
“Flagella effect” of thermally activated delayed fluorescence emitter dominating the efficiency of non-doped solution-processed OLEDs†
Guimin
Zhao
,
Yuheng
Lou
,
Renjie
Ji
,
Qiyin
Ran
,
Haowen
Chen
,
Wenwen
Tian
,
Wei
Jiang
 * and
Yueming
Sun
* and
Yueming
Sun
Jiangsu Province Hi-Tech Key Laboratory for Bio-Medical Research, Jiangsu Engineering Laboratory of Smart Carbon-Rich Materials and Device, School of Chemistry and Chemical Engineering, Southeast University, Nanjing, Jiangsu 211189, China. E-mail: jiangw@seu.edu.cn
First published on 16th November 2023
Abstract
Flexible chains have important research value in the field of organic optoelectronics and biomedicine because they can adjust the functionalization of organic molecules in the aggregated state. Herein, by regulating the length and mold of the flexible chain attached to a thermally activated delayed fluorescence (TADF) core, we propose the “flagella effect” of solution-processed emitters for non-doped organic light-emitting diodes. In the single-molecular state, the emission behavior of these molecules remained consistent because of the unified TADF core; however, the aggregated films showed significantly different emission characteristics. A longer alkyl chain improved the photoluminescence quantum yield (PLQY) of the pristine film, but blocked the carrier transport and balance. Alternatively, a shorter alkyl chain matched the carrier mobility but sacrificed the PLQY and color quality. Accordingly, the maximum external quantum efficiency (EQEmax) of the molecule with a hexane chain was 18.7%, which was about 2 times that of the molecule with an ethane chain and approaching 80 times that of the molecule with a dodecane chain. Besides, compared with homologous alkyl chains, the molecule with multi-oxygen chains only achieved an EQEmax of 2.1%, which is attributed to the large degree of crystallization and aggregation of the multi-oxygen chains in the pristine film, as confirmed by atomic force microscopy measurement.
1. Introduction
It is well known that bacteria with a tiny volume are the simplest and indispensable creatures on Earth, making up a rich and colorful microcosm.1 Bacterial flagella, acting as a vital component of the bacterial cell (vibrio, bacillus and individual coccus), are slender, wavy curved, and hairy filamentous organelles that are responsible for driving the movement of bacterial bodies on surfaces or in liquids.2,3 Different bacteria have different flagellar structures, where the number of flagellates can be as small as one to two or as many as hundreds. The presence of flagella greatly enriches and enhances the biological functions of individual bacteria. Similarly, as the smallest unit of organic functional materials, an organic molecule at the microscopic level is similar to a bacterium. Accordingly, if the flexible chain of organic molecules is modified like flagella on their periphery through simple molecular engineering, it is expected that the function of the molecule can be adjusted and the optimal performance of organic materials can be realized in the aggregate state.π-Conjugated molecules are widely used in organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs), organic photovoltaics (OPVs) and other fields because of the diversity and variability of their molecular structure.4–8 However, due to the large conjugated system and rigidity of organic materials, their preparation and purification are generally difficult, further hindering their application in solution-processed devices.9,10 Thus, to solve this dilemma, flexible chains with high flexibility as solubilizing fragments have been extensively employed to modify molecules to improve their solution processability.11–21 Burn et al. reported that 2-ethylhexyl surface groups acted as solubilizing groups for branched thermally activated delayed fluorescent (TADF) emitters to realize a maximum external quantum efficiency (EQEmax) of close to 10% for solution-processed OLEDs.22 Besides, flexible chains can encapsulate the central organic chromophore to improve the emission efficiency. For example, Shao et al. reported that an n-butoxy-encapsulated dendritic TADF emitter achieved an EQEmax of up to 20.6% for solution-processed OLEDs, which was 2.0 times that of its tert-butyl-encapsulated counterparts.23 In fact, there are also many unexpected regulatory effects by flexible chains on the molecular conformation, morphology, arrangement and orientation of organic materials in thin films.24–30 Wang et al. developed a clever side-chain engineering to facilitate the nonradiative thermal deactivation-involved photothermal property in the aggregate state.31 Li et al. regulated the arrangement of organic dyes with different alkyl chains on the surface of TiO2, which is beneficial for the light-harvesting and inhibition of electron recombination, leading to an improved OPV performance.32 However, to date, although numerous studies on flexible chains have been reported and some mature mechanisms on solid-state films have been proposed for organic semiconductors, systematic research on the effect of flexible chains in OLEDs is rare and needs further exploration.
Herein, to investigate the effect of the length and mold of flexible chains on the physical properties and device performance of solution-processed OLEDs, we rationally designed a series of TADF-active molecules bearing alkyl chains with different lengths (ethane to dodecane) and oligo(ethylene glycol) (OEG) chains. These molecules exhibited the same emission behavior in toluene solution, illustrating that the TADF emission was retained. However, interestingly, these molecules showed different photophysical performances in thin films depending on the flexible chain. In the case of the materials with different alkyl chain lengths, the longer the alkyl chain, the better the protection of the TADF emission core, thus improving the solid-state PLQY from 41% to 63%, but simultaneously a longer alkyl chain also hindered the carrier transport and balance. Meanwhile, a shorter alkyl chain maintained the carrier mobility and balance but