
Bioorthogonal chemistry-based prodrug strategies for enhanced biosafety in tumor treatments: current progress and challenges
Yongchao
Yao†
ac,
Ying
Chen†
b,
Chang
Zhou†
a,
Quanzhi
Zhang
a,
Xun
He
 a,
Kai
Dong
a,
Chengli
Yang
a,
Bingyang
Chu
a and
Zhiyong
Qian
*a
a,
Kai
Dong
a,
Chengli
Yang
a,
Bingyang
Chu
a and
Zhiyong
Qian
*a
aDepartment of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: anderson-qian@163.com
bThe State Key Laboratory of Functions and Applications of Medicinal Plants & School of Pharmaceutical Sciences, Guizhou Medical University, University Town, Guian New District, Guizhou 550025, China
cPrecision Medicine Translational Research Center (PMTRC), West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China
First published on 18th September 2024
Abstract
Cancer is a significant global health challenge, and while chemotherapy remains a widely used treatment, its non-specific toxicity and broad distribution can lead to systemic side effects and limit its effectiveness against tumors. Therefore, the development of safer chemotherapy alternatives is crucial. Prodrugs hold great promise, as they remain inactive until they reach the cancer site, where they are selectively activated by enzymes or specific factors, thereby reducing side effects and improving targeting. However, subtle differences in the microenvironments between tumors and normal tissue may still result in unintended cytotoxicity. Bioorthogonal reactions, known for their selectivity and precision without interfering with natural biochemical processes, are gaining attention. When combined with prodrug strategies, these reactions offer the potential to create highly effective chemotherapy drugs. This review examines the safety and efficacy of prodrug strategies utilizing various bioorthogonal reactions in cancer treatment.
 Yongchao Yao | Yongchao Yao completed his PhD in Chemistry in 2019 under the supervision of Prof. Shiyong Zhang at Sichuan University. In 2020, he joined the laboratory of Prof. Zhiyong Qian for postdoctoral research at the same University. Dr Yao has published over 20 papers on international journals including Nat. Synth., Chem, Adv. Mater., Angew. Chem. Int. Ed., Chem. Sci., Chem. Mater., Small, J. Mater. Chem. A/B, Chem. Commun., Acta Biomater., Chem. Eng. J., J. Controlled Release, Asian J. Pharm. Sci., J. Energy Chem. and so on. Now his research activity primarily focuses on the rational design of functional nanostructures toward applications for medical devices, micro/nano biosensors, biochips, and electrochemical high-throughput drug synthesis etc. |
 Ying Chen | Ying Chen obtained her PhD degree from Sichuan University in China in 2020. She then joined the School of Pharmaceutical Sciences in Guizhou Medical University as an associate professor. Her current research mainly focuses on the development of novel drug delivery systems for cancer therapy through the ferroptosis or cuproptosis pathway. |
 Chang Zhou | Chang Zhou obtained her PhD degree from Sichuan University in China in 2012. He subsequently completed standardized specialist training and advanced AO Trauma training and has been working in orthopedics ever since. He has participated in several research projects funded by the National Natural Science Foundation, focusing on minimally invasive fracture surgery techniques and bone defect research. He has also been invited to present at the AO Trauma Asia Pacific Conference, SICOT World Congress of Orthopaedics, and other international conferences. |
 Zhiyong Qian | Zhiyong Qian received his PhD degree in Organic Chemistry from Chengdu Institute of Organic Chemistry, Graduate School of Chinese Academy of Sciences. After finishing his postdoctoral training on nano-biomaterials at Sichuan University, he joined the State Key Laboratory of Biotherapy at Sichuan University as a full professor. His major research interest is on biomaterials for cancer therapy and tissue engineering. Professor Qian has published over 100 papers in international journals including Science Advances, Advanced Materials, ACS Nano, Nano Today, Advanced Functional Materials, and so on. Meanwhile, he has developed “Injectable Docetaxel Polymer Micelles” and “Injectable Paclitaxel Polymer Micelles” approved with CFDA and transferred NDA to pharmaceutical industry for clinical trials. He has served as an Associate Editor of Signal Transduction and Targeted Therapy, Chinese Chemical Letters and Journal of Biological Engineering, and Editorial Board Member of several International Journals. |
1. Introduction
Cancer continues to be a leading cause of mortality worldwide, despite significant improvements in patient survival rates.1 To further enhance and refine existing therapies, cancer treatment strategies are continually evolving through basic and clinical research. Among these strategies, chemotherapy remains the most traditional and widely used approach for cancer treatment.2–4 However, the clinical application of chemotherapy is hindered by severe side effects, primarily resulting from the non-targeted delivery and inherent toxicity of conventional drugs. These limitations often lead to treatment failure and impede further advancements in chemotherapy.5–7 Therefore, innovative treatments are needed to improve therapeutic efficacy in both basic research and clinical applications.8–10 Prodrug strategies have gained prominence in anticancer therapy for their ability to minimize toxicity during conversion and achieve site-specific activation or release in response to stimuli within the tumor microenvironment,11,12 such as tumor-specific enzymes, disulfide levels, reactive oxygen species (ROS), pH, or exogenous stimuli.13,14 Similar to traditional chemotherapy drugs, prodrugs mainly come in two types of dosage forms:15 (1) prodrugs based on small molecules, including gemcitabine,16,17 doxorubicin,18 cisplatin,19 paclitaxel,20 cabazitaxel,21 and chlorambucil;22 (2) prodrugs based on nanocarriers,23 such as micelles,24–29 liposomes,30–33 nanogels,34 polymers,35,36 dendrimers37,38 and silica nanocapsules,39 which improve tumor accumulation through the enhanced permeability and retention effect.40Despite significant progress, traditional prodrug (TP) strategies in cancer treatment still face limitations that hinder their application, including low tumor-specific accumulation and significant interference in prodrug activation due to subtle differences between the microenvironments of tumors and normal tissues.41–45 Based on click chemistry, bonding and cleavage chemical reactions were developed for use under biological conditions, known as bioorthogonal chemistry. This approach is fast, precise, and causes minimal interference with biochemical reactions.46–49 Consequently, a new strategy has emerged that replaces the drug activation process of traditional methods with bioorthogonal chemistry, significantly addressing the limitations of conventional prodrugs. This new approach is referred to as bioorthogonal chemistry-based prodrugs (BCPDs). In the TP strategy, drugs reach tumor tissues either actively or passively and are then activated or released by tumor-specific stimuli, but this approach carries the risk of off-target effects or toxicity.50–52 In contrast, for BCPDs, tumor-specific stimuli are not involved in drug activation but are used to mark the tumor tissue.53,54 Briefly, targeting and activation in TP strategies occur continuously, whereas BCPDs involve a preliminary marking step followed by an uninterrupted biochemical prodrug activation process. Additionally, the TP strategy may be affected by insufficient tumor stimuli, leading to insensitivity or even off-target effects.43,55,56 In contrast, BCPDs can avoid these issues through an efficient catalytic mark process in the tumor.52,57,58 BCPDs show significant potential in overcoming these limitations by enabling precise activation and reducing tumor heterogeneity, making them more effective than traditional TP strategies41,59–62 (Table 1). BCPDs also offer excellent kinetics, biocompatibility, and interference-free advantages in native biological processes. Various bioorthogonal reaction strategies, such as copper-mediated azide–alkyne cycloaddition (CuAAC),63–72 palladium-induced depropargylation,73–81 ruthenium-induced dealkylation,82 inverse electron demand Diels–Alder (IEDDA) click reactions,83–94 and ligation reactions based on aldimine condensation,95 have been employed to develop BCPDs. Although BCPDs represent an optimized prodrug strategy with outstanding features, they are still not perfect and require further refinement. This review explores the strengths and weaknesses of BCPDs and discusses their potential in cancer treatment. The development of highly efficient and safer BCPDs that respond specifically to the tumor microenvironment holds immense promise for clinical applications. However, current BCPD strategies often encounter challenges due to the staggered administration of the prodrug and activator, which limits their clinical translation.69 To overcome these limitations and achieve concise and compatible designs in line with biological systems, there is a growing need for prodrug systems that employ traceless bioorthogonal reactions triggered by endogenous stimuli.61 Such an ideal approach would eliminate the need for separate administration of the prodrug and activator, enabling precise control over drug release within the target tissue. Additionally, it is crucial to minimize the low atom economy associated with cleavage reactions and reduce unnecessary metabolic burden in these strategies to ensure the safety and efficacy of BCPD systems.61,96
| Targeting and activation conditions | TP strategies | Ref. | BCPD strategies | Ref. |
|---|---|---|---|---|
| Tumor-specific enzymes | Single stimuli cannot achieve precise targeting. Designing multi-stimuli responsive prodrugs is quite challenging. Additionally, insufficient stimulus sources may lead to inadequate drug activation. | 97–99 | A multi-stimuli responsive mark system can be easily established, offering greater flexibility for marker design and improving mark precision. Additionally, the structural design of BCPDs does not need to consider the tumor microenvironment, and the activation of prodrugs is not affected by the tumor microenvironment. | 100–102 |
| Disulfide levels | 103–108 | 101 and 109 | ||
| ROS | 110–112 | 113 | ||
| pH | 114–116 | 117–121 | ||
| Exogenous stimuli | Designing exogenous stimuli-responsive prodrugs is challenging. | 111 and 122 | Exogenous stimuli are used for the precise delivery of markers, with virtually no restrictions on prodrugs. | 123 and 124 |
In this review, we outline the key principles for designing BCPDs, with a focus on several important considerations: (1) prioritize the use of non-toxic and degradable catalysts whenever possible. The selection of the catalyst is critical for BCPD activation, and using non-toxic, degradable options minimizes the risk of adverse side effects and ensures safe elimination from the body. (2) When the use of a metal catalyst is unavoidable, it is essential to design materials that facilitate rapid metabolism and minimize local toxicity. While metal catalysts offer unique benefits for bioorthogonal reactions, they can also pose toxicity risks. Implementing strategies to accelerate metabolism or encapsulate the catalyst within biocompatible materials can help mitigate these concerns. (3) Avoid staggered administration of the prodrug and trigger to minimize additional metabolic burden: Staggered administration of prodrugs and triggers can complicate dosing schedules and increase the metabolic burden on the body. Designing prodrug systems that allow simultaneous administration of both components improves patient compliance and reduces potential complications. (4) Incorporating traceless strategies for prodrug activation should be prioritized whenever possible: Traceless activation ensures the complete removal of activating groups or linkers after drug release, thereby minimizing side effects associated with prodrugs and enhancing treatment safety. In Table 2, we present a comprehensive summary of the prodrug activation system based on bioorthogonal reactions, highlighting their main advantages and potential limitations. Furthermore, this review provides an overview of the current landscape of BCPDs, categorized into three groups based on the underlying reaction mechanism: catalyst-mediated ligation reaction, catalyst-mediated cleavage reaction, and catalyst-free reaction (Scheme 1). It is important to note that the catalyst-free reaction category does not involve cleavage; instead, the two components of the prodrug activate each other. By emphasizing design strategies with a higher potential for clinical application, this review provides valuable insights and recommendations for developing safer BCPDs in future research.
