
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalised Al(III) metal organic frameworks for fluorescence sensing of nitroaromatic vapours†
Amina
Haj-Yahya
 a,
Dimitra
Kouskouki
a,
Antigoni G.
Margellou
a,
Evangelos K.
Andreou
b,
Gerasimos S.
Armatas
b and
Theodore
Lazarides
*a
a,
Dimitra
Kouskouki
a,
Antigoni G.
Margellou
a,
Evangelos K.
Andreou
b,
Gerasimos S.
Armatas
b and
Theodore
Lazarides
*a
aDepartment of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: tlazarides@chem.auth.gr
bDepartment of Materials Science and Engineering, University of Crete, Vassilika Vouton, 70013 Heraklion, Greece
First published on 14th May 2024
Abstract
The employment of fluorescence sensors provides a platform for rapid and efficient in-field detection of nitroaromatic compounds and is gaining increasing research ground. Herein, we report the synthesis and characterisation of three new fluorescent Al(III) MOFs, structurally analogous to MIL-53, with the assigned formula {Al(OH)(bdc)1−n(L-1)n}·xsolv (bdc2− = terephthalate; L-1 = 2-((benzyl)amino)-terephthalate). L-1 is a strongly fluorescent dicarboxylic ligand with a pendant π-electron rich aromatic group suitable for donor–acceptor interactions with electron-deficient nitroaromatic guests. Our MOFs show strong fluorescence quenching upon exposure to vapours of nitrobenzene, 1,3-dinitrobenzene, 4-nitrotoluene, and 2,4-dinitrotoluene. Additionally, we prepare and study MOF-polymer composites in the form of thin films that are strongly quenched in the presence of nitrobenzene vapours.
Introduction
Nitroaromatic compounds (NACs) are known for their highly explosive nature and, along with other highly nitrated compounds, are primary components of industrial explosives. They are widely used in military operations and terrorist acts and are found in many unexploded land mines worldwide. Furthermore, the extensive industrial use of NACs combined with their chemical stability has led to persistent contamination of soil and groundwater. Therefore, their constant monitoring in liquid and/or vapour phase is vital for environmental protection and population safety.1–4 To this day, the monitoring of NACs is mainly based on instrumental techniques (i.e. GC-MS, IMS, Raman) and on sniffer dogs for in-field detection. Despite their effectiveness, traditional analytical methods suffer from high cost, lack of portability and the requirement for highly trained personnel.5–8One new promising sensing technology is based on the detection of NACs by monitoring the luminescence response of sensory materials (changes in intensity, wavelength, quantum yield or lifetime of emission). Optical sensors are effective analytical tools due to their portability, facile visualisation and short response times.9–13 Most optical sensing devices used by the security industry today employ fluorescent conjugated polymers (CPs), which were first introduced for the detection of TNT by Swager and co-workers.14,15 The electron-deficient nature of NACs provides a convenient spectroscopic handle for their fluorimetric detection through mechanisms based on photoinduced electron and/or energy transfer. The conjugated polymer chain serves as an efficient transport medium for mobile excitons that sample many chromophore units along its length thus increasing their chances of encountering a site where an analyte is bound. This results in great signal amplification which increases the sensing sensitivity of CP sensors (often referred to as the “molecular-wire” effect).16–21
Luminescent metal organic frameworks (LMOFs) constitute one more class of materials which consist of interconnected chromophores and can produce enhanced signal upon interaction with analytes in an analogous way to CPs.22–26 MOFs are hybrid organic/inorganic crystalline materials that can be self-assembled from their corresponding metal ions or clusters and bridging organic ligands to form extended periodic and potentially porous structures. LMOFs provide some key advantages for optical sensing: they possess well-defined crystalline structures with surface functionality and tuneable functional sites that allow host–guest interactions between the framework and the analyte.27–30 Numerous LMOFs, mainly based on π-conjugated linkers, have been studied for the optical sensing of NACs and other electron-deficient targets.31–36 Although most research work has focused on detecting nitroaromatic analytes in solution, the detection of these chemicals in vapour phase is equally important.37–41
Herein, we report the fluorescence properties and sensing behaviour of a series of mixed linker Al(III) MOFs belonging to the MIL-53 structural type42 with the assigned formula {Al(OH)(bdc)1−n(L-1)n}·xsolv (bdc2− = terephthalate; L-1 = 2-((benzyl)amino)-terephthalate43). L-1 is a strongly fluorescent dicarboxylic acid with a pendant π-electron rich benzylamino aromatic group that fosters electron transfer processes between our MOFs and electron-deficient nitroaromatic guests. We study the fluorescence properties of these compounds that show selective strong emission quenching upon exposure to vapours of explosive-related NACs and are also capable of enhancing their fluorescence in the presence of electron-donating analytes.
Results and discussion
Synthesis and characterisation
In our previous work,43 we reported a Zr(IV) UiO-66 type MOF (Zr-1) based on the strongly fluorescent ligand 2-benzylaminoterephthalic acid (L-1). After acid activation, the protonated material pZr-1 can function as an ion exchange sensor for the anionic nitrophenols 2,4,6-trinitrophenol (TNP) and 2,4-dinitrophenol (DNP) in aqueous environment, with maximum quenching of ca. 90%. Zr-MOFs with amino-functionalised linkers have been reported to exhibit self-quenching phenomena due to the relative proximity of the chromophores within the matrix that allows inter-ligand charge transfer processes assisted by amino-groups.44 In an attempt to enhance the emission signal of our sensory materials, we decided to expand our research towards Al(III) MOFs that provide stable and flexible scaffolds with strong luminescence properties.We synthesised a series of fluorescent Al(III) MIL-53 type MOFs using terephthalic acid (H2bdc) and the fluorescent ligand L-1. As shown in Scheme 1 the preparation of Al-MOFs followed a modified solvothermal reaction methodology45–47 where AlCl3 and the respective ligands were heated at 120 °C for 24 h in H2O/DMF solvent mixtures within Teflon-lined stainless-steel autoclaves. The solid products were thoroughly soaked and washed with DMF to remove the residual reactants entrapped within the materials’ pores and, afterwards, the DMF molecules were exchanged by extensive soaking in MeOH with gentle heating (ca. 50 °C) to afford the final products.
 | ||
| Scheme 1 Schematic representation of the solvothermal synthetic procedure. | ||
Incorporation of different relative percentages of L-1 and H2bdc produced the following three MOFs: {Al(OH)(L-1)} (Al-1), {Al(OH)(bdc)0.5(L-1)0.5} (Al-0.5) and {Al(OH)(bdc)0.75(L-1)0.25} (Al-0.25). We confirmed the L-1 loading (mol% with respect to bdc) by 1H-NMR analysis on digested MOF samples in D2O/NaOD. We found that in general the determined L-1 loadings showed good agreement with ligand molar ratios in the reaction feed. The 1H-NMR spectra of digested MOFs and details on the methodology for determination of L-1 loadings can be found in ESI† (NMR spectroscopy & Fig. S1–S3, ESI†).
