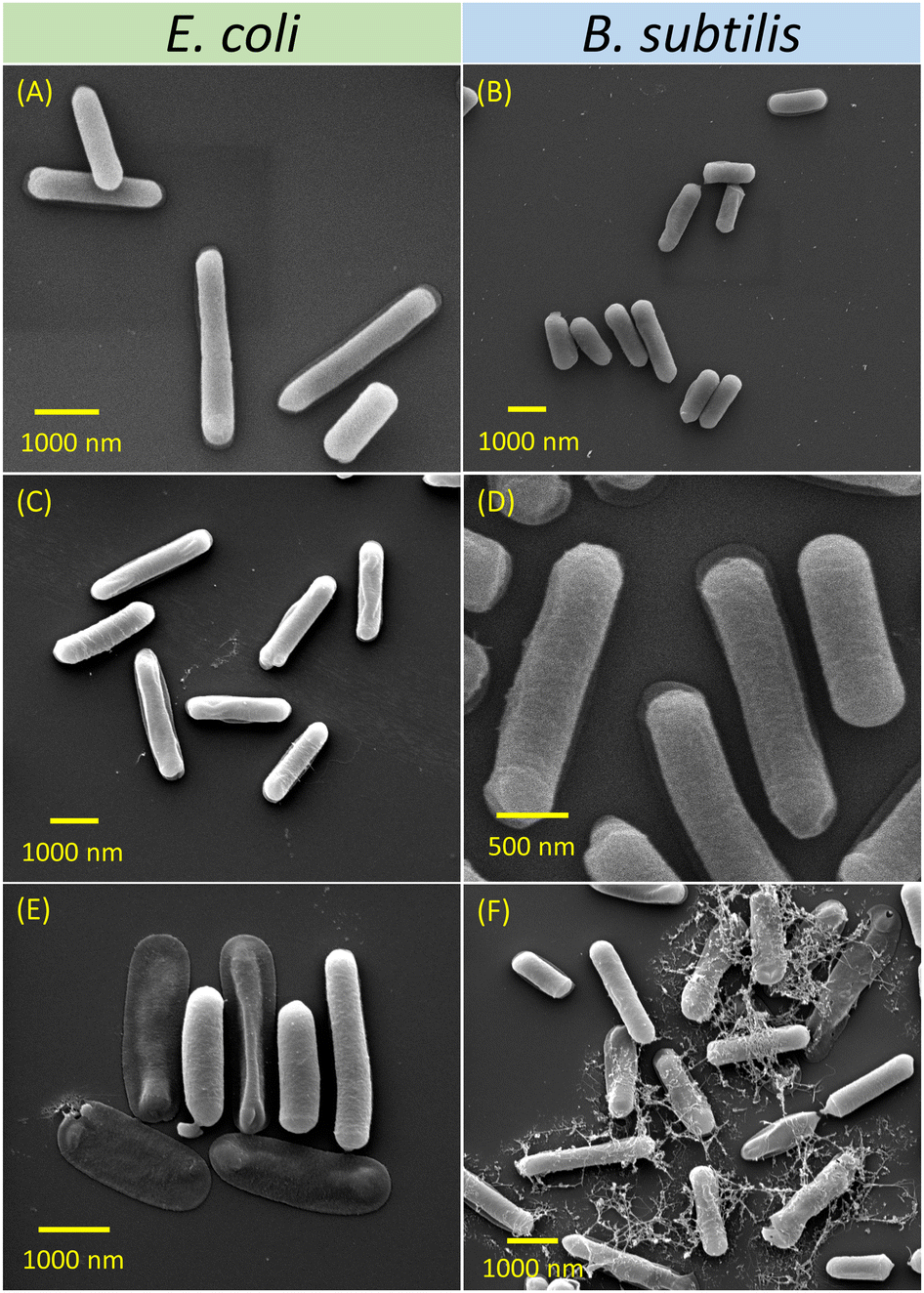 | ||
| Fig. 2 SEM images of untreated E. coli (A) and B. subtilis (B); E. coli incubated with P6-micelles (C), P7-micelles (E); and B. subtilis with P6-micelles (D), P7-micelles (F). Bacteria were incubated with P6-/P7-micelles (2500 μg mL−1) for 24 h prior to fixing. | ||
In studies with B. subtilis, we observed slightly shorter rod shapes of 1.0–2.5 μm (Fig. 2B). Due to the neutral hydrophilic shell, P6-micelles displayed no effect on B. subtilis inhibition as the bacteria still appeared long and were approximately 1.5–2.5 μm (Fig. 2D). However, when B. subtilis was incubated with P7-micelles, several distorted, ruptured, and irregular bacteria were observed (Fig. 2F). These results suggest cell membrane rupture, leading to the leakage of cytoplasmic contents, resulting in cell death. Such observations have also been reported by Tanganini et al., who utilized SEM to visualize the damage induced by self-assembled lignin NPs on B. subtilis, and similar deformed structures in bacterial cell morphology were noted, demonstrating the potential of NPs as antimicrobial agents.66
Drug loading into P6- and P7-micelles
The deformations observed in both E. coli and B. subtilis highlighted the potential of P7-micelles as promising alternatives to antibiotics. Additionally, their core–shell structure provides additional advantages beyond their intrinsic antimicrobial properties. This design enables encapsulation and controlled delivery of antibiotics, potentially offering additive or synergistic effects to enhance the overall antimicrobial efficacy of NPs. This feature presents a multifaceted approach in combatting microbial infections. To ascertain these advantages, CUR and TBF were first loaded into micelles from P6 and P7 at a polymer:drug ratio of 10:1. Drug loading capacity (DL%) and encapsulation efficiency (EE%) of CUR-loaded NPs were determined to be (i) 6.6% and 72.4% for P6-micelles (P6-CUR) and 3.4% and 36.9% for P7-micelles (P7-CUR, Table 2). We noted that the loading in P6-CUR was almost twice as much compared to P7-CUR. This disparity in CUR encapsulation may be due to a drop in pH when P7 was introduced into water during self-assembly (pH 4.3), which created an acidic environment that further suppressed the aqueous solubility of CUR and subsequently reduced its encapsulation.67 In contrast, the pH of P6 in water was 6.8, which helped prevent crystallization of CUR. NP sizes of P6 and P7-based micelles were examined by DLS and TEM (Fig. 3 and Fig. S22, ESI†). In general, hydrophobic drugs such as CUR may provide an additional driving force for self-assembly to give denser and smaller micelles, which was observed in both P6-CUR (45 nm) and P7-CUR (54 nm) compared to their blank counterparts.68ζ-Potential experiments revealed values of 3.4 mV (P6-CUR) and (7.14 mV) for P7-CUR. Due to the dehydration process, TEM revealed marginally smaller sizes than DLS, 44 nm (P6-CUR) and 52 nm (P7-CUR).
