
Enzyme/pH-sensitive nanoparticles based on poly(β-L-malic acid) for drug delivery with enhanced endocytosis†
Songyan
Guo
 *ab,
Youbei
Qiao
b,
Chaoli
Wang
b,
Yuming
Zhang
a,
Tiehong
Yang
b and
Hong
Wu
*b
*ab,
Youbei
Qiao
b,
Chaoli
Wang
b,
Yuming
Zhang
a,
Tiehong
Yang
b and
Hong
Wu
*b
aDepartment of Scientific Research, Shaanxi Provincial People's Hospital, Youyi West Street No. 256, Xi’an 710068, People's Republic of China. E-mail: spphgsy@126.com
bDepartment of Pharmaceutical Analysis, School of Pharmacy, Air Force Medical University, 169 West Changle Street, Xi’an 710032, People's Republic of China. E-mail: wuhong@fmmu.edu.cn
First published on 10th October 2024
Abstract
Nanoparticles (NPs) derived from branched copolymers of poly (β-L-malic acid) (PMLA) have been extensively investigated for drug delivery due to their high density of pendant carboxyl groups. This abundant functional group availability enhances their potential as effective drug delivery systems; however, the strong negative charge of PMLA poses a challenge in its uptake by cancer cells due to electrostatic repulsion. In this study, we developed novel enzyme- and pH-sensitive nanoparticles (EP-NPs) based on PMLA, demonstrating tumor-specific behavior and selective activation within tumor tissues. To enhance the cellular internalization of the nanoparticles, we incorporated transactivator of transcription (TAT). In summary, long-chain polyethylene glycol (PEG) was conjugated to PMLA to confer specificity to the TAT peptide. This was achieved using a tetrapeptide linker: alanine–alanine–asparagine–leucine (AANL), which serves as a substrate for legumain. Legumain is a highly conserved cysteine protease primarily found in lysosomes and blood vessels, initially discovered in legumes. It is markedly overexpressed in numerous solid tumors, as well as in endothelial cells and tumor-associated macrophages. The release of doxorubicin in tumor cells was sustained due to the low pH (5.0–5.5) and degradation of PMLA. The PEG modification optimized the particle size and shielded the nanoparticles from plasma proteins and detection by the reticuloendothelial system, thereby prolonging their long circulation time. Once the nanoparticles reached the tumor microenvironment, the AANL was cleaved by legumain, exposing the TAT peptide on the surface, which enhances cellular internalization. Both in vitro and in vivo efficacy studies demonstrated that these EP-NPs significantly inhibited tumor growth while exhibiting negligible systemic toxicity, thereby suggesting that the developed enzyme/pH-sensitive PMLA-based nanoparticle holds great promise as an anti-tumor drug delivery system.
1. Introduction
In recent years, nanoparticles (NPs) have been extensively studied to improve the selective delivery of drugs to the tumor site.1,2 NPs efficiently accumulate in solid tumors through passive or active targeting mechanisms to achieve the promotion of specific internalization of tumor cells.3 Passive targeting relies on the enhanced permeability and retention effect, a phenomenon discovered in 1986, which enables NPs to preferentially accumulate in tumor tissues due to their enhanced permeability and reduced clearance.4 This effect is based on abnormal features of blood vessels and the reduced lymphatic drainage in solid tumors, resulting in prolonged retention of NPs within the tumor mass and increased concentrations up to 10–50 times compared to normal tissues.5To develop effective nanoparticles, various biocompatible and biodegradable polymers have been evaluated as potential drug carriers.6 Among them, PMLA is an excellent candidate material due to its excellent biochemical properties.7 PMLA can be hydrolyzed to malic acid in vivo, and malic acid can be bio-assimilated and degraded to water and carbon dioxide through the Krebs cycle, which has significant biocompatibility and biodegradability.8 It also has carboxylic acid lateral functional groups, which can be used to chemically bind various molecules of interest such as drugs, targeting moieties, etc.9 However, the strong negative charge of PMLA increases the electrostatic repulsion with the negatively charged cell membrane, which lead to the low cellular uptake, and restricting its application as the drug carrier.10
To enhance controlled cellular internalization and drug release, numerous dual-trigger strategies have been developed. These approaches enable “smart” release by responding simultaneously or sequentially to two distinct physiological signals in the tumor microenvironment.11 Among them, enzyme-responsive nanodrug carriers for controlled release are rapidly developing. Enzymes are biological macromolecules with catalytic functions, playing key roles in both normal physiological processes and pathological progression.12–14 Legumain is a highly conserved lysosomal and vascular cysteine protease originally identified in legumes.15 It has specific substrate selectivity, cleaving peptides at the C-terminal side of asparagine residues.16 Legumain is chosen as a target for tumor therapy because its gene is significantly upregulated in various murine and human tumor tissues, while it is either absent or present at very low levels in the corresponding normal tissues.17 The function of legumain is to recognize a specific sequence (a tetrapeptide, AANL) in proteins and cleave the proteins to either activate or destroy them.15 Given its distinct expression pattern, legumain is an excellent candidate for designing anticancer drug carriers. Various legumain-based delivery systems have been developed and effectively utilized for this purpose.13,18–20 However, legumain-sensitive drug delivery systems using other promising materials are rarely reported.
While targeting ligands and stimuli-responsive elements in nanomedicines significantly enhances drug accumulation in tumors, inadequate cellular internalization remains a major challenge, particularly for macromolecular drugs such as proteins or RNA.21 To improve cellular uptake, other delivery patterns (cell-penetrating peptides, such as TAT) have been used to modify NPs, as they have been shown to be efficient in promoting translocation across the plasma membrane.22 However, the lack of selectivity in cell-penetrating peptides can lead to unwanted side effects, presenting a significant challenge.23 In response to these challenges, we designed a pH-sensitive drug delivery system based on PMLA, which overcomes the low cellular uptake due to the negative charge of PMLA with the TAT peptide. The long-chain PEG5000 linked with pH-sensitive hydrazone bonds was used to shield/deshield TAT peptides. Both in vitro and in vivo studies showed that the TAT peptide had good penetration into tumor cells. The TAT peptides had a good ability to enhance the cell membrane penetration of PMLA, used as a drug carrier. The selective specificity of TAT was further enhanced by pH-sensitive linkages associated with PEG shielding and deshielding.8 Nevertheless, the nanoconjugate had disadvantages such as poor redispersion and uneven particle size, which limited its use to improve anticancer efficacy.
In this study, we developed a nanoparticle system based on enzyme/pH-sensitive PMLA to enhance drug delivery, improve cellular uptake, and achieve controlled intracellular drug release. We achieved this by covalently conjugating pH-sensitive cis-aconitic anhydride-modified doxorubicin (CAD) to PMLA, resulting in an amphiphilic copolymer that readily forms polymeric nanoparticles. Additionally, we functionalized the nanoparticle surface with the cell-penetrating peptide TAT to enhance uptake by the target tumor cells. The final formulation was termed TAT–PEG2000–PMLA–CAD. The legumain-cleavable tetrapeptide (AANL) was used as a sensitive linker between PMLA and its long-chain PEG5000 block to serve as a steric shield for TAT in the nanoparticle (PEG5000–AANL–PMLA–CAD). The resulting pH/enzyme-sensitive nanoparticles exhibited a “Trojan horse” like behavior. During the extended circulation in the bloodstream under normal pH conditions, the PEGylated nanoparticle acted as a shield, effectively concealing the TAT peptide. This shielding strategy contributed to a prolonged circulation time, preventing the rapid clearance of the nanoparticle from the body. Upon reaching the tumor microenvironment, it was anticipated that the legumain enzyme, which is highly expressed extracellularly, would cleave the peptide linker. This cleavage event would lead to the exposure of the TAT peptide, facilitating enhanced intracellular penetration of the nanoparticles. Furthermore, the nanoparticles were engineered to release the conjugated drug rapidly in the endosomal/lysosomal, thereby enhancing the drug's therapeutic efficacy after cellular uptake. To our knowledge, this is the first study to introduce a biodegradable branched PMLA copolymer–drug conjugate nanoparticle as a legumain-sensitive system for cancer therapy. The structure and delivery strategy of this novel multifunctional PMLA-based nanoparticle system are depicted in Fig. 1.
 | ||
| Fig. 1 (A) The structure of nanomaterials. (B) The preparation of EP-NPs. (C) Illustration of the dual-sensitive properties, enzyme-sensitive deshielding TAT and pH-sensitive drug release. (D) Schematic illustration of the dual-sensitive DOX-loaded nanoparticle with the enzyme-sensitive function for effective tumor-targeted drug delivery, enhanced cellular uptake, and intracellular drug release. | ||
2. Materials and methods
2.1 Materials
PMLA was synthesized following our previous methodology.23 The molecular weight of PMLA was 10.79 kDa (Fig. S1, ESI†). We have confirmed the linearity of PMLA in our previous studies using FTIR.24 Doxorubicin hydrochloride (DOX·HCl) and cis-aconitic anhydride (CAA) were obtained from Aladdin (Shanghai). TAT–cysteine peptide (TATp–Cys) with one reactive thiol group and AANL peptide were sourced from Changxi Biotech (Shanghai). Legumain and anti-legumain were purchased from Wuhan Biological Company. NH2–PEG2000–MAL, mPEG2000–NH2, and mPEG5000–NH2 were purchased from Xi’an Ruixi Biological Technology Co., Ltd. N,N-Dicyclohexyl carbodiimide (DCC), N-hydroxy succinimide (NHS), and MTT were obtained from Dojindo (Japan). DAPI was purchased from Sigma-Aldrich. Fetal bovine serum (FBS), RPMI1640, and L15 medium were obtained from Hyclone Cell Culture and Bioprocessing (Thermo Scientific, USA). HPLC-grade acetonitrile and methanol were sourced from Merck (Darmstadt, Germany). Unless otherwise specified, other reagents were of general laboratory grade and purchased from Sigma (St. Louis, MO). Ultraviolet-visible (UV-vis) spectroscopy was performed using a Shimadzu Corporation instrument (Kyoto, Japan), and GPC analysis was conducted for characterization purposes.2.2 Cells and animals
MDA-MB-231 murine breast cancer cell lines and L929 were obtained from the Shanghai Institutes for Biological Sciences. Cells were grown in L15 and RPMI-1640 (Hyclone) supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin, 100 units mL−1, plus 100 μg mL−1 streptomycin) at 37 °C in a 5% CO2 atmosphere.Female BALB/c mice (18–20 g) were obtained from the Hubei Research Center of Laboratory Animals. Animals were maintained in a room at 22–24 °C with a light/dark cycle of 12/12 h and 55–60% relative humidity. Animals were fasted for 12 h, but were allowed free access to water before injection. 106 MDA-MB-231 cells were injected into the right foreleg of each 5-week-old female nude mouse subcutaneously. After one week, we divided the nude mice into 6 groups randomly, and each group consisted of 3 mice. Then each group received intravenous injection of drug-loaded nanoparticles, free DOX, and normal saline, respectively. After this, the tumor volumes were measured and calculated every three days with the formula W2 × L/2 (W, shorter diameter; L, longer diameter) (mm3). When the tumor diameter reached 1.5 cm, mice were sacrificed by intraperitoneal injection of 130 mg kg−1 pentobarbital. Then the tumors were removed and weighed. All animal experiments were carried out with the approval of the Medical Ethics Committee of Shaanxi Provincial People's Hospital (2023K-S215) and the Animal Care and Use Committee of Air Force Medical University(KY20212477-C-1).
2.3 Synthesis of CA-DOX (CAD)
CAD was synthesized by the ring-opening reaction between DOX and CAA using triethylamine as a catalyst. As shown in Scheme S1 (ESI†), DOX (50 mg) and CAA (50 mg) were dissolved in 10 mL of anhydrous dimethylformamide. The solution was stirred for 24 h at room temperature in the dark. The formed CAD was lyophilized. The chemical structures of CAD were characterized by 1H-NMR at 400 MHz (Bruker, Ettlingen, Germany) in D2O and MS-HPLC (Scheme S1, ESI†).2.4 Synthesis of Poly(β-benzyl malate) (PBM)
PBM was synthesized by ring-opening polymerization and starting from L-aspartic acid with a method similar to that previously described by our group.(Scheme S2, ESI†).25,262.5 Synthesis of PEG2000–PMLA–CAD (PPC)
DCC (22.9 mg, 0.12 mmol) and NHS (13.5 mg, 0.12 mmol) were added to the stirred solution of PBM (5 g, 0.1 mmol) in DMSO (20 mL). The reaction mixture was stirred for 12 h at room temperature. PEG2000–NH2 was then added to the solution and stirred for 12 h to give PEG2000–PBM. PEG2000–PMLA was obtained by hydrogenation of PBM–PEG2000. In addition, CAD was dissolved in 5 mL DMSO and stirred with 2-HEH for 12 h to synthesize CAD–NH2. Finally, the CAD–NH2 solution was added to the PMLA–PEG2000 solution and stirred for 24 h. The resulting PEG2000–PMLA–CAD was lyophilized and stored at 4 °C for further use (Scheme S3, ESI†).2.6 Synthesis of PEG5000–AANL–PMLA–CAD (PAPC)
The preparation of PEG5000–AANL–PMLA–CAD involved two steps: (i) synthesis of PEG5000–AANL and (ii) synthesis of PEG5000–AANL–PMLA–CAD (Scheme S4, ESI†). Briefly, in a typical procedure, PEG5000–NH2 (50.0 mg, 0.01 mmol) was first dissolved in 10 mL DMSO, followed by DCC (0.012 mmol) and NHS (0.012 mmol). After reacting for 12 h at room temperature, AANL was added to the mixture for another 12 h reaction. After that, the PEG5000–AANL was obtained. PBM was dissolved in 10 mL DMSO and reacted simultaneously with DCC and NHS. The PEG5000–AANL solution was then added to the PBM solution and stirred for 12 h. PMLA–AANL–PEG5000 was obtained by hydrogenation of PBM–AANL–PEG5000. In addition, CAD was dissolved in 5 mL DMSO and stirred with 2-HEH for 12 h to synthesize CAD–NH2. Finally, the CAD–NH2 solution was added to the PMLA–AANL–PEG5000 solution and stirred for 24 h. The resulting PEG5000–AANL–PMLA–CAD was lyophilized and stored at 4 °C for further use.2.7 Synthesis of TAT–PEG2000–PMLA–CAD (PPTC)
Briefly, NH2–PEG2000–Mal (20.0 mg, 0.01 mmol) was first dissolved in 3 mL DMSO followed by TAT (14.0 mg, 0.01 mmol). After the reaction at room temperature for 12 h, NH2–PEG2000–TAT was obtained. PBM was dissolved in 10 mL DMSO and reacted simultaneously with DCC and NHS. The NH2–PEG2000–TAT solution was then added to the PBM solution and stirred for 12 h. PMLA–PEG2000–TAT was obtained by hydrogenation of PBM–PEG2000–TAT. In addition, CAD was dissolved in 5 mL DMSO and stirred with 2-HEH for 12 h to synthesize CAD–NH2. Finally, the CAD–NH2 solution was added to the PMLA–PEG2000–TAT solution and stirred for 24 h. The resulting TAT–PEG2000–PMLA–CAD was lyophilized and stored at 4 °C for further use (Scheme S5, ESI†).2.8 Preparation and characterization of the nanoparticle
For the enzyme/pH-sensitive nanoparticles (EP-NPs), PAPC (1 mg), PPTC (0.1 mg) and PPC (8.9 mg) were added to ultrapure water (10 mL) to prepare the nanoparticles. The enzyme-sensitive behavior of EP-NPs was analyzed as follows. The nanoparticles were incubated with 0.15 mol L−1 Tris-HCl buffer (pH 7.4), which was shaken in a 37 °C water bath for 24 h. The mixture was then lyophilized. The pH-responsive and enzyme-sensitive behaviors of the EP-NPs were investigated as follows. 1H-NMR spectra were performed on a Bruker AV 400 NMR spectrometer in d-DMSO. Dynamic laser scattering (DLS) and transmission electron microscopy (TEM) measurements were performed as in our previous study.2.9 In vitro drug release
The drug release experiment was also tested according to our previous report with a slight modification. 10 mg of freeze-dried EP-NPs powder was dispersed in different media (2 mL) and then transferred to a dialysis bag (MWCO 1000) in a conical flask (containing 50 mL of release media). The separation media contained PBS (pH 7.4, 6.3 or 5.0). The shaker bed (100 rpm and 37 °C) was used to incubate the release media during the release study. At predetermined time intervals within 48 hours, 1 mL of the solution in the tube was removed for HPLC detection and replaced with 1 mL of fresh medium.2.10 PEG dissociation in the presence/absence of legumain
PEG5000–AANL–PMLA aqueous solution (5.0 mg mL−1, 500 μL) was added to 500 mL citric acid buffer (pH 6.3, containing 50 mM citric acid, 1 mM EDTA) and PBS (pH 7.4) with or without legumain (50 ng). The mixture was incubated at 37 C for different time points (0, 1, 2, 4 and 8 hours). The solutions were diluted with methanol (1.0 mL) and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes at room temperature. The supernatant was collected and diluted to 4.0 mL with deionized water. The resulting solution was transferred to an ultrafiltration centrifuge tube (MWCO 1000 Da), lyophilized, and stored at 4 °C for further GPC detection.
000 rpm for 10 minutes at room temperature. The supernatant was collected and diluted to 4.0 mL with deionized water. The resulting solution was transferred to an ultrafiltration centrifuge tube (MWCO 1000 Da), lyophilized, and stored at 4 °C for further GPC detection.
2.11 In vitro cytotoxicity assay
MDA-MB-231 and L929 cells were used for in vitro cytotoxicity assays. The cytotoxicity study of free DOX and dual-response nanoparticles with MDA-MB-231 cells and L929 cells. The concentrations of free DOX and DOX in nanoparticles ranged from 0.5 to 8 μg ml−1 when free DOX and dual-sensitive nanoparticles were co-cultured with MDA-MB-231 and L929 cells for 48 h.2.12 Immunohistochemistry
Paraffin sections were dewaxed, hydrated, and incubated overnight at 4 °C with the primary antibody. The primary anti-legumain antibody (AB clonal Technology, Wuhan, Hubei, China) was used at a 1:200 dilution. The next day, slides were then washed and incubated with the secondary antibody for one hour at 37 °C, followed by a one-minute incubation with 3,3′-diaminobenzidine and hematoxylin. The pictures were captured using a light microscope at the magnification of 200× and 400×.
2.13 Western blot
Cells were collected with PBS and centrifuged at 3000 g for 5 minutes at 4 °C. The pellets were resuspended with RIPA lysis and extraction buffer for 30 min at 4 °C, the total proteins (the supernatants) were collected after centrifuged at 12000g for 15 minutes. The protein concentration was measured using a BCA kit. Total proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, MA, USA) at a constant current of 200 mA for 2 hours. The membranes were then blocked with 5% non-fat milk for 1 hour. After this, the membranes were incubated with the primary antibody of anti-legumain antibody and anti-actin antibody at 4 °C overnight. The next day, after three washes with TBST, the membranes were incubated with secondary antibodies for 1 hour at room temperature and exposed. Images were captured using FluorChem HD2 (Alpha Innotech, San Francisco, CA, USA).
2.14 In vitro cellular uptake
The cellular uptake and distribution of nanoparticles in MDA-MB-231 cells and L929 cells were investigated by confocal laser scanning microscopy (CLSM) and flow cytometric analysis (FC), respectively. For CLSM, MDA-MB-231 cells and L929 cells were seeded in a 35-mm cell culture dish and were incubated at 37 °C with 5% CO2 for 24 h. DOX solution and nanoparticles (5 μg mL−1 DOX) were added, and the cells were incubated for 8 h at pH 7.4 and 6.3 within or without legumain, separately. After incubation, the sample solution was removed. Cells were rinsed with PBS (pH 7.4) at least thrice and fixed with 4% formaldehyde for 10 min. The remaining liquid was dried completely. VECTASHIELD mounting medium with DAPI (5 μg mL−1) was added to prevent the fading of fluorescence and dye nuclei blue. Fluorescence was observed using the confocal laser scanning microscope (CLSM) at 405 nm for DAPI and 488 nm for DOX.For flow cytometric analysis, cells were cultured in 6-well plates at 5 × 105 cells per well for 24 h. Cells were trypsinized and collected by centrifugation at 1200 rpm for 3 min. Media containing DOX-loaded nanoparticles (DOX: 5 μg mL−1) at different pH values were added to the cells, and incubation was performed for 8 h. The fluorescence was then measured using a flow cytometer (BD FACS Calibur) at predetermined times.
2.15 In vivo tumor growth inhibition study
The tumor-bearing mice were randomly divided into six groups (n = 3 for each group) and were treated by intravenous injection with saline, free DOX, EP-NPs, P-NPs, E-NPs and C-NPs, respectively. The injected amount for different nanoparticles was 200 μL saline with DOX dose of 5 mg kg−1 body weight, and administered every 3 days. Tumor size and body weight were measured every day after treatment. Tumor samples from sacrificed mice were collected and stained with hematoxylin and eosin (H&E) for histological examination. Then, each section was visualized under light microscopy.2.16 Statistical analysis
For the comparative analysis of continuous variables across multiple groups, statistical significance was evaluated using one-way analysis of variance (ANOVA) with Tukey post-hoc testing, employing GraphPad Prism 9 software (GraphPad Software, CA). The disparity between the two groups was assessed through Student's t-test. Data were presented as the mean value ± standard deviation (s.d.) from three independent experiments, and each experiment was replicated at least three times. The sample sizes were specified within each figure or figure legend. Statistical significance was denoted by *p < 0.05, **p < 0.01, and ***p < 0.001.3. Results and discussion
3.1 Synthesis and characterization of copolymers
Linear PMLA polymers have gained increasing attention as carriers in cancer therapy. The fusion of biodegradable PMLA–PEG copolymer–drug conjugates with AANL peptide in the main chain has opened up new possibilities for the development of safe and efficient PMLA-based drug delivery vehicles. In this study, we aimed to design a nanoparticle based on a branched PMLA–PEG copolymer–DOX conjugate as a nanodrug delivery system (NDDS) for breast cancer therapy. The synthetic flexibility of PMLA copolymers has proven effective when combined with anticancer drugs. Furthermore, the branched architecture of the functional nanoparticle offers nanoscale advantages, including high accumulation in tumor tissues. In addition, the nanoparticles demonstrate enhanced stability and convenience compared to nanoconjugates. Importantly, these nanoparticles possess stimulus-responsive and pH-sensitive drug release properties, which significantly enhance their therapeutic efficacy.In our previous studies, DOX was directly conjugated to PMLA via an amide bond, and the resulting nanoconjugates exhibited pH-sensitive drug release patterns.25 However, it's uncertain whether the released drug was free DOX or not; the pH-sensitive phenomenon was probably caused by the hydrolysis of PMLA in an acidic environment. In this study, DOX was conjugated to PMLA using a pH-sensitive bond, allowing for the targeted release of DOX specifically within the endosome or lysosome following cellular uptake. Fourier transform infrared spectroscopy (FTIR) analysis (Fig. S2A, ESI†) revealed the disappearance of peaks at 1250 cm−1 and 3520 cm−1, corresponding to H–N–H vibrations. As shown in Fig. S2B (ESI†), Peaks 6.52 ppm and 6.21 ppm correspond to the protons of –COCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) in the cis- and trans-forms of CAD, respectively. The emergence of a peak at 1547 cm−1, characteristic of CAD, indicated the presence of amide bond stretching vibrations. Additionally, the successful synthesis of CAD was confirmed by the presence of a peak at 701 in the mass spectrometry (MS) analysis (Fig. S2C, ESI†).
