
Unveiled effects of the methylammonium chloride additive on formamidinium lead halide: expediting carrier injection from the photoabsorber to carrier transport layers through spontaneously modulated heterointerfaces in perovskite solar cells†
Naoyuki
Nishimura
 *a,
Ranjan Kumar
Behera
a,
Ryuzi
Katoh
b,
Hiroyuki
Kanda
a,
Takurou N.
Murakami
a and
Hiroyuki
Matsuzaki
*a
*a,
Ranjan Kumar
Behera
a,
Ryuzi
Katoh
b,
Hiroyuki
Kanda
a,
Takurou N.
Murakami
a and
Hiroyuki
Matsuzaki
*a
aNational Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: naoyuki-nishimura@aist.go.jp; hiroyuki-matsuzaki@aist.go.jp
bCollege of Engineering, Nihon University, Koriyama, Fukushima 963-8642, Japan
First published on 29th May 2024
Abstract
Perovskite solar cells (PSCs) based on a narrow-bandgap formamidinium lead halide (FAPbI3) photoabsorber have garnered substantial attention owing to their high photovoltaic (PV) performances. Extensive studies have established that the introduction of methylammonium chloride (MACl) significantly improves the quality of the FAPbI3 bulk, and hence, this method has been commonly employed. Upon heating the photoabsorber's precursor film, the incorporated MACl facilitates the crystalline growth of FAPbI3, simultaneously volatilizing and dissipating from the perovskite layer. However, not only the photoabsorber's bulk quality but also heterointerfaces between the photoabsorber and carrier transport materials importantly contribute to the PV performances. Paradoxically, the MACl effects on FAPbI3 heterointerfaces have been sparingly explored and consequently remain elusive. Herein, the effects of MACl on these heterointerfaces are unveiled by time-resolved photoluminescence spectroscopy and time-resolved microwave conductivity. The MACl additive accelerates carrier injection from FAPbI3 to the carrier transport materials presumably owing to the occurrence of spontaneous modulation of the heterointerfaces. In particular, at the heterointerface between FAPbI3 and the titanium oxide (TiO2) electron transport layer, an emissive interlayer, that is, a chloride-containing wide-bandgap FA1−xMAxPbI3−yCly interlayer is formed spontaneously, which most likely has multiple advantages: hole blocking and facilitation of electron injection from FAPbI3 to TiO2 without additional hole trapping, leading to the observed PV performance enhancement. Consequently, the present results provide novel insights into the effects of the widely employed MACl additive on the FAPbI3 photoabsorber, thereby propelling the further advancement of PSCs. Furthermore, this study demonstrates effective investigation of carrier dynamics with regard to the heterointerfaces, which is challenging, and will promote the development of the materials science field.
Introduction
Perovskite solar cells (PSCs) have been intensively studied as promising candidates for next-generation photovoltaic (PV) systems, owing to their potential for scalable production and high PV performances. This surge of interest in PSCs was triggered by the demonstration of a relatively high power conversion efficiency (PCE) approaching 10% for PSCs based on a methylammonium lead halide (MAPbI3) photoabsorber in 2012.1,2 Over the past decade, PSCs have undergone significant development by employing various techniques, including modifications to the composition of perovskite photoabsorbers, transitioning from the archetypal MAPbI3 to A-site modulated perovskites.3–11One of the most prominent compositions of perovskite photoabsorbers is formamidinium lead halide (FAPbI3), distinguished by its narrow bandgap energy, relatively closely aligning with the ideal bandgap energy of single-junction PV (≈1.4 eV).5 Although FAPbI3 was initially reported as a photoabsorber in the early stages of perovskite development in 2013,5 challenges in film quality, pertaining to crystallinity and crystal phase control, impeded its progress. However, recent progress has successfully addressed this issue, establishing FAPbI3 as a major photoabsorber.5–27 Notably, the addition of alkylammonium chloride, typically methylammonium chloride (MACl), to the perovskite precursor has proven instrumental in achieving high-quality FAPbI3 formation.6–12 Upon heating the perovskite precursor film with the MACl additive, MACl facilitates crystal growth of FAPbI3 and simultaneously volatilizes, contributing to improvements in film quality.6–21 The MACl–FAPbI3 system has become a widely acknowledged system, contributing to recent record-breaking PCE achievements.11
Meanwhile, PSCs consist of multiple layers (standard configuration from the top: metal conductor/hole transport material (HTM)/perovskite photoabsorber/electron transport material (ETM)/transparent conductor1,2 (e.g., Au/Spiro-OMeTAD/FAPbI3/TiO2/FTO: Fig. 1)). Consequently, PSC performance depends on the quality of the photoabsorber layer as well as on carrier injection from the photoabsorber to neighboring layers (i.e., carrier transport layers). The manipulation of heterointerfaces, specifically the HTM/perovskite and perovskite/ETM interfaces, assumes a pivotal role in enhancing PV performances. Despite the established use of the MACl additive for improving FAPbI3 bulk quality, the effects of MACl on heterointerfaces have been sparingly explored and consequently remain elusive. This is attributable to the inherent difficulty in differentiating MACl effects on the FAPbI3 bulk from those on heterointerfaces. Thus, investigation of the nature of heterointerfaces independently of the bulk is imperative for the continued advancement of PSCs and the broader field of materials science.
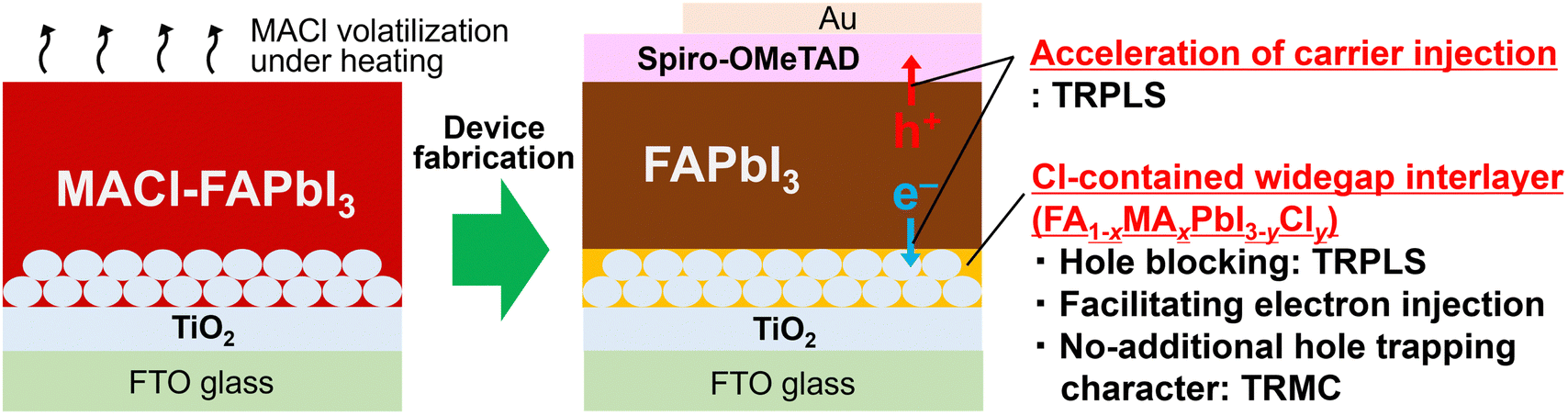 | ||
| Fig. 1 Schematic illustration of unveiled effects of MACl: spontaneously modulated heterointerfaces facilitating carrier collection. | ||
This study elucidates the effects of the predominant MACl additive on carrier dynamics, specifically involving heterointerfaces in PSCs, employing time-resolved photoluminescence spectroscopy (TRPLS) and time-resolved microwave conductivity (TRMC). TRPLS measurements reveal two key features: (i) MACl addition accelerates carrier injection from FAPbI3 to carrier transport layers and (ii) MACl induces the spontaneous formation of a chlorine (Cl)-containing wide-bandgap interlayer, FA1−xMAxPbI3−yCly, at the FAPbI3/TiO2 heterointerface. This FA1−xMAxPbI3−yCly interlayer presumably functions as a hole-blocking layer and as an electron-injection facilitator. In addition, TRMC measurements indicate that the formed interlayer does not act as an additional hole trap, dispelling concerns about potential carrier trapping effects owing to the presence of the additional FAPbI3/FA1−xMAxPbI3−yCly heterointerface. In summary, this study reveals that MACl addition is advantageous not only for enhancing FAPbI3 bulk quality but also for expediting carrier injection from FAPbI3 to carrier transport materials through spontaneously modulated heterointerfaces (Fig. 1), thereby offering insights pivotal to the ongoing development of PSCs.