| Samples | P6-CUR | P7-CUR | P6-TBF | P7-TBF |
|---|---|---|---|---|
| a Drug loading (DL%) and encapsulation efficiencies (EE%) obtained by UV-Vis. b Hydrodynamic diameter (DH) and zeta potential (ζ) obtained by DLS. | ||||
| DL%a | 6.6 ± 0.1 | 3.4 ± 0.6 | 6.6 ± 0.8 | 8.7 ± 0.6 |
| EE%a | 72.4 ± 1.3 | 36.9 ± 6.7 | 72.7 ± 8.8 | 95.7 ± 6.1 |
| D H (nm) | 45 ± 13 | 54 ± 7 | 134 ± 10 | 115 ± 11 |
| ζ (mV) | 3.4 ± 0.6 | 7.1 ± 0.7 | 4.2 ± 1.6 | 16.0 ± 10.0 |
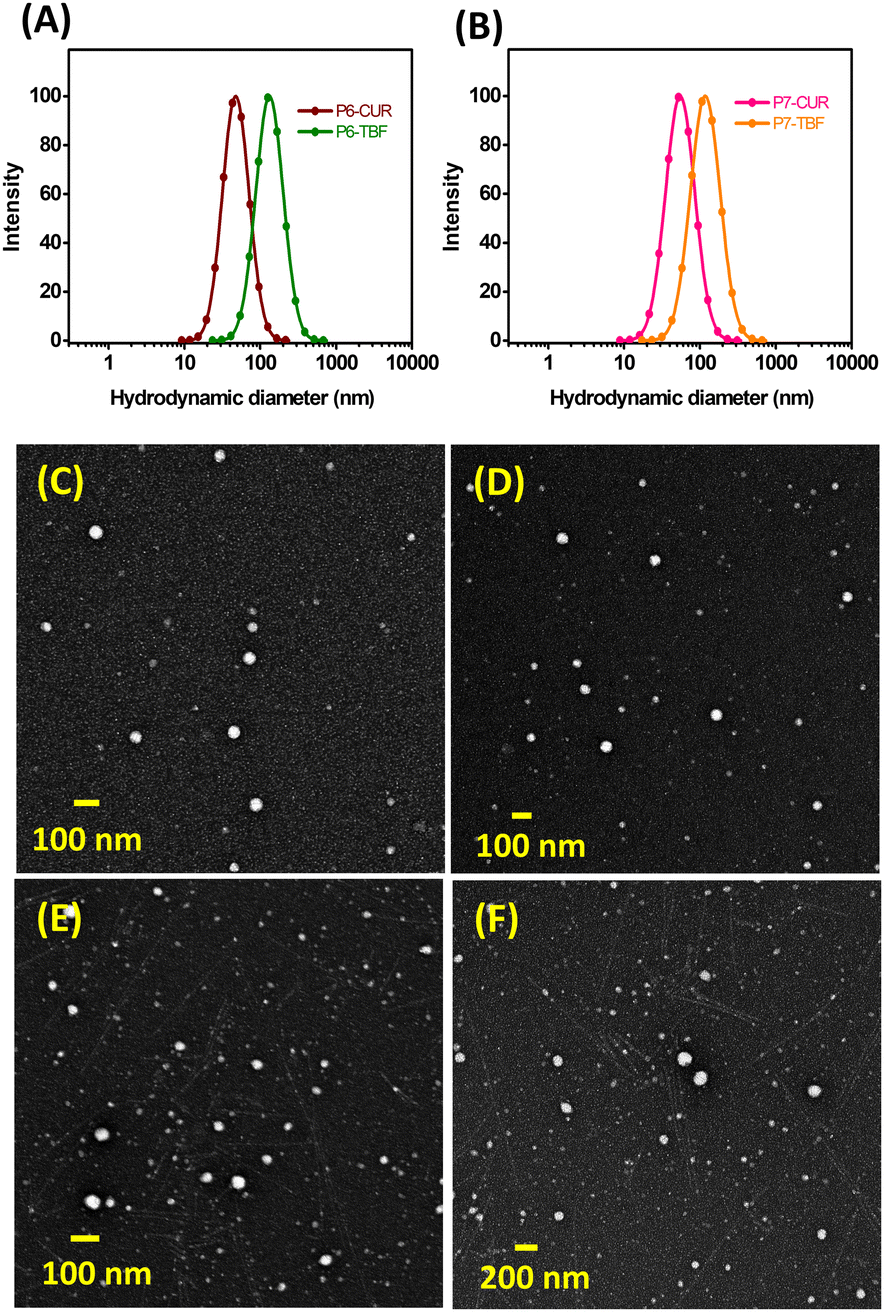 | ||
| Fig. 3 DLS analyses of P6-CUR and P6-TBF (A), and P7-CUR and P7-TBF (B). TEM images of P6-CUR (C), P6-TBF (D), P7-CUR (E), and P7-TBF (F). | ||
DL and EE for TBF-encapsulated self-assemblies were found to be (i) 6.6% and 72.7% for P6-micelles (P6-TBF) and (ii) 8.7% and 95.7% for P7-micelles (P7-TBF), suggesting better encapsulation of TBF than CUR for P7. This is, to our knowledge, the first study examining TBF loading into miktoarm polymer based micellar assemblies. DLS showed particle sizes of 134 nm for P6-TBF and 115 nm for P7-TBF, and ζ-potential values were determined to be 4.17 mV (P6-TBF) and 16.04 mV (P7-TBF). TEM revealed smaller particle sizes of 70 nm and 74 nm for P6-TBF and P7-TBF, respectively, due to dehydration effects during sample preparation.
Drug release studies from P6- and P7-micelles
Drug release from nanoformulations was evaluated under simulated physiological conditions (0.01 M PBS, pH 7.4, 37 °C). We noted that the introduction of positively charged corona did not impede drug release significantly. As depicted in Fig. 4A, CUR release over time was marginally faster from P7-micelles. The most significant differences were observed in the early hours of drug release (0–8 h), where CUR release was higher from P6-CUR than cationic P7-CUR NPs; beyond 8 h, the cumulative differences periodically diminished. For instance, within the first hour, the release from P6-CUR was 27%, whereas it was slightly slower at 19% for P7-CUR. At 2 h, the release was at 38% for P6-CUR and 29% for P7-CUR; 4 h, 45% for P6-CUR and 37% with P7-CUR; at 8 h, 51% for P6-CUR and 51% for P7-CUR. These differences were, however, quite miniscule, and suggest that the cationic shell did not cause any significant delay in CUR release. In the subsequent time period, CUR release from P7-CUR was found to be slightly faster than P6-CUR: 24 h: 59% for P6-CUR, and 66% for P7-CUR; 48 h: 75% for P6-CUR and 74% for P7-CUR; and finally 72 h: 80% for P6-CUR and 83% for P7-CUR.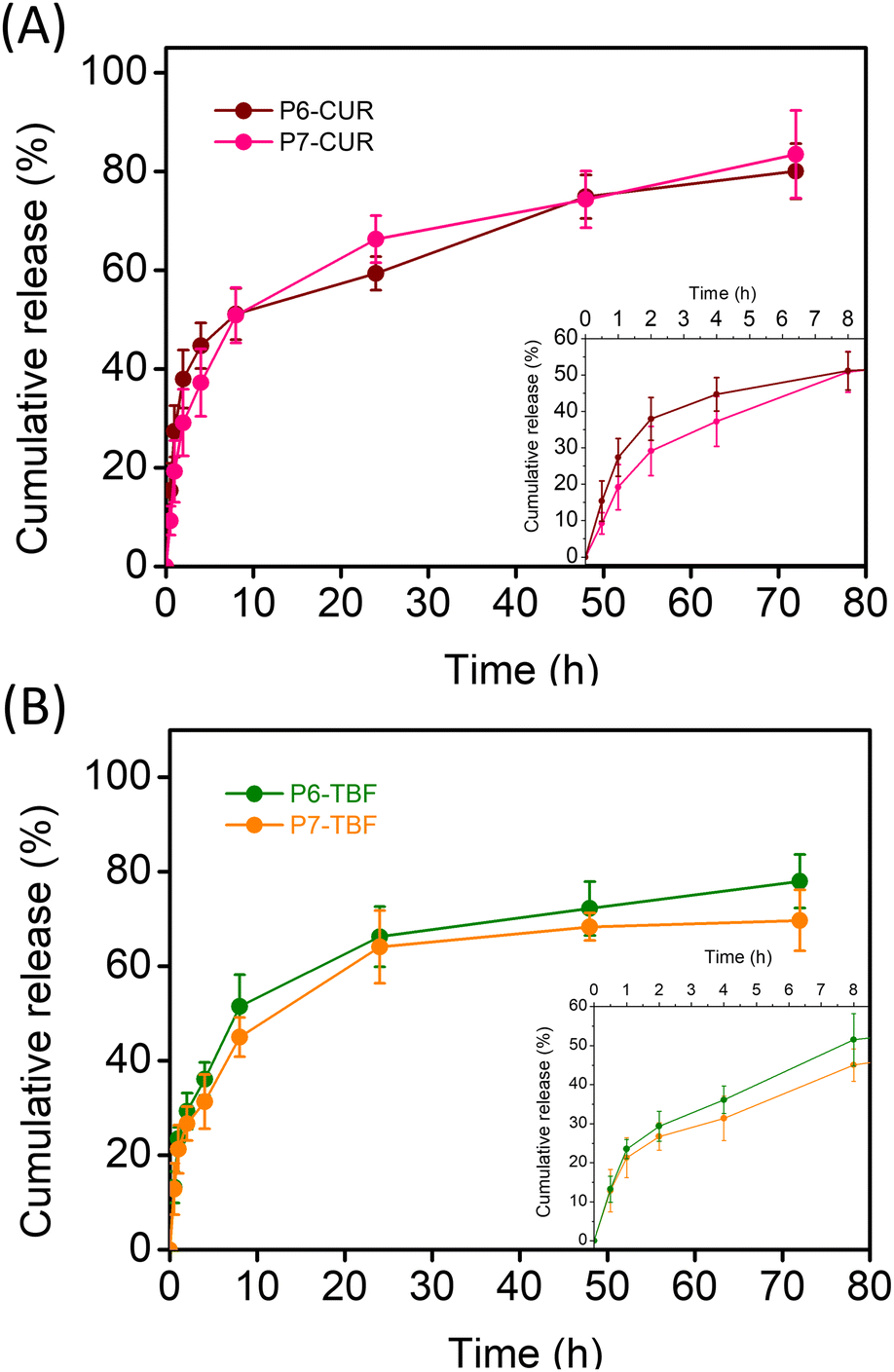 | ||
| Fig. 4 (A) CUR release from P6-CUR and P7-CUR; (B) TBF release from P6-TBF and P7-TBF. All studies were conducted at 37 °C in 1× PBS buffer (pH 7.4, +1% v/v Tween 80). Error bars represent a standard deviation of n = 3. | ||
For TBF-loaded micelles, P6-TBF exhibited slightly faster release rates than P7-TBF over the observed time period (Fig. 4B). The disparity in release rates between the two systems generally increased in the first 8 h. For example, 1 h: TBF release was 23% for P6-TBF and 21% for P7-TBF; 2 h, 29% for P6-TBF and 27% for P7-TBF, indicating a trend of slightly faster release of P6-TBF. This trend continued with 36% release for P6-TBF and 31% for P7-TBF at 4 h, 52% for P6-TBF and 64% for P7-TBF at 8 h. From 8 h, minimal differences were noted between P6-TBF and P7-TBF. Specifically, at 24 h, the release was 66% for P6-TBF and 64% for P7-TBF; at 48 h, 72% for P6-TBF and 68% for P7-TBF; and finally at 72 h, 78% for P6-TBF and 70% for P7-TBF. These results further highlight that the positively charged shell in P7-micelles did not interfere with the release of varied drugs with different pharmacokinetics. It also suggests that the multifunctional P7-micelles are promising candidates not only for their intrinsic antimicrobial properties but also as effective antibiotic delivery vehicles, showcasing their versatility and potential in combating microbial infections.
Drug release kinetic studies