Powder X-ray diffraction (PXRD) patterns of pristine samples, shown in Fig. 1, demonstrate that all three materials possess crystalline frameworks and are in good agreement with the diffraction pattern of NH2–MIL-53(as).46 To further validate this, we used the PXRD data of the MOFs with higher loadings of L-1, Al-1 and Al-0.5, to apply Le Bail (structureless) refinements that showed adequate fitting to the diffraction pattern of the open-pore structure of MIL-53,48 with no extra diffraction peaks appearing and agreement factors calculated to ≤10 (Fig. S12 and S13, ESI†).
 | ||
| Fig. 1 PXRD patterns of NH2–MIL-53(as)46 (black) and pristine Al-1 (red), Al-0.5 (blue), Al-0.25 (green). | ||
The metal secondary building unit (SBU) of the MIL-53 framework48 comprises of an infinite rod of apex-sharing zig-zag chains of AlO4(OH)2 octahedra, with four carboxylic O atoms occupying the equatorial plane and the bridging OH− ligands occupying the axial positions. Each chain is connected to four neighbouring inorganic chains by bridging terephthalate ligands, thus forming one-dimensional rhomboidal channels running along the crystallographic a-axis (Fig. 2). In our modified frameworks Al-chains are interconnected by terephthalate or L-1 linkers with the benzylamine side-groups projecting inside the rhomboidal channels (Scheme 1), as determined by Manos and co-workers for an analogous MOF containing 2-picolylamine side groups.49 It has been well reported that MIL-53 displays a large breathing effect corresponding to about 50% volume change upon transitioning from low temperature closed-pore monoclinic structure (MIL-53lt) to high temperature open-pore orthorhombic structure (MIL-53ht).48,50,51 Incorporation of ligand L-1 with a bulky benzyl side group in the MIL-53 framework introduces steric hindrance effects that do not allow contraction of the pores, resulting in our materials conforming to the open-pore form.
 | ||
| Fig. 2 (a) The structure of the parent framework MIL-53 in its open form as viewed along the a axis. (b) View of the metal SBU of MIL-53 showing the infinite zig-zag rod of apex-sharing AlO4(OH)2 octahedra and how they are connected to the rest of the framework through the terephthalate bridging ligands (generated using published data42). | ||
We observe that even though the PXRD pattern of Al-0.25 is dominated by the same pattern as the other two materials, it displays two additional diffraction peaks. We hypothesise that competition between two different crystalline phases arises during the crystallisation process thereby resulting in separate crystalline regions within the framework. Le Bail analysis showed that the PXRD data of Al-0.25 are better fitted to a NH2–MIL-53 Al(III) MOF showing a monoclinic distortion52 (Fig. S14, ESI†).
The IR spectra of all compounds show similar characteristic bands (Fig. S5, ESI†). We observe a broad band at 3658 cm−1 due to O–H stretching of the bridging OH groups from the AlO4(OH)2 chains and the N–H stretch band of the secondary amine group can be seen at 3404 cm−1. The strong absorption bands at ca. 1575 and 1402 cm−1 are attributable to the asymmetric and symmetric stretching vibrations for the μ2-coordinated –COO− groups of L-1. As the L-1 to bdc2− ratio decreases, two new bands emerge, shown more distinctly for Al-0.25, at ca. 1595 and 1443 cm−1 attributed to μ2-coordinated –COO− groups of bdc2−.53,54 Finally, the characteristic low-energy bands of Al–O modes can be seen at ca. 600 and 470 cm−1.55
Chemical and thermal stability
As will be described in following sections, MOF employment in vapour sensing applications requires prior treatment of the sensing materials with water or methanol. We performed PXRD analyses to examine the stability of our samples towards these solvents. In a typical process, the studied sample was treated with water or methanol for 24 h at room temperature and was subsequently isolated via centrifugation and dried at 50 °C for 24 h to afford a fine powder. The PXRD patterns for each sample after water and methanol treatment (Fig. S8, ESI†) are identical to the PXRD patterns of pristine materials confirming that the overall framework retains its crystallinity and remains unaltered.Thermal stability was examined by thermogravimetric (TG) analysis under air flow on pristine and H2O-exchanged samples (Fig. 3 and Fig. S15, ESI†). In overview, all TG curves show a two-step weight loss, starting with loss of guest molecules followed by linker elimination and structural decomposition that leaves an amorphous residue of Al2O3, as confirmed by IR spectroscopy (Fig. S7, ESI†).
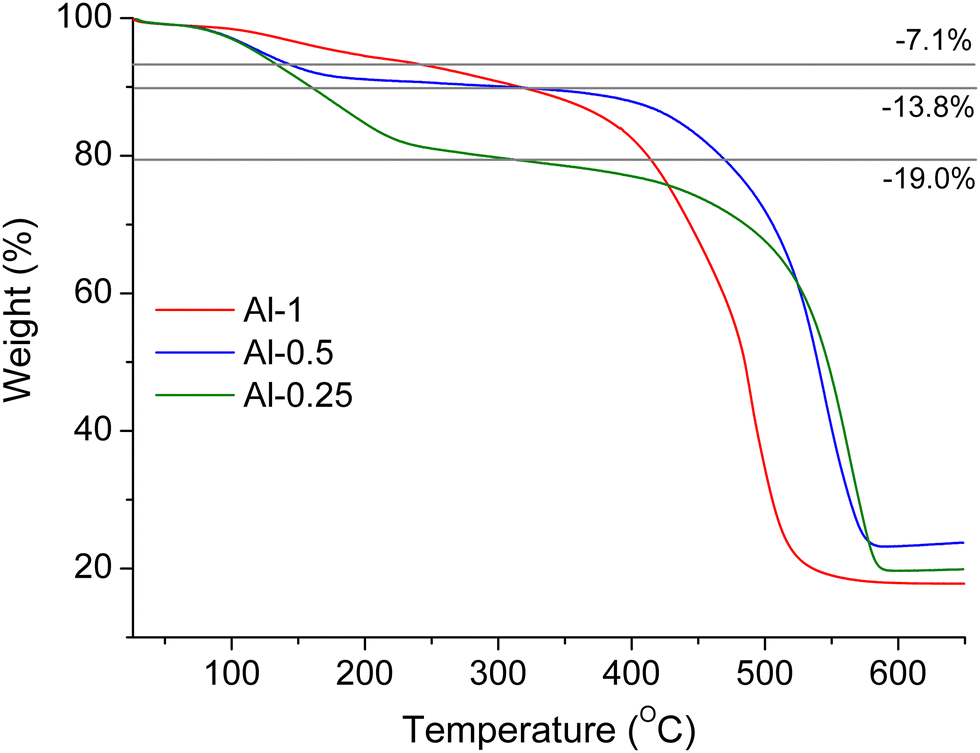 | ||
| Fig. 3 TGA curves for pristine Al-1 (red), Al-0.5 (blue) and Al-0.25 (green) under air flow. | ||
For pristine materials, the first weight loss step at ca. 100–250 °C corresponds to removal of DMF molecules. The second weight loss step due to framework collapse occurs at ca. 350–450 °C. The decomposition temperatures observed for our materials are lower compared to the parent material MIL-53 (∼500 °C)48 but similar to the thermal stability reported for amino-functionalised samples.46 Interestingly, the mass loss due to DMF elimination is greater for lower L-1 loadings, indicating that a smaller percentage of the functionalised ligand provides bigger capacity for DMF molecules inside the framework's pores. Furthermore, the loading amount of L-1 is inversely correlated to the temperature at which the framework collapses, so thermal stability follows the ascending order: Al-1 < Al-0.5 < Al-0.25. Water-exchanged samples show similar TG curves to the pristine compounds, where the initial weight loss due to dehydration is observed up to 100 °C and structural decomposition occurs at 380–480 °C following the same trend described above.
In addition to the TG analysis, we performed temperature dependent X-ray powder diffraction (TDPXRD) measurements on samples of Al-1 and Al-0.5 (Fig. S10 and S11, ESI†) that were heated from room temperature up to 600 °C in steps of 100 °C. The TDPXRD data show that diffraction patterns remain unaltered up to 400 °C after which point the intensity of the reflections decreases due to the framework decomposition resulting in amorphous Al2O3. We do not observe considerable changes in 2θ angles or appearance of new diffraction peaks suggestive of temperature-induced breathing behaviour56 which further demonstrates that the materials adopt an orthorhombic crystallisation phase with no breathing processes involved.
BET surface area and porosity
To evaluate the porosity and specific surface area, we performed N2 adsorption–desorption measurements on MeOH exchanged samples that afforded type I isotherms with narrow hysteresis upon desorption (Fig. 4).57 Surface area calculations following the Brunauer–Emmett–Teller (BET) model revealed limited porosity for our compounds compared to non-functionalised MIL-53 materials. We observe a slight increase in surface area for half loaded Al-0.5 (87 m2 g−1) compared to Al-1 (35 m2 g−1) and a more substantial increase for Al-0.25 (243 m2 g−1). In agreement with TG analysis, N2 sorption measurements also confirm that lowering of the L-1 to bdc2− ratio allows for more void space in the pores. Moreover, the extended porosity of Al-0.25 might also be associated with the coexistence of different crystalline phases within its framework. | ||
| Fig. 4 Nitrogen sorption isotherms at 77 K of Al-1 (red), Al-0.5 (blue) and Al-0.25 (green) after treatment with MeOH and overnight evacuation at 150 °C. | ||
Photophysical properties
As we described in our previous work,43 ligand L-1 in MeOH solution produces a broad emission signal (Fig. 5) with maximum at 455 nm (λexc = 360 nm), a relatively high quantum yield of 53% and an emission lifetime of 11 ns (Fig. S20, ESI†). | ||
| Fig. 5 Normalised emission spectra of L-1 in MeOH (black) (λexc = 360 nm), Al-1 (red), Al-0.5 (blue), Al-0.25 (green) (λexc = 390 nm). | ||
Fluorescence measurements on solid powder samples of our Al-MOFs exhibit turquoise fluorescence upon selective ligand excitation at 390 nm with a similar emission profile that consists of a broad band with maximum at ca. 475 nm, slightly red-shifted for higher L-1 loadings (Fig. 5). It is noticeable that Al-0.25 produces a narrower emission band more similar to that observed for L-1 in solution compared to the other two materials. These observations indicate that there is a degree of interchromophoric interaction within the frameworks which increases at higher L-1 loading.