| Catalysts or prodrug strategy | Activation method | Main advantages | Possible limitation | Ref. |
|---|---|---|---|---|
| DNAzyme encapsulated bimetallic MOFs (Cu, Zn) | CuAAC | Tumor enrichment of the catalyst enabled by DNAzymes for synergistic gene therapy in the presence of Zn | Toxicity of the metal | 68 |
| DNA-templated ultrasmall Cu nanoparticles | CuAAC | Tumor enrichment of the catalyst enabled by DNAzymes, and more effective valence conversion of the Cu catalyst induced by DNAzyme | Toxicity of the metal and potential metabolic burden | 70 |
| Ultrasound induced Cu@poly (acrylic acid) nanocomplexes | CuAAC | The valence conversion of Cu effectively controlled and enhanced through the application of ultrasound, providing a promising approach for optimizing the catalytic activity | Toxicity of the metal | 72 |
| ZIF-90@ATP aptamers | Induce endogenous Cu production for CuAAC | Prodrug activation without the exogenous Cu catalyst largely avoiding side effects of Cu | Promising | 71 |
| Gold resin | Au-induced depropargylation | Transition metal catalysts (TMCs) with higher biocompatibility | Toxicity of the metal | 125 |
| Pd resin | Pd-induced depropargylation | Reducing the toxicity by heterogeneous palladium chemistry | Toxicity of the metal | 126 |
| Nano encapsulated Pd | Pd-induced depropargylation | Combination treatment with the nanoencapsulated prodrug and palladium compound | Toxicity of the metal | 77 |
| Pd and Fe nanorobots | Magnetically driven Pd-induced depropargylation | Increasing the enrichment of Pd by magnetic induction, and precisely activated by responding to the tumor microenvironment | Toxicity of the metal | 127 |
| Exosome encapsulated Pd nanosheets | Pd-induced depropargylation | More effective tumor enrichment by cancer-derived exosomes | Toxicity of the metal | 79 |
| Ru based ZnS nanoparticles | Ru-induced deallylation | The use of ZnS nanoparticles to enhance the biocompatibility of Ru-based compounds | Toxicity of the metal | 82 |
| Nontoxic Pt complexes | Pt induced cleavage reaction of N-propargyl | Metal catalysts with high biocompatibility avoiding the inherent toxicity of the catalyst | Promising | 128 |
| Nontoxic Pt complexes | Pt-derived cleavage reactions of O/N-propargyl | Metal catalysts with high biocompatibility and effectiveness | Promising | 80 |
| EISA (enzyme-instructed supramolecular self-assembly) for mediated prodrug intracellular accumulation | IEDDA | Effective tumor enrichment of the catalyst by EISA | Metabolic burden | 86 |
| EISA strategy | IEDDA | Effective tumor enrichment of the Tz group by EISA | Metabolic burden | 129 |
| Micelles based on tetrazine | IEDDA | Effective tumor enrichment of catalyst by nano-effects | Metabolic burden | 91 |
| Fragments were designed as active drugs | IEDDA | The fragments serving as a therapeutic agent | Promising | 130 |
| Fragments were designed as active drugs | IEDDA | The fragments serving as therapeutic gas | Promising | 131 |
| ROS induced prodrug activation | Ligation reaction based on aldimine condensation | ROS triggered prodrug activation without any fragment | Promising | 95 |
| Nitroreductase induced prodrug activation | Ligation reaction based on aldimine condensation | Enzyme triggered prodrug activation without any fragment | Promising | 132 |
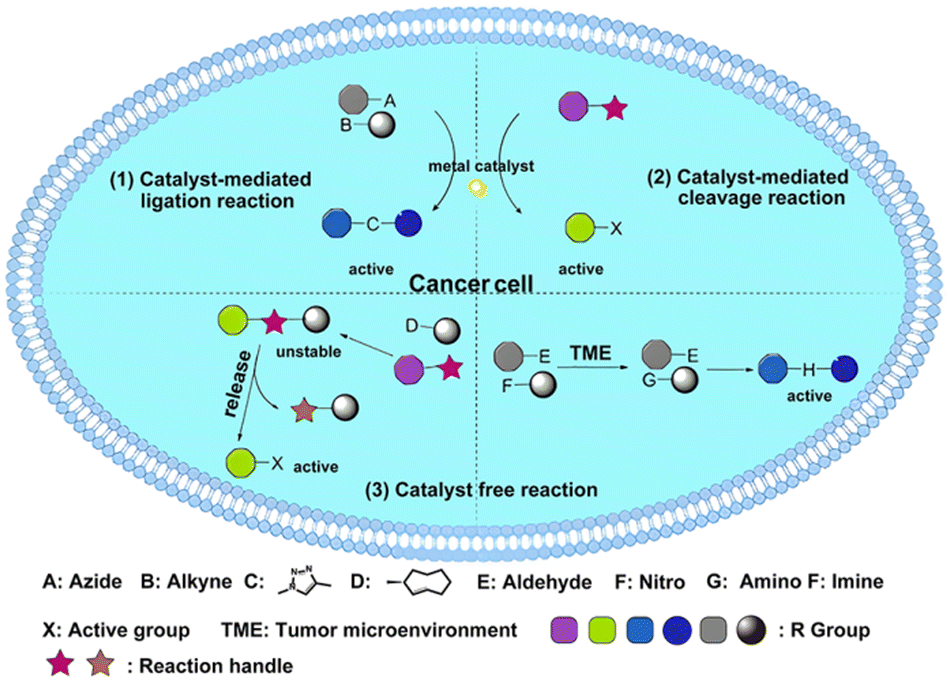 | ||
| Scheme 1 The three main types of BCPDs: (1) catalyst-mediated ligation reaction; (2) catalyst-mediated cleavage reaction; (3) catalyst-free reaction. | ||
2. Bioorthogonal chemistry for biomedical applications
Bioorthogonal chemistry has garnered significant recognition across various fields in recent decades.133,134 It includes ligation and cleavage reactions that can occur within biological environments without interfering with the natural metabolic processes of living organisms.135 Bioorthogonal reactions employ chemical groups such as azide, tetrazine, alkyne, or olefin, which are typically absent from biological systems. These specialized groups demonstrate high selectivity and efficiency in binding to their corresponding counterparts, resulting in reactions with high yields. Consequently, bioorthogonal chemistry has found extensive applications in numerous areas. For example, it allows for the precise modification of proteins, enabling researchers to introduce specific modifications or tags for purposes such as studying protein functions or tracking protein localization.136,137 In the field of cellular engineering, bioorthogonal chemistry plays a crucial role in manipulating and engineering cells to develop novel therapies, diagnostic tools, and biosensors.133 Moreover, bioorthogonal chemistry is also utilized in prodrug activation, where inactive drug compounds are designed to be selectively activated within the target site, minimizing off-target effects and optimizing therapeutic efficacy.129,138–141 Overall, bioorthogonal chemistry offers a versatile toolkit of reactions that can be employed in a wide range of biological applications. Its ability to perform specific and efficient chemical transformations within complex biological systems has opened up new possibilities for research, diagnostics, and therapeutic interventions.The chemical modification of proteins through bioorthogonal reactions has become a powerful tool, enabling diverse and complex operations, and has garnered increasing attention.142 Researchers have developed various strategies to achieve efficient and site-specific protein labeling using bioorthogonal chemistry. In one study, Peng and colleagues143 successfully performed site-specific fluorescence labeling of intracellular proteins using IEDDA cycloaddition reactions. They employed this approach to dynamically detect the behavior of interferon-inducible transmembrane protein 3 (IFITM3) in living cells, overcoming the limitations of fluorescent protein markers due to IFITM3's small size (Fig. 1A). By introducing bioorthogonal chemistry, they not only addressed the drawbacks of fluorescent protein tags but also expanded the possibilities for site-specific imaging of intracellular proteins in live cells. Shi and coworkers144 developed a fluorescent probe based on dibenzoazacyclooctyne (DBCO) for protein labeling. They achieved this through acid-promoted rearrangement and silver-catalyzed amide condensation reactions (Fig. 1B). The introduction of DBCO on the probe enabled switchable click reactivity toward amino groups on proteins in the presence of silver catalysts. Although the use of silver catalysts may limit the applicability to some extent, this design holds potential for practical protein modification and fluorescence labeling, as any protein containing amino acids can be labeled. Li and colleagues145 reported a visible light-triggered protein modification method based on phenanthrenequinone and vinyl ether bioorthogonal reactions (Fig. 1C). The reaction handle demonstrated stability in biological environments, and were inert to multiple nucleophilic species. This method enabled spatially controlled labeling of bovine serum albumin and lysozyme, as well as labeling of cell surfaces, employing a catalyst-free bioorthogonal reaction triggered by visible light. The artificial chemical reaction handles in bioorthogonal chemistry allowed for precise regioselective protein labeling. In another study by Li et al.,146 a photoredox catalysis-based protein modification method was developed (Fig. 1D). They selectively modified the target protein at tyrosine residues, as tyrosine exhibited reaction potential in photoredox chemistry and was less abundant in the protein. The modified protein was then introduced with a dialdehyde tag via photocatalysis to create a bioorthogonal reaction handle through aldimine condensation, enabling site-selective protein labeling or functionalization. These studies highlight the versatility and potential of bioorthogonal chemistry in protein modification. They offer valuable insights into developing novel strategies for site-specific labeling, expanding protein imaging capabilities, and achieving regioselective modifications.
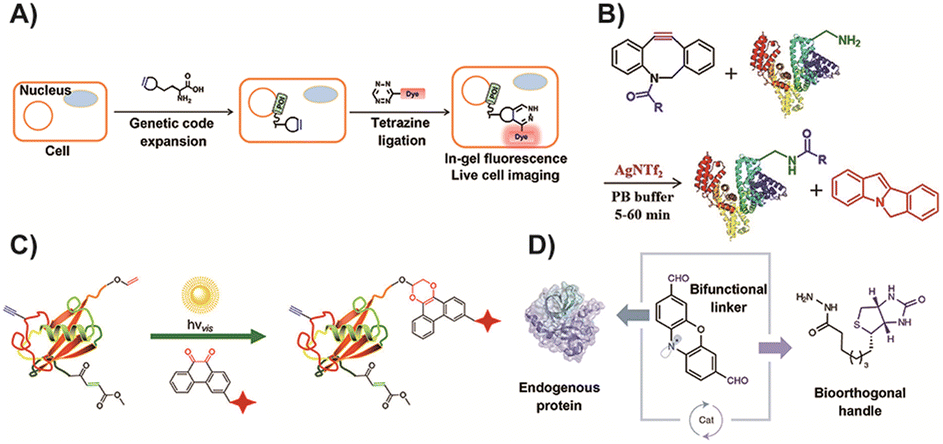 | ||
| Fig. 1 (A) Fluorescence labeling of protein by IEDDA cycloaddition reactions. Copied with permission.143 Copyright 2016, American Chemical Society; (B) protein fluorescence labeling using the silver-catalyst aldimine condensation reaction. Copied with permission144 Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (C) light triggered protein modification by the SPAAC reaction. Copied with permission145 Copyright 2018, American Chemical Society; (D) aldimine condensation based protein modification method. Copied with permission146 Copyright 2021, Springer Nature. | ||
Bioorthogonal chemistry has also demonstrated its potential in driving the transformation of chemical groups on cell membranes and within cells, enabling the detection and functionalization of cellular behavior.147 Hao and colleagues148 utilized bioorthogonal reaction handles to mark cells and induce changes in the behavior of human bone marrow-derived mesenchymal stem cells through the IEDDA cycloaddition reaction (Fig. 2A). This modular approach allows for the modulation of cell behavior, making it applicable to cell engineering and tissue modeling. In the context of cell membrane modification, Zhang and colleagues developed a cell membrane platform capable of in situ generating dendrimers using the design of IEDDA reactions (Fig. 2B).149 This strategy enables the in situ synthesis of artificial organelles within cellular systems, which can contribute to future research on the control of cellular behaviors. Furthermore, the behaviors of membrane-located proteins or biomolecules, such as voltage-gated ion channels regulated by membrane potential, can be controlled.150 Liu and coworkers reported the detection of membrane potential using a fluorescence voltage indicator based on the IEDDA reaction (Fig. 2C).151 This approach allows for voltage imaging in the far-red spectrum by binding organic fluorophores to cell membrane proteins. The introduction of voltage indicators into cell detection enhances the sensitivity of in vivo detection, enabling further study of membrane potential.
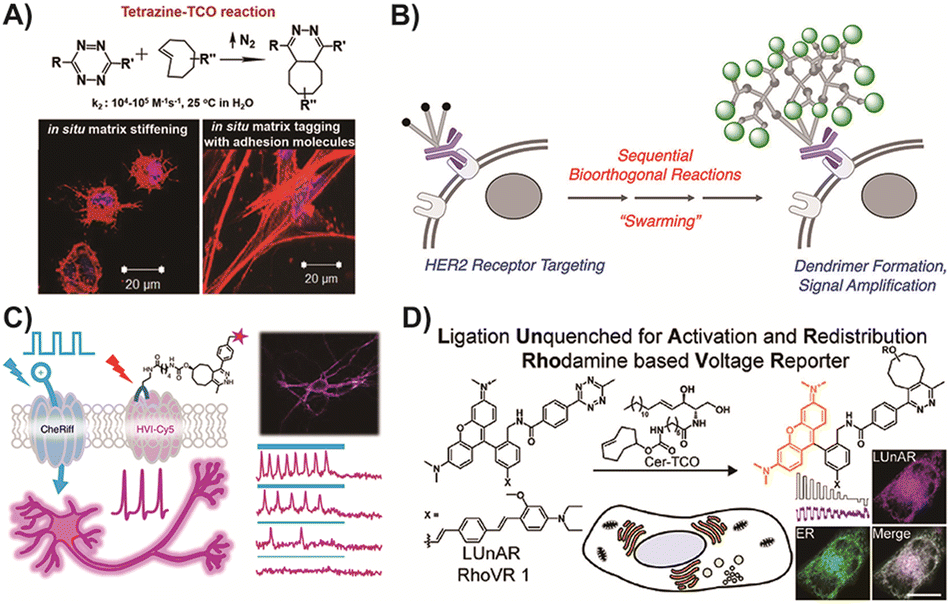 | ||
| Fig. 2 (A) The fluorescence labeling of hMSCs by the IEDDA cycloaddition reaction. Copied with permission148 Copyright 2018, American Chemical Society; (B) the artificial dendrimer modification of cellular system. Copied with permission149 Copyright 2020, American Chemical Society; (C) the first voltage indicator using the IEDDA cycloaddition reaction. Copied with permission151 Copyright 2021, Springer Nature; (D) the usage of voltage indicator in mechanism verification. Copied with permission152 Copyright 2022, American Chemical Society. | ||
Similarly, Klier and colleagues utilized the IEDDA cycloaddition reaction to target subcellular membranes of living cells, specifically detecting the endoplasmic reticulum potential in response to changes in the plasma membrane potential (Fig. 2D).153 This provided direct confirmation of the relationship between endoplasmic reticulum potential and plasma membrane potential. Beyond the applications mentioned, bioorthogonal chemistry has proven valuable in prodrug activation. It holds the potential to enhance the selectivity and targeting of TP prodrugs. In the following section, we will explore the application of bioorthogonal reactions in prodrugs for tumor treatment in greater detail.
3. Bioorthogonal chemistry-based prodrugs (BCPDs) for antitumor treatment
In recent years, in vivo prodrug activation using bioorthogonal chemistry has emerged as a promising approach for cancer therapy. This strategy offers several advantages, including high efficiency, sensitivity, and low toxicity, making it a strong candidate for clinical applications.15,154–156 However, several challenges remain to be addressed.91 The use of TMCs, which can be inherently toxic, and the metabolic burden caused by small molecular fragments after prodrug activation present significant limitations.69,95,132 Additionally, the complexity and customization required for traditional drugs create further obstacles in optimizing prodrugs through bioorthogonal reactions. Designing and synthesizing prodrugs with bioorthogonal handles necessitates careful consideration of their chemical properties, stability, and compatibility with specific bioorthogonal reactions. Moreover, developing analytical techniques and imaging methods to monitor prodrug activation in real-time is crucial for evaluating the effectiveness of BCPDs and optimizing treatment regimens. In this section, we will provide an overview of recent developments in BCPDs for cancer therapy.3.1. Catalyst-mediated ligation reaction
CuAAC is a widely used catalyst-mediated ligation reaction in bioorthogonal chemistry, known for its short activation time and high yields in physiological environments. It is facilitated by Cu(I) ions as catalysts, promoting the reaction between alkynes and nitrogen-containing compounds such as azides to form 1,2,3-triazole compounds. It is worth noting that copper (Cu) is the third most abundant transition metal in the human body, with an average content of about 100 milligrams in adults, and is crucial for a range of biological processes.157 In the spirit of avoiding the use of exogenous catalysts to reduce potential side effects and complexity, and to improve the biocompatibility and safety of biological reactions, utilizing endogenous copper as a catalyst for CuAAC may be a promising approach. Under physiological conditions, free Cu(I) and Cu(II) cations exhibit significant cytotoxicity due to the rapid production of ROS through redox cycles and a tendency to replace Zn(II) and Fe(II)/(III) cations from soft metal binding sites such as iron sulfur clusters.158 Therefore, the availability of Cu(II)/(II) cations within cells is strictly regulated by a large amount of excessive chelated thiols, such as the endogenous antioxidant GSH.159 Currently, there are few examples of using endogenous copper for CuAAC reactions. Most research focuses on how to more effectively utilize exogenous copper catalysts to achieve efficient and controlled reactions. However, with a deeper understanding of copper metabolism in tumor cells, more effective methods may be developed in the future to harness endogenous copper for bioorthogonal reactions.Despite the inherent toxicity of Cu metal limiting its clinical application, it remains the most reliable and commonly used method in basic research.160 To address this limitation, Wang and colleagues68 devised a therapeutic nanoplatform by encapsulating therapeutic DNAzyme into a Cu and Zn-based metal–organic framework (MOF) for drug activation and gene therapy (Fig. 3A). In this design, the DNAzyme's targeting ability to human early growth response-1 facilitated the enrichment of Cu(II) in cancer cells, which was subsequently reduced to Cu(I) by sodium ascorbate, a precursor. This reduction triggered the CuAAC reaction, leading to the production of the prodrug. Simultaneously, Zn(II) activated the DNAzyme for gene silencing, thereby enhancing gene therapy. The bimetallic MOF nanoparticle served not only as the delivery vehicle for DNAzyme but also as the catalyst for CuAAC prodrug activation. The results of the prodrug system demonstrated the synergistic therapy's efficacy in both avoiding the side effects of transition metals and effectively inhibiting tumor metastasis. Additionally, the nanoscale catalyst was not only easily enriched in tumor sites but also more readily metabolized compared to free Cu. Therefore, the potent tumor targeting ability of nanoparticles could mitigate the toxic and side effects associated with transition metals to a certain extent. In a similar approach, You and colleagues70 employed the DNAzyme's targeting ability to accurately deliver Cu into tumor cells, enabling the activation of a prodrug with high catalytic activity specifically within the cancer cells. Distinct from the previous study, this strategy facilitated the generation of Cu(I) for in situ prodrug activation through DNAzyme-induced valence conversion of ultrasmall Cu nanoparticles exposed to endogenous hydrogen peroxide in cancer cells. Additionally, the DNAzyme with cancer cell-targeting ability could generate cancer-killing radical species, leading to synergistic therapy (Fig. 3B). However, further research is required to determine whether the residual Cu after activation can be effectively metabolized or if it might potentially cause local toxicity in other tissues.
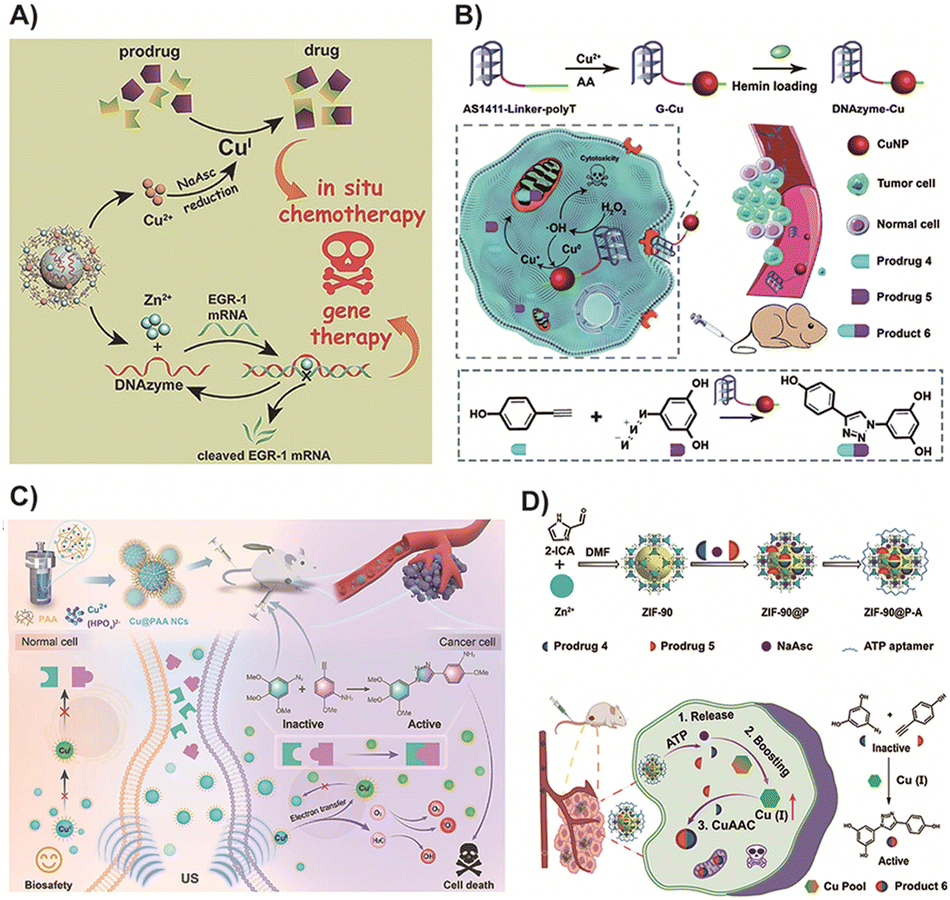 | ||
| Fig. 3 (A) DNAzyme based prodrug activation and gene therapy. Copied with permission68 Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (B) DNAzyme-augmented bioorthogonal catalytic prodrug for synergistic cancer therapy. Copied with permission70 Copyright 2022, The Royal Society of Chemistry; (C) mechanism and treatment process of poly (acrylic acid)-modified Cu nanocomplexes in cancer therapy. Copied with permission72 Copyright 2023, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (D) scheme of the endogenous enhancement and drug activation for cancer therapy. Copied with permission71 Copyright 2023, American Chemical Society. | ||
In another study, Xia and colleagues72 developed unique Cu nanocomplexes modified with poly(acrylic acid) that exhibited distinct features compared to previously reported Cu nanoparticles. This nanocomplex design allowed for reversible valence interconversion between Cu(II) and Cu(I) under exogenous ultrasound stimulation (Fig. 3C). Importantly, these Cu nanoparticles demonstrated excellent biocompatibility, as the potential toxicity of Cu(I) to normal tissues was mitigated by a non-spontaneous Fenton-like reaction, which was limited by the availability of oxygen. Furthermore, the poly(acrylic acid)-modified Cu nanocomplexes possessed dispersity and ultra-small size, enabling their rapid digestion and effective elimination from the body through the urinary system, thereby enhancing their biosafety. This approach rendered Cu a safer catalyst and provided insights for the design of ultrasound-mediated bioorthogonal chemistry prodrugs. However, the successful application of this strategy in complex biological contexts remains challenging due to safety concerns associated with the inherent toxicity of transition metals. Therefore, exploring the prodrug activation of CuAAC without the need for exogenous transition metals represents an intriguing direction for future research.
A recent strategy reported by Zhu and colleagues71 aimed to minimize the side effects of transition metals. They designed an adenosine triphosphate (ATP) aptamer-functionalized metal–organic framework (MOF) nanoparticle that could produce an endogenous catalyst for bioorthogonal reactions in situ. This approach enabled the elevation of endogenous Cu(I) levels without the need for an exogenous Cu catalyst, thereby accelerating the CuAAC reaction and facilitating the production of an effective anticancer drug at the tumor sites (Fig. 3D). Specifically, within the tumor cells, the nanocomposite responded to high concentrations of ATP, leading to the release of prodrugs and sodium ascorbate. Sodium ascorbate was used to enhance the Cu(I) species in situ by converting Cu(II) to Cu(I). This strategy allowed for prodrug activation through endogenous Cu, avoiding the side effects associated with exogenous Cu usage. Moreover, it facilitated the delivery of prodrugs in sufficient amounts and an optimized proportion to tumor tissues for on-demand release. Selecting and treating a tumor model with high endogenous Cu ion concentration will maximize the effectiveness of the aforementioned prodrug strategy. Specifically, in patients with breast cancer, lung cancer, bladder cancer, and other cancers, Cu levels in both serum and tumors are significantly elevated.161
Moreover, it is worth noting that the targeting of CuAAC-based prodrugs primarily relied on enriching the catalyst at tumor sites. However, to further enhance the enrichment of the catalyst in tumor sites, future designs should explore not only Cu nanosizing but also other extracellular stimuli methods. These methods can include the use of specific targeting ligands, responsive nanoparticles, or external triggers that can enhance the accumulation of the catalyst in tumor tissues. By improving the localization of the catalyst, prodrug activation can be more precise and efficient, leading to enhanced therapeutic outcomes. However, the toxicity or potential biological activity of various parts of the prodrug lacks clinical practice, presenting further hurdles in drug application. Its transition to clinical practice as a drug requires consideration of its economic viability, patient acceptance, and unknown biochemical activities.
3.2. Catalyst-mediated cleavage reaction
Prodrugs based on catalyst-mediated cleavage reactions often involve the incorporation of an active drug and its protective cage, which results in the production of small molecule fragments upon activation. This approach has inherent drawbacks in terms of atom economy, as it leads to the generation of unnecessary metabolic byproducts and possible risks of biotransformation of toxic metabolites. Additionally, the use of TMCs in these prodrug systems can pose localized safety hazards.61,96 Increasing the accumulation of TMCs, such as Pd, Au, or Ru species, specifically in tumor tissues has been explored. One effective approach is the utilization of microscale or nanoscale delivery systems, which can facilitate the local enrichment of the catalyst in the tumor microenvironment while minimizing its distribution in normal tissues. By achieving a higher concentration of the catalyst in tumor tissues, the potential side effects on normal tissues can be reduced. Furthermore, enhancing the metabolism of the catalysts by the liver or facilitating their elimination through the urinary system can also contribute to improving biosafety.55 Overall, strategies that focus on achieving local enrichment of TMCs in tumor tissues, along with enhanced metabolism or elimination from the body, are important for improving the safety and efficacy of catalyst-mediated prodrugs. Continued research in these areas will contribute to the development of more efficient and safer prodrug systems for clinical applications.72Pd-based nano-catalysts or micro-catalysts have been utilized in various studies. For instance, Weiss and colleagues employed micro-sized Pd0-amino resins73 for gemcitabine prodrug activation in tumor therapy (Fig. 4A).162 In this research, the gemcitabine drug was caged using an N-propargyloxycarbonyl group, which is highly susceptible to heterogeneous Pd catalysts, resulting in the formation of prodrugs. Cellular experiments demonstrated that neither the prodrug nor the catalyst exhibited cytotoxicity when cultured separately, indicating their relative safety towards normal cells. Additionally, this strategy broadened the applicability of amino-containing drugs by modulating their pharmacodynamics and pharmacokinetics. However, it is important to note that the large size of the Pd0-amino resins used in this study poses limitations in terms of body fluid transport capacity and tumor enrichment. In order to enhance the effectiveness of the prodrug delivery system, further improvements are required. Strategies such as optimizing the size and surface properties of the catalyst particles or incorporating them into nanoscale carriers can enhance their transportability and tumor accumulation capacity, leading to improved therapeutic outcomes. Recently, Zhang entrapped Pd catalyst into Au nanostructures named nanozymes and injected in situ into tumor tissue for prodrug activation, avoiding TMC's toxicity (Fig. 4B).163 Nanozymes could also function independently in tumor therapy, thereby offering an economical and effective option due to their simple structure.164,165 These in situ injected nanozymes avoid possible destruction by the bloodstream and remain at the solid tumor for at least a week after a single injection, due to its structure. Therefore, while Pd-based micro-catalysts have shown promise in prodrug activation, efforts should be directed towards refining their size and transport properties to maximize their potential in tumor therapy. This would enable better delivery and targeted activation of prodrugs, leading to enhanced efficacy and reduced toxicity to normal tissues.
 | ||
| Fig. 4 (A) Scheme of Pd0-resin mediated prodrug activation. Copied with permission162 Copyright 2014, American Chemical Society; (B) scheme of Pd nanozyme in situ injection for better enrichment long-term retention. Copied with permission163 Copyright 2022, Elsevier Inc. | ||
In order to achieve efficient tumor accumulation, Miller and colleagues77 employed a nano-encapsulated Pd compound74 to mediate alloxylcarbamate cleavage and facilitate the release of the activated drug. By utilizing the enhanced permeability and retention (EPR) effect, the catalyst and prodrugs could effectively target tumor cells together, and the presence of Pd nanoparticles enhanced the activation process (Fig. 5A). This approach enabled targeted drug delivery and activation, improving the overall efficacy of the treatment. Recently, 2D black phosphorus nanosheets were developed to increase the surface-to-volume ratio, thereby improving the catalytic activity of Pd (Fig. 5B).166 Besides, this carrier enhanced solubility, the long-term stability and degradability in physiological environments preventing them from accumulating in vivo and improving the biosafety of Pd.
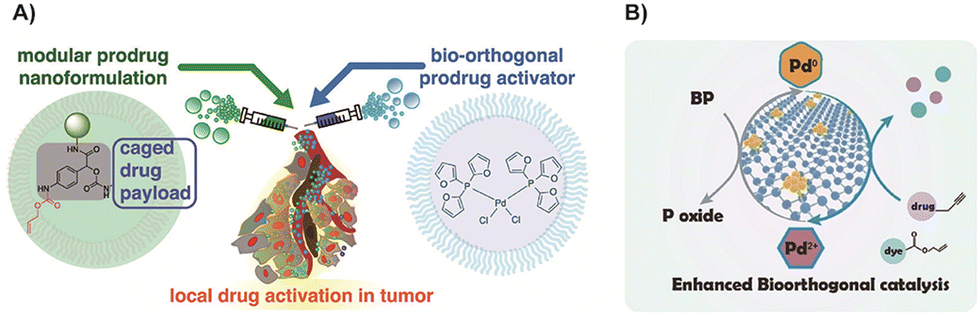 | ||
| Fig. 5 (A) Scheme of prodrug uncaging using Pd nanoparticles. Copied with permission77 Copyright 2018, American Chemical Society; (B) scheme of 2D black phosphorus nanosheets to load Pd with better reactivity. Copied with permission166 Copyright 2023, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
To further enhance the enrichment of TMCs in tumor tissues, Hoop and co-workers designed a magnetically driven Pd and Fe nanorobot.167 This nanorobot could specifically target tumor sites through external stimulation and activate a 5-fluorouracil (5-FU) prodrug for cancer therapy (Fig. 6A). This innovative strategy allows TMCs to be directed to specific locations, reducing their inherent toxicity to normal tissues. The introduction of transition metal nanorobots into prodrug activation represents a significant advancement in cancer therapy. Overall, these studies demonstrate the potential of Pd-based catalysts, whether in nano-encapsulated form or integrated into nanorobots, to target specific sites and improve the enrichment of catalysts in tumor tissues. This approach minimizes the toxicity to normal tissues and establishes a new direction for utilizing TMCs in prodrug activation for cancer therapy.
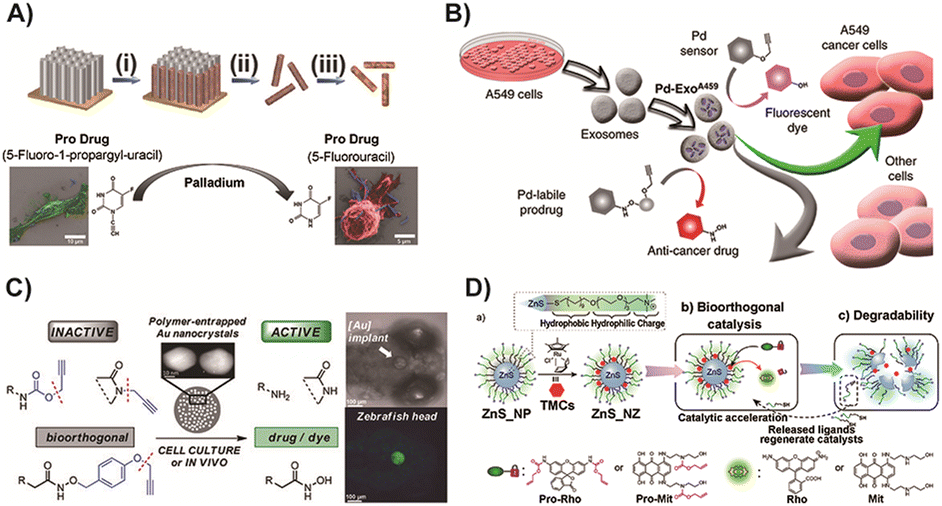 | ||
| Fig. 6 (A) Synthesis of Pd and Fe nanorobots and the scheme of 5-FU prodrug activation. Copied with permission167 Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (B) CO-mediated Pd nanosheet catalyst loaded with exosomes for prodrug activation in cancer target therapy. Copied with permission79 Copyright 2019, Springer Nature; (C) scheme of gold nanoparticle triggered prodrug and fluorescent dye. Copied with permission125 Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (D) Ru based ZnS nanozymes with enhanced efficiency for cancer therapy. Copied with permission82 Copyright 2022, American Chemical Society. | ||
Furthermore, Pd nanosheets combined with cancer-derived exosome vesicles, as reported by Sancho-Albero and colleagues, were used to enhance enrichment of catalysts at tumor sites.79 These exosomal vesicles, which naturally secrete compounds, possess unique characteristics such as prolonged circulation, low immunogenicity, and selectivity for progenitor cells. By utilizing exosomes as carriers, Pd nanosheets can be delivered to tumor sites with improved selectivity and targeting (Fig. 6B). This exosome-directed catalyst prodrug therapy enables more precise and targeted treatment. In addition to Pd catalysts, gold-resins have also been investigated for triggering prodrugs through catalyst-mediated cleavage reaction. These gold-resins, which consist of gold nanoparticles immobilized on a resin support, offer unique properties that make them suitable for BCPD activation. Pérez-López and colleagues125 reported the use of gold nanoparticle catalysis for locally controlled activation of prodrugs (Fig. 6C). This system demonstrated the ability to achieve localized and controlled release of a fluorescent dye in the brain of zebrafish. This approach not only expanded the application of transition metals in BCPD activation but also enabled fluorescence detection in the target area, which can be valuable for diagnosis and treatment. However, it is important to note that the biomedical applications of transition metal-catalyzed bioorthogonal processes are still limited, especially in the complex environment of the human body. Safety considerations remain a significant concern.78 Further research is necessary to address these challenges and ensure the safe and effective use of TMCs in biomedical applications.
Recently, Zhang and colleagues82 designed a platform using ZnS combined with Ru catalysis for prodrug dealkylation activation (Fig. 6D). One key advantage of this system is its better biodegradability, which helps to avoid the risk of particle accumulation in the body. The biodegradation of the nano-catalyst leads to the release of thiolate surface ligands, which in turn accelerates the activation of the prodrug. This approach offers the potential for safe application in living systems, providing localized therapeutics with minimal adverse effects. The development of such bioorthogonal reactions using TMCs has shown promise, and researchers are actively working to optimize these strategies to enhance their safety profile. The control of toxicity associated with transition metals remains an important area of focus for future research in the context of clinical treatments. By continuing to refine and improve the design of these catalyst-mediated prodrug activation systems, it may be possible to achieve safer and more effective therapies in the future.
For prodrugs that utilize metal catalysts, the design concept is simplified, allowing for more extensive applications. As mentioned earlier, the introduction highlighted the effective mitigation of the inherent toxicity associated with transition metals in the catalyst component of these prodrugs. An intriguing and promising direction is the use of metals with lower toxicity or higher biocompatibility as catalysts. Apart from gold (Au) as mentioned previously, there are ongoing studies exploring the use of platinum (Pt) in this context, although Pt is rarely used as a catalyst for bioorthogonal reactions and is not widely reported in prodrugs.168 While Pt-based compounds, such as cisplatin, have found clinical applications in cancer treatment,169 the use of cisplatin as a catalyst for BCPDs is rare. This approach holds potential for enhancing drug efficacy while reducing the toxic side effects associated with transition metals. Recently, Oliveira and colleagues128 developed a metal-mediated bond cleavage reaction for drug activation using Pt complexes (K2PtCl4 or cisplatin) for the first time (Fig. 7A). They introduced pentynoyl tertiary amide and N-propargyl reaction handles to drug molecules (monomethyl auristatin E and 5-FU) to form prodrugs. The prodrug system successfully underwent decaging in a cellular environment, releasing active drugs under the catalysis of Pt salts. This innovative approach achieved activation of BCPDs during cisplatin chemotherapy, improving drug efficacy while reducing the inherent toxicity associated with transition metals in the context of BCPDs. However, it is important to note that this strategy has currently been demonstrated primarily in in vitro systems. The instability of platinum complexes under biological conditions poses a challenge to their application. Nonetheless, this work opens up avenues for research on Pt-mediated decaging reactions and holds promise for future advancements in the field. Subsequently, Sun and colleagues80 developed Pt-mediated cleavage reactions of O/N-propargyl structures for BCPD activation using a nitric oxide (NO) donor (Fig. 7B). This activation system was tested with various compounds including a gas donor, 5-FU, a fluorophore, and tyrosine. Importantly, the Pt catalyst remained stable as Pt(IV) in normal cells, which did not activate the prodrug. However, upon entering tumor cells, Pt(IV) was reduced to Pt(II), activating the prodrug system and releasing NO by cleaving the propargyl bonds, leading to tumor cell apoptosis. This strategy intelligently utilized the configuration transition of Pt to selectively activate the prodrug, thereby avoiding the inherent toxicity associated with transition metals and improving the selectivity of the prodrugs. However, it should be noted that the prodrug system is also currently limited to in vitro studies, and further investigations are needed for in vivo applications.
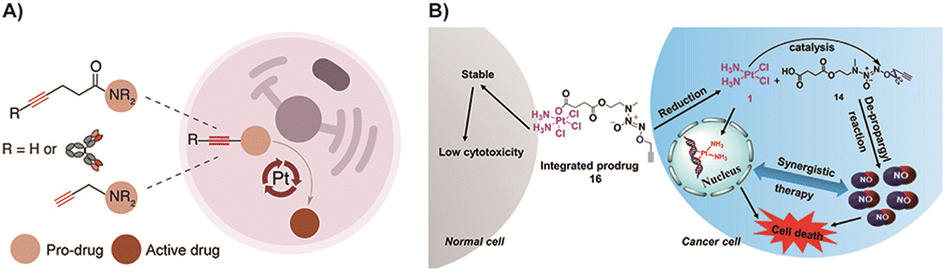 | ||
| Fig. 7 (A) Scheme of the Pt mediated bond cleavage reaction. Copied with permission128 Copyright 2020, American Chemical Society; (B) scheme of Pt mediated bond cleavage with different trigger activities in normal cells or tumor cells. Copied with permission80 Copyright 2020, American Chemical Society. | ||
Moreover, the strategy of catalyst-mediated cleavage reactions is well-suited for protecting multiple active drug groups. Unlike the CuAAC-based prodrug strategy, which is primarily limited to modifying structures similar to resveratrol, this approach has broader applicability to various drugs, showing potential for clinical use.74,79,167 For example, recent advancements include the development of new Pd-activated prodrug types, as highlighted by Dal Forno.170 Existing methods have mainly focused on cleaving C–O or C–N bonds, which restrict their use to drugs containing amino or hydroxyl groups. Dal Forno introduced a novel approach involving the decaging of an ortho-quinone prodrug via palladium-mediated C–C bond cleavage.
3.3. Catalyst-free reaction
In recent years, while catalyst-mediated prodrug systems have demonstrated improved biological safety, concerns remain regarding the potential risks associated with the use of catalysts in complex biological systems.69,95,132 Consequently, researchers have increasingly focused on developing strategies to eliminate the need for catalysts in BCPD systems. This approach not only addresses the safety concerns but also simplifies the design and application of prodrugs, making them more versatile and accessible.Another important aspect that researchers have been addressing is the issue of poor atom economy resulting from the formation of molecular fragments through intermolecular charge transfer during ligation reactions.86,129 In order to enhance the efficiency of prodrug activation and minimize wasteful byproducts, the development of traceless reaction modes has garnered significant attention. A traceless reaction refers to a process where the activation of the prodrug leads to the release of the active drug without leaving behind any molecular fragments or remnants. This approach ensures maximum atom economy and eliminates the need for subsequent removal or metabolism of residual fragments, thereby enhancing the overall efficiency and therapeutic potential of BCPDs. Moreover, the exploration of catalyst-free BCPDs represents an exciting area of research with significant potential for clinical applications. By eliminating the risks associated with catalyst use and improving atom economy through traceless reaction modes, these prodrugs offer enhanced biological safety, simplified design principles, and expanded versatility.
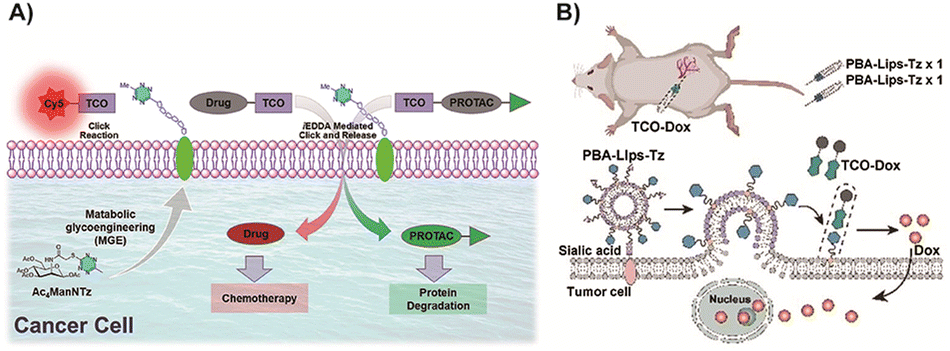 | ||
| Fig. 8 (A) Building tetrazine-functionalized chemical receptors for biorthogonal targeting, activation, and delivery of drugs to cancer cells. Copied with permission171 Copyright 2022, Elsevier Inc.; (B) scheme of tumor cell membrane modification using special liposomes for better prodrug enrichment. Copied with permission172 Copyright 2024, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In recent research, the utilization of nanomedicines in the field of BCPDs based on IEDDA reactions has provided promising advancements. Suehiro and colleagues91 developed Tz-functionalized amphiphilic block copolymers that can self-assemble into micelles in aqueous solutions. These nanomedicines efficiently convert trans-cyclooctene-functionalized prodrugs into their active anticancer drug forms through an IEDDA reaction, enabling in vivo prodrug activation and effective inhibition of tumor growth without significant systemic toxicity (Fig. 9A). Intracellular self-assembly of prodrugs offers an additional benefit by enhancing drug accumulation within target cells. Yao and colleagues86 proposed the strategy of enzyme-instructed supramolecular self-assembly (EISA) for cancer therapy (Fig. 9B). This approach facilitates the selective accumulation of Tz groups within tumor cells, allowing for precise uncaging and induction of cancer cell death upon reaction with the bioorthogonal handle of the prodrug. The EISA strategy offers excellent spatiotemporal control and can effectively screen tumor cells. However, it should be noted that the production of molecule fragments from Tz after activation may increase the metabolic burden, emphasizing the need to improve atom economy in future research. Building upon this strategy, Yao and colleagues122 designed a similar prodrug in which active drugs were caged with TCO (Fig. 9C). Through EISA-mediated intracellular accumulation, this prodrug could activate multiple drugs simultaneously via IEDDA cycloaddition reactions. This approach expands the selection space of active drugs compared to previous strategies. While the selectivity and practicality of using multiple drugs have been significantly improved, the issue of drug fragmentation remains a challenge that needs to be addressed.
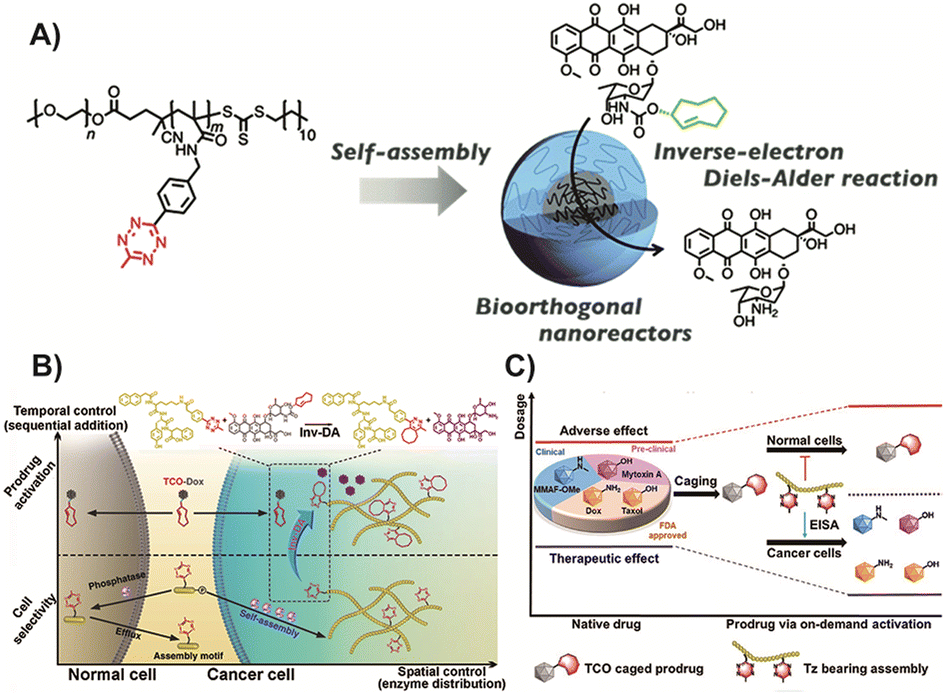 | ||
| Fig. 9 (A) TCO based micelles enriched in tumor cells and could catalyze any prodrug in cancer cells. Copied with permission91 Copyright 2022, The Royal Society of Chemistry; (B) Scheme of EISA based prodrug for cancer precise enrichment. Copied with permission86 Copyright 2018, Springer Nature; (C) Scheme of enlarging the therapeutic window of typical drugs via the process of EISA-mediated on-demand prodrug activation. Copied with permission129 Copyright 2021, Elsevier Inc. | ||
In summary, the integration of nanomedicines into the field of BCPDs based on IEDDA reactions offers tremendous potential for advancing targeted cancer therapy. The work by Suehiro and Yao, and their respective colleagues, demonstrates the successful utilization of Tz-functionalized block copolymers and EISA strategies for in vivo prodrug activation and tumor growth inhibition. However, further research is necessary to address issues related to nanomedicine stability, biocompatibility, clinical translation, and optimizing the atom economy to maximize the therapeutic potential of these innovative approaches.
Indeed, by carefully designing these fragments to exhibit therapeutic activity, it becomes possible to optimize atom utilization and avoid unnecessary metabolic burden. Neumann and colleagues130 reported a novel strategy where the released fragments in the process were designed to function as drugs themselves (Fig. 10A). These two prodrugs could activate each other upon meeting through an IEDDA reaction. In this way, the Tz structure that would be considered as excess metabolic waste in other reports still retained miR21 inhibition ability after the reaction. In this work, Tz was used as a protecting group for bioactive pyridazines, introducing a novel concept of catalyst-free traceless prodrug activation by IEDDA reaction. It is important to note that no animal treatment model was established in this study, and further research is needed to investigate the mode of administration and overall treatment effects. Nevertheless, this approach presents a promising concept in the field of BCPD activation. Similarly, Ji and colleagues131 proposed a distinct structure that releases CO, active drugs, and fluorescent products upon activation, showcasing an approach that optimizes atom economy by effectively utilizing each product. The molecular fragments generated in this process can serve multiple purposes, including fluorescence, and the released gas can be employed for gas therapy in tumor treatments, particularly advantageous for combination therapy (Fig. 10B). However, the absence of animal treatment models does limit the comprehensive safety evaluation of this strategy. Nevertheless, it exhibits tremendous promise for future applications in cancer treatment. By maximizing the utilization of every atom in the reaction, this strategy introduces new avenues for the clinical application of BCPDs.
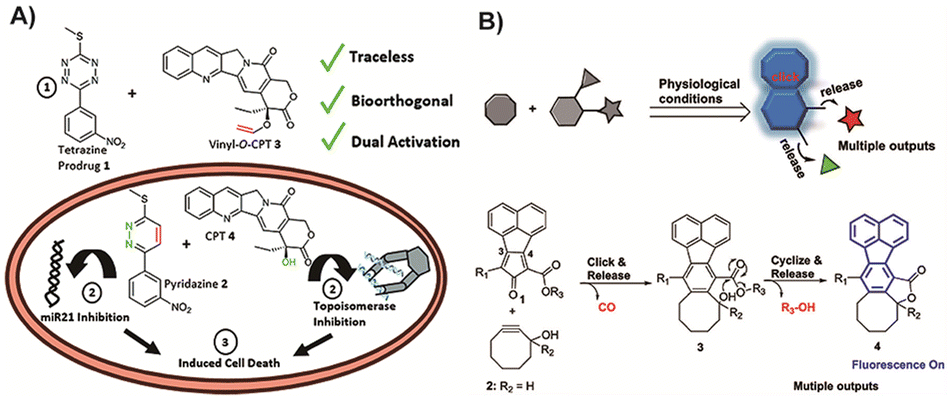 | ||
| Fig. 10 (A) Traceless strategy of IEDDA whose fragment was also designed as an active drug after activation. Copied with permission130 Copyright 2018, The Royal Society of Chemistry; (B) the strategy of using the fragment as a fluorescence molecule and then using CO generated in the process as an active drug for cancer treatment. Copied with permission131 Copyright 2019, American Chemical Society. | ||
Ji and coworkers also designed a special structure which could release CO and active drugs as well as fluorescence products upon activation. This strategy of making the best use of every product improves the atom economy. Consequently, molecular fragments could also perform some functions, fluorescence in this work, and compared with previous work,173 the gas released during this process also could be used as gas therapy for tumor treatments, which was invaluable for combination therapy (Fig. 10B). However, the lack of animal treatment models does affect the final comprehensive safety evaluation results with this strategy, but this strategy is indeed very promising for future applications in cancer treatment. This strategy of making full use of any atom in the reaction could provide more possibilities for clinical applications of BCPDs. It is important to acknowledge that molecular design can pose a significant challenge in the context of the IEDDA reaction, potentially restricting the range of drugs that can be employed and limiting the scope of diseases that can be targeted. Moreover, the requirement for separate or alternate administration of the two components of the prodrugs can present a barrier to their clinical application. These factors emphasize the necessity for further research and development to overcome these challenges and fully harness the potential of BCPDs in clinical settings.69
Compared to the previous two strategies, the prodrug based on the IEDDA reaction offers the advantage of modifying multiple drugs to a certain extent without requiring metal catalysts. However, it still faces the challenge of effectively bringing the two parts together to react in a suitable location. The two reaction handles will immediately react upon contact, hence the co-administration mode still results in losses due to early drug activation and potential toxicity issues arising from drug activation, necessitating a change in administration strategy.69 The sequential administration model can clearly solve this problem effectively, a concept fully achievable in basic research. However, patient compliance poses a significant challenge in clinical research. An effective approach to address this issue often involves the use of nanocarriers. Nanocarriers can not only assist in isolating the two reaction handles but also utilize the passive targeting capabilities of nanocarriers or active targeting molecules on the carrier surface to achieve precise drug delivery and controlled release of prodrugs. Another noteworthy point is the atomic economy of this strategy, which may not be able to fully utilize resources and could impose additional metabolic burden. However, in clinical research, demonstrating the biological activity of the molecular fragment is also crucial.
This strategy holds the most promising prospects for clinical application because it combines the advantages of biorthogonal prodrug design without the concerns of transition metal toxicity associated with other strategies. However, its application is somewhat limited by the chosen administration method. The inability of this strategy to deliver drugs in a single dose also restricts its clinical potential. Nevertheless, by designing the two-part prodrug with different reaction handles at a nano scale69 or exploring alternative drug delivery methods, such as buccal mucosal administration, this strategy remains promising for clinical use.
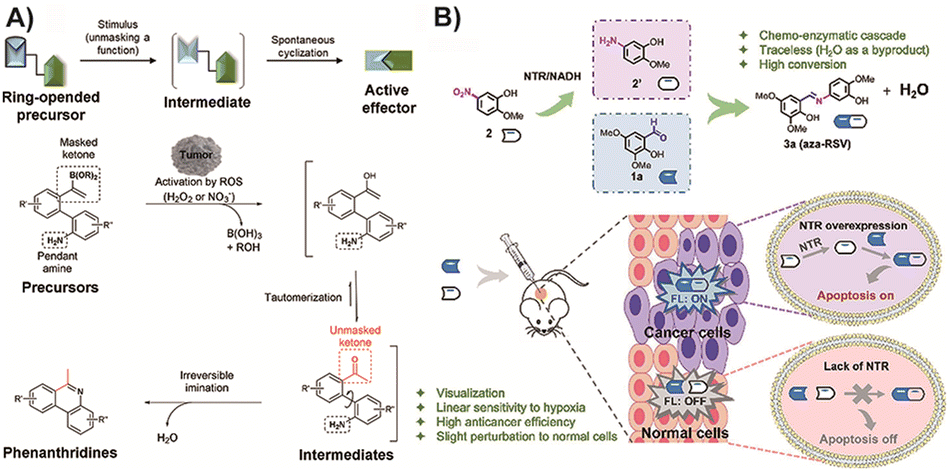 | ||
| Fig. 11 (A) Prodrug response to ROS, then click linking via aldimine condensation and inducing tumor cell death. Copied with permission95 Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (B) two parts of the prodrug meeting in tumor cells, one of them responded to nitroreductase and then clicked to turn into drug analogues to kill cancer cells. Copied with permission132 Copyright 2022, Elsevier Inc. | ||
In a recent study by Hu and colleagues,132 they followed a similar concept and designed a prodrug consisting of two parts that can respond to nitroreductase, an enzyme overexpressed in tumor tissue. When these parts meet in tumor cells, they link to generate drug analogues of pterostilbene, with water as the only by-product. Notably, the in situ synthesized product within tumor cells also exhibits certain fluorescence characteristics, showing potential for integrated diagnosis and treatment (Fig. 11B). Additionally, a solid tumor model was established to provide evidence of in situ prodrug activation, although further research is needed to optimize the administration model for better practical application. Overall, this multifunctional molecule prodrug demonstrates impressive responsiveness, toxicity effects, and fluorescence capabilities for cancer imaging. However, the potential toxicity of the precursors may limit its clinical application. It is important to acknowledge that aldimine condensation reaction-based BCPDs are still in the proof-of-concept stage, with limited research progress toward clinical application and primarily focused on in situ administration. Nevertheless, this strategy holds promise as a future research direction for traceless BCPD treatment.
The aldehyde amine condensation reaction exhibits the characteristics of a bioorthogonal reaction, but its reaction rate is inferior to that of transition metal-catalyzed biorthogonal reactions.132 The prodrugs activated are predominantly based on the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC bonds, giving them similar biological activity to styrene analogs.174 In contrast to the CuAAC reaction-based stilbene analog prodrug system, this method operates without a catalyst and offers improved biosafety. Additionally, for stilbene analogs that have already been divided into two fragments, shielding the reaction handle can enable them to respond to the tumor environment, allowing simultaneous drug delivery. Therefore, this strategy does not require the delivery and controlled release of a nanocarrier to avoid undesired reactions in unnecessary locations. However, further design and research are necessary to mitigate the toxicity and biological activity associated with the prodrug itself. Moreover, its structural analogs can only exhibit biological activity similar to resveratrol, which limits its efficacy in broad-spectrum anticancer treatments. The efficacy of reaction handle shielding, activation, and prodrug activation efficiency still requires exploration. Determining whether this approach can be extended to other bioactive molecules needs further investigation and study.
N and CC bonds, giving them similar biological activity to styrene analogs.174 In contrast to the CuAAC reaction-based stilbene analog prodrug system, this method operates without a catalyst and offers improved biosafety. Additionally, for stilbene analogs that have already been divided into two fragments, shielding the reaction handle can enable them to respond to the tumor environment, allowing simultaneous drug delivery. Therefore, this strategy does not require the delivery and controlled release of a nanocarrier to avoid undesired reactions in unnecessary locations. However, further design and research are necessary to mitigate the toxicity and biological activity associated with the prodrug itself. Moreover, its structural analogs can only exhibit biological activity similar to resveratrol, which limits its efficacy in broad-spectrum anticancer treatments. The efficacy of reaction handle shielding, activation, and prodrug activation efficiency still requires exploration. Determining whether this approach can be extended to other bioactive molecules needs further investigation and study.
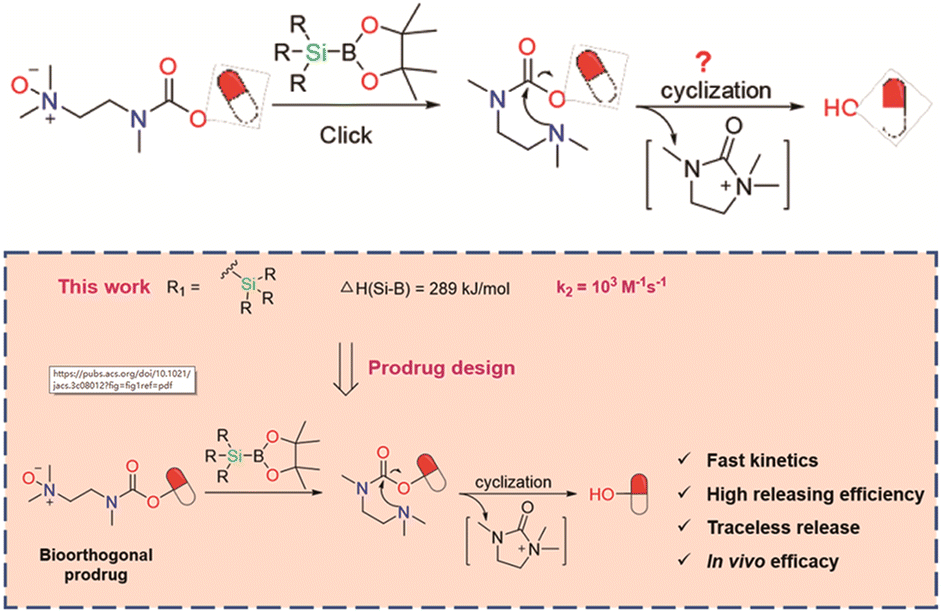 | ||
| Fig. 12 Scheme of new bioorthogonal decaging reaction based on the reaction of boronic acid and N-oxide with better reactivity. Copied with permission175 Copyright 2023, American Chemical Society. | ||
Although the excellent treatment effects and controllable toxic side effects of these bioorthogonal chemistry-based prodrugs have been demonstrated primarily in basic research, there are still challenges to overcome in translating these findings to the complex internal environment of the human body. The human body presents numerous biological barriers, including immune responses, enzymatic degradation, and variability in tumor microenvironments. Further research is needed to optimize the design of prodrugs, improve their stability, enhance their circulation time, and ensure their effective delivery to tumor sites while minimizing off-target effects. Additionally, thorough studies are required to assess the long-term safety, biocompatibility, and potential toxicity of these prodrugs in preclinical and clinical settings. By addressing these challenges, bioorthogonal chemistry-based prodrugs have the potential to become valuable tools in cancer therapy, providing targeted and efficient treatment options with reduced side effects.
4. Conclusion and perspectives
In conclusion, BCPDs present a powerful strategy for drug delivery, leveraging the many advantages of bioorthogonal chemistry. However, challenges such as the use of catalysts and the metabolic burden due to low atom economy remain limiting factors. This review offers a comprehensive overview of BCPDs, highlighting both their historical context and recent advancements. The focus has been on the safety of prodrug strategies and the current knowledge regarding their application in tumor treatment. Bioorthogonal reactions were categorized based on the usage of catalysts: catalyst-mediated ligation reactions, catalyst-mediated cleavage reactions, and catalyst-free reactions. The advantages and strategies associated with these reactions were discussed in detail, along with possible directions for improvement. In terms of safer administration, several principles were identified: (1) use non-toxic and degradable catalysts whenever possible. (2) If the use of a catalyst is unavoidable, ensure rapid metabolism through material design or loading. (3) Avoid staggered administration of the prodrug and trigger. (4) Utilize traceless strategies after prodrug activation, if feasible. Overall, the future direction of biorthogonal prodrug development may involve catalyst-free and traceless reaction models. By minimizing the reliance on catalysts and implementing traceless strategies, the limitations related to the catalyst usage and metabolic burden can be addressed. This approach holds promise for advancing the field of BCPDs and improving their safety and efficacy.Data availability
The data and information presented in this review are based on previously published studies, which are cited throughout the article.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (32201090) and the Guizhou Provincial Basic Research Program (Natural Science, ZK[2023]303).References
- J. Zou, H. Bai, L. Zhang, Y. Shen, C. Yang, W. Zhuang, J. Hu, Y. Yao and W. Hu, J. Mater. Chem. B, 2024, 12, 8523–8542 RSC.
- N. André, M. Carré and E. Pasquier, Nat. Rev. Clin. Oncol., 2014, 11, 413–431 CrossRef PubMed.
- Y. Yao, X. Dai, Y. Tan, Y. Chen, C. Liao, T. Yang, Y. Chen, Y. Yu and S. Zhang, Adv. Healthcare Mater., 2020, 10, 2001430 CrossRef PubMed.
- Y. Yao, Y. Chen, Y. Liu, Y. Zhu, Y. Liu and S. Zhang, Langmuir, 2019, 35(17), 5871–5877 CrossRef CAS PubMed.
- Y. Yao, D. Xu, C. Liu, Y. Guan, J. Zhang, Y. Su, L. Zhao, F. Meng and J. Luo, RSC Adv., 2016, 6, 97684–97693 RSC.
- J. H. Patil, J. K. Patel, U. A. Shah, P. O. Patil, A. S. Chaudhari and H. H. Goswami, Nano Biomed. Eng., 2024 DOI:10.26599/NBE.2024.9290078.
- Y. Yao, H. Xu, C. Liu, Y. Guan, D. Xu, J. Zhang, Y. Su, L. Zhao and J. Luo, RSC Adv., 2016, 6, 9082–9089 RSC.
- Q. Chen, Y. Pei, K. Tang and M. G. Albu-Kaya, Collagen Leather, 2023, 5, 20 CrossRef CAS.
- S. Zhou, Y. Li, Q. Wu and C. Gong, MedComm: Biomater. Appl., 2024, 3, e70 Search PubMed.
- Y. Li, Y. Dong, X. Zhou and K. Fan, MedComm: Biomater. Appl., 2023, 2, e36 CAS.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- A. B. Cook and P. Decuzzi, ACS Nano, 2021, 15, 2068–2098 CrossRef CAS PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, Chem. Sci., 2021, 12, 1668–1674 RSC.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- M. Maiti, S. A. Yoon, Y. Cha, K. K. Athul, S. Bhuniya and M. H. Lee, Chem. Commun., 2021, 57, 9614–9617 RSC.
- Z. Zhao, X. Tao, Y. Xie, Q. Lai, W. Lin, K. Lu, J. Wang, W. Xia and Z.-W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202202855 CrossRef CAS PubMed.
- E. Maurits, M. J. van de Graaff, S. Maiorana, D. P. A. Wander, P. M. Dekker, S. Y. van der Zanden, B. I. Florea, J. J. C. Neefjes, H. S. Overkleeft and S. I. van Kasteren, J. Am. Chem. Soc., 2020, 142, 7250–7253 CrossRef CAS PubMed.
- D. Qi, L. Xing, L. Shen, W. Sun, C. Cai, C. Xue, X. Song, H. Yu, H. Jiang, C. Li, Q. Jin and Z. Zhang, Chin. Chem. Lett., 2022, 33, 4595–4599 CrossRef CAS.
- P. Thapa, M. Li, M. Bio, P. Rajaputra, G. Nkepang, Y. Sun, S. Woo and Y. You, J. Med. Chem., 2016, 59, 3204–3214 CrossRef CAS PubMed.
- T. Liu, H. Zou, J. Mu, N. Yu, Y. Xu, G. Liu, X. Liang and S. Guo, Chin. Chem. Lett., 2021, 32, 1751–1754 CrossRef CAS.
- M. Ni, W.-J. Zeng, X. Xie, Z.-L. Chen, H. Wu, C.-M. Yu and B.-W. Li, Chin. Chem. Lett., 2017, 28, 1345–1351 CrossRef CAS.
- X. Dong, R. K. Brahma, C. Fang and S. Q. Yao, Chem. Sci., 2022, 13, 4239–4269 RSC.
- W. Mao, D. Mao, F. Yang and D. Ma, Chem. – Eur. J., 2019, 25, 2272–2280 CrossRef CAS PubMed.
- M. Cong, G. Xu, S. Yang, J. Zhang, W. Zhang, D. Dhumal, E. Laurini, K. Zhang, Y. Xia, S. Pricl, L. Peng and W. Zhao, Chin. Chem. Lett., 2022, 33, 2481–2485 CrossRef CAS.
- Y. Liu, S. Jiang, W. Mao, P. Li, F. Zhou and D. Ma, Chin. Chem. Lett., 2022, 33, 209–212 CrossRef CAS.
- T. Liu, H. Zou, J. Mu, X. Zhang, G. Liu, N. Yu, B. Yuan, X. Yuan, X. Liang and S. Guo, Chin. Chem. Lett., 2023, 34, 108135 CrossRef CAS.
- S. Wang, K. Yu, Z. Yu, B. Zhang, C. Chen, L. Lin, Z. Li, Z. Li, Y. Zheng and Z. Yu, Chin. Chem. Lett., 2023, 34, 108184 CrossRef CAS.
- X. Wu, M. Liu, C. Zheng, Y. Wang, Y. Zheng, Y. Qian, Z. Liao, G. Fang and J. Shen, Chin. Chem. Lett., 2023, 34, 107590 CrossRef CAS.
- Q. Zhang, Y. Chen, R. Lu, Y. Yao, C. Li, Y. Yu and S. Zhang, J. Mater. Chem. B, 2020, 8, 2719–2725 RSC.
- G. F. Boafo, Y. Shi, Q. Xiao, K. T. Magar, M. Zoulikha, X. Xing, C. Teng, E. Brobbey, X. Li, X. Jiang, X. Wang, Y. Yang, S. Kesse and W. He, Chin. Chem. Lett., 2022, 33, 4600–4604 CrossRef CAS.
- Z. Li, Q. Xu, X. Lin, K. Yu, L. Lin, Y. Liu, Z. Yu, T. Liu and D. Luo, Chin. Chem. Lett., 2022, 33, 1875–1879 CrossRef CAS.
- H. Huang, C. Dong, M. Chang, L. Ding, L. Chen, W. Feng and Y. Chen, Exploration, 2021, 1, 50–60 CrossRef PubMed.
- Y. Lu, D. Jia, X. Ma, M. Liang, S. Hou, W. Qiu, Y. Gao, P. Xue, Y. Kang and Z. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 8940–8951 CrossRef CAS PubMed.
- T. Nishimura, Y. Sasaki and K. Akiyoshi, Adv. Mater., 2017, 29, 1702406 CrossRef PubMed.
- Y. Song, D. Li, J. He, M. Zhang and P. Ni, Chin. Chem. Lett., 2019, 30, 2027–2031 CrossRef CAS.
- S. da Silva Santos, E. Igne Ferreira and J. Giarolla, Molecules, 2016, 21, 686 CrossRef PubMed.
- X. Hu, Z. Chai, L. Lu, H. Ruan, R. Wang, C. Zhan, C. Xie, J. Pan, M. Liu, H. Wang and W. Lu, Adv. Funct. Mater., 2019, 29, 1807941 CrossRef.
- H. Xiang, Y. Wu, X. Zhu, M. She, Q. An, R. Zhou, P. Xu, F. Zhao, L. Yan and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 11449–11461 CrossRef CAS PubMed.
- Y. Yao, C. Li, F. Liu, P. Zhao, Z. Gu and S. Zhang, Phys. Chem. Chem. Phys., 2019, 21, 10477–10487 RSC.
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed.
- X. Ji, Curr. Top. Med. Chem., 2021, 21, 2155–2156 CrossRef CAS PubMed.
- M. Chang, F. Gao, D. Pontigon, G. Gnawali, H. Xu and W. Wang, J. Am. Chem. Soc., 2023, 145, 14155–14163 CrossRef CAS PubMed.
- H. Liu, M. Ji, P. Xiao, J. Gou, T. Yin, H. He, X. Tang and Y. Zhang, Asian J. Pharm. Sci., 2024, 100922 CrossRef PubMed.
- H. Zheng, H. Wu, D. Wang, S. Wang, D. Ji, X. Liu, G. Gao, X. Su, Y. Zhang and Y. Ling, Eur. J. Med. Chem., 2024, 116457 CrossRef CAS PubMed.
- G. Singh, R. Singh, G. Singh, J. Stanzin, H. Singh, G. Kaur and J. Singh, RSC Adv., 2024, 14, 7383–7413 RSC.
- V. K. Tiwari, M. K. Jaiswal, S. Rajkhowa and S. K. Singh, Click Chem., Springer, 2024, pp. 175–203 Search PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- X. Pei, Z. Luo, L. Qiao, Q. Xiao, P. Zhang, A. Wang and R. A. Sheldon, Chem. Soc. Rev., 2022, 51, 7281–7304 RSC.
- A. Najjar, A. Najjar and R. Karaman, Molecules, 2020, 25, 884 CrossRef CAS PubMed.
- C. Chen, Y. Yang, Z. Wang, H. Li, C. Dong and X. Zhang, J. Med. Chem., 2023, 66, 8428–8440 CrossRef CAS PubMed.
- Q. Fu, S. Shen, P. Sun, Z. Gu, Y. Bai, X. Wang and Z. Liu, Chem. Soc. Rev., 2023, 52, 7737–7772 RSC.
- T. Sun and C. Jiang, Adv. Drug Delivery Rev., 2023, 196, 114773 CrossRef CAS PubMed.
- A. Shinde, S. B. Prasad, D. A. Srinivasarao, S. Shah, P. Famta, P. Khairnar, G. Pandey, G. Vambhurkar, A. Hedaoo and R. Kumar, Appl. Mater. Today, 2024, 38, 102162 CrossRef.
- Y. Ma, Y. Zhou, J. Long, Q. Sun, Z. Luo, W. Wang, T. Hou, L. Yin, L. Zhao and J. Peng, Angew. Chem., Int. Ed., 2024, 136, e202318372 CrossRef.
- Y.-L. Ma, S. Yan, X.-J. Xu, H. Cao and R. Wang, Chin. Chem. Lett., 2024, 35, 108645 CrossRef CAS.
- J. Chen, Y. Yao, X. Mao, Y. Chen and F. Ni, J. Drug Targeting, 2024, 1–26 CrossRef PubMed.
- P. Adhikari, G. Li, M. Go, D. Mandikian, H. Rafidi, C. Ng, S. Anifa, K. Johnson, L. Bao and H. Hernandez Barry, J. Am. Chem. Soc., 2024, 146, 19088–19100 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- B. Li, P. Liu, H. Wu, X. Xie, Z. Chen, F. Zeng and S. Wu, Biomaterials, 2017, 138, 57–68 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. Am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212–14214 RSC.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS PubMed.
- J. Huang, L. Wang, P. Zhao, F. Xiang, J. Liu and S. Zhang, ACS Catal., 2018, 8, 5941–5946 CrossRef CAS.
- Z. Wang, J. Niu, C. Zhao, X. Wang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2021, 60, 12431–12437 CrossRef CAS PubMed.
- L. Wang, P. Jing, J. Tan, C. Liao, Y. Chen, Y. Yu and S. Zhang, Biomaterials, 2021, 273, 120823 CrossRef CAS PubMed.
- Y. You, H. Liu, J. Zhu, Y. Wang, F. Pu, J. Ren and X. Qu, Chem. Sci., 2022, 13, 7829–7836 RSC.
- J. Zhu, Y. You, W. Zhang, F. Pu, J. Ren and X. Qu, J. Am. Chem. Soc., 2023, 145, 1955–1963 CrossRef CAS PubMed.
- L. Xia, M. Chen, C. Dong, F. Liu, H. Huang, W. Feng and Y. Chen, Adv. Mater., 2023, 35, 2209179 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- S. Alonso-de Castro, E. Ruggiero, A. Ruiz-de-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. López-Gallego and L. Salassa, Chem. Sci., 2017, 8, 4619–4625 RSC.
- S. Alonso-de Castro, A. L. Cortajarena, F. López-Gallego and L. Salassa, Angew. Chem., Int. Ed., 2018, 57, 3143–3147 CrossRef CAS PubMed.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814–12826 CrossRef CAS PubMed.
- Z. Du and X. Qu, Nat. Catal., 2019, 2, 837–838 CrossRef CAS.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS PubMed.
- T. Sun, T. Lv, J. Wu, M. Zhu, Y. Fei, J. Zhu, Y. Zhang and Z. Huang, J. Med. Chem., 2020, 63, 13899–13912 CrossRef CAS PubMed.
- Z. Chen, H. Li, Y. Bian, Z. Wang, G. Chen, X. Zhang, Y. Miao, D. Wen, J. Wang, G. Wan, Y. Zeng, P. Abdou, J. Fang, S. Li, C.-J. Sun and Z. Gu, Nat. Nanotechnol., 2021, 16, 933–941 CrossRef CAS PubMed.
- X. Zhang, S. Lin, R. Huang, A. Gupta, S. Fedeli, R. Cao-Milan, D. C. Luther, Y. Liu, M. Jiang, G. Li, B. Rondon, H. Wei and V. M. Rotello, J. Am. Chem. Soc., 2022, 144, 12893–12900 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- E. Jimenez-Moreno, Z. Guo, B. L. Oliveira, I. S. Albuquerque, A. Kitowski, A. Guerreiro, O. Boutureira, T. Rodrigues, G. Jimenez-Oses and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 243–247 CrossRef CAS PubMed.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. ten Hoeve, H. M. Janssen, A. H. A. M. van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1484 CrossRef PubMed.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- K. Neumann, A. Gambardella, A. Lilienkampf and M. Bradley, Chem. Sci., 2018, 9, 7198–7203 RSC.
- X. Xie, B. Li, J. Wang, C. Zhan, Y. Huang, F. Zeng and S. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 41875–41888 CrossRef PubMed.
- H. Li, J. Conde, A. Guerreiro and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2020, 59, 16023–16032 CrossRef CAS PubMed.
- R. Dzijak, J. Galeta, A. Vázquez, J. Kozák, M. Matoušová, H. Fulka, M. Dračínský and M. Vrabel, JACS Au, 2021, 1, 23–30 CrossRef CAS PubMed.
- F. Suehiro, S. Fujii and T. Nishimura, Chem. Commun., 2022, 58, 7026–7029 RSC.
- Y. Wang, G. Shen, J. Li, W. Mao, H. Sun, P. Feng and H. Wu, Org. Lett., 2022, 24, 5293–5297 CrossRef CAS PubMed.
- L. Liu, D. Zhang, M. Johnson and N. K. Devaraj, Nat. Chem., 2022, 14, 1078–1085 CrossRef CAS PubMed.
- W. Kuba, B. Sohr, P. Keppel, D. Svatunek, V. Humhal, B. Stöger, M. Goldeck, J. C. T. Carlson and H. Mikula, Chem. – Eur. J., 2023, 29, e202203069 CrossRef CAS PubMed.
- H. Maslah, C. Skarbek, C. Gourson, M. A. Plamont, S. Pethe, L. Jullien, T. Le Saux and R. Labruere, Angew. Chem., Int. Ed., 2021, 60, 24043–24047 CrossRef CAS PubMed.
- Y. Li, Z. Lou, H. Li, H. Yang, Y. Zhao and H. Fu, Angew. Chem., Int. Ed., 2020, 59, 3671–3677 CrossRef CAS PubMed.
- G. Xu and H. L. McLeod, Clin. Cancer Res., 2001, 7, 3314–3324 CAS.
- N. Schellmann, P. M. Deckert, D. Bachran, H. Fuchs and C. Bachran, Mini-Rev. Med. Chem., 2010, 10, 887–904 CrossRef CAS PubMed.
- G. Y. Li, Z. W. Song, Y. Ru, J. Zhang, L. X. Luo, W. Yang, H. Wu, H. B. Jin, X. W. Bao, D. Wei, Z. Yan, H. J. Qu, Z. Zhu, X. Xue and G. Zhou, Exploration, 2023, 3, 20220141 CrossRef CAS PubMed.
- Y. Zhang, L. Zhang, W. Wang, Q. Deng, M. Liu, Z. Zhu, H. Liu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2023, 62, e202306395 CrossRef CAS PubMed.
- Z. Gao, Q. Shao, J. Xing, Y. Liang, F. Meng, J. Chen, W. He, Y. Li and B. W. Sun, Angew. Chem., Int. Ed., 2024, e202409849 Search PubMed.
- X. Zhang, Y. Liu, M. Jiang, J. A. Mas-Rosario, S. Fedeli, R. Cao-Milan, L. Liu, K. J. Winters, C.-M. Hirschbiegel, A. Nabawy, R. Huang, M. E. Farkas and V. M. Rotello, Chem. Sci., 2024, 15, 2486–2494 RSC.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450–6456 CrossRef CAS PubMed.
- C. Deng, M. Zheng, S. Han, Y. Wang, J. Xin, O. Aras, L. Cheng and F. An, Adv. Funct. Mater., 2023, 33, 2300348 CrossRef CAS PubMed.
- B. Chen, K. Guo, X. Zhao, Z. Liu, C. Xu, N. Zhao and F. J. Xu, Exploration, 2023, 3, 20220140 CrossRef CAS PubMed.
- N. Wang, Y. Liu, D. Peng, Q. Zhang, Z. Zhang, L. Xu, L. Yin, X. Zhao, Z. Lu and J. Peng, Adv. Healthcare Mater., 2024, 2401646 CrossRef PubMed.
- W. Wang, X. He, X. Wang, T. Zhao, O. Muraoka, G. Tanabe, W. Xie, T. Zhou, L. Xing, Q. Jin and H. Jiang, Chin. Chem. Lett., 2024, 35, 108656 CrossRef CAS.
- C. Deng, J. Zhang, F. Hu, S. Han, M. Zheng, F. An and F. Wang, Small, 2024, 2400667 CrossRef PubMed.
- L. Wang, P. Jing, J. Tan, C. Liao, Y. Chen, Y. Yu and S. Zhang, Biomaterials, 2021, 273, 120823 CrossRef CAS PubMed.
- J. Tan, P. Jing, X. Xiao, Y. Liao, C. Liao and S. Zhang, Sci. China: Chem., 2023, 66, 2654–2663 CrossRef CAS.
- Y. Liu, H. Wang, M. Ding, W. Yao, K. Wang, I. Ullah, E. Bulatov and Y. Yuan, Nano Lett., 2024, 24, 8741–8751 CrossRef CAS PubMed.
- H. An, C. Yang, Z. Jiang, J. Yuan, Z. Qiu, L. Chen, X. Chen, M. Huang, L. Huang, H. Lin, B. Cheng, H. Liu and Z. Yu, Chin. Chem. Lett., 2024, 35, 109134 CrossRef CAS.
- J. Wang, W. Cao, W. Zhang, B. Dou, X. Ding, M. Wang, J. Ma and X. Li, J. Med. Chem., 2024, 67, 8296–8308 CrossRef CAS PubMed.
- Y. Ding, C. Wang, Y. Ma, L. Zhu, B. Lu, Y. Wang, J. Wang, T. Chen, C.-M. Dong and Y. Yao, Acta Biomater., 2022, 143, 381–391 CrossRef CAS PubMed.
- J. C. Ho and C.-Y. Chen, Macromol. Chem. Phys., 2024, 225, 2300386 CrossRef CAS.
- P. Sun, Z. Li, D. Zhang, W. Zeng, Y. Zheng, L. Mei, H. Chen, N. Gao and X. Zeng, Chin. Chem. Lett., 2024, 35, 108346 CrossRef CAS.
- L. Zuo, J. Ding, C. Li, F. Lin, P. R. Chen, P. Wang, G. Lu, J. Zhang, L.-L. Huang and H.-Y. Xie, Chem. Sci., 2020, 11, 2155–2160 RSC.
- Z. Zhao, Q. Zong, J. Li, M. Jiang, K. Wang and Y. Yuan, Chem. Commun., 2023, 59, 3878–3881 RSC.
- Y. Sun, H. Zhao, F. Pu, H. Liu, L. Chen, J. Ren and X. Qu, Adv. Healthcare Mater., 2024, 2400899 CrossRef PubMed.
- K. Wang, M. Jiang, T. Li, Y. Liu, Q. Zong, Q. Xu, I. Ullah, Y. Chen, W. Xue and Y. Yuan, Adv. Mater., 2024, 36, 2402322 CrossRef CAS PubMed.
- Q. Wang, X. Li, Z. Cao, W. Feng, Y. Chen and D. Jiang, J. Am. Chem. Soc., 2024, 146, 8228–8241 CrossRef CAS PubMed.
- J. Zheng, H. Ge, D. Zhou, Q. Yao, S. Long, W. Sun, J. Fan, J. Du and X. Peng, Adv. Mater., 2023, 35, 2308205 CrossRef CAS PubMed.
- C. Liu, J. Niu, T. Cui, J. Ren and X. Qu, J. Am. Chem. Soc., 2022, 144, 19611–19618 CrossRef CAS PubMed.
- X. Yan, K. Li, T.-Q. Xie, X.-K. Jin, C. Zhang, Q.-R. Li, J. Feng, C.-J. Liu and X.-Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202318539 CrossRef CAS PubMed.
- A. M. Perez-Lopez, B. Rubio-Ruiz, V. Sebastian, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaria and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- M. Hoop, A. S. Ribeiro, D. Rösch, P. Weinand, N. Mendes, F. Mushtaq, X.-Z. Chen, Y. Shen, C. F. Pujante, J. Puigmartí-Luis, J. Paredes, B. J. Nelson, A. P. Pêgo and S. Pané, Adv. Funct. Mater., 2018, 28, 1705920 CrossRef.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. de Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869–10880 CrossRef CAS PubMed.
- Q. Yao, S. Gao, C. Wu, T. Lin and Y. Gao, Biomaterials, 2021, 277, 121119 CrossRef CAS PubMed.
- K. Neumann, A. Gambardella, A. Lilienkampf and M. Bradley, Chem. Sci., 2018, 9, 7198–7203 RSC.
- X. Ji, R. E. Aghoghovbia, L. K. C. De La Cruz, Z. Pan, X. Yang, B. Yu and B. Wang, Org. Lett., 2019, 21, 3649–3652 CrossRef CAS PubMed.
- L. Hu, B. Li, Y. Liao, S. Wang, P. Hou, Y. Cheng and S. Zhang, Bioorg. Chem., 2022, 129, 106167 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- E. Jalali and J. S. Thorson, Curr. Opin. Biotechnol., 2021, 69, 290–298 CrossRef CAS PubMed.
- E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710–7729 RSC.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 CrossRef CAS PubMed.
- C. Adam, A. M. Pérez-López, L. Hamilton, B. Rubio-Ruiz, T. L. Bray, D. Sieger, P. M. Brennan and A. Unciti-Broceta, Chem. – Eur. J., 2018, 24, 16783–16790 CrossRef CAS PubMed.
- S. Alonso-de Castro, A. Terenzi, S. Hager, B. Englinger, A. Faraone, J. C. Martinez, M. S. Galanski, B. K. Keppler, W. Berger and L. Salassa, Sci. Rep., 2018, 8, 17198 CrossRef PubMed.
- J. M. Fairhall, J. C. Camilli, B. H. Gibson, S. Hook and A. B. Gamble, Bioorg. Med. Chem., 2021, 46, 116361 CrossRef CAS PubMed.
- X. Zhang, R. F. Landis, P. Keshri, R. Cao-Milan, D. C. Luther, S. Gopalakrishnan, Y. Liu, R. Huang, G. Li, M. Malassine, I. Uddin, B. Rondon and V. M. Rotello, Adv. Healthcare Mater., 2021, 10, e2001627 CrossRef PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS PubMed.
- T. Peng and H. C. Hang, J. Am. Chem. Soc., 2016, 138, 14423–14433 CrossRef CAS PubMed.
- W. Shi, F. Tang, J. Ao, Q. Yu, J. Liu, Y. Tang, B. Jiang, X. Ren, H. Huang, W. Yang and W. Huang, Angew. Chem., Int. Ed., 2020, 59, 19940–19944 CrossRef CAS PubMed.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed.
- B. X. Li, D. K. Kim, S. Bloom, R. Y. C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed.
- J. B. Pawlak, G. P. P. Gential, T. J. Ruckwardt, J. S. Bremmers, N. J. Meeuwenoord, F. A. Ossendorp, H. S. Overkleeft, D. V. Filippov and S. I. van Kasteren, Angew. Chem., Int. Ed., 2015, 54, 5628–5631 CrossRef CAS PubMed.
- Y. Hao, J. Song, A. Ravikrishnan, K. T. Dicker, E. W. Fowler, A. B. Zerdoum, Y. Li, H. Zhang, A. K. Rajasekaran, J. M. Fox and X. Jia, ACS Appl. Mater. Interfaces, 2018, 10, 26016–26027 CrossRef CAS PubMed.
- Y. Zhang, M. Üçüncü, A. Gambardella, A. Baibek, J. Geng, S. Zhang, J. Clavadetscher, I. Litzen, M. Bradley and A. Lilienkampf, J. Am. Chem. Soc., 2020, 142, 21615–21621 CrossRef CAS PubMed.
- Y. Xu, P. Zou and A. E. Cohen, Curr. Opin. Chem. Biol., 2017, 39, 1–10 CrossRef CAS PubMed.
- S. Liu, C. Lin, Y. Xu, H. Luo, L. Peng, X. Zeng, H. Zheng, P. R. Chen and P. Zou, Nat. Chem., 2021, 13, 472–479 CrossRef CAS PubMed.
- P. E. Z. Klier, A. M. M. Gest, J. G. Martin, R. Roo, M. X. Navarro, L. Lesiak, P. E. Deal, N. Dadina, J. Tyson, A. Schepartz and E. W. Miller, J. Am. Chem. Soc., 2022, 144, 12138–12146 CrossRef CAS PubMed.
- P. E. Z. Klier, A. M. M. Gest, J. G. Martin, R. Roo, M. X. Navarro, L. Lesiak, P. E. Deal, N. Dadina, J. Tyson, A. Schepartz and E. W. Miller, J. Am. Chem. Soc., 2022, 144, 12138–12146 CrossRef CAS PubMed.
- R. Walther, J. Rautio and A. N. Zelikin, Adv. Drug Delivery Rev., 2017, 118, 65–77 CrossRef CAS PubMed.
- W.-C. Geng, J. L. Sessler and D.-S. Guo, Chem. Soc. Rev., 2020, 49, 2303–2315 RSC.
- H. Han, H. Wang, P. Jangili, M. Li, L. Wu, Y. Zang, A. C. Sedgwick, J. Li, X. He, T. James and J. S. Kim, Chem. Soc. Rev., 2023, 52, 879–920 RSC.
- J. Osredkar and N. Sustar, J. Clin. Toxicol., 2011, s3, 1, DOI:10.4172/2161-0495.S3-001.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- A. Steinbrueck, A. C. Sedgwick, J. T. Brewster, K.-C. Yan, Y. Shang, D. M. Knoll, G. I. Vargas-Zúñiga, X.-P. He, H. Tian and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 3726–3747 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- Y. Luo, X. He, Q. Du, L. Xu, J. Wang, W. Zhang, Y. Zhong, D. Guo, Y. Liu and X. Chen, Exploration, 2024 DOI:10.1002/EXP.20230134.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, J. Doungchawee, L. J. Castellanos-Garcia, K. N. Sikora, T. Jeon, R. Goswami, S. Fedeli, A. Gupta, R. Huang, C. M. Hirschbiegel, R. Cao-Milan, P. K. D. Majhi, Y. A. Cicek, L. Liu, D. J. Jerry, R. W. Vachet and V. M. Rotello, J. Controlled Release, 2023, 357, 31–39 CrossRef CAS PubMed.
- G. Tang, J. He, J. Liu, X. Yan and K. Fan, Exploration, 2021, 1, 75–89 CrossRef PubMed.
- B. Du, D. Li, J. Wang and E. Wang, Adv. Drug Delivery Rev., 2017, 118, 78–93 CrossRef CAS PubMed.
- M. Rong, J. Liu, Z. Sun, T. Li, Y. Li, C. Jiang and L. Lu, Angew. Chem., Int. Ed., 2023, 62, e202216822 CrossRef CAS PubMed.
- M. Hoop, A. S. Ribeiro, D. Rösch, P. Weinand, N. Mendes, F. Mushtaq, X.-Z. Chen, Y. Shen, C. F. Pujante, J. Puigmartí-Luis, J. Paredes, B. J. Nelson, A. P. Pêgo and S. Pané, Adv. Funct. Mater., 2018, 28, 1705920 CrossRef.
- W. Wang, X. Zhang, R. Huang, C. M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed.
- A. Gutiérrez-González, P. Destito, J. R. Couceiro, C. Pérez-González, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 16059–16066 CrossRef PubMed.
- G. M. Dal Forno, E. Latocheski, A. Beatriz Machado, J. Becher, L. Dunsmore, A. L. St John, B. L. Oliveira, C. D. Navo, G. Jimenez-Oses, R. Fior, J. B. Domingos and G. J. L. Bernardes, J. Am. Chem. Soc., 2023, 145, 10790–10799 CrossRef CAS PubMed.
- J. Chen, P. Ji, G. Gnawali, M. Chang, F. Gao, H. Xu and W. Wang, Acta Pharm. Sin. B, 2022, 13, 2736–2746 CrossRef PubMed.
- Y. Ma, Y. Zhou, J. Long, Q. Sun, Z. Luo, W. Wang, T. Hou, L. Yin, L. Zhao, J. Peng and Y. Ding, Angew. Chem., Int. Ed., 2024, 63, e202318372 CrossRef CAS PubMed.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- Y. Matsuno, Y. Atsumi, M. Alauddin, M. M. Rana, H. Fujimori, M. Hyodo, A. Shimizu, T. Ikuta, H. Tani, H. Torigoe, Y. Nakatsu, T. Tsuzuki, M. Komai, H. Shirakawa and K. I. Yoshioka, Sci. Rep., 2020, 10, 5388 CrossRef PubMed.
- Z. Yan, Y. Pan, G. Jiao, M. Xu, D. Fan, Z. Hu, J. Wu, T. Chen, M. Liu, X. Bao, H. Ke and X. Ji, J. Am. Chem. Soc., 2023, 145, 24698–24706 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |