
A critical revelation of lithium ferromanganese phosphate (LMFP) performance in a Mn-rich cathode for Li-ion batteries using Fe equivalents to occupy a Mn site†
Ruifen
Yang
ab,
Longjiao
Chang
 *ab,
Shaohua
Luo
*cd,
Xiaolong
Bi
ab,
Wei
Yang
ab,
Kedi
Cai
ef,
Anlu
Wei
ab and
Zenglei
Hou
ab
*ab,
Shaohua
Luo
*cd,
Xiaolong
Bi
ab,
Wei
Yang
ab,
Kedi
Cai
ef,
Anlu
Wei
ab and
Zenglei
Hou
ab
aSchool of Chemical and Material Engineering, Bohai University, Jinzhou, 121013, Liaoning, China. E-mail: jsz337@sina.com
bLiaoning Key Laboratory of Engineering Technology Research Center of Silicon Materials, Jinzhou, 121013, Liaoning, China
cKey Laboratory of Dielectric and Electrolyte Functional Material Hebei Province, Qinhuangdao, 066004, Hebei, China. E-mail: tianyanglsh@163.com
dSchool of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao, 066004, Hebei, China
eCollege of Chemistry and Materials Engineering, Bohai University, Jinzhou 121000, China
fLiaoning Engineering Technology Research Center of Supercapacitor, Bohai University, Jinzhou, 121013, China
First published on 15th March 2024
Abstract
The modification of a polyanionic positive electrode material LiMnPO4 by transition metal doping was experimentally studied on the basis of carbon coating in order to address the drawbacks of low electronic conductivity and charge–discharge performance. A hydrothermal procedure was successfully used to create a Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, and 1/4) cathode material, and its performance was evaluated. After comparing the material's electrochemical performance at different doping concentrations, the ideal Fe content was found to be x = 1/4. The Li(Mn1−xFex)PO4/C sample exhibits excellent electrochemical performance, discharge ability of 141.6 mA h g−1, 100 cycles at 1C rate, capacity retention ratio of 97.88%, and the best kinetic characteristics. First principles calculations showed that the Li(Mn1−xFex)PO4/C material has the smallest band gap, and the total state density is closest to the Fermi level; thus, more electrons can be used to transition to the conduction band, which is the reason for its excellent discharge ability. The introduction of Fe reduces the ELF function value near the original Mn position and decreases electron localization and the diffusion energy barrier, which is very conducive to electron movement and improves the diffusion kinetics of lithium ions in a doping system.
1 Introduction
At a time when oil and other conventional energy sources are increasingly scarce around the world, there is an exigent need for the effective development and use of sustainable energy, such as solar, wind and tidal energy.1 Nevertheless, these new sources of energy are unstable and constant; thus, they need to be converted into electricity before they can be exported, which has motivated research on rechargeable batteries. Lithium-ion batteries (LIBs) have become the dominant technology in the field of man-carried electronic equipment and electromobiles owing to their high energy density, excellent cycling steadiness, poor self-discharge characteristics and environmental friendliness.2,3 High energy density lithium-ion batteries have been associated with an array of issues in recent years, including deteriorating battery life and the coexistence of electrochemical side reactions that can compromise electrode integrity in harsh settings. The design of cathode materials for LIBs is fraught with numerous significant obstacles.4,5 Therefore, enhancing the nature of lithium-ion batteries via the design of new and emerging materials and doping materials has emerged as a research hotspot. As can be seen from Fig. 1,6 there is a great potential for polyanionic cathode materials: PO43− series electrodes with high reaction potential are more suitable for large-scale energy storage systems. However, the mass and charge transport retardation of LiMnPO4 severely hinders its application.7–9 For many years, researchers have been trying to promote the property of the material. While combining conductive elements and decreasing the material's size can improve the LiMnPO4 material's electrochemical performance, it has minimal effect on the material's electron–ion transport capabilities since its crystal properties remain largely unaffected.10–13 Elemental doping can fundamentally elevate the performance of LiMnPO4 materials, and one of the most widely used methods is the doping of equivalent or covalent cations at Li or transition metal Mn sites.14,15 Theoretical calculations suggest that Li site doping can reduce the electrochemical performance of LiMnPO4 by blocking the conduction pathway, which in turn can cause damage to the electrochemical performance of LiMnPO4. Therefore, the doping of the Li site is a matter of controversy.16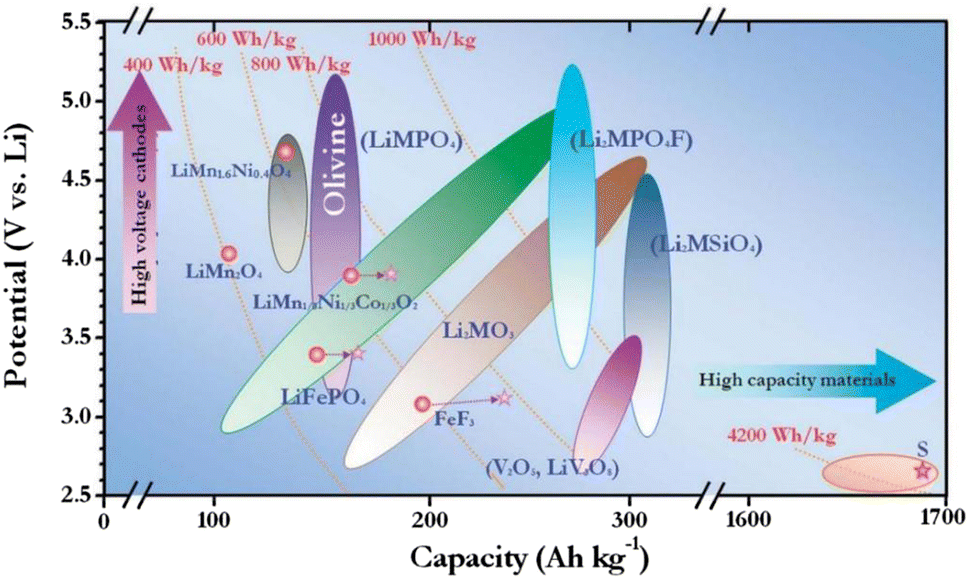 | ||
| Fig. 1 Schematic diagram of the cathode material potentials of the Li-ion battery versus capacity, where M = Fe, Mn, Co, Ni, etc. | ||
The electrochemical properties of lithium manganese phosphate can be effectively improved by doping manganese with various metal elements. These methods can cause dispersed metal ions LiMnPO4 to provide conducting bridges, which can substantially enhance the electronic conductivity of LiMnPO4 as well as the charge and discharge properties of materials. Among these, the inclusion of iron can not only solidify the material's structure and increase its energy density but it also provides the material two platforms for charging and discharging, which enhances the material's response kinetics.17,18 For this reason, in recent years, research has been more concentrated on Li(Mn1−xFex)PO4 composite materials. Han et al. prepared spherical or annular secondary particles of Li(Mn1−xFex)PO4/C (x = 0.2, 0.5, 0.8) using a modified polyol reaction to enhance the electrochemical performance of the LiMnPO4 material.19 LiMn0.8Fe0.2PO4/C is the one that shows the highest performance. It delivers a high discharge capability of 161 mA h g−1 at 0.05C and a capacity retention rate of 80.4% for 900 cycles at a discharge rate of 0.5C. In addition, 90% and 83% of its original capacity can be maintained at 45 °C and 55 °C after 100 cycles, respectively. Wang et al. selectively grew iron-doped Mn3O4 nanoparticles onto GO for the first time through controlled hydrolysis and solvothermal reaction with Li+ and PO4− to generate Li(Mn1−xFex)PO4 single crystal nanomaterials, displaying a high specific capacity and unprecedented high power.20 At high discharge rates of 20C and 50C, the stable capacity of the material was respectively 132 mA h g−1 and 107 mA g−1, and 85% and 70% at 0.5C (155 mA g−1). Li+ can diffuse rapid in the y-direction of [100], setting the stage for complex hybrid materials design and engineering and advanced energy storage with graphene.
Because iron and manganese are transition metals and occupy the same lattice in the material, the properties are more similar and can be highly compatible; thus, Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, 1/4) materials can be formed.21–24 In this paper, based on carbon coating, the doping modification of the transition metal potential of the positive polyanionic LiMnPO4 material is investigated experimentally. Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, 1/4) electrode was successfully prepared the by hydrothermal method, and its performance was tested. The electrochemical performances of the materials with different doping amounts were compared, and the best Fe content was selected. Simultaneously, in order to better understand and support experimental phenomenon, first principles were applied to explain the reasons for the differences in electrochemical properties from the electronic structure.
2 Experiment and calculation
2.1 The synthesis of the Li(Mn1−xFex)PO4/C cathode
All of the drug solutions employed in this research were of analytical reagents, and all of them were planned using deionized water. The technological process is shown in Fig. 2, and the detailed operation steps can be summarized as follows.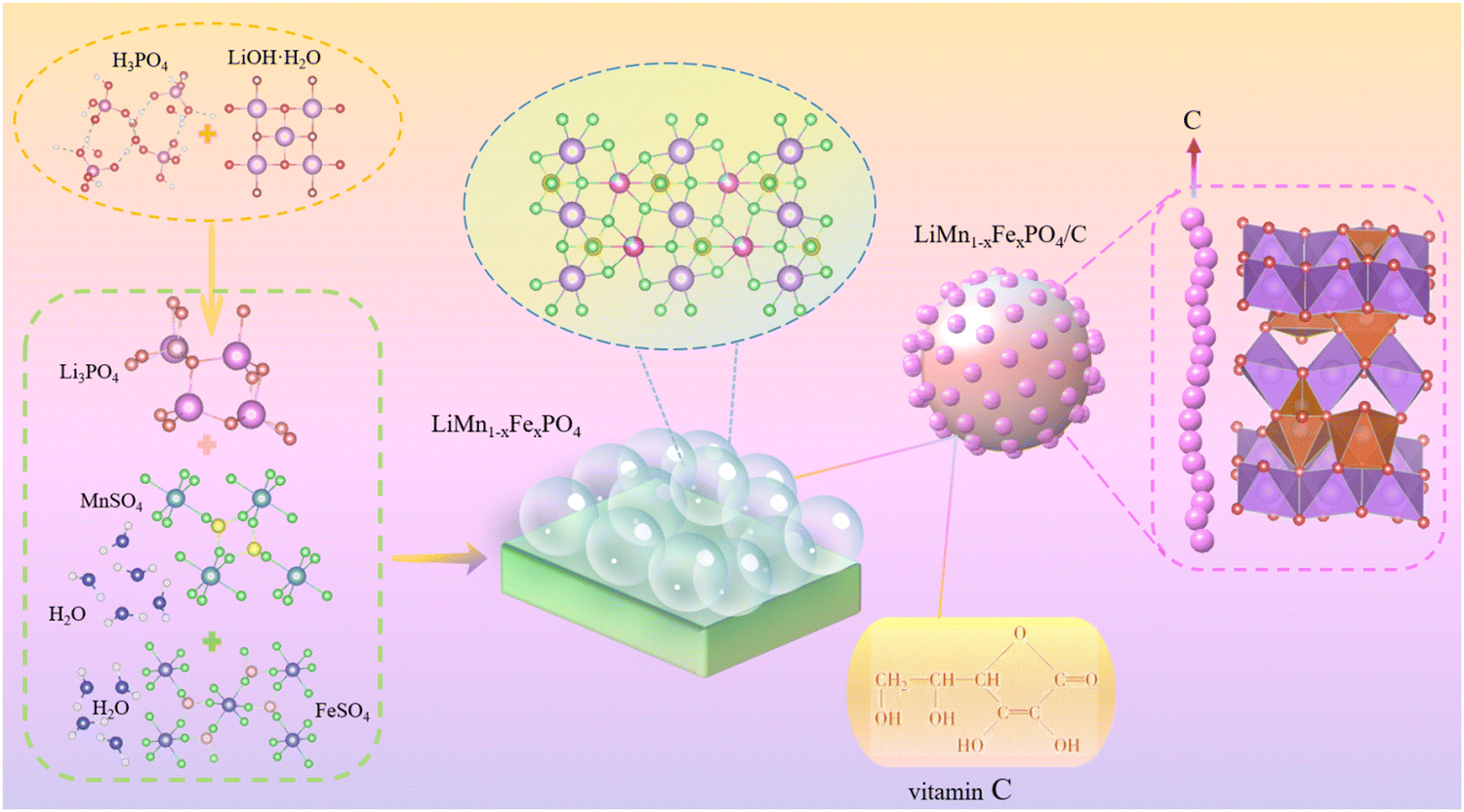 | ||
| Fig. 2 The plot of the Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, 1/4) composite material. | ||
First, LiOH·H2O crystal of a certain mass was weighed according to the ratio of the quantities of matter in the composite Li(Mn1−xFex)PO4/C. A 200 mL solution of LiOH·H2O was prepared and placed in a constant temperature collecting heating stirrer to heat up to a certain temperature. For the chemical reaction, 50 mL of the H3PO4 standard solution was mixed in the warm LiOH·H2O liquor, and the rate of acceleration of the drop was controlled until the white precipitate was fully precipitated. Li3PO4 grey powder was obtained after standing at room temperature for 1 h, followed by washing, frying, sieving, and calcination.