Results and discussion
Three distinct types of PSCs were fabricated, each based on different photoabsorbers: (i) MACl–FAPbI3 with n-octylammonium iodide (OAI) passivation,9,22,28–30 (ii) pristine MACl–FAPbI3, and (iii) pristine FAPbI3 without MACl. While the comparison between (i) and (ii) confirms the effects of OAI passivation, the comparative analysis between (ii) and (iii) reveals the effects of the MACl additive: not only the improvement of FAPbI3 bulk quality but also the spontaneous modulation of the heterointerfaces in PSCs (Fig. 1).Table 1 provides the PV parameters of the cells used in the subsequent TRPLS measurements. In the reverse scan, the PCEs were determined to be 20.9%, 16.9%, and 13.5% for cells based on MACl–FAPbI3 with OAI passivation, pristine MACl–FAPbI3, and pristine FAPbI3 without MACl, respectively. Please refer to the ESI† for additional details, including statistical analysis of PV performance and other characterization methods.
| Sample | Scan | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|
| OAI treated MACl–FAPbI3 | Reverse | 25.5 | 1.12 | 0.73 | 20.9 |
| Forward | 25.4 | 1.09 | 0.67 | 18.6 | |
| Pristine MACl–FAPbI3 | Reverse | 24.1 | 0.97 | 0.73 | 16.9 |
| Forward | 23.8 | 0.90 | 0.68 | 14.5 | |
| Pristine FAPbI3 without MACl | Reverse | 22.7 | 0.93 | 0.64 | 13.5 |
| Forward | 22.6 | 0.90 | 0.54 | 11.1 |
This unequivocally confirms that the addition of MACl into FAPbI3 substantially improved the PV performance,6–9 with further enhancement observed with OAI passivation,9,22 aligning with findings of previous reports. The X-ray diffraction (XRD) patterns (Fig. S4, ESI†) and the resultant larger grain size observed in the top view of scanning electron microscopy (SEM) images (Fig. S5, ESI†) suggest that the MACl addition likely improved the bulk quality31,32 of FAPbI3 with a little change in their bandgap energies: both have approximately 1.53 eV (Fig. S6, ESI†). Furthermore, TRMC measurement indicates enhancement of carrier mobility of FAPbI3 when using the MACl additive (Fig. S7, ESI†). Consequently, the enhanced PV performance is attributed, in part, to the improved FAPbI3 bulk, a phenomenon consistent with previous reports.6–9 However, an additional contributing factor is the alteration of heterointerfaces pertinent to carrier collection, which will be discussed further.
To investigate the properties of the PSCs used in this study and to differentiate between bulk and heterointerface properties, TRPLS measurements were conducted. A comparative analysis between (i) the perovskite monolayer sample and (ii) the solar cell offers insights into the kinetics of carrier injection from the perovskite layer to the carrier transport materials. The PL lifetimes of (i) the perovskite monolayer samples serve as indicators of the carrier lifetime within each perovskite layer. The decay kinetics of the perovskite monolayer over the quartz substrate can be expressed by following relation:33–35
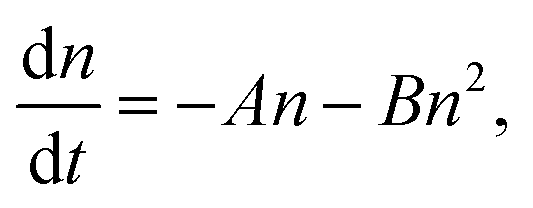 | (1) |
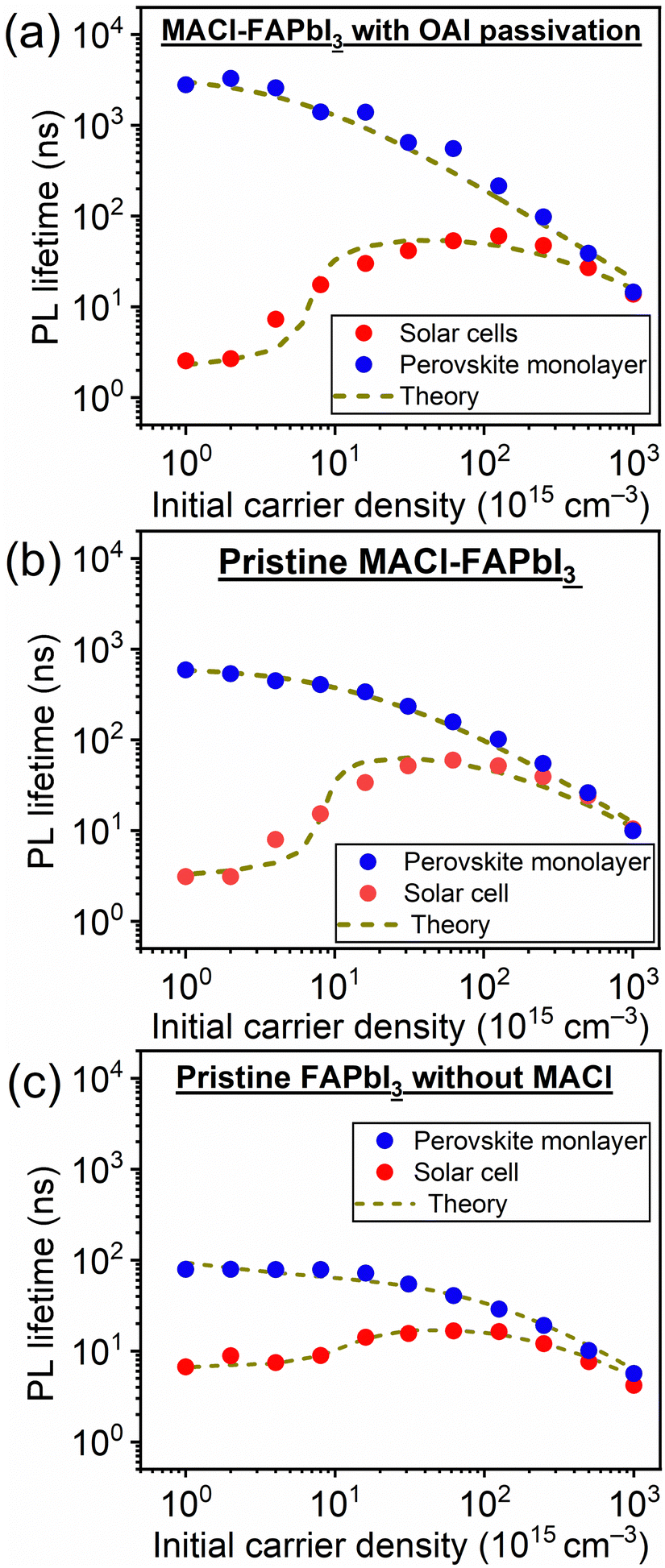 | ||
| Fig. 2 Excitation power dependence of the PL lifetime of samples comprising (a) MACl–FAPbI3 with OAI passivation, (b) pristine MACl–FAPbI3, and (c) pristine FAPbI3 without MACl additive. | ||
Meanwhile, the extent of carrier decay in (ii) solar cells is expressed as follows:33,34
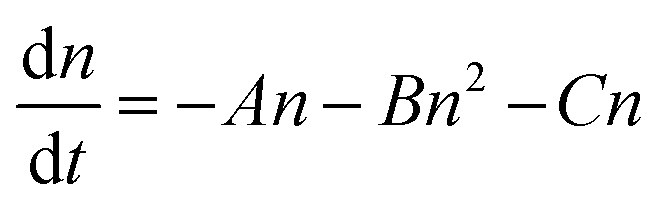 | (2) |
These features based on eqn (1) and (2) were observed in the practical PL lifetime measurements, and the trends were different among the employed photoabsorber layers, indicating each carrier dynamics involving FAPbI3 bulk quality and carrier injection from perovskite to carrier transport layers. Fig. 2 illustrates the excitation-power dependence of the PL lifetimes of both the solar cells and the corresponding perovskite monolayer on quartz substrate samples.33–35 The PL lifetimes are defined as the time from the initial PL intensity to 1/e of that intensity, and the initial carrier densities per unit volume are calculated using excitation power densities per unit area and the absorption coefficient of FAPbI3 at 650 nm36 (see ESI† for more details). For the monolayer sample, when subjected to weak excitations, the dominance of single-carrier trapping and/or recombination (A) is evident, resulting in prolonged PL lifetimes, particularly in passivated samples, reaching the microsecond order. However, with stronger excitation, the