
Design and synthesis of a shikimoyl-functionalized cationic di-block copolypeptide for cancer cell specific gene transfection†
Abinash
Padhy‡
 a,
Pritam
Das‡
bc,
Namita S.
Mahadik
bc,
Sidharth
Panda
a,
Mahammad
Anas
d,
Sabyasachi
Das
a,
Rajkumar
Banerjee
*bc and
Sayam
Sen Gupta
*a
a,
Pritam
Das‡
bc,
Namita S.
Mahadik
bc,
Sidharth
Panda
a,
Mahammad
Anas
d,
Sabyasachi
Das
a,
Rajkumar
Banerjee
*bc and
Sayam
Sen Gupta
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia, West Bengal 741246, India. E-mail: sayam.sengupta@iiserkol.ac.in
bDepartment of Oils, Lipids Science and Technology, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007, India. E-mail: banerjee@iict.res.in
cAcademy of Scientific and Innovative Research (AcSIR), CSIR-HRDC Campus, Ghaziabad 201002, India
dSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India
First published on 15th August 2024
Abstract
Targeted and efficient gene delivery systems hold tremendous potential for the improvement of cancer therapy by enabling appropriate modification of biological processes. Herein, we report the design and synthesis of a novel cationic di-block copolypeptide, incorporating homoarginine (HAG) and shikimoyl (LSA) functionalities (HDA-b-PHAGm-b-PLSAn), tailored for enhanced gene transfection specifically in cancer cells. The di-block copolypeptide was synthesized via sequential N-carboxyanhydride (NCA) ring-opening polymerization (ROP) techniques and its physicochemical properties were characterized, including molecular weight, dispersity, secondary conformation, size, morphology, and surface charge. In contrast to the cationic poly-L-homoarginine, we observed a significantly reduced cytotoxic effect of this di-block copolypeptide due to the inclusion of the shikimoyl glyco-polypeptide block, which also added selectivity in internalizing particular cells. This di-block copolypeptide was internalized via mannose-receptor-mediated endocytosis, which was investigated by competitive receptor blocking with mannan. We evaluated the transfection efficiency of the copolypeptide in HEK 293T (noncancerous cells), MDA-MB-231 (breast cancer cells), and RAW 264.7 (dendritic cells) and compared it with commonly employed transfection agents (Lipofectamine). Our findings demonstrate that the homoarginine and shikimoyl-functionalized cationic di-block copolypeptide exhibits potent gene transfection capabilities with minimal cytotoxic effects, particularly in cancer cells, while it is ineffective for normal cells, indicative of its potential as a promising platform for cancer cell-specific gene delivery systems. To evaluate this, we delivered an artificially designed miRNA-plasmid against Hsp90 (amiR-Hsp90) which upon successful transfection depleted the Hsp90 (a chaperone protein responsible for tumour growth) level specifically in cancerous cells and enforced apoptosis. This innovative approach offers a new avenue for the development of targeted therapeutics with an improved efficacy and safety profile in cancer treatment.
Introduction
Gene therapy stands out as a leading approach for addressing genetic disorders. However, a significant obstacle to its application lies in efficiently delivering exogenous genetic material into target cells. While viral vectors are commonly employed for this purpose, they are beset by clinical limitations such as immunogenicity, short shelf-life, and low cargo capacity.1,2 This necessitates exploration into alternative avenues for achieving secure and effective gene therapies, particularly through the utilization of polymer-based vehicles.3–14Among non-viral vectors, polymeric vectors present an enticing option due to their capacity to create versatile functionalities with customizable structures and by incorporating targeted ligands.15 Notably, poly(ethylene imine) (PEI) and poly(L-lysine) (PLL) have been among the earliest structures investigated by researchers due to their ability to condense genetic material and subsequently deliver it, in addition to their commercial availability.16,17 Numerous reports have highlighted diverse polymeric architectures and designs capable of fulfilling the task of gene delivery.3,4,18 Several groups have also focused on developing biodegradable polymeric gene delivery vectors, including cell-penetrating peptides, amphiphilic peptides, and polypeptide systems.19–21 Pioneering work by Reineke and colleagues has demonstrated the efficacy of glycopolymer-based systems in delivering therapeutic nucleic acids, offering both biocompatibility and enhanced stabilization compared to conventional cationic polymeric systems.22 These glycopolymeric systems play a crucial role in targeting various cells and tissues, specifically the ones expressing carbohydrate receptors.23–25 Since cancerous cells are known to overexpress glycan receptors,26–28 such glycan-decorated polymers and nanocarriers can also be used to shuttle therapeutic cargo to cancer cells.29–32 One notable receptor in this context is the mannose receptor (CD206 or MRC1) found on MDA-MB-231 breast cancer cells33–36 and antigen-presenting cells (APCs).37 Gras-Masse and co-workers have demonstrated that mannose mimicking quinic and shikimic acids are also effective ligands for the mannose receptor of dendritic cells.38,39 Chaudhuri and co-workers have achieved dendritic cell-based genetic immunization using these ligands.40–44 Additionally, recent findings by Sen Gupta and colleagues have demonstrated the nuclear-specific internalization of shikimoylated-polypeptides, highlighting their potential for targeted gene delivery.45 Building upon these advancements, we aim to exploit overexpressed mannose receptors on breast cancer cells and macrophagic cells to design a glycopolypeptide-based gene delivery vector.
In this study, our objective is to design biocompatible and biodegradable glycopolypeptide-based delivery carriers capable of delivering genetic materials and transfecting specific cells of interest. We anticipate that these carriers could serve as efficient vehicles for delivering gene editing systems like CRISPR–Cas9, thereby enabling the correction of malfunctioning genomes to treat genetic disorders. To achieve this, we have integrated several essential components into our system: (i) a positively charged block for nucleic acid complexation, (ii) hydrophobic chain ends to ensure the stability of the polyplex system, and (iii) a mannose receptor-targeting shikimoyl poly-glycan moiety that has demonstrated superior specificity for nuclear internalization compared to mannose congeners.
To this end, we successfully synthesized AB-type di-block cationic glycopolypeptides and meticulously characterized these structures. For the complexation of anionic nucleic acids, we opted for a poly-homoarginine block bearing a guanido moiety as the cationic segment. While various arginine-containing peptides46,47 have been explored for gene delivery applications, the utilization of homoarginine remains relatively limited. The incorporation of an additional methylene spacer has been shown to enhance the flexibility of the pendant guanido group, potentially augmenting nucleic acid binding capabilities.48 In our previous endeavor, we developed homoarginine polypeptides to generate pH-responsive supra-amphiphilic nanoparticles marred by a multi-step, low-yielding synthetic approach.49
To enhance targeting specificity, we included a shikimoylated-polypeptide block, leveraging its superior activity and specificity for glycan receptors compared to mannose congeners.40,45 The ability of shikimoyl moieties to induce endosomal escape and shuttle to the nuclear region45 further augments their suitability for our goal of utilizing them as gene delivery systems. Subsequently, the synthesized di-block copolypeptides were combined with eGFP-expressing plasmid DNA to form polyplexes, which were then screened for their toxicity prior to in vitro delivery assays. The cellular internalization and transfection abilities of these polyplexes were assessed across HEK 293T, MDA-MB-231, and RAW 264.7 cell lines. The incorporation of the shikimoylated-polypeptide block reduced overall cytotoxicity compared to homoarginine polypeptides and demonstrated excellent selectivity in trafficking genetic materials into cells.
Experimental section
Materials and methods
Details of the chemicals, instruments, synthesis of all monomeric components, and characterization methods are provided in the ESI.†Synthesis of HDA-poly(N,N′-di(Boc)-L-homoarginine)m (10)
Polymerization of N,N′-di(Boc)-L-homoarginine-NCA (HAG-NCA) compound 5 was carried out using high-vacuum techniques. In a Schlenk tube inside a glovebox, 200 mg of 5 (0.48 mmol) was dissolved in 2 mL of 1,2-dichloroethane (1, 2-DCE), and to this hexadecylamine (HDA) (0.5 M stock solution in 1,2-DCE, 1/m eq. to NCA monomer) was added as the initiator. The sealed tube was taken out from the glovebox, and the Schlenk tube was kept in an ice bath before applying the vacuum. After applying the vacuum, the reaction mixture was sealed and stirred at room temperature. The progress of the polymerization reaction was monitored using IR spectroscopy by comparing the intensities of the anhydride stretches at 1776 and 1847 cm−1 of the parent NCA monomer. Generally, the polymerization reactions were completed within 24 h. Aliquots were removed after completion of the polymerization reaction for GPC analysis. Finally, the solvent was removed under reduced pressure from the reaction mixture. The resulting residue was redissolved in DCM, and the polymer was precipitated out by the addition of diethyl ether. The precipitated polymer was collected by centrifugation and dried to afford HDA-poly(ε-N,N′-di(Boc)-L-homoarginine)m10 as a white solid product (yield 160 mg, ∼80% yield).Synthesis of HDA-poly(L-homoarginine)m (HDA-PHAGm) (11)
10 was dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM:TFA (100 mg mL−1) and subject to vigorous stirring at room temperature for 6 h. The volatiles were removed using high vacuum to afford a crude solid product. The crude product was purified by precipitation with MeOH/diethyl ether thrice. Then, it was dissolved in DI water and transferred into dialysis tubing (3.5 kDa cutoff). The polymers were dialyzed against DI water for three days, changing the water once every 2 h for the first day and then thrice every day. Dialyzed polymers were lyophilized to yield HDA-poly(L-homoarginine)m as a white fluffy solid (∼80% yield).
:1 DCM:TFA (100 mg mL−1) and subject to vigorous stirring at room temperature for 6 h. The volatiles were removed using high vacuum to afford a crude solid product. The crude product was purified by precipitation with MeOH/diethyl ether thrice. Then, it was dissolved in DI water and transferred into dialysis tubing (3.5 kDa cutoff). The polymers were dialyzed against DI water for three days, changing the water once every 2 h for the first day and then thrice every day. Dialyzed polymers were lyophilized to yield HDA-poly(L-homoarginine)m as a white fluffy solid (∼80% yield).