It is important to mention that our Al(III) MOFs, show much higher fluorescence quantum yields compared to their Zr(IV) counterparts. We found that the emission quantum yields of solid samples of Al-1, Al-0.5 and Al-2.25 range between 15% and 22% (Fig. S21–S23, ESI†), more than ten times higher than the quantum yield determined for Zr-1 (ca. 1%, Fig. S24, ESI†)43 which is based on the same chromophore (L-1). The low emission intensities of Zr(IV) MOFs of the UiO structural type have been attributed to self-quenching phenomena associated with intra-framework excimer formation and inter-ligand energy transfer.58 Additionally, it has been suggested that the Zr(IV) clusters can offer an electron transfer quenching pathway through transient formation of Zr(III)59 even though theoretical and spectroscopic evidence suggest no appreciable direct ligand-to-metal-charge-transfer (LMCT) character in the excited state of 2-aminoterephtahlate-bridged Zr(IV) MOFs.60,61 Since similar inter-ligand quenching phenomena as those found in Zr(IV) MOFs can be at play in our Al(III) MOFs, we propose that the increased emission intensity of the latter primarily stems for the presence of non-redox active Al(III) which precludes electron transfer quenching pathways. Interestingly, among our materials, Al-0.5 is the most efficient emitter with a quantum yield of 22.5% whereas for Al-1 and Al-0.25 we determined quantum yields of 16.2% and 15.5%, respectively. We hypothesise that this small difference in quantum yield values can be interpreted on the basis of the relatively close proximity of the chromophores within the fully loaded material Al-1, that can lead to self-quenching phenomena. Concerning Al-0.25, we can attribute the somewhat lower fluorescence quantum yield to the lower concentration of emitting chromophores.
The materials showed comparable results for time-resolved emission studies (Fig. S25–S27, ESI†) displaying emission decays that could be satisfactorily fitted to three exponential decay functions. As presented in detail in Table 1, there are two clearly dominant components of ca. 5 ns and ca. 10 ns and a faster component of ca. 1 ns with only minor contribution (1–6%). All time constants lie within the fluorescence range and are thus indicative of ligand-based singlet excited states. The observation of multiexponential decays is common in solid-state measurements because, in contrast to studies in dilute solution where the chromophores exist in the same average environment, inhomogeneities within a solid, such as between chromophores in the bulk and those close to the surface of the crystallites, can lead to different rates of radiative and non-radiative relaxation.62 It is worth noting that the longer fluorescence lifetime component (9.5–10.0 ns) shows good agreement with the fluorescence lifetime of L-1 (11 ns). Therefore, the shorter components can be attributed to quenching processes possibly due to close interactions between chromophores in the solid state. These observations are in line with the observed differences in the steady-state emission profiles of the three Al(III) MOFs (vide supra).
| τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | χ 2 | ||||
|---|---|---|---|---|---|---|---|
| Al-1 | 0.9 ± 0.3 | 1.3% | 5.5 ± 0.2 | 58.6% | 10.0 ± 0.3 | 40.0% | 1.011 |
| Al-0.5 | 0.8 ± 0.2 | 1.7% | 4.3 ± 0.1 | 56.8% | 9.4 ± 0.2 | 41.5% | 1.169 |
| Al-0.25 | 1.0 ± 0.9 | 5.9% | 4.7 ± 0.2 | 52.7% | 9.7 ± 0.2 | 41.4% | 1.101 |
Detection of nitroaromatic vapours
Several MOF-based optical sensors have been reported in recent years for the detection of nitroaromatic explosives in the vapour phase. A collection of representative examples can be found in Table S3, ESI.† Most sensory materials reported thus far respond to a group of NACs rather than a single specific target, the main challenge being the extremely low vapour pressures of nitroaromatics. In the case of TNT, the development of sensing applications usually targets its manufacturing precursors and byproducts.63–65 In order to evaluate the sensing ability towards NACs, we studied the fluorescence response of our materials under exposure to equilibrated vapours of nitrobenzene (NB), 1,3-dinitrobenzene (1,3-DNB), 4-nitrotolune (4-NT) and 2,4-dinitrotoluene (2,4-DNT). A noteworthy feature of our MOFs is that the λexc used in sensing experiments lies in lower energy than the absorption regions of targeted analytes (Fig. S28, ESI†), thereby offering better avoidance of inner filter-effects caused by nitroaromatic compounds which absorb in the blue region.In a typical procedure, MOF powder was drop-casted on a microscope cover glass slide and the loaded glass was heated at 120 °C for 24 h. We tested thermal treatment in a range of temperatures (100–180 °C) and opted for a mild activation temperature of 120 °C to ensure that the overall framework retains its crystallinity, and the functionalised ligand remains unaltered (Fig. S1 and S8, ESI†) while the activated MOF shows sufficient sensing performance. Prior to sensing experiments, each sample's emission spectrum was recorded multiple times in the span of 30 minutes to ensure signal stability (Fig. S19, ESI†). Afterwards, the glass was carefully placed within a quartz cuvette containing the analyte and emission spectra were recorder after a specified exposure time (Scheme 2a). The integrated fluorescence intensity data before and after exposure were used for the determination of quenching percentages. A more detailed report on the experimental process can be found in ESI.†
 | ||
| Scheme 2 Schematic representation of (a) the vapour detection setup (b) the possible donor–acceptor interactions between the pore or external surfaces of Al-MOFs and nitroaromatic analytes. | ||
During preliminary detection studies, we concluded that the fully functionalised MOF Al-1 shows slower sensing response compared to the mixed-linker materials (Fig. 6), so we shifted our focus to Al-0.5 and Al-0.25. To examine the sorption capacity of our materials towards NB, we performed TG and 1H-NMR analysis on samples that had been exposed to analyte vapours for a span of 3 days to ensure sorption saturation. The TG curves of NB loaded materials (Fig. S16, ESI†) show an expected two-step weight loss, where the first weight loss step consists of considerably higher mass loss percentages compared to H2O treated samples and extends to 200–250 °C due to the loss of guest analyte molecules occurring at higher temperatures than dehydration. Consistently to what we described for thermogravimetric analysis and surface area measurements; mass loss due to NB elimination is greater for samples with lower L-1 loadings that display greater sorption capacity. To provide further evidence for the interaction between NB and our Al(III) MOFs, we measured the 1H-NMR spectra of digested NB-loaded samples where we clearly observe three additional peaks corresponding to NB (Fig. S4, ESI†).
 | ||
| Fig. 6 Quenching percentage of the emission signals of Al-1, Al-0.5 and Al-0.25 after exposure to vapours of NB and 1,3-DNB for 15 minutes. | ||
In Fig. 7, we see the time-dependent fluorescence quenching of Al-0.5 and Al-0.25 upon continuous exposure to saturated analyte vapours. The quenching percentages appear to approach a plateau given enough exposure time with the order of quenching efficiency being NB > 4-NT > 1,3-DNB > 2,4-DNT. Notably, this order is not in accordance with the electron-withdrawing trend of these analytes and is instead directed by their vapour pressures and corresponding saturated vapour concentrations, given in Table 2. Liquid NB, which possesses the highest vapour pressure, is the strongest quencher producing as much as 85% and 81% quenching on the emission intensity of Al-0.5 and Al-0.25, respectively. While the vapour pressure of 4-NT is comparable to that of NB, its quenching efficiency is reduced possibly because of the presence of the electron-donating –CH3 group. 1,3-DNB and 2,4-DNT possess two electron-withdrawing –NO2 groups but their lower vapor pressures rank them weaker quenchers. We should note that due to the instant luminescence response of our materials in the presence of the analytes, especially in the case of NB, the detection dead time of our experimental process did not allow us to record the emission quenching to its full extent.
 | ||
| Fig. 7 Fluorescence quenching of Al-0.5 (a) and Al-0.25 (b) upon exposure to NB vapours and fluorescence quenching time graphs of Al-0.5 (c) and Al-0.25 (d) upon exposure to NB, 4-NT, 1,3-DNB and 2,4-DNT. | ||
| Vapour pressure (mm Hg) | Saturated concentration (ppm) | Temperature (°C) | |
|---|---|---|---|
| NB | 0.245 | 322.37 | 20–25 |
| 4-NT | 0.01 | 13.16 | 20–25 |
| 1,3-DNB | 0.0002 | 0.26 | 20–25 |
| 2,4-DNT | 0.000147 | 0.19 | 20–25 |
Given the lack of appreciable spectral overlap between the absorption of the analytes and the emissions of the Al(III) MOFs, the observed fluorescence quenching can be explained by a photoinduced electron transfer mechanism.43,66 The intraligand charge transfer character of the excited states of 2-amino terephthalic acid derivatives, makes them potentially good excited state electron donors for electron deficient systems such as nitroaromatic compounds.25 At the same time, the pendant benzyl side groups create a π-electron rich and hydrophobic microenvironment which provides a driving force for nitroaromatic molecules to diffuse within the MOF's cavities. Whilst the restrained available space within the MOFs’ pores limits the possibility of analyte encapsulation, the donor–acceptor interactions could take place between analytes and MOFs’ π-electron rich external surfaces, as depicted in Scheme 2. To ensure that the observed fluorescence quenching is indeed due to the MOF-analyte interaction, we performed an identical experiment for the detection of NB vapours on a solid sample of L-1 that produced no emission intensity changes (Fig. S32, ESI†). Likewise, titration experiments on a solution of L-1 in methanol did not afford any significant quenching upon addition of aliquots of analyte solution. On the other hand, experiments where a suspension of Al-1 was titrated with aliquots of an aqueous solution of 1,3-DNB showed clear emission quenching, albeit much less pronounced than that observed in gas phase experiments possibly due to partial blockage of the MOF-analyte interaction because of hydration effects (Fig. S33, ESI†). These experiments clearly demonstrate that the existence of L-1 within the porous structures of the Al-MOFs is the key factor which unlocks the potential for its interaction with nitroaromatic guests thereby leading to a detection signal.
To quantify and compare the gas sensing response of our materials, we performed exponential decay fit on the quenching data shown in Fig. 7(c) and (d) and the fitting results can be seen in Fig. S38 and S39, ESI.† In all cases, we observe an excellent fit for two exponential components that comprises of a fast component of 1–1.5 minute and a slow component of 10 minutes or higher. In general, the faster process has a greater contribution on the detection of NB and 4-NT whereas the slower process is predominant and reaches higher values of up to 30 minutes for the less volatile analytes 1,3-DNB and 2,4-DNT.
To obtain additional information on the nature of the quenching mechanism upon exposure to analytes, we performed nanosecond time-resolved fluorescence measurements on the Al(III) MOF samples before their exposure to analyte vapours and after the end of each sensing experiment. Details on the fitting results can be found in Table S2, ESI.† Interaction with the quenchers, especially NB, leads to a clear decrease in the values of all lifetime components accompanied with an increase (+10%) in the contribution of the fastest component. However, the degree of lifetime shortening is not in accordance with the observed decrease in fluorescence intensity as would be expected in the case of a fully dynamic quenching mechanism.67 Therefore, we theorise that static and dynamic quenching occur concurrently.58 The static portion of the observed quenching is predominant as it is facilitated by a combination of π–π interactions and hydrogen bonding between nitroaromatic guests and the benzylamino groups of L-1.68
To evaluate their reusability, the sensory materials were regenerated by heating at 120 °C for 24 h. Subsequent exposure to NB vapours resulted in quenching efficiencies of up to 79% for both materials (Fig. S34, ESI†). We observe that after one cycle of analyte exposure and re-activation the quenching efficiency values are slightly reduced, albeit the regenerated materials still exhibit sufficient quenching.
To investigate the sensing selectivity of our MOFs, we measured their emission response towards aliphatic analyte nitromethane and electron donating analytes benzene and toluene. Exposure to nitromethane, which is smaller in size but lacks the aromatic ring which favours π–π interactions, produced quenching percentages of ≤20% (Fig. S35, ESI†). In contrast, in the presence of electron-rich benzene and toluene both MOFs exhibit signal enhancement (Fig. S36 and S37, ESI†). Al-0.25 exposed to toluene reaches a potent enhancement percentage of ca. 100% after just 20 minutes of exposure. Moreover, in the case of toluene we observe a red-shift of up to 10 nm for Al-0.5 that could be associated with exciplex formation between MOF and analyte.38
The fabrication of MOF films is a crucial step towards incorporating the sensory materials in optical devices for practical in-field applications.69–72 In an initial effort to prepare fluorescent films using samples of Al-0.5 and Al-0.25, we mixed ca. 1 g of a gel containing 1.5%wt of a cellulose based polymer (hydroxyethyl cellulose, HEC) with 10 mg of MOF powder and the resulting paste was applied to cover glass slides to produce MOF–polymer composites Al-0.50@HEC and Al-0.25@HEC. PXRD patterns confirm that our MOFs retain their crystallinity within the composite materials (Fig. S9, ESI†). The polymer is visible on the samples’ IR spectra, where the hydroxyl groups produce a characteristic strong broad band centred around 3400 cm−1. Additionally, the C–H stretch bands can be seen at around 2870 cm−1 and the C–O stretch band of the anhydroglucose units can be seen at ca. 1050 cm−1 (Fig. S6, ESI†).73
The loaded glasses were heated at 120 °C for 24 h and were used in fluorescent measurements following the same procedure described above. Quenching results upon exposure to NB vapours can be seen in Fig. 8. The MOF films display strong emission quenching, comparable to quenching results calculated for the corresponding MOF powders. Among the studied materials, Al-0.25@HEC provides the most efficient sensing system, reaching quenching of 89% in 25 minutes.
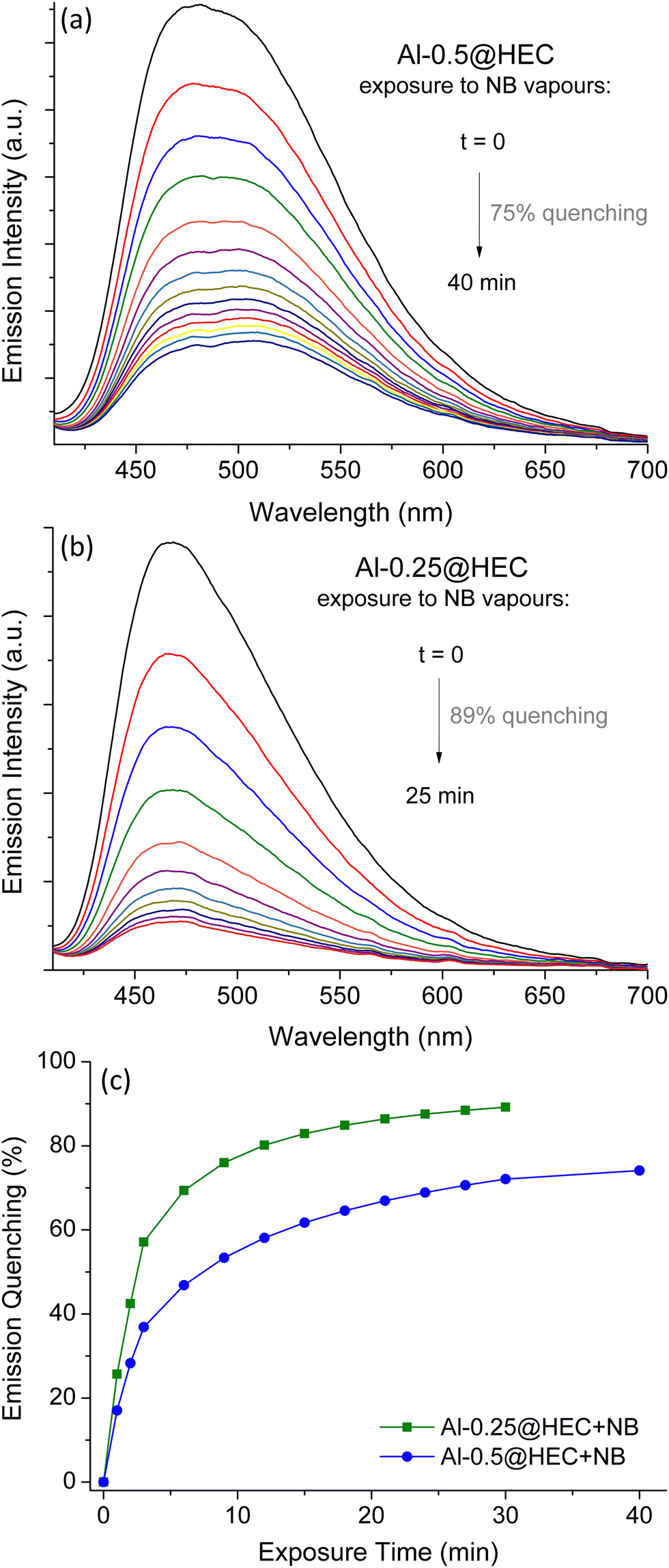 | ||
| Fig. 8 Fluorescence quenching of Al-0.5@HEC (a) and Al-0.25@HEC (b) upon exposure to NB vapours. (c) Fluorescence quenching time graphs of Al-0.5@HEC (blue) and Al-0.25@HEC (green) upon exposure to NB. | ||
As mentioned above, several MOFs designed for the detection of nitroaromatic vapours have been reported in previous years. Some representative examples are given in Table 3. Most examples from the current literature report transition metal MOFs (mainly Zn, Cd, Pb) along with a number of MOFs based on alkaline earth metals (Mg) and lanthanides (Eu, Tb, Dy). To the best of our knowledge, our materials are the only studied MOFs based on aluminium, a cheap and abundant metal that is environmentally benign and, thus, suitable for environmental remediation and monitoring applications. We also show that Al-MOFs possess high thermal and hydrolytic stability and retain their sensing efficiency when combined with a polymer binder to form thin films. These features render our sensory materials a useful platform for the possible development of in-field detection systems.
| MOF | Analyte | Quenching (%) | λ exc (nm) | λ em (nm) | Ref. |
|---|---|---|---|---|---|
| [Zn2(bpdc)2(bpee)]·2DMF | DMNB, 2,4-DNT | 85, 84 | 320 | 420 | 75 |
| [Zn2(TCPPE)] | NB, 2,4-DNT | >80 | 365 | 535 | 76 |
| [Cd3(L)(bipy)2·4DMA]n | NB | >90 | 314 | 381 | 77 |
| [Tb4(L)6(H2O)8] | Various NACs | Up to 94 | 360 | 543 | 78 |
| {Al(OH)(L-1)} | Various NACs | Up to 89 | 360 | 475 | This work |
| {Al(OH)(bdc)0.5(L-1)0.5} | |||||
| {Al(OH)(bdc)0.75(L-1)0.25} | |||||
Conclusions
We have presented the preparation and study of a series of fluorescent Al(III) MOFs: {Al(OH)(L-1)} (Al-1), {Al(OH)(bdc)0.5(L-1)0.5} (Al-0.5) and {Al(OH)(bdc)0.75(L-1)0.25} (Al-0.25), based on terephthalate (bdc2−) and the strongly fluorescent ligand L-1. L-1 carries a pendant π-electron rich benzylamino group that forms a favourable environment for donor–acceptot interactions with electron-deficient nitroaromatic guests. Powder X-ray diffraction studies combined with temperature dependent X-ray powder diffraction measurements showed that the materials are structurally analogous to MIL-53 and adopt an open pore form, due to steric hindrance introduced by the bulky side group of L-1 that does not allow pore contraction. Our MOFs retain crystallinity after treatment with water and methanol and show thermal stability comparable to what has been reported for other MIL-53 type MOFs. Exposure of the sensory materials to nitroaromatic vapours produced strong quenching effects with quenching percentages of up to 85% and the order of quenching efficiency being NB > 4-NT > 1,3-DNB > 2,4-DNT, in accordance with the analytes’ corresponding vapour pressures. The sensing response of mixed-linker materials Al-0.5 and Al-0.25 was superior to that of the fully functionalised Al-1, an effect most possibly attributed to the formers' increased porosity. A preliminary study of MOF-polymer composites in the form of thin films showed that, in the presence of NB vapours, the materials exhibit a strong quenching performance of up to 89% in 25 minutes. Research on fluorescent metal–organic frameworks for the detection of nitroaromatic compounds is ongoing in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “1st Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment” (Project Number: 3371).Notes and references
- K. S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- D. R. S. Lima, M. L. S. Bezerra, E. B. Neves and F. R. Moreira, Rev. Environ. Health, 2011, 26, 101–110 CAS.
- P. Kovacic and R. Somanathan, J. Appl. Toxicol., 2014, 34, 810–824 CrossRef CAS PubMed.
- C. Zhu, H. Huang and Y. Chen, Environ. Pollut., 2022, 307, 119570 CrossRef CAS PubMed.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS.
- E. K. Oztekin, D. J. Burton and D. W. Hahn, Appl. Spectrosc., 2016, 70, 676–687 CrossRef CAS PubMed.
- D. A. Olley, E. J. Wren, G. Vamvounis, M. J. Fernee, X. Wang, P. L. Burn, P. Meredith and P. E. Shaw, Chem. Mater., 2011, 23, 789–794 CrossRef CAS.
- E. S. Forzani, D. Lu, M. J. Leright, A. D. Aguilar, F. Tsow, R. A. Lalesias, Q. Zhang, J. Lu, J. Li and N. Tao, J. Am. Chem. Soc., 2009, 131, 1390–1391 CrossRef CAS PubMed.
- K. J. Albert and D. R. Walt, Anal. Chem., 2000, 72, 1947–1955 CrossRef CAS PubMed.
- M. E. Germain and M. J. Knapp, J. Am. Chem. Soc., 2008, 130, 5422–5423 CrossRef CAS PubMed.
- M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321–5322 CrossRef CAS.
- S. W. Thomas III, J. P. Amara, R. E. Bjork and T. M. Swager, Chem. Commun., 2005, 4572–4574 RSC.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- A. Narayanan, O. P. Varnavski, T. M. Swager and T. Goodson III, J. Phys. Chem. C, 2008, 112, 881–884 CrossRef CAS.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS PubMed.
- Q. Zhang, C. Zhang, L. Cao, Z. Wang, B. An, Z. Lin, R. Huang, Z. Zhang, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 5308–5315 CrossRef CAS PubMed.
- S. A. Diamantis, A. Margariti, A. D. Pournara, G. S. Papaefstathiou, M. J. Manos and T. Lazarides, Inorg. Chem. Front., 2018, 5, 1493–1511 RSC.
- A. Sharma, D. Kim, J.-H. Park, S. Rakshit, J. Seong, G. H. Jeong, O.-H. Kwon and M. S. Lah, Commun. Chem., 2019, 2, 39 CrossRef.
- S. S. Rajasree, J. Yu, S. M. Pratik, X. Li, R. Wang, A. S. Kumbhar, S. Goswami, C. J. Cramer and P. Deria, J. Am. Chem. Soc., 2022, 144, 1396–1406 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 1979, 2013(341), 1230444 Search PubMed.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- G.-L. Yang, X.-L. Jiang, H. Xu and B. Zhao, Small, 2021, 17, 2005327 CrossRef CAS PubMed.
- D.-G. Cai, T.-F. Zheng, S.-J. Liu and H.-R. Wen, Dalton Trans., 2024, 53, 394–409 RSC.
- J. H. Lee, S. Kang, J. Y. Lee, J. Jaworski and J. H. Jung, Chem. Eur. J., 2013, 19, 16665–16671 CrossRef CAS PubMed.
- F. Zhang, Y. Wang, T. Chu, Z. Wang, W. Li and Y. Yang, Analyst, 2016, 141, 4502–4510 RSC.
- N. Xu, Q. Zhang and G. Zhang, Dalton Trans., 2019, 48, 2683–2691 RSC.
- C. Yu, X. Sun, L. Zou, G. Li, L. Zhang and Y. Liu, Inorg. Chem., 2019, 58, 4026–4032 CrossRef CAS PubMed.
- H. Xu, F. Zhong, F. Chen, T.-X. Luan, P. Li, S. Xu and J. Gao, J. Mater. Chem. C, 2022, 10, 7469–7475 RSC.
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem. – Eur. J., 2014, 20, 2276–2291 CrossRef CAS PubMed.
- Z. Hu, S. Pramanik, K. Tan, C. Zheng, W. Liu, X. Zhang, Y. J. Chabal and J. Li, Cryst. Growth Des., 2013, 13, 4204–4207 CrossRef CAS.
- S. Pramanik, Z. Hu, X. Zhang, C. Zheng, S. Kelly and J. Li, Chem. – Eur. J., 2013, 19, 15964–15971 CrossRef CAS PubMed.
- M. Jurcic, W. J. Peveler, C. N. Savory, D. O. Scanlon, A. J. Kenyon and I. P. Parkin, J. Mater. Chem. A, 2015, 3, 6351–6359 RSC.
- D. Liu, X. Liu, Y. Liu, Y. Yu, F. Chen and C. Wang, Dalton Trans., 2014, 43, 15237–15244 RSC.
- H. Zhao, L. Di, S.-W. Wang, J.-J. Zhang, Z. Liu, W.-J. Fang, S.-Q. Liu, J. Ni and X.-D. Song, Sens. Actuators, B, 2021, 328, 129025 CrossRef CAS.
- G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 2976–2977 RSC.
- A. Chatz-Giachia, A. E. Psalti, A. D. Pournara, M. J. Manos, C. Pappa, K. Triantafyllidis and T. Lazarides, J. Mater. Chem. C, 2022, 10, 12307–12315 RSC.
- M. Gutiérrez, F. Sánchez and A. Douhal, Chem. – Eur. J., 2016, 22, 13072–13082 CrossRef PubMed.
- T. K. Trung, P. Trens, N. Tanchoux, S. Bourrelly, P. L. Llewellyn, S. Loera-Serna, C. Serre, T. Loiseau, F. Fajula and G. Férey, J. Am. Chem. Soc., 2008, 130, 16926–16932 CrossRef CAS PubMed.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48, 3057–3064 CrossRef CAS PubMed.
- X. Cheng, A. Zhang, K. Hou, M. Liu, Y. Wang, C. Song, G. Zhang and X. Guo, Dalton Trans., 2013, 42, 13698–13705 RSC.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- D. Evangelou, A. Pournara, C. Tziasiou, E. Andreou, G. S. Armatas and M. J. Manos, Inorg. Chem., 2022, 61, 2017–2030 CrossRef CAS PubMed.
- O. Weser and V. Veryazov, Front. Chem., 2017, 5 Search PubMed.
- J. Wieme, K. Lejaeghere, G. Kresse and V. Van Speybroeck, Nat. Commun., 2018, 9, 4899 CrossRef CAS PubMed.
- J. M. Chin, E. Y. Chen, A. G. Menon, H. Y. Tan, A. T. S. Hor, M. K. Schreyer and J. Xu, CrystEngComm, 2013, 15, 654–657 RSC.
- M. Mihaylov, K. Chakarova, S. Andonova, N. Drenchev, E. Ivanova, A. Sabetghadam, B. Seoane, J. Gascon, F. Kapteijn and K. Hadjiivanov, J. Phys. Chem. C, 2016, 120, 23584–23595 CrossRef CAS.
- Z. Jia, M. Jiang and G. Wu, J. Chem. Eng., 2017, 307, 283–290 CrossRef CAS.
- C. Djordjević, Spectrochim. Acta, 1961, 17, 448–453 CrossRef.
- N. Reimer, B. Gil, B. Marszalek and N. Stock, CrystEngComm, 2012, 14, 4119–4125 RSC.
- D. V. Patil, P. B. S. Rallapalli, G. P. Dangi, R. J. Tayade, R. S. Somani and H. C. Bajaj, Ind. Eng. Chem. Res., 2011, 50, 10516–10524 CrossRef CAS.
- M. Gutiérrez, R. Navarro, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2017, 19, 16337–16347 RSC.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- J. G. Santaclara, F. Kapteijn, J. Gascon and M. A. van der Veen, CrystEngComm, 2017, 19, 4118–4125 RSC.
- T. Lazarides, T. L. Easun, C. Veyne-Marti, W. Z. Alsindi, M. W. George, N. Deppermann, C. A. Hunter, H. Adams and M. D. Ward, J. Am. Chem. Soc., 2007, 129, 4014–4027 CrossRef CAS PubMed.
- S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
- A. K. Chaudhari, S. S. Nagarkar, B. Joarder and S. K. Ghosh, Cryst. Growth Des., 2013, 13, 3716–3721 CrossRef CAS.
- J. Qin, B. Ma, X.-F. Liu, H.-L. Lu, X.-Y. Dong, S.-Q. Zang and H. Hou, J. Mater. Chem. A, 2015, 3, 12690–12697 RSC.
- L. Liu, X. Chen, J. Qiu and C. Hao, Dalton Trans., 2015, 44, 2897–2906 RSC.
- V. Balzani, P. Ceroni and J. Alberto, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, Weinheim, 2014 Search PubMed.
- L. Liu, X. Chen, J. Qiu and C. Hao, Dalton Trans., 2015, 44, 2897–2906 RSC.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- F. G. Moscoso, J. Almeida, A. Sousaraei, T. Lopes-Costa, A. M. G. Silva, J. Cabanillas-Gonzalez, L. Cunha-Silva and J. M. Pedrosa, J. Mater. Chem. C, 2020, 8, 3626–3630 RSC.
- N. Panagiotou, F. G. Moscoso, T. Lopes-Costa, J. M. Pedrosa and A. J. Tasiopoulos, Inorg. Chem. Front., 2022, 9, 4850–4863 RSC.
- A. Abramova, N. Couzon, M. Leloire, P. Nerisson, L. Cantrel, S. Royer, T. Loiseau, C. Volkringer and J. Dhainaut, ACS Appl. Mater. Interfaces, 2022, 14, 10669–10680 CrossRef CAS PubMed.
- K. Wang and L. Ye, Polym.-Plast. Technol. Mater., 2010, 49, 807–811 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 2014, Boca Raton, 95th, revised edn Search PubMed.
- A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed.
- X.-G. Liu, H. Wang, B. Chen, Y. Zou, Z.-G. Gu, Z. Zhao and L. Shen, Chem. Commun., 2015, 51, 1677–1680 RSC.
- F.-Y. Yi, Y. Wang, J.-P. Li, D. Wu, Y.-Q. Lan and Z.-M. Sun, Mater. Horiz., 2015, 2, 245–251 RSC.
- X. Wang, J.-L. Li, C. Jiang, P. Hu, B. Li, T. Zhang and H.-C. Zhou, Chem. Commun., 2018, 54, 13271–13274 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00862f |
| This journal is © The Royal Society of Chemistry 2024 |