contribution of bimolecular recombination (B) to carrier decay becomes more pronounced, consequently shortening the PL lifetime down to several nanoseconds.33,34 In solar cells, carrier injection (C) is an additional factor contributing to carrier decay. Under weak excitation, carrier injection predominates, leading to a short PL lifetime within several nanoseconds. In contrast, stronger excitation slows down carrier injection due to the accumulation of carriers at both the interfaces of the perovskite/carrier transport materials and within the bulk of the carrier transport materials. As a result, moderate excitation power yields a longer PL lifetime compared to weak excitation. Ultimately, under further strong excitation, bimolecular recombination (B) becomes dominant, resulting in similar PL lifetime values for monolayer samples and solar cells under the strongest excitation (an initial carrier density of 1018 cm−3).33,34
Moreover, the kinetics parameters were quantitatively determined by fitting the excitation power dependence of the PL lifetime33,34 (see ESI† for fitting details). Table 2 presents the calculated kinetics parameters. According to eqn (1) and (2), smaller values of A and B indicate fewer carrier losses in the FAPbI3 photoabsorber bulk, while a larger value of C represents faster carrier injection from FAPbI3 to carrier-transport layers. A notable observation is the significant reduction of the A value by OAI passivation from 1.6 × 106 s−1 to 1.5 × 105 s−1 (a one-order decrease), while B and C values remain almost unchanged. This trend suggests that the primary role of OAI passivation is the suppression of defects inducing single-carrier trapping, thereby enhancing PV performance: an outcome consistent with prior reports.9,22,28–30
| Sample | A (s−1) | B (cm3 s−1) | C (s−1) |
|---|---|---|---|
| MACl–FAPbI3 with OAI passivation | 1.5 × 105 | 2.4 × 10−11 | 2.5 × 108 |
| Pristine MACl–FAPbI3 | 1.6 × 106 | 3.5 × 10−11 | 3.0 × 108 |
| Pristine FAPbI3 without MACl | 7.7 × 106 | 6.4 × 10−11 | 1.1 × 108 |
In contrast, a comparison between pristine MACl–FAPbI3 and MACl-free FAPbI3 samples showed trends different from those observed with/without OAI passivation, thereby revealing the effects of the MACl additive. The MACl addition not only reduced the A and B parameters, with A decreasing from 7.7 × 106 s−1 to 1.6 × 106 s−1 and B decreasing from 6.4 × 10−11 cm3 s−1 to 3.5 × 10−11 cm3 s−1, indicating an improvement in FAPbI3 bulk quality consistent with previous reports on MACl effects.6–9 However, C values, pertaining to carrier injection, also underwent significant changes with MACl addition. The C value increased from 1.1 × 108 s−1 to 3.0 × 108 s−1 (an approximately three-fold increase), accelerating carrier injection from FAPbI3 to carrier transport materials. This significant change in the C value strongly suggests that MACl addition not only affects the FAPbI3 bulk but also influences the heterointerfaces between FAPbI3 and carrier transport materials.
The PL spectra offer insights into changes at a heterointerface with the addition of MACl. Fig. 3 illustrates the time-resolved PL spectra of solar cells with and without MACl when excited from the FTO side. The PL spectra were segregated based on the time scale: the initial (0–5 ns) and the later part (30–50 ns). In the absence of MACl, minimal changes were observed in the PL spectra within the specified time range (Fig. 3a). On the contrary, for the sample with MACl, a notable spectral alteration emerged between the time intervals; a PL spectrum resembling that of the MACl-free counterpart, peaking at around 810 nm, was observed in the later time scale (30–50 ns), whereas in the initial time scale (0–5 ns), a blue-shifted PL was evident (Fig. 3b). The central photon energy of the deconvoluted blue-shifted PL was estimated to be 1.62 eV (765 nm; Fig. 3c), marking an approximately 90 meV increase compared with the primary PL (Fig. S11: 1.53 eV, 811 nm, ESI†). The absence of blue-shifted PL with excitation from the HTM side for the solar cell suggested its attribution to the ETM side (Fig. S12a, ESI†). Moreover, the sample of the MACl–FAPbI3 perovskite monolayer on a quartz sample did not exhibit the blue-shifted PL (Fig. S12b, ESI†), leading to the conclusion that the observed short lifetime and blue-shifted PL originated from emission at the FAPbI3/TiO2 heterointerface, modulated by the presence of MACl.
