
PVA/KGM dual network hydrogels doped with carbon nanotube-collagen corona as flexible sensors for human motion monitoring
Xingzhong
Cao
 ,
Tingxiang
He
,
Jinqi
Sui
,
Yihan
Yan
,
Xiang
Liu
,
Leipeng
Liu
and
Shenghua
Lv
*
,
Tingxiang
He
,
Jinqi
Sui
,
Yihan
Yan
,
Xiang
Liu
,
Leipeng
Liu
and
Shenghua
Lv
*
College of Bioresources Chemical and Materials Engineering, Shaanxi University of Science and Technology, Xi’an 710021, China. E-mail: lvsh@sust.edu.cn
First published on 30th January 2024
Abstract
Hydrogel strain sensors with features such as frost resistance, high biocompatibility, and volume stability have shown irreplaceable advantages in many fields. Herein, a “part assembly” strategy is proposed in which carbon nanotubes and collagen are designed as two “parts” that are assembled to obtain collagen protein corona (PC) and introduced into the polyvinyl alcohol (PVA)/konjac gum (KGM) composite hydrogel interior, preparing a double network composite hydrogel of PVA/KGM/PC. The first network structure is formed by the interaction of PVA and glycerol, and the second network structure is formed by the interaction of the KGM itself. The prepared PVA/KGM/PC composite hydrogel has good mechanical properties, frost resistance, high sensitivity, and a wide range of strain responses, with strains of 226% and 222.5% at room temperature and −20 °C, respectively. It can monitor not only the human arm, knee bend, and other large ranges of motion but also the human heartbeat, pulse and other small ranges of physiological activities. In addition, the Gauge factor of the hydrogel can reach 3.38. The PVA/KGM/PC composite hydrogel containing collagen corona prepared in this study can be extensively used as a flexible strain sensor in emerging fields, such as medical monitoring, intelligent robotics, and wearable devices, providing a new strategy for optimizing the preparation process of hydrogels.
1. Introduction
In recent years, flexible sensors have been deeply stimulated by the rapid development of emerging industrial applications in medical monitoring devices, intelligent robots, wearable equipment, and energy storage materials.1–3 They have attracted considerable attention from researchers due to their ability to sensitively capture subtle changes in the external world.4,5 Flexible sensors are composed of a combination of active materials and flexible substrate designs that can convert various external stimuli into signals that can be detected, giving them flexibility and sensation similar to those of human skin.Today, the design of flexible sensors requires novel structures and suitable materials, mainly including the selection of active materials and flexible substrates.6 Commonly used flexible substrate materials include polyimide (PI),7 polyether ether ketone (PEEK),8 polyether ether sulfone (PES),9 polycarbonate, poly(vinyl naphthalene) (PEN), hydrogel,10 and polyethylene terephthalate (PET).11 However, the thermal stability, transparency, electrical conductivity and mechanical elasticity of the materials should be considered before practical application,12,13 and the selected material should meet the performance requirements of flexible and sensitive sensing.6,7 Hydrogel, as a special class of solid-like materials with a three-dimensional network structure, has demonstrated irreplaceable advantages in the field of flexible sensors owing to its remarkable properties such as transparency, flexibility, extensibility, viscoelasticity and biocompatibility simultaneously.2,14 Therefore, researchers generally prepare high-performance flexible sensing hydrogels by selecting suitable materials and applying them to flexible sensors.15–17 At present, the common materials used for the preparation of hydrogels are natural polymers and synthetic polymers. Natural polymers are primarily cellulose,18 starch,19 chitosan,20 and konjac gum (KGM), whereas synthetic polymers are primarily polyacrylamide,21 poly(dimethylsiloxane) (PDMS),22 poly(enedipyridone) (PVP),23 and polyvinyl alcohol (PVA). The advantage of hydrogels prepared by natural polymers is that they come from a wide range of sources and have a low price, while their disadvantage is that their mechanical strength is poor,24 which cannot meet the performance requirements of flexible sensors for flexible substrates. The hydrogels prepared by synthetic polymers generally have the advantages of good strength and volume stability, but they have the disadvantages of low biocompatibility25 and certain toxicity to the human body.26 Therefore, the preparation of composite hydrogels using a mixture of natural polymers and synthetic polymers can not only solve the defects of poor basic performance but also improve the biocompatibility of hydrogels.27 For this purpose, we focused specifically on two water-soluble polymers: KGM and PVA. KGM is a natural polymer polysaccharide, and it can not only form its independent network structure but also has many excellent properties, such as gelation and antibacterial properties.28 Although PVA is a highly biocompatible synthetic polymer, it has good mechanical properties, such as high strength and elasticity. Composite hydrogels prepared with KGM and PVA may have good strength and elasticity,29 which can better meet the current performance requirements for hydrogels such as flexible sensors.30–32
The active material is another important factor that affects the performance and applications of flexible sensors,6 and it must have a sensitive ability to sense, convert and transmit signal.33 Commonly used active materials are carbon family nanoparticles, such as graphene, graphene oxide (GO) and carbon nanotubes (CNTs), and non-transition metal oxides, such as nano zinc oxide and nano tin oxide.34 CNTs show unique advantages owing to their excellent electrical conductivity and special tubular structure.35
Currently, nano active materials are mainly dispersed uniformly in hydrogels through physical dispersion or chemical reactions to exhibit sensitive sensing properties.36 However, most nano active nanomaterials have strong hydrophobicity, which causes them to repel each other after entering the hydrophilic network system of the hydrogel,37 thereby seriously affecting the performance of hydrogel sensor.38,39 Therefore, improving the hydrophilicity of nano active materials has become the main problem to be solved. At present, the hydrophilicity of nano active materials is primarily improved by chemical modification,40 such as introducing some hydrophilic groups (–OH, –COOH, etc.) on the surface or edges of nano active materials by oxidation41 or etching.42 However, there are some special nano active materials (gold nanoparticles and silver nanoparticles) that are difficult to modify and need to rely on a more sophisticated process.43,44 Therefore, the dispersion of nano active nanomaterials into hydrogel through a simple and effective way is the focus of this study.
The discovery of protein corona (PC) provides a new way to solve the dispersion problem of active nanomaterials in hydrogels. The PC refers to a structure formed by protein assembly induced by nanomaterials entering the biological environment (blood, serum culture, and protein solution).45 It was first proposed by Dawson et al. in 2006.46 They found that when nanoparticles are ingested into organisms, they adsorb proteins on their surfaces to form nanoparticle-PC core–shell complexes, which are called protein corona.46 In a study of the protein corona, researchers found that the endogenous nanoparticle protein corona formed by grilled salmon in human serum protein can considerably reduce the glycolysis metabolism of normal rat kidney cells, indicating that it can lead to the recovery of oxidative phosphorylation levels.47 According to further studies, the protein corona formed in grilled salmon can reduce nanoparticle-induced mitochondrial damage.48 The protein corona structure formed by roast beef in human serum proteins relieves the swelling of cells induced by lysosomes and inhibits the reduction of the mitochondrial membrane potential caused by nanoparticles.49 These studies have shown that nanoparticles can form protein corona in the presence of some proteins. With the rapid development of nanoscience engineering, researchers have been inspired by the protein corona to obtain new ideas; it can reduce the surface energy of nanoparticles, which considerably affects the biological effects, compatibility and toxicity of nanoparticles, and has an important impact on the absorption, dispersion, energy conversion and signal transmission of nanoparticles in biological systems or environmental systems.50,51 Therefore, the method of preparing protein corona has become a new strategy to change the interface properties of nanoparticles, which has important applications in targeted drug design, intelligent sensing materials and signal transmission.
Inspired by the preparation method and properties of PC, the preparation and performance of PVA/KGM composite strain sensing hydrogel containing protein corona formed by CNT-collagen were studied in this work. CNTs were introduced into the double-penetrating network structure system of PVA/KGM composite hydrogel using a PC solution as the medium. It overcame the problem of poor compatibility of CNTs in hydrogels faced by traditional methods and achieved uniform penetration of CNTs in the three-dimensional network structure in a low-cost manner, which considerably improved the performance of the prepared flexible sensor. The effect of the assembly conditions of the PC on the properties of the strain sensing hydrogels was investigated in this research project, particularly their sensing performance under low strain. The research results are expected to provide a new strategy for the introduction of nano-fillers.