Then, crystals of MnSO4·H2O and FeSO4·H2O of a certain mass were weighed according to the ratio of the quantities of material in the Li(Mn1−xFex)PO4/C composite material. Both crystals were dissolved in a 36 mL mixed solution of PEG400–H2O with the off-white powder Li3PO4 synthesized, where the ratio of the volumes of PEG400–H2O was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. After stirring, the resulting mixture was transferred to a 50 mL PTFE-lined reactor and reactor at 160 °C for 9 hours. The powder Li(Mn1−xFex)PO4 was obtained by cooling the solution to room temperature, washing, vacuum drying and sieving.
:2. After stirring, the resulting mixture was transferred to a 50 mL PTFE-lined reactor and reactor at 160 °C for 9 hours. The powder Li(Mn1−xFex)PO4 was obtained by cooling the solution to room temperature, washing, vacuum drying and sieving.
Lastly, the Li(Mn1−xFex)PO4 powder was thoroughly mixed with vitamin C to a certain percentage, and the mixed slurry was stirred in a ball mill for 3 hours (adding 20 mL anhydrous ethanol). The precursor powder was obtained by drying the mixed slurry in an oven at 80 °C for 8 h. The Li(Mn1−xFex)PO4/C composites were prepared by sintering in a tube at 550 °C for 3 hours in nitrogen atmosphere.
:1:1. After the resulting slurry was evenly coated on the aluminium foil, in order to enhance the uniformity and density of the electrode, the slurry was extruded onto the electric roller. The slurry was allowed to dry in a vacuum oven at 120 °C for 8 h. The stack of CR2032 buttons was assembled in a glove box filled with high-purity argon using Celgard 2400 as the diaphragm with the electrode material prepared as the positive electrode and the Li metal as the opposing electrode. The electrolyte has a design and a ratio of 1.0 M LiPF6 in EC:DMC:EMC = 1:1:1 vol%. A CT2001A LAND battery tester was used to measure the charge–discharge and cycling properties under constant current for the 2.5–4.5 V voltage range. Impedance and CV tests were performed at Shanghai Chenhua Electrochemistry Workstation (CHI660E).
2.2 Computing method
All crystal structure plots were drawn in the VESTA software package, and the computational process was performed using the VASP software package. The geometry, lattice vectors and atomic coordinates of the initial crystal of Li(Mn1−xFex)PO4/C were optimized using the conjugate gradient (CG) method until the forces acting on each relaxation atom were less than 0.05 eV Å−1. The energy of the optimized crystal structure was calculated, including the state density, energy band and population of the material. The SCF convergence standard for electronic self-consistent calculation was 1.0 × 10−8 eV atom−1, and the calculation converges completely when the energy change of the system is below the set of convergence values.The exchange correlation energy was handled in the pin-polarized generalize-gradient-approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) formula. Considering the correlation of electrons in Mn-3d and Fe-3d orbitals in Li(Mn1−xFex)PO4 cells, a Hubbard-like correction (GGA+U) was added to the total energy functional for correction, the effective U values of Mn and Fe were set respectively at 3.9 eV and 5.3 eV, and the cutoff kinetic energy of the wave function in the plane wave was set at 520 eV. In order to fully converge the stress, the Monkhorst–Pack method was used to integrate the Brillouin region. A 2 × 2 × 2 k grid was used for structural relaxation, and a 4 × 4 × 4 k grid was used for energy minimization calculation.
3 Results and discussion
3.1 Phase and structural characterization
Fig. 3(a) exhibits the X-ray diffraction (XRD) patterns of Li(Mn1−xFex)PO4/C composites with different Fe doping amounts. As can be seen from the figure, under the four addition amounts, the materials all have the same obvious diffraction peak as the pure phase, and all of them correspond to all the spectral lines in the JCPDF standard card (33-0803). Therefore, the introduction of Fe does not significantly affect the XRD pattern of the material and does not alter the main phase structure of the LiMnPO4 material. Each Li(Mn1−xFex)PO4/C electrode diffraction peak shape is sharp, indicating good crystallinity.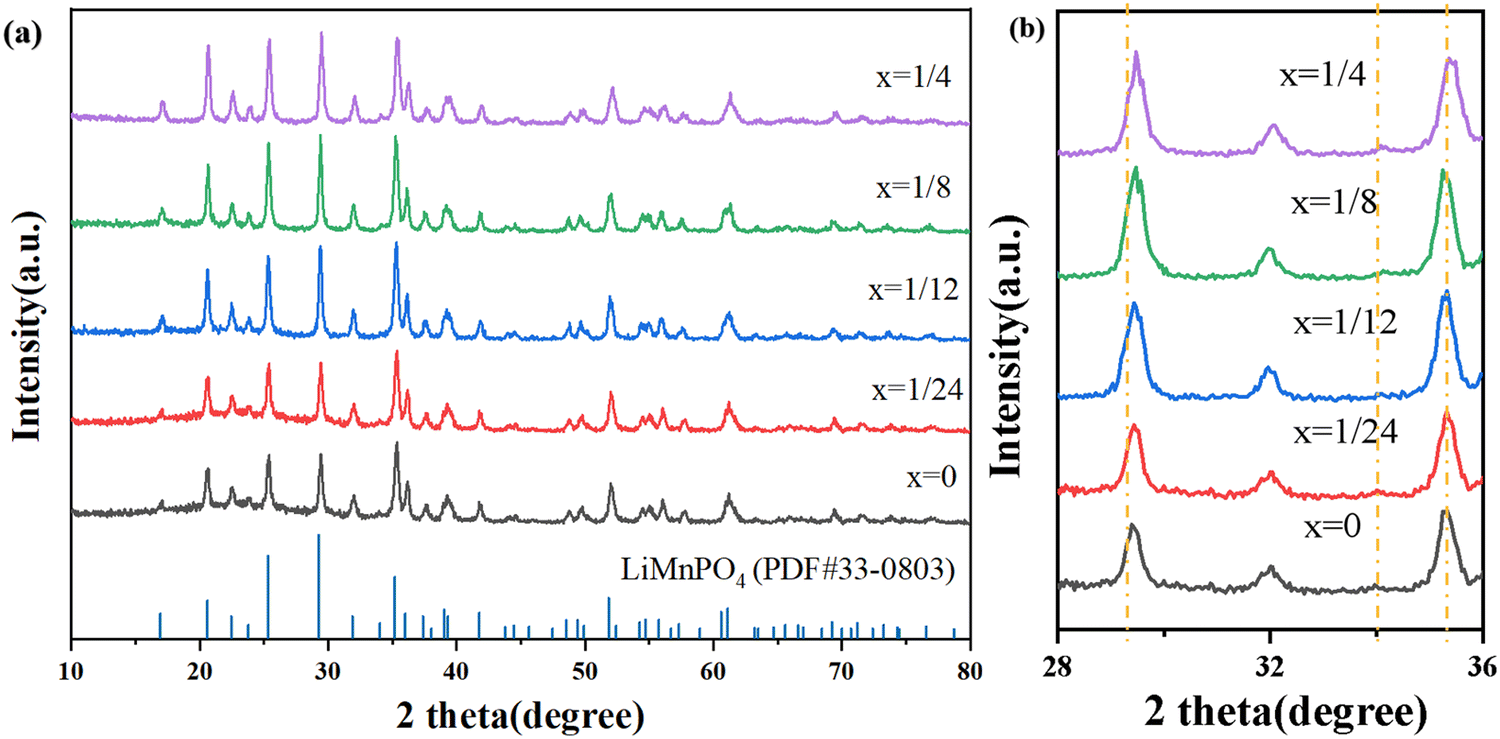 | ||
| Fig. 3 The XRD patterns of Li(Mn1−xFex)PO4/C materials. | ||
It can be obtained from the magnification in Fig. 3(b) that in the range 2θ = 28–36°, the diffraction peaks of materials with Fe doping amount of x = 1/24, 1/12, 1/8, and 1/4 have small shifts successively, indicating that Fe elements have successfully entered the LiMnPO4 lattice, and the offset increases as the amount of Fe doping is increased. The fact that there isn’t a C diffraction peak in the diagram indicates that the carbon layer that was created during the experiment has good dispersion and is an amorphous phase devoid of independent crystal formation. Therefore, the synthesized material has a single crystalline phase, namely, the olivine structure under the Pnma space group. In order to determine the actual composition of the Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, 1/4) samples, ICP was used for characterization, as shown in Table 1.
| x | Li | Mn | Fe | P | O | C |
|---|---|---|---|---|---|---|
| 0 | 4.07 | 32.58 | 0 | 18.28 | 37.83 | 7.13 |
| 1/24 | 4.11 | 31.15 | 1.41 | 18.31 | 37.84 | 7.03 |
| 1/12 | 4.16 | 29.41 | 2.59 | 18.35 | 37.86 | 7.09 |
| 1/8 | 4.13 | 28.39 | 4.17 | 18.29 | 37.75 | 7.15 |
| 1/4 | 4.09 | 24.26 | 8.36 | 18.27 | 37.79 | 7.08 |
The XRD patterns of the Li(Mn1−xFex)PO4/C samples were further refined according to the Rietveld method, as evidenced from Fig. 4. The least square method was used to fit the diffraction peak of the sample Li(Mn1−xFex)PO4/C, and the lattice parameters of the sample were obtained (see Table S1, ESI†). It was found that they were basically unchanged within the measurement error range. The lattice parameters of the Fe-doped sample showed little change compared to pure LiMnPO4/C, suggesting that Li(Mn1−xFex)PO4/C fundamentally maintained the crystal structure in accordance with the initial LiMnPO4/C material. As the Fe doping is increased, the value of c/a increases, indicating that the doping reduces the cation mixing and LiMn3/4Fe1/4PO4/C is optimal. Since Mn2+ has a larger ionic radius than Fe2+, the size of the cell should theoretically decrease when Fe2+ replaces Mn2+. From the data in Table S1 (ESI†), the volume of the Li(Mn1−xFex)PO4/C system gradually decreases with the increase in doping, which provides evidence that the crystal structure of the Li(Mn1−xFex)PO4/C material with Fe dopant sites has been produced.
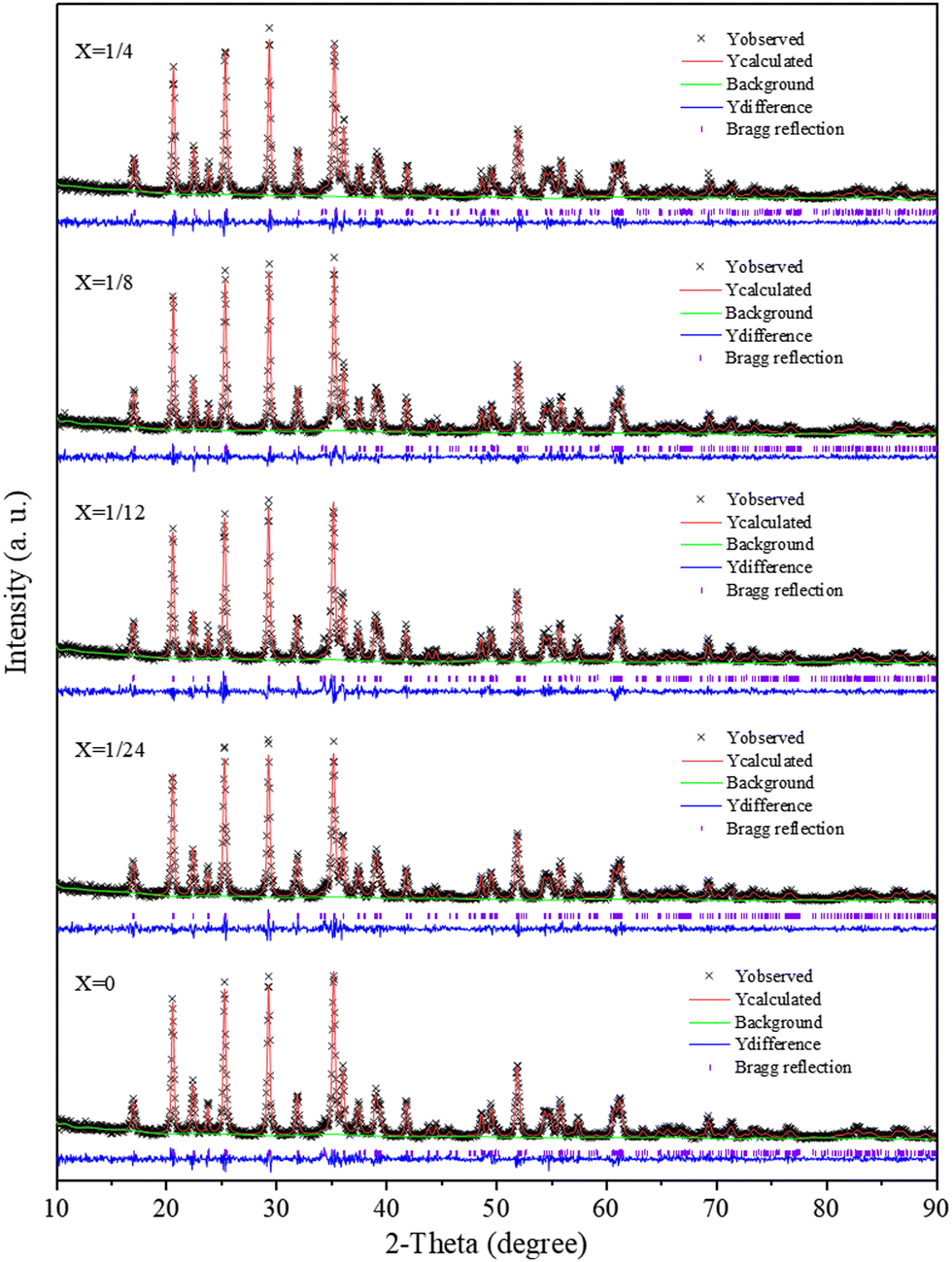 | ||
| Fig. 4 The Rietveld patterns of Li(Mn1−xFex)PO4/C materials. | ||
The two phase mechanism of Li+ insertion and removal in the main structure during the charging and discharging of the Li(Mn1−xFex)PO4 positive electrode material can be represented by the equation below. The radius of the Fe ion is smaller than that of Mn ion; thus, the introduction of Fe reduces the lattice spacing and helps to boost the diffusion channel of lithium-ion in LiMnPO4.
| charge: Li(Mn1−xFex)PO4 − yLi − ye− → y(Mn1−xFex)PO4 + (1 − y)Li(Mn1−xFex)PO4 |
| discharge: (Mn1−xFex)PO4 + yLi + ye− → yLi(Mn1−xFex)PO4 + (1 − y)Li(Mn1−xFex)PO4 |
In order to gain a detailed understanding of the effect of Fe introduction on the crystal construction of LiMnPO4, the Li(Mn1−xFex)PO4 doping system of LiMnPO4 expanded supercell was established using the VESTA modeling software. In order to prevent doping atom aggregation, the model was constructed using the random dispersion doping principle, with the proportions of Fe and Mn set at 1:23, 1:11, 1:7, and 1:3 (Fig. S1, ESI†). Table S2 (ESI†) shows the mean bond length of Li–O, Mn–O, P–O and Fe–O bonds of atoms near the center of the Li(Mn1−xFex)PO4 crystal and the changes in the O–P–O bond angles that make up PO4 tetrahedron. After Fe doping, P–O bond length and O–P–O bond angle do not change significantly, indicating that the main structure of the crystal composed of PO4 tetrahedron changes little, and Li(Mn1−xFex)PO4 still retains the same skeleton structure of the lattice as that of the raw material LiMnPO4, with a good structural stability. In contrast, Mn–O bonding is shorter than that of Fe–O bonding; a slight change in the lattice constant after doping may be caused by this. Changes in the bond length and bond angle cause the orthogonal structure to shrink inward or outward, which can change the spread of Li+, thereby affecting the electrochemical performance of the material in question. Therefore, doping may improve the electronic conductivity of the LiMnPO4 electrode, and only doping LiMnPO4 with appropriate Fe concentration can enhance the electrochemical performance of the material. Compared with the undoped Li–O bond, the bond length increases after doping, stating that the formed binding energy between lithium ion and oxygen ion decreases after doping, which favors the de-intercalation of Li+ and, to a certain extent, the spread rate of Li+ is increased. With an Fe doping level of 1/4, the Li–O of the biggest sample, LiMn3/4Fe1/4PO4, is more favorable to lithium-ion de-intercalation in principle. This lowers the activation energy involved in the insertion and removal of lithium ions and effectively increases the electrical conductivity.
The process of Li+ migration can be regarded as an inverse process of Li vacancy migration; when calculating Li+ migration in VASP software, the formation energy of Li vacancy (denoted as E1) should be calculated first, and then Li ion should be placed in the middle position of the Li migration process to find the maximum formation energy E2 through transition state search, and then the diffusion barrier ΔE = E1 − E2, which is the difference in formation energy between the two calculations. Studies have shown that when lithium ions in the olivine structure LiMnPO4 (Pnma) are diffused along the b axis, the migration barrier in the diffusion process is the smallest,25 and its diffusion path is a 1-dimensional channel.
To gain insight into the charge transfer in the bonding process of Li(Mn1−xFex)PO4 materials’ binding process after Fe doping, Table S3 (ESI†) shows the average net charge of atoms in the expanded Li(Mn1−xFex)PO4 system. The initial valence electron configurations of Li, O, P, Fe and Mn atoms in the material are as follows: 2s1, 2s22p4, 3s23p3, 3d64s2, 3d54s2; the outermost electron numbers of Li, O, P, Mn and Fe elements are respectively taken as 1, 6, 5, 7 and 8. The net charge numbers of Li, P, Mn and O in the Li(Mn1−xFex)PO4 are close to those of the initial LiMnPO4 material at each doping amount. It is obvious that the net charge number of Fe atom is larger than that of Mn atom in Fe doped material, indicating that Fe atom transfers more electrons. The atom that forms a covalent connection with O will have a higher covalent component and a larger net charge (Mulliken atomic charges) due to the larger charge transfer. It can be observed from Table S3 (ESI†) that the average net charge of P element in LiMn3/4Fe1/4PO4 was the highest at 2.5872 (e), indicating that it formed the most stable P–O bond in the Li(Mn1−xFex)PO4 system.
Fig. 5(a) renders the total XPS spectra of LiMn3/4Fe1/4PO4/C particles, confirming the presence of the elements Mn, Fe, P, O, C and Li elements in the sample, which is in agreement with the analysis of the EDS face scan energy spectrum analysis below. The characteristic peak corresponding to the C-1s map in Fig. 5(b) verifies the existence of C in the sample. Fig. 5(b) show the corresponding characteristic peaks in the P-2p and O-1s maps. P-2p peaks at about 133.3 eV and O-1s peaks near 529.3 eV and 533.25 correspond to lattice oxygen and surface oxygen, where surface oxygen is formed by oxygen vacancies 1s. The lattice O of the orbital interacts with the P element of the 2p orbital to form the PO43− ion.
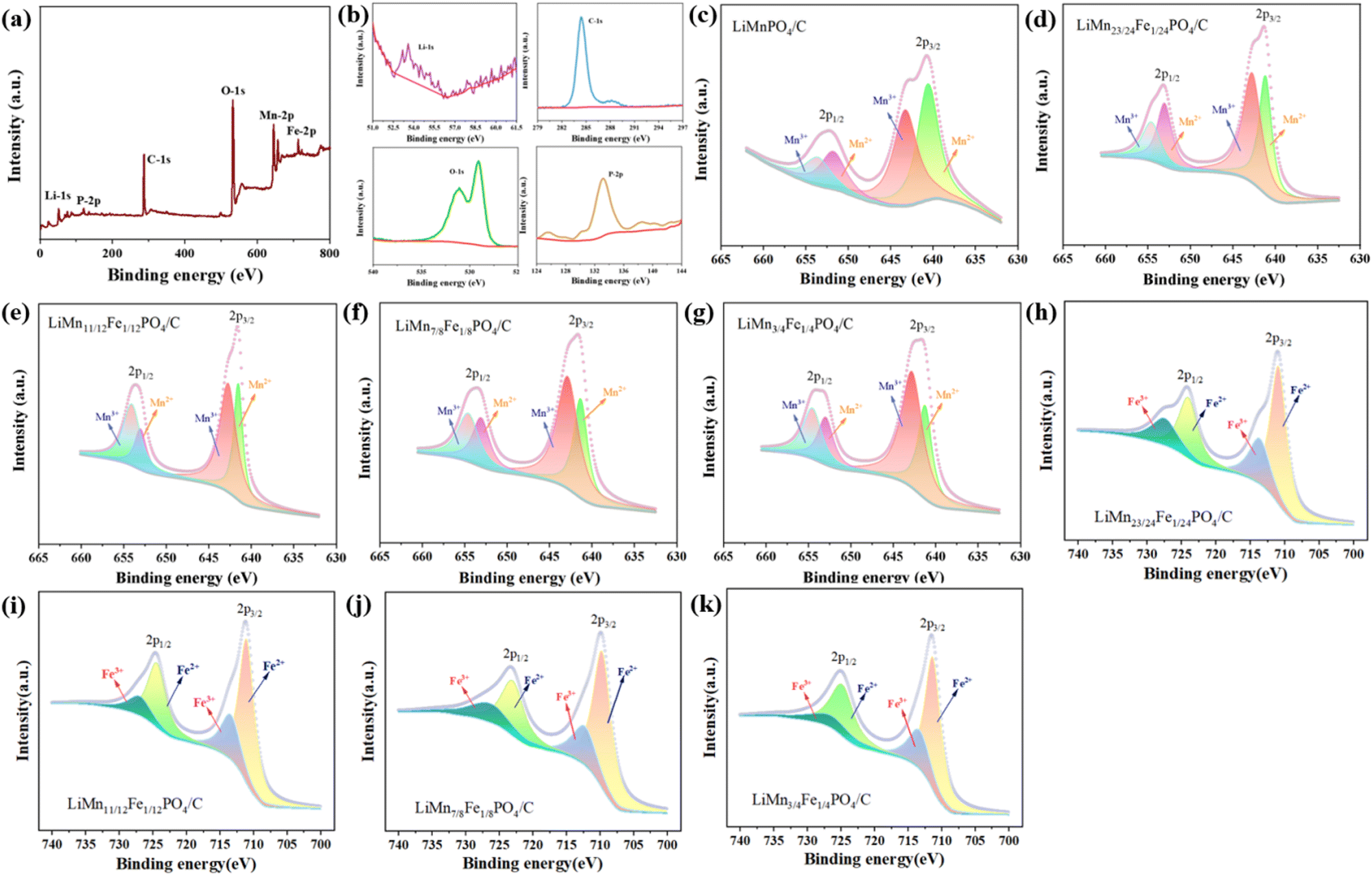 | ||
| Fig. 5 XPS spectra of LiMn3/4Fe1/4PO4/C composites: (a) total energy spectrum; (b) the Li element in the 1s orbital, the C element in the 1s orbital, the P element in the 2p orbital and the O element in the 1s orbital; (c)–(g) Mn 2p orbital XPS profiles of the Li(Mn1−xFex)PO4/C sample; (h)–(k) Fe 2p orbital XPS profiles of the Li(Mn1−xFex)PO4/C sample. | ||
Mn3+ is a d4 configuration with a high spin t32g(↑) e1g(↑) state and is a typical Jahn–Teller active ion, which has also been found in other transition metal chemical compounds such as LiMnO2 and LiNiO2.26 The Jahn–Teller influence can bring about the bulk and cell deformation of the electrode and ultimately cause the electrode to deteriorate mechanically.27 Moreover, Mn3+ will cause Li(Mn1−xFex)PO4/C crystal particles to become relatively large; nevertheless, too much Mn3+ will cause O defects, which will alter the material's electrochemical properties. Hence, the existence of Mn3+ ions in Li(Mn1−xFex)PO4/C materials is detrimental to the material stability in the process of charge and discharge.
To further analyse the form of Mn presence and chemical state in the Li(Mn1−xFex)PO4/C composite electrodes, fine-spectrum XPS tests were carried out on samples with different Fe abundances in the Mn-2p region. As can be seen in Fig. 5(c)–(g), the hybrid characteristic peaks of Mn3+ and Mn2+ are well separated on Mn-2p3/2 and Mn-2p1/2 orbits. Using the XPSPEAK41 software, the least square method is used to perform peak splitting fitting of the Mn-2p spectral peaks on the surface. In the XPS spectrum, Mn-2p3/2 and Mn-2p1/2 orbitals have been used to simulate two characteristic peaks, which indicate that Mn exists in the shape of Mn3+ and Mn2+. As can be seen in Fig. 5(h)–(k), the hybrid characteristic peaks of Fe2+ and Fe3+ are well separated on Fe-2p3/2 and Fe-2p1/2 orbitals. Peak splitting fitting of the Fe-2p spectral peaks on the surface is carried out using the least square approach and the XPSPEAK41 program. Fe-2p3/2 and Fe-2p1/2 orbitals have been employed to simulate two distinctive peaks in the XPS spectrum that take the forms of Fe3+ and Fe2+, respectively, and which serve as indicators that Fe is present. In the Li(Mn1−xFex)PO4/C material, the energy of the orbital is less than that of the pure phase LiMnPO4/C. Subsequent investigation showed that doping altered the peak area and half-peak width of Mn3+ and Mn2+, which is considered to have caused variations in the relative concentrations of Mn3+ and Mn2+ in the Li(Mn1−xFex)PO4/C electrode. The content of Mn2+ was estimated by the ratio of the peak areas of Mn2+/(Mn2+ + Mn3+) in the Mn 2p3/2 and Mn 2p1/2. It is not difficult to see that the Mn2+ decreased content gradually increases with increasing amount of Fe doping. Mn2+ makes the relatively large Li(Mn1−xFex)PO4/C crystal particles. Thus, the higher the iron content, the smaller the volume of the material, which is accordance with the further refinement of XRD in Fig. 4. As a result, the LiMn3/4Fe1/4PO4/C material performs excellently.
The SEM topography and the EDS of the Li(Mn1−xFex)PO4/C are depicted in Fig. 6. The morphology of the Li(Mn1−xFex)PO4/C system is primarily spherical accumulation, with numerous small particles attached above, forming materials with different grain sizes. The spherical Li(Mn1−xFex)PO4/C electrode has a large specific superficial area, which can offer more electrochemistry activated sites for the fast de-intercalation of Li+, effectively diminishing the concentration polarization. There was no significant change in the grain size with the increase in Fe content. The four synthetic samples have similar grain shapes and are spheroid and small in size. The complete removal of Li+ from the electrode material and the full release of material capacity are ensured by the fine dispersion of the lithium positive material, which additionally enables the material to fully permeate the electrolyte and ensures its wettability. Careful observation states that the crystalline size of the LiMn3/4Fe1/4PO4/C material seems to be finer, and the fine uniform electrode material makes for the rapid removal of lithium ions and the improvement of electrochemical performance. Moreover, it is observed that the material has a multistage layered structure consisting of micropores and macrospores, which can supply enough cushion area for the change of volume produced by the lithium insertion and removal process, improve the structure stability of material and thus promote its cyclic stability. As can be seen from Fig. S4 (ESI†), manganese, iron, oxygen, phosphorus, and carbon on the surface of the material are evenly distributed throughout the measured samples and are in agreement with the SEM morphology of the material. Additionally, when Fe doping increases, Mn content also gradually drops, and both elements’ atomic percentages are essentially consistent with the doping ratio. Roughly speaking, the atomic percentage of each element (At%) is line with the ratio of LiMn3/4Fe1/4PO4/C; in particular, the ratio of manganese and iron is synthesized in strict accordance with 3:1. It is also possible to expose LiMn3/4Fe1/4PO4/C composite to be tightly and evenly enclosed by amorphous carbon layers of formless construction. A homogeneous carbon layer is formed by the carbonation of ascorbic acid during high temperature roasting. The carbon layer can direct more electrons into the periphery of the active material, allowing it to smoothly enter the lithium-ion entrapment process. More importantly, the dispersed amorphous C layers at the periphery of the LiMn3/4Fe1/4PO4 grains are interconnected to form a three-dimensional network structure, which can effectively inhibit the agglomeration of LiMn3/4Fe1/4PO4 grains, facilitate fast electron transport, and allow the ability of the electrode material to be completely released.
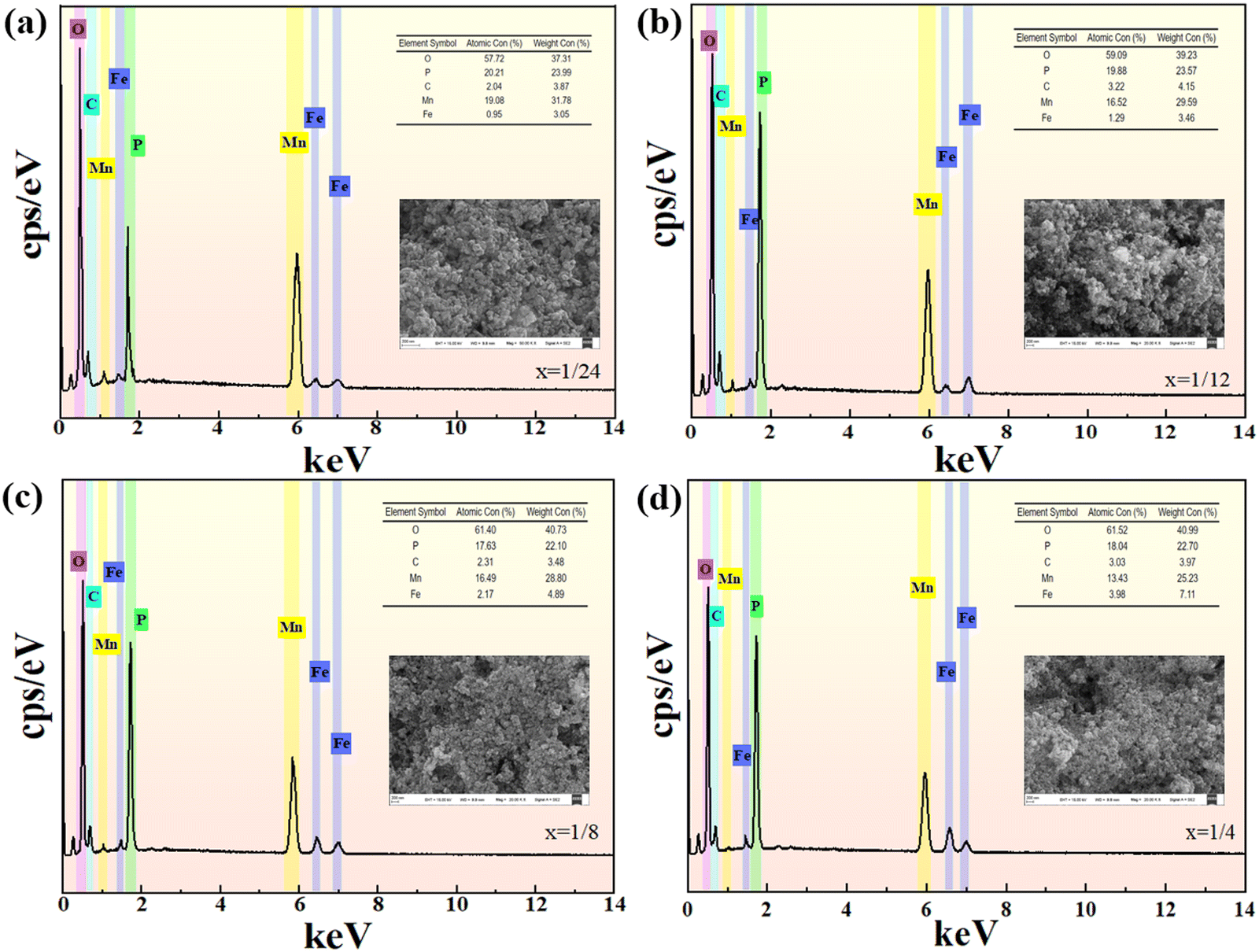 | ||
| Fig. 6 The SEM images and EDS spectra of Li(Mn1−xFex)PO4/C. | ||
The micro-crystal structure of the LiMn3/4Fe1/4PO4/C cathode material is displayed in Fig. 7. The transmission electron microscopy (TEM) images of the grains demonstrate that they are polished on the particle surface and have high crystallinity (Fig. 7(a)). Fig. 7(b) depicts an image created using the fast Fourier transform (FFT). LiMn3/4Fe1/4PO4/C has an approximate plane spacing (adjacent lattice fringe spacing) of 3.73 Å, which is equal to the d values of the (101) plane on the LMFP material standard card. LiMn3/4Fe1/4PO4/C has clear and continuous lattice fringes and smooth grain edges, which correspond to the XRD results and represent the sample's good clarity. The area of the particles used in SAED analysis is depicted in Fig. 7(c). The selected region electron diffraction illustrates both the polycrystalline nature of the fabricated cathode materials and the decrease in the ring diffraction patterns of the polycrystalline samples. The hkl graphs of the subsequent LMFP composites, respectively, correlate to the observed notable ring patterns: (101), (200/121), (211), and (312).
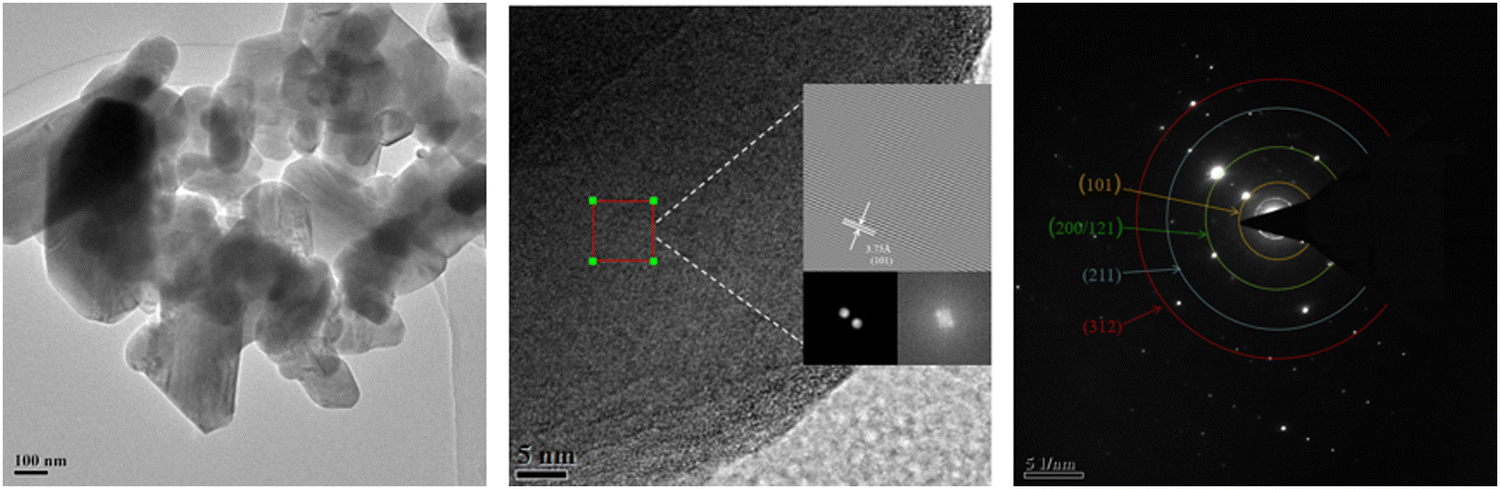 | ||
| Fig. 7 (a) TEM image, (b) HRTEM image, and (c) SAED pattern of LiMn3/4Fe1/4PO4/C cathode materials. | ||
3.2 Electrochemical performance
Fig. 8(a) states the initial charge–discharge curve of the Li(Mn1−xFex)PO4/C composite material at 1C, which is charged to a voltage of 4.5 V, then discharged to 2.5 V. Below this voltage, the charging and discharging platforms close to 4.2 V and 4.0 V respectively correspond to the lithium insertion and removal processes and to the oxidation and reduction potential of Mn2+/Mn3+, accompanied by the valence state change of the manganese ion. The platform capacity improves inch by inch with the enhancement of the iron doping content. As can be described in the figure, each material displays a better specific discharge capability. The discharge capacities of LiMn3/4Fe1/4PO4/C, LiMn7/8Fe1/8PO4/C, LiMn11/12Fe1/12PO4/C, LiMn23/24Fe1/24PO4/C and LiMnPO4/C are respectively 141.6, 135.7, 131.1, 123.8 and 113.3 mA h g−1, and these values are higher than that of the pure phase material, indicating that introducing a proper amount of Fe can indeed enhance the electrochemical performance of the material. This is because the redox reaction has good reversibility, which means that it has good dynamic characteristics and greatly enhances the specific discharge capacity at a high voltage. It also has minimal polarization phenomenon when the material acts as the electrode. Finally, the underlying discrepancy between the charge and discharge plateaus of the electrodes at this doping amount is minimal. Furthermore, the presence of Fe also enhances the spread of Li+, thereby significantly enhancing the conductivity of the material. It is worth mentioning that the LiMn3/4Fe1/4PO4/C sample has a more stationary and longer discharge voltage plateau with the best discharging capacity, indicating the excellent rate performance potential.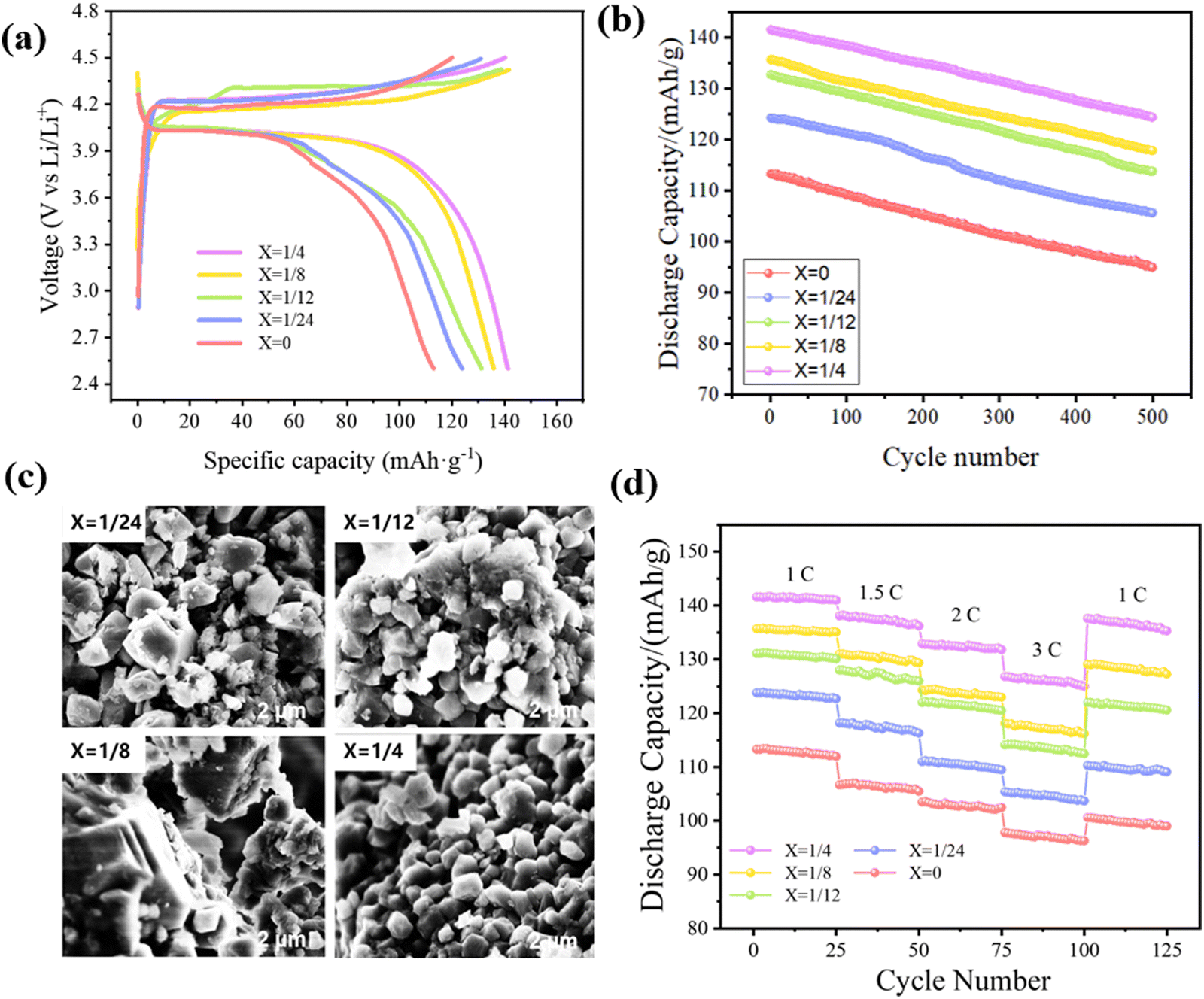 | ||
| Fig. 8 (a) The initial charge and discharge curves of Li(Mn1−xFex)PO4/C at 1C. (b) The cycling performance of the Li(Mn1−xFex)PO4/C sample at 1C. (c) SEM images of Li(Mn1−xFex)PO4/C after 500 cycles. (d) The rate capability of the Li(Mn1−xFex)PO4/C sample. | ||
500 cycle tests were performed on Li(Mn1−xFex)PO4/C electrode materials with different amounts of Fe doping at 1C, as evidenced from Fig. 8(b). During the cycle, the specific discharge ability of each electrode was mildly reduced. As the addition of Fe increases, the specific discharge ability of the material progressively increases. The presence of Fe has improved the intrinsic electroconductibility of Li(Mn1−xFex)PO4/C materials; thus, the recycling properties of materials have also been improved. After 500 cycles of charge and discharge, the discharge capacities of LiMnPO4/C, LiMn23/24Fe1/24PO4/C, LiMn11/12Fe1/12PO4/C, LiMn7/8Fe1/8PO4/C and LiMn3/4Fe1/4PO4/C were 107.0, 118.4, 127.0, 132.2 and 138.6 mA h g−1, and the capacity retention rates were 94.44%, 95.64%, 96.87%, 97.42% and 97.88%, respectively. Clearly, LiMn3/4Fe1/4PO4/C has superb cyclic properties and a high invertible capacity, and the attenuation of capacity is minimal. In addition, it can be seen from Fig. 8(c) that after 500 battery cycles, the particles of x = 1/4 of the material are relatively complete and uniform, indicating that the LiMn3/4Fe1/4PO4/C material structure is more stable.
The rate performance of the electrode is critical for LIBs. The rate performance curve for the Li(Mn1−xFex)PO4/C cathode materials is described in Fig. 8(d). Each sample is within the 2.5–4.5 V voltage range. The power was then sequentially loaded at 1C, 1.5C, 2C, and 3C ratios for 25 weeks, and then it went back to the original 1C ratio. As illustrated in the diagram, with the gradual improvement in the discharging rate, the discharging capacity of the Li(Mn1−xFex)PO4/C electrode is a step-like attenuation, which is a common phenomenon of LIB. After 125 weeks of rate testing, the recovery capacities of LiMn3/4Fe1/4PO4/C, LiMn7/8Fe1/8PO4/C, LiMn11/12Fe1/12PO4/C, LiMn23/24Fe1/24PO4/C and LiMnPO4/C is respectively 135.3, 127.3, 120.6, 109.1, and 99.0 mA h g−1 at 1C. As a consequence, the LiMn3/4Fe1/4PO4/C electrode performs noticeably better than other materials under the same circumstances, with better structural integrity and reversibility, and it displays a reasonably excellent discharge capacity at any discharge current density. This is due to the fact that the proper doping of Fe enhances the electron/ionic electroconductibility of the material, promotes redox activity, and then alleviates the structural slumping created by the instability of the removal phase of Mn3/4Fe1/4PO4/C in the charging–discharging process of the LiMn3/4Fe1/4PO4/C nano-electrode. It is worth mentioning that the doping amount of Fe is higher than that of pure LiMnPO4/C material even at 1/24, thus confirming that the entrance of Fe can indeed enhance the discharge capability of the material.
In order to understand the reason why transition metal Fe doping improves electrochemistry, we start from the electronic construction of the Li(Mn1−xFex)PO4 system, taking the comparison of the amount of occupied states near Fermi as the starting point and making the analysis of the total state density (TDOS) of spin polarized supercell systems and the wave state density (PDOS) of Li-s, Li-p, Fe-4s, Fe-3p, Fe-3d, Mn-4s, Mn-3p, Mn-3d, P-2s, P-2p, O-2s and O-2p in Li(Mn1−xFex)PO4 materials.
Fig. 9(a) exhibits the total state density of the Li(Mn1−xFex)PO4 system at each doping amount. From the figure, it can be observed that the total state density shifts to the low energy in the presence of iron doping, resulting in the conduction band passing through the Fermi appearance at the bottom, which in turn results in an increase in the number of conduction bands. Since the number of valence electrons is equal to the integral of the horizontal and vertical axes with respect to the density of states, the larger the supercell, the greater the total number of valence electrons, and hence the larger the ordinate. In this paper, the supercell with each doping amount contains 192 atoms, which is a large model; thus, the vertical coordinates are −250–250 states per eV. The Li(Mn1−xFex)PO4 system of total state density data including energy, spin up states and spin down states, by looking for energy values near the Fermi level (0), an energy difference between a maximum occupied state (HOMO) and a minimum unoccupied state (LUMO) can be used to determine a material band gap at each doping amount. Li(Mn1−xFex)PO4 material bandgaps of x = 1/24, 1/12, 1/8 and 1/4 are 3.586 eV, 3.525 eV, 3.515 eV and 3.503 eV, respectively. The band gap is related to the difficulty of electron transition in the material: the smaller the band gap, the less energy is needed for electron transition, and more easily the electron transition will be; thus, the conductivity can be summarized as x = 0 < x = 1/24 < x = 1/12 < x = 1/8 < x = 1/4. This may be due to the fact that the conductivity of Fe should be better than that of Mn; thus, the more Fe in the system, the more the occupied states near the Fermi level.
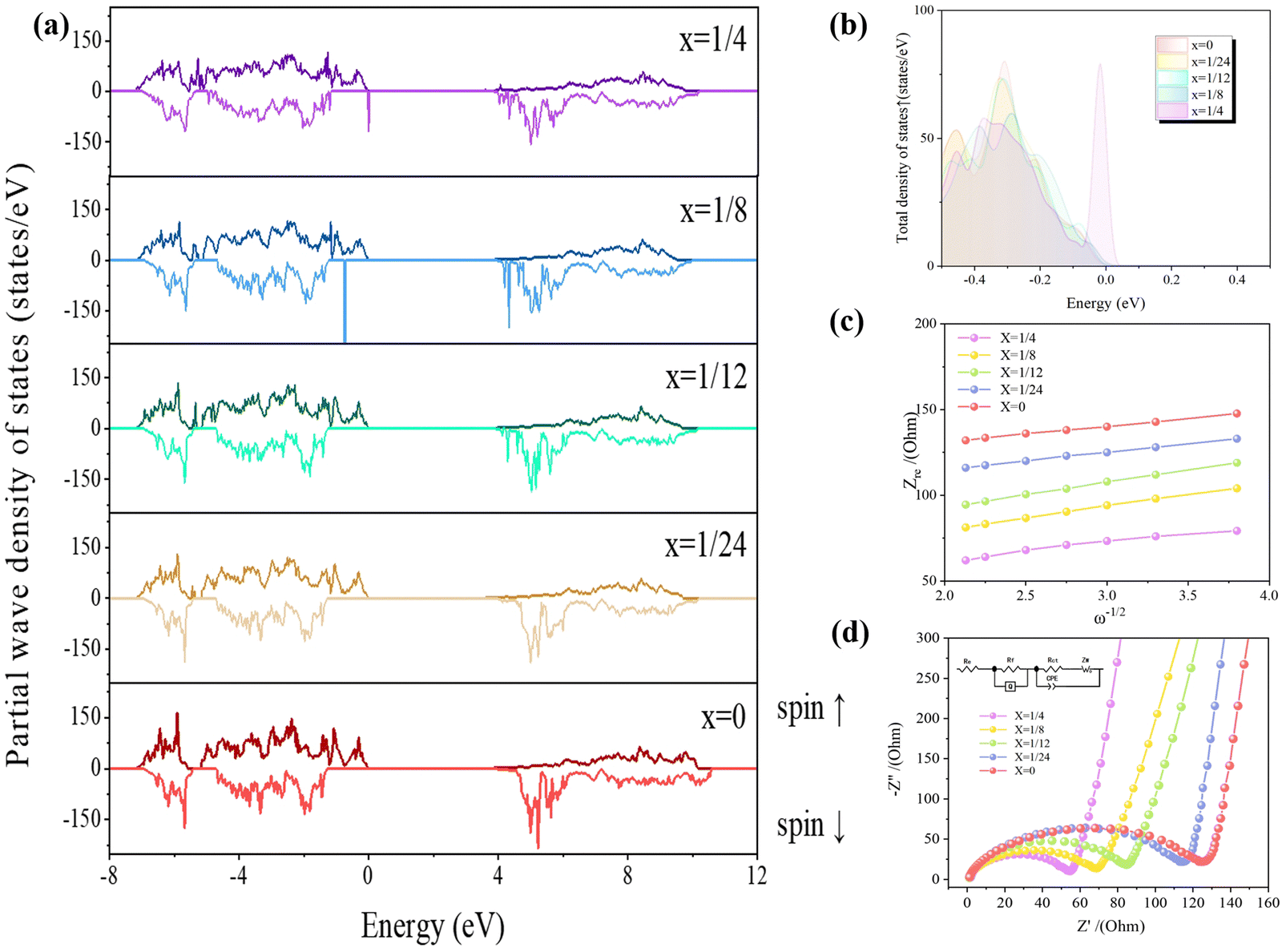 | ||
| Fig. 9 (a) Total density of states map of the Li(Mn1−xFex)PO4 system (drawing from the comparison of the amount of occupied states near the Fermi level). (b) Comparison of total density of states (TDOS) of Li(Mn1−xFex)PO4 systems near the Fermi level. (c) Nyquist impedance spectroscopy of Li(Mn1−xFex)PO4/C cathode materials (illustration shows the corresponding equivalent circuit). (d) Linear matching of low frequency between Zre and ω−1/2 to Li(Mn1−xFex)PO4/C. | ||
In order to illustrate the optimal doping amount of the Li(Mn1−xFex)PO4 system, Fig. 9(a) has been detailed to give a comparison of the total state density in the spin up near the Fermi level [−0.5 eV, 0.0 eV], as shown in Fig. 9(b). According to the thickness of states near the Fermi level, the best doping quantity can be judged theoretically. The closer the electrons are to the Fermi level, the easier they transition, the more occupied states they have, and the better their conductivity. From Fig. 9(b), it can be seen intuitively that [0.0 eV] corresponds to Fermi level, moving from [0.0 eV] to the negative direction; the material that is encountered first will indicate the material that has more occupied states. It can be found that the total state density of the LiMn3/4Fe1/4PO4 crystal is closest to the Fermi level [0.0 eV], indicating that more electrons in the valence band of the material can be used to transition to the conduction band at this doping amount; thus, its electrical conductivity is the best.
Fig. S4 (ESI†) shows the split-wave state density of atoms in the s, p and d orbitals in LiMnPO4, it can be seen that the total state density is mainly contributed by the Mn-3d state sum, O-2p state, P-2p state, Li-p state and Li-s state, while the Mn-3p state, Mn-4s state, P-2s state and O-2s state hardly contribute to the total state density. The valence states are the recombination of the d orbitals of manganese and p orbitals of oxygen. The low guidance state is mainly the Mn-d state, while the high guidance state is mainly the P-p and O-p states. Most of the spins in the Mn-d state are fully filled but only a small number of spins in the Mn-d state exist at the leading edge. The resonance between the Mn-3d and P-2p orbitals forms stable covalent bonds of Mn–O and P–O. The s orbitals of the O and P atoms are located in the low energy region. In addition, the valence states near Fermi level [−0.50 eV, 0.00 eV] are mainly Mn-3d states (corresponding to low-energy bonding orbital t2 state) and O-2p states, while the conduction bands [2.35 eV, 5.49 eV] are mainly Mn-3d states (corresponding to high-energy bonding orbital e state) and Li-p states. The oxidation state of manganese atom in LiMnPO4 is trivalent, and the high-spin channel has 4 electrons with an electron configuration of (t2g)3(eg)1. When the LIB is in the process of charging or discharging, the oxidation state of manganese ion alternates between Mn3+ and Mn4+. The energy spectrum of Mn-3d is between 8 eV, smaller than the Fermi level, and about 12 eV, larger than the Fermi level. In the final analysis, the position of the transition metal Mn-3d state primarily determines the electrical conductivity of the LiMnPO4 crystal. As a result, this is a significant source for thinking about the idea of using Fe transition metal element in the site doping of the LiMnPO4 crystal in order to enhance the electrical conductivity of the LiMnPO4 material. To get a better understanding of the contribution of Fe doping to the electronic construction of the crystal, we respectively calculated the density of atomic states in the material with Fe doping amount of 1/24, 1/12, 1/8 and 1/4, as shown in Fig. S5–S8 (ESI†), respectively. In the Li(Mn1−xFex)PO4 doping system, we can see that the crystal electron structures are mainly the Fe-3d, Mn-3d, O-2p, P-2p, and P-2s orbitals, showing strong localization. Li-s states and Li-p states have the weakest contribution to the valence band. After Fe replaces some Mn in LiMnPO4, the Fe-3d state appears at the maximum valence band and pushes the unoccupied Mn-3d spin down band to move to the low energy region. Part of the state density of the Fe-3d orbital is still occupied by electrons at the Fermi level; thus, Fe mainly exists in the form of Fe2+ in the Li(Mn1−xFex)PO4 system. The presence of the doped element Fe induces additional d states in the band gap of pure phase LiMnPO4; the Fe-d orbital of Li(Mn1−xFex)PO4 produces a few spins at the edge of the conduction band, which is responsible for the reduction of the LiMnPO4 band gap. When Fe is doped, a brand-new electronic valence state appears between the energy bands, which decreases the band gap and induces a new electron state in Fe-3d.
In the Li(Mn1−xFex)PO4 system, the initial valence electron configurations of lithium, oxygen, phosphorus, iron and manganese atoms are 2s1, 2s22p4, 3s23p3, 3d64s2 and 3d54s2, respectively. As can be seen from Fig. S5–S8 (ESI†), the energy band occupied by Mn-3d electrons in the samples with different doping amounts is divided into two parts, one is from −6.0 eV to the Fermi level, and the other is from 4 eV to 8.0 eV. The band occupied by Fe-3d electrons is divided into two parts, one near −5 eV and the other between 4 eV and 6.0 eV. The bands occupied by the O-2p electrons are concentrated between −7.0 eV and the Fermi level; the energy bands of P-2p and P-2s are mainly between −7.0 eV and −5.0 eV. In the Li(Mn1−xFex)PO4 doped system, the valence states of Li, O, P, Mn and Fe are respectively +1, −2, +5, +3 and +2. The outermost electron configurations of Fe and Mn are respectively 3d64s2 and 3d54s2, and their d electron numbers are respectively 6 and 5. All five d electrons of Mn are filled in the spin up part, while Fe has five d electrons in the spin up part and one d electron in the spin down part, and both Mn3+ and Fe2+ exist in high spin states.
In the Li(Mn1−xFex)PO4 doped system, spin-up and spin-down electronic states appear in both the valence band and conduction band. With the increase in the doping amount, due to the increasing contribution of the Fe-3d orbital, the spin-down electron state density peak of the Fe-3d orbital valence band near the Fermi level [−0.50 eV, 0.00 eV] increases. The peak value of the electron state density of the Fe-3d orbital conduction band [4.38 eV, 6.24 eV] is also increasing, and the change is increasingly obvious. The Mn-3d state density peak in the Li(Mn1−xFex)PO4 doping system shows a little downward trend as doping increases, but there is essentially no change in the state density of other electrons or the comparable electron state density prior to doping.
To sum up, in LiMnPO4 materials, all O-2p electrons participate in the valence band formation near the Fermi level in different degrees, the conduction electrons near the Fermi level are composed of Mn-3d and O-2p hybrid electrons, and Mn-3d plays a major role. In LiMn1−xFexPO4, the conduction electrons near the Fermi level are composed of Fe-3d, Mn-3d and O-2p hybrids, Fe-3d and Mn-3d play a major role, but only O-2p electrons near Fe-3d atoms are hybridized, which further explained that the energy gap of Li(Mn1−xFex)PO4 materials is determined by Fe-3d and Mn-3d; there is O-2p electron and Fe-3d, Mn-3d electron hybridization phenomenon. In doped materials, electrons near the Fermi level are primarily made up of Fe-3d electrons.
Electrochemical impedance spectroscopy (EIS) can dynamically monitor the reaction of Li+ and the diffusion of Li+; it is one of the most important measuring tools for the analysis of the reaction dynamics of embedded Li+. Fig. 9(c) reveals the Nyquist diagram of Li(Mn1−xFex)PO4/C positive material after 25 circulations of charge and discharge at 1C. Every sample consists of diagonal lines in the low-frequency domain and a concave semicircle in the high-frequency domain. Electrochemical polarization occurs in the high-frequency space, which is controlled by the reaction dynamic of the electrode, corresponding to the transfer of charge. The low-frequency region is dominated by the spread course of reagents or products in the electrode reactivity, and the concentration polarity phenomenon appears.28
In the Zsimpwin program, the following equivalent circuit is used to fit the original data.
| Re + (Rf‖Q) + [(Rct + Zw‖CPE)] | (1) |
The Li(Mn1−xFex)PO4/C positive material consists of numerous interfaces and boundary of the phase. Strictly speaking, the centre of the high frequency half-circle in the Nyquist diagram is located beneath the Z′ axis, i.e., it becomes an arc of the circle, and there is a phenomenon of a decrease in the semicircle. The reason of half-circle rotation is concerned with the inhomogeneity of the electrode/electrolyte interface, which is most likely because the roughness of the electrode surface causes a change in the capacitance of the double layer, which leads to the fact that the charge shift resistor at points in the plane is no longer a number.34 The Ohm impedance Re of LiMn3/4Fe1/4PO4/C has the smallest intercept in the Z′ direction at high frequency. Furthermore, the semicircular diameter of this material is also the smallest in the high-frequency zone, which means that the LiMn3/4Fe1/4PO4/C material is hopeful for breaking through the dynamic limit and has the best charge transfer capacity so as to obtain bigger discharging level and better rate ability.35
All corresponding data obtained through equivalent circuit matching method are given in Table S4 (ESI†). The Re values of each positive electrode are all low, manifesting that the electrolyte (1.0 M LiPF6 in EMC:DMC:EC = 1:1:1 vol%) can well realize the transfer of Li+ between the positive and negative active materials. As can be seen, for all Li(Mn1−xFex)PO4/C positive terminals, the Rct are much larger; thus, the Rct is a significant index to evaluate the charge shift resistance. In addition, a smaller Rct can speed up the electron transfer and the insertion/removal within the interface between the active material and the electrolyte. It is obvious that the order of Rct values of diversiform materials is LiMnPO4/C < LiMn23/24Fe1/24PO4/C < LiMn11/12Fe1/12PO4/C < LiMn7/8Fe1/8PO4/C < LiMn3/4Fe1/4PO4/C, consistent with the order of cycle and rate performance.
All composite samples have linear slopes greater than 45° in the low frequency space, which suggests that they have a certain capacity, which is also a feature of fast Li+ in-electrode diffusion. It can be seen from Fig. 9(c) that the Warburg component gradient of the SEI spectrum of LiMn3/4Fe1/4PO4/C is large at low frequencies, and it is noted in Table S4 (ESI†) that the electrode interface of the LiMn3/4Fe1/4PO4/C electrode has the highest double layer capacitance, which indicates that its electrochemical capacitance is higher, resulting in the highest Li+ conduction capacity in its interior.36 The analysis results are in agreement with the charge/discharge curve in Fig. 8(a), which proves to be a significant factor for improving the discharge ability.
The spread coefficient of Li+ (DLi+) may be computed in terms of the following eqn (2),37 and the calculation results are shown in Table S5 (ESI†). The results exhibit that the LiMn3/4Fe1/4PO4/C cathode electrode has the largest DLi+, an order of magnitude larger than other electrodes. Therefore, the LiMn3/4Fe1/4PO4/C material is more conducive to the removal and embedding of Li+.
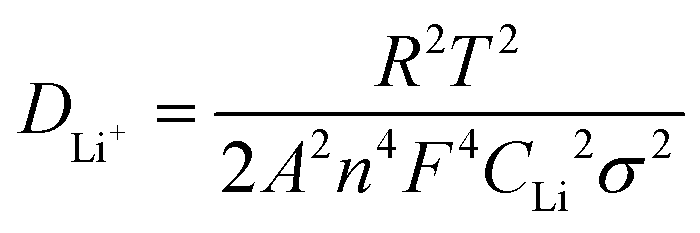 | (2) |
In the above equation, R – ideal gas constant, whose value is 8.314 J (mol K)−1; T – Kelvin temperature, which is equal to 298 Kelvin; A – surface area of the electrode surface active material, A = 0.64 cm2 in the current test; n – the quantity of electrons transferred in oxidation or reduction reactions, n = 1 in this experiment; F – Faraday's constant, which is 96485 C mol−1; CLi – concentration of lithium ion in the material of the electrode, CLi = 0.0227 mol cm−3 in the current test; σ – Warburg's impedance factor, determined by eqn (3), is in accordance with the gradient of Zre in Fig. 9(d).
| Zre = Re + Rf + Rct + σω−0.5 | (3) |
In addition, the exchange current density I0 in eqn (4) is also an important parameter for describing the dynamics features, and it can be employed to depict the electrochemical activity of the electrode material,38 as shown in Table S5 (ESI†). The results showed that the exchange current density of LiMnPO4/C was enhanced by Fe doping, and the cathode electrode of LiMn3/4Fe1/4PO4/C was found to have the highest I0. This is due to the fact that Fe has a higher conductivity than Mn; the more Fe in the system, the more occupied states are near the Fermi level, wherein the grain limit resistance and the charge shift resistance can be reduced effectively, the lithium ions can be transported quickly, and the exchange current density can be increased, which positively impacts the specific capacitance of the sample. To sum up, Fe = 1/4 is the ideal doping amount for preparing lithium ion battery Li(Mn1−xFex)PO4/C cathode.
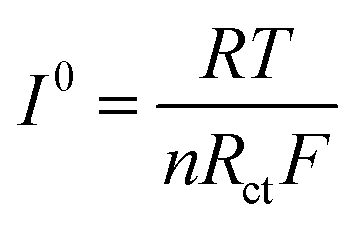 | (4) |
In the above equation, R – the ideal gas constant, whose value is 8.314 J (mol K)−1; T – Kelvin temperature, which is equal to 298 Kelvin; n – the quantity of electrons shifted in oxidation/reduction reactions, n = 1 in this experiment; Rct – Li+ charge shift resistance between the active electrode material and the electrolyte; F – Faraday's constant, which is 96485 C mol−1.
Fe introduction will increase the number of energy levels in the conduction band of the LiMnPO4 material. It is discovered that the width of the band gap may be effectively reduced based on the differentiation between the conductor and the insulator as well as the energy band conclusion; the more closely the material is aligned with the conductor, the narrower the band gap and the better the conductivity. The band structure shown in Fig. S9 (ESI†) explains why the Fe-doped Li(Mn1−xFex)PO4 system has excellent electrical conductivity. In x = 1/24, 1/12, 1/8 and 1/4 doping gradients, the introduction of Fe decreases the conduction band of LiMnPO4 and the valence band shifts to the Fermi level, resulting in the phenomenon that the band gap of LiMnPO4 decreases gradually. Therefore, the addition of a few Fe atoms to replace Mn atoms in the intrinsic system of LiMnPO4 can increase the electrical conductivity. The band gaps of Li(Mn1−xFex)PO4 of x = 1/24, 1/12, 1/8 and 1/4 are respectively 3.585 eV, 3.523 eV, 3.519 eV and 3.505 eV, which are basically consistent with the results of the total state density map of the system, as shown in Fig. 9(a). When Fe is 1/4, the gap width is the smallest; thus, 1/4 is the optimum concentration. In addition, when x = 1/4, the energy band fluctuation around the Fermi step of the doped material is obvious, and the number of energy bands around the Fermi step is large, showing that the effective mass of electrons in these energy bands is small, and the degree of non-localization is large, the atomic orbitals composed of this energy band have a good ability to accommodate electrons, which contributes to the transport of charge carriers; thus, the LiMn3/4Fe1/4PO4 crystal has good electrical conductivity.
The charge density difference is the difference between the superposition of the electron density of a crystal and the electron density of a neutral atom, it is beneficial to visualize the bonding properties. In Fe-doped systems, electrons are redistributed around the doped atoms. As depicted in Fig. 10(a), this paper compares the change in the electron density in space between Li2FePO4 and Li2Fe23/24Mn1/24PO4 materials after de-lithiation.
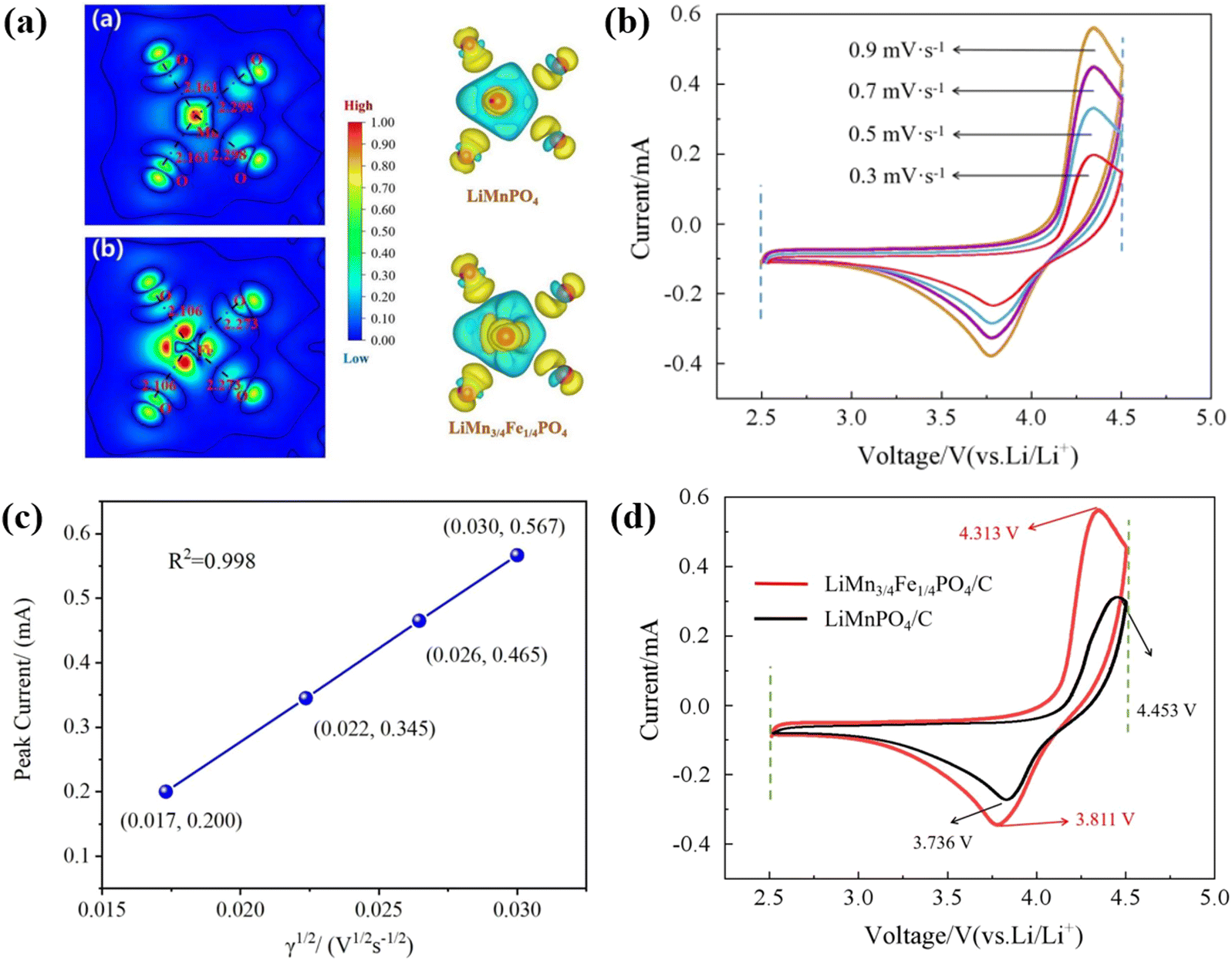 | ||
| Fig. 10 Iso-surface 3D contours of the charge density difference in the de-lithiation process and planar graphs centered on Fe or Mn atom (a) between LiMnPO4 and MnPO4 and (b) between LiMn3/4Fe1/4PO4 and Mn3/4Fe1/4PO4. (b) CV profiles of the LiMn3/4Fe1/4PO4/C material at a scanning speed of 0.3, 0.5, 0.7, and 0.9 mV s−1. (c) Linear fitting of the square root of the peak redox current and scanning rate of the LiMn3/4Fe1/4PO4/C cathode. (d) CV profiles of the LiMn3/4Fe1/4PO4/C and LiMnPO4/C electrode at a sweep speed of 0.9 mV s−1. | ||
In the three-dimensional diagram to the right of Fig. 10(a), the light blue part shows the electron-loss region (charge consumption) and the yellow part shows the electron-enriched region (charge accumulation). The results show that O is a negative ion, while Mn and Fe are positive ions. Furthermore, there are clear d-orbital characteristics near Mn and Fe ions (the electron distribution is shaped like a cross), and clear p-orbital characteristics near O ions (the electron distribution is shaped like a dumbbell). This indicates that the O 2p state can efficaciously coincide with the Mn and Fe orbital to form a covalent bond, which is in line with previous research results. The iso-surfaces of Δ = ρ[LiMnPO4] − ρ[MnPO4] and Δ = ρ[LiMn3/4Fe1/4PO4] − ρ[Mn3/4Fe1/4PO4] are given on the left plane of Fig. 10(a). The shape of the iso-surface is similar to that of the d(x2 − y2) orbitals, indicating that the missing electrons are located respectively in the Mn-3d(x2 − y2) and Fe-3d(x2 − y2) orbitals. Both the atomic projection state density and the charge difference density reveal that the Mn ion in LiMnPO4 has a high spin d4 configuration and a high spin t32g(↑) e1g(↑) state. In addition, since the transition metal (TM) in LiMn3/4Fe1/4PO4 is located in the center of the octahedron, it is possible to introduce the three-dimensional orbital splitting of the Mn and Fe transition metal ions into t2g and eg eigenstates; thus, there are representative transition metal Mn(3d-t2g)3 and (3d-eg)2, Fe(3d-t2g)4 and (3d-eg)2 states.
The mechanism of the band gap decrease is further explored by analyzing the orbital distribution of charge density difference. In LiMnPO4 materials, the charge of the highest occupied molecular orbital (HOMO) comes from the hybridization between Mn (3d-t2g) and O2p, and the lowest occupied molecular orbital (LUMO) comes from the hybridization of Mn (3d-eg) and O 2p. In the case of Fe introduction, the HOMO orbital charge is mainly from TM (3d-t2g) and the O 2p orbital contributes a little charge, whereas the LUMO orbital charge is due to TM (3d-eg). The doping of Fe changes the hybridization pattern of Mn and O atoms at the original location and the distribution pattern of the electron cloud of the surrounding O atoms. In the process of de-lithiation, when part of Mn in the LiMnPO4 material is replaced by Fe, Mn–O is replaced by Fe–O bond, with the introduction of Fe, the length of TM–O bond at the transition metal (TM) position in the process of de-lithiation of the LiMn3/4Fe1/4PO4 material is shortened and becomes more stable.
Fig. 10(b) records the cyclic voltammetry (CV) curves of the LiMn3/4Fe1/4PO4/C cathode material at 0.3, 0.5, 0.7 and 0.9 mV s−1, the tested start-stop voltages are respectively 2.5 V and 4.5 V. It is evident that the oxidation peak shifts somewhat towards high potential, the reduction peak shifts slightly towards low potential, and the peak current and area increase with increasing scanning speed. At each scan speed, the curves have good symmetry and redox peaks are evident, which further verify that this material has the best dynamic characteristics, which is in agreement with the aforementioned charge–discharge capability, cycle performance, rate performance, and impedance analysis. In addition, it is pointed out that the oxidation peak of the material appears at about 4.33 V, corresponding to the oxidation reaction of Mn3+ → Mn4+, i.e., the Li+ removal process in the LiMn3/4Fe1/4PO4 olivine structure. The reduction peak of the material appears at about 3.86 V, corresponding to the reduction reaction of Mn4+ → Mn3+, i.e., the Li+ embedding process in the LiMn3/4Fe1/4PO4 olivine structure.
To further determine the diffusion coefficient of Li+ from the LiMn3/4Fe1/4PO4/C cathode electrode, the relation between the peak current and the square root of sweep speed in its CV curve was fitted, as illustrated in Fig. 10(c). This shows that there is a good linear relationship between the peak current and the square root of scanning rate, and the value of R2 is very close to 1, confirming that the redox reaction is controlled by diffusion in LiMn3/4Fe1/4PO4/C materials. The diffusion coefficient of lithium ions from the positive electrode LiMn3/4Fe1/4PO4/C can be calculated as 4.26 × 10−11 cm2 s−1, which is indicative of its excellent kinetic performance, based on eqn (5) and the slope in Fig. 10(c).
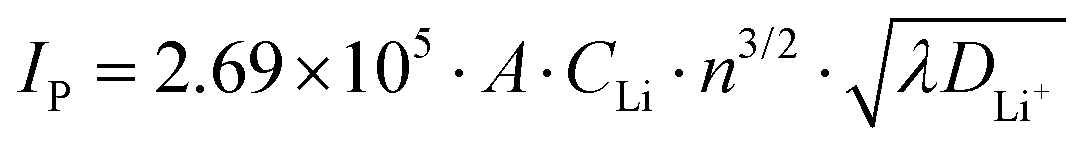 | (5) |
In the above equation, IP – peak current (mA); A – surface area of the electrode surface vibrant material, A = 0.64 cm2 in the current text; CLi – Concentration of Li+ in the material of the electrode, CLi = 0.0227 mol cm−3 in the current test; γ – scanning speed (mV s−1); DLi+ – lithium ion diffusion coefficient (cm2 s−1); n – the quantity of electrons transferred in oxidation or reduction reactions, n = 1 in this experiment.
Fig. 10(d) further shows CV curves of LiMn3/4Fe1/4PO4/C and LiMnPO4/C samples with scanning speeds of 0.9 mV s−1. Pure-phase LiMnPO4/C materials exhibit only fuzzy oxidation peaks due to the severe polarization phenomenon. It can be seen that the LiMn3/4Fe1/4PO4/C electrode showed significant redox peaks, with better symmetry and higher redox peaks, even at a high sweep velocity of 0.9 mV s−1. The latent difference between the positive and negative peaks of LiMn3/4Fe1/4PO4/C and LiMnPO4/C electrodes is respectively 0.502 V and 0.717 V. Therefore, this indicates that the Mn to Fe ratio of 3:1 lithium manganese iron phosphate (LMFP) material has better cyclic reversibility than the pure phase LiMnPO4 material. The intensity of the redox peak (especially the oxidation peak) of LiMn3/4Fe1/4PO4/C was also markedly larger than that of the pure LiMnPO4/C at the same high scanning speed of 0.9 mV s−1, which dramatically reduced electrode polarization and improved reaction kinetics and chemical reaction rates. It is evident that the space enclosed by the CV curve of the LiMn3/4Fe1/4PO4/C material is increased, effectively enriching the specific ability of the active material.
The electron local function (ELF) is an ideal tool for describing electron distribution in space, which is very important for clarifying the properties of bonding and ion diffusion and can be used to explain the internal factors of the enhancement of lithium-ion diffusion in the CV curve. Research theory shows that the ELF value is always positive and between 0 and 1, and the closer to 0, the lower the degree of electron localization. There is little difference in the local charge between different Fe doping levels. Actually, the only things that can explain the issue in the Li(Mn1−xFex)PO4 system are a pure phase LiMnPO4 and the crystal comparison following the local doping of Fe at the same site.
Fig. 11 shows the spatial distribution of charge density of Li(Mn1−xFex)PO4 materials; this figure shows the comparison of local charge density of LiMnPO4 material along the (100) crystal plane before and after doping, revealing the charge change of the LiMnPO4 crystal before and after Fe doping. In the figure, the electron density on the isosurface of electron density is 0.1 e Å−3, and the redder the scale on the left, the larger the charge density, i.e., “Low → High” represents “low electron density → completely delocalized electron density → completely localized electron density”. Obviously, doping has a very significant effect on the charge. The localization of Fe-3d and Mn-3d electrons can be seen from the electron density profile at the doping point. After doping, the distribution range of the electron density contour in the doping site is wider than that before doping, and the electrical conductivity is better. After Fe doping, the local charge density around the original Mn site decreases, indicating that Fe atom has a stronger reducing effect on the localization of surrounding electrons than Mn. From the ELF value of doping site (Fe), it can be observed that compared with LiMnPO4 of pure phase, the existence of Fe element can reduce the ELF function value near the original Mn position; this indicates that the localization of electrons is reduced and the diffusion energy barrier is reduced, which is advantageous for electron motion and improves the diffusion rate of lithium ions in the doping system. Therefore, the more Fe content, the better, which further verified the previous conclusion that LiMn3/4Fe1/4PO4 had the best performance.
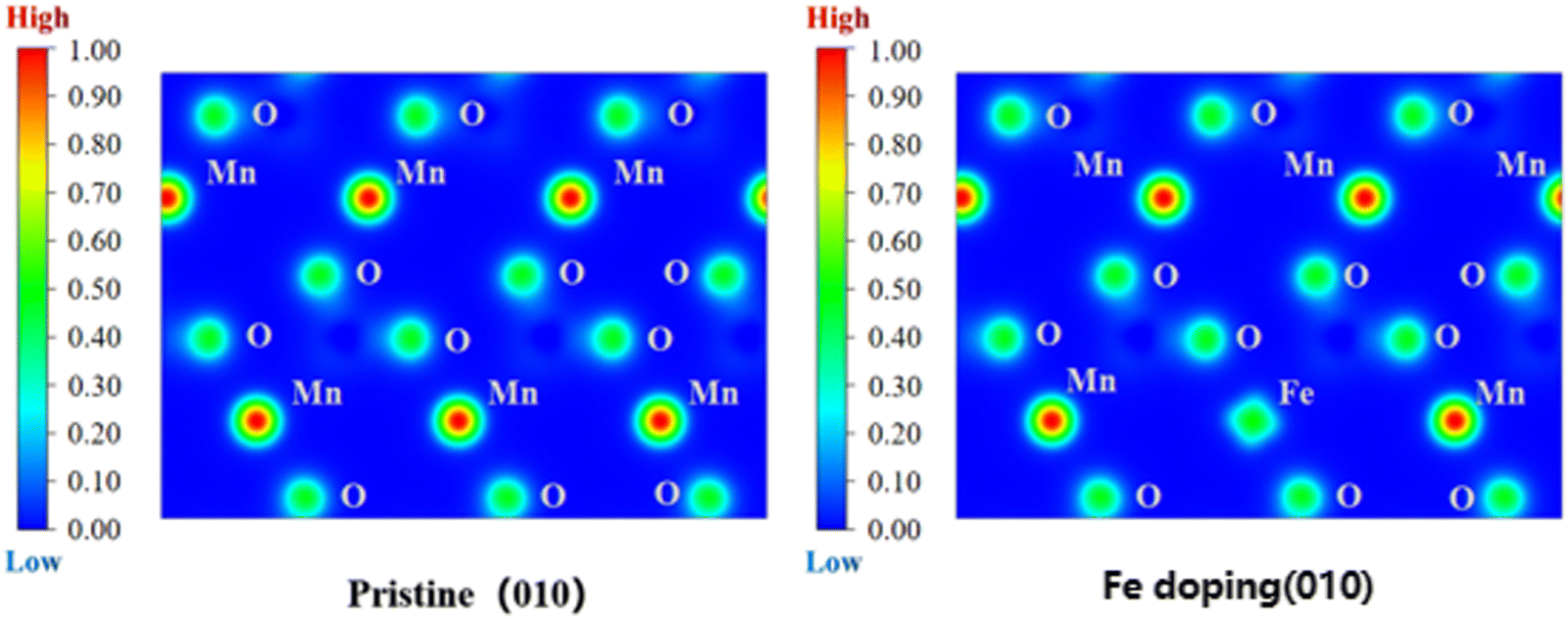 | ||
| Fig. 11 Comparison of local charge density before and after Fe doping. | ||
4 Conclusion
In this paper, a high performance Li(Mn1−xFex)PO4/C (x = 0, 1/24, 1/12, 1/8, 1/4) positive electrode was successfully prepared through the hydrothermal method, which successfully solved the problem of poor intrinsic performance of pure phase material LiMnPO4. The conclusions are as follows.XRD, XPS and EDS tests showed that Fe elements successfully entered the LiMnPO4 lattice and formed the Li(Mn1−xFex)PO4/C cathode material. The SEM images show that the samples are mainly spherical and attached with many small agglomeration particles. With the addition of Fe doping, the c/a value increases, indicating that doping reduces the mixing of cations; the LiMn3/4Fe1/4PO4/C material has the greatest c/a value. The transmission electron microscopy (TEM) images of the grains demonstrate that they are polished on the particle surface and have high crystallinity. The intrinsic quality of the high performance of the LiMn3/4Fe1/4PO4/C positive electrode is explained by the simulation of the electron structure and charge density. The energy difference between the highest HOMO and lowest LUMO in the total density of states (TDOS) can be used to determine the energy value near the Fermi level. This energy difference indicates that the band gaps of the LiMn3/4Fe1/4PO4/C material are x = 1/24, 1/12, 1/8, and 1/4, respectively, and are 3.586 eV, 3.525 eV, 3.515 eV, and 3.503 eV, which is essentially consistent with the band calculation.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51874079, 51804035, 52104307), Natural Science Foundation of Liaoning Province (no. 2019-ZD-0507), Natural Science Foundation of Hebei Province (no. E2018501091), the Fundamental Research Funds for the Central Universities (no. N172302001, N182312007, N182304015), Qinhuangdao City University student of Science and Technology Innovation and Entrepreneurship Project (no. PZB1810008T-46, PZB1810008T-14), the Training Foundation for Scientific Research of Talents Project, Hebei Province (no. A2016005004), Hebei Province Higher Education Science and Technology Research Project (no. QN2017403), Department of Education Projects of Liaoning Province (no. LQ2020012), Open Research Subject of Key Laboratory of Dielectric and Electrolyte Functional Material Hebei Province (no. 9081200122006), Support Program of Young Top Talent of of Liaoning Province (no. XLYC2007197), Hebei Key Laboratory of Dielectric and Electrolyte Functional Material, Northeastern University at Qinhuangdao (no. HKDEFM2023201), Department of Education Projects of Liaoning Province (no. JYTM20231612), Performance subsidy fund for Key Laboratory of Dielectric and Electrolyte Functional Material Hebei Province (no. 22567627H).References
- F. Wang, J. D. Harindintwali and Z. Yuan, et al., Technologies and perspectives for achieving carbon neutrality, Innovation, 2021, 2(4), 100180 CAS.
- Y. Qiu and F. Jiang, A review on passive and active strategies of enhancing the safety of lithium-ion batteries, Int. J. Heat Mass Transfer, 2022, 184, 122288 CrossRef CAS.
- L. A. Román-Ramírez and J. Marco, Design of experiments applied to lithium-ion batteries: A literature review, Appl. Energy, 2022, 320, 119305 CrossRef.
- N. Piao, X. Gao and H. Yang, et al., Challenges and development of lithium-ion batteries for low temperature environments, ETransportation, 2022, 11, 100145 CrossRef.
- P. U. Nzereogu, A. D. Omah and F. I. Ezema, et al., Anode materials for lithium-ion batteries: A review, Appl. Surf. Sci. Adv., 2022, 9, 100233 CrossRef.
- V. Aravindan, J. Gnanaraj and Y. S. Lee, et al., LiMnPO4–A next generation cathode material for lithium-ion batteries, J. Mater. Chem. A, 2013, 1(11), 3518–3539 RSC.
- H. Ji, G. Yang and H. Ni, et al., General synthesis and morphology control of LiMnPO4 nanocrystals via microwave-hydrothermal route, Electrochim. Acta, 2011, 56(9), 3093–3100 CrossRef CAS.
- K. Zaghib, M. Trudeau and A. Guerfi, et al., New advanced cathode material: LiMnPO4 encapsulated with LiFePO4, J. Power Sources, 2012, 204, 177–181 CrossRef CAS.
- X. L. Pan, C. Y. Xu and D. Hong, et al., Hydrothermal synthesis of well-dispersed LiMnPO4 plates for lithium ion batteries cathode, Electrochim. Acta, 2013, 87, 303–308 CrossRef CAS.
- H. Zhang, Y. Gong and J. Li, et al., Selecting substituent elements for LiMnPO4 cathode materials combined with density functional theory (DFT) calculations and experiments, J. Alloys Compd., 2019, 793, 360–368 CrossRef CAS.
- J. Zheng, L. Ni and Y. Lu, et al., High-performance, nanostructure LiMnPO4/C composites synthesized via one-step solid state reaction, J. Power Sources, 2015, 282, 444–451 CrossRef CAS.
- J. Han, J. Yang and Z. Xu, et al., Dramatic improvement in high-rate capability of LiMnPO4 nanosheets via crystallite size regulation, J. Alloys Compd., 2022, 894, 162510 CrossRef CAS.
- J. Han, J. Yang and H. Lu, et al., Effect of synthesis processes on the microstructure and electrochemical properties of LiMnPO4 cathode material, Ind. Eng. Chem. Res., 2022, 61(22), 7451–7463 CrossRef CAS.
- F. A. Vásquez and J. A. Calderón, Vanadium doping of LiMnPO4 cathode material: correlation between changes in the material lattice and the enhancement of the electrochemical performance, Electrochim. Acta, 2019, 325, 134930 CrossRef.
- V. Ragupathi, P. Panigrahi and G. S. Nagarajan, Enhanced electrochemical performance of nanopyramid-like LiMnPO4/C cathode for lithium–ion batteries, Appl. Surf. Sci., 2019, 495, 143541 CrossRef CAS.
- J. Ma, Y. Li and N. S. Grundish, et al., The 2021 battery technology roadmap, J. Phys. D: Appl. Phys., 2021, 54(18), 183001 CrossRef CAS.
- C. Wang, S. Li and Y. Han, et al., Assembly of LiMnPO4 nanoplates into microclusters as a high-performance cathode in lithium-ion batteries, ACS Appl. Mater. Interfaces, 2017, 9(33), 27618–27624 CrossRef CAS PubMed.
- I. Seo, B. Senthilkumar and K. H. Kim, et al., Atomic structural and electrochemical impact of Fe substitution on nano porous LiMnPO4, J. Power Sources, 2016, 320, 59–67 CrossRef CAS.
- H. Xu, J. Zong and F. Ding, et al., Effects of Fe2+ ion doping on LiMnPO4 nanomaterial for lithium ion batteries, RSC Adv., 2016, 6(32), 27164–27169 RSC.
- H. Wang, Y. Yang and Y. Liang, et al., LiMn1−xFexPO4 nanorods grown on graphene sheets for ultrahigh-rate-performance lithium ion batteries, Angew. Chem., Int. Ed., 2011, 123(32), 7502–7506 CrossRef.
- K. Y. Zhang, Z. Y. Gu and E. H. Ang, et al., Advanced polyanionic electrode materials for potassium-ion batteries: Progresses, challenges and application prospects, Mater. Today, 2022, 54, 189–201 CrossRef CAS.
- J. Z. Guo, Z. Y. Gu and M. Du, et al., Emerging characterization techniques for delving polyanion-type cathode materials of sodium-ion batteries, Mater. Today, 2023, 221–244 CrossRef.
- M. Y. Wang, X. X. Zhao and J. Z. Guo, et al., Enhanced electrode kinetics and properties via anionic regulation in polyanionic Na3+xV2(PO4)3−x(P2O7)x cathode material, Green Energy Environ., 2022, 7(4), 763–771 CrossRef CAS.
- Z. Y. Gu, Y. L. Heng and J. Z. Guo, et al., Nano self-assembly of fluorophosphate cathode induced by surface energy evolution towards high-rate and stable sodium-ion batteries, Nano Res., 2023, 16(1), 439–448 CrossRef CAS.
- N. H. Kwon, H. Yin and T. Vavrova, et al., Nanoparticle shapes of LiMnPO4, Li+ diffusion orientation and diffusion coefficients for high volumetric energy Li+ ion cathodes, J. Power Sources, 2017, 342, 231–240 CrossRef CAS.
- C. A. Marianetti, D. Morgan and G. Ceder, First-principles investigation of the cooperative Jahn-Teller effect for octahedrally coordinated transition-metal ions, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(22), 224304 CrossRef.
- A. Mukhopadhyay and B. W. Sheldon, Deformation and stress in electrode materials for Li-ion batteries, Prog. Mater. Sci., 2014, 63, 58–116 CrossRef CAS.
- F. Cheng, H. Wang and Z. Zhu, et al., Porous LiMn2O4 nanorods with durable high-rate capability for rechargeable Li-ion batteries, Energy Environ. Sci., 2011, 4(9), 3668–3675 RSC.
- S. Liu, L. Xiong and C. He, Long cycle life lithium ion battery with lithium nickel cobalt manganese oxide (NCM) cathode, J. Power Sources, 2014, 261, 285–291 CrossRef CAS.
- Q. Zhang, C. Yan and Y. Meng, et al., Hierarchical mesoporous Li2FeSiO4/C sheaf-rods as a high-performance lithium-ion battery cathode, J. Alloys Compd., 2018, 767, 195–203 CrossRef CAS.
- L. Li, E. Han and C. Mi, et al., The effect of Ni or Pb substitution on the electrochemical performance of Li2FeSiO4/C cathode materials, Solid State Ionics, 2019, 330, 24–32 CrossRef CAS.
- C. M. Doherty, R. A. Caruso and B. M. Smarsly, et al., Hierarchically porous monolithic LiFePO4/carbon composite electrode materials for high power lithium ion batteries, Chem. Mater., 2009, 21(21), 5300–5306 CrossRef CAS.
- W. Zheng, X. Huang and Y. Ren, et al., Porous spherical Na3V2(PO4)3/C composites synthesized via a spray drying-assisted process with high-rate performance as cathode materials for sodium-ion batteries, Solid State Ionics, 2017, 308, 161–166 CrossRef CAS.
- S. K. Martha, J. Nanda and G. M. Veith, et al., Electrochemical and rate performance study of high-voltage lithium-rich composition: Li1. 2Mn0. 525Ni0. 175Co0. 1O2, J. Power Sources, 2012, 199, 220–226 CrossRef CAS.
- H. Gao, L. Wang and Y. Zhang, et al., Tartaric acid assisted synthesis of Li2FeSiO4/C: Effect of carbon content on the electrochemical performance of Li2FeSiO4/C for lithium ion batteries, Powder Technol., 2014, 253, 638–643 CrossRef CAS.
- P. Gao, C. Zhang and G. Wen, Equivalent circuit model analysis on electrochemical impedance spectroscopy of lithium metal batteries, J. Power Sources, 2015, 294, 67–74 CrossRef CAS.
- X. Z. Liao, Z. F. Ma and Q. Gong, et al., Low-temperature performance of LiFePO4/C cathode in a quaternary carbonate-based electrolyte, Electrochem. Commun., 2008, 10(5), 691–694 CrossRef CAS.
- C. Hwang, T. Kim and J. Shim, et al., Fast ultrasound-assisted synthesis of Li2MnSiO4 nanoparticles for a lithium-ion battery, J. Power Sources, 2015, 294, 522–529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00049h |
| This journal is © The Royal Society of Chemistry 2024 |