intermolecular aggregation decreased the PLQY and redshifted the emission. Consequently, the non-doped solution-processed OLEDs based on the molecule with a hexane chain exhibited an EQEmax of 18.7%, which was almost 2 times that of the molecule with an ethane chain, and nearly 80 times that of the molecule with a dodecane chain. In addition, a low EQEmax of 2.1% was achieved for the OEG chain-based solution-processed OLEDs, which was less than one-seventh of that with the homologous alkyl-chain molecule. Atomic force microscopy measurement showed that the large degree of crystallization and aggregation of multi-oxygen chains destroyed the morphology in the thin film, resulting in a large surface roughness of 1.87 nm. Hence, this work inspires us to consider not only the solubility and solid-state PLQY, but also the carrier transport in the TADF emitter for solution-processed OLEDs. Meanwhile, this study opens a new avenue for exploring the “flagella effect” of flexible chains in the film state.
2. Results and discussion
2.1 Synthesis and characterization
According to the literature, the introduction of flexible alkyl chains in molecules increases their solubility and facilitates the solution processability of OLEDs.33–37 However, the systematic study of the behavior and role of flexible chains in OLEDs remains a challenge, especially in the aggregated state. Given the excellent TADF emission of (2s,3r,4r)-2,3,4,5,6-penta(9H-carbazol-9-yl)benzonitrile (5CzBN),38–40 in this work, alkyl chains with different lengths, ranging from ethane, propane, hexane, heptane, and nonane to dodecane, were introduced in the 5CzBN core to obtain six TADF compounds, which were denoted as 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod, respectively (Fig. 1). The OEG chains feature hydrophilicity, high polarity, high flexibility and ionic conductivity, which also have been widely used as side chains in conjugated polymers for OPV and OFET.41–44 Therefore, it is worth exploring in depth whether these characteristics can be adopted in OLEDs like other fields of optoelectronics. Accordingly, the alkyl chain was replaced with an OEG chain to obtain the 5CzBN-OEG molecule. By adjusting the length and mold of the flexible chain, we aimed to study the discrepancy in the emissive behavior and morphology in the thin-film state.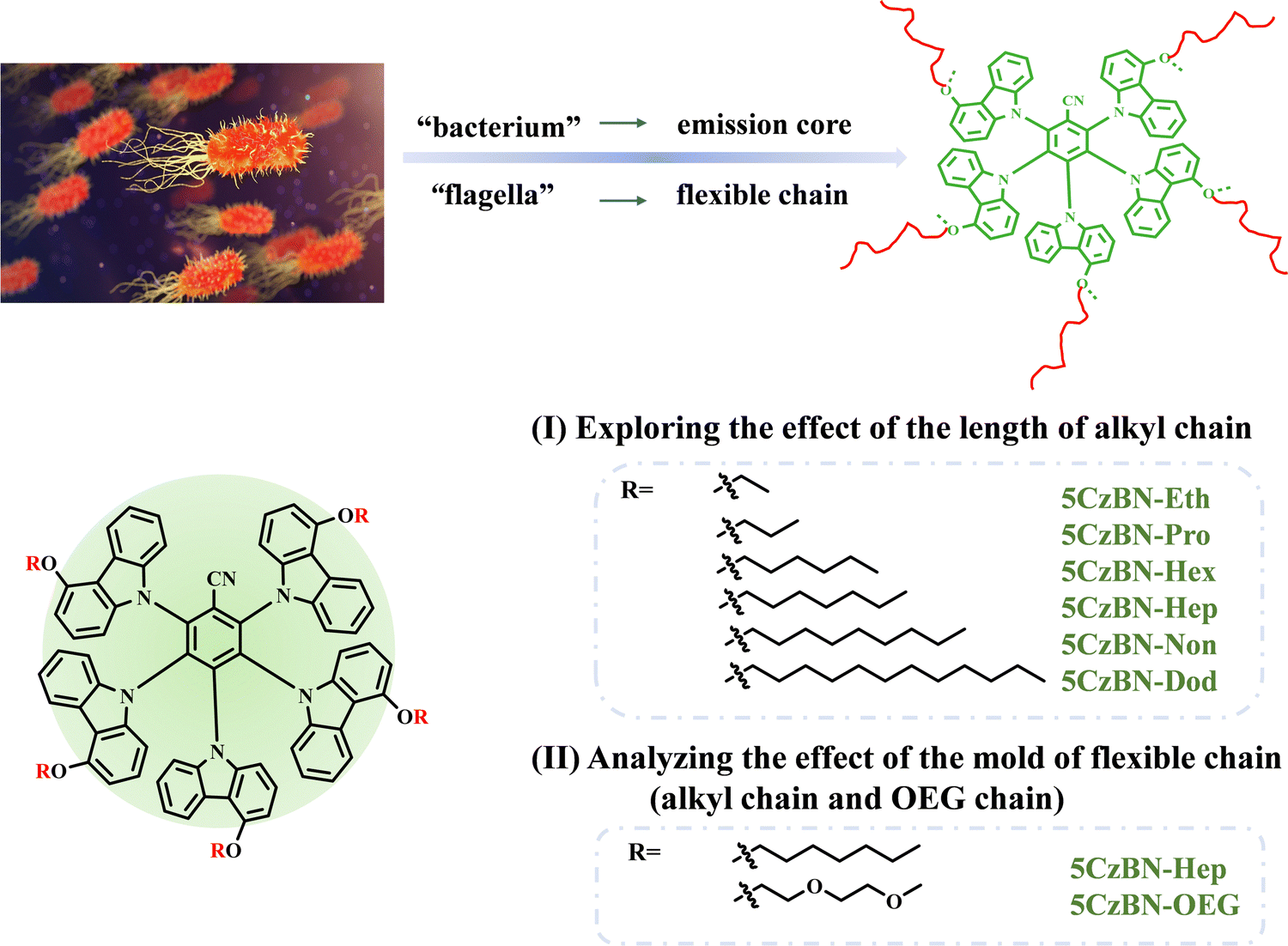 | ||
| Fig. 1 Molecular design strategy of TADF emitters. | ||
The detailed synthesis of 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hep, 5CzBN-OEG, 5CzBN-Non and 5CzBN-Dod is presented in Scheme S1, ESI,† while that of 5CzBN-Hex was shown in our previous work.45 These new molecules were obtained via the two-step reaction of Williamson ether synthesis and nucleophilic substitution in considerable yield. These materials were purified by column chromatography, and then recrystallization. It is worth mentioning that because the carbon atoms are replaced by oxygen atoms, the polarity of the eluent used for column chromatography of 5CzBN-OEG was much higher than that for column chromatography of 5CzBN-Hep. The molecular structures of the new molecules were fully characterized via1H NMR, 13C NMR, high-resolution mass spectrometry and MALDI-TOF mass spectrometry (Fig. S1–S12, ESI†), which provided a good match with the targeted structure. In addition, all the emitters possessed excellent solubility in commonly used organic solvents, such as toluene, dichloromethane, chloroform, and tetrahydrofuran. This endows the corresponding materials with good solubility during solution processing.
2.2 Thermal and morphological properties
As one of the primary methods for the fabrication of OLED devices, solution technology requires materials with high thermal stability. Consequently, the thermal stability of the emitters was evaluated via thermogravimetric analysis and differential scanning calorimetry (DSC) under a nitrogen atmosphere, as shown in Fig. 2a and b, respectively. All the compounds exhibited high thermal decomposition temperatures (Td, corresponding to 5% weight loss) in the range of 386–413 °C, indicating their high degree of thermal stability. Moreover, DSC measurements were carried out from 20–200 °C at a heating rate of 10 °C min−1. These materials exhibited apparent peaks with a glass transition temperature (Tg) in the measurement temperature range, which gradually decreased with an increase in the alkyl chain length. In the case of 5CzBN-Eth, its Tg was 183 °C, whereas that observed for 5CzBN-Dod was 64 °C, which shows that its molecular structure was more flexible. However, a lower Tg can limit the improvement in the efficiency of OLEDs.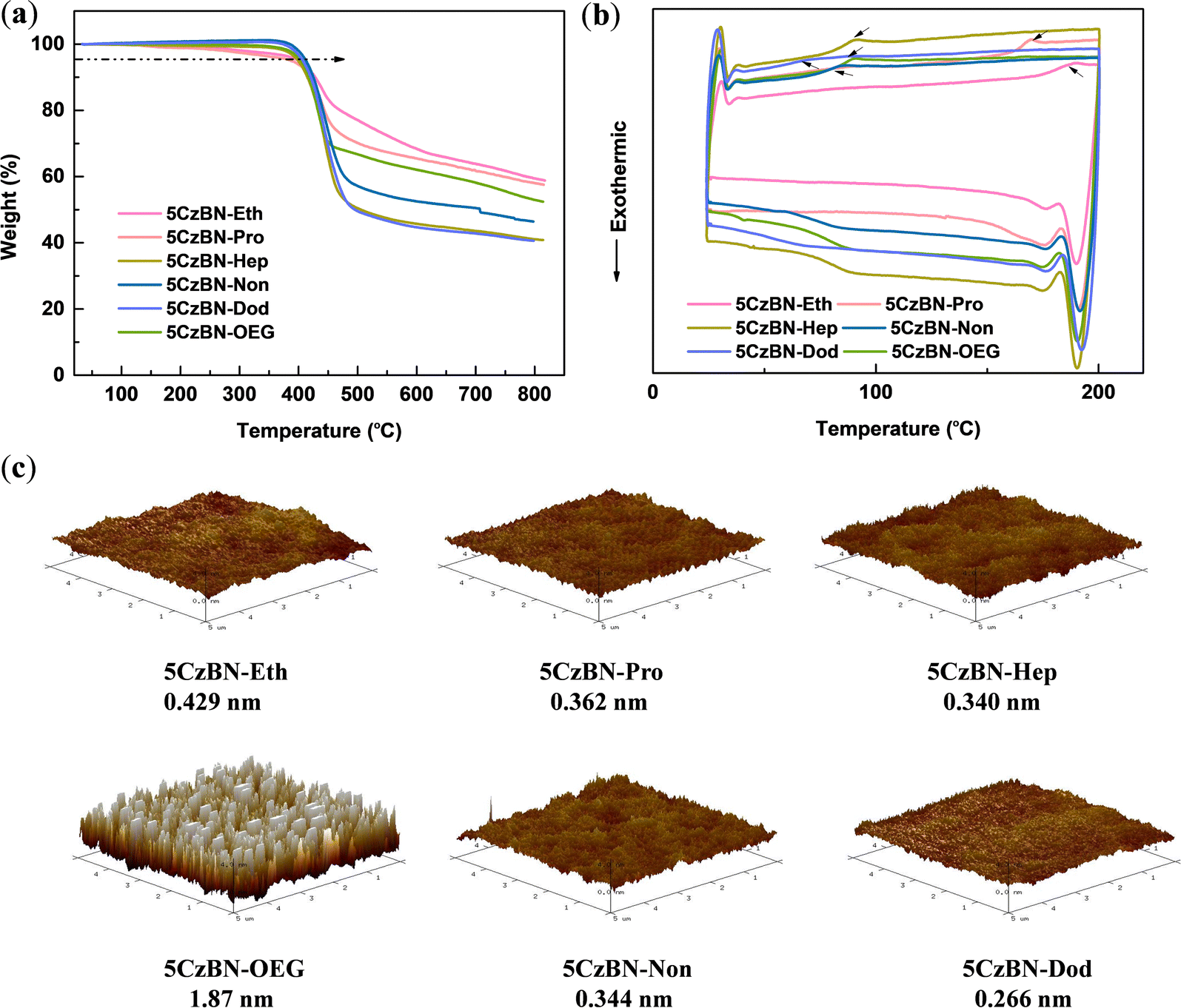 | ||
| Fig. 2 (a) TGA curves and (b) DSC curves of 5CzBN derivatives recorded at a heating rate of 10 °C min−1. (c) AFM topographic images and surface roughness of the solution-processed films of 5CzBN derivatives. | ||
The morphology of pristine films is critical in the performance of non-doped solution-processed OLEDs. Poor film quality can lead to leakage current, traps or defects, which is not conducive to the efficient use of excitons. Accordingly, atomic force microscopy (AFM) was employed to analyze the uniformity and flatness of the spin-coated films. As shown in Fig. 2c, smooth films without any pinholes or cracks for compounds with alkyl chains were observed, and all the root-mean-square (RMS) values of surface roughness were less than 0.5 nm. A longer the alkyl chain was beneficial to obtain a smoother film. The RMS values of 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod were 0.429 nm, 0.362 nm, 0.340 nm, 0.344 nm and 0.266 nm, respectively. However, the film based on 5CzBN-OEG showed remarkable crystallinity with an RMS value of 1.87 nm, which is about 6-fold that of 5CzBN-Hep. This can be attributed to the fact that the high-flexibility OEG chain easily induces the molecular backbone to form tighter packing patterns and triggers intermolecular aggregation. These results suggest that the OEG chain results in a poor morphology, which can affect the solution-processable devices.