in the cis- and trans-forms of CAD, respectively. The emergence of a peak at 1547 cm−1, characteristic of CAD, indicated the presence of amide bond stretching vibrations. Additionally, the successful synthesis of CAD was confirmed by the presence of a peak at 701 in the mass spectrometry (MS) analysis (Fig. S2C, ESI†).
The nanoparticle was formed with three copolymers. The obtained PAPC, PPTC and PPC were identified by 1H NMR and FTIR (Fig. S3–S5, ESI†). The drug conjugation efficiencies of PAPC, PPTC and PPC were 24.3%, 21.2% and 26.8% (w/w), respectively. As shown in Fig. S3 (ESI†), the signals of PAPC at 2.95 (g) and 5.38 (g) ppm were assigned to methylene and methane protons of PMLA, respectively. The signals at 3.53 ppm were assigned to the protons of –CH2– of PEG. The signals at 3.72(b) and 0.82–1.25(c, d, e and f) ppm were assigned to AANL. The signals at 4.19 and 7.93 ppm were assigned to the benzene and methyl protons of CAD, respectively. The 1H-NMR spectra indicated the successful synthesis of the polymer PAPC, PPC and PPTC.
3.2 Synthesis and characterization of nanoparticles
The dynamic light scattering (DLS) measurements revealed that the average particle sizes of the DOX-loaded nanoparticles were 129.7 ± 3.5 nm for EP-NPs, 116.9 ± 2.6 nm for P-NPs, 143.1 ± 3.2 nm for E-NPs, and 132.0 ± 4.2 nm for insensitive C-NPs (Fig. 2A–C and Table S1, ESI†). TEM micrographs confirmed the spherical morphology of both nanoparticle types. The apparent mean diameters observed through TEM were approximately 70 nm. The reason why the sizes obtained via DLS were slightly larger than those determined by TEM is that the TEM measurement detected the diameter of the bare nanoparticles, while the diameter measured by DLS is the hydrodynamic diameter, which contains the solvated ions and organic layers.27 The reasonable diameter of prodrug nanoparticles is about 100 nm, which achieves excellent selective accumulation in tumor areas through enhanced permeability and retention effects.28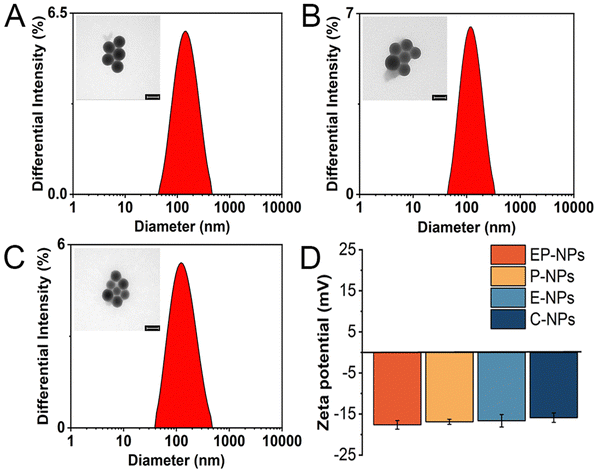 | ||
| Fig. 2 The DLS and TEM results of nanoparticles (A) EP-NPs (B) E-NPs and (C) P-NPs. Scale bar is 100 nm. (D) The zeta potential of nanoparticles. | ||
As shown in Fig. 2D and Table S1 (ESI†), the zeta potential of these nanoparticles (EP-NPs, P-NPs, E-NPs, C-NPs) was −17.63 ± 1.06 mV, −16.89 ± 0.63 mV, −16.65 ± 1.51 mV and −15.89 ± 1.15 mV, respectively. These results indicated that there is no significant difference between each groups, as the only differences among the nanoparticles were in the enzyme-sensitive peptides and pH-sensitive bonds, with the rest of the structure being essentially the same. The zeta potential of nanomaterials showed that the zeta potential of nanoparticles coated with PEG and containing DOX was closer to neutral compared with PMLA (−31 mV). The zeta potential of PPTC is positive due to the positive charge carried by TAT peptides. (Fig. S6, ESI†). This highlights the significance of using long-chain PEG5000 to shield TAT, preventing non-selective membrane penetration during prolonged circulation in the body.29 Moreover, the stability of nanoparticle EP-NPs under different conditions was investigated. As shown in Fig. 3A–C, the particle size of EP-NPs in pH 5.0 PBS buffer significantly increased over 1, 2, and 3 hours, along with an increase in PDI. This was attributed to the cleavage of the cis-aconitic acid bond in EP-NPs, promoting the release of DOX and leading to decreased nanoparticle stability. After 3 hours, EP-NPs had begun to undergo self-degradation, and smaller particles (<10 nm) were detected by the particle size analyzer. Additionally, TEM results showed that the boundaries of the nanoparticles were unclear, leading to the dissociation of the nanoparticles. After 4 h, NPs could no longer be detected by the particle size analyzer, which may be due to the complete dissociation of NPs and self-degradation of PMLA (Fig. 3D). In contrast, the NPs in PBS buffer at pH 7.4 and RPMI 1640 cell culture medium remained very stable, with particle sizes of 137.2 nm and 150.3 nm, respectively, and a PDI increase of about 0.25 at 48 hours. This indicates that EP-NPs can maintain a relatively stable state under normal physiological conditions and quickly dissociate in the low pH environment inside the cell to release DOX after entering the cell (Fig. 3E).
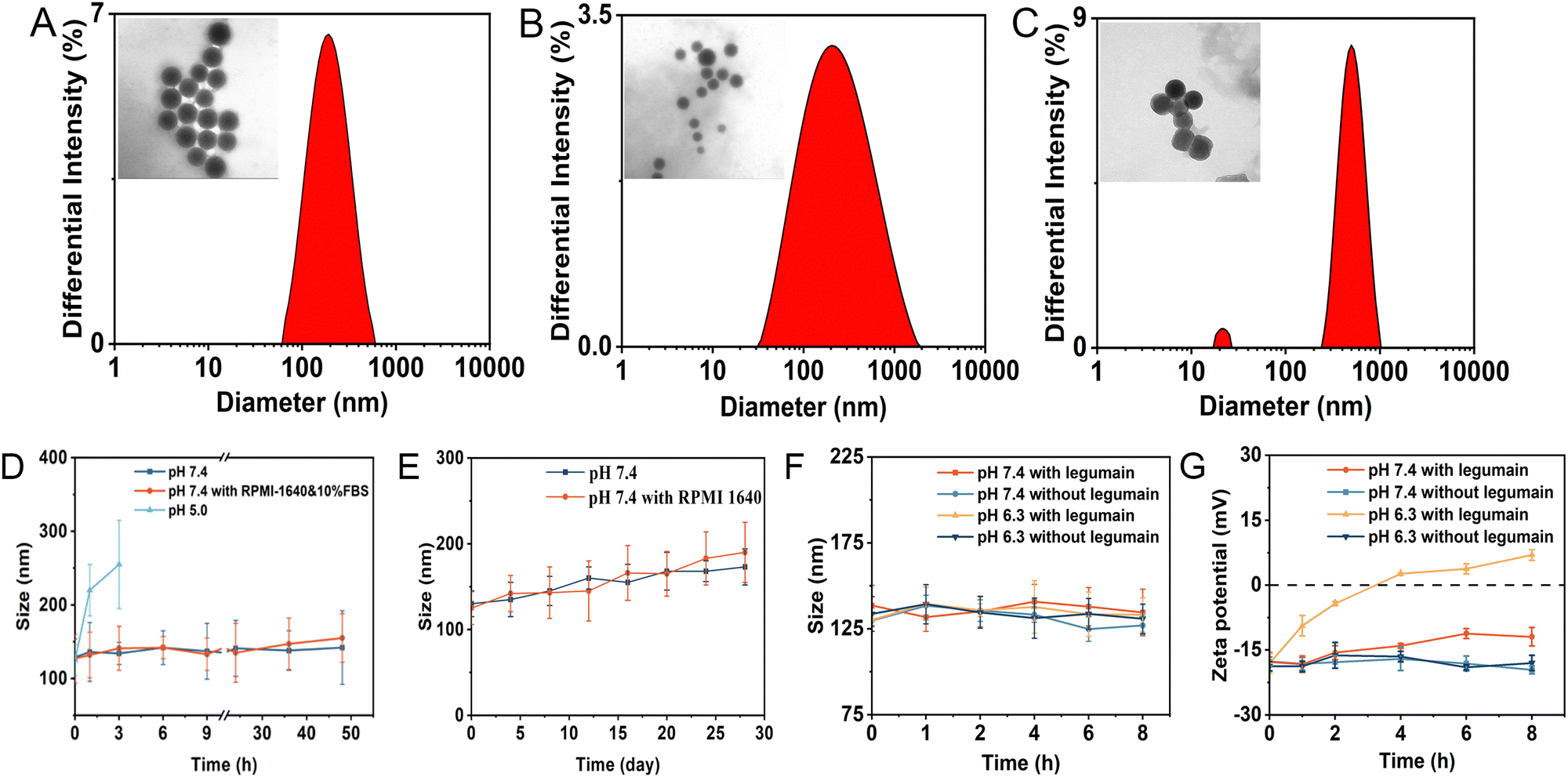 | ||
| Fig. 3 (A) The DLS and TEM results of EP-NPs at pH 5.0 for 1 h. (B) The DLS and TEM results of EP-NPs at pH 5.0 for 2 h. (C) The DLS and TEM results of EP-NPs at pH 5.0 for 3 h. Scale bar is 100 nm. (D) Changes in the particle size of EP-NPs when incubating at PBS (pH 7.4), RPMI 1640 medium containing 10% FBS and 5.0 for different time periods. (E) Changes in the particle size of EP-NPs upon the incubation with PBS (pH 7.4) and RPMI 1640 medium containing 10% FBS for 28 days. (F) The DLS results of EP-NPs which were incubated at pH 7.4 and 6.3 with or without legumain for different time periods. (G) The zeta potential changes of EP-NPs which was incubated at pH 7.4 and 6.3 with or without legumain for different time periods. | ||
The size of EP-NPs in the absence or presence of the legumain at pH 7.4 and 6.3 over 8 hours was investigated. As shown in Fig. 3F, the particle size of EP-NPs did not exhibit a significant difference in particle size in the presence or absence of legumain at different pH. Therefore, we expanded the particle size of the nanoparticles under different conditions. In the presence of legumain, EP-NPs showed a significant increase in potential at pH 6.3 (Fig. 3G). These data suggest that as the length of PEG increases, PEG forms a steric protective layer that neutralizes the surface charge of TAT. Under acidic conditions, legumain cleaves the AANL, leading to PEG deshielding, which exposes TAT and results in increased cell penetration of EP-NPs.
3.3 Drug release
It is well-known that the maleic acid derivatives can undergo degradation at mildly acidic pH values.30 In this study, we utilized a maleic acid amide derivative, cis-aconitic acid, to achieve pH-sensitive degradability for the controlled release of the drug. cis-Aconitic acid possesses one carboxyethyl and one hydrogen substituent, and exhibits favorable degradability under endosomal/lysosomal pH conditions. To investigate the release behavior of EP-NPs and demonstrate their pH sensitivity, in vitro DOX release profiles were assessed in PBS at pH 7.4, 6.3, and 5.0, representing the conditions found in normal physiological tissues, the tumor microenvironment, and intracellular endosomes/lysosomes, respectively. In this study, the cleavage of CAD was investigated firstly. As shown in Fig. 4A, the distinct peaks were observed for free DOX at an elution time of 4.06 min and CAD at an elution time of 1.62 min. Furthermore, when CAD was incubated at pH 5.0, significant degradation was observed with a half-life of approximately 1 h. The intensity of CAD peaks decreased, while the intensity of free DOX peaks significantly increased at pH 5.0. In contrast, no degradation was observed because the cis-aconitate bonds in CAD remained highly stable at pH 7.4 and 37 °C for 12 hours (Fig. 4B). These results suggest that the cis-aconitate bonds in CAD can be selectively cleaved under the endo/lysosomal conditions found in tumor cells, leading to the release of free DOX. In contrast, the cis-aconitate bond remains stable under normal physiological conditions and in the extracellular environment.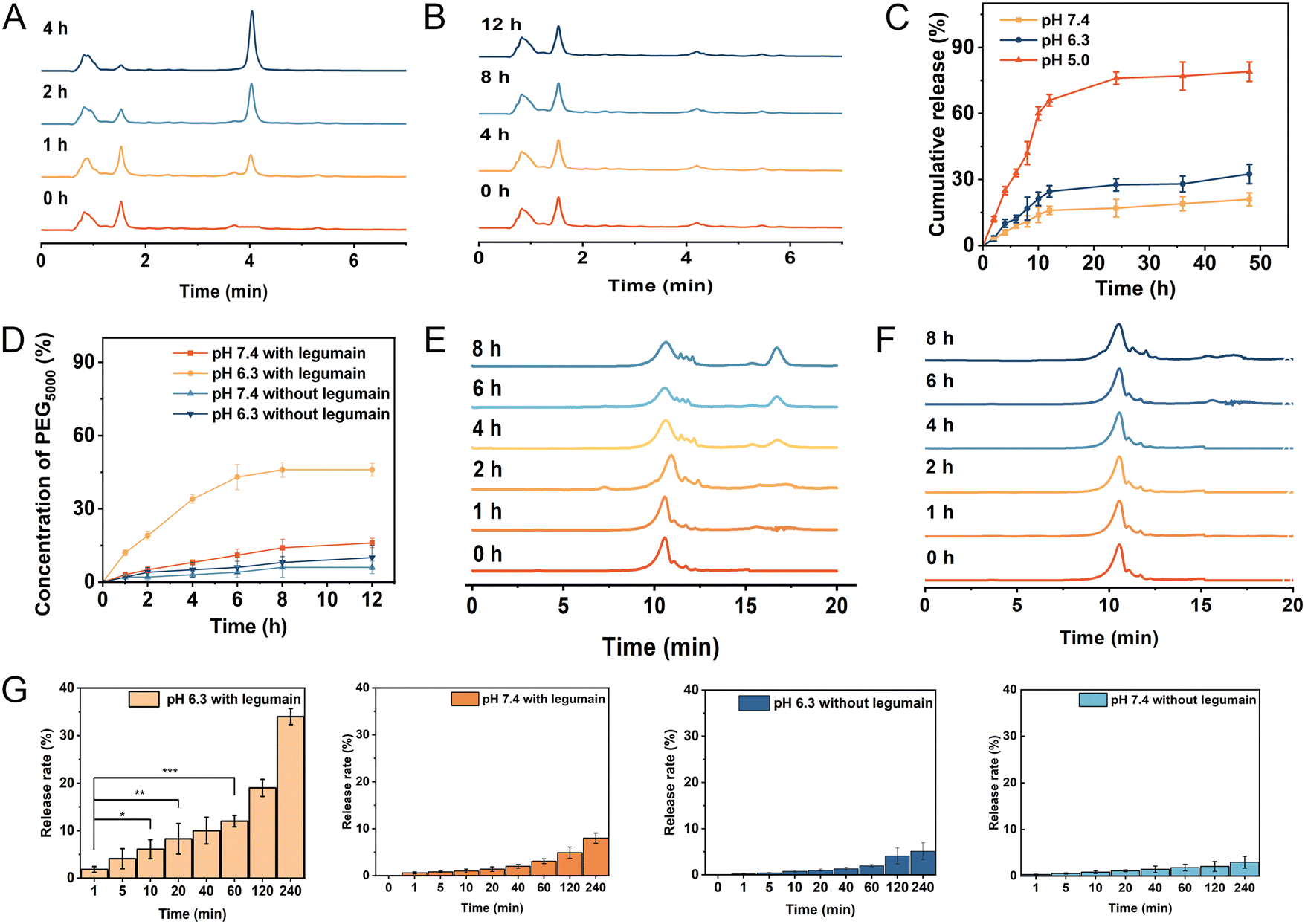 | ||
| Fig. 4 (A) Degradation of CAD incubated at PBS solution (pH 5.0) for different periods of time detected by HPLC. (B) Degradation of CAD incubated at PBS (pH 7.4) solution for different periods of time detected by HPLC. (C) Release profiles of DOX from the nanoparticles in PBS (pH 5.0, pH 6.3 or pH 7.4) at 37 °C. (D) Release profiles of PEG from the PEG–AANL–PMLA in PBS (pH 6.3 and pH 7.4 with legumain) at 37 °C. (E) Degradation of PEG–AANL–PMLA incubated at pH 6.3 PBS solution for different periods of time detected by GPC. (F) Degradation of PEG–AANL–PMLA incubated at pH 7.4 PBS solution for different periods of time detected by GPC. (G) Release profile of PEG5000–AANL–PMLA with or without legumain at different pH in 4 h. | ||
Next, the drug release of EP-NPs was observed. The EP-NPs exhibited minimal release at pH 7.4 and 6.3, releasing less than 14.3% and 19.5% of the loaded DOX, respectively (Fig. 4C). This finding suggests that EP-NPs possess an extended blood circulation time with minimal drug leakage. However, the cumulative release of DOX from EP-NPs significantly increased to over 83% when the pH dropped from 6.3 to 5.0. The observed burst release of EP-NPs at pH 5.0 can be attributed to the rapid cleavage of the acid-sensitive linkage in the acidic environment. This stimuli-responsive release profile of EP-NPs helps minimize drug loss during circulation while facilitating payload release at the target site, ultimately enhancing therapeutic efficacy and reducing potential side effects in vivo.
3.4 Legumain-triggered DePEGylation of EP-NPs