Synthesis of HDA-b-poly(N,N′-di(Boc)-L-homoarginine)m-b-poly(3,4,5-tri-acetyl shikimic-L-lysine)n (12)
In a reaction tube, N,N′-di(Boc)-L-homoarginine-NCA (HAG-NCA) compound 5 (150 mg, 0.36 mmol) was dissolved in 1.5 mL of DMF and a proton sponge (0.5 M stock solution in DMF, 0.25 eq. to NCA monomer) was added to it. After 5 min of stirring, hexadecylamine (0.5 M stock solution in DMF, 1/m eq. to NCA monomer) was added to it, and the reaction was stirred at room temperature. Generally, the polymerization reactions were completed within 36 hours. After complete consumption of the NCA monomer, 3,4,5-tri-acetyl shikimic-L-lysine-NCA (n eq. to the macro-initiator HDA-poly(N,N′-di(Boc)-L-homoarginine)m) (100 mg mL−1) in DMF was added followed by the addition of the proton sponge (0.5 M stock solution in DMF, 0.25 eq. to NCA monomer) as an additive. The progress of the polymerization reaction was monitored using IR spectroscopy by comparing the intensities of the anhydride stretches at 1778 and 1849 cm−1 of the parent NCA monomer. After completion, the reaction mixture was removed from the glove box, diluted with DCM, and worked up with water to remove water-soluble residue. The organic layer was combined and dried over anhydrous sodium sulfate. The organic part was evaporated to dryness, the resulting residue was redissolved in dichloromethane and the addition of diethyl ether precipitated out the polymer. The precipitated polymer was collected by centrifugation and dried to afford polypeptides in greater than 80% yield as a fluffy white compound.Fluorescent labeling of polypeptides with fluorescein-NHS ester (13)
To a solution of the protected polypeptide (50 mg mL−1 solution in dry THF) was added activated K2CO3 and FL-NHS (1.05 equivalents with respect to the polypeptide), and the reaction mixture was stirred overnight at room temperature. After the removal of the solid part by filtration, the solvent was removed from the crude reaction mixture and it was used for the deprotection of Boc and acetyl groups.Deprotection of HDA-b-poly(N,N′-di(Boc)-L-homoarginine)m-b-poly(3,4,5-tri-acetyl shikimic-L-lysine)n (HDA-b-PHAGm-b-PLSAn) (14)
Firstly, the Boc group deprotection of the polypeptides was performed with trifluoroacetic acid (TFA) and DCM (1:4). The requisite polymer was first dissolved in dry DCM (50 mg polypeptide per mL solution) followed by dropwise addition of TFA at 0 °C. The reaction mixture was stirred for 6 h at room temperature. Following the completion of the reaction, the reaction mixture was concentrated under reduced pressure and used as obtained for the next deprotection of acetyl groups. The Boc deprotected polypeptide was first dissolved in dry MeOH (50 mg mL−1 solution), followed by the addition of sodium methoxide (2 M) at 0 °C, upon which the pH of the reaction becomes basic (8–9). The reaction mixture was stirred for 4 h at room temperature. Following the completion of the reaction, the reaction was quenched by adding pre-washed amberlite H+ resin until the reaction mixture achieved a neutral pH. Following that, the mixture was filtered through a Whatman filter paper to remove the resin, and the filtrate was concentrated under reduced pressure. Following the final deprotection step, the solid residues were precipitated thrice with MeOH/diethyl ether to remove small molecule organic impurities. The solid residue was dissolved in deionized (DI) water and transferred to dialysis tubing (3.5 kDa molecular weight cutoff). The samples were dialyzed against DI water for 3 days, changing the water once every 2 h for the first day and then thrice daily to remove the impurities. Dialyzed polymers were lyophilized to yield respective block polypeptides as white fluffy solids.
The unlabelled polypeptides have been used for cytotoxicity assays, gene transfection, and quantification of gene transfection from FACS studies.
The fluorescently labeled polypeptides have been used for cellular internalization and quantification of internalization from FACS studies.
Formulation of polyplexes
All solutions were prepared by diluting components in nuclease-free water unless otherwise specified. Plasmid DNA solution was diluted to achieve a 100 ng μL−1 concentration. A polypeptide stock solution (1 mg mL−1) was obtained by dissolving lyophilized polymer powder into nuclease-free water. This polypeptide stock solution was then diluted to a concentration of 0.1 mg mL−1 or 0.5 mg mL−1 for polyplex formation. For the electrophoretic mobility shift assay (EMSA), the amount of DNA was kept constant at 200 ng (0.606 nmol of phosphates), and the amount of the polypeptide was varied to screen for the most efficient polyplex. Initially, 2 μL of pDNA was mixed with the requisite amount of polypeptide solution, vortexed for 30 s, and left to incubate for 45 min at room temperature on a dancing shaker prior to use. In other studies, the plasmid and polypeptide were mixed to achieve a particular N/P ratio and incubated to formulate the polyplex. This period of incubation will be hereafter referred to as the formulation time. The formulation time for each polyplex was kept constant at 45 min for all characterization and in vitro experiments.In vitro cytotoxicity assay
HEK 293T, MDA-MB-231, and RAW 264.7 cells were seeded in a flat-bottomed 96-well plate at a density of 5 × 103 cells per well in 1× DMEM containing 10% FBS. The plate was incubated at 37 °C with 5% CO2 for 18–24 h. Control (HDA-PHAGm) and di-block copolypeptides (HDA-b-PHAGm-b-PLSAn) were prepared and added to make a final concentration of 12.5, 25, 50, and 100 μg mL−1, respectively, in DMEM. Cells were incubated for 48 h at 37 °C with 5% CO2. After that, 12 μL of the solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (5 mg mL−1) was added to each well and further incubated at 37 °C with 5% CO2 for 3 h. At the end of incubation, the medium was aspirated from the wells, and 100 μL of DMSO:methanol (1:1 ratio) was added to dissolve the insoluble formazan crystals that formed. Cells were shaken rapidly on a microtiter plate shaker for ∼15 min until the dye was fully extracted from the cells and a homogeneous solution was formed. The absorbance was measured at 570 nm using a microtiter plate reader. The relative % cell viability was calculated using the equation: relative cell viability = (Atest/Acontrol), where Atest is the absorbance of the sample-treated cells and Acontrol is the absorbance of the untreated cells. Each absorbance was taken to be the mean of triplicate measurements (mean ± SD). The cell viability is represented as a percentage relative to untreated cells as a control.
Cellular internalization study using confocal microscopy
HEK 293T, MDA-MB-231, and RAW 264.7 cells were seeded in a 12-well plate at a density of 5 × 104 cells per well in DMEM containing 10% FBS. The plate was incubated at 37 °C with 5% CO2 for 18–24 h. After that, the incubation medium was replaced with the polypeptide or polyplex and incubated for 4 h at 37 °C with 5% CO2. Lysosomes were stained with 50 nM LysoTracker Red-DND (50 nM) in DMEM, and cells were incubated for 30 min at 37 °C with 5% CO2. After 30 min of incubation, cells were incubated with Hoechst 33342 (80 μM) for 10 minutes to stain the nuclei. Images were acquired using a Nikon, Japan confocal microscope by using a dry immersion 20× objective and were analyzed using the NIS Elements analysis (version 4.50) software.Cellular transfection study of polyplexes using confocal microscopy
For this study, HEK 293T, MDA-MB-231, and Raw 264.7 cells were plated at a density of 5 × 104 cells in each well of a 12-well plate in 1× DMEM complete medium containing 10% FBS. Cells were then incubated for 18–24 hours before the experiment at 37 °C with 5% CO2. After that, the polyplex was prepared in different charge ratios by mixing different amounts of the polypeptides with a constant amount of eGFP plasmid (1.5 μg plasmid DNA). Naked plasmid and Lipofectamine 2000 (commercial transfecting agent) were kept as negative and positive controls, along with an untreated group. The polyplex was prepared in serum-free medium (SFM) and added to the cells. After 4 hours, complete medium was added to each well with polyplex in SFM and incubated for 24 hours. After 24 hours, this medium was replaced with fresh, complete DMEM and incubated for 48 hours. In another experiment, to verify the transfection efficiency in the presence of DMEM, the formulated polyplex was added to the cells by diluting with complete DMEM, which was subsequently replaced with fresh medium after 24 hours and incubated for a further 48 hours. After these specific hours, the treatment medium of each well was changed, washed with cold PBS, and observed under a confocal microscope (Nikon, Japan) at 20× magnification. The images obtained were analyzed using NIS Elements analysis (version 4.50) software.Cytotoxicity assay after transfection of amiR-Hsp90 (artificial miRNA-plasmid against Hsp90)
For this study, HEK 293T and MDA-MB-231 cells were plated in a 96-well plate at a density of 5 × 103 cells per well in 1× DMEM containing 10% FBS. Cells were then incubated for 18–24 hours before starting the experiment at 37 °C with 5% CO2. The highest charge ratios (polypeptide:miRNA) of each di-block copolypeptide (also for the control polypeptide) were considered for polyplex preparation. The polyplex was prepared by mixing an appropriate amount of polypeptide with a constant amount of artificial miRNA-plasmid against Hsp90 (amiR-Hsp90) developed in a previous report by Banerjee and co-workers.50 Here naked polypeptide was also kept as a control along with an untreated group. First the polyplex was prepared in 50 μL serum free medium (SFM) and then incubated in a 37 °C incubator for 20 minutes. After this, the polyplex was added to the cells and incubated for 4 hours. After 4 hours, double the amount of complete DMEM (150 μL) was added to each well having the polyplex in SFM and incubated for 24 hours. After 24 hours, this medium was replaced with fresh complete DMEM and again incubated for 48 hours and 96 hours in separate plates. After specific hours of treatment, 12 μL of MTT solution (5 mg mL−1) was added to each well and further incubated for 3 hours. At the end of the incubation, the medium was discarded from each well and 100 μL of DMSO:methanol (1:1 ratio) was added to dissolve the insoluble formazan crystals that formed. Cells were shaken rapidly on a microtiter plate shaker for ∼15 min until a homogeneous solution is formed. The absorbance was measured at 570 nm using a microtiter plate reader and relative % cell viability was calculated. Each absorbance was taken to be the mean of triplicate measurements (mean ± SD). The cell viability is represented as a percentage relative to untreated cells as a control.
Results and discussion
Synthesis and characterization of the homoarginine and shikimoyl-functionalized protected di-block copolypeptide [HDA-b-PHAGm-b-PLSAn]
The ring-opening polymerization (ROP) of N-carboxy anhydrides (NCA) stands as a well-established technique for synthesizing polypeptides from amino acid-based monomers. Nevertheless, the synthesis of poly-L-arginine (PAG) and poly-L-homoarginine (PHAG) via their respective NCA poses a major challenge, impeding their integration into block copolypeptides.51 Previous endeavors by Deming and colleagues showcased the synthesis of poly-L-homoarginine through post-polymerization modification of poly-L-lysine, employing 3,5-dimethyl-1-pyrazolylformaminidium nitrate subsequent to the polymerization of transition-metal-initiated α-amino acid N-carboxyanhydride.52 Similarly, our group pursued the synthesis of poly-L-arginine via di-Cbz-protected arginine NCA polymerization,53 albeit encountering limitations such as modest yields and degradation of the polypeptide backbone under harsh deprotection conditions of the Cbz group (HBr/AcOH). In addressing these challenges, we explored an alternative approach for synthesizing homoarginine (HAG) derivatives by reacting tri-Boc-guanidine (TBG) with the ε-position of L-lysine (α-N-Cbz-α′-OBn-L-lysine-NH2)49 (synthetic methodology given in Fig. S1, ESI‡). However, the low yield and intricate nature of synthesizing α-N-Cbz-α′-OBn-L-lysine-NH2,54 coupled with the inefficiency of TBG as a guanidylating agent, prompted the development of a more straightforward route for producing the HAG-NCA monomer.In this study, we introduce a novel synthetic methodology for preparing HAG-NCA monomers, achieving higher yields, and employing simpler separation techniques (Fig. 1). By introducing the guanidine group into the 9-BBN L-lysine complex through a single-step reaction with N,N′-di(Boc)-S-methylisothiourea followed by 9-BBN deprotection, we improved the overall yield of N,N′-di-Boc-L-homoarginine while minimizing synthetic steps and purification procedures (Fig. 1). Subsequently, the obtained homoarginine monomer underwent cyclization via a previously reported technique to yield HAG-NCA monomers, which were subsequently characterized through 1H NMR, 13C NMR, and FT-IR spectroscopy (see Fig. S2 for FT-IR and see the NMR section in the ESI‡). Similar techniques were employed to synthesize the shikimic acid-functionalized L-lysine NCA (LSA-NCA) monomer.45 This systematic approach not only addresses the challenges associated with synthesizing HAG-NCA monomers but also lays the groundwork for the production of poly-glycan mimicking block-copolypeptides with improved efficiency and precision.
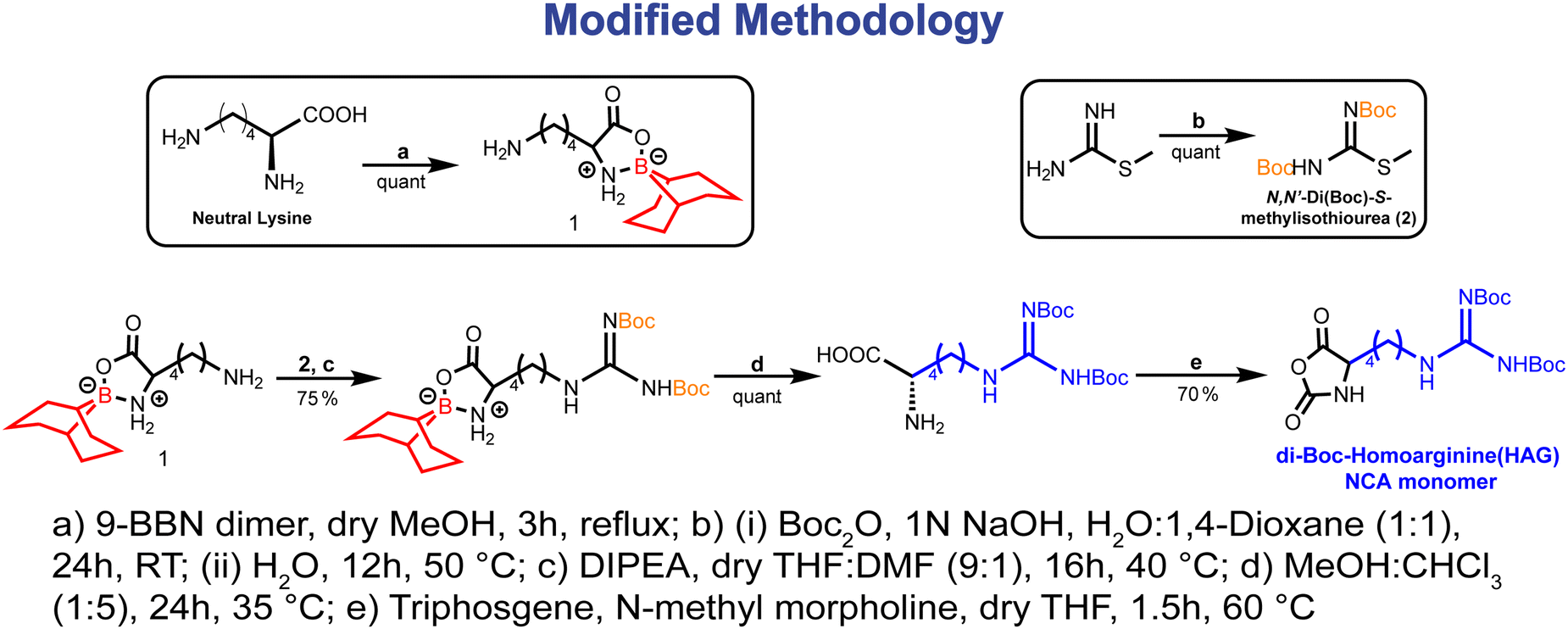 | ||
| Fig. 1 Modified methodology for the synthesis of HAG-NCA monomer. | ||
The synthesis of protected di-block copolypeptides was conducted through a process involving two sequential ring-opening polymerizations (ROPs) of N-carboxyanhydrides (NCA) with minimal purification steps (Fig. 2a). Initially, polymerization of homoarginine-NCA (HAG-NCA) was initiated by hexadecylamine (HDA) in the presence of a proton sponge in dry N,N-dimethylformamide (DMF). This choice of initiator not only facilitated a more efficient ROP but also introduced hydrophobicity into the system, potentially aiding in the self-assembly of the copolypeptides. Although the ROP of HAG-NCA in DMF exhibited sluggish kinetics, the utilization of DMF favored living polymerization of the first block, thus controlling the generation of block-copolypeptides. The addition of the proton sponge (0.25 eq.) as an additive further enhanced this process, leading to the completion of HAG-NCA polymerization (100 mg mL−1) within 48 hours.
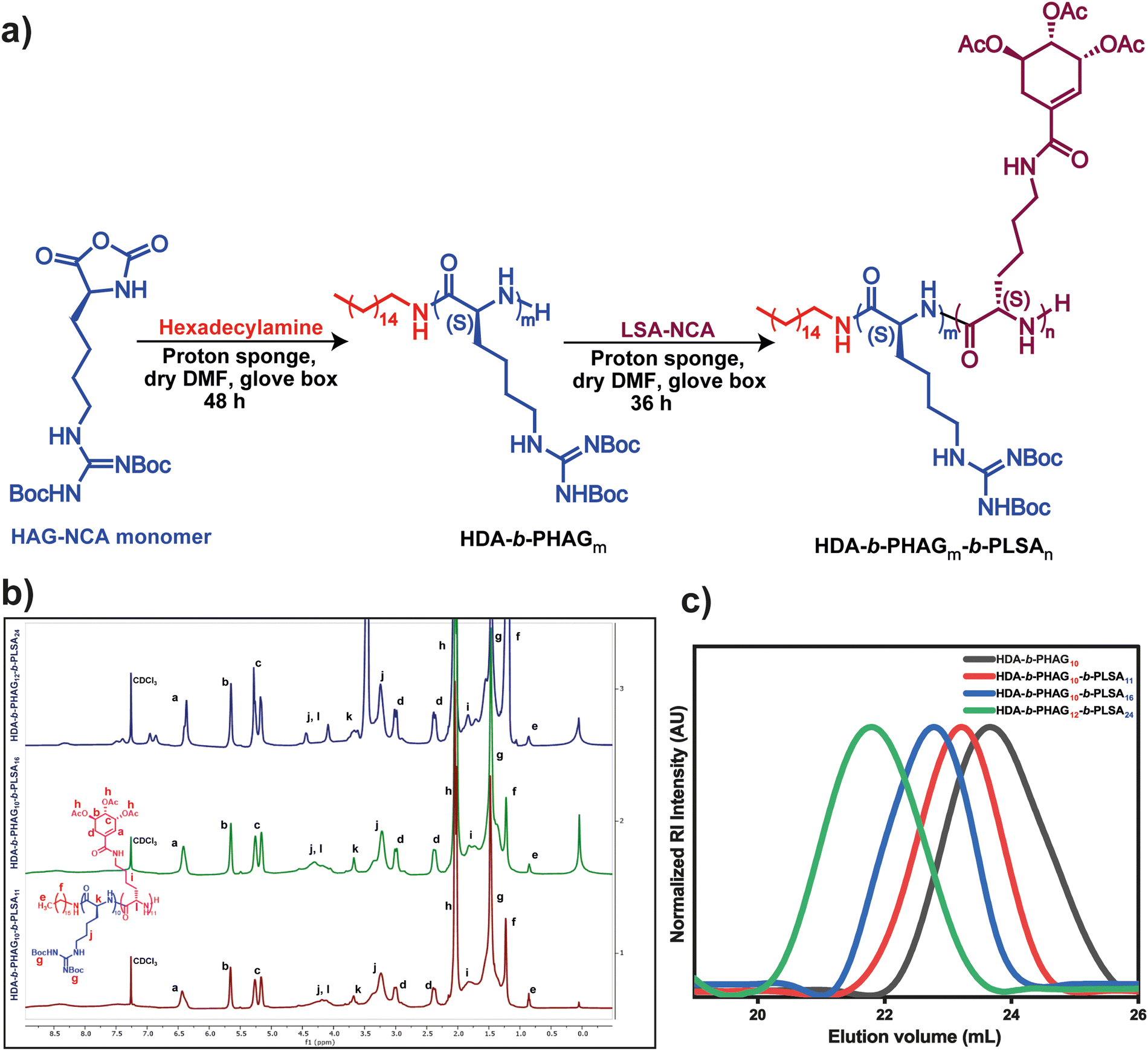 | ||
| Fig. 2 (a) Synthetic scheme of protected HDA-b-PHAGm-b-PLSAn di-block copolypeptides, (b) 1H NMR spectra of the protected HDA-b-PHAGm-b-PLSAn di-block copolypeptides, (c) SEC traces of the protected polypeptides (0.025 M LiBr in DMF as the eluent at 45 °C). | ||
The progress of polymerization was monitored via FT-IR spectroscopy, observing the disappearance of anhydride stretching bands. Upon the complete consumption of the HAG-NCA monomer, the shikimic acid-functionalized L-lysine-NCA (LSA-NCA) monomer (100 mg mL−1) in dry DMF was introduced into the macroinitiator, and the reaction mixture was stirred for an additional 36 hours to conclude the reaction. Manipulation of the monomer equivalents allowed for the generation of three distinct di-block copolypeptides with varying chain lengths, designated as HDA-b-PHAG10-b-PLSA11, HDA-b-PHAG10-b-PLSA16, and HDA-b-PHAG12-b-PLSA24. The degree of polymerization of these synthesized copolypeptides was evaluated through careful 1H NMR analysis (Fig. 2b) (see the NMR section in the ESI†) with the relative intensity of the peak at 0.86 ppm due to the characteristic proton present in the initiator hexadecylamine (alkane–CH3), with the proton peaks of the homoarginine moiety at 3.7 ppm (chiral proton peak of amino acid) and at 1.47 ppm (–Boc group), and those of the shikimoyl moiety present in the polypeptide chain at 6.4, 5.65, and 5.2 ppm. Subsequent analysis through size-exclusion chromatography (SEC) unveiled monomodal molecular weight distribution curves (Fig. 2c) exhibiting reasonably narrow dispersity (Đ = Mw/Mn) values ranging from ∼1.13 to 1.22 (Table 1), indicative of living controlled polymerization. The molecular weights deduced from SEC analysis and 1H NMR spectroscopy closely aligned with the targeted monomer-to-initiator ratios, further validating our precise control over polymer chain length during the polymerization process.
| S. no. | Polypeptide | M n (×10−3 gm mol−1) | Dispersitya (Đ) | Yieldb (%) |
|---|---|---|---|---|
| a Calculated from size exclusion chromatography in DMF containing 0.025 M LiBr as an eluent at 45 °C. b Total isolated yield of the polypeptides. | ||||
| 1 | HDA-b-PHAG10 | 3.54 | 1.21 | 80 |
| 2 | HDA-b-PHAG10-b-PLSA11 | 5.1 | 1.22 | 85 |
| 3 | HDA-b-PHAG10-b-PLSA16 | 7.69 | 1.13 | 80 |
| 4 | HDA-b-PHAG12-b-PLSA24 | 12.63 | 1.22 | 85 |
Deprotection and characterization of the di-block copolypeptide [HDA-b-PHAGm-b-PLSAn]
Obtaining a fully deprotected di-block copolypeptide involved a sequential deprotection process, starting with the removal of the Boc group from the guanidine moiety of the homoarginine (HAG) block, followed by the deprotection of the acetyl group from the shikimoyl moiety of the LSA block. Initially, the Boc group was removed from the HAG block by treatment with a trifluoroacetic acid (TFA)/dichloromethane (DCM) mixture (1:4) at room temperature for 12 hours. Subsequently, the acetyl group of the shikimoyl moiety was deprotected using sodium methoxide (NaOMe) in dry methanol (MeOH) at room temperature for 6 hours (see details in the Experimental section) (Scheme S1, ESI‡). The resulting polypeptides were purified by dialysis against deionized (DI) water and buffer, followed by freeze-drying to obtain fully deprotected polypeptides in the form of white amorphous powders for further characterization. Confirmation of Boc and acetyl group deprotection was achieved through 1H NMR spectroscopy. Notably, the absence of proton peaks at 1.47 and 2.04 ppm in the 1H NMR spectra indicated successful deprotection of the Boc group from the HAG block and the acetyl group from the LSA block, respectively (see Fig. S3 and the NMR section in the ESI‡).
Circular dichroism (CD) spectroscopy was employed to assess the secondary conformation of the di-block copolypeptides in aqueous media at pH 7.4. Previous studies indicated that poly-L-homoarginine exhibited a random coil conformation,49 while shikimoylated-polypeptides displayed an α-helical conformation.45 In the current investigation, the CD spectra revealed that the di-block copolypeptides adopted a random coil conformation, evidenced by a peak in the ∼205–210 nm range (Fig. S4, ESI‡). This observation suggests that the inclusion of the shikimoylated glycopolypeptide block did not influence the secondary conformation of the homoarginine block. The random coil conformation of the block copolypeptides is attributed to the ionic charge repulsion of the guanidine side chains.47,55
Furthermore, the di-block copolypeptides were characterized via transmission electron microscopy (TEM), dynamic light scattering (DLS), and zeta potential measurements. TEM imaging revealed a spherical morphology of the polypeptide nanoparticles exhibiting a small diameter of approximately ∼20 nm due to the presence of the hexadecyl hydrophobic block (Fig. S5(a), ESI‡). DLS analysis further corroborated this observation, indicating a narrow size distribution (Fig. S5(b), ESI‡). Additionally, zeta potential measurements demonstrated that these polypeptide nanoparticles possessed cationic character on their surface, with zeta potential values ranging from (+) 34.7 ± 6.7 mV to (+) 44 ± 7.7 mV (refer to Table S1, ESI‡).
Characterization of polyplexes
The fully deprotected di-block copolypeptides were solubilized in nuclease-free water, subsequently mixed with plasmid DNA solution, vigorously vortexed, and incubated for 45 minutes to yield polyplex formulations (Scheme 1). To understand the binding kinetics of polypeptide–pDNA complexation, the electrophoretic mobility shift assay (EMSA) (Fig. S6, ESI‡) was performed. In EMSA, agarose gel electrophoresis was conducted utilizing different charge ratios (N/P) of polyplexes, juxtaposed with plasmid DNA alone as a control. | ||
| Scheme 1 Schematic of the polyplex formulation. | ||
Herein, N/P is defined as the ratio of moles of cationic nitrogen (N) in the polypeptide system to the moles of anionic phosphate (P) in the nucleic acid material. Notably, for HDA-b-PHAG10-b-PLSA11 and HDA-b-PHAG10-b-PLSA16, complexation was initiated at N/P 4:1, with complete retardation observed at a ratio of 7:1.
This observation aligns with the anticipated behavior, given the similar zeta potential values of both di-block copolypeptides, which were approximately (+) ∼35 mV. Conversely, for HDA-b-PHAG12-b-PLSA24, which exhibited a higher positive charge ((+) 44 mV), complexation commenced at N/P 2:1, reaching full retardation at 4:1 (refer to Fig. S6, ESI‡). Following the determination of optimal N/P ratios for complexation, polyplexes were formulated at a 10:1 charge ratio, and their morphologies and sizes were evaluated through cryo-TEM, AFM, and DLS analyses. The morphologies of the polyplexes appeared distorted and spherical in dried-state TEM images, indicative of alterations from their native state (refer to Fig. S7, ESI‡). However, cryo-TEM imaging revealed well-defined structures in spherical polyplexes, a characteristic also observed in AFM imaging (refer to Fig. 3). Cryo-TEM preserves the native hydrated state of the polyplexes, resulting in larger observed sizes compared to dried-state imaging. Specifically, the polyplex sizes ranged from ∼273 to 375 nm in cryo-TEM, consistent with AFM images and hydrodynamic diameter values obtained from DLS experiments (refer to Table 2). Conversely, polyplex sizes ranged from ∼130 to 200 nm in dried-state TEM imaging (refer to Fig. S7, ESI‡), illustrating the impact of sample preparation methods on observed morphologies and sizes.
 | ||
| Fig. 3 Characterization of polyplexes using cryo-TEM and AFM imaging. | ||
| S. no. | Polyplexes | Charge ratio (N/P) | Diametera (Dh, nm) | PDIa | Diameterb (Dh, nm) | PDIb | Zeta potentialc (ζ) |
|---|---|---|---|---|---|---|---|
| a Determined by DLS measurement in water. b Determined by DLS measurement in 100 mM phosphate buffer of pH ∼ 7.4. c Determined by zeta potential measurement in water. | |||||||
| 1 | HDA-b-PHAG10-b-PLSA11 | 4 | ∼104 | 0.21 | ∼107 | 0.33 | (−) 7.60 ± 4.6 |
| 7 | ∼124 | 0.21 | ∼394 | 0.31 | (+) 8.77 ± 3.8 | ||
| 10 | ∼140 | 0.24 | ∼420 | 0.25 | (+) 17.9 ± 5.3 | ||
| 2 | HDA-b-PHAG10-b-PLSA16 | 4 | ∼145 | 0.34 | ∼164 | 0.37 | (−) 7.34 ± 6.7 |
| 7 | ∼171 | 0.22 | ∼447 | 0.24 | (+) 4.77 ± 4.0 | ||
| 10 | ∼184 | 0.23 | ∼512 | 0.30 | (+) 13.4 ± 4.3 | ||
| 3 | HDA-b-PHAG12-b-PLSA24 | 2 | ∼169 | 0.20 | ∼183 | 0.24 | (−) 4.22 ± 4.1 |
| 4 | ∼191 | 0.17 | ∼561 | 0.35 | (+) 5.50 ± 5.2 | ||
| 6 | ∼243 | 0.32 | ∼685 | 0.23 | (+) 12.0 ± 3.5 | ||
| 10 | ∼1038 | 0.57 | ∼1113 | 0.31 | (+) 14.4 ± 6.5 | ||
In vitro cytotoxicity assay
Previously, we evaluated the cytotoxicity of poly-L-homoarginine and its corresponding supra-amphiphilic nanoparticles (NPs) in vitro using an MTT assay with MDA-MB-231 cells.49 The cationic poly-L-homoarginine exhibited dose-dependent cytotoxicity against the aggressive cancerous MDA-MB-231 cells, rendering it unsuitable for gene delivery applications. Given the potential of the shikimoyl moiety to act as a sheath, akin to numerous other glycopolypeptides, we postulated that its inclusion could mitigate the cytotoxic effects observed with the cationic HAG. To illustrate this, cytotoxicity investigations were carried out in three distinct cell lines, namely, HEK 293T (a normal human embryonic kidney cell line), MDA-MB-231 (a highly aggressive breast cancer cell line), and RAW 264.7 (a macrophage cell line).This selection aimed to elucidate variations in the behavior of the di-block copolypeptides, considering the varying expression levels of the mannose receptor (CD206 or MRC1) across these cell lines (low expression in HEK 293T and high expression in MDA-MB-231 and RAW 264.7).33–37
As a comparative benchmark against the di-block copolypeptides, we further synthesized a homopolypeptide, HDA-b-PHAG10, and assessed its biocompatibility with the aforementioned cell lines. The control polypeptide HDA-b-PHAG10 exhibited some level of cytotoxicity in all tested cell lines (40–75% reduction in viable cells), as evidenced by Fig. 4A–C. Conversely, the di-block copolypeptides demonstrated no cytotoxic effects in any of the tested cell lines even upon treatment with concentrations up to ∼100 μg mL−1 of the polypeptides, as depicted in Fig. 4D–F. More than 80% of cells were viable for treating di-block copolypeptides in the case of all the cell lines. Thus, the incorporation of the shikimoyl-functionalized glycopolypeptide block alongside the cationic homoarginine block effectively alleviated this cytotoxicity concern. Consequently, the integration of the glycopolypeptide block rendered these polypeptides promising candidates for gene delivery applications, necessitating further comprehensive investigation.
 | ||
| Fig. 4 Cytotoxicity studies using the MTT assay for the treatment of HDA-b-PHAGm control polypeptides in HEK 293T (A), MDA-MB-231 (B), and RAW 264.7 (C) cells and HDA-b-PHAGm-b-PLSAn di-block copolypeptides in HEK 293T (D), MDA-MB-231 (E), and RAW 264.7 (F) cells. | ||
Cellular internalization studies
Our prior investigations into the intracellular dynamics of cationic poly-L-homoarginine have revealed that, post-uptake by cells, the polypeptide remains dispersed throughout the cytoplasm, a behavior consistent with other cationic polypeptides.49 In contrast, the shikimoyl-functionalized glycopolypeptide demonstrated a distinctive pattern of nuclear-specific internalization, evading endolysosomal entrapment, unlike other sugar moieties such as galactose and mannose.45 Motivated by the prospect of delivering genetic materials precisely into the nucleus, we engineered di-block copolypeptides comprising cationic homoarginine and neutral shikimoyl sugar moieties.To probe the cellular uptake of these constructs, uptake studies were conducted utilizing fluorescein-tagged di-block copolypeptides (Fl_HDA-b-PHAGm-b-PLSAn) across three cell lines over a 4-hour incubation period, with a subsequent examination of their intracellular distribution using confocal microscopy (Fig. 5). Imaging revealed a predominantly cytoplasmic distribution of the di-block copolypeptides post-internalization, albeit with sporadic instances of colocalization with the nucleus (albeit nonspecific) (Fig. S8–S10, ESI‡). Notably, the shikimoyl-functionalized homo-polypeptides exhibited a distinct trajectory, initially evading the endolysosomal pathways before translocating into the nucleus, likely facilitated by interaction with importin-α/β1 receptors on the nuclear pore complex (NPC).45 The concept of the virtual gate model of nuclear transport suggests that molecules of larger size must pay a greater entropic cost to traverse the nuclear pore.56 Although the augmented positive charge density of the cationic di-block copolypeptides facilitates endosomal escape, their increased size impedes nuclear entry, as evidenced by polyplex internalization (Fig. 6), predominantly observed within the cytoplasmic domain post-4-hour uptake. Notably, in the case of the polyplexes, their larger size and reduced positive charge relative to native polypeptides resulted in comparatively diminished internalization, a phenomenon corroborated by confocal microscopy images (Fig. S8–S10, ESI‡) and flow cytometry analyses (Fig. S11, ESI‡).
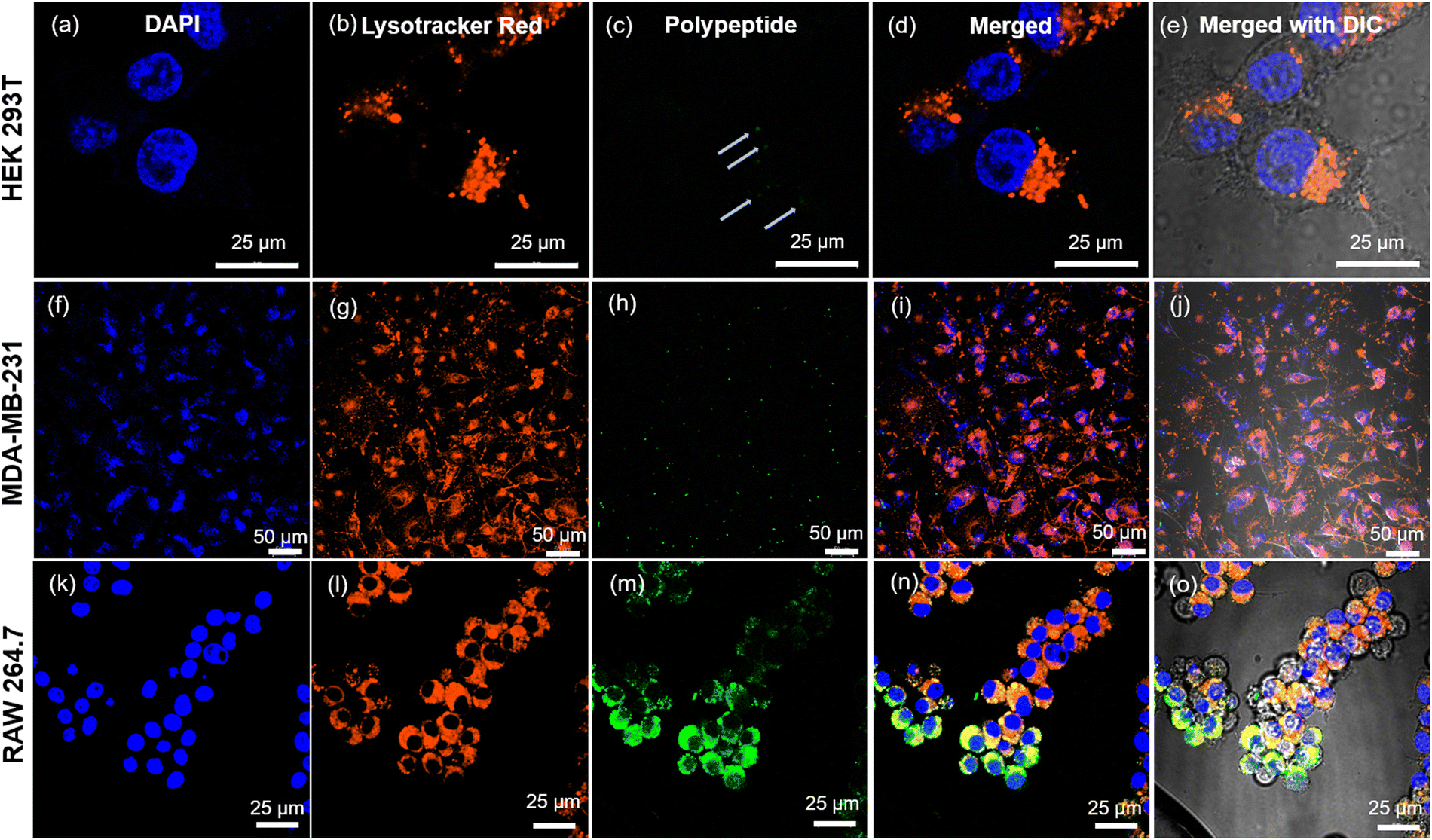 | ||
| Fig. 5 Cellular internalization studies using confocal microscopy for the treatment of Fl_HDA-b-PHAGm-b-PLSAn di-block copolypeptides on HEK 293T, MDA-MB-231, and RAW 264.7 cells for 4 h. HEK-293T, MDA-MB-231, and RAW 264.7 cells were cultured for 4 h with Fl_HDA-b-PHAGm-b-PLSAn (12 μg mL−1) in 1× DMEM and then stained with LysoTracker Red (50 nM) (for staining lysosomes) for 30 min and Hoechst 33342 (80 μM) (for staining nuclei) for 10 min. The cells were probed by confocal microscopy. HEK 293T: images (a)–(e), MDA-MB-231: (f)–(j), and RAW 264.7: (k)–(o) (where (e), (j) and (o) are merged with bright field images). | ||
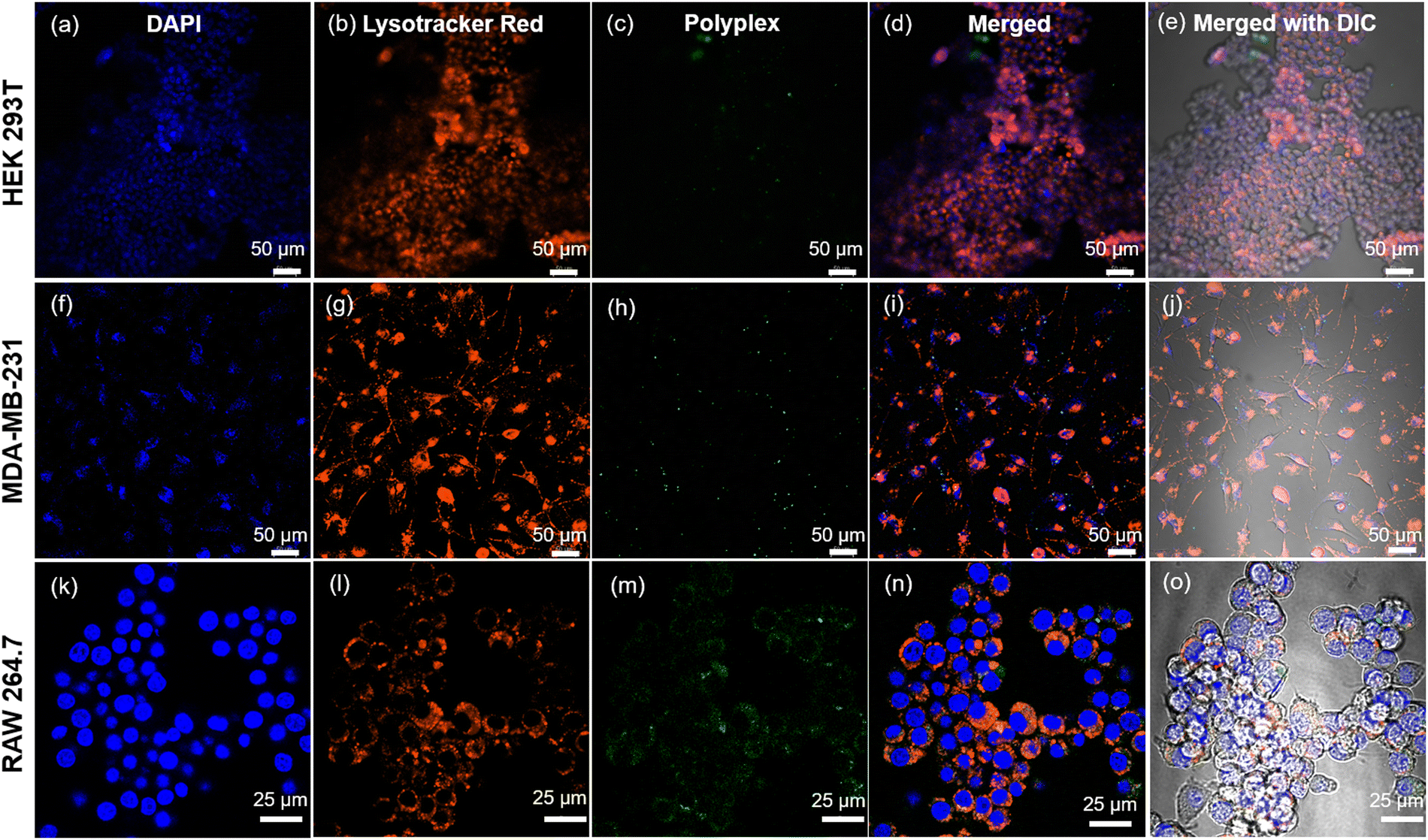 | ||
| Fig. 6 Cellular internalization studies using confocal microscopy for the treatment of polyplexes (complex of plasmid DNA and Fl_HDA-b-PHAGm-b-PLSAn di-block copolypeptide) on HEK 293T, MDA-MB-231, and RAW 264.7 cells for 4 h. HEK-293T, MDA-MB-231, and RAW 264.7 cells were cultured for 4 h with polyplexes (12 μg mL−1) in 1× DMEM and then stained with LysoTracker Red (50 nM) (for staining lysosomes) for 30 min and Hoechst 33342 (80 μM) (for staining nuclei) for 10 min. The cells were then probed by confocal microscopy: (a)–(e) HEK 293T, (f)–(j) MDA-MB-231, and (k)–(o) RAW 264.7 (where (e), (j) and (o) are merged with bright field images). | ||
Our investigations further delineated lower internalization of the control polypeptide (HDA-b-PHAG10) than di-block copolypeptides upon treating even with a higher amount (Fig. S12–S14, ESI‡). The observed varying mechanisms of internalization across different cell lines indicate that di-block copolypeptides enter cells via receptor-mediated endocytosis. In contrast, control polypeptides might enter via the cationic fusion-mediated or passive diffusion-mediated pathway. Specifically, internalization of the di-block copolypeptide was markedly lower in the HEK 293T normal cell line for both polypeptides and polyplexes, whereas significantly higher internalization was observed in MDA-MB-231 and RAW 264.7 cells, which overexpress the specific glycan receptors, for both constructs. These insights into the polypeptide and polyplex internalization underscore the potential of cationic di-block copolypeptides as carriers of genetic materials, with the inclusion of shikimoyl-functionalized glycopolypeptides conferring selectivity in genome delivery to specific cell populations.
Investigation of receptor-mediated endocytosis
Subsequently, we sought to elucidate whether the glycopolypeptide block engages with cell surface mannose receptors to facilitate entry via receptor-mediated endocytosis. To this end, cells were pre-treated with mannan, a natural ligand for mannose receptors, for 30 minutes, followed by treatment with fluorescein-tagged di-block copolypeptides (Fl_HDA-b-PHAGm-b-PLSAn) (100 μg) and a fluorescein-tagged control homopolypeptide (Fl_HDA-b-PHAGm) (100 μg). Remarkably, a discernible reduction in internalization was observed for MDA-MB-231 and RAW 264.7 cells upon mannan pre-treatment, as depicted in Fig. 7, which was also corroborated perfectly with flow cytometry analysis (Fig. S15B and C, ESI‡). Conversely, internalization levels remained unaffected in HEK 293T cells, underscoring the involvement of cell surface mannose receptors in the internalization of polypeptides and polyplexes (Fig. S15A, ESI‡). Concurrently, minimal alterations in internalization were noted for control polypeptides across all cell lines (Fig. S12–S14, ESI‡), suggesting that cationic poly-L-homoarginine likely enters cells via membrane fusion or passive diffusion-mediated uptake. This was also supported by FACS analysis (Fig. S15D–F, ESI‡). However, these findings also suggest the shikimoyl-functionalized glycopolypeptide block is not only mitigating the cytotoxicity of cationic di-block copolypeptides but also facilitating cell entry via mannose receptor-mediated endocytosis, thereby enabling the delivery of genetic material into the cytoplasm of specific target cells.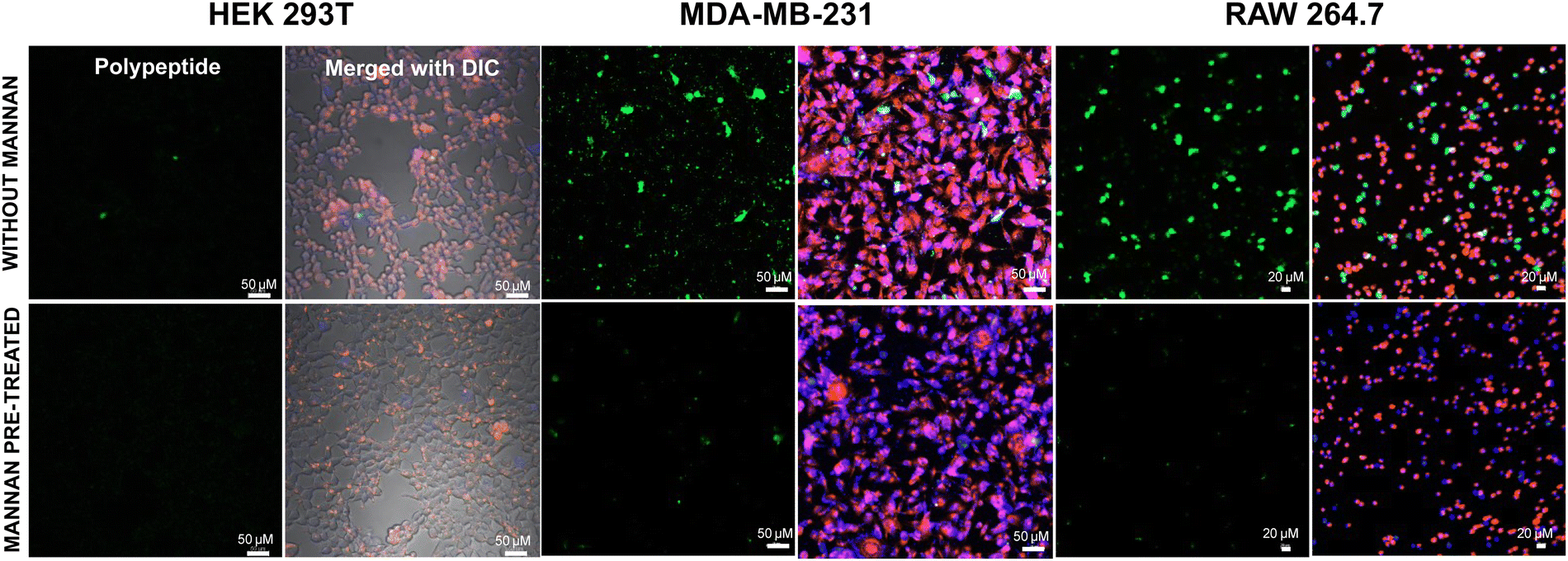 | ||
| Fig. 7 Cellular internalization studies using confocal microscopy for the treatment of the Fl_HDA-b-PHAGm-b-PLSAn di-block copolypeptide before and after mannan treatment. | ||
Transfection studies
Following the internalization studies, we proceeded to conduct gene transfection experiments across all cell lines under investigation. The polyplex (with unlabelled HDA-b-PHAGm-b-PLSAn and eGFP plasmid DNA) formulation was prepared in serum-free media (see details in the Experimental section) and subsequently administered to the cells for a 4-hour duration at 37 °C with 5% CO2. Following incubation, the cells were allowed to culture for 48 hours, after which confocal microscopy was employed for imaging. Notably, minimal transfection was observed in the noncancerous HEK 293T cells (Fig. S16, ESI‡), potentially attributed to their low internalization resulting from diminished cell surface mannose receptors. Conversely, despite the pronounced internalization observed in RAW 264.7 cells, achieving transfection in dendritic cells proved challenging, likely due to sluggish decomplexation of plasmid DNA from polyplexes, potentially leading to polyplex clearance or degradation (data not provided).In contrast, the cancerous MDA-MB-231 cells exhibited notably high transfection rates, as evidenced by confocal microscopy and flow cytometry analyses (Fig. 8 and Fig. S17, ESI‡). Intriguingly, an enhancement in transfection efficiency was observed with an increase in the degree of polymerization of the LSA block from 11 to 16 (Fig. 8G–L for HDA-b-PHAG10-b-PLSA11 and Fig. 8M–R for HDA-b-PHAG10-b-PLSA16), culminating in maximal transfection efficiency observed for HDA-b-HAG12-b-LSA24 (Fig. 8S–X) for HDA-b-PHAG12-b-PLSA24. This was also well supported by FACS analysis (Fig. S17, ESI‡), which claimed HDA-b-PHAG12-b-PLSA24 (among the three cationic di-block copolypeptides) to be the best-performing polypeptide for transfection aided by both the enhanced positive charge and repeating unit of LSA blocks. We further performed the transfection experiments in the presence of serum proteins in MDA-MB-231 cancer cell lines. Confocal microscopy and flow cytometry analysis were performed after 48 h of incubation (Fig. S19 and S20, ESI‡). In this case, we also observed a successful transfection of the eGFP plasmid. However, the effectiveness of the polypeptides in affecting transfection is reduced in the presence of serum proteins. Again, in the presence of serum, HDA-b-PHAG12-b-PLSA24 is the most effective, and HDA-b-PHAG10-b-PLSA11 is the least effective polypeptide for transfection.
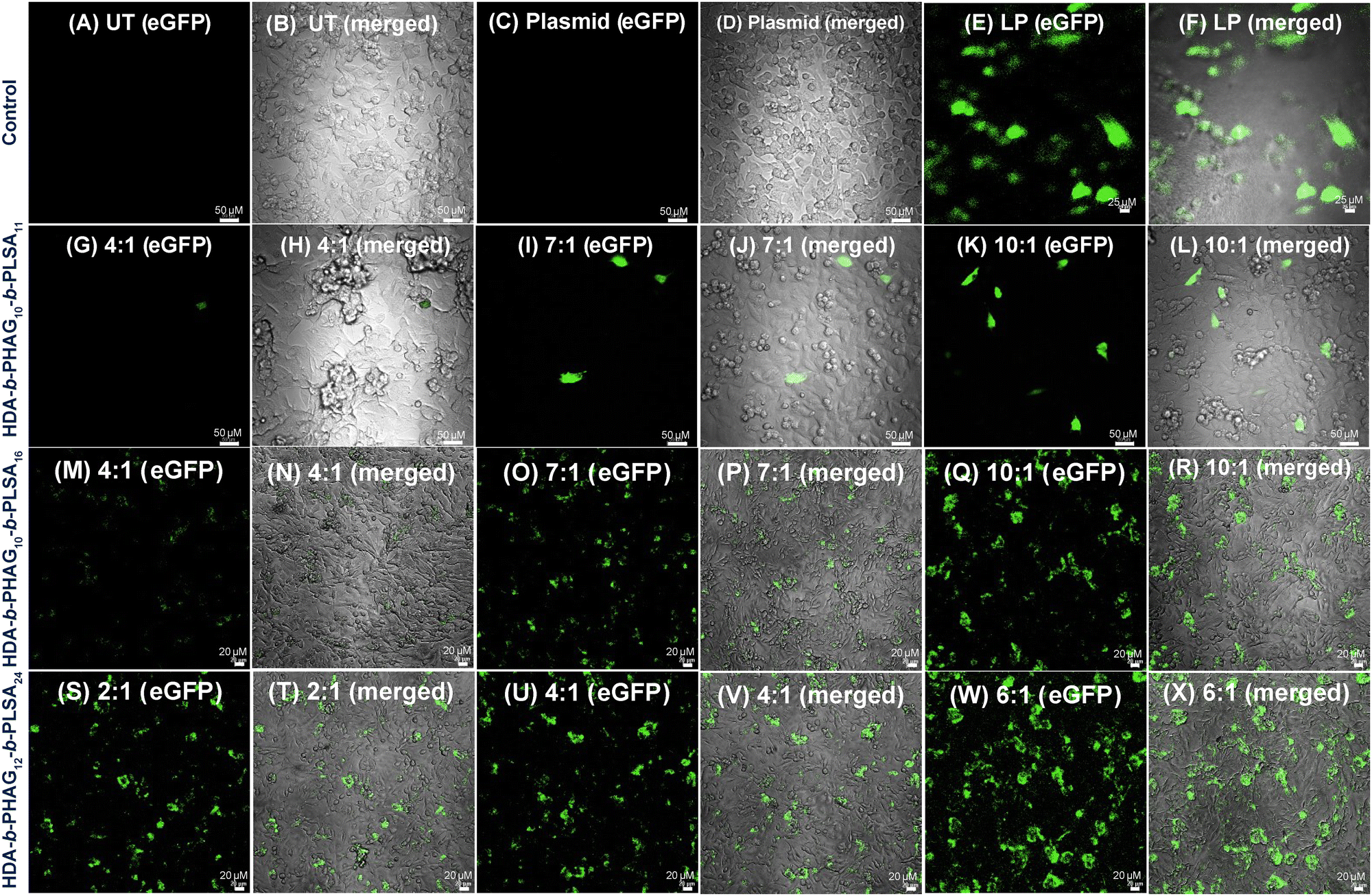 | ||
| Fig. 8 Transfection experiment (eGFP expression) in MDA-MB-231 cells for the treatment of polyplexes at different charge ratios. Treatment groups are taken as HDA-b-PHAG10-b-PLSA11 (G–L), HDA-b-PHAG10-b-PLSA16 (M–R), HDA-b-PHAG12-b-PLSA24 (S–X). Control groups are taken as untreated (A) and (B), plasmid (C) and (D), and Lipofectamine 2000 (E) and (F). | ||
These findings underscore the potential of leveraging homoarginine and shikimoyl-functionalized glycopolypeptides for achieving selective cancer cell transfection, a feat that remains elusive for commercial transfecting agents such as Lipofectamine. In contrast to Lipofectamine treatment, which yielded similar levels of transfection across HEK 293T and MDA-MB-231 cell lines (Fig. S17C and S18B, ESI†), indicating a lack of selectivity, the shikimoyl-functionalized cationic di-block copolypeptides exhibited notable selectivity in transfection, favoring the breast cancer cell line over the normal cell line.
Cytotoxicity assay after transfection of the amiR-Hsp90 plasmid
Following successful selective eGFP transfection in MDA-MB-231 cancerous cells, we investigated the transfection efficiency of these block co-polypeptides by delivering large anti-cancer plasmids. We chose to deliver a fragile and less stable RNA plasmid, amiR-Hsp90, which is responsible for depleting the level of Hsp90. Heat shock protein, often known as Hsp90, is a molecular chaperone protein that is responsible for maturing different proteins, including kinases and transcription factors, and preserving their functional integrity. In most cases, Hsp90 is overexpressed in several cancers, such as breast cancer (MDA-MB-231). Thus, the inhibition of the Hsp90 level by transfecting the anti-Hsp90-plasmid specifically in cancer cells can serve as a good chemo-therapeutic approach. In this context, Banerjee and co-workers have shown that glucocorticoid receptor (GR)-targeted cationic liposome–amiR-Hsp90 lipoplex formulation can be used to target cancer cells selectively and deplete the Hsp90 level, which ultimately resulted in the cancer cell specific cytotoxicity as well as induction of apoptosis in tumour mass and significant tumour growth inhibition.50In this study, we have prepared a polyplex of the amiR-Hsp90 plasmid with various di-block copolypeptides (HDA-b-PHAGm-b-PLSAn) and the control homopolypeptide (HDA-b-PHAGm). Both these polyplexes were added to the cancerous MDA-MB-231 and non-cancer HEK 293T cell lines. As expected, the highest transfection of amiR-Hsp90 and the subsequent cytotoxic effect was observed with HDA-b-HAG12-b-LSA24, which could be due to a higher cationic charge as well as a higher number of repeating units of the LSA block. Improved Hsp90 inhibition and significant cytotoxicity observed specifically in cancer cells are the subsequent outcomes of the successful amiR-Hsp90 transfection. In Fig. 9, it is clearly evident that the HDA-b-HAG12-b-LSA24–amiR-Hsp90 polyplex mediated cytotoxicity is cancer cell specific and no such effects were observed in non-cancer cells (until a maximum of 96 hours).
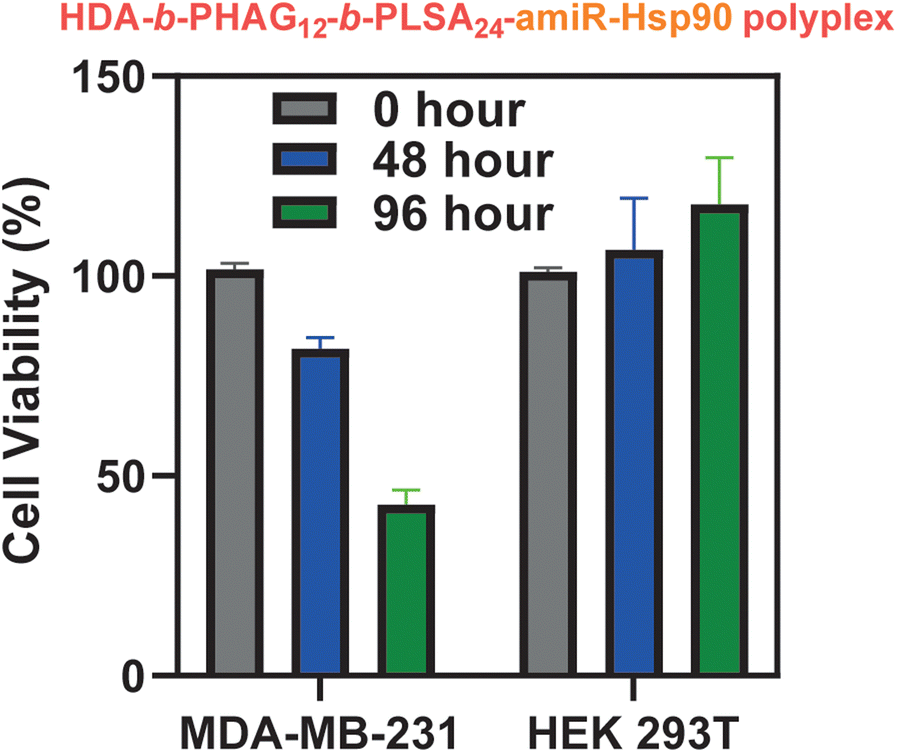 | ||
| Fig. 9 Cytotoxicity effect after transfection of the amiR-Hsp90 plasmid in MDA-MB-231 and HEK 293T cells using HDA-b-HAG12-b-LSA24. | ||
The other two di-block copolypeptides also achieved some level of toxicity in MDA-MB-231 cells, but were less effective as compared to HDA-b-HAG12-b-LSA24 (Fig. S19, ESI‡). The control homopolypeptide HDA-b-HAG10 showed some cytotoxicity for both MDA-MB-231 and HEK 293T cells, but since no specificity was observed, the effect could be attributed to the cytotoxic effect of the naked polypeptides (data not provided). All these data strongly indicated that the homoarginine and shikimoyl-functionalized glycopolypeptides along with the amiR-Hsp90 plasmid construct can be effectively used to induce selective cytotoxicity in cancer cells and hence can be used for anti-cancer therapies.
Conclusions
In summary, our study underscores the strategic design and synthesis of AB-type di-block copolypeptides comprising cationic homoarginine and shikimoyl-functionalized glycopolypeptide blocks, aimed at mitigating cytotoxicity and enhancing selectivity for gene delivery applications. Initially, we tackled the challenge of synthesizing poly-L-homoarginine, which proved arduous via conventional N-carboxyanhydride (NCA) polymerization methods. Through innovative synthetic strategies, including the use of guanidine groups and NCA modification techniques, we successfully synthesized HAG-NCA monomers with improved yields and simplified procedures. Subsequent characterization and polymerization efforts yielded well-defined di-block copolypeptides with controlled molecular weights and narrow dispersity, indicative of controlled polymerization processes. Our comprehensive characterization efforts, spanning spectroscopic analysis, size-exclusion chromatography (SEC), and morphological studies, confirmed the successful synthesis and characterization of the di-block copolypeptides. These analyses provided insights into their structural properties, including morphology, size, and dispersity, essential for understanding their behavior in biological systems.Further investigations into the cellular uptake and internalization mechanisms of the di-block copolypeptides revealed their potential for targeted gene delivery applications. Confocal microscopy studies elucidated the preferential localization of these copolypeptides within the cytoplasm, facilitated by the endosomal escape aided by the cationic nature of the di-block copolypeptide. Importantly, our findings demonstrated enhanced internalization and transfection efficiency in cancerous MDA-MB-231 cells, highlighting the selectivity and potential therapeutic utility of these copolypeptides for cancer gene therapy. Crucially, our studies also revealed the role of mannose receptors in mediating the cellular internalization of the di-block copolypeptides, underscoring the significance of receptor-mediated endocytosis in their uptake mechanism. Pre-treatment with mannan, a natural ligand for mannose receptors, attenuated the internalization of the copolypeptides in cancerous and macrophage cell lines, further supporting the involvement of mannose receptor-mediated pathways. Overall, our findings provide valuable insights into the design and synthesis of biocompatible and selective gene delivery vectors, offering potential applications in targeted cancer therapy. The successful development and characterization of these di-block copolypeptides underscore their promise as alternatives to conventional transfecting agents, with enhanced selectivity and reduced cytotoxicity. Future studies will focus on optimizing the design and formulation of these copolypeptides for improved gene delivery efficiency and therapeutic efficacy in preclinical cancer models.