2.3 Theoretical calculation
To explore the influence of flexible chains on the geometric and electronic structures of these compounds, the distributions of their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were simulated by density functional theory (DFT) at the B3LYP/6-31g(d) level. The HOMOs and LUMOs of these emitters are well separated, where their HOMOs are mainly located on the electron-rich carbazole moieties and LUMOs on the electron-deficient benzonitrile moiety, implying the typical characteristic of TADF emitters (Fig. 3). The optimized ground-state configurations adopt a divergent branch shape, with large torsion angles in the range of 63°–66° between the five carbazole moieties and benzonitrile unit. This leads to largely minimized spatial overlap between the HOMO and LUMO with small singlet–triplet energy difference (ΔEST) values of 0.13–0.14 eV (Table S1, ESI†), which satisfies the prerequisite for efficient up-conversion through the endothermic reverse intersystem crossing (RISC) process. The calculated optical band gaps (Eg) are in the range of 3.20–3.22 eV. Also, nearly identical HOMO and LUMO energy levels were found in these 5CzBN derivatives, which is consistent with their CV curves, indicating that the length and type of flexible chains are not correlated with the frontier molecular orbital distributions in the single molecular state. It is noteworthy that the dipole moment of 5CzBN-OEG is 6.0096 Debye, which is much larger than that of its alkyl chain counterparts. This phenomenon can be attributed to its exposed methoxy group, which matches the polarity of the above-mentioned eluent. This is also in accordance with the previously reported larger polarity of the OEG chain.46 Time-dependent DFT was also employed to calculate the excited-state energy level. Fig. S13 (ESI†) shows the hole and particle of the first excited singlet state (S1), which are distributed in the donor and acceptor components, respectively, revealing the typical charge transfer (CT) character. Meanwhile, the hole and particle of the first excited triplet state (T1) display a mixed state of local excited state (LE) and CT characteristic, favoring the promotion of RISC.47 With an increase in the alkyl chain length, the molecule structure becomes more spatially expanded where the length, width, and height increase from 21 Å, 19 Å, and 11 Å for 5CzBN-Eth to 43 Å, 40 Å and 11 Å for 5CzBN-Dod, respectively, as shown in Fig. S14 (ESI†). The molecular radius is extended from 12 Å for 5CzBN-Eth to 24 Å for 5CzBN-Dod. Given that the T1 exciton of these compounds is only distributed in the 5CzBN core, the alkyl chains with gradually increasing length act as inert components to expand their molecular size and enlarge their intermolecular distances, which can protect the emissive core and increase the PLQY. Comparing the molecular sizes of 5CzBN-Hep and 5CzBN-OEG, their negligible difference demonstrates that the peripheral flexible chains do not affect the spatial extension of the single molecule. Here, although the solid-state emissive efficiency is expected to be enhanced, the charge carrier mobility has to been considered due to the overlarge molecular size from the outer alkyl chain to the 5CzBN core, and the interrupted π-conjugation, which will be discussed in device fabrication.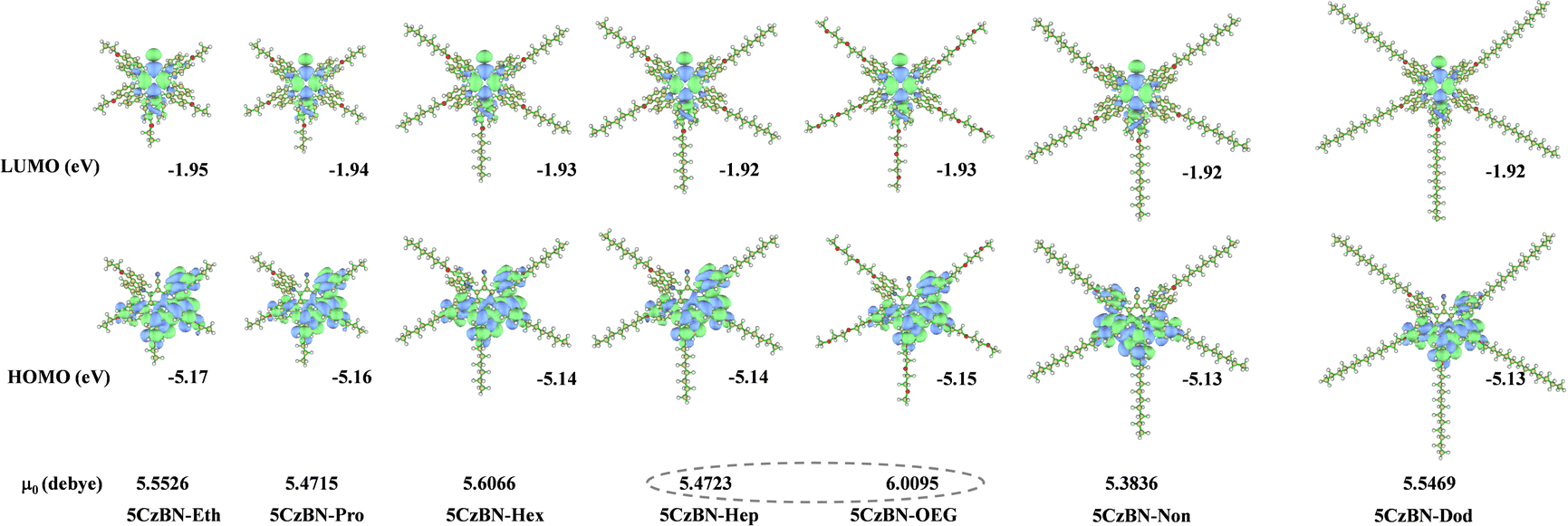 | ||
| Fig. 3 Optimized S0 geometries, twisted dihedrals between donor and acceptor units, calculated HOMO–LUMO energies, distributions, band gap energy, excited-state energy levels, and energy difference of 5CzBN derivatives at the B3LYP/6-31G(d) level in the gas phase. | ||
Additionally, the HOMO energy levels of the emitters were determined by analyzing the electrochemical behavior in their cyclic voltammetry (CV) curves in dichloromethane solution, as presented in Fig. S15 (ESI†) and Table 1. The CV curves of these materials have similar oxidation potentials, indicating that the centers of the electrochemical oxidation processes are not affected by the peripheral flexible chain. Consequently, the EHOMO was calculated according to the following equation: EHOMO = −(Eox + 4.30) eV, which was in the range of −5.25 eV to −5.27 eV for all the materials (Table 1). This suitable and shallow EHOMO is beneficial for the effective injection of holes from the anode to the emitting layer.
| Compound | T d /Tgb (°C) | λ em (nm) | Φ PL (%) | τ p /τdf (ns/μs) | K r (106 s−1) | K nr,T (105 s−1) | K ISC (107 s−1) | K RISC (106 s−1) |
|---|---|---|---|---|---|---|---|---|
| a T d was measured by TGA (corresponding to 5% weight loss). b T g was measured by DSC. c The emission peak in solution-processed solid film at 300 K. d Absolute PLQY evaluated using an integrating sphere measured in solution-processed solid film at 300 K. e Prompted (PF). f Delayed fluorescence (DF) lifetime. g The rate constant of radiative transition of singlet exciton. h The rate constant of non-radiative transition of triplet exciton. i The rate constant of intersystem crossing. j The rate constant of reverse intersystem crossing. | ||||||||
| 5CzBN-Eth | 400/183 | 524 | 41 | 30/1.53 | 6.8 | 3.8 | 2.6 | 0.83 |
| 5CzBN-Pro | 386/165 | 520 | 43 | 30/1.63 | 5.7 | 4.2 | 2.8 | 1.1 |
| 5CzBN-Hex | 397/116 | 507 | 55 | 27/1.59 | 2.9 | 3.1 | 3.4 | 4.1 |
| 5CzBN-Hep | 399/85 | 507 | 57 | 35/1.82 | 2.7 | 2.5 | 2.6 | 3.1 |
| 5CzBN-Non | 413/79 | 502 | 59 | 32/1.76 | 2.9 | 2.6 | 2.9 | 3.4 |
| 5CzBN-Dod | 406/64 | 499 | 63 | 31/1.92 | 3.0 | 2.1 | 2.9 | 3.3 |
| 5CzBN-OEG | 404/85 | 517 | 42 | 24/1.77 | 11.0 | 4.3 | 3.5 | 0.56 |
2.4 Photophysical properties
The UV-vis absorption spectra of these molecules in toluene solution (10−5 mol L−1) exhibit identical absorption spectra, i.e., strong absorption bands in the range of 200 nm–400 nm and a broad low-energy absorption in the range of 400 nm–450 nm, corresponding to the local π–π* and n–π* transitions and the intramolecular charge transfer (CT) of the molecular conjugated backbone from the carbazole donor to the benzonitrile acceptor moiety, respectively (Fig. 4a–g). Consequently, the experimental optical band gap (Eg) is 2.77 eV for all the compounds. According to the Eg, the ELUMO of all the materials is in the range of −2.48 eV to −2.51 eV (Table 1). Additionally, the absorption spectra of these compounds are independent of the polarity in different solvents ranging from toluene to dichloromethane (Fig. S16, ESI†). Meanwhile, consistent emission behaviors of these compounds were observed in toluene solution (10−5 mol L−1), in which the emission peaks were stable at 489 nm. Their S1 energy level is the same, which was calculated to be 2.81 eV using the onset of photoluminescence (PL) spectra. Different from the absorption spectra, the PL spectra (Fig. S17, ESI†) showed a remarkable bathochromic shift with an increase in the solvent polarity, which demonstrates the strong CT characteristic of S1. To detect the ΔEST in diluted solution, the phosphorescent spectra were recorded at 77 K in toluene (10−5 mol L−1), which also exhibit identical bands and characteristic vibrational profiles and show a mixture of LE and CT natures. This experimental result is also consistent with the theoretical calculation. The corresponding energy levels of T1 and ΔEST were determined to be 2.71 eV and 0.10 eV, respectively. This small ΔEST suggests the efficient up-conversion process and underlying TADF property. Importantly, the same absorption and emission spectra of the seven compounds in toluene solution imply that the different flexible chains do not affect the electronic structure of the molecular backbone in the isolated state.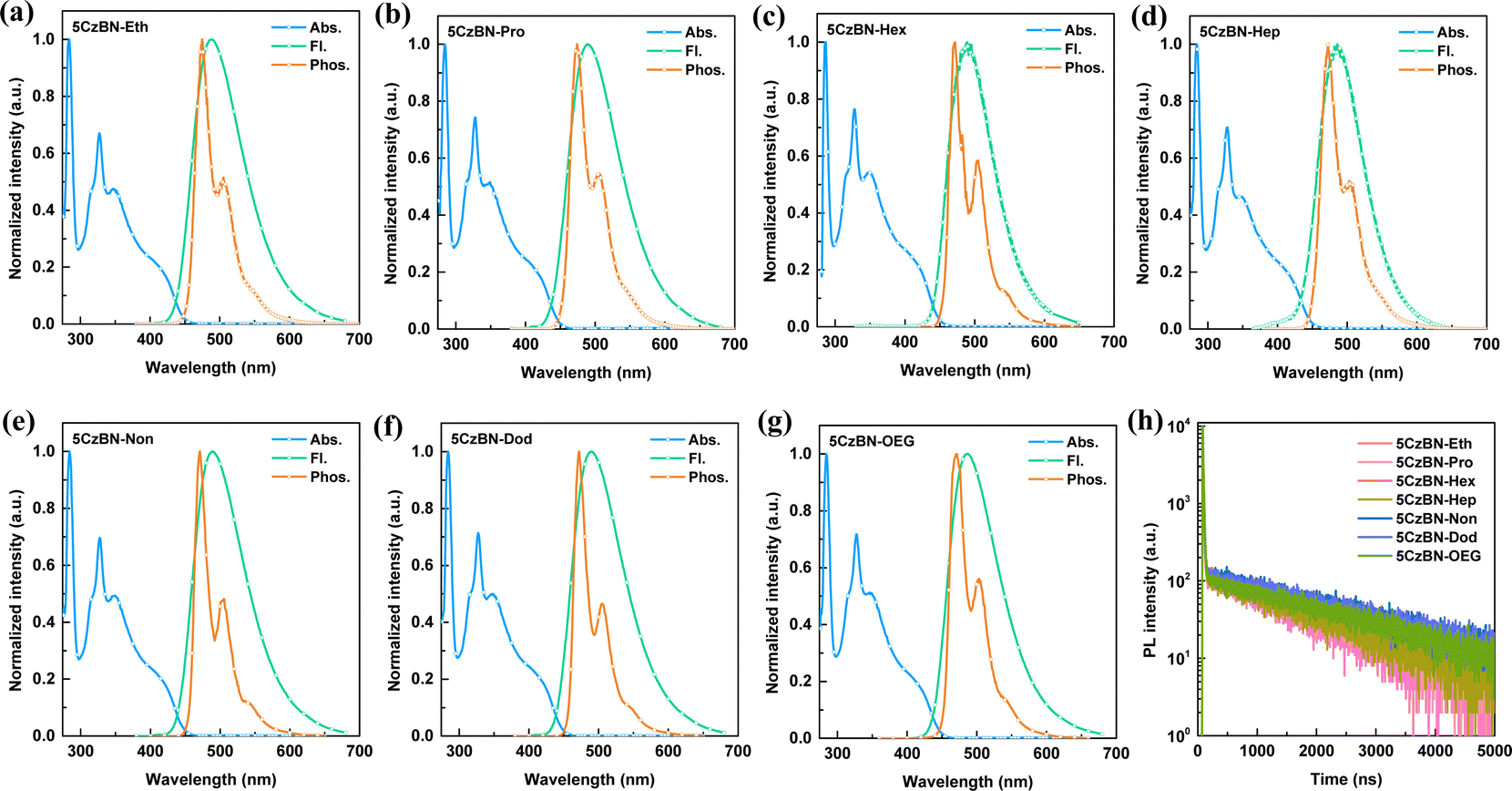 | ||
| Fig. 4 Ultraviolet-visible (UV-vis) absorption, photoluminescence (PL) and phosphorescent spectra of (a) 5CzBN-Eth, (b) 5CzBN-Pro, (c) 5CzBN-Hex, (d) 5CzBN-Hep, (e) 5CzBN-Non, (f) 5CzBN-Dod and (g) 5CzBN-OEG in toluene solution with a concentration of 10−5 M. (h) Transient PL decay curves of 5CzBN derivatives in toluene solution after N2 bubbling for 10 min with a laser of 375 nm. | ||
To study the existence of delayed fluorescence, the transient PL decay spectra and PLQY of these compounds were measured in the toluene (10−5 mol L−1). Before degassing, the delayed component (0.28 μs) in the PL decay curves was absent, whereas obvious second-order exponential decay with a prompt lifetime (τp = 9 ns) and prolonged delayed lifetime (τd = 2.40 μs) upon 10 min of N2 bubbling were observed, demonstrating the TADF characteristics (Fig. 4h and Fig. S18, ESI†). The PLQY remarkably increased from 13% to 87%, after 10 min of N2 bubbling to remove the dissolved O2 molecules in toluene solution. Furthermore, the delayed lifetime or PLQY in degassed toluene showed basically the same values, which again serves to emphasize the earlier point that the emission behavior of these materials is not influenced by the different flexible chains in the isolated state. Additionally, all these compounds show obvious aggregation-induced emission (AIE) behavior in tetrahydrofuran/water mixture solvent, and the phenomenon of bathochromic shift and hypochromatic shift represent the twisted intramolecular charge transfer (TICT) state and formation of nanoaggregates, respectively (Fig. S19, ESI†).48 When the water fraction increased to 60–70%, nanoaggregates were formed, which protected the triplet excitons from oxygen to up-convert to singlet excitons, emitting delayed fluorescence through the RISC process. A similar mechanism of AIE was reported in previous works.21,49 This is also consistent with the obvious enhancement in PLQY in degassed toluene after isolating oxygen.
Unlike toluene solution, compared with 5CzBN-Hep, a redshift in the absorption spectra in thin film was observed for 5CzBN-OEG (Fig. S20, ESI†). Also, a similar redshift also occurred in the fluorescence spectra, where peaks were observed at 507 nm and 517 nm for 5CzBN-Hep and 5CzBN-OEG, respectively (Fig. 5a). The more flexible OEG chains easily reduce the π–π stacking distance, resulting in strong interaction in the molecular backbone. Considering that these 5CzBN derivatives have the same molecular backbone, the redshifted absorption spectra and emission spectra are ascribed to the strong interaction in the conjugated molecular backbone in the solid state. Besides, the emission peaks of 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod in thin film are located at 524, 520, 507, 507, 502 and 499 nm, respectively. To further analyze the change in PL behavior from dispersed to aggregated state, the PL spectra in the doped PMMA films were measured with different doped ratios ranging from 1 wt% to 100 wt% (Fig. S21, ESI†). A redshift of 48 nm, 46 nm, 31 nm, 31 nm, 26 nm and 23 nm was observed for 1 wt% to 100 wt%, whereas 27 nm, 25 nm, 10 nm, 9 nm, 6 nm and 3 nm from 10 wt% to 100 wt% for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod, respectively (Fig. 5b). The diminished redshifted emission indicates that longer alkyl chains help to reduce the aggregation and interaction between the molecules. A redshift of 37 nm and 31 nm was observed from 1 wt% to 100 wt% and a redshift of 20 nm and 9 nm from 10 wt% to 100 wt% for 5CzBN-OEG and 5CzBN-Hep, respectively. The analysis suggests that the OEG chain, compared with the alkyl chain, resulted in greater intermolecular aggregation, which is well matched with the molecular dynamic simulation.
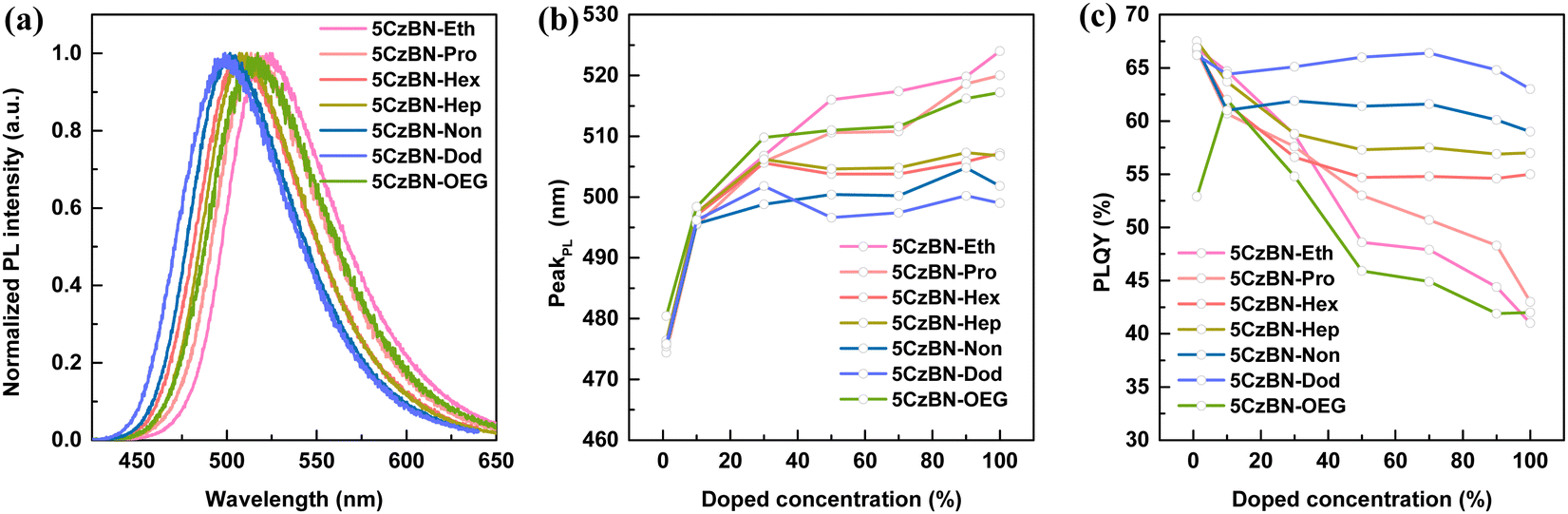 | ||
| Fig. 5 (a) PL spectra of neat films, (b) PL peak vs. doped concentration curves in PMMA matrix, and (c) PLQY values vs. doped concentration curves in PMMA matrix at 298 K (excitation wavelength: 375 nm). | ||
Besides the PL spectra, the PLQYs in the doped films with different concentrations showed significant changes. Specifically, with an increase in the doping ratio, all the molecules exhibited declined PLQY values, following the order of 5CzBN-Eth > 5CzBN-Pro > 5CzBN-OEG > 5CzBN-Hex > 5CzBN-Hep > 5CzBN-Non > 5CzBN-Dod (Fig. 5c). The PLQYs in the thin film were 41%, 43%, 55%, 57%, 42%, 59% and 63% for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-OEG, 5CzBN-Non and 5CzBN-Dod, respectively. This indicates that a too short alkyl chain length not only sacrifices the PL behavior of the emission core, but also leads to a reduction in PLQY due to molecular aggregation. Additionally, the more flexible OEG chain also lowers the PLQY because of the detrimental molecular aggregation. This fact can also be illustrated by the transient PL decay spectra of the doped films (Fig. 6). Primarily, whether the doped concentration was 1 wt% or 100 wt%, the transient PL decay spectra always presented an obvious bi-exponential decay containing a short prompt lifetime (ns) and longer delayed components (μs), again emphasizing the TADF property of these materials. It should be noted that the delayed lifetime change significantly with an increase in the doping proportion. When the doping ratio was 1 wt%, the delayed lifetimes were the longest, which were 3.35 μs, 3.45 μs, 3.68 μs, 3.73 μs, 3.86 μs, 3.65 μs, and 3.10 μs for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-OEG, 5CzBN-Non and 5CzBN-Dod, respectively. When the doping concentration changed from 10 wt% to 100 wt%, the compounds with a short alkyl chain and OEG chain showed a continuous shortened τd, following the order of 5CzBN-Eth > 5CzBN-Pro > 5CzBN-OEG > 5CzBN-Hex > 5CzBN-Hep > 5CzBN-Non > 5CzBN-Dod. Particularly, it is worth mentioning that 5CzBN-Non and 5CzBN-Dod showed a weak change in delayed lifetime, demonstrating that the longer the alkyl chain, the better the protection of excitons.50 According the prompt and delayed lifetimes and the PLQY values in thin film, the calculated rate constants (kRISC) of the molecules with a longer alkyl chain are 4.1 × 106 s−1, 3.1 × 106 s−1, 3.4 × 106 s−1 and 3.3 × 106 s−1 for 5CzBN-Hex, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod, respectively, which are much higher than that of 5CzBN-Eth (0.83 × 106 s−1), 5CzBN-Pro (1.1 × 106 s−1) and 5CzBN-OEG (0.56 × 106 s−1). This is probably because the stronger intermolecular interaction in 5CzBN-Eth, 5CzBN-Pro and 5CzBN-OEG quenches the excitons, resulting in a significantly lower delayed proportion. It is also important that the remarkably large rate constants of the non-radiative transition of the triplet exciton (knr,T) are 3.8 × 105 s−1 for 5CzBN-Eth, 4.2 × 105 s−1 for 5CzBN-Pro and 4.3 × 105 s−1 for 5CzBN-OEG, which are consistent with the obvious intermolecular interaction and exciton quenching. The corresponding parameters are summarized in Table 1. All these results imply that a TADF emitter with a shorter alkyl chain and OEG chain will display a poor device performance.
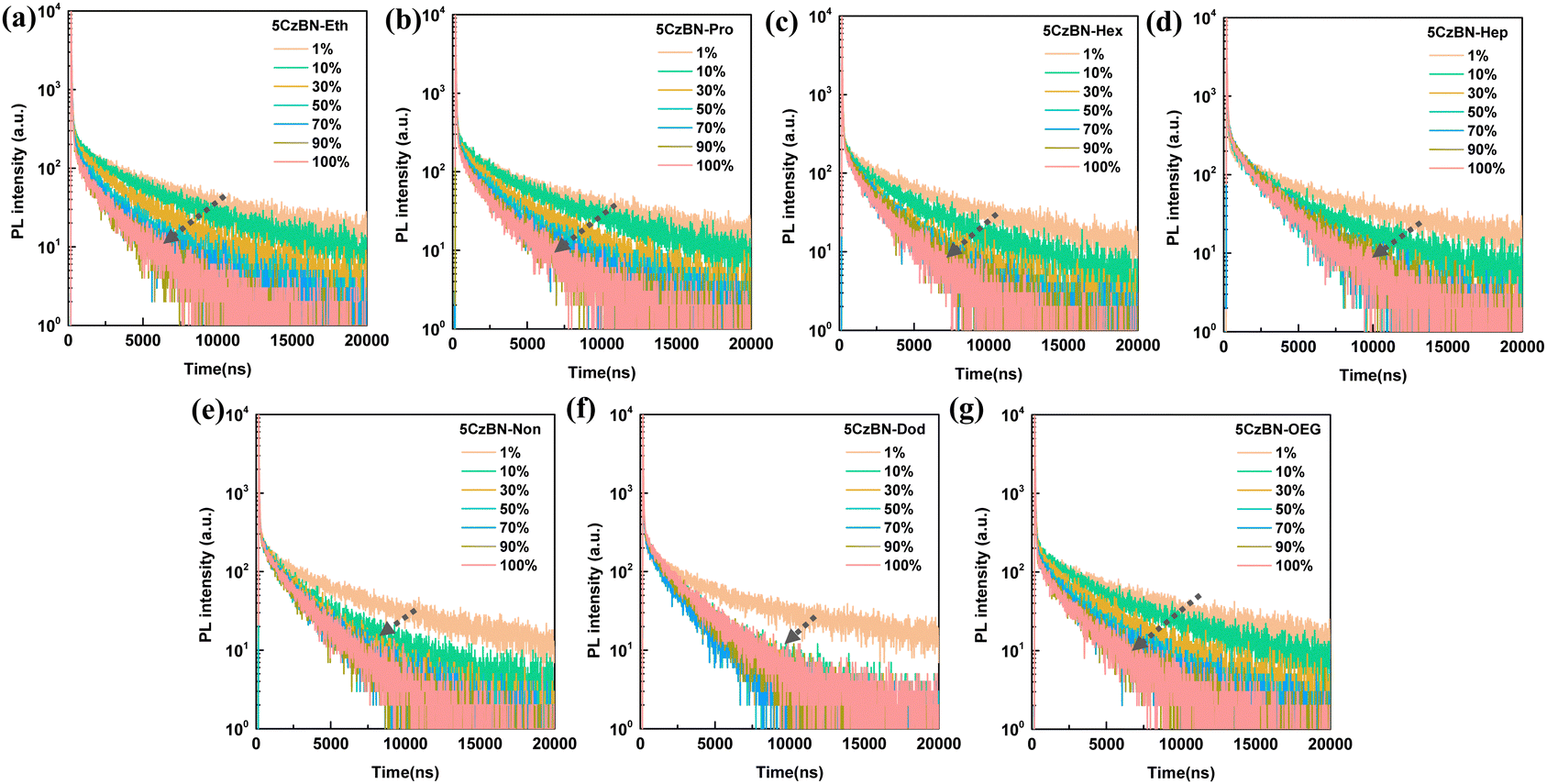 | ||
| Fig. 6 PL decay curves of PMMA-doped films of (a) 5CzBN-Eth, (b) 5CzBN-Pro, (c) 5CzBN-Hex, (d) 5CzBN-Hep, (e) 5CzBN-Non, (f) 5CzBN-Dod and (g) 5CzBN-OEG at the excitation wavelength of 375 nm at room temperature. | ||
2.5 Device characterization
Encouraged by the differentiated photophysical behaviors of the emitters in thin film, we were interested in the device performance of non-doped OLEDs. Accordingly, we fabricated a series of solution-processable devices with the configuration of ITO/PEDOT:PSS (40 nm)/emitting layer (EML, 40 nm)/POT2T (30 nm)/Cs2CO3 (1 nm)/Al (100 nm), in which poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) and Cs2CO3 served as the hole-injection layer, 2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine (POT2T) was used as the electron-transporting layer (ETL), and ITO and Al acted as the anode and cathode, respectively. The EMLs were 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-OEG, 5CzBN-Non and 5CzBN-Dod, respectively. The corresponding energy-level diagram and structure of the fabricated OLEDs are shown in Fig. 7a. To certify the rationality of the all-solution process to prepare OLED devices, the solvent resistance of these materials was explored based on the variation in their absorption intensity before and after spin-rinsing with isopropyl alcohol, which is required as a solvent for the electron-transport layer during the solution process, as displayed in Fig. S22 (ESI).† In the case of 5CzBN-Eth and 5CzBN-Pro with a short length of alkyl chain, their absorption intensity decreased by more than 30%, which is derived from the poor packaging of their molecular structure. When recrystallizing these two compounds, we also found that they could be dissolved in alcohol solvents, which are disadvantageous to the efficiency of fully solution-processed devices. In contrast to 5CzBN-Hep, the absorption intensity of 5CzBN-OEG showed a more obvious decline before and after rinsing, which is attributed to the OEG chain endowing this molecule with higher polarity.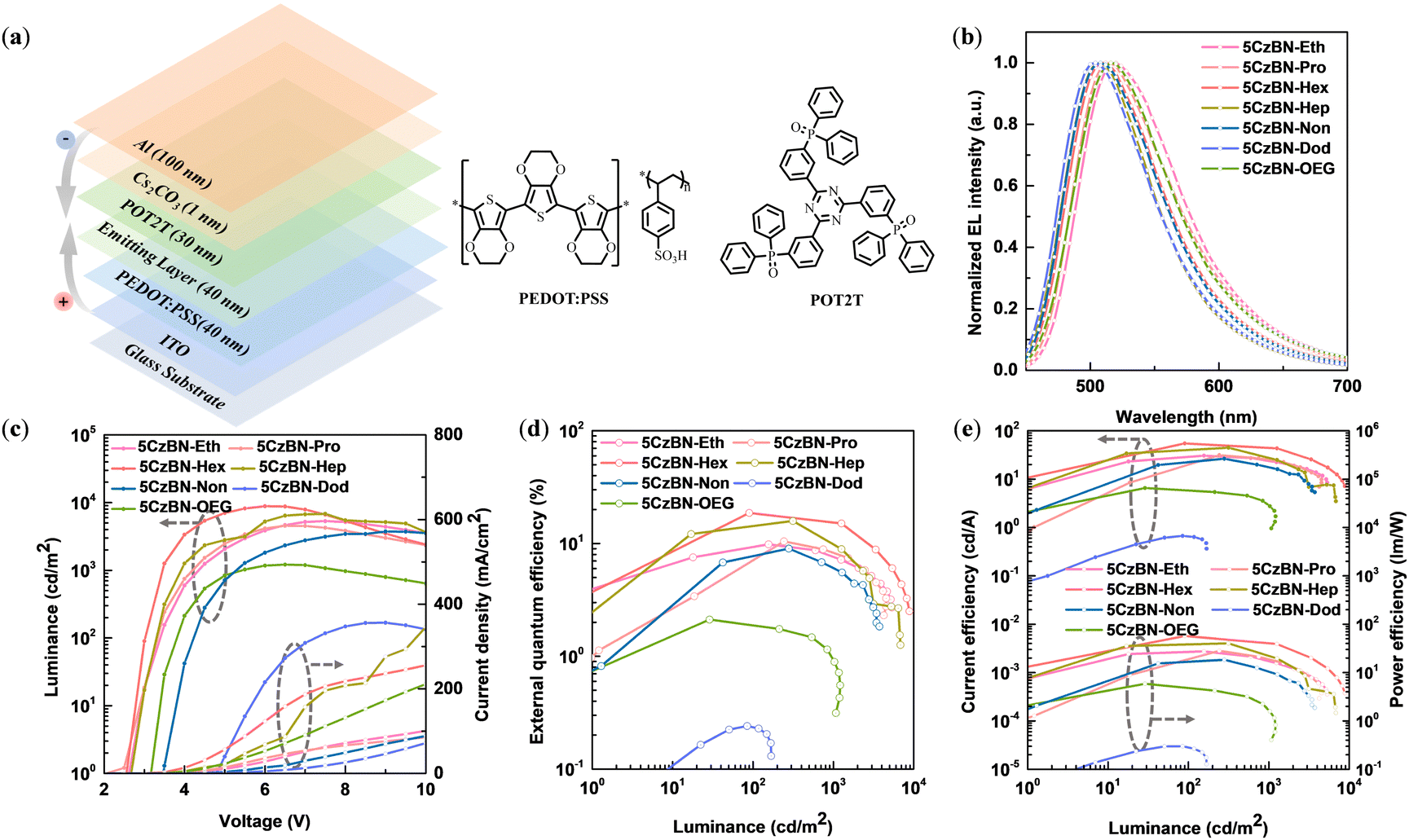 | ||
| Fig. 7 (a) Device structure and energy diagram. (b) Electroluminescence (EL) spectra. (c) Current density–voltage–luminance (J–V–L) characteristics. (d) External quantum efficiency versus luminance (EQE–L) plots of the fully solution-processed devices. (e) Current efficiency–luminance–power efficiency (CE–L–PE) plots of the fully solution-processed devices. | ||
As exhibited in Fig. 7b, the single electroluminescence (EL) peaks of the emitters are located at 520 nm, 516 nm, 508 nm, 508 nm, 516 nm, 506 nm and 506 nm for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-OEG, 5CzBN-Non and 5CzBN-Dod, respectively. The redshift in the EL spectra from 506 nm for 5CzBN-Dod to 520 nm for 5CzBN-Eth is consistent with the PL spectra, which is ascribed to the encapsulation of the longer alkyl chain, weakening the molecular interaction. Similarly, when comparing the EL spectra of 5CzBN-Hep and 5CzBN-OEG, a redshift of 8 nm was observed, which also indicates the stronger intermolecular interaction of the OEG chain under an electric field. In the current density–voltage–luminance curves of these emitters (Fig. 7c), the turn-on voltage (Von) was recorded. The Von value increased with the extension of the alkyl chain, which was 2.6 V, 2.5 V, 2.9 V, 2.9 V, 3.5 V and 4.6 V for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep, 5CzBN-Non and 5CzBN-Dod, respectively. Also, the Von of 5CzBN-OEG was 0.2 V higher than that of 5CzBN-Hep. The obvious contrast in Von reflects the difference in the injection barrier of the EML in the non-doped devices. The maximum luminance (Lmax) values of the devices were 5332 cd m−2, 4538 cd m−2, 8853 cd m−2, 6838 cd m−2 and 3447 cd m−2 for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep and 5CzBN-Non, respectively. However, the Lmax value of the 5CzBN-Dod-based device was only 168 cd m−2. Correspondingly, the EQEmax was 9.8%, 10.4%, 18.7%, 15.9%, and 9.0% for 5CzBN-Eth, 5CzBN-Pro, 5CzBN-Hex, 5CzBN-Hep and 5CzBN-Non, respectively; however, the device based on 5CzBN-Dod only achieved an EQEmax of 0.24% (Fig. 7d). It should be noted that the EQEmax of the 5CzBN-Hex-based device was 13.1% in our previous report,45 which increased to 18.7% by optimizing the thickness of the spin-coated ETL. The corresponding EQEmax was 2.1% for the 5CzBN-OEG-based device, which was only less than one-seventh that of the 5CzBN-Hep-based device. The current density of 5CzBN-OEG was significantly higher than that of the alkyl-chain molecules, which is attributed to the smaller π–π distance with the OEG chains, but the low luminance contributes to poor EL efficiency. The current efficiency-luminance-power efficiency curves are shown in Fig. 6e, and all the device performance data are summarized in Table 2. This huge difference in EL performance prompted us to further analyze the origin.
| Emitting layer | ELa (nm) | V on (V) | L max (cd m−2) | CEmaxd (cd A−1) | PEmaxe (lm W−1) | EQEmaxf (%) | CIEg (x, y) |
|---|---|---|---|---|---|---|---|
| a The stable EL emission spectrum at 10 V. b Turn-on voltage at the luminescence of 1 cd m−2. c Maximum luminance. d Maximum current efficiency. e Maximum power efficiency. f Maximum external quantum efficiency. g The Commission International de L’Eclairage (CIE) coordinates at 10 V. | |||||||
| 5CzBN-Eth | 520 | 2.6 | 5332 | 30.5 | 27.4 | 9.8 | (0.33, 0.57) |
| 5CzBN-Pro | 516 | 2.5 | 4538 | 31.4 | 28.2 | 10.4 | (0.30, 0.55) |
| 5CzBN-Hex | 508 | 2.9 | 8853 | 54.7 | 57.3 | 18.7 | (0.28, 0.54) |
| 5CzBN-Hep | 508 | 2.9 | 6838 | 44.7 | 40.1 | 15.9 | (0.28, 0.53) |
| 5CzBN-Non | 506 | 3.5 | 3447 | 26.3 | 18.4 | 9.0 | (0.26, 0.53) |
| 5CzBN-Dod | 506 | 4.6 | 168 | 0.67 | 0.30 | 0.24 | (0.25, 0.51) |
| 5CzBN-OEG | 516 | 3.1 | 1215 | 6.5 | 5.9 | 2.1 | (0.30, 0.56) |
According to the computational formula of EQEmax, the charge balance factor (γ) also plays a decisive role in the device performance. Then, for analyzing the influence of the flexible chain on the carrier charge transport, we measured the carrier-only devices using the space-charge-limited current method, with the configuration of ITO|PEDOT:PSS (40 nm)|EML (40 nm)|MoO3 (20 nm)|Al (100 nm) and ITO|Al (50 nm)|EML (40 nm)|POT2T (30 nm)|Cs2CO3 (1 nm)|Al (100 nm) for the hole-only and electron-only devices, respectively. As shown in Fig. S23 (ESI†), with a voltage of 2–6 V, the matching degree of electron and hole mobility followed the order of 5CzBN-Eth > 5CzBN-Pro > 5CzBN-Hex > 5CzBN-Hep > 5CzBN-Non > 5CzBN-Dod, and 5CzBN-Hep > 5CzBN-OEG. With an increase in the length of the alkyl chain, the electron mobility rapidly enhanced, which is attributed to the increased content of inert component and decreased content of conjugated molecular backbone. Consequently, the poor carrier balance together with the longer alkyl chain increased Von, as shown in Fig. 7c.