In this study, long chain PEG5000 was linked to PMLA through an enzyme-sensitive linker (AANL), enabling the controlled release of PEG5000 in response to the tumor microenvironment. The cleavage efficiency of PEG5000 needed to be investigated. As shown in Fig. 4D, approximately 30% of PEG was cleaved after a 4-hour reaction in the presence of 100 ng mL−1 of legumain at pH 6.3. The cleavage rate subsequently decreased, resulting in a 45% removal of PEG after 8 h of cleavage. In contrast, the presence of legumain did not induce the cleavage of the copolymer at pH 7.4 because legumain is only active in acidic environments.31 In Fig. 4E, PEG exhibited a peak at an elution time of 17.1 min, while PMLA–AANL–PEG showed a peak at an elution time of 11.7 min. Upon incubation of PMLA–AANL–PEG with legumain at pH 7.4 and pH 6.3, significant degradation was observed. The intensity of the PMLA–PEG peaks decreased, while the intensity of the PEG peaks significantly increased in the presence of legumain at pH 6.3. However, no degradation was observed, and the AANL moiety within PMLA–AANL–PEG remained stable in the presence of legumain at pH 7.4 and 37 °C for 8 hours (Fig. 4F). This finding suggests that the PEG surface on the nanoparticles can be efficiently cleaved by legumain in an acidic environment. The release rate of PEG5000–AANL–PMLA without legumain at 240 minutes was not significantly different in the confidence interval. Meanwhile, the release rate of PEG5000–AANL–PMLA with legumain at pH 7.4 also had no significant difference. In contrast, the release rate of PEG5000–AANL–PMLA with legumain at pH 6.3 was highly significantly different in the confidence interval at 240 minutes (Fig. 4G).3.5 Legumain expression level at the tumor sites
Legumain expression levels were examined in tumor-bearing mice tissues of MDA-MB-231 and MDA-MB-231 cells. As shown in Fig. 5A, the results of this analysis showed that legumain expression is highly upregulated in tumor-bearing mice tissues and MDA-MB-231 cells through IHC. Moreover, the legumain expression levels of the MDA-MB-231 group were compared with the L929 group by western blot analysis. The results showed that legumain was more stably expressed in MDA-MB-231 cells, but was lowly expressed in L929 cells (Fig. 5B). | ||
| Fig. 5 (A) Immunohistochemical staining of legumain in paraffin-embedded breast cancer tissues(left 200×, right 400×). (B) Expression of legumain validated by western blot analysis of breast tumor samples (MDA-MB-231) and mouse fibroblasts cell samples (L929). (C) Confocal microscopic images of cellular internalization of EP-NPs by MDA-MB-231 and L929 cells incubated at pH 7.4 and 6.3 for 8 h. DAPI (blue) was used to stain cell nucleus. The scale bar is 60×. Flow cytometry analysis of cellular uptake of EP-NPs by MDA-MB-231 cells and L929 cells at pH 7.4 and pH 6.3 conditions for 8 h. | ||
3.6 In vitro cellular uptake evaluation
Cellular uptake behavior was investigated through confocal laser scanning microscopy (CLSM) and flow cytometry analysis in both a breast cancer cell line (MDA-MB-231) and a normal cell line (L929) following an 8-hour incubation with the developed NPs at pH 7.4 or 6.3. Given that DOX possesses intrinsic fluorescence, it served as a fluorescent probe for CLSM observation. The cell nuclei were stained with DAPI (blue) to facilitate subcellular visualization. As shown in Fig. 5C, the overlap between DOX and the nucleus (DAPI) is due to the fact that DOX acts on the nucleus by passive diffusion. The cellular uptake of EP-NPs and E-NPs was higher compared to that of P-NPs and C-NPs in the MDA-MB-231 cell line. This enhanced cellular uptake in MDA-MB-231 cells was more pronounced than L929, potentially due to the overexpression of legumain in MDA-MB-231 cells. This suggests that the legumain-triggered dePEGylation process led to the exposure of TAT. Specifically, due to the insensitive chemical bond between the nanocarrier and the antitumor drug, the E-NPs are internalized by the cells through TAT and trapped in subcellular compartments, such as endosomes and lysosomes. The tumor microenvironment is characterized by a low pH range (4.0–6.5), which provides optimal catalytic activity for acidic proteases such as legumain.14,32 In the MDA-MB-231 cell line, E-NPs demonstrated a slightly higher cellular uptake due to TAT exposure, while the uptake of EP-NPs was the highest. Furthermore, the cellular uptake of EP-NPs was significantly enhanced at pH 6.3. Moreover, the higher uptake of PNP and CNP at pH 6.3 compared to pH 7.4 is due to the PEG5000 being attached to the PMLA terminal group via a non-enzyme-sensitive amide bond. Based on our previous study, the amide bond can undergo slight cleavage under acidic conditions.33 Additionally, combining the drug release experiments with previous findings, it is evident that PMLA also undergoes slight degradation in acidic conditions, leading to DOX release and subsequent passive diffusion into the cells.34 These factors contribute to the slightly enhanced cellular uptake of PNP and CNP.Cellular uptake was further evaluated quantitatively by flow cytometry. The average fluorescence intensity in all these cells after EP-NPs and E-NPs treatment was significantly improved (p < 0.05) compared to that of the cells treated with P-NPs and C-NPs at pH 6.3. In addition, the average fluorescence intensity of MDA-MB-231 after EP-NPs treatment was significantly higher (p < 0.05) than that of L929 at pH 7.4 or 6.3, which was consistent with the CLSM observations.
3.7 In vitro cytotoxicity study
The in vitro cytotoxic effects of EP-NPs and free DOX on MDA-MB-231 and L929 were evaluated using the MTT colorimetric assay at pH 6.3 and 7.4 because it is known that tumor cells develop unique microenvironments, such as slightly acidic (pH 4.0–6.0) and a low pH (4.0–6.5) ensures optimal catalytic activity of legumain.28 The MDA-MB-231 and L929 cell lines were selected in this study because the MDA-MB-231 cells expressed high levels of active legumain and the L929 cells had only weak expression of active legumain in vitro (Fig. 5B).32 The cytotoxicity results were shown in Fig. 6A–D and Table S2 (ESI†). The IC50 of EP-NPs against MDA-MB-231 cells at pH 7.4 (27.52 μg mL−1) was much higher than that at pH 6.3 (1.08 μg mL−1). In addition, the IC50 of EP-NPs against MDA-MB-231 cells was much lower than that of and the L929 cells (9.12 μg mL−1) at pH 6.3. Furthermore, the IC50 of E-NPs against MDA-MB-231 cells was lower than that of P-NPs and C-NPs, although not significantly different from L929 cells at pH 6.3. This result suggested that legumain plays a role in the intracellular uptake of E-NPs, but the presence of an insensitive amide linker prevents a large amount of DOX from being released in endosomes and lysosomes. | ||
| Fig. 6 Cytotoxicity of free DOX and various nanoparticles against MDA-MB-231 cells at different concentrations of DOX under (A) pH 6.3 and (B) pH 7.4 for 48 h. Cytotoxicity of free DOX and various nanoparticles against L929 cells at different concentrations of DOX under (C) pH 6.3 and (D) pH 7.4 for 48 h. Cytotoxicity of nanomaterials and various nanoparticles against MDA-MB-231 cells at different concentrations under (E) pH 7.4 and (F) pH 6.3 for 48 h. Cytotoxicity of nanomaterials and various nanoparticles against L929 cells at different concentrations under (G) pH 7.4 and (H) pH 6.3 for 48 h. | ||
The cytotoxicity profiles of different DOX formulations suggested that the combination of enzyme-sensitive and pH-sensitive components in the nanoparticles had a synergistic effect on cytotoxicity. Additionally, the cytotoxicity of polymers was also evaluated (PMLA–AANL–PEG, PMLA–PEG–TAT, and PEG–PMLA). Fig. 6E–H showed that the cytotoxic effect of different polymer concentrations (ranging from 0.1 to 1000 μg mL−1) on MDA-MB-231 cells and L929 was negligible, indicating the good biocompatibility and safety of the polymers at pH 7.4.
3.8 In vivo tumor growth inhibition study
The in vivo antitumor activity of free DOX, EP-NPs, E-NPs, P-NPs and C-NPs was shown in Fig. 7. Tumor volume rapidly increased in normal saline-treated tumor-bearing mice. EP-NPs, E-NPs, P-NPs and C-NPs obviously delayed the tumor growth. Compared with free DOX, EP-NPs, E-NPs and P-NPs exhibited the most potent antitumor activity in vivo. | ||
| Fig. 7 The antitumor effect of nanocomplex on nude mice bearing MDA-MB-231 cells subcutaneously. (A) Schematic representation of the protocols of antitumor study in nude mice. (B) Photographs of dissected tumor tissues and dissected tumor tissues from the nude mice, respectively. (C) The weight of dissected tumor tissues and dissected tumor tissues from the nude mice. (D) The variation profiles of tumor volumes during injection. (E) Tumor volume change curves of the mice in the above treatment groups, the average tumor growth curves. (F) The variation profiles of body weights during injection. (G) H&E examination of the tumors from mice bearing MDA-MB-231 mammary tumors after 28-day injection. Data was given as mean ± SD (n = 3). The scale bar is 100 μm. | ||
At the conclusion of the treatment, we measured the weight of each tumor (Fig. 7B–E), which corroborated the data on tumor growth inhibition. As shown in Fig. 7D, the tumor volume of the saline-treated group increased rapidly within 21 days, whereas E-NPs and P-NPs modestly inhibited tumor growth. In contrast, EP-NPs showed the highest tumor inhibitory effect. The mice injected with free DOX showed slight inhibition of tumor growth for 6 days. In addition, body weight changes were measured in all groups of mice as an indicator of systemic toxicity; severe systemic toxicity was observed in animals treated with free DOX. The death of mice was observed on day 6 after the free DOX treatment. However, the weight of the other four groups showed no significant effect during the study period, suggesting that the nanoparticles could reduce the side effects of the drugs (Fig. 7F).
Furthermore, immunohistochemical staining with hematoxylin and eosin (H&E) examination of tumor sections (Fig. 7G) revealed that the PBS and C-NPs groups exhibited minimal induction of tumor cell apoptosis, while the other three nanoparticle treatments resulted in significant apoptotic activity in tumor cells. Among them, EP-NPs demonstrated the most pronounced apoptotic effect.
4 Conclusion
In conclusion, we have successfully developed dual-sensitive nanoparticles, EP-NPs, based on PMLA, exhibiting effective drug delivery, enhanced cellular uptake, and intracellular drug release. The nanoparticle demonstrates prolonged circulation under normal physiological conditions (pH 7.4). In the tumor microenvironment, legumain-sensitive deshielding of the PEG–AANL layer resulted in the exposure of TAT-conjugated polymer nanoparticles, thereby enhancing cellular internalization. Upon internalization, the nanoparticle can escape from the endosomes/lysosomes of tumor cells to trigger drug release and ensure nuclear delivery of anticancer drugs. The NPs exhibit high tumor accumulation and excellent inhibition of tumor growth in both cancer cells and xenograft models.Overall, we believe that this novel drug delivery system holds great potential for improved tumor targeting and enhanced intracellular uptake. Moreover, this system demonstrates versatility in its compatibility with small molecules, chemotherapeutics, and proteins. Therefore, this multifunctional platform can optimize anti-tumor therapeutic strategies and potentially improve the prognosis of cancer patients.
Abbreviations
| AANL | Alanine–alanine–asparagine–leucine |
| AEP | Asparaginyl endopeptidase |
| CAA | cis-Aconitic anhydride |
| CAD | cis-Aconitic anhydride-modified doxorubicin |
| CLSM | Confocal laser scanning microscopy |
| C-NPs | Control nanoparticles(non enzyme/pH-sensitive) |
| DCC | Dicyclohexylcarbodiimide |
| DLS | Dynamic light scattering |
| DMSO | Dimethyl sulfoxide |
| DOX | Doxorubicin |
| E-NPs | Enzyme-sensitive nanoparticles |
| EP-NPs | Enzyme/pH-sensitive nanoparticles |
| GPC | Gel permeation chromatography |
| HPLC | High performance liquid chromatography |
| NHS | N-Hydroxysuccinimide |
| NPs | Nanoparticles |
| PAPC | PEG5000–AANL–PMLA–CAD |
| PEG | Polyethylene glycol |
| PMLA | Poly β-malic acid |
| PBM | Poly(β-benzyl malate) |
| P-NPs | pH-sensitive nanoparticles |
| PPC | PEG2000–PMLA–CAD |
| PPTC | TAT–PEG2000–PMLA–CAD |
| TAT | Transactivative transcription protein |
| TEM | Transmission electron microscope |
Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge support from the National Natural Science Foundation of China (Grant No. 32171388), the Key Research & Development Program of Shaanxi (2022ZDLSF05-17), Natural Science Basic Research Program of Shaanxi (No. 2024JC-YBQN-0990), Health Research Projects of Shaanxi Province (No. 2022D-049) and Science and Technology Talents Support Program of Shaanxi Provincial People's Hospital (No. 2021JY-04, 2023BJ-02), and Research Incubation Fund of Shaanxi Provincial People's Hospital (2023YJY-01).References
- Y. Zaiki, A. Iskandar and T. W. Wong, Biotechnol. Adv., 2023, 67, 108200 CrossRef.
- S. Puri, M. Mazza, G. Roy, R. M. England, L. Zhou, S. Nourian and J. Anand Subramony, Adv. Drug Delivery Rev., 2023, 200, 114962 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 Search PubMed.
- R. Sun, J. Xiang, Q. Zhou, Y. Piao, J. Tang, S. Shao, Z. Zhou, Y. H. Bae and Y. Shen, Adv. Drug Delivery Rev., 2022, 191, 114614 CrossRef PubMed.
- A. S. Piotrowski-Daspit, A. C. Kauffman, L. G. Bracaglia and W. M. Saltzman, Adv. Drug Delivery Rev., 2020, 156, 119–132 CrossRef PubMed.
- X. Huang, L. Xu, H. Qian, X. Wang and Z. Tao, J. Nanobiotechnol., 2022, 20, 295 CrossRef PubMed.
- S. Guo, Q. Zhou, T. Yang, Y. Qiao, L. Fan, C. Wang and H. Wu, J. Biomed. Nanotechnol., 2018, 14, 1039–1051 CrossRef CAS PubMed.
- J. Zhang, D. Chen, G. Liang, W. Xu and Z. Tao, Trends Biochem. Sci., 2021, 46, 213–224 CrossRef PubMed.
- Q. Zhou, Y. Hou, L. Zhang, J. Wang, Y. Qiao, S. Guo, L. Fan, T. Yang, L. Zhu and H. Wu, Theranostics, 2017, 7, 1806–1819 CrossRef PubMed.
- X. Wang, C. Li, Y. Wang, H. Chen, X. Zhang, C. Luo, W. Zhou, L. Li, L. Teng, H. Yu and J. Wang, Acta Pharm. Sin. B, 2022, 12, 4098–4121 CrossRef PubMed.
- R. Salve, P. Kumar, W. Ngamcherdtrakul, V. Gajbhiye and W. Yantasee, Mater. Sci. Eng., C, 2021, 124, 112084 CrossRef PubMed.
- Y. Li, Y. Niu, J. Zhu, C. Gao, Q. Xu, Z. He, D. Chen, M. Xu and Y. Liu, Nanoscale, 2020, 12, 2673–2685 RSC.
- D.-S. Liang, W.-P. You, F.-F. Zhu, J.-H. Wang, F. Guo, J.-J. Xu, X.-L. Liu and H.-J. Zhong, Colloids Surf., B, 2023, 226, 113283 CrossRef PubMed.
- J. Vandooren, G. Opdenakker, P. M. Loadman and D. R. Edwards, Adv. Drug Delivery Rev., 2016, 97, 144–155 CrossRef PubMed.
- S. Ishii, Methods Enzymol., 1994, 244, 604–615 CAS.
- Q. Qi, O. Obianyo, Y. Du, H. Fu, S. Li and K. Ye, J. Med. Chem., 2017, 60, 7244–7255 CrossRef CAS.
- J. Zhan, J. Zhong, S. Ma, W. Ma, Y. Wang, Z. Yu, Y. Cai and W. Huang, Chem. Commun., 2020, 56, 6957–6960 RSC.
- H. Jin, Y. He, P. Zhao, Y. Hu, J. Tao, J. Chen and Y. Huang, Theranostics, 2019, 9, 265–278 CrossRef CAS.
- G. Chen, W. Xiong, Z. Gu, Y. Gao, J. Hou, L. Long, H. Wang, A. M. Asrorov, B. Muhitdinov, Q. Xu and Y. Huang, Int. J. Biol. Macromol., 2022, 223, 1485–1494 CrossRef CAS PubMed.
- L. Cohen, Y. D. Livney and Y. G. Assaraf, Drug Resistance Updates, 2021, 56, 100762 CrossRef CAS PubMed.
- P. Boisguérin, K. Konate, E. Josse, E. Vivès and S. Deshayes, Biomedicines, 2021, 9, 583 CrossRef.
- M. M. Khan, N. Filipczak and V. P. Torchilin, J. Controlled Release, 2021, 330, 1220–1228 CrossRef PubMed.
- Y. Qiao, C. Wang, B. Liu, Y. Peng, H. Meng, T. Yang, Q. Zhou, S. Guo and H. Wu, J. Biomed. Nanotechnol., 2019, 15, 28–41 CrossRef.
- Y. Qiao, X. Duan, L. Fan, W. Li, H. Wu and Y. Wang, J. Polym. Res., 2014, 397 CrossRef.
- Z. Yu, H. Ren, Y. Zhang, Y. Qiao, C. Wang, T. Yang and H. Wu, Molecules, 2021, 26, 7169 CrossRef PubMed.
- V.-D. Doan, T. L. Phan, V. T. Le, Y. Vasseghian, L. O. Evgenievna, D. L. Tran and V. T. Le, Chemosphere, 2022, 286, 131894 CrossRef PubMed.
- D. Sun, J. Ding, C. Xiao, J. Chen, X. Zhuang and X. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 21202–21214 CrossRef PubMed.
- Y. Qiao, C. Zhan, C. Wang, X. Shi, J. Yang, X. He, E. Ji, Z. Yu, C. Yan and H. Wu, J. Mater. Chem. B, 2020, 8, 8527–8535 RSC.
- J.-Z. Du, H.-J. Li and J. Wang, Acc. Chem. Res., 2018, 51, 2848–2856 CrossRef CAS PubMed.
- M. Poreba, Biol. Chem., 2019, 400, 1529–1550 CrossRef CAS PubMed.
- Z. Zhang, M. Xie and K. Ye, Expert Opin. Ther. Targets, 2016, 20, 1237–1245 CrossRef CAS PubMed.
- S. Zheng, Y. Cai, Y. Hong, Y. Gong, L. Gao, Q. Li, L. Li and X. Sun, Drug Delivery, 2022, 29, 1764–1775 CrossRef CAS PubMed.
- Q. Zhou, T. Yang, Y. Qiao, S. Guo, L. Zhu and H. Wu, Int. J. Nanomed., 2015, 10, 1941–1952 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01681e |
| This journal is © The Royal Society of Chemistry 2024 |