To investigate the mechanism of drug release, we assessed the release curves to several standard mathematical models. These models offer insights into whether drug release is primarily diffusion-based or dissolution-driven, which can facilitate the formulation of pharmaceutics and predict drug bioavailability in the body.69,70 We evaluated the zero order, first order, Higuchi, and Korsmeyer–Peppas models by comparing the correlation coefficients (R2) (Table 3 and Fig. S23–S26, ESI†). For CUR release from P6 micelles, the Higuchi model demonstrated the best fit, yielding an R2 and a kH value of 0.914 and 12.075, respectively. This result suggests that drug release was governed by Fickian diffusion. P7-CUR also exhibited the best fit with the Higuchi model; however, corresponding results from the Korsmeyer–Peppas model yielded comparatively good R2 values of 0.950 compared to 0.951. This evaluation is indicative that drug release was controlled by more than one process (i.e., anomalous transport), which was further confirmed as the n value was 0.586. In general, spherical particles are said to follow anomalous transport when 0.43 < n < 0.85.71 The Korsmeyer–Peppas model was also the most suited to describe delivery of TBF from both P6- and P7-micelles, with the highest R2 values of 0.957 and 0.964, respectively. TBF release from P6 micelles was driven by non-Fickian diffusion (n = 0.454), whereas TBF delivery from P7-micelles was diffusion controlled (n ≤ 0.43).| Model | Parameter | P6-CUR | P7-CUR | P6-TBF | P7-TBF |
|---|---|---|---|---|---|
| Zero-order | k 0 (mg h−1) | 0.903 | 0.987 | 0.907 | 0.825 |
| R 2 | 0.654 | 0.744 | 0.714 | 0.699 | |
| First-order | k 1 (h−1) × 10−3 | 27.6 | 31.6 | 29.5 | 28.8 |
| R 2 | 0.268 | 0.339 | 0.313 | 0.312 | |
| Korsmeyer–Peppas | k K–P (h−n) | 24.362 | 16.669 | 19.738 | 18.919 |
| n | 0.417 | 0.586 | 0.454 | 0.418 | |
| R 2 | 0.909 | 0.950 | 0.957 | 0.964 | |
| Higuchi | k H (mg1/3 h−1/2) | 12.075 | 11.491 | 11.116 | 10.264 |
| R 2 | 0.914 | 0.951 | 0.939 | 0.935 |
Evaluation of antibacterial properties of CUR and TBF loaded P7-micelles
We were intrigued in exploring potential antibacterial properties of our intrinsically active NPs which were loaded with small molecule antimicrobial agents, and focused our investigation on P7-CUR and P7-TBF for this purpose. CUR is known for its ability to inhibit bacterial growth by interfering with membrane proteins and disrupting cellular processes.41,72 It has been shown that CUR can easily permeate bacterial membranes, rendering bacteria more susceptible to antibiotics. E. coli inhibition studies with free CUR are presented in Fig. 5A. CUR exhibited concentration-dependent antimicrobial activity against E. coli: concentrations of CUR at 200–300 μg mL−1 were found to be much more potent in inhibiting E. coli growth, while lower concentrations of 50–150 μg mL−1 displayed reduced efficacy.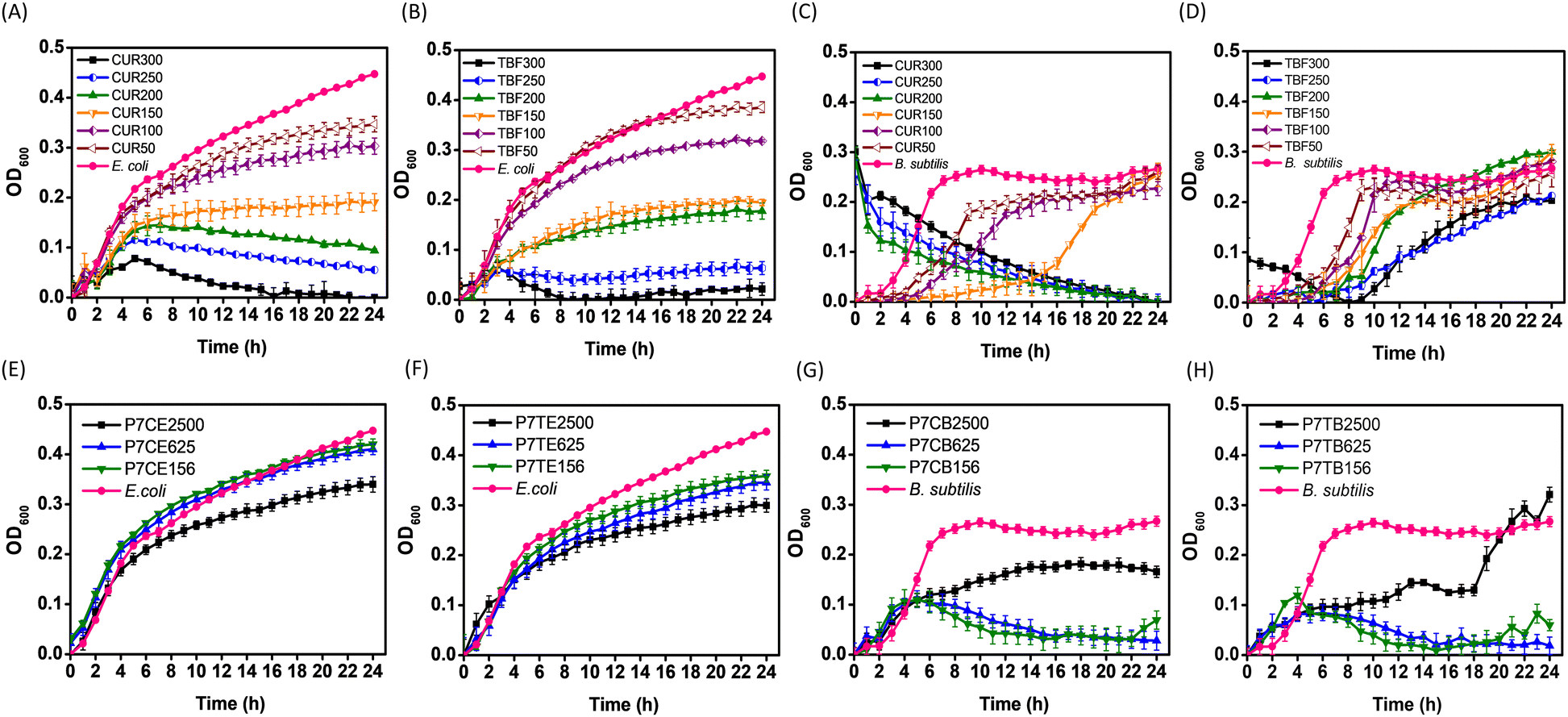 | ||
| Fig. 5 Growth curves of E. coli treated with CUR (A) and TBF (B); B. subtilis treated with CUR (C) and TBF (D); E. coli treated with P7-CUR (E) and P7-TBF (F); and B. subtilis treated with P7-CUR (G) and P7-TBF (H). Graph legends: P7C represents P7-CUR whereas P7T represents P7-TBF. Letter following sample represents bacterial strains, while the numbers that follow suit represent concentration of micellar solutions (e.g., P7CE2500 contains 2500 μg mL−1 of P7-CUR added to E. coli, P7TB2500 contains 2500 μg mL−1 of P7-TBF added to B. subtilis). For additional concentrations tested, see ESI† (Fig. S27–S30). | ||
As noted above, empty P7-micelles showed intrinsic antibacterial activity against E. coli only at higher concentrations (2500 μg mL−1, Fig. 1G). Inhibition of E. coli with P7-CUR nanoformulations became prominent after 16 h at concentrations as low as 156–625 μg mL−1 (Fig. 5E), which contained only 5.7 to 23.0 μg mL−1 of CUR. When P7-CUR concentrations were increased to 1250 to 2500 μg mL−1 (containing 46.1 to 92.2 μg mL−1 of encapsulated CUR), E. coli growth was inhibited as early as 3 h. These results clearly suggest that (i) P7-CUR shows much better efficacy against E. coli than empty P7-micelles, which show intrinsic antibacterial activity only at concentrations of 2500 μg mL−1 or higher; and (ii) CUR encapsulated in P7-micelles is effective in the onset of E. coli growth inhibition with concentrations of encapsulated CUR lower or comparable to free CUR. The onset of inhibition with P7-CUR is related to the gradual diffusion and delivery of CUR from P7-CUR. These findings highlight the advantages offered by P7-micelles as nanocarriers: intrinsic biocidal activity, coupled with the delivery of a potent lipophilic antibacterial agent. By leveraging on these properties, P7-CUR collectively provides better efficacy in impeding E. coli growth and offers a promising approach for combating bacterial infections.
Subsequently, we analysed the growth curves of E. coli upon treatment with TBF and P7-TBF. The reported mode of action of TBF involves inhibition of squalene epoxidase that is responsible for converting squalene to squalene epoxide, a key precursor in ergosterol biosynthesis.73,74 As ergosterol synthesis is impeded, the integrity of the cell membrane is compromised and subsequently weakened. In addition, intracellular accumulation of squalene results in rapid cell death, as it is toxic to fungal cells.75 TBF showed concentration-dependent antimicrobial activity against E. coli (Fig. 5B), with concentrations of TBF at 250–300 μg mL−1 proving to be more efficient in inhibiting E. coli growth than lower concentrations (50–200 μg mL−1).
The inhibition of E. coli with P7-TBF was also found to be concentration dependent (Fig. 5F), with an onset of inhibition at 4 h, which increased with an increase in concentration of 156–2500 μg mL−1 of P7-TBF. The time-related inhibitory effect was particularly pronounced at the highest concentration of 2500 μg mL−1, which contained 240 μg mL−1 of TBF. Comparatively, at P7-TBF concentrations of 156–625 μg mL−1 (containing 15–60 μg mL−1 of TBF), lower amounts led to lesser TBF-induced activity. As noted above for CUR, the inhibitory effect of P7-TBF on E. coli growth is relatively higher than empty P7-micelles (without encapsulated TBF); and the onset of inhibition from encapsulated TBF in P7-TBF is related to its time-dependent release. These results demonstrate once again the potential dual-action applications of these nanoformulations: direct intrinsic bactericidal activity and timely release of antimicrobial agents.
As for E. coli, CUR showed a concentration-dependent inhibitory activity against B. subtilis (Fig. 5C). Concentrations of CUR at 200–300 μg mL−1 were found to effectively arrest the growth of B. subtilis, whereas CUR at lower concentrations (50–150 μg mL−1) only slowed down bacterial growth. At 50 μg mL−1 of CUR, B. subtilis exhibited a logarithmic growth phase between 0–9 h, followed by a relatively stationary phase persisting until 24 h. With an increase in concentration to 100 μg mL−1, the logarithmic phase extended to 14 h before transitioning into the stationary phase; at 150 μg mL−1, the logarithmic phase was further prolonged to 24 h, with no discernible stationary phase observed.
Inhibition by P7-CUR against B. subtilis was also concentration-dependent (Fig. 5G). With P7-CUR concentrations in the range of 156–625 μg mL−1, which contained 5.7–23.0 μg mL−1 of CUR, B. subtilis exhibited exponential growth in the first 4 h before entering the death phase. This earlier onset of the death phase clearly suggests that P7-CUR is more effective at inhibiting B. subtilis growth than P7-micelles, in which cells entered the death phase only at 16 h. It was also evident that CUR encapsulated in P7-micelles effectively inhibited B. subtilis growth at concentrations lower than free CUR (Fig. 5C and G). Additionally, at lower P7-CUR concentrations, nanoformulations were found to be more efficient at inhibiting the growth of B. subtilis than E. coli. This disparity in efficacy may be attributed to structural differences in the bacterial cell membrane, as reported by Ngwabebhoh et al.76 As noted for empty P7-micelles, a higher concentration of P7-CUR (2500 μg mL−1, containing 92 μg mL−1 of CUR) was not as effective in inhibiting the growth of B. subtilis. This suggests that at higher concentrations, NPs likely coat the external bacterial surface, which prevents cell leakage and reduces its potency.63 It leads to additive effects of P7-CUR being observed only at lower concentrations. Our findings suggest that the combined efficacy of P7-micelles and CUR against B. subtilis can be optimally realized at lower concentrations of drug-loaded antibacterial formulation (P7-CUR).
In a similar study with TBF and P7-TBF on B. subtilis, it was noted that free TBF efficacy was also concentration-dependent (Fig. 5D). Between 50 and 100 μg mL−1, B. subtilis showed a logarithmic growth phase between 0–9 h, followed by a relatively stationary phase persisting until 24 h. Upon increasing the concentration to 150–250 μg mL−1, the logarithmic phase extended to 24 h, resulting in no discernible stationary phase; at 300 μg mL−1, TBF effectively impedeed B. subtilis growth from the onset until 8 h, beyond which it entered the logarithmic phase.
In contrast, lower concentrations of P7-TBF were more efficient against B. subtilis (Fig. 5H). With P7-TBF concentrations of 156–625 μg mL−1 (containing 15–60 μg mL−1 of TBF), the bacteria experienced a logarithmic growth in the first 4 h. Subsequently, the optical density at 600 nm gradually decreased, indicating a reduction in the population of B. subtilis in the presence of P7-TBF. In comparison to empty P7-micelles, for which cells entered the death phase at 16 h, the earlier onset of the death phase suggests that P7-TBF is more effective at inhibiting the growth of B. subtilis, and at encapsulated TBF concentrations lower than that of free TBF (Fig. 5D and H). However, at 2500 μg mL−1 (containing 240 μg mL−1 of encapsulated TBF), the effectiveness of P7-TBF in inhibiting the growth of B. subtilis diminished when compared to blank P7-micelles. Similar to P7-CUR, it is possible that higher concentrations of nanoformulations formed a protective coating on the external bacterial surface, subsequently preventing the leakage of cell contents and reducing its efficacy. Therefore, additive effects of P7-micelles and TBF were observed only at lower concentrations. Nevertheless, inhibition seen at lower concentrations highlights the potential of encapsulating TBF into intrinsically antimicrobial P7-micelles to impede B. subtilis growth, providing promising avenues for utilizing P7-micelles as dual-action formulations to inhibit bacterial growth and deliver therapeutic agents.
Evaluation of antibacterial properties of CUR and TBF loaded P6-micelles
We subsequently examined efficacy of the loaded antibacterial cargo in P6 nanoparticles that do not have any inherent bactericidal efficacy of their own (Fig. S31–S35, ESI†). These formulations will undergo time-based release of bactericides, CUR and TBF, as demonstrated in P7 nanoparticles. At a concentration of 156 μg mL−1, P6-CUR (containing 11.3 μg mL−1 of encapsulated CUR) showed negligible inhibitory effects against E. coli. With 312 μg mL−1 of P6-CUR (containing 22.6 μg mL−1 of encapsulated CUR), inhibition of E. coli growth was observed after 15 h. Treatment with higher P6-CUR concentrations (625–2500 μg mL−1, with CUR levels from 45 to 181 μg mL−1) resulted in increased efficiency in inhibiting E. coli which began right from the outset. The results show that P6 micelles act as nanocarriers of CUR, and P6-CUR exhibits inhibition against E. coli which is due to the released CUR from the P6 formulation. We noted that despite containing almost twice the amount of encapsulated CUR, P6-CUR's inhibitory effectiveness was lower than that of P7-CUR, as noted by the higher OD600 observed over time. This is due to a lack of intrinsic antibacterial activity in P6. It shows that the combined effect of cationic surface and time-release of bactericidal cargo (in P7-CUR) help enhance inhibitory efficacy in combating bacterial infections. Additionally, as the concentration of CUR released over time increased, the inhibitory effect observed also multiplied, as evidenced by the disparity in OD600 readings between treated and untreated E. coli samples. This effect becomes more pronounced with time. For instance, the difference in OD600 readings between 2 h (OD600 = 0.044 for treated and 0.068 for untreated) and 8 h (OD600 = 0.210 for treated and 0.262 for untreated) and 24 h (OD600 = 0.324 for treated and 0.447 for untreated) gradually increases due to release and accumulation of CUR to impede E. coli growth (27% of CUR released at 2 h, 45% at 8 h, and 59% at 24 h).The growth curves of E. coli following treatment with P6-TBF are shown in Fig. S31B (ESI†). The inhibition of E. coli by P6-TBF was also found to be concentration-dependent, intensifying with an increase from 156 to 2500 μg mL−1. The inhibitory effect was particularly notable at the highest concentration of 2500 μg mL−1, which contained 182 μg mL−1 of TBF. Similar to observations with P6-CUR, the inhibitory effect of P6-TBF on E. coli growth is attributed to the release of the drug from the nanoformulations. The inhibitory effect observed also increased as TBF release increased from 24% at 2 h, to 36% at 8 h, and to 66% at 24 h. The disparity in OD600 readings between 2 h (OD600 = 0.052 for treated and 0.068 for untreated) and 8 h (OD600 = 0.210 for treated and 0.262 for untreated) and 24 h (OD600 = 0.324 for treated and 0.447 for untreated) shows the effect of TBF concentration in impeding E. coli growth. Additionally, the relatively higher OD600 in P6-TBF compared to P7-TBF underscores the significance of surface charge in achieving the dual-action applications of these nanoformulations.
The analysis of growth curves of B. subtilis treated with P6-CUR also revealed a decrease in OD600 compared to untreated bacterial samples, attributed to the antibacterial activity of the released CUR (Fig. S31C, ESI†). The inhibition by P6-CUR against B. subtilis was concentration-dependent. Lower concentrations of P6-CUR were more effective at inhibiting B. subtilis growth compared to E. coli due to structural differences between the two bacteria. With P6-CUR concentrations ranging from 156 to 625 μg mL−1, containing 11.3–45.3 μg mL−1 of CUR, B. subtilis exhibited exponential growth in the first 7 h before entering the death phase.
In the treatment of B. subtilis with various concentrations of P6-TBF (Fig. S31D, ESI†), at 156 to 625 μg mL−1 (containing 11.4–45.4 μg mL−1 of TBF), bacteria exhibited logarithmic growth in the initial 8 h, followed by a subsequent reduction in the population of B. subtilis. At 2500 μg mL−1 (with 182 μg mL−1 of encapsulated TBF), the efficacy of P6-TBF in inhibiting B. subtilis growth was slightly enhanced.
SEM analyses of bacteria
SEM was used to gain visual insights into the morphology of both E. coli and B. subtilis after exposure to CUR, TBF, P7-CUR, and P7-TBF (Fig. 6). Upon exposure to CUR, E. coli retained their rod-like shapes, measuring approximately 2–3 μm in length (Fig. 6A). However, closer inspection of the images revealed much rougher surfaces compared to untreated E. coli, which are indicative of the disruptive effects of CUR on membrane integrity. Similarly, treatment with P7-CUR also resulted in wrinkly surfaces (Fig. 6B), affirming the inhibitory efficacy of P7-CUR against the growth of E. coli. | ||
| Fig. 6 SEM images of E. coli incubated with CUR (A), P7-CUR (B); TBF (C), P7-TBF (D) and B. subtilis incubated with CUR (E), P7-CUR (F), TBF (G), P7-TBF (H). Bacteria were incubated with samples (2500 μg mL−1) for 24 h prior to fixing. | ||
In E. coli incubated with TBF (Fig. 6C), the most noticeable difference compared to untreated E. coli was the elongated structures, with lengths ranging from 3–7 μm. Notably, a slight pinch was also seen in the middle of the bacterial cells, similar to those reported by Rivas-Marin et al. in Gemmata obscuriglobus.77 These abnormalities suggest that TBF likely slowed down the division and growth of E. coli cells. Non-uniform surfaces were also observed which suggested membrane damage. E. coli exposed to P7-TBF resulted in similar non-uniform structures that were approximately 3–8 μm (Fig. 6D). These observations suggest effective inhibition of the growth of E. coli induced by P7-TBF, complementing the results obtained from the growth curve measurements.
The morphology of B. subtilis incubated with varied formulations was subsequently evaluated. Treatment with CUR resulted in deformed, perforated structures, accompanied by debris likely originating from dead bacteria (Fig. 6E). These observations suggest a weakened bacterial cell membrane and a more pronounced effect of CUR on B. subtilis compared to E. coli. The absence of an outer membrane in B. subtilis may allow less obstructed diffusion of CUR, which could lead to an improvement in bacterial inhibition. Similarly, when B. subtilis was incubated with P7-CUR, analogous misshaped, punctured structures were observed, clearly suggesting the effectiveness of P7-CUR in impeding B. subtilis growth (Fig. 6F). The observed morphological deformations collectively provide evidence for the disruptive impact of P7-TBF on B. subtilis, further emphasizing its potential in addressing microbial infections.
The morphology of B. subtilis exposed to TBF is shown in Fig. 6G, and it shows several interesting observations such as wrinkled surfaces, shrunken bacterial contents, and ruptured bacteria. These results suggest damage to the bacterial membrane upon TBF exposure. Moreover, when B. subtilis was treated with P7-TBF (Fig. 6H), the bacterial membrane appeared to be significantly damaged as no distinct boundaries could be observed. In addition, we observed several ruptured structures, all of which suggested the effectiveness of P7-TBF to inhibit the growth of B. subtilis. These collectively highlight the potent antimicrobial activity of P7-TBF to disrupt bacterial membranes and impede bacterial growth effectively. In 2017, Mukherjee et al. synthesized a leucine-based poly(2-hydroxyethyl methacrylate) for the treatment of B. subtilis.78 They noted fusion of B. subtilis cell membrane upon polymer treatment, which caused a surface collapse in the bacterial cells. Ding et al. also reported a quinine-based quarternized polymer that induced the adhesion of individual B. subtilis cells, revealing the potent effect of their polymer on bacterial cells.79
Evaluation of toxicity toward human endothelial cells
We exposed human umbilical vein endothelial cells (HUVEC) to TBF, CUR, P7-CUR, and P7-TBF and evaluated the post-exposure viability of the cells using an MTT assay. As can be observed in Fig. 7, the inclusion of both CUR and TBF into NPs significantly decreased the toxicity of free CUR and TBF. These effects were significant at CUR and TBF concentrations as low as 4.7 and 50 μg mL−1, respectively. P7-TBF toxicity was not different than that of P7 unloaded nanoparticles. Although the improvement in viability was significant for CUR incorporated into P7-CUR, the cell viability after the exposure of P7-CUR could not be restored to the level measured for HUVEC cells exposed to unloaded P7-micelles as shown in Fig. 7A. The latter may be due to limitations of the MTT assay, as it is sensitive to confounding factors such as differences in mitochondrial function, pH etc.80 These could be mitigated by in vivo evaluation of the lowest concentration for efficacy of P7-CUR. This difference between P7-CUR and P7-TBF may be related to the higher encapsulation efficiency of TBF into P7 NP and/or due to the observed drug release kinetics and biophysical characteristic differences between the two types of loaded NPs.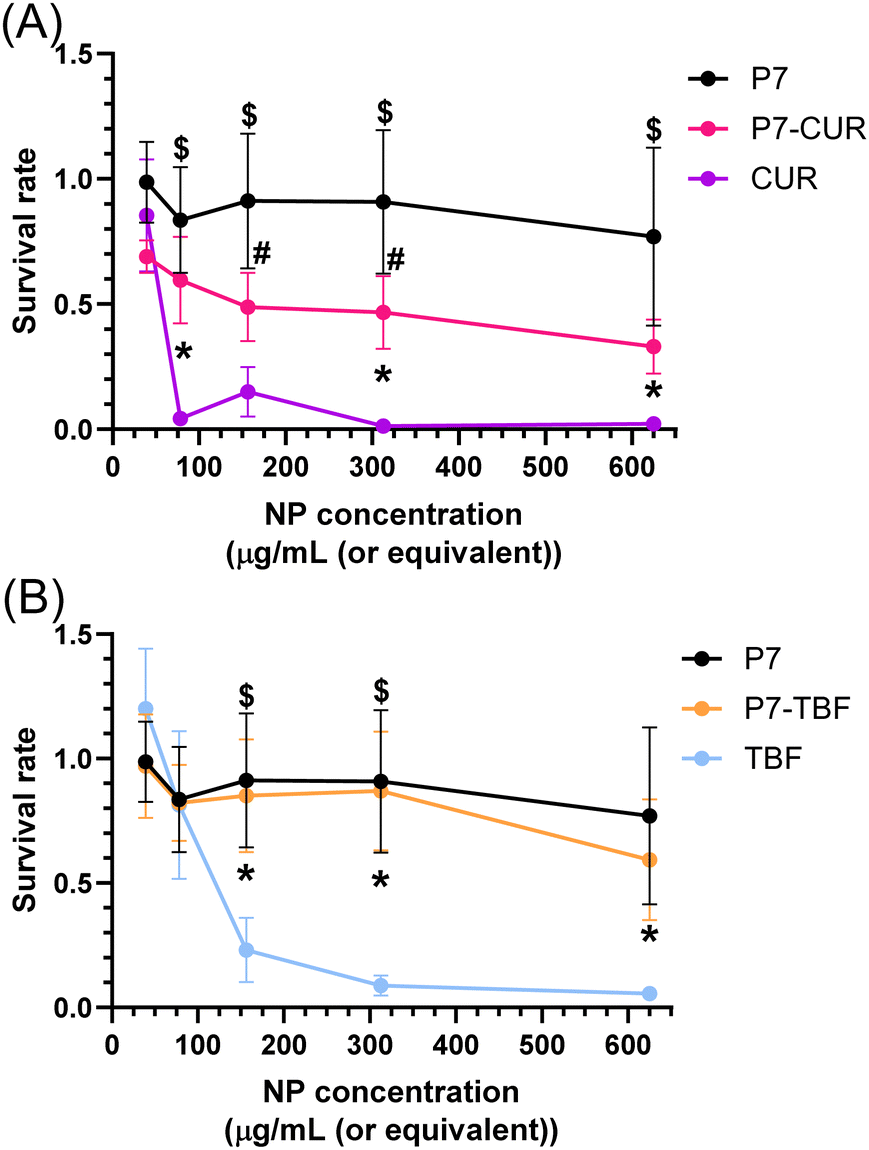 | ||
| Fig. 7 MTT assessment of HUVEC cell viability following exposure to different concentrations of P7-micelles (A and B), P7-CUR and CUR (A), P7-TBF and TBF (B). Cells were exposed to different concentrations of nanoparticles using two-fold dilutions of P7-micelles, P7-CUR, and P7-TBF ranging from 625 to 39.1 μg mL−1 or their loading concentrations of CUR and TBF. After 24 h exposure, cell proliferation was assessed using a MTT assay. Survival rates were normalized against unexposed cells. Data are expressed as the average of 5 biological replicates +/− SD. Symbols are presented for p < 0.05 using two-way ANOVA with Tuckey multiple comparison post-hoc test. $ = P7-micelles vs. CUR/TBF, # = P7-micelles vs. P7-CUR/TBF, * = P7-CUR/TBF vs. CUR/TBF. | ||
Conclusions
Cationic NPs offer a promising multifunctional platform in the ongoing battle against antimicrobial resistance. We have capitalized on the dual functionality of such NPs, which not only inhibit bacterial growth through their cationic shell but also serve as vehicles for the targeted delivery of antimicrobial agents. A pivotal aspect of our strategy involves the synthesis of miktoarm polymers with cationic amine moieties (P7), achieved through a meticulous combination of arm- and core-first methods. The polymer self-assembles into well-defined micelles with a positively charged hydrophilic shell, thus imparting intrinsic antimicrobial properties to the NPs. Cationic P7-micelles are particularly effective against the Gram-positive bacterium, B. subtilis, which could be attributed to favourable interactions between the positively charged amines of P7-micelles and the oppositely charged bacteria membrane. However, P7-micelles are less effective against the Gram-negative bacterium, E. coli, possibly due to an additional lipopolysaccharide layer that impedes penetration. Nanoformulations reported in this study extend beyond their intrinsic antimicrobial properties, as they also serve as proficient nanocarriers to deliver antimicrobial agents such as CUR and TBF. P7-CUR and P7-TBF exhibit inhibitory activity against E. coli at higher concentrations, whereas their effectiveness against B. subtilis is optimized at lower concentrations. The improved cell viability observed for P7-TBF makes its profile almost ideal for further in vivo efficacy assessment. This versatility highlights the potential of P7-micelles as a multifaceted approach to combat microbial infections. By leveraging their intrinsic antimicrobial properties and adeptness as efficient nanocarriers for antimicrobial agents, cationic NPs offer an advantageous avenue in the quest to address antimicrobial resistance.Author contributions
Conceptualization, formal analysis, methodology, resources, visualization, writing-original draft, writing – review & editing: H. W. Y, S. M. A. O, G. T.-J., E. R., J.-C. T., T. G. M. vdv and A. K; investigation: H. W. Y., S. M. A. O., G. T.-J., and L. S. R. C.; funding acquisition and supervision: A. K.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council of Canada under grant number RGPIN-2023-03565 (A. K.). H. W. Y. thanks the Fonds de Recherche du Québec Nature et technologies for the Doctoral research scholarship. H. W. Y. also thanks DrawBioMed for the tutorial on bacterial illustration presented in Scheme 1.References
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. Clotaire Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Duong Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Young Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- K. Yusuf, V. Sampath and S. Umar, Int. J. Mol. Sci., 2023, 24, 3110 CrossRef CAS PubMed.