Author contributions
SSG and AP designed the work. SSG and RKB supervised the work. AP and PD contributed equally to the work. AP synthesized and characterized all the monomers and polypeptides and conducted all the other experiments. AP and NM initiated the biological studies. PD performed all the in vitro experiments. MA performed the SEC analysis. SP and SD helped in monomer synthesis. The manuscript was written by AP and edited by PD, SP, SSG, and RKB. All the authors have given approval for the final version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Conflicts of interest
The authors declare no competing financial conflicts.Acknowledgements
AP acknowledges the Prime Minister's Research Fellows (PMRF) scheme for fellowship. PD, NM, and SD acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, for fellowship. MA acknowledges DST, India, and IACS for fellowship. SP acknowledges KVPY for fellowship. AP acknowledges Mr Devyan Das and Dr Sonia Agrawal for helping with eGFP plasmid DNA isolation. AP and SP acknowledge Dr Basudeb Mondal for his valuable insights during the initiation of the project. AP and SSG acknowledge Prof. Tarun Kumar Mandal, IACS, Kolkata, for providing a GPC facility. Some images have been made using BioRender. SSG acknowledges the DST-Nano mission (grant no. DST/NM/NB/2018/16). We acknowledge IISER Kolkata and IICT Hyderabad for the research environment and facilities. IICT assigned manuscript no. IICT/Pubs./2024/170.Notes and references
- J. C. M. van der Loo and J. F. Wright, Hum. Mol. Genet., 2016, 25, R42–R52 CrossRef CAS.
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor, K. Chawla, S. Chattopadhyay, D. Chandra, N. Chilukuri and V. Betapudi, Front. Oncol., 2019, 9, 297 CrossRef.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS PubMed.
- L. Ke, P. Cai, Y.-L. Wu and X. Chen, Adv. Ther., 2020, 3, 1900213 CrossRef CAS.
- C. Van Bruggen, D. Punihaole, A. R. Keith, A. J. Schmitz, J. Tolar, R. R. Frontiera and T. M. Reineke, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32919–32928 CrossRef CAS.
- Y. Zhang, A. Satterlee and L. Huang, Mol. Ther., 2012, 20, 1298–1304 CrossRef CAS.
- C. Pichon, L. Billiet and P. Midoux, Curr. Opin. Biotechnol, 2010, 21, 640–645 CrossRef CAS PubMed.
- U. R. Dahiya, S. Mishra, S. Chattopadhyay, A. Kumari, A. Gangal and M. Ganguli, ACS Omega, 2019, 4, 20547–20557 CrossRef CAS.
- S. K. Lai, D. E. O'Hanlon, S. Harrold, S. T. Man, Y.-Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1482–1487 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- N. Oh and J.-H. Park, ACS Nano, 2014, 8, 6232–6241 CrossRef CAS PubMed.
- L. K. Medina-Kauwe, J. Xie and S. Hamm-Alvarez, Gene Ther., 2005, 12, 1734–1751 CrossRef CAS PubMed.
- A. K. Ikramy, K. Kentaro, A. Hidetaka and H. Hideyoshi, Pharmacol. Rev., 2006, 58, 32 CrossRef PubMed.
- P. Midoux, G. Breuzard, P. J. Gomez and C. Pichon, Curr. Gene Ther., 2008, 8, 335–352 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- F. E. Farber, J. L. Melnick and J. S. Butel, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1975, 390, 298–311 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- Z. Kang, Q. Meng and K. Liu, J. Mater. Chem. B, 2019, 7, 1824–1841 RSC.
- C. K. Chen, P. K. Huang, W. C. Law, C. H. Chu, N. T. Chen and L. W. Lo, Int. J. Nanomed., 2020, 15, 2131–2150 CrossRef CAS.
- T. J. Thomas, H. A. Tajmir-Riahi and C. K. S. Pillai, Molecules, 2019, 24, 3744 CrossRef CAS PubMed.
- C. Van Bruggen, J. K. Hexum, Z. Tan, R. J. Dalal and T. M. Reineke, Acc. Chem. Res., 2019, 52, 1347–1358 CrossRef CAS.
- Y. P. Borguet, S. Khan, A. Noel, S. P. Gunsten, S. L. Brody, M. Elsabahy and K. L. Wooley, Biomacromolecules, 2018, 19, 1212–1222 CrossRef CAS PubMed.
- A. Sizovs, L. Xue, Z. P. Tolstyka, N. P. Ingle, Y. Wu, M. Cortez and T. M. Reineke, J. Am. Chem. Soc., 2013, 135, 15417–15424 CrossRef CAS PubMed.
- Y. Gao, X. Luan, J. Melamed and I. Brockhausen, Cells, 2021, 10, 1252–1282 CrossRef CAS.
- L. Wells, K. Vosseller and G. W. Hart, Science, 2001, 291, 2376–2378 CrossRef CAS PubMed.
- H. Yamaguchi, Trends Glycosci. Glycotechnol., 2002, 14, 139–151 CrossRef.
- C. Bonduelle, J. Huang, T. Mena-Barragán, C. Ortiz Mellet, C. Decroocq, E. Etamé, A. Heise, P. Compain and S. Lecommandoux, Chem. Commun., 2014, 50, 3350–3352 RSC.
- D. Pati, A. Y. Shaikh, S. Das, P. K. Nareddy, M. J. Swamy, S. Hotha and S. S. Gupta, Biomacromolecules, 2012, 13, 1287–1295 CrossRef CAS PubMed.
- B. Pandey, N. Patil, G. Bhosle, A. Ambade and S. Gupta, Bioconjugate Chem., 2018, 30, 633–646 CrossRef.
- B. Mondal, B. Pandey, N. Parekh, S. Panda, T. Dutta, A. Padhy and S. Sen Gupta, Biomater. Sci., 2020, 8, 6322–6336 RSC.
- B. Mondal, A. Padhy, S. Maji, A. Gupta and S. Sen Gupta, Biomater. Sci., 2023, 11, 1810–1827 RSC.
- Z. Ye, Q. Zhang, S. Wang, P. Bharate, S. Varela-Aramburu, M. Lu, P. H. Seeberger and J. Yin, Chem. – Eur. J., 2016, 22, 15216–15221 CrossRef CAS PubMed.
- D. Brevet, M. Gary-Bobo, L. Raehm, S. Richeter, O. Hocine, K. Amro, B. Loock, P. Couleaud, C. Frochot, A. Morère, P. Maillard, M. Garcia and J.-O. Durand, Chem. Commun., 2009, 1475–1477, 10.1039/B900427K.
- Y.-H. Tang, H.-C. Lin, C.-L. Lai, P.-Y. Chen and C.-H. Lai, Biosens. Bioelectron., 2018, 116, 100–107 CrossRef CAS.
- H.-Y. Li, H.-C. Lin, B.-J. Huang, A. Z. Kai Lo, S. Saidin and C.-H. Lai, Langmuir, 2020, 36, 11374–11382 CrossRef CAS PubMed.
- A. K. Azad, M. V. Rajaram and L. S. Schlesinger, J. Cytol. Mol. Biol., 2014, 1(1), 5 Search PubMed.
- C. Grandjean, G. Angyalosi, E. Loing, E. Adriaenssens, O. Melnyk, V. Pancré, C. Auriault and H. Gras-Masse, ChemBioChem, 2001, 2, 747–757 CrossRef CAS.
- P. Chenevier, C. Grandjean, E. Loing, F. Malingue, G. Angyalosi, H. Gras-Masse, D. Roux, O. Melnyk and L. Bourel-Bonnet, Chem. Commun., 2002, 2446–2447 RSC.
- R. Srinivas, P. P. Karmali, D. Pramanik, A. Garu, Y. Venkata Mahidhar, B. K. Majeti, S. Ramakrishna, G. Srinivas and A. Chaudhuri, J. Med. Chem., 2010, 53, 1387–1391 CrossRef CAS PubMed.
- R. Srinivas, A. Garu, G. Moku, S. B. Agawane and A. Chaudhuri, Biomaterials, 2012, 33, 6220–6229 CrossRef CAS PubMed.
- A. Garu, G. Moku, S. K. Gulla and A. Chaudhuri, Mol. Ther., 2016, 24, 385–397 CrossRef CAS PubMed.
- C. Voshavar, R. C. R. Meka, S. Samanta, S. Marepally and A. Chaudhuri, J. Med. Chem., 2017, 60, 1605–1610 CrossRef CAS PubMed.
- R. R. Meka, S. Mukherjee, C. R. Patra and A. Chaudhuri, Nanoscale, 2019, 11, 7931–7943 RSC.
- B. Mondal, N. S. Mahadik, R. Banerjee and S. Sen Gupta, ACS Macro Lett., 2022, 11, 289–295 CrossRef CAS.
- T. Okuda, A. Sugiyama, T. Niidome and H. Aoyagi, Biomaterials, 2004, 25, 537–544 CrossRef CAS PubMed.
- H. Tang, L. Yin, K. H. Kim and J. Cheng, Chem. Sci., 2013, 4, 3839–3844 RSC.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS.
- K. Praveen, S. Das, V. Dhaware, B. Pandey, B. Mondal and S. S. Gupta, ACS Appl. Bio Mater., 2019, 2, 4162–4172 CrossRef CAS.
- S. K. Pore, A. Choudhary, B. Rathore, A. Ganguly, P. Sujitha, C. Ganesh Kumar, S. B. Agawane, J. M. Kumar, V. Scaria, B. Pillai and R. Banerjee, Biomaterials, 2013, 34, 6804–6817 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS PubMed.
- M. Kar, N. Tiwari, M. Tiwari, M. Lahiri and S. S. Gupta, Part. Part. Syst. Charact., 2013, 30, 166–179 CrossRef CAS.
- S. Wiejak, E. Masiukiewicz and B. Rzeszotarska, Chem. Pharm. Bull., 1999, 47, 1489–1490 CrossRef CAS.
- R. Zhang, N. Zheng, Z. Song, L. Yin and J. Cheng, Biomaterials, 2014, 35, 3443–3454 CrossRef CAS.
- M. P. Rout, J. D. Aitchison, M. O. Magnasco and B. T. Chait, Trends Cell Biol., 2003, 13, 622–628 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01233j |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |