
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the power of liquid chromatography in examining a library of degradable poly(2-oxazoline)s in nanomedicine applications†
Ekaterina
Tsarenko
ab,
Natalie E.
Göppert
ab,
Philipp
Dahlke
 c,
Mira
Behnke
ab,
Gauri
Gangapurwala
ab,
Baerbel
Beringer-Siemers
ab,
Lisa
Jaepel
ab,
Carolin
Kellner
ab,
David
Pretzel
ab,
Justyna A.
Czaplewska
ab,
Antje
Vollrath
ab,
Paul M.
Jordan
bc,
Christine
Weber
ab,
Oliver
Werz
bc,
Ulrich S.
Schubert
*abd and
Ivo
Nischang
*abde
c,
Mira
Behnke
ab,
Gauri
Gangapurwala
ab,
Baerbel
Beringer-Siemers
ab,
Lisa
Jaepel
ab,
Carolin
Kellner
ab,
David
Pretzel
ab,
Justyna A.
Czaplewska
ab,
Antje
Vollrath
ab,
Paul M.
Jordan
bc,
Christine
Weber
ab,
Oliver
Werz
bc,
Ulrich S.
Schubert
*abd and
Ivo
Nischang
*abde
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de; ivo.nischang@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cDepartment of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich Schiller University Jena, Philosophenweg 14, 07743, Jena, Germany
dHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstr. 12-14, 07743 Jena, Germany
eHelmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 17th October 2024
Abstract
A library of degradable poly(2-alkyl-2-oxazoline) analogues (dPOx) with different length of the alkyl substituents was characterized in detail by gradient elution liquid chromatography. The hydrophobicity increased with increased side chain length as confirmed by a hydrophobicity row, established by reversed-phase liquid chromatography. Those dPOx were cytocompatible and formed colloidally stable nanoparticle (NP) formulations with positive zeta potential. Dynamic light scattering (DLS) revealed that dPOx with increased hydrophobicity tended to form NPs with increased sizes. NPs created from the most hydrophobic polymer, degradable poly(2-nonyl-2-oxazoline) (dPNonOx), showed tendency for aggregation at pH 5.0, and in the presence of protease in solution, in particular for NPs formulated without surfactant. Liquid chromatography revealed enzymatic degradation of dPNonOx NPs, clearly demonstrating the disappearance of polymer signals and the appearance of hydrophilic degradation products eluting close to the chromatographic void time. The degradation process was confirmed by 1H NMR spectroscopy. dPNonOx NPs containing the anti-inflammatory drug BRP-201 as payload reduced 5-lipoxygenase activity in human neutrophils. Thereby, composition analysis of the resultant NPs, including drug quantification, was also enabled by liquid chromatography. The results indicate the importance of a detailed analysis of the final polymer-based NP formulations by a multimethod approach, including, next to standard applied techniques such as DLS/ELS, the underexplored potential of liquid chromatography. The latter is demonstrated to resolve a fine structure of solution composition, together with an assessment of possible degradation pathways and is versatile in determining hydrophobicity/hydrophilicity of polymer materials. Our study underscores the power of liquid chromatography for characterization of soft matter drug carriers.
Introduction
Poly(2-oxazoline)s (POx) are a class of biocompatible synthetic polymers known since the late 1960s.1–4 Their biomedical applications have been thoroughly discussed.5 The functionalities and modification of such polymers can be well-controlled during the cationic ring opening polymerization (CROP) of substituted 2-oxazoline monomers resulting in a polymer of tunable tailor-made properties such as molar mass and overall hydrophilicity/lipophilicity of the material.6,7 Hydrophilic POx, i.e., poly(2-methyl-2-oxazoline) and poly(2-ethyl-2-oxazoline) (PEtOx) are often compared with the most extensively used polymer in the pharmaceutical industry – poly(ethylene glycol) (PEG).8 The so-called “stealth behavior” of those POx has been reported being similar to PEG. This means poor recognition for the PEGylated/POxylated nanoscale drug delivery systems (DDS) by the immune system and prolongation of drug circulation with delayed clearance from the bloodstream through metabolic pathways.9–11 According to hydrodynamic and light scattering studies in solution, the family of POx can be tailored to properties that enable their utilization as a suitable replacement for the widely applied PEG.12 Such replacement appears desirable due to the reported anti-PEG antibodies, hypersensitivity, and allergic reaction to the treatment with PEG-containing nanomedicines.13 Such reactions have even been reported among patients who have never been treated with PEG-containing therapeutics before. This is not surprising since PEGs are ubiquitously abundant in many cosmetics and dietary supplements.9,14,15Several studies reveal the application of amphiphilic POx in DDS, e.g., in the form of micelles or nanoparticles (NPs), for the targeted delivery of anti-inflammatory active pharmaceutical ingredients (APIs).8,16,17 However, one of the drawbacks of POxylated systems, similar to PEGylated systems, is the lack of biodegradability. Ultimately, this would be desirable, once the nanomedicine reached the target in the body.5 The hydrolysis of POx requires harsh acidic conditions and high temperatures. Both are far from the optimum in living organisms. Such hydrolysis results in linear poly(ethylene imine) (PEI) chains and low molar mass saturated carboxylic acids as degradation products.18
One mean to overcome such drawback is to chemically modify the main polymer chain, e.g., by incorporating amide groups into an original PEI backbone. This could tailor an increased biodegradability potential. To address this, Göppert et al. established a series of degradable analogues of POx via a sequence of postpolymerization reactions.19 Firstly, PEtOx was hydrolyzed under acidic conditions to yield linear PEI. Subsequently, partial oxidation of the PEI chain led to statistically distributed glycine moieties, resulting in poly(ethylene imine-co-glycine), an oxidized form of the original PEI (further referred to as oxPEI). In the last step, the oxPEI backbone was functionalized with acyl chlorides to yield a library of poly(2-n-alkyl-2-oxazoline-stat-glycine)s (further referred to as dPOx) with a varying alkyl side chain length. In total, nine new dPOx copolymers were created with the following alkyl side chains: methyl (dPMeOx), ethyl (dPEtOx), n-propyl (dPPropOx), n-butyl (dPButOx), n-pentyl (dPPentOx), n-hexyl (dPHexOx), n-heptyl (dPHeptOx), n-octyl (dPOctOx), and n-nonyl (dPNonOx). The reaction scheme as well as the schematic representation of the chemical structures of the final library is shown in Fig. 1.
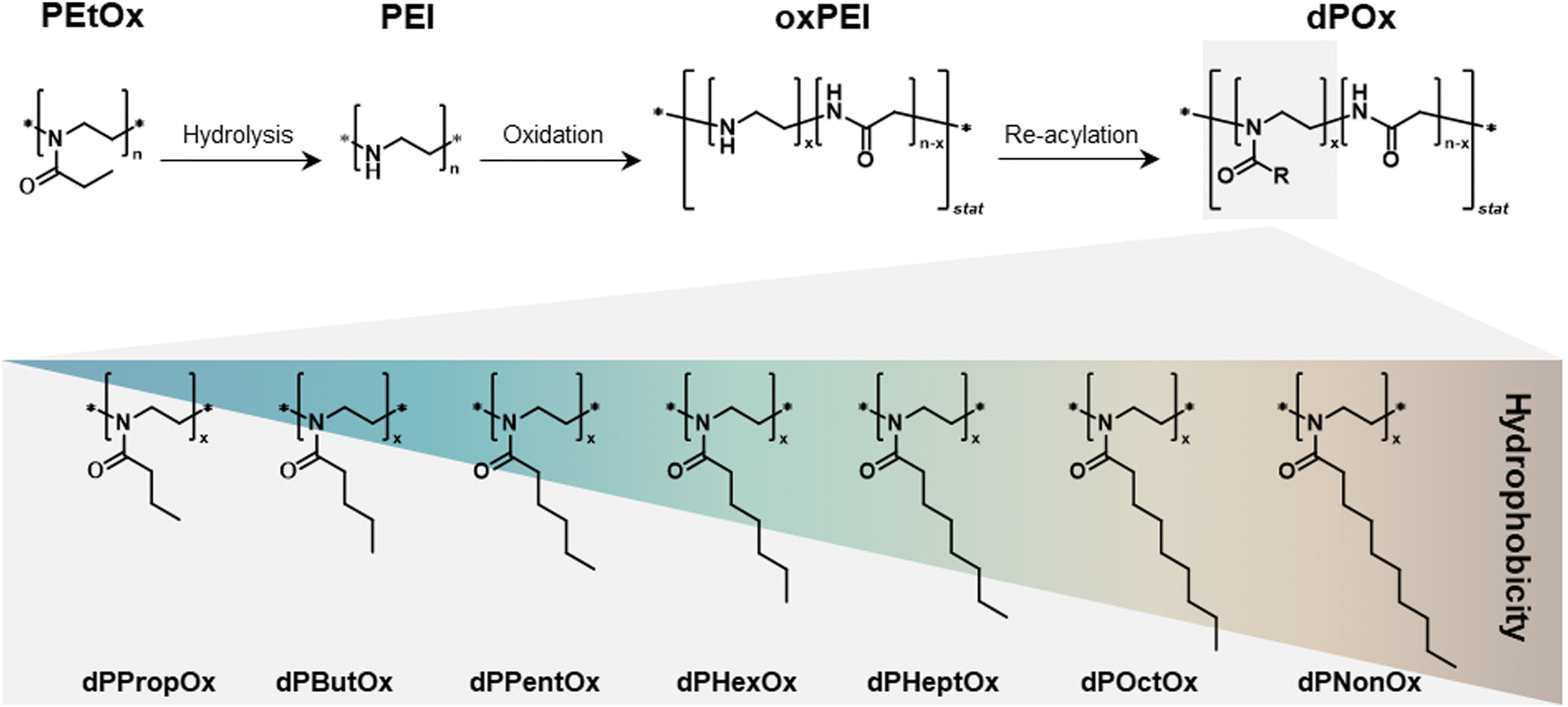 | ||
| Fig. 1 Schematic representation of the synthesis route toward a degradable poly(2-n-alkyl-2-oxazoline-stat-glycine) (dPOx) library of polymers with alkyl side chains of different length. Further details on the synthetic procedures are published elsewhere.19 | ||
The structure of the obtained statistical copolymers imposes difficulties on the characterization of such materials. Analysis by size exclusion chromatography (SEC) was hampered by poor solubility of the homologous dPOx in a common solvent for all the polymers, i.e., dPOx, oxPEI, and PEI.19 Characterization by mass spectrometry (MS) techniques led to spectra of very high complexity. This could originate from overlapping charge states (in electrospray ionization, ESI-MS) or from unrepresentative mass spectral patterns obtained by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for copolymers of high dispersity.20,21 The well-known mass discrimination effects also occurred,22,23 and several samples could simply not be ionized.
Liquid chromatography that utilizes the specific interactions of analytes with the chromatographic surface under retentive conditions (as opposed to the hydrodynamic volume-based separation principle of SEC) can be considered a standard technique for quality control of novel small molar mass therapeutics or therapeutic proteins and conjugates when establishing manufacture procedures in the pharmaceutical industry.24 Synthetic polymer chromatography, on the other hand, is by far more challenging. The possibility for the polymer to interact in various ways with the chromatographic column materials has resulted in a range of model studies, but a characterization of samples of “real” applicative interest is rarely performed.25,26 Elution of synthetic macromolecules is influenced by several factors and strongly dependent on their molar mass, composition, end-group functionalities, and polymer architecture.27,28 Furthermore, synthetic polymers are inherently disperse in terms of their molar mass and potentially show an additional variation in their composition. This complicates understanding and optimizing chromatographic behavior significantly.29
In the present study, we demonstrate how liquid chromatography can play a significant role in the characterization of novel materials, both copolymers and NPs, designed for biomedical applications. Firstly, the library of dPOx was characterized by gradient elution liquid chromatography to establish how the side chain length influences the dPOx elution behavior and how it is connected to polymer hydrophobicity. The separation of polymers in gradient elution liquid chromatography is driven by several factors such as partition, size exclusion, and adsorption/desorption. Among others, it can be tuned by the mobile phase composition, i.e., by gradual increase of mobile phase elution strength.30 Gradient elution liquid chromatography has already been used for characterization and separation of hydrophilic and hydrophobic POx in solution.31–34 The reversed-phase (RP) monolithic silica column utilized here as stationary phase has previously demonstrated appreciable performance in characterization of PEGs and POx tailored with different α- and ω-end groups.25,35,36 Also, the unique hierarchical porous silica rod structure containing macro- and mesopores confined by the chromatographic surface enabled the polymeric NP composition analysis. This included isocratic determination of the drug content with high efficiency and, simultaneously, gradient elution of polymer content within one chromatographic run.37
In addition to establishing of chromatographic conditions suited for the library of dPOx, the current study takes the next step, i.e., the use of the materials for NP formulation and encapsulation of an anti-inflammatory drug. Important aspects describing the NP properties, are accompanied by appropriate chromatographic characterization. In this study, the dPOx polymers were tested for their impact on cell viability and a screening of NP formulation ability was performed. Furthermore, the NPs based on the most hydrophobic polymer, dPNonOx, were formulated to investigate the influence of surfactant on NP stability. NP stability in different media was investigated by dynamic light scattering (DLS). The enzymatic degradation of dPNonOx NPs was also monitored by liquid chromatography and proton nuclear magnetic resonance (1H NMR) spectroscopy. Here, liquid chromatography was utilized for monitoring macromolecular integrity as well as for NP composition analysis of drug-loaded dPNonOx NPs including the drug loading quantification. For this, the anti-inflammatory small molecule BRP-201 was encapsulated in dPNonOx NPs by nanoprecipitation. This API is known to suppress the formation of pro-inflammatory lipid mediators.38 To complete the study, the biological activity of BRP-201-loaded dPNonOx NPs was evaluated in human neutrophils to demonstrate the applicability of dPNonOx as carrier material.
Experimental section
Materials
The synthesis of the dPOx statistical copolymers is described elsewhere.19 Standard characterization data of those materials are provided in Table S1 (ESI†). BRP-201 was synthesized according to an established procedure.39 Fig. S1 (ESI†) shows a schematic representation of the chemical structure of both the drug BRP-201 and dPOx. Acetone (99+%, extra pure) was purchased from Acros Organics (Geel, Belgium). Poly(vinyl alcohol) (PVA, partially hydrolyzed poly(vinyl acetate): Mowiol 4–88, Mw 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and dimethylsulfoxide (DMSO, anhydrous ≥99.9%), sodium acetate, acetic acid, and proteinase K from Tritirachium album were obtained from Sigma-Aldrich (Darmstadt, Germany). The acetate buffer was prepared using sodium acetate (≥99%, ACS reagent; Sigma-Aldrich) and glacial acetic acid (ACS, Reag. Ph. Eur.; VWR). The buffer pH value was adjusted using 1 M solutions of HCl (37%, analytical reagent grade; Fischer Scientific) and NaOH (≥99%; Roth). Purified water was received from a Barnstead™ GenPure™ xCAD Water Purification System from Thermo Scientific (Waltham, MA, USA) and was used in all stages of NP preparation, purification, and characterization studies. Deuterium oxide (D2O) was purchased from Eurisotop (Saint-Aubin, France). LC-MS grade acetonitrile (CH3CN) and water (H2O) as well as formic acid (FA, ≥99.9%) were purchased from VWR (Darmstadt, Germany).
000 g mol−1) and dimethylsulfoxide (DMSO, anhydrous ≥99.9%), sodium acetate, acetic acid, and proteinase K from Tritirachium album were obtained from Sigma-Aldrich (Darmstadt, Germany). The acetate buffer was prepared using sodium acetate (≥99%, ACS reagent; Sigma-Aldrich) and glacial acetic acid (ACS, Reag. Ph. Eur.; VWR). The buffer pH value was adjusted using 1 M solutions of HCl (37%, analytical reagent grade; Fischer Scientific) and NaOH (≥99%; Roth). Purified water was received from a Barnstead™ GenPure™ xCAD Water Purification System from Thermo Scientific (Waltham, MA, USA) and was used in all stages of NP preparation, purification, and characterization studies. Deuterium oxide (D2O) was purchased from Eurisotop (Saint-Aubin, France). LC-MS grade acetonitrile (CH3CN) and water (H2O) as well as formic acid (FA, ≥99.9%) were purchased from VWR (Darmstadt, Germany).
Liquid chromatography
The elution behavior as well as hydrophobicity/hydrophilicity of polymers was studied by liquid chromatography using an UltiMate™ 3000 Rapid Separation (RS) UHPLC chromatographic system (Thermo Fisher Scientific, Waltham, MA, USA). For that, a monolithic Chromolith® High Resolution RP-18 endcapped (100 × 4.6 mm) column from Merck KGaA (Darmstadt, Germany) was used as a stationary phase. The utilized flow rate was 1 mL min−1. The column oven temperature was set to 35 °C and the autosampler temperature was set to 17 °C. Elution was monitored by two detectors: a charged aerosol detector (Corona™ Veo™ RS, CAD) and a diode array detector (DAD). The CAD was used as a universal detector. The data were collected at 5 Hz acquisition frequency with the nebulizer tempered at 45 °C. Simultaneously, the DAD was operated at 290 nm (polymer absorbance maximum) and 312 nm (BRP-201 absorbance maximum). The mobile phase consisted of CH3CN and H2O and linear gradient elution was applied. The starting conditions, i.e., 20/80 (%, v/v) CH3CN/H2O, were kept constant for 1 min, afterward the CH3CN content was linearly increased from 20% to 100% in 10 min, followed by a hold at 100% for 9 min. After that, the CH3CN content was decreased back to 20% in 4 min and the column was re-equilibrated to the initial conditions for 6 min prior to the next injection. The total run time was 30 min.The polymers were dissolved at a concentration of 1 mg mL−1 in mixtures of CH3CN and 0.1% (v/v) aqueous formic acid at the following ratios: PEtOx, PEI, oxPEI, dPMeOx, dPEtOx, dPPropOx, dPButOx at 50/50 (%, v/v), dPPentOx and dPHexOx at 75/25 (%, v/v), and dPHeptOx, dPOctOx and dPNonOx at 100% CH3CN.
For drug loading determination in dPNonOx NPs, the elution conditions were modified to allow for efficient separation of BRP-201 and polymer according to a previously developed protocol.37 The final method comprised an isocratic hold at 85/15 (%, v/v) CH3CN/H2O for 5 min to allow elution of the drug, followed by a gradient toward 100% CH3CN in 2 min to elute the dPNonOx. The CH3CN content was held constant for 8 min and then decreased back to 85% in 0.5 min. The column was equilibrated for 4.5 min before the next injection. The total run time was 20 min, and the flow rate was set to 1.5 mL min−1.
The lyophilized NP samples were dissolved in 200 μL DMSO and sonicated for 1 min at room temperature. Afterward, 800 μL 85/15 (%, v/v) CH3CN/H2O was added to the solution and the sonication was repeated. For calibration, BRP-201 was dissolved in DMSO to obtain a stock solution of 1000 μg mL−1. The BRP-201 stock solution was diluted to a series of concentrations: 60, 50, 20, 10, and 5 μg mL−1. The final solvent composition in the calibration standards was 200 μL DMSO and 800 μL 85/15 (%, v/v) CH3CN/H2O.
In all the measurements, the injection volume was 10 μL. Prior to the analyses, all samples were filtered through a hydrophobic 0.45 μm pore size polytetrafluoroethylene (PTFE) filter (AppliChrom, Oranienburg, Germany). Chromatographic data were processed using the Thermo Scientific™ Dionex™ Chromeleon™ 7.2 SR5 Chromatography Data System software.
Nanoparticle formulation
The screening of NP formation of all dPOx, the formulation of dPNonOx NPs with and without surfactant PVA as well as formulation of dPNonOx NPs containing the drug BRP-201 was performed utilizing the batch nanoprecipitation method. The detailed procedures are described in Section S2.1 of the ESI.†Dynamic light scattering (DLS) and electrophoretic light scattering (ELS)
The NP size (hydrodynamic diameter dh, Z-average), polydispersity index (PDI), and zeta potential (ζ) were determined by DLS and ELS measurements using a Zetasizer Nano ZS and Zetasizer Ultra (Malvern Panalytical Ltd., Malvern, UK). Prior to the measurements, a volume of 10 or 100 μL of the NP dispersions were diluted 1:10 or 1:100 using purified water. DLS was measured in polystyrene cuvettes (Brand GmbH + Co KG, Wertheim, Germany) with the following settings, unless stated otherwise: measurement temperature 25 °C, five runs of each sample with 30 s of equilibration time and 30 s of acquisition time. ELS measurements were performed in capillary cell cuvettes DTS1070 (Malvern Panalytical Ltd, Malvern, UK).
In vitro evaluations of potential cytotoxic effects of dPOx polymers in L929 fibroblasts
The potential cytotoxicity of the investigated polymers was evaluated using the mouse fibroblast cell line L929 (CLS, Eppelheim, Germany) and the commercial PrestoBlue™ Assay (Thermo Fisher Scientific, Waltham, MA, USA), which measures the metabolic activity of cells as an indirect marker of cell viability. This assay works with a cell-permeable resazurin-based solution that functions as a cell viability indicator by using the reducing power of living cells to quantitatively measure the proliferation of cells. For cell culture, the Dulbecco's Modified Eagle Medium (DMEM) was supplemented with 10% fetal calf serum, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin (Biochrom, Berlin, Germany) and used as culture media. Cells were routinely kept at 37 °C in a humidified atmosphere containing 5% CO2. Cells were plated at a density of 104 cells per well in 96-well plates and grown for 24 h. After 24 h, culture media was replaced by fresh media containing the dissolved polymers (hydrophobic polymers which were not directly soluble in cell culture media, i.e., dPPropOx, dPButOx, dPPentOx, dPHexOx, dPHeptOx, dPOctOx, dPNonOx, were prepared as a DMSO stock solution which was then further diluted in cell culture media in a ratio of at least 1:10) with varying concentrations from 0.1 to 100 μg mL−1. Control cells were incubated with fresh culture medium or culture media containing the respective DMSO amount in case of the hydrophobic polymers. After additional 24 h of incubation, the media were removed and cells were incubated for 45 min at 37 °C with fresh media containing 10% PrestoBlue™ reagent. The resulting fluorescence intensity of the conditioned media was measured at λEx = 560 nm/λEm = 590 nm. Samples displaying a cell viability value higher than 70% of that of non-treated cells on the same plate were considered as non-cytotoxic according to ISO 10993-5. Data are expressed as mean ± standard deviation (SD) of the measurements performed in six technical replicates with at least three independent determinations.
NP stability in different storage media
The stability of dPNonOx NPs formulated with and without surfactant PVA was investigated by DLS in different storage media: in 50 mM acetate buffer (pH 5.0) and in presence of proteinase K. For that, the NP suspension (initial concentration of 1 mg mL−1) was incubated at 37 °C with 50 mM acetate buffer (pH 5.0) or with a proteinase K solution (2 mg mL−1 in water) in 1:2 mass ratio (particle:proteinase K). The mean count rate (in kilocounts per seconds (kcps)) as well as NP size were monitored at a fixed attenuator setting of 6. The mean count rate of each sample at the beginning of the measurements was kept around 500 kcps. For data interpretation, the initial count rate was set to 100% and the count rate values collected over time are represented in percentage.
In vitro degradation of dPNonOx NPs
The degradation behavior of dPNonOx NPs over longer period of time was investigated by proton nuclear magnetic resonance (1H NMR) spectroscopy and liquid chromatography. For that, the NP batch dPNonOx (without PVA) at an initial concentration of 2 mg mL−1 was incubated with a proteinase K solution in water at 37 °C in mass ratios of 1:2 and 1:4 (particle:proteinase K) for over 68 days.
After 0, 10, 20, 30, and 68 days after the experiment start, aliquots of 5 mL were taken and lyophilized. 1H NMR spectra were measured on a Bruker AC 300 MHz spectrometer at room temperature using D2O as a solvent. Chemical shifts (δ) are given in parts per million (ppm) using the residual non-deuterated solvent resonance signal for referencing the chemical shift.
After the NMR measurements, the samples were lyophilized once more. For the liquid chromatography measurements, the lyophilized aliquots were dissolved in 50/50 (%, v/v) CH3CN/H2O and sonicated for 3 min. Prior to the analysis, the samples were filtered through a 0.45 μm pore size PTFE filter (AppliChrom GmbH, Germany).
Cell isolation and cell culture of human neutrophils
The peripheral venous blood of healthy human adult donors (females and males between the ages of 18 and 65) that received no anti-inflammatory treatment for the past ten days was used to prepare leukocyte concentrates. The ethical committee of the University Hospital Jena approved the protocol. All procedures were conducted in accordance with the applicable guidelines and regulations, as described in a previous standard protocol.40 To isolate neutrophils, the leukocyte concentrates were mixed with dextran from Leuconostoc spp. After sedimentation of erythrocytes, the supernatant was centrifuged on a lymphocyte separation medium (Histopaque®-1077, Sigma Aldrich, Darmstadt, Germany). Hypotonic lysis using water removed contaminating erythrocytes from the neutrophils. The pelleted neutrophils fraction was washed twice in ice-cold phosphate-buffered saline pH 7.4 (PBS) and finally resuspended in PBS.Determination of 5-lipoxygenase (5-LOX) activity in neutrophils
The effects on 5-LOX activity in human neutrophils were evaluated by pre-incubating cells (5 × 106 in 1 mL) for 15 min at 37 °C in PBS containing 0.1% glucose and 1 mM CaCl2 with vehicle (PBS), NPs (empty or containing 0.01, 0.03 or 0.1 μM of BRP-201) or free 0.3 μM BRP-201.41 Cells were then stimulated with 2.5 μM Ca2+-ionophore A23187 (Cayman, Ann Arbor, USA) for 10 min, and the incubation was stopped with 1 mL ice-cold methanol containing 200 ng mL−1 prostaglandin B1. Solid phase extraction was used to isolate the formed 5-LOX products (LTB4, trans-LTB4, epi-trans-LTB4, and 5-HETE), which were separated and analyzed by reversed-phase liquid chromatography as described previously.42In vitro evaluations of potential influence on cellular membrane integrity of dPOx NPs in human neutrophils
A CytoTox 96® non-radioactive cytotoxicity assay kit was used to assess the release of lactate dehydrogenase (LDH) for cell integrity analysis. The supernatants after incubation of the cells (1 × 106 neutrophils) were centrifuged at 400 × g for 5 min at 4 °C and diluted to appropriate LDH concentrations. A NOVOstar microplate reader (BMG LABTECH GmbH, Offenburg, Germany) was used to measure the absorbance at 490 nm. The cell integrity was determined according to the manufacturer's guidelines.Results and discussion
Liquid chromatography of dPOx library
In a previous study, nine new dPOx copolymers were synthesized: dPMeOx, dPEtOx, dPPropOx, dPButOx, dPPentOx, dPHexOx, dPHeptOx, dPOctOx, and dPNonOx.19 Rationally, one may anticipate that the polymers differ in material hydrophobicity dependent on alkyl side chain length (Fig. 1). To demonstrate this, the elution behavior of the homologous series of dPOx was investigated by gradient elution liquid chromatography. A linear gradient from lower (20%, v/v) to higher (100%, v/v) CH3CN content in the mobile phase was utilized. As the utilized stationary phase was silica modified with octadecyl (C18) alkyl chains, separation is expected to follow reversed-phase chromatographic mode, where the retention time can be related to sample polarity.The elution behavior of dPOx was monitored via universal CAD (Fig. 2A) and DAD at 290 nm (Fig. 2B). The elution traces in CAD revealed numerous signals, which indicates the high dispersity of the copolymer samples. When comparing the polymer elution trace with a solvent blank (Fig. S2, ESI†), for each dPOx the signal intensity of the elution trace increased compared to the solvent blank injection. Most likely, the elution pattern of dPOx represents a widely distributed peak with more narrow resolved peaks on top, referring to polymer species of either a certain repeating unit composition or chain length. For each dPOx, the most prominent peak group in the elugram shifted toward longer retention with the increase of alkyl side chain length from dPMeOx to dPNonOx, according to an expected hydrophobicity increase (Fig. 2A). The elution trace recorded via DAD (Fig. 2B) showed mostly one prominent peak for each elugram, however the trend of increased retention time with the longer alkyl side chain is still evident.
 | ||
| Fig. 2 Elution traces of dPOx recorded by (A) CAD and (B) DAD at 290 nm. (C) Example of overlaid CAD and DAD elution traces for dPButOx. Numbers 1, 2, 3 indicate three peaks that match in their retention time, irrespective which detector was used. Overlaid elugrams of other dPOx are present in Fig. S3 (ESI†). (D) Hydrophobicity row for the dPOx library presented as retention time of peaks from CAD and DAD traces (peaks 1, 2, 3) as a function of side chain length (expressed in number of carbon atoms). The non-characteristic peak indicated in (A) with a “*” was observed in all runs irrespective of which sample was analyzed and was not used for further characterization and data interpretation. | ||
Based on these findings, a hydrophobicity row of the dPOx library was established. For that, the dPOx elution traces monitored via CAD and DAD were overlaid (Fig. 2C and Fig. S3, ESI†). The retention times of the most intense three matched peaks from DAD and CAD were plotted as a function of side chain length (Fig. 2D). The expected decrease of the materials’ hydrophilicity is seen in a direct increase in retention time as all the dPOx materials feature the same degree of polymerization (DP). Similar observations were made with non-degradable POx recently.34
Overall, the dPMeOx and dPEtOx demonstrated hydrophilic properties as they eluted with the dead volume (∼1.5 mL). At the same time, they produced a second broad low-intensity peak around 5 min (whose retention time was used in the established hydrophobicity row in Fig. 2D). dPNonOx was the copolymer with the longest retention time (>14 min), therefore representing the most hydrophobic polymer in the series.
In vitro cytotoxicity of dPOx
The potential cytotoxicity of the dPOx library was tested via a PrestoBlue™ assay using L929 cell line (Fig. S4 and S5, ESI†). Water-soluble dPMeOx and dPEtOx remained non-toxic up to concentrations of 6 mg mL−1 and 3 mg mL−1, respectively (Fig. S4B and S4C, ESI†). Although their compatibility toward the cell line is not as high as that of the non-degradable PEtOx featuring the same DP value (Fig. S4A, ESI†),11 such concentrations exceed the range typically used in pharmaceutical applications.43 The diminished cytocompatibility might be due to the presence of non-acylated ethylene imine units within the polymer backbone due an incomplete re-acylation in the last synthesis step (compare Fig. 1).The more hydrophobic dPOx, featuring very limited or no water solubility, were tested up to polymer concentrations of 100 μg mL−1 by dilution of a polymer solution in DMSO in the cell culture media (Fig. S5, ESI†). At 100 μg mL−1, the safest cytotoxicity profile was found for dPPropOx and dPNonOx (Fig. S5H, ESI†). At lower concentrations, all dPOx remained non-cytotoxic.
Formulation ability of dPOx
Envisaging the encapsulation of a hydrophobic API, we selected the non-water soluble dPOx, i.e., dPPropOx to dPNonOx, for further investigations regarding their ability to form NPs in aqueous media via nanoprecipitation. In nanoprecipitation, a polymer is dissolved in a water-miscible solvent. Then, the polymer is precipitated into an aqueous phase under defined conditions. After the precipitation, the organic solvent is usually evaporated, leaving the NP suspension in water under constant stirring. Based on the subsequent application, several parameters including the initial polymer concentration in formulation and the presence of a surfactant, e.g., PVA, can be varied to influence the final particle size and colloidal stability.44For screening the ability of dPOx to form NPs in water, the nanoprecipitation was implemented without surfactants to explore the influence of the alkyl chain length on the NP properties. For that, all dPOx were dissolved in acetone at varying concentrations from 1 to 20 mg mL−1 and were injected into water. The NP characteristics such as hydrodynamic diameter (dh, Z-average), PDI, and zeta potential were obtained by DLS/ELS (Table S2 and Fig. S6, S7, ESI†). All dPOx formed NPs with sizes below 200 nm, even at the highest initial polymer concentration of 20 mg mL−1. The PDI values stayed below 0.15 in all cases, except for NPs prepared from acetone solutions of 1 mg mL−1, indicating narrow size distributions which is beneficial for consistent behavior of NPs.45
By comparison of the final NP size (dh, Z-average) in dependence of the side chain length, several trends can be discerned. Firstly, the initial polymer concentration influenced the hydrodynamic diameter of the obtained NPs, i.e., a higher concentration resulted in larger NPs. This finding has been reported before with different polymer-based carrier materials.46,47 Secondly, increase in the side chain length from dPPropOx to dPNonOx resulted in a larger hydrodynamic diameter of the obtained NPs, except for an initial polymer concentration of 1 mg mL−1. For such low concentrations, NP formation appeared less controlled as indicated by the systematically increased PDI values and apparently lower zeta potential values for all NPs. Higher zeta potential values are usually preferred as they evidence the electrostatic repulsion between the NPs leading to slower aggregation of NP in suspension.48
Zeta potentials for all formulated dPOx NPs displayed positive values in a range from 15 to 45 mV, depending on the utilized initial copolymer concentration. Most likely, either the glycine moieties or residual non-functionalized ethylene imine units present in dPOx cause the positive surface charge. Similar findings have been reported recently for NPs formulated from amphiphilic block copolymers comprising hydrophilic dPOx blocks.21
Stability and in vitro degradation of dPNonOx NPs
Apparently, all hydrophobic dPOx seemed to be suitable for NP formation. As dPNonOx tended to feature the best cytocompatibility, this material was selected for further studies. It has previously been found that the addition of surfactants is favorable for the formation of API-loaded NPs even if the blank NPs can be formulated well without the use of such.49 To investigate the influence of a surfactant on the NP properties, unloaded NP were prepared with and without applying PVA in the aqueous phase used during nanoprecipitation. PVA is partially hydrolyzed poly(vinyl acetate), that possesses both hydrophobic acetate as well as hydrophilic hydroxyl groups and, thus, can reduce the surface tension of hydrophobic NP materials dispersed in aqueous media.50,51 This leads to effective prevention of aggregation and increase in colloidal stability.The presence of the surfactant did not influence the main NP properties, as dPNonOx NPs formulated with and without PVA centered about the same size and zeta potential values (Table S3 and Fig. S8, ESI†).
The stability of dPNonOx NPs formulated with and without PVA was investigated in acetate buffer (50 mM, pH 5.0) and under proteinase K action at a mass ratio of 1:2 (particle:proteinase K) by DLS (Fig. 3). After 14 days of incubation at 37 °C both dPNonOx NPs were stable in aqueous suspension (Fig. 3A). Acidic conditions influenced the stability of the dPNonOx NPs: a decrease of the mean count rate and increase of the apparent NP size pointed toward NP aggregation (Fig. 3B). The aggregation was delayed when PVA was present as a surfactant and set in after three days. In contrast, NPs without surfactant revealed an immediate decrease of the count rate.
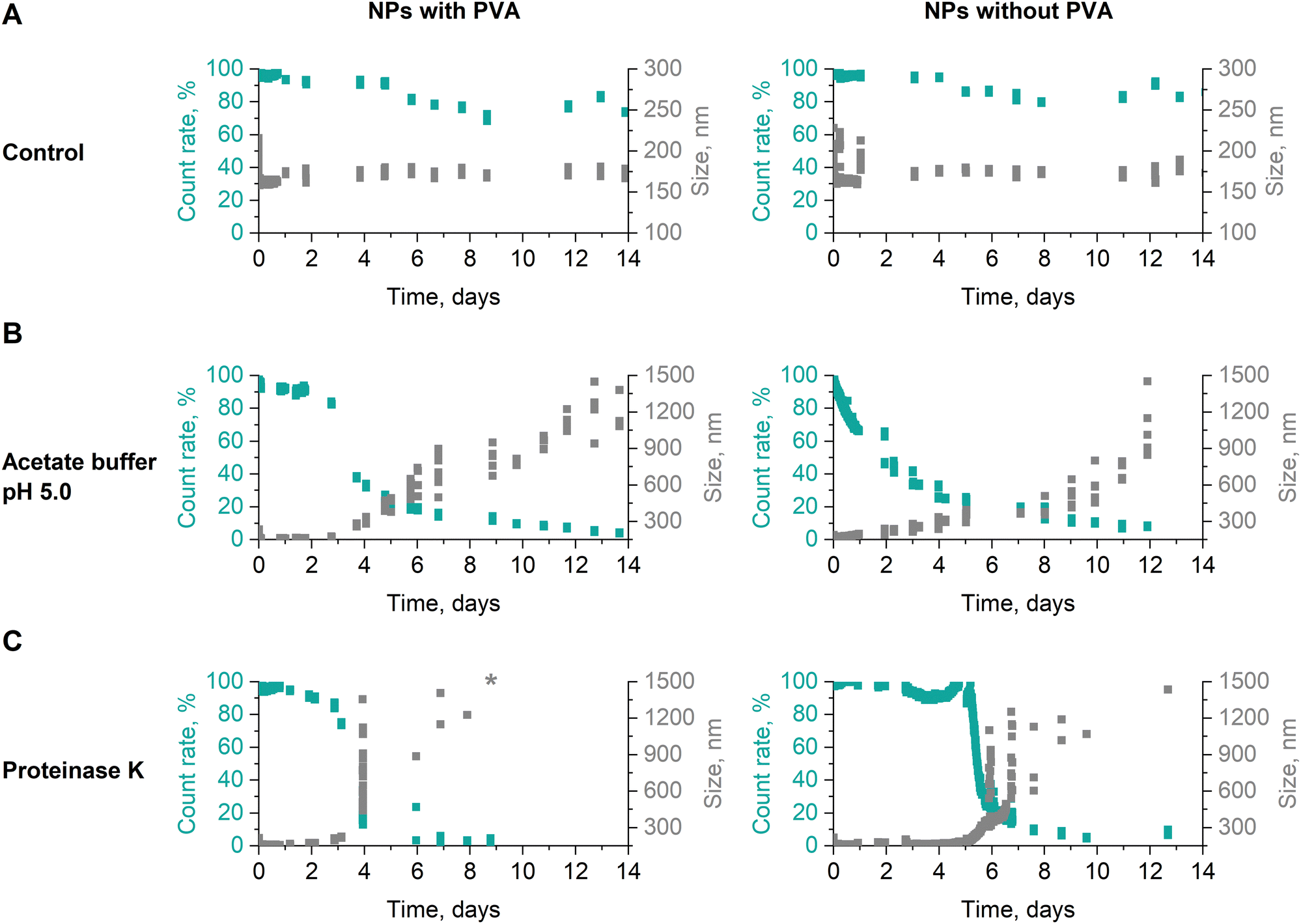 | ||
| Fig. 3 Stability of dPNonOx NPs formulated with and without the surfactant PVA. The NPs were kept at 37 °C (A) in water as a control as well as (B) were incubated in the 50 mM acetate buffer (pH 5.0) or (C) mixed with the enzyme proteinase K in a mass ratio 1:2 (particle:proteinase K). NP size and count rate were monitored by DLS. The size value indicated with a “*” exceeds the size axis range. | ||
Proteinase K belongs to the class of proteases, is capable of degrading peptide bonds, and has previously been reported to boost the biodegradation of polyesters and polyesteramides.52,53 Incubated with proteinase K, dPNonOx NPs with PVA were stable for the first three days (Fig. 3C). On the fourth day of incubation at 37 °C, the count rate suddenly decreased. Although occurring likewise for NPs without PVA, the observation was delayed for one day. However, the decrease in count rate was again accompanied by an increase in overall apparent particle size.
Although DLS is a relatively fast and straightforward method for NP size determination, it often results in over-estimated sizes due to domination of the scattering behavior by larger particles or aggregates.54 Therefore, DLS cannot distinguish between aggregation effects and degradation of the NPs. Hydrodynamic techniques such as analytical ultracentrifugation (AUC) and field-flow fractionation (FFF) are known for more precise size determination of colloidal systems.55 They, however, cannot reveal the chemical changes in the materials.
To obtain information regarding the products of the enzymatic degradation, techniques such as liquid chromatography or NMR spectroscopy are required. For such investigations, the dPNonOx NPs without PVA were incubated with proteinase K at 37 °C for 68 days. Then, lyophilized samples were investigated using the gradient elution liquid chromatography method described above (Fig. 4). The elution of dPNonOx NPs stored with the enzyme was monitored by CAD (Fig. 4A). Taking into consideration that dPNonOx is a highly disperse copolymer obtained through a multi-step synthesis procedure, a multiplicity of signals was observed in the elugram of the dissolved NPs, similarly to the polymer elution traces (Fig. 2A).
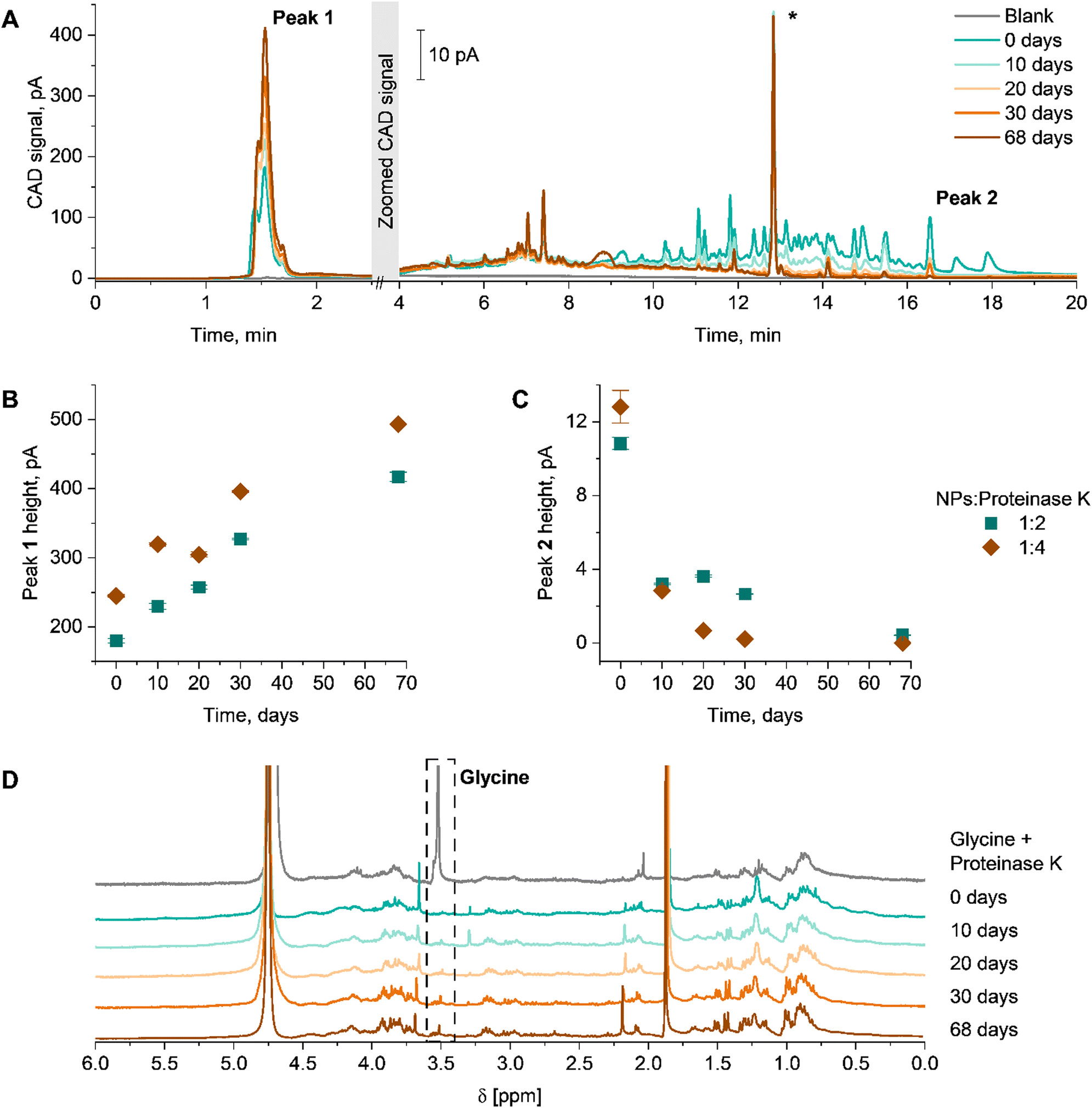 | ||
| Fig. 4 Enzymatic degradation of dPNonOx NPs under proteinase K action in a mass ratio 1:2 (particle:proteinase K). (A) Elugrams of dPNonOx NP suspension stored with proteinase K over 68 days and recorded by CAD. The non-characteristic peak indicated with a “*” was observed in all runs irrespective of which sample was analyzed and was not used for further characterization and data interpretation. Degradation of dPNonOx NPs is illustrated via pseudo-kinetic degradation plots established via (B) Peak 1 height and (C) Peak 2 height as a function of time in days. The error bars were calculated from three repetitive injections of each sample. (D) Overlay of 1H NMR spectra (300 MHz, D2O) of the lyophilized dPNonOx NP suspension stored with proteinase K in a mass ratio 1:2 (particle:proteinase K). The individual normalized spectra are stacked for clarity. The dashed black box indicates the glycine singlet at 3.7 ppm. | ||
To illustrate the degradation process, the change of signal intensity of the characteristic peaks was closely monitored. Peak heights were used to indicate the progression of the hydrolysis (Fig. 4B and C). The intensity of the very first peak having a particular fine structure (∼1.5 min, Peak 1 in Fig. 4A) increased over time. Preliminary experiments also showed non-retained behavior of proteinase K on the column and under present chromatographic conditions (Fig. S9, ESI†). The increased abundance of the signal close to the void time (Fig. 4B) demonstrates an increase of hydrophilic products present in the NP suspension, originating from enzymatic degradation of dPNonOx. Indeed, the expected degradation products of dPNonOx include hydrophilic low molar mass amines, decanoic acid, and glycine (Fig. S10, ESI†).56
The elugrams demonstrate the decrease of the dPNonOx signal intensity in the range between 10 and 20 min of elution time with increase of incubation time, which correlates to a lower amount of dPNonOx present in the suspension. The height of the dPNonOx characteristic peak (∼16.5 min, Peak 2 in Fig. 4A, that was used for the dPOx hydrophobicity row in Fig. 2D) constantly decreased upon prolonged proteinase K treatment (Fig. 4C), clearly demonstrating the degradation of dPNonOx.
Twice the amount of proteinase K led to a faster degradation of the material. That was evidenced by the faster drop of signal intensity between 10 and 20 min already after 10 days (Fig. S11A, ESI†) as well as by the faster decreasing height of Peak 2 and faster increasing height of Peak 1 (Fig. 4B and C).
The enzymatic degradation of dPNonOx NPs formulated without PVA was proven by 1H NMR spectroscopy as well (Fig. 4D and Fig. S11B, ESI†). The singlet appearing at 3.7 ppm can be assigned to the methylene protons of the glycine that is formed during the dPNonOx backbone hydrolysis (Fig. S10, ESI†). However, a clear assignment of further degradation products in the spectra was hampered due to various signals arising from the proteinase K.
All in all, the results highlight the degradation of dPNonOx NPs not only through NP aggregation in suspension, but also through hydrolysis of the polymer backbone leading to the formation of hydrophilic degradation products.
BRP-201-loaded dPNonOx NPs
NPs formed from dPNonOx demonstrated promising properties to be used as DDS, as they showed low cytotoxicity and degraded under proteinase K action with potential for sustained drug release. To investigate dPNonOx NPs as a carrier material, the anti-inflammatory drug BRP-201 was chosen as it inhibits 5-lipoxygenase (5-LOX)-activating protein (FLAP) in the 5-LOX pathway of arachidonic acid metabolism leading to reduced formation of pro-inflammatory leukotrienes.38,41 Low water solubility of BRP-201 and strong non-specific protein binding, leading to fast elimination from the organism, evidence the necessity of a carrier material to improve its bioavailability and pharmacokinetics.57Hence, dPNonOx NPs were loaded with BRP-201 by co-nanoprecipitation. For formulation, the drug dissolved in DMSO was mixed with polymer dissolved in acetone and the solution was added to the aqueous phase containing PVA to obtain drug-loaded NPs in water. As the initial screening had shown that dPNonOx can be formulated from acetone at polymer concentrations of 5 and 20 mg mL−1, we kept those high concentrations, because the drug loading often increases along with the polymer concentration.58 After evaporation of the solvent and purification of the particles via ultrafiltration, the loading capacity (LC, %) values were determined by liquid chromatography.
For the composition analysis, the elution of re-dissolved lyophilized aliquots of empty dPNonOx NPs as well as dPNonOx NPs loaded with BRP-201 was monitored using CAD and DAD (Fig. 5). The measurement conditions comprised an isocratic hold at 85/15 (%, v/v) CH3CN/H2O to allow the elution of the small drug molecule with high efficiency, followed by increase of CH3CN content to ensure complete elution of the polymer. The universal detection via CAD resulted in an elution pattern with many resolved species, which is consistent with observations made during the dPOx library characterization (Fig. 5A).
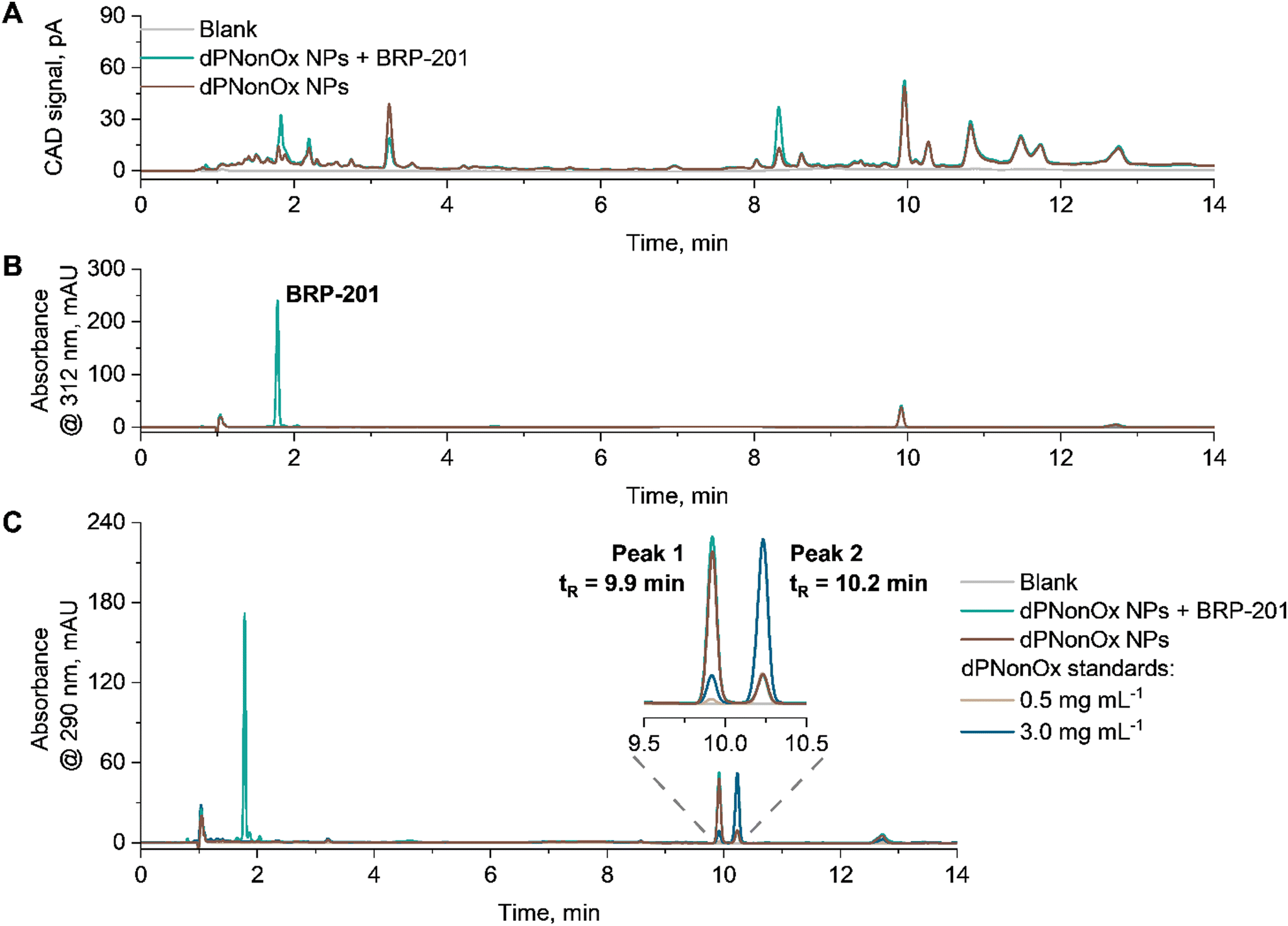 | ||
| Fig. 5 Composition analysis of an exemplified batch of drug-loaded dPNonOx NPs formulated with an initial polymer concentration of 20 mg mL−1. Overlaid elution traces of solvent as well as empty and BRP-201-containing dPNonOx NPs recorded by (A) CAD and (B) DAD at 312 nm. Peak at 1.8 min refers to BRP-201. (C) Overlaid elugrams of empty and BRP-201-containing dPNonOx NPs as well as dPNonOx standards (0.5 and 3.0 mg mL−1) recorded by DAD at 290 nm. Solvent composition in all the samples: 200 μL DMSO and 800 μL 85/15 (%, v/v) CH3CN/H2O. Measurement conditions: flow rate 1.5 mL min−1, isocratic hold for 5 min at 85% (v/v) of CH3CN in the mobile phase followed by a linear gradient of CH3CN (from 85% (v/v) to 100%) in 2 min. | ||
Monitoring the elution of dPNonOx NPs by DAD at the polymer absorbance maximum, i.e., 290 nm, revealed a difference in polymer elution patterns for dPNonOx used in NP formulation and untreated dPNonOx (further referred to as standard) (Fig. 5C). The signal intensity of the two main peaks in the elugram indicated as Peak 1 and Peak 2 in Fig. 5C were reversed (Table S4 and Fig. S12, ESI†). The same signal ratios for empty and drug-loaded dPNonOx NPs demonstrate that the encapsulation of the drug in dPNonOx NPs did not affect the polymer elution pattern. It hence appears likely that the change in elution pattern resulted from the formulation process, which involved solvents, sample centrifugation, lyophilization, and sonication procedures. Further optimization of the formulation process and screening of the resultant NPs by liquid chromatography may, in the future, resolve the issue and allow the quantification of the polymer content in the final formulations.
At the same time, the BRP-201 component was monitored by DAD at 312 nm (Fig. 5B). Its content was determined using a calibration curve that was linear from 5 to 60 μg mL−1 (Fig. S13, ESI†). The elution patterns of three aliquots of dPNonOx NPs containing BRP-201 were repeatable (Fig. S14, ESI†) and the average amount of BRP-201 determined within one formulation batch revealed a coefficient of variation below 3% (Table S5, ESI†).
When three independent formulations were prepared, LC values of 1.5 ± 0.2% (for 5 mg mL−1 initial polymer concentration) and 1.8 ± 0.1% (for 20 mg mL−1 initial polymer concentration) were determined via the developed liquid chromatography protocol (Table S6 and Fig. S15C, ESI†). The LC values are similar to the values previously reported for other nanocarriers.57,59,60 Varying the initial polymer concentration during formulation resulted in NPs with hydrodynamic diameters of 170 ± 30 nm for 5 mg mL−1 and 220 ± 60 nm for 20 mg mL−1, respectively (Fig. S15A, ESI†). Encapsulation of the cargo did not affect the particle size at a significant level for both concentrations, as seen in the overlapping size distributions of loaded and empty NPs (Fig. S16, ESI†). However, it resulted in a decrease of the NP zeta potential, particularly in case of an initial polymer concentration of 20 mg mL−1 (Fig. S15B, ESI†). This might be due to the migration of glycine or residual ethylene imine moieties from the particle surface to form hydrogen bonds with drug molecules. Such hydrogen bonds are believed to contribute to successful encapsulation of BRP-201 into dPNonOx NPs.
Inhibition of 5-LOX activity and toxicity of BRP-201-loaded dPNonOx NPs
Neutrophils, as a primary source of pro-inflammatory leukotrienes, strongly express 5-LOX and its helper protein FLAP. In this aspect, neutrophils were chosen to test the capability of the encapsulated FLAP inhibitor BRP-201 to inhibit the 5-LOX product formation.61 BRP-201-containing dPNonOx NPs were pre-incubated with neutrophils for 15 min and then stimulated by calcium ionophore A23187 (2.5 μM) for 10 min to investigate the FLAP-dependent formation of pro-inflammatory 5-LOX products.In accordance with recently published data, free BRP-201, at a concentration of 0.3 μM, significantly inhibited 5-LOX product formation.62 At the same drug concentration, loaded NPs performed in a similar fashion. BRP-201-loaded NPs were also effective at a concentration of 0.03 μM, but showed only a minor effect at 0.01 μM (Fig. 6). Inhibition of cellular 5-LOX product formation is another evidence for the assumption that the drug is successfully released from the NPs.
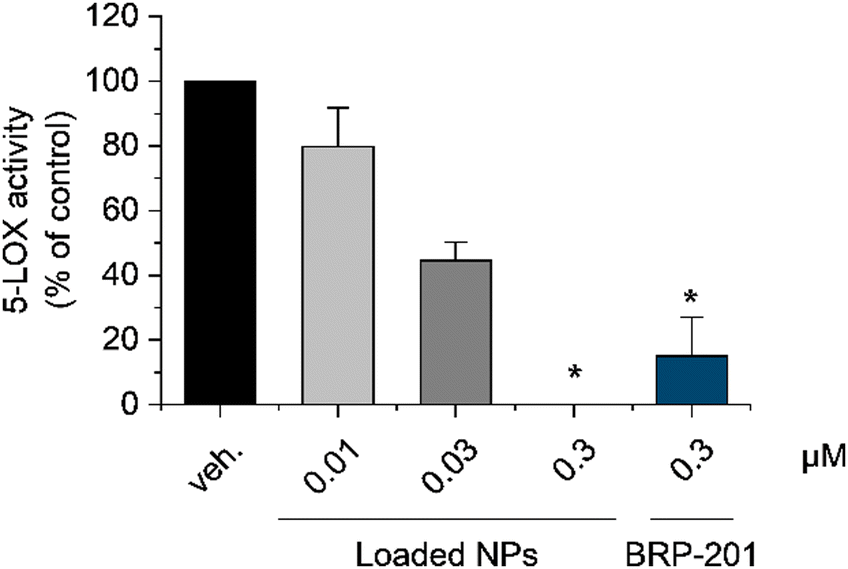 | ||
| Fig. 6 Inhibition of 5-LOX product formation by encapsulated BRP-201 in dPNonOx NPs in neutrophils. Human primary neutrophils were preincubated for 15 min at 37 °C with PBS (vehicle, veh.), free 0.3 μM BRP-201 or BRP-201-loaded dPNonOx NPs containing 0.01, 0.03 or 0.3 μM BRP-201, respectively, and then stimulated with 2.5 μM A23187. After 10 min, the reaction was stopped, and 5-LOX products were extracted via solid phase extraction (SPE) and analyzed by reversed phase liquid chromatography. Values are given as 5-LOX products (LTB4, trans-LTB4, epi-trans-LTB4, and 5-HETE) in percentage of control. For statistical analysis matched one-way ANOVA with Tukey's multiple comparisons test was used; *p < 0.05. | ||
The unloaded NPs inhibited 5-LOX product formation as well, at least at a concentration of 9.3 mg L−1, which corresponds to the NP concentration of drug-loaded dPNonOx NPs containing 0.3 μM of BRP-201 (Fig. S17, ESI†). However, membrane integrity in human neutrophils remained intact after 3 h of incubation with unloaded NPs as well as untreated dPNonOx, displaying no cytotoxic effect (Fig. S18, ESI†).
Conclusions
Degradable analogues of POx (dPOx) containing glycine units in the polymer backbone were characterized by gradient elution liquid chromatography. The findings reveal the shift of elution time with elongation of polymer side chain length in dPOx. Those dPOx were cytocompatible and formed colloidally stable nanoparticle (NP) formulations with positive zeta potentials. More hydrophobic dPOx tended to form larger NPs. NPs formulated with and without surfactant PVA from the most hydrophobic polymer in the library, dPNonOx, were stable in water at 37 °C over 14 days of incubation time. However, the presence of surfactant increased NP lifetime in acidic media (pH 5.0, 50 mM acetate buffer). At the same time, NPs formulated without PVA appeared to be more stable under enzymatic degradation with proteinase K. The degradation of NPs under the action of proteinase K was monitored over 68 days by liquid chromatography and 1H NMR spectroscopy and revealed the formation of glycine and low molar mass degradation products over time, proving the polymer decomposition.The small molecule BRP-201 was successfully encapsulated in the dPNonOx NPs and NP composition analysis, including drug loading, was performed by liquid chromatography. Even though the quantification of the polymer content in the formulation was not possible, the findings highlight liquid chromatography being a powerful tool for screening of the final formulations for quality control purposes. Chromatography is highly sensitive to compositional and structural changes of the utilized materials, which is reflected in a sensitive and quantitative change of the elution fingerprint of these disperse systems. Drug-loaded dPNonOx NPs demonstrated no cytotoxic effect and encapsulated BRP-201 led to the inhibition of 5-LOX product formation in neutrophils, thereby confirming the drug release from degradable NPs. The dPNonOx itself revealed a pharmacological effect that is of interest for further investigations.
At the same time, the study highlights once again liquid chromatography being a universal technique to detect the chemical structural changes at all stages of material development. That is of great importance for formulation strategies and translation of results to the pharmaceutical industry where not only the quantity of the drug, but also a compositional study of intact formulations is essential for future quality control requirements.
Author contributions
Ekaterina Tsarenko (E. T.) – conceptualization, characterization of polymers and NPs by liquid chromatography, visualization and writing – the original draft, review & editing. Natalie E. Göppert (N. E. G.) – synthesis of all polymers and 1H NMR measurements, writing – review & editing. Philipp Dahlke (P. D.) – 5-LOX activity and cytotoxicity assays of NPs, writing – review & editing. Mira Behnke (M. B.) – DLS measurements, writing – review & editing. Gauri Gangapurwala (G. G.) – NP formulation, writing – review & editing. Baerbel Beringer-Siemers (B. B. S.) – NP formulation, DLS measurements, writing – review & editing. Lisa Jaepel (L. J.) – liquid chromatography measurements of drug-loaded NPs, writing – review & editing. Carolin Kellner (C. K.) – cytotoxicity assay of all polymers, writing – review & editing. David Pretzel – supervision of C. K., writing – review & editing. Justyna A. Czaplewska – synthesis of BRP-201, writing – review & editing. Antje Vollrath – supervision of G. G., M. B., B. B. S., writing – review & editing. Paul M. Jordan – supervision of P. D., writing – review & editing. Christine Weber – conceptualization, supervision of N. E. G., writing – the original draft, review & editing. Oliver Werz – funding acquisition, writing – review & editing. Ulrich S. Schubert – funding acquisition, writing – review & editing. Ivo Nischang – conceptualization, funding acquisition, project administration, supervision of E. T., writing – the original draft, review & editing.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Additional data generated during and/or analyzed during the current study are available from the authors on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project number 316213987 – SFB 1278 “PolyTarget” (projects A01, A04, Z01) and the German Federal Ministry for Economic Affairs and Climate Action (BMWK, project BaseLipid, 16LP401003). The UHPLC system was funded by the DFG – 438688627. I. N. acknowledges funding by the DFG – 471397362.References
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci. A-1 Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed. Engl., 1966, 5, 875–888 CrossRef CAS PubMed.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- T. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- C. Englert, J. C. Brendel, T. C. Majdanski, T. Yildirim, S. Schubert, M. Gottschaldt, N. Windhab and U. S. Schubert, Prog. Polym. Sci., 2018, 87, 107–164 CrossRef CAS.
- S. Jana and R. Hoogenboom, Polym. Int., 2022, 71, 935–949 CrossRef CAS.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- V. R. De La Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- A. Mero, G. Pasut, L. D. Via, M. W. M. Fijten, U. S. Schubert, R. Hoogenboom and F. M. Veronese, J. Controlled Release, 2008, 125, 87–95 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- M. Grube, M. N. Leiske, U. S. Schubert and I. Nischang, Macromolecules, 2018, 51, 1905–1916 CrossRef CAS.
- W.-A. Chen, D.-Y. Chang, B.-M. Chen, Y.-C. Lin, Y. Barenholz and S. R. Roffler, ACS Nano, 2023, 17, 5757–5772 CrossRef CAS PubMed.
- Y. Zhao, C. Wang, L. Wang, Q. Yang, W. Tang, Z. She and Y. Deng, Eur. J. Pharm. Biopharm., 2012, 81, 506–513 CrossRef CAS PubMed.
- Y. Ma, Q. Yang, L. Wang, X. Zhou, Y. Zhao and Y. Deng, Eur. J. Pharm. Sci., 2012, 45, 539–545 CrossRef CAS PubMed.
- E. Vlassi, A. Papagiannopoulos and S. Pispas, Eur. Polym. J., 2017, 88, 516–523 CrossRef CAS.
- P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS.
- L. Tauhardt, K. Kempe, K. Knop, E. Altuntaş, M. Jäger, S. Schubert, D. Fischer and U. S. Schubert, Macromol. Chem. Phys., 2011, 212, 1918–1924 CrossRef CAS.
- N. E. Göppert, M. Kleinsteuber, C. Weber and U. S. Schubert, Macromolecules, 2020, 53, 10837–10846 CrossRef.
- H. C. M. Byrd and C. N. McEwen, Anal. Chem., 2000, 72, 4568–4576 CrossRef CAS PubMed.
- N. E. Göppert, A. Vollrath, L. M. Stafast, S. Stumpf, B. Schulze, S. Hoeppener, C. Weber and U. S. Schubert, RSC Appl. Polym., 2024, 2, 184–195 RSC.
- M. W. F. Nielen, Mass Spectrom. Rev., 1999, 18, 309–344 CrossRef CAS.
- M. S. Engler, S. Crotty, M. J. Barthel, C. Pietsch, U. S. Schubert and S. Böcker, Rapid Commun. Mass Spectrom., 2016, 30, 1233–1241 CrossRef CAS PubMed.
- S. B. Ganorkar and A. A. Shirkhedkar, Rev. Anal. Chem., 2017, 36 Search PubMed.
- N. Engel, M. Dirauf, J. A. Czaplewska, I. Nischang, M. Gottschaldt and U. S. Schubert, R. Soc. Open Sci., 2024, 11, 231008 CrossRef CAS PubMed.
- D. Ilko, S. Puhl, L. Meinel, O. Germershaus and U. Holzgrabe, J. Pharm. Biomed. Anal., 2015, 104, 17–20 CrossRef CAS PubMed.
- T. Chang, Recent Progress in Separation of Macromolecules and Particulates, American Chemical Society, 2018, ch. 1, vol. 1281, pp. 1–17 Search PubMed.
- E. Uliyanchenko, S. van der Waals and P. J. Schoenmakers, Polym. Chem., 2012, 3, 2313–2335 RSC.
- A. M. Striegel, TrAC, Trends Anal. Chem., 2020, 130 Search PubMed.
- W. J. Staal, Chromatographia, 2022, 85, 1–5 CrossRef CAS.
- A. Chojnacka, K. Kempe, H. C. van de Ven, C. Englert, R. Hoogenboom, U. S. Schubert, H.-G. Janssen and P. Schoenmakers, J. Chromatogr. A, 2012, 1265, 123–132 CrossRef CAS PubMed.
- O. Sedlacek, O. Janouskova, B. Verbraeken and R. Hoogenboom, Biomacromolecules, 2019, 20, 222–230 CrossRef CAS PubMed.
- J. Kim, V. Beyer and C. R. Becer, Macromolecules, 2022, 55, 10651–10661 CrossRef CAS.
- K. Mint, J. P. Morrow, N. M. Warne, X. He, D. Pizzi, S. Z. O. Shah, G. K. Pierens, N. L. Fletcher, C. A. Bell, K. J. Thurecht and K. Kempe, Polym. Chem., 2024, 15, 2662–2676 RSC.
- M. Brunzel, T. C. Majdanski, J. Vitz, I. Nischang and U. S. Schubert, Polymers, 2018, 10 Search PubMed.
- M. Brunzel, M. Dirauf, M. Sahn, J. A. Czaplewska, N. Fritz, C. Weber, I. Nischang and U. S. Schubert, J. Chromatogr. A, 2021, 1653, 462364 CrossRef CAS PubMed.
- E. Tsarenko, U. S. Schubert and I. Nischang, Anal. Chem., 2023, 95, 565–569 CAS.
- B. Wang, L. Wu, J. Chen, L. Dong, C. Chen, Z. Wen, J. Hu, I. Fleming and D. W. Wang, Signal Transduction Targeted Ther., 2021, 6, 94 CrossRef CAS PubMed.
- E. Banoglu, E. Çelikoğlu, S. Völker, A. Olgaç, J. Gerstmeier, U. Garscha, B. Çalışkan, U. S. Schubert, A. Carotti, A. Macchiarulo and O. Werz, Eur. J. Med. Chem., 2016, 113, 1–10 CrossRef CAS PubMed.
- P. M. Jordan, J. Gerstmeier, S. Pace, R. Bilancia, Z. Rao, F. Börner, L. Miek, Ó. Gutiérrez-Gutiérrez, V. Arakandy, A. Rossi, A. Ialenti, C. González-Estévez, B. Löffler, L. Tuchscherr, C. N. Serhan and O. Werz, Cell Rep., 2020, 33, 108247 CrossRef CAS PubMed.
- C. Kretzer, P. M. Jordan, R. Bilancia, A. Rossi, T. Gür Maz, E. Banoglu, U. S. Schubert and O. Werz, J. Inflammation Res., 2022, 15, 911–925 CrossRef CAS PubMed.
- O. Werz, E. Bürkert, B. Samuelsson, O. Rådmark and D. Steinhilber, Blood, 2002, 99, 1044–1052 CrossRef CAS PubMed.
- A. Zielińska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef PubMed.
- B. Shkodra-Pula, A. Vollrath, U. S. Schubert and S. Schubert, Colloids for Nanobiotechnology – Synthesis, Characterization and Potential Applications, 2020, pp. 233–252 Search PubMed.
- J. W. Hickey, J. L. Santos, J.-M. Williford and H.-Q. Mao, J. Controlled Release, 2015, 219, 536–547 CrossRef CAS PubMed.
- M. Homs, G. Calderó, M. Monge, D. Morales and C. Solans, Colloids Surf., A, 2018, 536, 204–212 CrossRef CAS.
- A. Vollrath, C. Kretzer, B. Beringer-Siemers, B. Shkodra, J. A. Czaplewska, D. Bandelli, S. Stumpf, S. Hoeppener, C. Weber, O. Werz and U. S. Schubert, Polymers, 2021, 13, 2557 CrossRef CAS PubMed.
- L. Wu, J. Zhang and W. Watanabe, Adv. Drug Delivery Rev., 2011, 63, 456–469 CrossRef CAS PubMed.
- O. Mert, S. K. Lai, L. Ensign, M. Yang, Y.-Y. Wang, J. Wood and J. Hanes, J. Controlled Release, 2012, 157, 455–460 CrossRef CAS PubMed.
- B. Shkodra-Pula, C. Grune, A. Traeger, A. Vollrath, S. Schubert, D. Fischer and U. S. Schubert, Int. J. Pharm., 2019, 566, 756–764 CrossRef CAS PubMed.
- B. Song and C.-W. Cho, J. Pharm. Invest., 2024, 54, 249–266 CrossRef CAS.
- J. A. Mótyán, F. Tóth and J. Tőzsér, Biomolecules, 2013, 3, 923–942 CrossRef PubMed.
- A. Bher, Y. Cho and R. Auras, Macromol. Rapid Commun., 2023, 44, 2200769 CrossRef CAS PubMed.
- F. Quattrini, G. Berrecoso, J. Crecente-Campo and M. J. Alonso, Drug Delivery Transl. Res., 2021, 11, 373–395 CrossRef CAS PubMed.
- G. Cinar, J. I. Solomun, P. Mapfumo, A. Traeger and I. Nischang, Anal. Chim. Acta, 2022, 1205, 339741 CrossRef CAS PubMed.
- H. C. Haas, N. W. Schuler and R. L. Macdonald, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 3143–3158 CrossRef CAS.
- C. Kretzer, B. Shkodra, P. Klemm, P. M. Jordan, D. Schröder, G. Cinar, A. Vollrath, S. Schubert, I. Nischang, S. Hoeppener, S. Stumpf, E. Banoglu, F. Gladigau, R. Bilancia, A. Rossi, C. Eggeling, U. Neugebauer, U. S. Schubert and O. Werz, Cell. Mol. Life Sci., 2022, 79 Search PubMed.
- A. Zeb, M. Gul, T.-T.-L. Nguyen and H.-J. Maeng, J. Pharm. Invest., 2022, 52, 683–724 CrossRef CAS.
- M. Behnke, A. Vollrath, P. Dahlke, F. P. Larios, M. Chi, E. Tsarenko, P. M. Jordan, C. Weber, M. Dirauf, J. A. Czaplewska, B. Beringer-Siemers, S. Stumpf, C. Kellner, C. Kretzer, S. Hoeppener, I. Nischang, M. Sierka, C. Eggeling, O. Werz and U. S. Schubert, Mater. Today Chem., 2024, 35, 101848 CrossRef CAS.
- J. Ismail, L. C. Klepsch, P. Dahlke, E. Tsarenko, A. Vollrath, D. Pretzel, P. M. Jordan, K. Rezaei, J. A. Czaplewska, S. Stumpf, B. Beringer-Siemers, I. Nischang, S. Hoeppener, O. Werz and U. S. Schubert, Pharmaceutics, 2024, 16, 187 CrossRef CAS PubMed.
- M. Werner, P. M. Jordan, E. Romp, A. Czapka, Z. Rao, C. Kretzer, A. Koeberle, U. Garscha, S. Pace, H.-E. Claesson, C. N. Serhan, O. Werz and J. Gerstmeier, FASEB J., 2019, 33, 6140–6153 CrossRef CAS PubMed.
- Z. T. Gür, B. Çalışkan, U. Garscha, A. Olgaç, U. S. Schubert, J. Gerstmeier, O. Werz and E. Banoglu, Eur. J. Med. Chem., 2018, 150, 876–899 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01812e |
| This journal is © The Royal Society of Chemistry 2024 |