According to the literature, compared with the alkyl chain, the OEG chain is inclined to form more compact π–π stacking in the conjugated molecular backbone because more the flexible OEG chain provides smaller steric hindrance for π–π stacking.44 Thus, to quantitatively evaluate the influence of the alkyl chain and multi-oxygen chain on the intermolecular interactions, the aggregation structure of 5CzBN-Hep and 5CzBN-OEG was further simulated by molecular dynamics. As shown in Fig. 8, the box is filled with 50 molecules of 5CzBN-Hep and 5CzBN-OEG, respectively. The innermost 5CzBN-OEG has a more bent arrangement of flexible chains, indicating that the OEG chain has a more flexible feature. This great flexibility can result in a smaller π–π stacking distance. According to the energy of the AC box and single molecule, the van der Waals interaction energies of 5CzBN-Hep and 5CzBN-OEG with surrounding organic molecules were calculated to be −29.8 kJ mol−1 and −76.0 kJ mol−1, respectively. The larger negative value of the van der Waals interaction energy for 5CzBN-OEG indicates that it has a much stronger intermolecular interaction with the surrounding organic molecules than 5CzBN-Hep, which easily lead to the tendency to self-aggregate.51,52 This dynamic simulation also matches the morphology of the thin film, as confirmed by AFM measurement, which can explain the poor efficiency of the 5CzBN-OEG-based device.
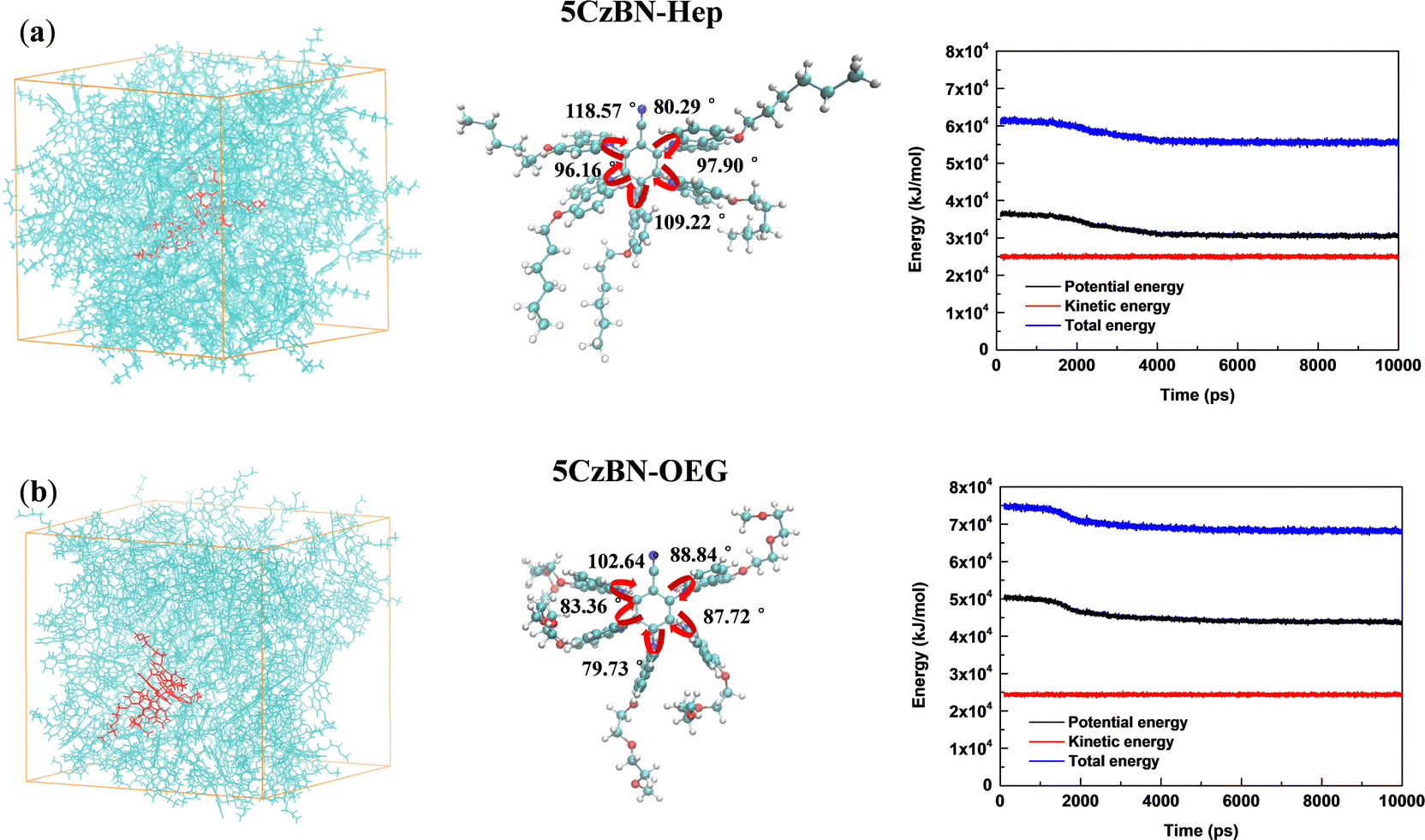 | ||
| Fig. 8 Molecular dynamics simulation results of (a) 5CzBN-Hep and (b) 5CzBN-OEG system with an AC (amorphous cell) box filled with 50 molecules. The separate molecule selected from the box of the 5CzBN-Hep and 5CzBN-OEG system. | ||
Although the enhanced PLQY values from 5CzBN-Hex to 5CzBN-Dod were confirmed in the solid-state film, the remarkable reduction in EQEmax could originate from the imbalance in carrier transport. However, with the relatively matched balance of carrier mobility, the corresponding efficiency declined from 5CzBN-Hex to 5CzBN-Eth, which is due to the depressed PLQY value stemming from the inefficient encapsulation-induced molecular aggregation. Besides, when the alkyl chain is replaced by the OEG chain, the PLQY is inferior, but the poor spin-coated morphology could be the dominant reason for the low efficiency of the fully solution-processed OLEDs. Hence, a near 2-fold improvement in EQEmax from 9.8% for 5CzBN-Eth to 18.7% for 5CzBN-Hex, 80-fold improvement from 0.24% for 5CzBN-Dod to 18.7% for 5CzBN-Hex, and 8-fold improvement from 2.1% for 5CzBN-OEG to 15.9% for 5CzBN-Hep were realized by simple regulation of the length and type of flexible chain. This result also makes it clear that the carrier mobility balance and PLQY of the thin film, as well as the morphology should be systematically considered to improve the device efficiency, especially for non-doped solution-processed devices.
3 Conclusion
In summary, inspired by bacterial flagella, we developed a series of solution-processed TADF emitters by regulating the length of the alkyl chain or substituting an OEG chain for the alkyl chain. All the compounds exhibited the similar emission behavior of the TADF core in diluted toluene solution. As expected, the photophysical performance of solid-state films showed a significant dependence on the flexible chain. Specifically, a longer alkyl chain length could provide better encapsulation and protection of the TADF core to improve the solid-state PLQY; however, it impeded the carrier mobility and balance. Alternatively, shorter alkyl chains facilitated the balance of carrier transport but caused a decrease in the PLQY and bathochromic shift due to the obvious intermolecular aggregation. Thus, the EQEmax of 5CzBN-Hex was 18.7% for the fully solution-processed OLEDs, which was about 2 times that of 5CzBN-Eth and approaching 80 times that of 5CzBN-Dod. In addition, for 5CzBN-OEG, its poor morphology induced by the large degree of crystallization and aggregation of multi-oxygen chains in thin film ultimately resulted in a low EQEmax of 2.1%, which was less than one seventh of 5CzBN-Hep. This work enlightens us to comprehensively consider the key factor (morphology, PLQY and γ) affecting the performance of solution-processed devices. More importantly, this study provides a reference for the systematic study of regulating the flexible chains to tune the behavior in thin film.Author contributions
Guimin Zhao – data curation, methodology, conceptualization, validation, roles/writing – original draft, and writing - review & editing. Yuheng Lou – investigation. Renjie Ji – resources. Qiyin Ran – software. Haowen Chen and Wenwen Tian – data curation. Wei Jiang – visualization, and supervision. Yueming Sun – funding acquisition.Conflicts of interest
All authors declare no competing financial interests.Acknowledgements
The authors are grateful for the grants from the National Natural Science Foundation of China (22135004 and 52203214) and Natural Science Foundation of Jiangsu Province (BK20231433). The authors are also thankful for the support from the Open Fund of the Key Lab of Organic Optoelectronics & Molecular Engineering committee.References
- B. K. X. Ho, B. Azahari, M. F. B. Yhaya, A. Talebi, C. W. C. Ng, H. A. Tajarudin and N. Ismail, Sustainability, 2020, 12, 9468 CrossRef CAS.
- S. Nakamura and T. Minamino, Biomolecules, 2019, 9, 279 CrossRef CAS.
- C. N. W. Chun, H. A. Tajarudin, N. Ismail, B. Azahari, M. M. Z. Makhtar and L. K. Yan, Sustainability, 2020, 13, 21 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- F. Liu, S. Bi, X. Wang, X. Leng, M. Han, B. Xue, Q. Li, H. Zhou and Z. Li, Sci. China: Chem., 2019, 62, 739–745 CrossRef CAS.
- W. Sheng, Y.-Q. Zheng, Q. Wu, K. Chen, M. Li, L. Jiao, E. Hao, J.-Y. Wang and J. Pei, Sci. China: Chem., 2020, 63, 1240–1245 CrossRef CAS.
- J. Tian, L. Fu, Z. Liu, H. Geng, Y. Sun, G. Lin, X. Zhang, G. Zhang and D. Zhang, Adv. Funct. Mater., 2019, 29, 1807176 CrossRef.
- X. Xu, Y. Zhao and Y. Liu, Small, 2023, 19, e2206309 CrossRef.
- T.-W. Lee, T. Noh, H.-W. Shin, O. Kwon, J.-J. Park, B.-K. Choi, M.-S. Kim, D. W. Shin and Y.-R. Kim, Adv. Funct. Mater., 2009, 19, 1625–1630 CrossRef CAS.
- H. J. Cheon, Y. S. Shin, N. H. Park, J. H. Lee and Y. H. Kim, Small, 2022, 18, e2107574 CrossRef.
- T. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- Y. Xie and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 575–584 CrossRef CAS.
- Y. Zou, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef.
- X. Liu, B. He, A. Garzón-Ruiz, A. Navarro, T. L. Chen, M. A. Kolaczkowski, S. Feng, L. Zhang, C. A. Anderson, J. Chen and Y. Liu, Adv. Funct. Mater., 2018, 28, 1801874 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2013, 26, 604–615 CrossRef.
- X. Tian, K. Shoyama, B. Mahlmeister, F. Brust, M. Stolte and F. Wurthner, J. Am. Chem. Soc., 2023, 145, 9886–9894 CrossRef CAS.
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. B. Dias, Z. Ren, S. Yan and M. R. Bryce, Angew. Chem., Int. Ed., 2022, 61, e202115140 CrossRef CAS.
- X. Ban, W. Jiang, T. Lu, X. Jing, Q. Tang, S. Huang, K. Sun, B. Huang, B. Lin and Y. Sun, J. Mater. Chem. C, 2016, 4, 8810–8816 RSC.
- X. Ban, T. Zhou, K. Zhang, Q. Cao, F. Ge, D. Zhang, P. Zhu, Z. Liu, Z. Li and W. Jiang, Chem. Eng. J., 2022, 441, 135898 CrossRef CAS.
- D. Zhou, S. Wu, G. Cheng and C.-M. Che, J. Mater. Chem. C, 2022, 10, 4590–4596 RSC.
- D. Liu, M. Zhang, W. Tian, W. Jiang, Y. Sun, Z. Zhao and B. Z. Tang, Aggregate, 2022, 3, e164 CrossRef CAS.
- C. S. K. Ranasinghe, A. Thamarappalli, J. Jang, M. Gao, M. Koodalingam, P. L. Burn, E. V. Puttock and P. E. Shaw, Org. Electron., 2022, 105, 106500 CrossRef CAS.
- Z. Ma, Y. Wan, W. Dong, Z. Si, Q. Duan and S. Shao, Chin. Chem. Lett., 2021, 32, 703–707 CrossRef CAS.
- G. Zhao, Y. He, Z. Xu, J. Hou, M. Zhang, J. Min, H.-Y. Chen, M. Ye, Z. Hong, Y. Yang and Y. Li, Adv. Funct. Mater., 2010, 20, 1480–1487 CrossRef CAS.
- X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886–4887 CrossRef CAS PubMed.
- J. Zhang, L. Tan, W. Jiang, W. Hu and Z. Wang, J. Mater. Chem. C, 2013, 1, 3200 RSC.
- M. Yasa, T. Depci, E. Alemdar, S. O. Hacioglu, A. Cirpan and L. Toppare, Renewable Energy, 2021, 178, 202 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2015, 138, 173 CrossRef.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS.
- J. Yang, H. Gao, Y. Wang, Y. Yu, Y. Gong, M. Fang, D. Ding, W. Hu, B. Z. Tang and Z. Li, Mater. Chem. Front., 2019, 3, 1391–1397 RSC.
- S. Song, Y. Zhao, M. Kang, Z. Zhang, Q. Wu, S. Fu, Y. Li, H. Wen, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2107545 CrossRef CAS.
- J. Wang, S. Liu, Z. Chai, K. Chang, M. Fang, M. Han, Y. Wang, S. Li, H. Han, Q. Li and Z. Li, J. Mater. Chem. A, 2018, 6, 22256–22265 RSC.
- D. Chen, F. Tenopala-Carmona, J. A. Knoller, A. Mischok, D. Hall, S. Madayanad Suresh, T. Matulaitis, Y. Olivier, P. Nacke, F. Giesselmann, S. Laschat, M. C. Gather and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202218911 CrossRef CAS PubMed.
- G. Zhao, R. Zhou, G. Zhang, H. Chen, D. Ma, W. Tian, W. Jiang and Y. Sun, J. Mater. Chem. C, 2022, 10, 5230–5239 RSC.
- Y. Li, G. Xie, S. Gong, K. Wu and C. Yang, Chem. Sci., 2016, 7, 5441–5447 RSC.
- D. Sun, E. Duda, X. Fan, R. Saxena, M. Zhang, S. Bagnich, X. Zhang, A. Kohler and E. Zysman-Colman, Adv. Mater., 2022, 34, e2110344 CrossRef.
- D. Sun, R. Saxena, X. Fan, S. Athanasopoulos, E. Duda, M. Zhang, S. Bagnich, X. Zhang, E. Zysman-Colman and A. Kohler, Adv. Sci., 2022, 9, e2201470 CrossRef.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef.
- D. Liu, J. Wei, W. Tian, W. Jiang, Y. Sun, Z. Zhao and B. Z. Tang, Chem. Sci., 2020, 11, 7194–7203 RSC.
- S. Torabi, F. Jahani, I. Van Severen, C. Kanimozhi, S. Patil, R. W. A. Havenith, R. C. Chiechi, L. Lutsen, D. J. M. Vanderzande, T. J. Cleij, J. C. Hummelen and L. J. A. Koster, Adv. Funct. Mater., 2015, 25, 150 CrossRef CAS.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357–4363 CrossRef CAS.
- B. Meng, J. Liu and L. Wang, Polym. Chem., 2020, 11, 1261–1270 RSC.
- X. Chen, Z. Zhang, Z. Ding, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 10376–10380 CrossRef CAS.
- G. Zhao, D. Liu, P. Wang, X. Huang, H. Chen, Y. Zhang, D. Zhang, W. Jiang, Y. Sun and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202212861 CrossRef CAS.
- M. Shao, Y. He, K. Hong, C. M. Rouleau, D. B. Geohegan and K. Xiao, Polym. Chem., 2013, 4, 5270 RSC.
- H. Noda, H. Nakanotani and C. Adachi, Sci. Adv., 2018, 4, eaao6910 CrossRef.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS.
- Y. Jin, Q. C. Peng, S. Li, H. F. Su, P. Luo, M. Yang, X. Zhang, K. Li, S. Q. Zang, B. Z. Tang and T. C. W. Mak, Natl. Sci. Rev., 2022, 9, nwab216 CrossRef CAS PubMed.
- G. Zhao, R. Zhou, G. Zhang, H. Chen, X. Wang, Z. Zhang, W. Tian, W. Jiang and Y. Sun, Adv. Opt. Mater., 2023, 11, 2203065 CrossRef CAS.
- X. Peng, K. Gao, W. Qiu, Z. Xu, M. Li, W. Xie, J. Yang, D. Li, K. Liu and S. J. Su, Adv. Opt. Mater., 2023, 2300868 CrossRef CAS.
- H. Shen, F. Sun, X. Zhu, J. Zhang, X. Ou, J. Zhang, C. Xu, H. H. Y. Sung, I. D. Williams, S. Chen, R. T. K. Kwok, J. W. Y. Lam, J. Sun, F. Zhang and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 15391–15402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03484d |
| This journal is © The Royal Society of Chemistry 2024 |