- L. Huang and K. Crothers, Respirology, 2009, 14, 474–485 CrossRef PubMed.
- A. Van de Louw, A. Mirouse, O. Peyrony, V. Lemiale and E. Azoulay, Semin. Respir. Crit. Care Med., 2019, 40, 498–507 CrossRef PubMed.
- T. Franquet and P. Domingo, Radiol. Clin. North Am., 2022, 60, 507–520 CrossRef PubMed.
- WHO fungal priority pathogens list to guide research, development and public health action. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO Search PubMed.
- R. Orrell-Trigg, M. Awad, S. Gangadoo, S. Cheeseman, Z. L. Shaw, V. K. Truong, D. Cozzolino and J. Chapman, Analyst, 2024, 149, 1597–1608 RSC.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS.
- J. Loree and S. L. Lappin, Bacteriostatic Antibiotics, https://www.ncbi.nlm.nih.gov/books/NBK547678/, (accessed October 15, 2023).
- P. Patel, H. R. Wermuth, C. Calhoun and G. A. Hall, Antibiotics, https://www.ncbi.nlm.nih.gov/books/NBK535443/.
- J. Tits, B. P. A. Cammue and K. Thevissen, Int. J. Mol. Sci., 2020, 21, 8873 CrossRef CAS PubMed.
- K. You, B. Gao, M. Wang, X. Wang, K. C. Okoro, A. Rakhimbekzoda and Y. Feng, J. Mater. Chem. B, 2022, 10, 1005–1018 RSC.
- L. Yu, C. Lin, Z. Zheng, Z. Li and X. Wang, Colloids Surf., B, 2016, 145, 392–400 CrossRef CAS PubMed.
- A. Nimmagadda, X. Liu, P. Teng, M. Su, Y. Li, Q. Qiao, N. K. Khadka, X. Sun, J. Pan, H. Xu, Q. Li and J. Cai, Biomacromolecules, 2017, 18, 87–95 CrossRef CAS.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- H. C. Parkin, S. T. G. Street, B. Gowen, L. H. Da-Silva-Correa, R. Hof, H. L. Buckley and I. Manners, J. Am. Chem. Soc., 2024, 146, 5128–5141 CrossRef CAS PubMed.
- T. J. Cuthbert, B. Hisey, T. D. Harrison, J. F. Trant, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2018, 57, 12707–12710 CrossRef CAS PubMed.
- H. Kuday, N. C. Süer, A. Bayır, B. Aksu, A. Hatipoğlu, M. M. Güncü, İ. Acaroğlu Degitz, M. Gallei and T. Eren, ACS Appl. Polym. Mater., 2021, 3, 6524–6538 CrossRef CAS.
- X. Sun, P. Cao, X. Bai, Y. He, P. Song and R. Wang, Macromolecules, 2023, 56, 9498–9508 CrossRef CAS.
- Z. Hou, Y. V. Shankar, Y. Liu, F. Ding, J. L. Subramanion, V. Ravikumar, R. Zamudio-Vázquez, D. Keogh, H. Lim, M. Y. F. Tay, S. Bhattacharjya, S. A. Rice, J. Shi, H. Duan, X.-W. Liu, Y. Mu, N. S. Tan, K. C. Tam, K. Pethe and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2017, 9, 38288–38303 CrossRef CAS.
- W. Shen, P. He, C. Xiao and X. Chen, Adv. Healthcare Mater., 2018, 7, 1800354 CrossRef.
- L. Liu, K. C. Courtney, S. W. Huth, L. A. Rank, B. Weisblum, E. R. Chapman and S. H. Gellman, J. Am. Chem. Soc., 2021, 143, 3219–3230 CrossRef CAS PubMed.
- N. Pasquier, H. Keul, E. Heine, M. Moeller, B. Angelov, S. Linser and R. Willumeit, Macromol. Biosci., 2008, 8, 903–915 CrossRef CAS PubMed.
- C. Jiang, J. Chen, Z. Li, Z. Wang, W. Zhang and J. Liu, Expert Opin. Drug Delivery, 2019, 16, 363–376 CrossRef CAS.
- Z. A. I. Mazrad, C. A. Choi, Y. M. Kwon, I. In, K. D. Lee and S. Y. Park, ACS Appl. Mater. Interfaces, 2017, 9, 33317–33326 CrossRef CAS.
- J. J. Virgen-Ortíz, J. C. S. dos Santos, Á. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- Z. Fan, K. H. L. Po, K. K. Wong, S. Chen and S. P. Lau, ACS Appl. Nano Mater., 2018, 1, 1811–1818 CrossRef CAS.
- W. Liu, W. Pei, M. Moradi, D. Zhao, Z. Li, M. Zhang, D. Xu and F. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 18794–18805 CrossRef CAS.
- B. Heli, G. R. Younes, K. Arguindeguy and A. Ajji, Carbohydr. Polym. Technol. Appl., 2024, 7, 100457 CAS.
- N. Fattahi, L. Gorgannezhad, S. F. Masoule, N. Babanejad, A. Ramazani, M. Raoufi, E. Sharifikolouei, A. Foroumadi and M. Khoobi, Adv. Colloid Interface Sci., 2024, 325, 103119 CrossRef CAS.
- V. Keshavarz, M. Kazemi, B. Khalvati, F. Zare, A. Dehshahri and H. Sadeghpour, Biotechnol. Prog., 2024, 40, e3443 CrossRef CAS.
- M. Liu, J. Li and B. Li, Langmuir, 2018, 34, 1574–1580 CrossRef CAS PubMed.
- M. Gultekinoglu, B. Kurum, S. Karahan, D. Kart, M. Sagiroglu, N. Ertaş, A. Haluk Ozen and K. Ulubayram, Mater. Sci. Eng., C, 2017, 71, 1166–1174 CrossRef CAS PubMed.
- M. Gultekinoglu, Y. Tunc Sarisozen, C. Erdogdu, M. Sagiroglu, E. A. Aksoy, Y. J. Oh, P. Hinterdorfer and K. Ulubayram, Acta Biomater., 2015, 21, 44–54 CrossRef CAS PubMed.
- V. Lotocki, H. Yazdani, Q. Zhang, E. R. Gran, A. Nyrko, D. Maysinger and A. Kakkar, Macromol. Biosci., 2021, 21, 2000305 CrossRef CAS PubMed.
- Z. Zhu, G. Jeong, S.-J. Kim, I. Gadwal, Y. Choe, J. Bang, M.-K. Oh, A. Khan and J. Rao, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2391–2396 CrossRef CAS.
- P. S. L. Manasa, A. D. Kamble and U. Chilakamarthi, ACS Omega, 2023, 8, 34868–34878 CrossRef CAS.
- T. Rudrappa and H. P. Bais, J. Agric. Food Chem., 2008, 56, 1955–1962 CrossRef CAS.
- N. Dogra, R. Choudhary, P. Kohli, J. D. Haddock, S. Makwana, B. Horev, Y. Vinokur, S. Droby and V. Rodov, J. Agric. Food Chem., 2015, 63, 2557–2565 CrossRef CAS PubMed.
- H. Gunes, D. Gulen, R. Mutlu, A. Gumus, T. Tas and A. E. Topkaya, Toxicol. Ind. Health, 2016, 32, 246–250 CrossRef CAS PubMed.
- D. Zheng, C. Huang, H. Huang, Y. Zhao, M. R. U. Khan, H. Zhao and L. Huang, Chem. Biodiversity, 2020, 17, e2000171 CrossRef CAS PubMed.
- S. S. Hettiarachchi, Y. Perera, S. P. Dunuweera, A. N. Dunuweera, S. Rajapakse and R. M. G. Rajapakse, ACS Omega, 2022, 7, 46494–46500 CrossRef CAS.
- R. Jagannathan, P. M. Abraham and P. Poddar, J. Phys. Chem. B, 2012, 116, 14533–14540 CrossRef CAS.
- G. Garcia-Effron, A. Gomez-Lopez, E. Mellado, A. Monzon, J. L. Rodriguez-Tudela and M. Cuenca-Estrella, J. Antimicrob. Chemother., 2004, 53, 1086–1089 CrossRef CAS PubMed.
- Web Annex A. World Health Organization Model List of Essential Medicines – 23rd List, 2023. In: The selection and use of essential medicines 2023: Executive summary of the report of the 24th WHO Expert Committee on the Selection and Use of Essential Medicines, 24–28 April 2023. Geneva: World Health Organization; 2023 (WHO/MHP/HPS/EML/2023.02). Licence: CC BY-NC-SA 3.0 IGO.
- S. M. AbdelSamie, A. O. Kamel, O. A. Sammour and S. M. Ibrahim, Eur. J. Pharm. Sci., 2016, 88, 91–100 CrossRef CAS PubMed.
- D. Łagowski, S. Gnat, A. Nowakiewicz, M. Osińska and M. Dyląg, Infection, 2020, 48, 889–897 CrossRef.
- G. Arjmand and M. R. Haeri, Adv. Biomed. Res., 2022, 11, 90 CrossRef CAS PubMed.
- J. D. Friedl, V. Nele, G. De Rosa and A. Bernkop-Schnürch, Adv. Funct. Mater., 2021, 31, 2103347 CrossRef CAS.
- R. Pawar, A. Pathan, S. Nagaraj, H. Kapare, P. Giram and R. Wavhale, Polym. Adv. Technol., 2023, 34, 3296–3316 CrossRef CAS.
- H. W. Yong, M. Ferron, M. Mecteau, T. Mihalache-Avram, S. Lévesque, É. Rhéaume, J.-C. Tardif and A. Kakkar, Biomacromolecules, 2023, 24, 4064–4077 CrossRef CAS.
- H. W. Yong and A. Kakkar, Macromol. Biosci., 2021, 21, 2100105 CrossRef CAS PubMed.
- S. A. A. Rizvi and A. M. Saleh, Saudi Pharm. J., 2018, 26, 64–70 CrossRef PubMed.
- C. Deng, X. Chen, H. Yu, J. Sun, T. Lu and X. Jing, Polymer, 2007, 48, 139–149 CrossRef CAS.
- S. Zhang, J. Zou, F. Zhang, M. Elsabahy, S. E. Felder, J. Zhu, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 18467–18474 CrossRef CAS.
- W. Scarano, P. de Souza and M. H. Stenzel, Biomater. Sci., 2015, 3, 163–174 RSC.
- H. Su, F. Wang, W. Ran, W. Zhang, W. Dai, H. Wang, C. F. Anderson, Z. Wang, C. Zheng, P. Zhang, Y. Li and H. Cui, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4518–4526 CrossRef CAS PubMed.
- M. Haktaniyan and M. Bradley, Chem. Soc. Rev., 2022, 51, 8584–8611 RSC.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- Z. Breijyeh, B. Jubeh and R. Karaman, Molecules, 2020, 25, 1340 CrossRef CAS PubMed.
- M. Tavakolian, M. Okshevsky, T. G. M. van de Ven and N. Tufenkji, ACS Appl. Mater. Interfaces, 2018, 10, 33827–33838 CrossRef CAS PubMed.
- E.-H. Westman, M. Ek, L.-E. Enarsson and L. Wågberg, Biomacromolecules, 2009, 10, 1478–1483 CrossRef CAS PubMed.
- S.-H. Lim and S. M. Hudson, Carbohydr. Res., 2004, 339, 313–319 CrossRef CAS PubMed.
- K. Divya, S. Vijayan, T. K. George and M. S. Jisha, Fibers Polym., 2017, 18, 221–230 CrossRef CAS.
- S. Chakraborti, A. K. Mandal, S. Sarwar, P. Singh, R. Chakraborty and P. Chakrabarti, Colloids Surf., B, 2014, 121, 44–53 CrossRef CAS PubMed.
- I. C. Tanganini, C. H. M. Camargos, J. C. Jackson, C. A. Rezende, S. R. Ceccato-Antonini and A. F. Faria, RSC Sustainability, 2024, 2, 459–474 RSC.
- M. Kharat, Z. Du, G. Zhang and D. J. McClements, J. Agric. Food Chem., 2017, 65, 1525–1532 CrossRef CAS PubMed.
- R. Basak and R. Bandyopadhyay, Langmuir, 2013, 29, 4350–4356 CrossRef CAS PubMed.
- in Strategies to Modify the Drug Release from Pharmaceutical Systems, ed. M. L. Bruschi, Woodhead Publishing, 2015, ch. 5, pp. 63–86 Search PubMed.
- M. P. Paarakh, P. A. Jose, C. M. Setty and G. V. Peterchristoper, IJPRT, 2018, 8, 12–20 Search PubMed.
- M.-H. Hsiao, K.-H. Lin and D.-M. Liu, Soft Matter, 2013, 9, 2458–2466 RSC.
- P. Tyagi, M. Singh, H. Kumari, A. Kumari and K. Mukhopadhyay, PLoS One, 2015, 10, e0121313 CrossRef PubMed.
- M. Nowosielski, M. Hoffmann, L. S. Wyrwicz, P. Stepniak, D. M. Plewczynski, M. Lazniewski, K. Ginalski and L. Rychlewski, J. Chem. Inf. Model., 2011, 51, 455–462 CrossRef CAS PubMed.
- N. S. Ryder, Br. J. Dermatol., 1992, 126, 2–7 CrossRef PubMed.
- A. K. Gupta, B. Elewski, W. S. Joseph, S. R. Lipner, C. R. Daniel, A. Tosti, E. Guenin and M. Ghannoum, Mycoses, 2024, 67, e13683 CrossRef CAS PubMed.
- F. Asabuwa Ngwabebhoh, S. Ilkar Erdagi and U. Yildiz, Carbohydr. Polym., 2018, 201, 317–328 CrossRef CAS PubMed.
- E. Rivas-Marin, S. Stettner, E. Y. Gottshall, C. Santana-Molina, M. Helling, F. Basile, N. L. Ward and D. P. Devos, Nat. Commun., 2019, 10, 2916 CrossRef PubMed.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2017, 2, 1633–1644 CrossRef CAS PubMed.
- H.-H. Ding, M.-H. Zhao, L. Zhai, J.-B. Zhen, L.-Y. Sun, J.-Z. Chigan, C. Chen, J.-Q. Li, H. Gao and K.-W. Yang, Polym. Chem., 2021, 12, 2397–2403 RSC.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, Int. J. Mol. Sci., 2021, 22, 12827 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, synthesis of P1–P5, 1H and 13C NMR spectra of P1–P7, MALDI-TOF spectrum, GPC traces, FT-IR spectra, TEM analyses, CMC graphs, kinetic analyses, and growth curve analyses (other concentrations). See DOI: https://doi.org/10.1039/d4tb01200c |
| This journal is © The Royal Society of Chemistry 2024 |