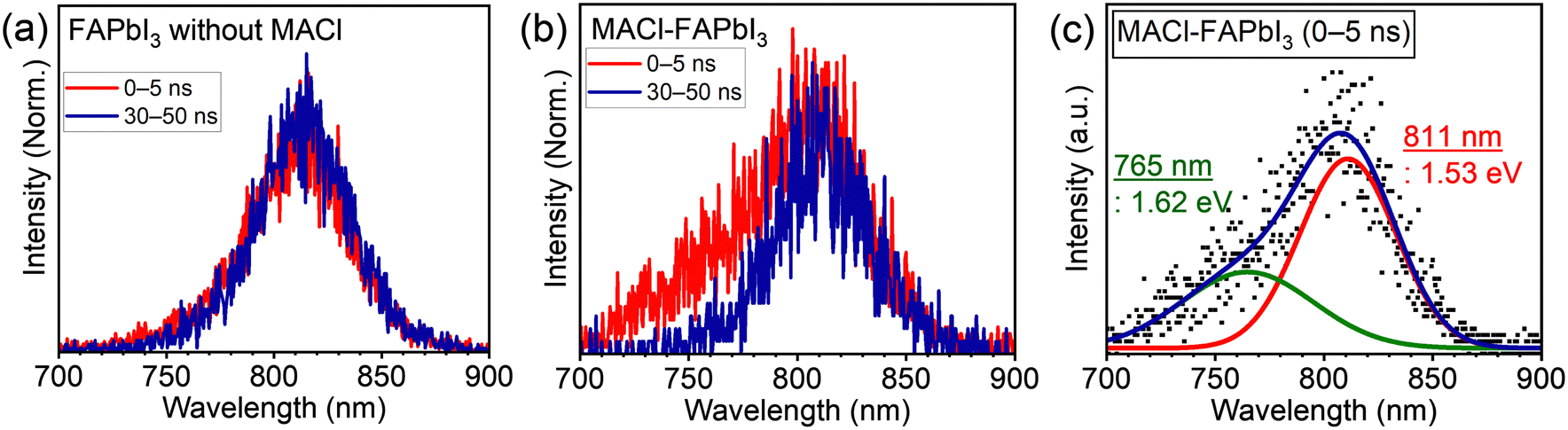 | ||
| Fig. 3 PL spectra of solar cells consisting of (a) FAPbI3 without MACl and (b) the MACl–FAPbI3 photoabsorber divided by time scales of 0–5 ns and 30–50 ns. (c) Deconvoluted spectra of the MACl–FAPbI3 cell in the initial time scale (0–5 ns). | ||
The anticipated modulation of the FAPbI3/TiO2 heterointerface and the resulting blue-shifted PL observed in Fig. 3c, attributed to the Cl composition derived from MACl, prompted a comprehensive compositional depth analysis. Secondary ion mass spectroscopy (SIMS) results depicted in Fig. 4 showcase FAPbI3 with and without MACl samples using TiO2/FTO substrates, distinguished by the presence of Pb and I, Ti, and Sn for FAPbI3, TiO2, and FTO, respectively. In the MACl-containing sample, a significantly heightened Cl intensity exceeding 1000 counts was observed in the TiO2 region, approximately ten-fold higher than that in the perovskite regime. This observation unequivocally indicates the segregation of Cl from MACl at the FAPbI3/TiO2 heterointerface, forming a Cl-containing interlayer, presumed to be FAPbI3−yCly.
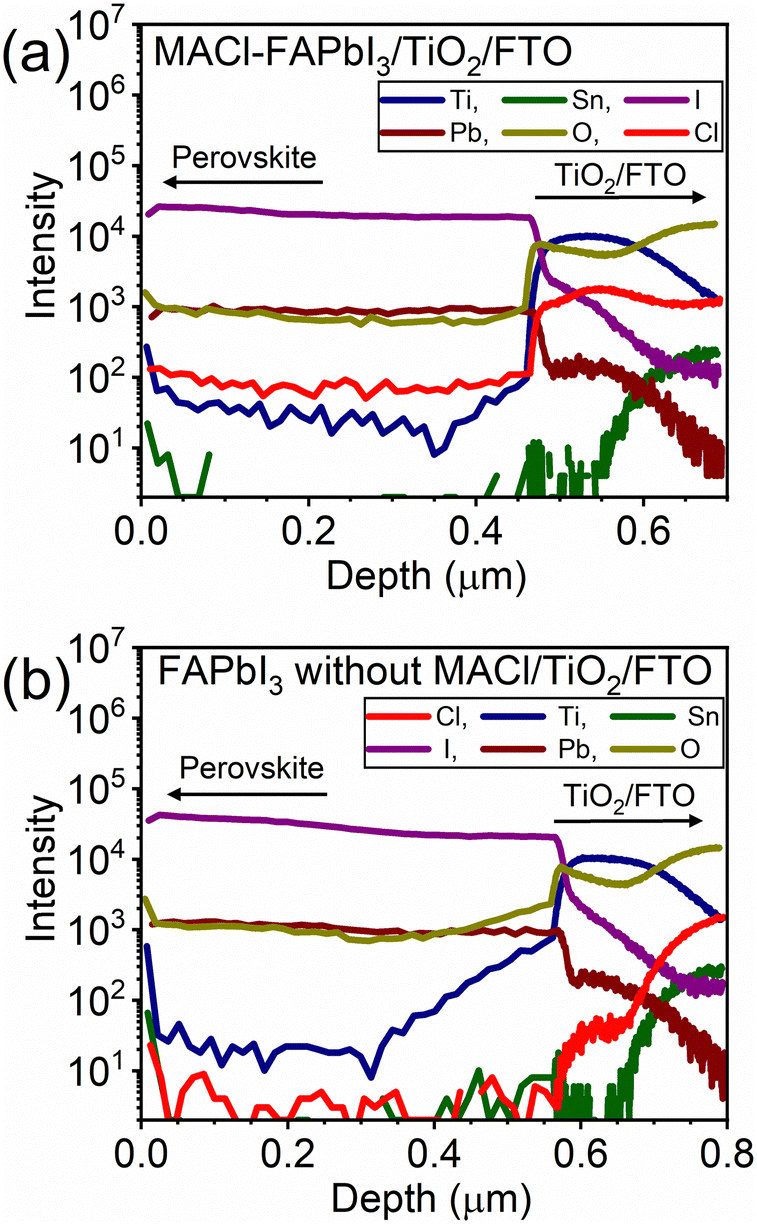 | ||
| Fig. 4 Compositional depth analysis of (a) MACl–FAPbI3/TiO2/FTO and (b) FAPbI3 without MACl/TiO2/FTO. | ||
Considering the bandgaps of MAPbCl3 (3.11 eV)37,38 and FAPbI3 (1.53 eV; Fig. S6 and S11, ESI†), and a linear bandgap relation between them was premised, 5 mol% Cl composition in the perovskite halogen (i.e., FAPbI2.85Cl0.15) could be roughly estimated, based on the blue-shifted PL energy of 1.62 eV, as observed in Fig. 3c. The estimated Cl content of 5 mol% may be high with respect to the FA cation in the A-site of the perovskite regarding crystal structure distortion. Therefore, a small portion of MA cations from MACl can be contained in the FAPbI3−yCly interlayer (FA1−xMAxPbI3−yCly), thus mitigating crystal distortion.8,10 Consequently, owing to the high Cl content at the FAPbI3/TiO2 interface, the observed blue-shifted PL shown in Fig. 3c is most likely attributed to the spontaneously formed FA1−xMAxPbI3−yCly interlayer.
Furthermore, the depth of the high-Cl-regime correlated well with the high-Ti region on the measured scale, suggesting a thin thickness for the FA1−xMAxPbI3−yCly interlayer. This assumed thin thickness implies that a robust chemical interaction between the Cl− anion and the TiO2 surface induced the formation of the FA1−xMAxPbI3−yCly interlayer, likely not limited by the diffusion of Cl− anions in the perovskite precursor film. In other words, the relatively distant position of the Cl− contained interlayer from the gas phase, which is unfavorable for volatilization, is unlikely the primary reason for interlayer formation. Instead, the chemical interaction between Cl− and TiO2 is presumed to be the primary factor contributing to the formation of the FA1−xMAxPbI3−yCly interlayer. Hence, this spontaneous modulation of the perovskite/TiO2 interface with the Cl content derived from MACl would occur for both the mesoporous TiO2 ETMs and planar TiO2 ETMs,39–43 taking advantage of the chemical interaction.
Even for the sample without MACl, a detectable Cl signal, up to 50 counts, was observed in the TiO2 region (Fig. 4b). This weak Cl signal originates from the m-TiO2 utilized in this study, as confirmed by SIMS measurements on a TiO2/FTO reference sample (Fig. S13, ESI†). This implies a trace amount of Cl contamination in the production process of m-TiO2. Although the employed m-TiO2 does contain some Cl, the quantity is significantly lower (over 200 times less) than that of the spontaneously formed FA1−xMAxPbI3−yCly interlayer (Fig. 4). Hence, the presence of Cl in m-TiO2 negligibly influences the PV performance of samples without MACl.
The presence of FA1−xMAxPbI3−yCly is deemed advantageous for carrier collection, attributed to two key aspects: (i) its nature as a hole-blocking layer and (ii) the strong interfacial coupling between TiO2 and FA1−xMAxPbI3−yCly, facilitating efficient electron injection. The (i) hole-blocking nature is based on the assumption of energy alignment within the wide-bandgap interlayer (Fig. 5). Since halogen anions contribute more to the valence band (VB) of the perovskite than the conduction band (CB),44 the formed FA1−xMAxPbI3−yCly interlayer likely possesses a deeper VB top compared to FAPbI3, with a marginal change in the CB bottom energy. The short PL lifetime of the FA1−xMAxPbI3−yCly interlayer indicates rapid hole transfer from FA1−xMAxPbI3−yCly to FAPbI3. This short lifetime also implies that holes cannot access the FA1−xMAxPbI3−yCly interlayer from FAPbI3 (i.e., hole transfer from FAPbI3 to the FA1−xMAxPbI3−yCly interlayer cannot proceed), as PL occurs only when both electrons and holes coexist in a material. This hole-blocking feature aligns with the assumed energy gap of VB tops between FAPbI3 and FA1−xMAxPbI3−yCly, differing by up to 90 meV, a substantial difference from room temperature energy (kT ≈ 26 meV). Therefore, the wide-bandgap FA1−xMAxPbI3−yCly interlayer likely functioned as a hole-blocking layer, contributing favorably to charge separation (Fig. 5). We here note that the hole leaking from FAPbI3 to FTO potentially occurs via physical pinholes in the TiO2 layer45–51 and/or via defect states in TiO2 shallower than the VB top of TiO2.39,52,53 Therefore, the presence of the FA1−xMAxPbI3−yCly interlayer possessing the VB deeper than FAPbI3 is advantageous for blocking the hole leaking regardless of the deeper VB top of TiO2 than that of FAPbI3.
 | ||
| Fig. 5 Illustration of the functions of the wide-bandgap FA1−xMAxPbI3−yCly interlayer formed between FAPbI3 and TiO2. | ||
Regarding (ii) the strong interfacial coupling, certain density functional theory calculations indicate that interfacial Cl in perovskites results in robust electron coupling and chemical bonding to TiO2.54,55 Specifically, the partial density of states calculations illustrate that Cl incorporation into a perovskite material strengthens the interfacial coupling of CBs between the unoccupied titanium d-orbital in TiO2 and the lead p-orbital in a perovskite.54 Enhanced CB coupling is advantageous for more efficient electron injection (Fig. 5). Consequently, the formed FA1−xMAxPbI3−yCly interlayer likely contributes to the accelerated carrier collection observed in Fig. 2.
The potential concern that the introduction of an additional heterointerface might act as a carrier trap led to an investigation into the carrier-trap nature of the Cl-contained FAPbI3/TiO2 interlayer by TRMC.23,56–62 We have recently proposed TRMC as an effective means to probe the carrier-trap nature of heterointerfaces in PSCs.23 After carrier injection from the perovskite to a carrier transport material, TRMC detects the lifetime of the remaining carriers in the perovskite, considering that the carrier mobility of carrier transport materials is negligible compared to perovskite materials23,59 (see ESI† for details). Fig. 6 illustrates TRMC signal decays of the MACl–FAPbI3 monolayer on a quartz sample and the FAPbI3–MACl/TiO2 bilayer on a quartz sample. Here, we note that for the excitation conditions shown in Fig. 6, weak excitation power, where bimolecular recombination (B in eqn 1) becomes negligible and single-carrier trapping and/or recombination (A in eqn 1) dominates the carrier lifetime, was employed (see Fig. S14; excitation power dependence of TRMC decays, ESI†). In the short time scale of the TRMC signal shown in Fig. S15 (ESI†), the MACl–FAPbI3/TiO2 bilayer sample exhibited a steep decay corresponding to electron injection from perovskite to TiO2 in the early stage, but the monolayer sample did not. In light of this electron injection, consequently, single and double exponential fittings were employed for the analysis of the long-time range TRMC decay for the monolayer and bilayer samples, respectively (Fig. 6b and c). Table 3 details each lifetime based on the fitting.
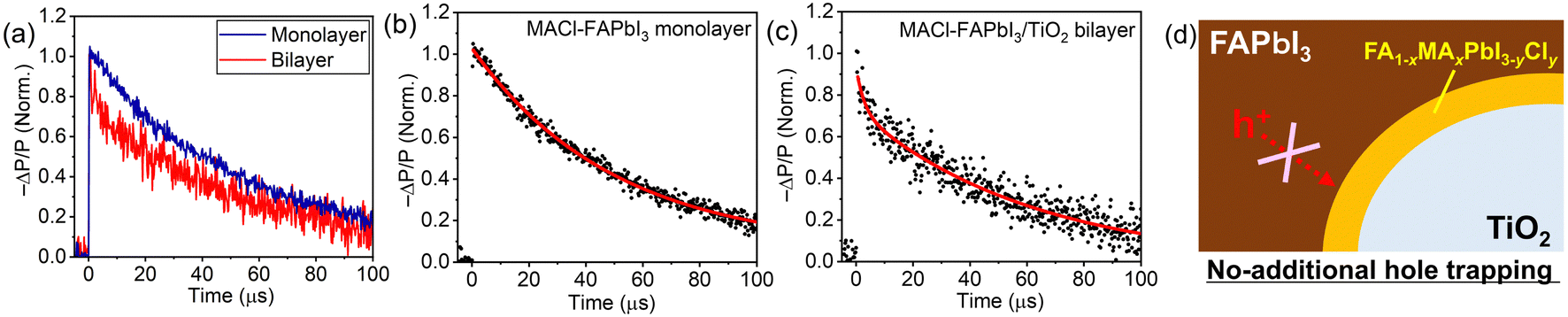 | ||
| Fig. 6 (a) TRMC signal decays of the MACl–FAPbI3 monolayer and MACl–FAPbI3/TiO2 bilayer samples; fitting curves for the (b) monolayer sample and (c) bilayer sample; and (d) illustration of no-additional hole-trapping nature of the FA1−xMAxPbI3−yCly interlayer. | ||
| Sample | τ 1 (μs) | τ 2 (μs) | τ average (μs) |
|---|---|---|---|
| MACl–FAPbI3 monolayer | 50 ± 1 | — | 50 ± 1 |
| MACl–FAPbI3/TiO2 bilayer | 2.7 ± 1.1 | 62 ± 7 | 57 ± 2 |
The MACl–FAPbI3 monolayer sample exhibited a lifetime (τ) of 50 ± 1 μs. Meanwhile, the MACl–FAPbI3/TiO2 bilayer sample showed a short lifetime (τ1) of 2.7 ± 1.1 μs and a long lifetime (τ2) of 62 ± 7 μs, resulting in an average lifetime (τaverage) of 57 ± 2 μs with each component weight in the fitting of A1 = 0.08 and A2 = 0.92. The longer lifetime of the τ2 component of the bilayer sample relative to the monolayer lifetime τ implies that the FA1−xMAxPbI3−yCly interlayer might suppress hole traps over TiO2. However, the similar values of average lifetimes of the monolayer and bilayer samples indicate that the FA1−xMAxPbI3−yCly interlayer does not act as an additional hole trap (Fig. 6d). Consequently, the spontaneously formed FA1−xMAxPbI3−yCly interlayer with the MACl additive is not detrimental to the hole lifetime; yet, it likely facilitates carrier collection.
Conclusions
In this study, the effects of MACl, a widely employed additive for a narrow-bandgap FAPbI3 photoabsorber, are unveiled through the application of time-resolved spectroscopies for investigating carrier dynamics of PSCs. While the MACl additive is recognized for its role in enhancing FAPbI3 bulk quality, our investigation, employing the excitation power dependence of TRPLS, has unveiled its additional effectiveness in expediting carrier injection from FAPbI3 to carrier transport materials. (Fig. 1)Furthermore, through a combination of TRPLS and compositional depth analysis (SIMS), it was observed that the MACl additive results in the formation of a Cl-containing wide-bandgap interlayer (FA1−xMAxPbI3−yCly: ≈1.62 eV) at the FAPbI3/TiO2 interface, with a short lifetime of several nanoseconds. The FA1−xMAxPbI3−yCly interlayer is proposed to serve two key functions: (i) acting as a hole-blocking layer advantageous for charge separation due to its deep VB top and (ii) facilitating electron injection from FAPbI3 to TiO2 owing to the strong interfacial coupling between their CBs. (Fig. 5) The TRMC measurements indicate that the formed FA1−xMAxPbI3−yCly interlayer does not exhibit additional hole-trapping characteristics (Fig. 6c), suggesting that it is not detrimental to the PV performances rather presumably accelerating carrier collection (Fig. 1).
In conclusion, our findings demonstrate that the MACl additive, beyond its role in improving FAPbI3 bulk quality, spontaneously modulates heterointerfaces and accelerates carrier injection from FAPbI3 to carrier transport materials. This work provides valuable insights into the effects of the commonly used MACl additive on a promising FAPbI3 photoabsorber, contributing to the ongoing development of PSCs. Furthermore, the effective investigation of heterointerfaces performed in this study using time-resolved spectroscopies serves as a crucial demonstration, paving the way for the development of an effective means to explore multi-layered devices for solar energy conversion, including solar PV systems, and contributing to the advancement of the broad field of materials science.
Experimental section
Solar cell fabrication
PSC fabrication was achieved by following conventional methods with minor adjustments,22,23 as detailed in the ESI.† Initially, mesoporous TiO2 (m-TiO2)/compact layer TiO2 (c-TiO2)/FTO substrates were prepared using spray pyrolysis for c-TiO2 and spin-coating for m-TiO2. Subsequently, the FAPbI3 perovskite layer was deposited on the TiO2/FTO substrates by spin-coating in a dry air atmosphere. The FAPbI3 precursor solution, comprising synthesized FAPbI3 powder (with 40 mol% MACl for the FAPbI3 powder), was prepared by dissolving it in a mixed solution of N,N-dimethylformamide and dimethyl sulfoxide. The substrate was spin-coated with the precursor solution at 6000 rpm for 50 s and promptly heated at 423 K for 10 min. During the spin-coating process, 1 mL of chlorobenzene was introduced after spinning for 10 s. In certain samples, OAI passivation over the as-prepared FAPbI3 layer was implemented to suppress perovskite surface defects. This involved spin-coating an OAI/isopropyl alcohol solution over the FAPbI3 layer, followed by heating at 373 K for 5 min. The deposition of the HTM was accomplished through spin-coating a solution containing Spiro-OMeTAD and additives. Finally, a 200 nm thick Au conductor layer was deposited via thermal evaporation.PV performance measurement
Current–voltage curves were systematically acquired using a source meter (R6243, ADVANTEST) and a solar simulator (XIL-05B100KP, Seric Co.), calibrated with a Si-reference cell to generate AM1.5G illumination (100 mW cm−2). The current–voltage scans were conducted at a constant speed of 100 mV s−1, concurrently exposed to simulated sunlight. Each experimental condition encompassed measurements on over 10 samples, from which the most optimal specimens were selected. Subsequently, the solar cell properties were computed based on the averages with standard deviations derived from the chosen samples.TRPLS measurements
TRPLS measurements35 were conducted on the solar cells and perovskite quartz monolayer samples prepared utilizing the same methodology that was employed for the solar cells using a streak camera (Hamamatsu Photonics, StreakScope C4334) equipped with a monochromator. A Ti:sapphire laser with a regenerative amplifier (Spectra-Physics, Solstice; a central wavelength of 800 nm, a pulse width of 100 fs, a pulse energy of 3.5 mJ per pulse, and a repetition rate of 1 kHz) was used as a light source. Part of the output from the laser was used for the excitation of an optical parametric amplifier (OPA; Spectra-Physics, TOPAS Prime). The output from the OPA at a wavelength of 650 nm was used as the excitation light, unless stated otherwise. The samples were excited from the glass side. The excitation light was filtered with a long-pass color glass filter (>700 nm, R70) and only the PL from the sample was detected by the measurement apparatus. The irradiated area of the excitation light at the sample position was evaluated using a beam profiler (Newport LBP2-HR-VIS2). All the measurements were conducted in air at room temperature (∼297 K).TRMC measurements
TRMC measurements for MACl–FAPbI3 on a quartz monolayer sample and MACl–FAPbI3/TiO2 on a quartz bilayer sample were executed within custom-designed apparatus. Both samples were subjected to OAI passivation. The excitation source employed was the second harmonic (532 nm) of a Nd3+:YAG laser (VN-200-10-ST, VM-TIM). Further details regarding the experimental configuration can be found elsewhere.56,58Author contributions
Naoyuki Nishimura: conceptualization, project administration, investigation, resources, formal analysis, writing – original draft, writing – reviewing and editing; Ranjan Kumar Behera: formal analysis, investigation, writing – reviewing and editing; Ryuzi Katoh: investigation, writing – reviewing and editing; Hiroyuki Kanda: writing – reviewing and editing; Takurou N. Murakami: funding acquisition, writing – reviewing and editing; and Hiroyuki Matsuzaki: investigation, formal analysis, conceptualization, project administration, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is based on results obtained from a project, JPNP21016, commissioned by the New Energy and Industrial Technology Development Organization (NEDO).References
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Gratzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Env. Sci., 2016, 9, 1989–1997 RSC.
- M. Saliba, T. Matsui, K. Domanski, J. Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J. P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- T. M. Koh, K. Fu, Y. Fang, S. Chen, T. C. Sum, N. Mathews, S. G. Mhaisalkar, P. P. Boix and T. Baikie, J. Phys. Chem. C, 2013, 118, 16458–16462 CrossRef.
- Z. Wang, Y. Zhou, S. Pang, Z. Xiao, J. Zhang, W. Chai, H. Xu, Z. Liu, N. P. Padture and G. Cui, Chem. Mater., 2015, 27, 7149–7155 CrossRef CAS.
- F. Xie, C.-C. Chen, Y. Wu, X. Li, M. Cai, X. Liu, X. Yang and L. Han, Energy Env. Sci., 2017, 10, 1942–1949 RSC.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Gratzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- L. Bi, Q. Fu, Z. Zeng, Y. Wang, F. R. Lin, Y. Cheng, H. L. Yip, S. W. Tsang and A. K. Jen, J. Am. Chem. Soc., 2023, 145, 5920–5929 CrossRef CAS PubMed.
- J. Park, J. Kim, H. S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- H. Min, M. Kim, S. U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- E. Choi, J. W. Lee, M. Anaya, A. Mirabelli, H. Shim, J. Strzalka, J. Lim, S. Yun, M. Dubajic, J. Lim, J. Seidel, R. E. Agbenyeke, C. G. Kim, N. J. Jeon, A. M. Soufiani, H. H. Park and J. S. Yun, Adv. Ener. Mater., 2023, 13, 2301717 CrossRef CAS.
- K. A. Elmestekawy, B. M. Gallant, A. D. Wright, P. Holzhey, N. K. Noel, M. B. Johnston, H. J. Snaith and L. M. Herz, ACS Energy Lett., 2023, 8, 2543–2551 CrossRef CAS PubMed.
- X. Huang, F. Cao, S. Zhan, Q. Feng, M. Zhu, Z. Su, X. Gao, J. Yin, J. Li, N. Zheng and B. Wu, Joule, 2023, 7, 1556–1573 CrossRef CAS.
- Y. Kim, G. Kim, E. Y. Park, C. S. Moon, S. J. Lee, J. J. Yoo, S. Nam, J. Im, S. S. Shin, N. J. Jeon and J. Seo, Energy Env. Sci., 2023, 16, 2226–2238 RSC.
- S. Ryu, B. Gil, B. Kim, J. Kim and B. Park, ACS Appl. Mater. Interfaces, 2023, 15(49), 56909–56917 CAS.
- D. Dastan, M. K. A. Mohammed, R. Sh Alnayli, S. M. Majeed, D. S. Ahmed, A. K. Al-Mousoi, R. Pandey, M. K. Hossain, S. Bhattarai, B. A. Al-Asbahi and M. F. Rahman, Langmuir, 2024, 40, 7560–7568 CrossRef CAS PubMed.
- J. Ge, R. Chen, Y. Ma, Y. Wang, Y. Hu, L. Zhang, F. Li, X. Ma, S. W. Tsang, J. You, A. K. Y. Jen and S. F. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319282 CrossRef CAS PubMed.
- K. Park, S. Tan, T. Kodalle, D. K. Lee, M. Abdelsamie, J. S. Park, J. H. Lee, S. K. Jung, J. H. Ko, N. G. Park, C. M. Sutter-Fella, Y. Yang and J. W. Lee, Adv. Mater., 2024, 36, e2307265 CrossRef PubMed.
- X. Wang, H. Huang, M. Wang, Z. Lan, P. Cui, S. Du, Y. Yang, L. Yan, Q. Zhang, S. Qu and M. Li, Adv. Mater., 2024, 36, e2310710 CrossRef PubMed.
- N. Nishimura, S. Mathew and T. N. Murakami, New J. Chem., 2023, 47, 4197–4201 RSC.
- N. Nishimura, H. Tachibana, R. Katoh, H. Kanda and T. N. Murakami, ACS Appl. Mater. Interfaces, 2023, 15, 44859–44866 CrossRef CAS PubMed.
- J. W. Yoo, E. Noh, J. Jang, K. S. Lee, J. Byeon, M. Choi, J. Im and S. I. Seok, Joule, 2023, 7, 797–809 CrossRef CAS.
- K. Huang, L. Chang, Y. Hou, W. Ji, T. Trân-Phú, A. D. Bui, A. O. Mayon, W. Wang, O. L. C. Lem, D. T. Nguyen, G. D. Tabi, L. Duan, Y. Liu, H. Shen, J. Yang, T. P. White, K. R. Catchpole, K. J. Weber and T. Duong, Adv. Ener. Mater., 2024, 2304073 CrossRef CAS.
- Y. Wang, S.-C. Chen, S. Tai, D. Wang, Y. Ma, J. Wu and M.-J. Lin, J. Mater. Chem. C, 2024, 12, 6540–6547 RSC.
- F. Feng, Y. Guan, F. Liu, C. Wu, H. Su, B. Wang, X. Zhang, Y. Liang, S. Cai, Y. Zhang, L. Xiao and S. Zheng, J. Mater. Chem. C, 2024, 12, 3410–3417 RSC.
- T. M. Koh, V. Shanmugam, X. Guo, S. S. Lim, O. Filonik, E. M. Herzig, P. Müller-Buschbaum, V. Swamy, S. T. Chien, S. G. Mhaisalkar and N. Mathews, J. Mater. Chem. A, 2018, 6, 2122–2128 RSC.
- H. Kim, S. U. Lee, D. Y. Lee, M. J. Paik, H. Na, J. Lee and S. I. Seok, Adv. Energy Mater., 2019, 9, 1902740 CrossRef CAS.
- S. Akin, B. Dong, L. Pfeifer, Y. Liu, M. Graetzel and A. Hagfeldt, Adv. Sci., 2021, 8, 2004593 CrossRef CAS PubMed.
- J. Y. Chun, B. G. Kim, W. Jang and D. H. Wang, Appl. Surf. Sci., 2022, 591, 22.153207 CrossRef.
- W. Jang, J. Lee, B. G. Kim, J. Lim, Z. Yang and D. H. Wang, Cryst. Growth Des., 2023, 23, 6916–6925 CrossRef CAS.
- T. Handa, D. M. Tex, A. Shimazaki, A. Wakamiya and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 954–960 CrossRef CAS PubMed.
- Y. Kanemitsu and T. Handa, Jpn. J. Appl. Phys., 2018, 57, 090101 CrossRef.
- Y. Pihosh, V. Nandal, R. Shoji, R. Bekarevich, T. Higashi, V. Nicolosi, H. Matsuzaki, K. Seki and K. Domen, ACS Energy Lett., 2023, 8, 2106–2112 CrossRef CAS.
- A. D. Wright, G. Volonakis, J. Borchert, C. L. Davies, F. Giustino, M. B. Johnston and L. M. Herz, Nat. Mater., 2020, 19, 1201–1206 CrossRef CAS PubMed.
- G. C. Papavassiliou and I. B. Koutselas, Synth. Met., 1995, 71, 1713–1714 CrossRef CAS.
- N. Kitazawa, Y. Watanabe and Y. Nakamura, J. Mater. Sci., 2002, 37, 3585–3587 CrossRef CAS.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. Garcia de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z. H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- E. Mosconi, E. Ronca and F. De Angelis, J. Phys. Chem. Lett., 2014, 5, 2619–2625 CrossRef CAS PubMed.
- Y. Ding, B. Ding, H. Kanda, O. J. Usiobo, T. Gallet, Z. Yang, Y. Liu, H. Huang, J. Sheng, C. Liu, Y. Yang, V. I. E. Queloz, X. Zhang, J. N. Audinot, A. Redinger, W. Dang, E. Mosconic, W. Luo, F. De Angelis, M. Wang, P. Dorflinger, M. Armer, V. Schmid, R. Wang, K. G. Brooks, J. Wu, V. Dyakonov, G. Yang, S. Dai, P. J. Dyson and M. K. Nazeeruddin, Nat. Nanotechnol., 2022, 17, 598–605 CrossRef CAS PubMed.
- N. Lalpour, V. Mirkhani, R. Keshavarzi, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and P. Gao, Sci. Rep., 2023, 13, 6368 CrossRef CAS PubMed.
- J. Liu, S. Li, Z. Qiu, Y. Liu, C. Qiu, W. Zhang, J. Qi, K. Chen, W. Wang, C. Wang, Z. Cui, Y. Su, Y. Hu, A. Mei and H. Han, Small, 2023, 19, e2300737 CrossRef PubMed.
- X. Pu, Q. Cao, J. Su, J. Yang, T. Wang, Y. Zhang, H. Chen, X. He, X. Chen and X. Li, Adv. Energy Mater., 2023, 13, 2301607 CrossRef CAS.
- S. Takahashi, S. Uchida, P. V. V. Jayaweera, S. Kaneko and H. Segawa, Sci. Rep., 2023, 13, 16068 CrossRef CAS PubMed.
- G. Giorgi, J. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. Lett., 2013, 4, 4213–4216 CrossRef CAS PubMed.
- Y. Wu, X. Yang, H. Chen, K. Zhang, C. Qin, J. Liu, W. Peng, A. Islam, E. Bi, F. Ye, M. Yin, P. Zhang and L. Han, Appl. Phys. Express, 2014, 7, 052301 CrossRef CAS.
- J. Choi, S. Song, M. T. Horantner, H. J. Snaith and T. Park, ACS Nano, 2016, 10, 6029–6036 CrossRef CAS PubMed.
- C. H. Ji, K. T. Kim and S. Y. Oh, RSC Adv., 2018, 8, 8302–8309 RSC.
- X. Guo, B. Zhang, Z. Lin, J. Ma, J. Su, W. Zhu, C. Zhang, J. Zhang, J. Chang and Y. Hao, Org. Electron., 2018, 62, 459–467 CrossRef CAS.
- M. T. Masood, S. Qudsia, M. Hadadian, C. Weinberger, M. Nyman, C. Ahlang, S. Dahlstrom, M. Liu, P. Vivo, R. Osterbacka and J. H. Smatt, Nanomaterials, 2020, 10, 10010181 CrossRef PubMed.
- S. Sandhu, C. Saharan, S. K. Buruga, S. A. Kumar, P. S. Rana, P. C. Nagajyothi and S. D. Mane, Ceram. Int., 2021, 47, 14665–14672 CrossRef CAS.
- Z. Dong, J. Wang, J. Men, J. Zhang, J. Wu, Y. Lin, X. Xie, J. Wang and J. Zhang, Inorg. Chem., 2024, 63, 5709–5717 CrossRef CAS PubMed.
- K.-M. Lee, W.-J. Lin, S.-H. Chen and M.-C. Wu, Org. Electron., 2020, 77, 105406 CrossRef CAS.
- S.-H. Chen, C.-M. Ho, Y.-H. Chang, K.-M. Lee and M.-C. Wu, Chem. Eng. J., 2022, 445, 136761 CrossRef CAS.
- R. Katoh, A. Furube, K.-i Yamanaka and T. Morikawa, J. Phys. Chem. Lett, 2010, 1, 3261–3265 CrossRef CAS.
- H. Oga, A. Saeki, Y. Ogomi, S. Hayase and S. Seki, J. Am. Chem. Soc., 2014, 136, 13818–13825 CrossRef CAS PubMed.
- S. Nakajima and R. Katoh, J. Mater. Chem. A, 2015, 3, 15466–15472 RSC.
- E. M. Hutter, J. J. Hofman, M. L. Petrus, M. Moes, R. D. Abellón, P. Docampo and T. J. Savenije, Adv. Energy Mater., 2017, 7, 1602349 CrossRef.
- M. L. Petrus, K. Schutt, M. T. Sirtl, E. M. Hutter, A. C. Closs, J. M. Ball, J. C. Bijleveld, A. Petrozza, T. Bein, T. J. Dingemans, T. J. Savenije, H. Snaith and P. Docampo, Adv. Energy Mater., 2018, 8, 1801605 CrossRef.
- N. Nishimura, H. Kanda, R. Katoh, A. Kogo and T. N. Murakami, J. Mater. Chem. A, 2024 10.1039/D4TA02036G.
- N. Nishimura, H. Tachibana, R. Katoh, H. Kanda and T. N. Murakami, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-ts5k7.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00843j |
| This journal is © The Royal Society of Chemistry 2024 |