2. Experimental
Materials
Polyvinyl alcohol (PVA), konjac gum (KGM), multi-walled carbon nanotubes (MWCNT), collagen peptides, anhydrous lithium chloride (LiCl), glycerol, and anhydrous ethanol. The water used was deionized.Preparation of the protein corona composite hydrogel
Synthesis of the protein corona. Note that 0.005 g of carbon nanotubes was added to the binary solvents of water and glycerol (5 g). Then, the mixture was sonicated for 30 min to make the CNTs evenly dispersed. Next, 0.4 g of collagen was added to the binary solvents of water/glycerol (10 g) and magnetically stirred for 20 min. After the collagen was completely dissolved, 1 ml of the CNT dispersion was added to the collagen solution and left to assemble for 60 min to obtain the protein corona (PC) solution. The ratio of water to glycerol in the above binary solvent is 4 to 1 (weight ratio). The preparation flow chart of the PC solution is shown in Fig. 1.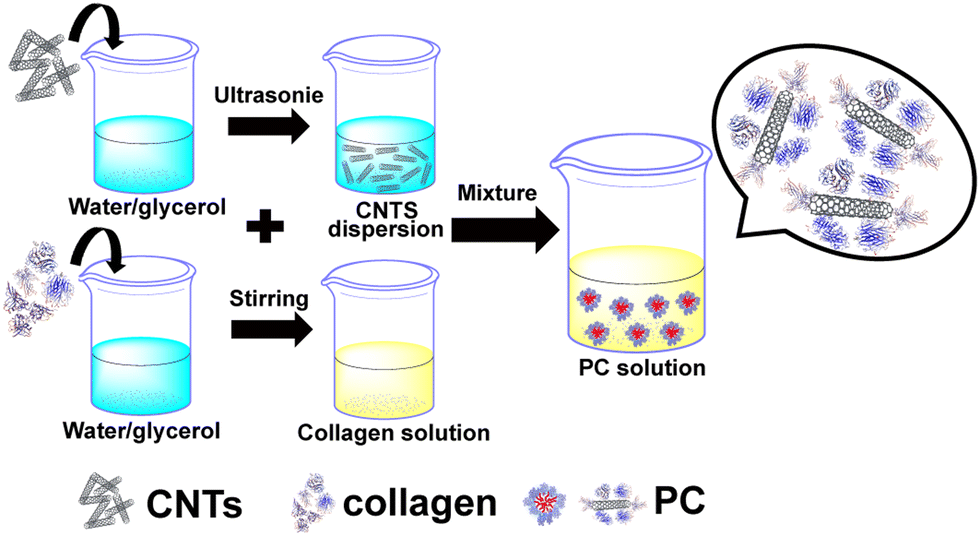 | ||
| Fig. 1 Preparation flow chart of a PC solution. | ||
Preparation of the PVA/KGM/PC composite hydrogel. We weighed 2.4 g of PVA into a three-necked flask containing water/glycerol (20 g), and the mixture was heated to 95 °C in an oil bath under stirring and then stirring was continued until PVA completely dissolved. The temperature then decreased to 60 °C. Then, 1 g of KGM was weighed into a binary solvent of water/glycerol (5 g) and magnetically stirred for 5 min to completely dissolve the KGM to obtain the KGM solution. Next, the prepared PC solution and KGM solution were added to a three-mouth flask and stirred for 2 h at 60 °C to obtain a composite hydrogel precursor solution containing PC. The composite was then poured into a mould and cooled to 25 °C. It was left to defoam and sealed with cling film. Then the composite hydrogel was freeze-thawed by freezing at −20 °C for 9 h and melting at 24 °C for 3 h. This freeze-thaw process is carried out three times. Finally, the freeze–thaw-completed hydrogels were soaked in LiCl solution to obtain a composite hydrogel containing PC, named PVA/KGM/PC hydrogel. The preparation flow chart of PVA/KGM/PC hydrogel is shown in Fig. 2.
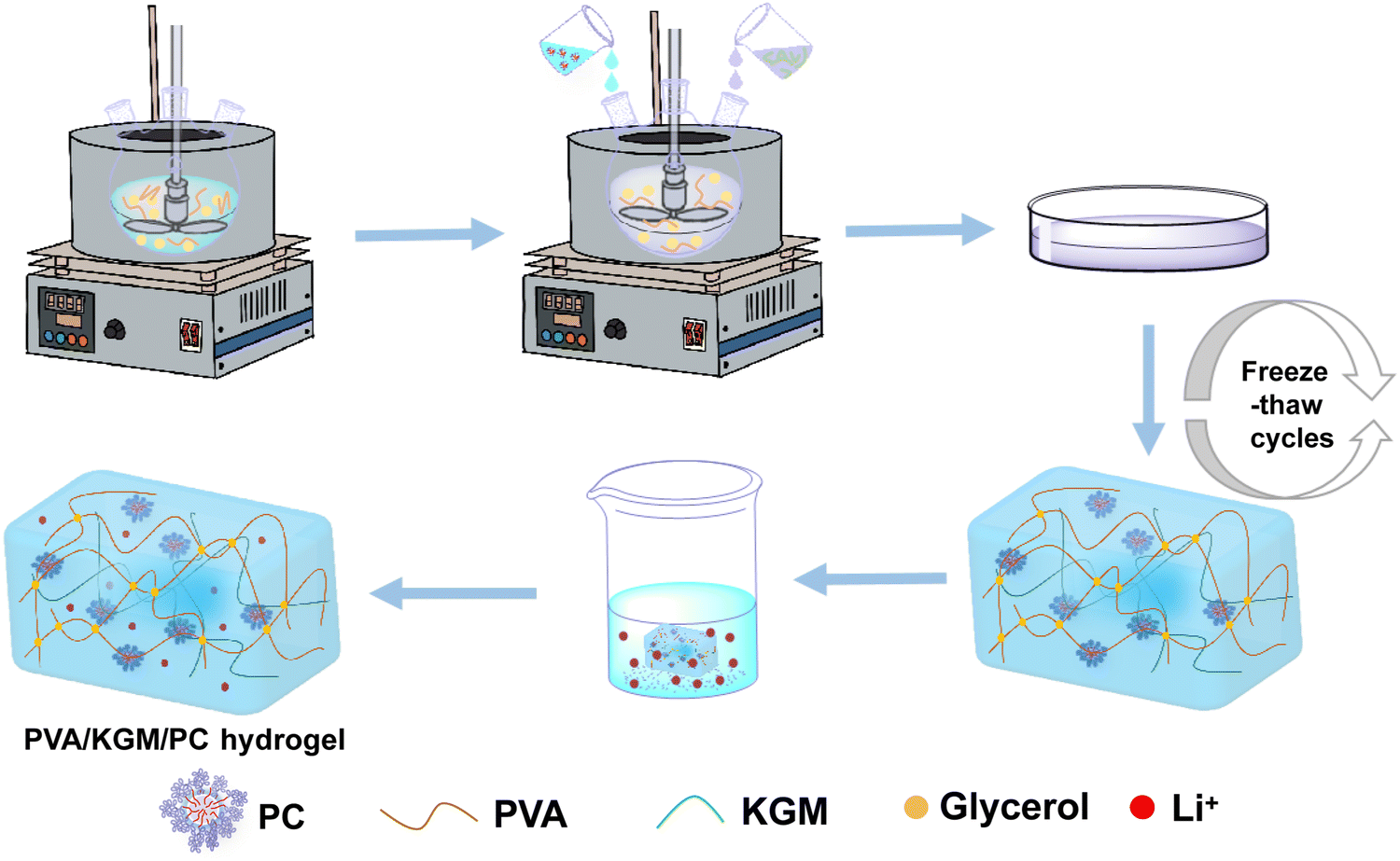 | ||
| Fig. 2 Preparation flow chart of PVA/KGM/PC hydrogel. | ||
Preparation of the PVA/KGM/PC composite hydrogel strain sensors: the prepared composite hydrogel is cut into rectangular shapes and connected to the hydrogel strip with two copper wires. The hydrogel is used as an ionic conductor, and the copper wires are used as electrodes to prepare a resistive strain sensor.
Characterization
The conformational changes in collagen before and after PC formation were evaluated using circular dichroism spectroscopy. The assembly situation of PC was characterized by transmission electron microscopy (TEM, FEI Tenai G2 F20 S-TWIN).The microstructure of the different sections of the PVA/KGM/PC composite hydrogel was observed using scanning electron microscopy (SEM, S-4800). The samples were sprayed with a gold coating for about 60 s before testing.
The elongation-at-break and tensile strength of the PVA/KGM/PC composite hydrogel were tested at 25 °C and −20 °C using a servo material multi-functional high- and low-temperature control tester (AI-7000-NGD, Dongguan, China) at a speed of 100 mm min−1, respectively. The samples were composed of a 2 × 35 mm dumbbell cutter, and an average value of three repeated tests was taken for each sample.
The rheology test was performed using a rotating rheometer (DHR-1, TA, America) at a fixed strain of 0.1%. The dynamic storage modulus and loss modulus of PVA/KGM/PC composite hydrogels were tested at an angular velocity of 10 rad s−1 from 25 °C to −20 °C and from 0.1 to 100 rad s−1 at 25 °C. The diameter of the fat plate used in the experiments was 40 mm. The size of the samples was 16.5 mm in diameter and 2 mm in thickness.
The conductivity of the PVA/KGM/PC composite hydrogel was described using a four-probe tester (RTS-9, Guangzhou, China) with a sample diameter of 15 mm and a thickness of 1 mm. In this work, Li+ was introduced into the PVA/KGM/PC composite hydrogel 3D network system by applying the solvent replacement method. Conductivity (σ, S cm−1) was calculated using the following equation:
| σ = d/(R × A) | (1) |
The sensing performance was characterized by making a rectangle of PVA/KGM/PC composite hydrogel clamped on a tensile testing machine, and tested at 25 °C and −20 °C at 0.4 mm s−1. The hydrogel was then connected to an LCR meter (TH2829A, Shenzhen, China) with copper wires at both ends. The change in resistance of the hydrogel was recorded in real time, and the relative rate of change in resistance was calculated using the following equation:
| ΔR/R0 = (R − R0)/R0 × 100 | (2) |
The sensitivity of the hydrogel sensing performance is expressed by the Gauge factor (GF), which is calculated using the following formula:
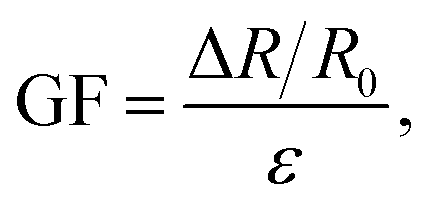 | (3) |
The hydrogel sensor is fixed to each part of the body through copper tape, and the two ends are connected to the LCR meter through copper wires to achieve real-time monitoring of human movement through the resistance changes recorded by the computer.
3. Results and discussion
Characterization of PC and PVA/KGM/PC
The CNT-based PC was obtained by applying the “part assembly” strategy, and the precursor solution of the PVA/KGM/PC composite hydrogel was prepared using the one-pot method. The double network structure of PVA/KGM/PC composite hydrogels was obtained using the freeze–thaw cycle and immersion method. The internal intermolecular forces are shown in Fig. 3a. The results show that the first network structure is formed by hydrogen bonding between PVA and glycerol forms,52 and the second network structure is formed by hydrogen bonding between KGM molecular chains.53 The PC is distributed in the cross-linked system of the PVA/KGM composite hydrogel. The structural concept schematic of the PC is shown in Fig. 3b. The “CNT parts” are used as a matrix to assemble collagen to its surface. The hydrophobic inner cavities of collagen and CNTs are bonded by hydrophobic interactions. The hydrophilic part of collagen is exposed to the surface to form hydrogen bonding interactions with –OH on PVA chains and KGM chains. PC is more adapted to the hydrophilic system of the hydrogel inside and can enhance the mechanical properties of the hydrogel.54Fig. 3c and d display the particle size distribution of CNTs and collagen, respectively, where the particle size of CNTs is mainly concentrated at around 100 nm and the particle size of collagen is around 8 nm. This meets the requirement of a “part” assembly strategy and is consistent with the conclusion reached by Wang et al.55Fig. 3 also that smaller collagen can be assembled on the surface of CNTs, which is consistent with the above PC structure schematic diagram.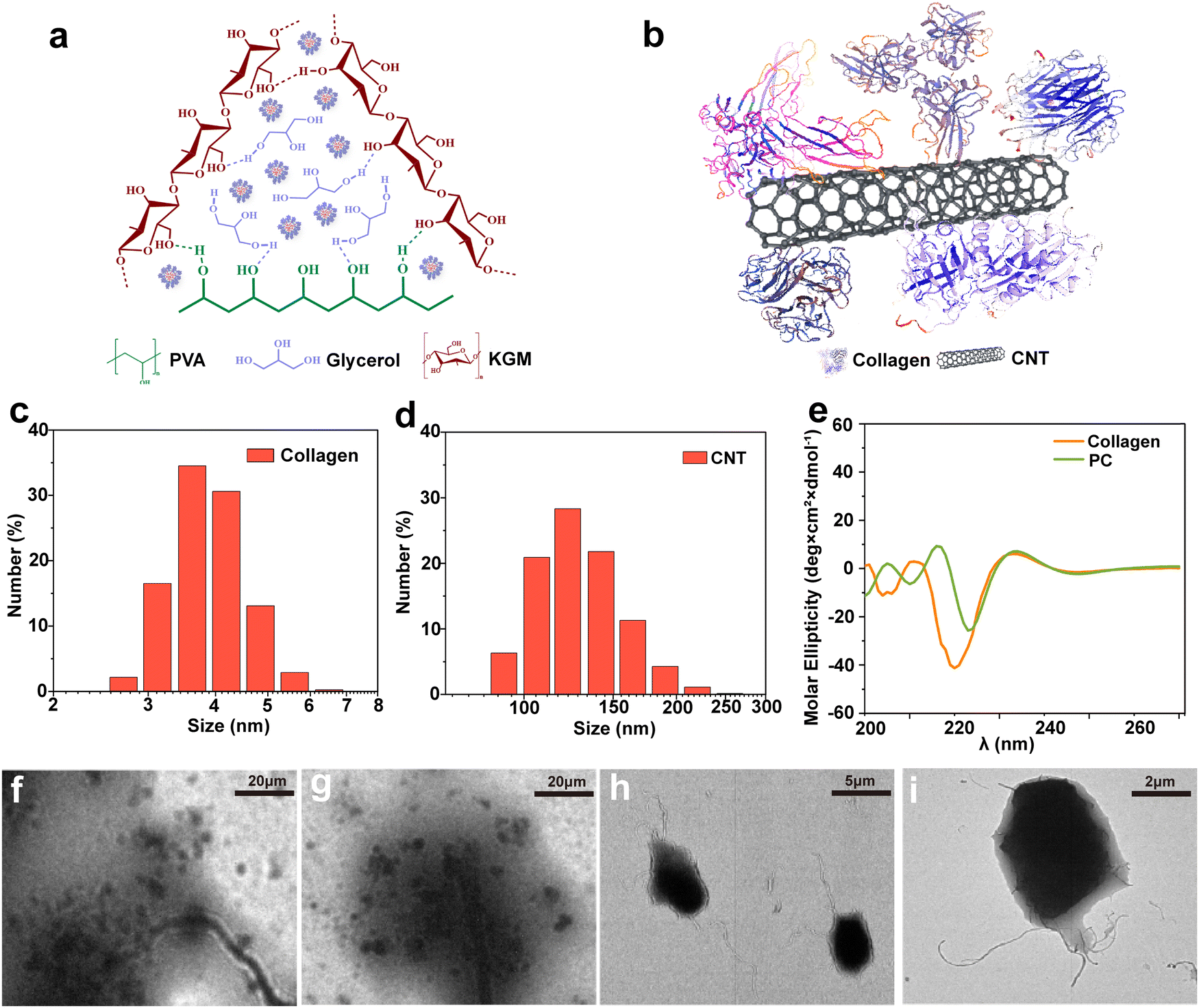 | ||
| Fig. 3 (a) Schematic of intermolecular forces within PVA/KGM/PC composite hydrogel. (b) Schematic of the PC structure. (c) Particle size distribution of collagen. (d) Particle size distribution of CNTs. (e) Circular dichroism of collagen and PC. (f–i) TEM images of PC at different magnifications. | ||
The effect of the assembly of PC on the conformation of collagen was evaluated using circular dichroism spectroscopy, as shown in Fig. 3e. The negative peak was observed at 208 and 198 nm, and the small and broad positive peak at 222 nm showed the α-helix structure and the irregularly curled structure of collagen before assembly, respectively.56,57 However, when collagen was assembled on the surface of CNTs to form PC, the negative peak at 208 nm considerably changed. The intensity of the peak weakened and was accompanied by the appearance of a red shift. This indicates that the formation of PC reduced the proportion of α-helix in collagen and changed its hydrophobicity.58 The positive peak at 222 nm did not considerably change. This indicates that the formation of PC did not affect the irregularly coiled structure of collagen. However, this indicates the successful assembly of PC. As depicted in Fig. 3f–i, the morphology of PC was characterized by TEM, and the CNTs were observed with the collagen assembled on its surface at different magnifications, confirming the conjecture of PC structure and illustrating the successful assembly of PC.
Microstructure characterization of PVA/KGM/PC hydrogel
The number and robustness of pores affect ionic motion and the range of external forces that can be withstood. If the pores are not strong enough to collapse when deformation occurs, the internal forces will be uneven, thus affecting mechanical stability.59,60 The three-dimensional pore-like structure of the PVA/KGM/PC composite hydrogel was observed by freeze-drying the composite hydrogel sample and scanning the fracture cross-section in different planes using SEM. Fig. 4(a), (b) and (c) show the side profile, oblique profile, and flat profile of PVA/KGM/PC composite hydrogel, respectively. A large number of dense pore structures can be observed in these structures. The pore structures are of different sizes owing to the interpenetration of the dual network, which indicates that there are a large number of channels inside the PVA/KGM/PC composite hydrogel for ion transport. The stability of the dual network prevents the pores from collapsing easily during the deformation of the hydrogel, which is a better approach to bear the external force applied to it. Thus, the dual network hydrogel system gives it a large mechanical strength and better volume stability.61 It also improves electrical conductivity as a conductor and sensitivity as a sensor. In addition, it shows that the introduction of PC does not negatively affect the number and stability of the pore structure. | ||
| Fig. 4 SEM images of PVA/KGM/PC composite hydrogel freeze-dried samples after brittle fracture in different planes: (a) side profile; (b) oblique profile; and (c) flat profile. | ||
Properties of the PVA/KGM/PC hydrogel
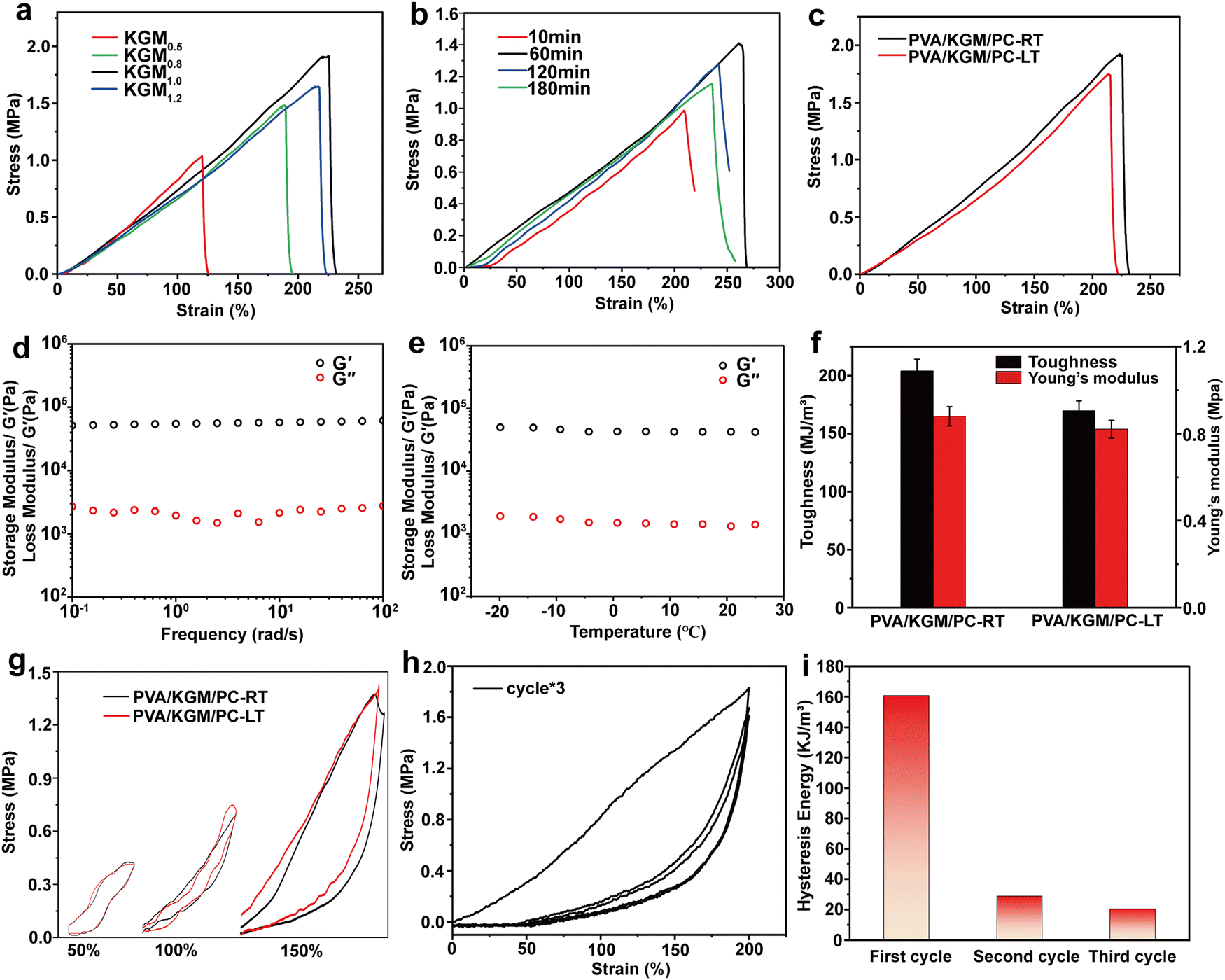 | ||
| Fig. 5 Mechanic properties of PVA/KGM/PC composite hydrogels. (a) Stress–strain curves with different KGM contents. (b) Stress–strain curves with different PC assembly times. (c) Stress–strain curves at 25 °C and −20 °C. (d) Storage modulus (G′) and loss modulus (G′′) at different frequencies. (e) Storage modulus (G′) and loss modulus (G′′) at different temperatures. (f) Toughness and Young's modulus diagram at 25 °C and −20 °C. (g) Single extension-shrinkage cyclic curves at different strains at 25 °C and −20 °C. (h) Three continuous cyclic stretching and releasing. (i) The corresponding hysteresis energy during various loading cycles. | ||
The effect of assembly times of PC on the strain and stress of the hydrogel is shown in Fig. 5b. When the assembly time was increased from 10 to 120 min, the elongation-at-break and stress of the hydrogel showed a gradual increase and reached a maximum of 271% and 1.41 MPa at 120 min, respectively. When the assembly time exceeds 120 min, the elongation-at-break and its stress decreases. This behavior may be because with the increase in PC assembly time, the content of assemblable collagen on the surface of CNTs gradually tends to saturate.64 Within 120 min, collagen is continuously assembled on the surface of CNTs so that it can be uniformly dispersed in the network system built by PVA and glycerol. In this process, the hydrophilic groups on the surface of PC form hydrogen bonding with PVA and KGM chains, which enhances the internal interaction system and gradually increases the mechanical properties of hydrogel.65,66 When the assembly time reaches 180 min, the collagen on the PC surface reaches saturation. The new collagen is adsorbed on the PC surface and causes agglomeration of the PC, preventing it from being uniformly dispersed in the network. The adsorbed collagen exposes more hydrophobic cavities because it cannot come into contact with the hydrophobic surface of the CNTs, which mutually repels the hydrogel interior, thus weakening its mechanical properties.67,68 Finally, a better formulation of KGM and PC assembly time in the preparation of composite hydrogel (KGM mass is 1 g, PC assembly time is 120 min) was determined by the above two single-factor tests. In subsequent tests, all PVA/KGM/PC composite hydrogels were prepared according to this formulation.
Frost resistance is an important evaluation index for hydrogel sensors. Excellent frost resistance can make them have a stable sensing performance in a harsh environment.69 In this work, the PVA/KGM/PC composite hydrogel was tested at 25 °C and −20 °C in the unidirectional tensile test and single-cycle tensile test, respectively, to characterize its stability in a low-temperature environment. The results are shown in Fig. 5c and g. In the unidirectional tensile test at 25 °C, it can be observed that the elongation-at-break and stress of the composite hydrogel can reach 226% and 1.9 MPa, respectively. In the −20 °C environment, its elongation-at-break and stress can still reach 222.5% and 1.5 MPa albeit there is a slight decrease, which reflects the excellent frost resistance of the composite hydrogel. From the single-cycle stretching experimental test of the PVA/KGM/PC composite hydrogel, it can be concluded that the loading–unloading curves at different strains show an obvious hysteresis loop. The energy dissipation of the hydrogel exhibits a non-linear growth trend.70 The hydrogel can still recover to 105% of its initial state at strains of 100% and 150% under continuous stretching-shrinking cycle tests without rest intervals. Its excellent recoverable performance has great potential in the field of flexible sensors. The loading–unloading curve of the composite hydrogel at −20 °C is similar to that at 25 °C. This indicates that it still maintains a stable energy dissipation state in a low-temperature environment, which reflects the excellent frost resistance of the composite hydrogel and shows good adaptability to achieve sensing performance in a low-temperature environment.71 In addition, the Young's modulus of the PVA/KGM/PC composite hydrogel was 0.91 and 0.82 MPa at 25 °C and −20 °C, respectively, and the maximum toughness reached 213.1 and 172.8 MJ m−3 (Fig. 5f). The reason for these mechanical property changes may be that the low-temperature environment weakened the hydrogen bonding system formed inside the hydrogel.72 Therefore, compared to room temperature, both showed a slight decrease.
Fig. 5d and e show the effect of frequency and temperature on the storage modulus (G′) and loss modulus (G′′) of PVA/KGM/PC composite hydrogel under different frequency and temperature changes, respectively. It can be observed that changes in G′ and G′′ show a linear trend regardless of whether frequency or temperature was used as a variable. It can also be observed that G′ is much higher than the G′′, indicating that it has good elasticity and certain strength,73 which meets the requirements of a flexible sensor. In addition, the G′ and G′′ of PVA/KGM/PC composite hydrogel maintain stable values with temperature change, which also reflects its excellent frost resistance.74 To explore the energy dissipation mechanism of the PVA/KGM/PC composite hydrogel after approaching the ultimate strain, three uninterrupted loading–unloading tests were performed at a 200% strain (Fig. 5h). The hysteresis energy was calculated for each cycle (Fig. 5i). After the first cycle, the hysteresis energy of the hydrogel rapidly dropped from 160.6 to 28.9 kJ m−3. Only 6% of the first stage was lost from the second cycle to the third cycle, indicating that the hydrogel underwent irreversible deformation after approaching the ultimate strain but could still maintain a certain stability. The curves of the second and third cycles basically remained the same. The unloading curves extremely overlapped. This indicates that the double network structure is severely damaged, and many hydrogen bonds are broken after the hydrogel is subjected to ultimate stretching,63,75 which leaves only part of the unbroken hydrogen bonds and the PC dispersed in them to provide energy for the second and third cycles and to ensure the subsequent stable energy dissipation,76 so that the double network structure can still play a certain sensing performance even after ultimate stretching. This reflects the strong destructive resistance of the PVA/KGM/PC composite hydrogel.
Sensing performance properties
To investigate the sensing performance and sensitivity of the PVA/KGM/PC composite hydrogel, strain sensors were prepared by connecting the two ends of the hydrogel with copper tape and wire. The rate of change of resistance and GF of the hydrogel were calculated according to eqn (2) and (3), respectively. The detection results of the sensing performance and sensitivity of the PVA/KGM/PC composite hydrogel are shown in Fig. 6. Fig. 6a and b show that the strain ranged from 0% to 200% with a relative resistance change of 580%. This indicates that it has a strain-dependent resistance property, and the change in the internal structure during stretching causes a resistive response.77 As the strain gradually increased from 0% to 200%, the hydrogel became narrower and longer, and the ion transport channels reduced, resulting in the paths becoming longer and the electric resistance becoming larger.78 Similarly, the same phenomenon occurred in the −20 °C environment. The relative resistance value reached 545% when the hydrogel strain increased from 0% to 200%. The behavior at −20 °C proves the extremely high adaptability of the hydrogel to a low-temperature environment. The sensitivity of the hydrogel sensor was then evaluated using the slope of the relative resistance change curve. The GFs were 3.11 and 2.82 at 25 °C and −20 °C, respectively, ranging from 0% to 50% strain. They were 3.38 and 2.03 in the range of 50–100% strain and changed to 2.81 and 2.30 in the range of 100–150%, respectively. They were 2.85 and 2.88 in the range of 150–200%. It can be seen that although the GF at 25 °C is higher than −20 °C in the strain range of 0–150%, the GF of the hydrogel in a low-temperature environment can still reach about 2–2.8, which reflects stable sensing performance and good sensitivity for ambient temperature. When the strain range is close to the limit, the hydrogel at −20 °C shows a higher sensitivity than that at 25 °C. This may be because the low-temperature environment caused the hysteresis phenomenon of the hydrogel under a large strain. The ion transport channels did not narrow rapidly, allowing more Li+ to pass through.79 The trend of relative resistance changes increased under the same strain, making it exhibit higher sensitivity.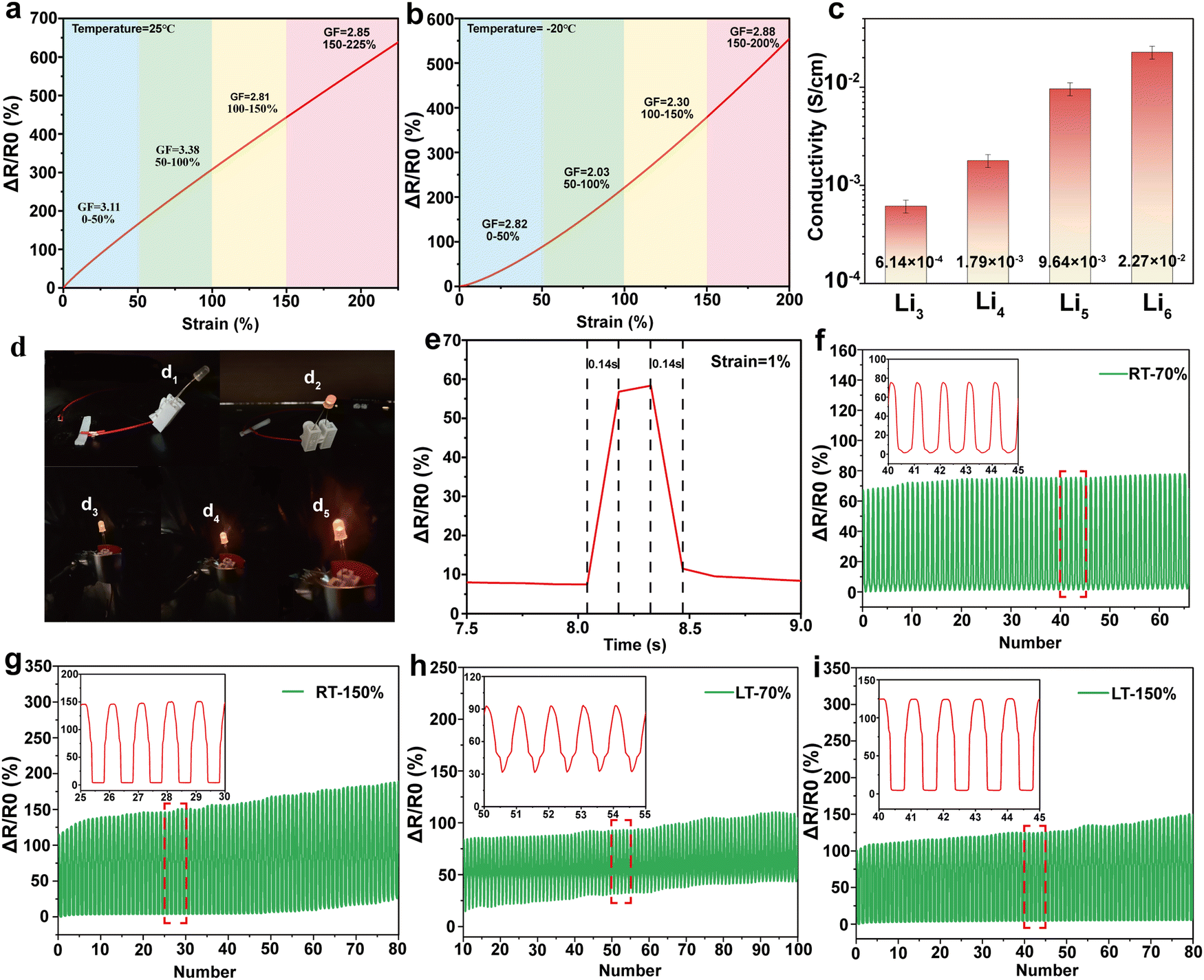 | ||
| Fig. 6 Sensing performance properties of PVA/KGM/PC composite hydrogel: (a) relative resistance change and gauge factor over a strain range of 0–200% at 25 °C. (b) Relative resistance change and gauge factor over a strain range of 0–200% at −20 °C. (c) Electrical conductivity with different Li+ immersion times. (d) Changes in LED brightness as a conductor. (e) Response time at 1% strain. (f) Relative resistance change curve over 100 stretch cycles at 70% strain at 25 °C. (g) Relative resistance change curve over 100 stretch cycles at 150% strain at 25 °C. (h) Relative resistance change curve over 100 stretch cycles at 70% strain at −20 °C. (i) Relative resistance change curve over 100 stretch cycles at 150% strain at −20 °C. | ||
Electrical conductivity is another important evaluation index in hydrogels as flexible sensors. Good electrical conductivity can confer a more comprehensive performance to hydrogels.80 In this work, Li+ was introduced into the PVA/KGM/PC composite hydrogel 3D network system using the solvent replacement method. The conductivity of the hydrogel is calculated using eqn (1). Fig. 6c shows the effect of the soaking times on the conductivity of the hydrogel. The results indicate that the conductivity increased from 6.14 × 10−4 S cm−1 to 2.27 × 10−2 S cm−1 with the increase in soaking time of Li2+ solution from 3 h to 6 h, an increase of two orders of magnitude. However, with a further increase in soaking time, the increase in conductivity was not obvious. The antifreeze property was significantly weakened. This may be attributed to the fact that with the increase in soaking time, the solvent replacement state inside the hydrogel approached dynamic saturation. After saturation, the Li+ solution began to destroy the hydrogen bonding system formed between PVA and glycerol, and replace glycerol, thus weakening the antifreeze property.63 Hence, the optimal soaking time was obtained as 6 h, which was used for the subsequent preparation of hydrogels.
Fig. 6d shows the change in LED lamp brightness under different stretching conditions of the PVA/KGM/PC composite hydrogel. Fig. 6d1 shows the lamp off when the circuit is not connected. Fig. 6d2 shows the lamp brightened after connecting the hydrogel to make the circuit connected. Fig. 6d3–d5 shows the change in LED lamp brightness during the recovery from the stretching state to the initial state in turn. The lamp gradually brightened, indicating that the hydrogel showed a positive correlation of resistance with strain during recovery from stretching. This again proves that the PVA/KGM/PC composite hydrogel has the characteristics of being a strain sensor.
Fig. 6e shows the response time of the PVA/KGM/PC composite hydrogel sensor at a strain of 1%. The results indicated that the resistance increases rapidly when the strain is applied. The response time is 0.14 s. When released, the resistance immediately decreases from a steady state and reaches the initial state within 0.14 s. This proves that the PVA/KGM/PC composite hydrogel can rapidly respond to small strain signals, which can be used to monitor the tiny activities of the human body.
To verify the stability of the hydrogel sensor during long periods of uninterrupted use, 100 cycles were performed at 25 °C and −20 °C, with 40 mm min−1 stretching speed at 70% and 150% strain, respectively (Fig. 6f–i). The results (calculated from eqn (2)) showed that the composite hydrogel exhibited a good repeatable response over 100 cycles at 25 °C and −20 °C. With 70% strain, the maximum relative resistance change basically remained a stable value. At 150% strain, although the relative resistance change showed a slight shift, it was maintained within a relatively stable range. This may be because the hydrogel under 70% strain undergoes reversible deformation, and its internal forces can be rapidly recovered before the next cycle. At a 150% strain, the hydrogel undergoes irreversible changes, and the internal hydrogen bonding cannot be recovered before the start of the next cycle, which requires a certain stagnation time.81 This is consistent with the conclusions drawn from the above mechanical properties. The result indicates that the hydrogel sensor shows superior stability in the strain range of 70% at both room temperature and low temperature and can perform stable sensing performance at a 150% large strain given a certain recovery time. This proves that the PVA/KGM/PC composite hydrogel has great potential as a flexible sensor for monitoring human activities.
Human motion monitoring
The above test results prove that the PVA/KGM/PC composite hydrogel exhibits excellent mechanical properties, high sensitivity, biocompatibility and fast response capability, and it is easy to prepare flexible strain sensors with different shapes to repeatedly identify various changes in the environment. The hydrogel was used as a strain gauge sensor to monitor various macroscopic body movements and subtle physiological signals.82,83For better demonstration, the hydrogel was cut into different sizes to detect various parts of the body. The rate of change in resistance in the test results are all calculated by applying eqn (2). The ability of the PVA/KGM/PC composite hydrogel sensor to monitor the fine physiological signals of the human body was first tested on the normal human pulse, heart beat and vocal cord vibration (Fig. 7a–c). Fig. 7a displays that the strain sensor can sensitively capture each pulse beat. From calculation, it conforms to the number of beats per minute under normal human conditions (60–100 times) regularity. Fig. 7c depicts that the resistance changes in accordance with heart beat regular fluctuations, proving that subtle signals can be accurately detected. As illustrated in Fig. 7b, when “Hello”, “shaanxi”, “Keji”, and “Daxue” were uttered, the vibration of the vocal folds caused the fluctuation of electric resistance to change. Simultaneously, the change in resistance was significantly higher when the stressed syllables, such as “Ni, Ha, Ke, and Da”, were uttered than the light syllables.84 The test results illustrate the ability of the PVA/KGM/PC hydrogel sensor to capture the weak physiological signals of the human body.
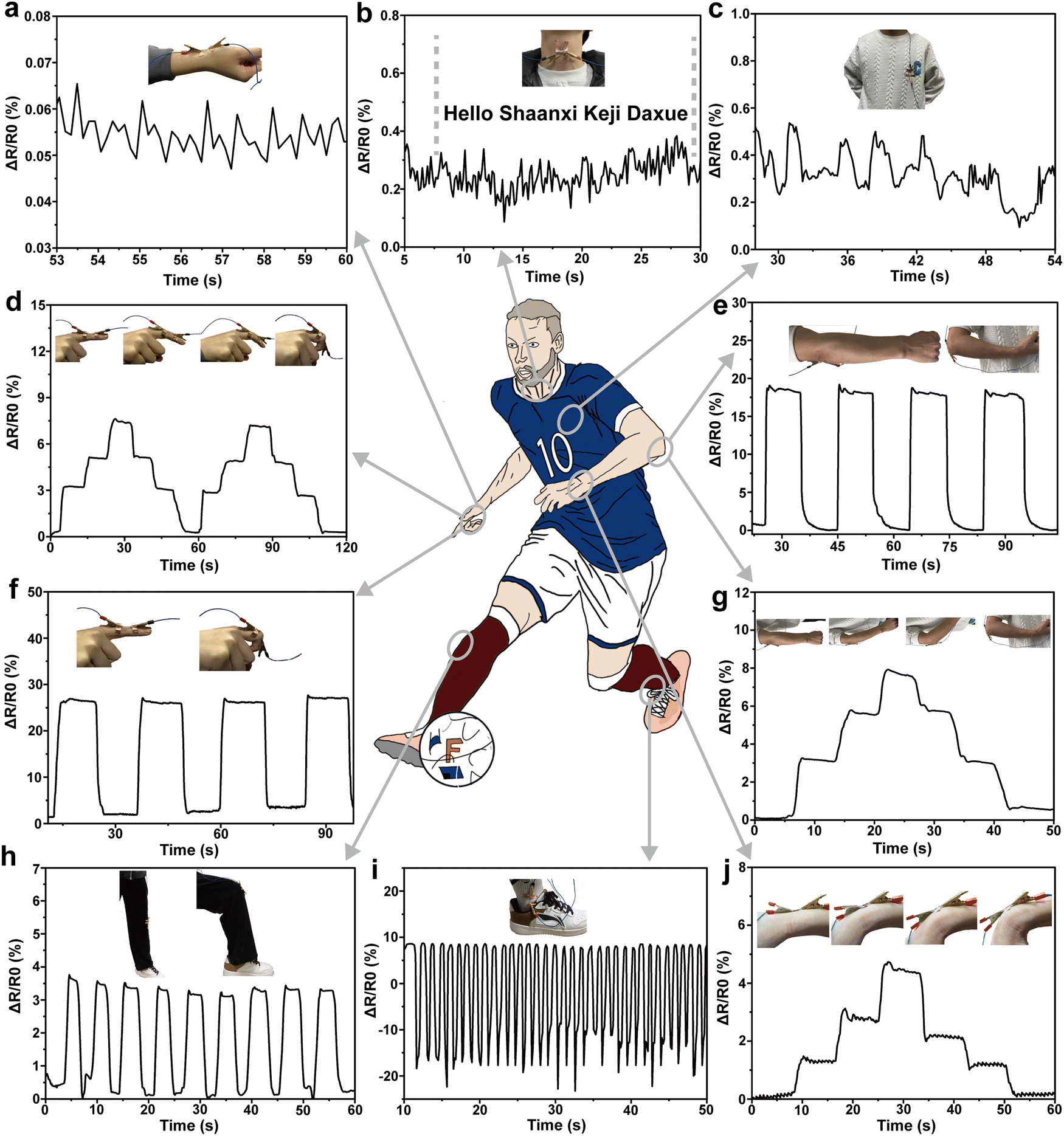 | ||
| Fig. 7 (a) Relative resistance change under a normal pulse beat. (b) Relative resistance change in vocal cord vibration when saying “Hello”, “Shaanxi”, “Keji”, and “Daxue”. (c) Relative resistance changes under a normal heartbeat. (d) and (f) Relative resistance change in fingers under different bending angles (0°, 30°, 60°, and 90°). (e) and (g) Relative resistance change in the arm at different bending angles (0°, 30°, 60°, and 90°). (h) Relative resistance change in the knee at different bending angles. (i) Relative resistance change during ankle movement. (j) Relative resistance change in the wrist under different bending angles (0°, 30°, 60°, and 90°). | ||
After that, the strain sensor was tested for macroscopic human motion monitoring, as shown in Fig. 7d and f, respectively. The hydrogel sensor was assembled on the index finger to monitor the changes in different bending cycles. The resistance change was 0 when the finger was straight. It showed a stepped pattern when the finger was bent to 30°, 60°, and 90°, which in turn indicates that there was a linear relationship between the change in resistance and the bending angle of the finger. When the finger performs a 0° to 90° bending cycle, the resistance change shows concave and convex trends, indicating that the sensor can accurately describe the bending change in the finger; specifically, different bending cycles show different trends.45 Similarly, it can be observed from the bending test of the wrist (Fig. 7j) that the hydrogel sensor can respond accurately to every angle of bending and pause in the process. In large-range strain human activity processes, such as arm bend (Fig. 7e and g), the hydrogel sensor also shows the same pattern as finger and wrist bending. The test under a wider range of strain (knee bending) was conducted with the results shown in Fig. 7h. It can be observed that reliable resistance changes were obtained, indicating that it can perform stable sensing performance for large-range strains. Fig. 7i shows the relative electric resistance change pattern when the ankle is active. The activity of the ankle includes the stretching and compression of the hydrogel sensor. It can be observed from the figure that the relative resistance shows a regular cyclic change from positive to negative values, which is consistent with the change in resistance with hydrogel stretching and contraction. This indicates that the hydrogel sensor can make a fast response when stretching and make an accurate response when compressing. Through the above monitoring tests on different parts of the human body, the PVA/KGM/PC composite hydrogel sensor shows excellent sensing performance. With tensile-shrinking two-way sensitive monitoring performance, and an extremely wide monitoring range from a small human heartbeat and pulse to a large knee bend, the sensor can give an accurate response. In addition, the excellent frost resistance enables it to show sensitive monitoring performance even in low-temperature environments, with wider application fields and better adaptability and has great potential as a flexible strain sensor in the field of human motion monitoring.85
4. Conclusion
This study successfully prepared a dual-network composite hydrogel containing PC using PVA/KGM through the “parts” assembly strategy and applied it as a flexible strain sensor for human motion monitoring. The PVA/KGM/PC composite hydrogel possesses good mechanical properties and frost resistance, exhibits excellent sensing performance and a high GF at 25 °C and −20 °C, and shows stronger environmental adaptability and stability compared to previous work while greatly reducing the preparation cost. In the human motion monitoring tests, the hydrogel sensors showed fast response capability from capturing weak physiological signals, such as the human heart, pulse, and vocal cords, to monitoring a large range of body motions, such as arm and leg bending, proving that the PVA/KGM/PC composite hydrogel has sensitive sensing performance and broad application prospects. This work is expected to provide new ideas for the introduction of nanofillers into polymer systems.Conflicts of interest
All authors declare that there is no conflict of interest.Acknowledgements
This work was supported by the General Project of the National Natural Science Foundation of China (21276152), Shaanxi Province Regional Innovation Capability Guidance Program Project (2021QFY04-04).References
- Y. Liu, L. Wang, Y. Mi, S. Zhao, S. Qi, M. Sun, B. Peng, Q. Xu, Y. Niu and Y. Zhou, J. Mater. Chem. C, 2022, 10, 13351–13371 RSC.
- X. Sun, F. Yao and J. Li, J. Mater. Chem. A, 2020, 8, 18605–18623 RSC.
- S. Sun, Y. Xu and X. Maimaitiyiming, Int. J. Biol. Macromol., 2023, 249, 125978 CrossRef CAS PubMed.
- Y. Guo, X. Wei, S. Gao, W. Yue, Y. Li and G. Shen, Adv. Funct. Mater., 2021, 31, 2203585 Search PubMed.
- Z. Li, F. Yin, W. He, T. Hang, Z. Li, J. Zheng, X. Li, S. Jiang and Y. Chen, Int. J. Biol. Macromol., 2023, 230, 123117 CrossRef CAS PubMed.
- E. Liu, Z. Cai, Y. Ye, M. Zhou, H. Liao and Y. Yi, Sensors, 2023, 23, 817 CrossRef CAS PubMed.
- M. Zhu, J. Hu, Q. Lu, H. Dong, D. D. Karnaushenko, C. Becker, D. Karnaushenko, Y. Li, H. Tang, Z. Qu, J. Ge and O. G. Schmidt, Adv. Mater., 2021, 33, 2007497 CrossRef CAS PubMed.
- Z. Pu, J. Tu, R. Han, X. Zhang, J. Wu, C. Fang, H. Wu, X. Zhang, H. Yu and D. Li, Lab Chip, 2018, 18, 3570–3577 RSC.
- Y. S. Moon, D. Kim, G. Lee, S. Y. Hong, K. K. Kim, S. M. Park and J. S. Ha, Carbon, 2015, 81, 29–37 CrossRef CAS.
- Y. Yi, M. Chiao, K. A. Mahmoud, L. Wu and B. Wang, J. Mater. Sci., 2022, 57, 8029–8038 CrossRef CAS.
- D. Wang, D. Zhang, P. Li, Z. Yang, Q. Mi and L. Yu, Nanomicro Lett., 2021, 13, 57 Search PubMed.
- Z. S. Nishat, T. Hossain, M. N. Islam, H. P. Phan, M. A. Wahab, M. A. Moni, C. Salomon, M. A. Amin, A. A. Sina, M. S. A. Hossain, Y. V. Kaneti, Y. Yamauchi and M. K. Masud, Small, 2022, 18, e2107571 CrossRef PubMed.
- Y. S. Rim, S. H. Bae, H. Chen, N. De Marco and Y. Yang, Adv. Mater., 2016, 28, 4415–4440 CrossRef CAS PubMed.
- R. Yin, D. Wang, S. Zhao, Z. Lou and G. Shen, Adv. Funct. Mater., 2020, 31, 2008936 CrossRef.
- W. Wang, S. Yang, K. Ding, L. Jiao, J. Yan, W. Zhao, Y. Ma, T. Wang, B. Cheng and Y. Ni, Chem. Eng. J., 2021, 425, 129949 CrossRef CAS.
- Y. Liang, L. Ye, X. Sun, Q. Lv and H. Liang, ACS Appl. Mater. Interfaces, 2019, 12, 1577–1587 CrossRef PubMed.
- H. Zhang, N. Tang, X. Yu, Z. Guo, Z. Liu, X. Sun, M.-H. Li and J. Hu, Chem. Eng. J., 2022, 430, 132779 CrossRef CAS.
- C. Chang and L. Zhang, Carbohydr. Polym., 2011, 84, 40–53 CrossRef CAS.
- C. Cui, Y. Jia, Q. Sun, M. Yu, N. Ji, L. Dai, Y. Wang, Y. Qin, L. Xiong and Q. Sun, Carbohydr. Polym., 2022, 291, 119624 CrossRef CAS PubMed.
- M. C. G. Pellá, M. K. Lima-Tenório, E. T. Tenório-Neto, M. R. Guilherme, E. C. Muniz and A. F. Rubira, Carbohydr. Polym., 2018, 196, 233–245 CrossRef PubMed.
- X. Hu, L. Feng, A. Xie, W. Wei, S. Wang, J. Zhang and W. Dong, J. Mater. Chem. B, 2014, 2, 3646–3658 RSC.
- N. Rodkate, U. Wichai, B. Boontha and M. Rutnakornpituk, Carbohydr. Polym., 2010, 81, 617–625 CrossRef CAS.
- N. Roy, N. Saha, T. Kitano and P. Saha, Carbohydr. Polym., 2012, 89, 346–353 CrossRef CAS PubMed.
- Z. Bao, C. Xian, Q. Yuan, G. Liu and J. Wu, Adv. Healthcare Mater., 2019, 8, 1900670 CrossRef PubMed.
- W. Tanan, J. Panichpakdee and S. Saengsuwan, Eur. Polym. J., 2019, 112, 678–687 CrossRef CAS.
- S. Bhatia, in Natural Polymer Drug Delivery Systems, 2016, ch. 3, pp. 95–118 DOI:10.1007/978-3-319-41129-3_3.
- A. Sionkowska, Prog. Polym. Sci., 2011, 36, 1254–1276 CrossRef CAS.
- S. Jiang, L. Shang, H. Liang, B. Li and J. Li, Food Hydrocolloids, 2022, 127, 107499 CrossRef CAS.
- S. G. Jin, Chem. – Asian J., 2022, 17, e202200595 CrossRef CAS PubMed.
- B. Ryplida, K. D. Lee, I. In and S. Y. Park, Adv. Funct. Mater., 2019, 29, 1903209 CrossRef.
- F. Lin, Z. Wang, Y. Shen, L. Tang, P. Zhang, Y. Wang, Y. Chen, B. Huang and B. Lu, J. Mater. Chem. A, 2019, 7, 26442–26455 RSC.
- Y. Li, H. Chengxin, J. Lan, B. Yan, Y. Zhang, L. Shi and R. Ran, Polymer, 2020, 186, 122027 CrossRef CAS.
- H. C. Ates, P. Q. Nguyen, L. Gonzalez-Macia, E. Morales-Narváez, F. Güder, J. J. Collins and C. Dincer, Nat. Rev. Mater., 2022, 7, 887–907 CrossRef PubMed.
- S. T. Han, H. Peng, Q. Sun, S. Venkatesh, K. S. Chung, S. C. Lau, Y. Zhou and V. A. L. Roy, Adv. Mater., 2017, 29, 1700375 CrossRef PubMed.
- Y. M. Chen, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 5990–5993 CrossRef CAS PubMed.
- M. T. Khorasani, A. Joorabloo, A. Moghaddam, H. Shamsi and Z. MansooriMoghadam, Int. J. Biol. Macromol., 2018, 114, 1203–1215 CrossRef CAS PubMed.
- E. Homede, M. Abo Jabal and O. Manor, Adv. Funct. Mater., 2020, 30, 2005486 CrossRef CAS.
- Q. Chang, M. A. Darabi, Y. Liu, Y. He, W. Zhong, K. Mequanin, B. Li, F. Lu and M. M. Q. Xing, J. Mater. Chem. A, 2019, 7, 24626–24640 RSC.
- P. K. Sharma, E. S. Kim, S. Mishra, E. Ganbold, R. S. Seong, Y. M. Kim, G. H. Jahng, H. Y. Rhee, H. S. Han, D. H. Kim, S. T. Kim and N. Y. Kim, Biosens. Bioelectron., 2022, 212, 114365 CrossRef CAS PubMed.
- F. J. Ibañez and F. P. Zamborini, Small, 2012, 8, 174–202 CrossRef PubMed.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- Z. Sun, Y. Yamauchi, F. Araoka, Y. S. Kim, J. Bergueiro, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima and T. Aida, Angew. Chem., Int. Ed., 2018, 57, 15772–15776 CrossRef CAS PubMed.
- Y. Xia, Y. Wu, T. Yu, S. Xue, M. Guo, J. Li and Z. Li, ACS Appl. Mater. Interfaces, 2019, 11, 21117–21125 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- G. Cui, L. Zhang, A. A. Zaky, R. Liu, H. Wang, A. M. Abd El-Aty and M. Tan, Int. J. Biol. Macromol., 2022, 216, 799–809 CrossRef CAS PubMed.
- Y. Song, H. Wang, L. Zhang, B. Lai, K. Liu and M. Tan, Food Funct., 2020, 11, 2358–2367 RSC.
- K. Liu, Y. Song and M. Tan, J. Agric. Food Chem., 2020, 68, 9789–9795 CrossRef CAS PubMed.
- G. Cui, W. Su and M. Tan, Compr. Rev. Food Sci. Food Saf., 2022, 21, 2002–2031 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- S. Shi, X. Peng, T. Liu, Y.-N. Chen, C. He and H. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS.
- Y. Zhou, R. Jiang, W. S. Perkins and Y. Cheng, Food Chem., 2018, 269, 80–88 CrossRef CAS PubMed.
- X. Liang, H. Li, J. Dou, Q. Wang, W. He, C. Wang, D. Li, J. M. Lin and Y. Zhang, Adv. Mater., 2020, 32, e2000165 CrossRef PubMed.
- S. Wang, K. Chen, L. Li and X. Guo, Biomacromolecules, 2013, 14, 818–827 CrossRef CAS PubMed.
- Y. You, L. Yang, H. Chen, L. Xiong and F. Yang, J. Agric. Food Chem., 2021, 69, 2306–2315 CrossRef CAS PubMed.
- R. Zhang, Y. Liu, X. Huang, M. Xu, R. Liu and W. Zong, Sci. Total Environ., 2018, 622–623, 306–315 CrossRef CAS PubMed.
- T. Zhang, M. Tang, Y. Yao, Y. Ma and Y. Pu, Int. J. Nanomed., 2019, 14, 993–1009 CrossRef CAS PubMed.
- Y. Zhu, J. Liu, T. Guo, J. J. Wang, X. Tang and V. Nicolosi, ACS Nano, 2021, 15, 1465–1474 CrossRef CAS PubMed.
- Y. Zhou, C. Wan, Y. Yang, H. Yang, S. Wang, Z. Dai, K. Ji, H. Jiang, X. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1806220 CrossRef.
- C. Hu, Y. Zhang, X. Wang, L. Xing, L. Shi and R. Ran, ACS Appl. Mater. Interfaces, 2018, 10, 44000–44010 CrossRef CAS PubMed.
- Y. Li, Y. Peng, J.-Y. Tian, S. Duan, Y. Fu, S. Zhang and M. Du, Colloids Surf., A, 2023, 670, 131577 CrossRef CAS.
- X. Liu, Z. Wu, D. Jiang, N. Guo, Y. Wang, T. Ding and L. Weng, Adv. Compos. Hybrid Mater., 2022, 5, 1712–1729 CrossRef CAS.
- S. Kihara, N. J. van der Heijden, C. K. Seal, J. P. Mata, A. E. Whitten, I. Koper and D. J. McGillivray, Bioconjugate Chem., 2019, 30, 1067–1076 CrossRef CAS PubMed.
- T. Zhu, J. Mao, Y. Cheng, H. Liu, L. Lv, M. Ge, S. Li, J. Huang, Z. Chen, H. Li, L. Yang and Y. Lai, Adv. Mater. Interfaces, 2019, 6, 1900761 CrossRef.
- W. Wang, Y. Zhang and W. Liu, Prog. Polym. Sci., 2017, 71, 1–25 CrossRef.
- L. Marichal, J. Degrouard, A. Gatin, N. Raffray, J. C. Aude, Y. Boulard, S. Combet, F. Cousin, S. Hourdez, J. Mary, J. P. Renault and S. Pin, Langmuir, 2020, 36, 8218–8230 CrossRef CAS PubMed.
- A. Bhogale, N. Patel, P. Sarpotdar, J. Mariam, P. M. Dongre, A. Miotello and D. C. Kothari, Colloids Surf., B, 2013, 102, 257–264 CrossRef CAS PubMed.
- H. Liu, X. Wang, Y. Cao, Y. Yang, Y. Yang, Y. Gao, Z. Ma, J. Wang, W. Wang and D. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 25334–25344 CrossRef CAS PubMed.
- K. Dey, S. Agnelli and L. Sartore, Micro, 2023, 3, 434–457 CrossRef.
- S.-N. Li, Z.-R. Yu, B.-F. Guo, K.-Y. Guo, Y. Li, L.-X. Gong, L. Zhao, J. Bae and L.-C. Tang, Nano Energy, 2021, 90, 106502 CrossRef CAS.
- T. Ke, L. Zhao, X. Fan and H. Gu, J. Mater. Sci. Technol., 2023, 135, 199–212 CrossRef CAS.
- R. Wang, W. Chi, F. Wan, J. Wei, H. Ping, Z. Zou, J. Xie, W. Wang and Z. Fu, ACS Appl. Mater. Interfaces, 2022, 14, 21278–21286 CrossRef CAS PubMed.
- C. Wang, K. Hu, C. Zhao, Y. Zou, Y. Liu, X. Qu, D. Jiang, Z. Li, M. R. Zhang and Z. Li, Small, 2020, 16, e1904758 CrossRef PubMed.
- X. Li, Q. Yang, Y. Zhao, S. Long and J. Zheng, Soft Matter, 2017, 13, 911–920 RSC.
- M. Wang, J. Zhou, X. Jiang, Y. Sheng, M. Xu and X. Lu, Eur. Polym. J., 2021, 146, 110257 CrossRef CAS.
- A. P. Sobha and S. K. Narayanankutty, Sens. Actuators, A, 2015, 233, 98–107 CrossRef CAS.
- Y. Sun, B. Liu, L. Liu and X. Yan, Adv. Funct. Mater., 2021, 32, 2109568 CrossRef.
- G. Chen, J. Huang, J. Gu, S. Peng, X. Xiang, K. Chen, X. Yang, L. Guan, X. Jiang and L. Hou, J. Mater. Chem. A, 2020, 8, 6776–6784 RSC.
- Z. Wang, Y. Cong and J. Fu, J. Mater. Chem. B, 2020, 8, 3437–3459 RSC.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- J. Yin, C. Lu, C. Li, Z. Yu, C. Shen, Y. Yang, X. Jiang and Y. Zhang, Composites, Part B, 2022, 230, 109528 CrossRef CAS.
- X. Di, Q. Ma, Y. Xu, M. Yang, G. Wu and P. Sun, Mater. Chem. Front, 2021, 5, 315–323 RSC.
- Q. Li, Y. Liu, D. Chen, J. Miao, C. Zhang and D. Cui, ACS Appl. Mater. Interfaces, 2022, 14, 51373–51383 CrossRef CAS PubMed.
- S. Liu, X. Tian, X. Zhang, C. Xu, L. Wang and Y. Xia, Chin. Chem. Lett., 2022, 33, 2205–2211 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |