
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic plasmonics: PEDOT nanoparticles are getting closer to the visible range†
Pierre
Bléteau
,
Sarra
Gam-Derouich
 *,
Xiaonan
Sun
and
Jean-Christophe
Lacroix
*
*,
Xiaonan
Sun
and
Jean-Christophe
Lacroix
*
Université Paris Cité, ITODYS, CNRS-UMR 7086, 15 rue Jean-Antoine de Baïf, 75205 Paris Cedex 13, France. E-mail: sarra.gam-derouich@u-paris.fr; lacroix@u-paris.fr
First published on 13th June 2024
Abstract
Most plasmonic materials use metallic nanoparticles (NPs) of gold or silver. Plasmonic behaviors are also possible to achieve in several organic materials such as carbon dots or conductive polymers (CPs). Here, we report the synthesis of poly(3,4-ethylenedioyothiophene) (PEDOT) arrays of NPs well organized on a surface by the combined use of e-beam lithography and electropolymerization. The as-grown NPs show plasmonic behavior, with small variations in localized surface plasmon resonance (LSPR) between 1200 and 1300 nm related to the differences in their diameters (90 nm, 120 nm, and 150 nm). The properties of the arrays were chemically tuned by treatment with acetic acid to enhance their conductivity, leading to a tremendous 300 nm blue shift of their optical extinction. The PEDOT NP array after this secondary doping shows an unprecedented LSPR maximum at 960 nm, close to the visible range.
Introduction
Plasmonics is a branch of photonics which uses nanostructured materials that support surface plasmons for the confinement and manipulation of light in matter at the subwavelength scale.1,2 Metallic (gold, silver and copper) nanoparticles (NPs) are the materials and nanostructures most used. They exhibit confined oscillations of their quasi-free electrons in the conduction bands. When the characteristic frequency of these oscillations coincides with that of the light excitation, a strong absorption occurs. This phenomenon is called localized surface plasmon resonance (LSPR) and for many metals it is observed in the visible and near-infrared range. This resonance induces a strong exaltation of the electromagnetic field1,2 very close to the NP structures and has been used in many enhanced spectroscopic techniques, in particular in the development of surface-enhanced Raman spectroscopy (SERS)2,3 and in nanosensors.2,4 Moreover, LSPR can occur at frequencies corresponding to typical electronic excitations of molecules which can lead to strong interactions between localized surface plasmons (LSPs) and molecular systems.5,6 This allows one to manipulate the other and makes it possible to develop active molecular plasmonic devices7–10 and plasmon-induced chemistry.11,12 Plasmonic materials can also yield various metamaterials with negative refractive indices, materials leading to imaging below the diffraction limits,13 chiral metamaterials,14,15 or even cloaks of invisibility for the most futurist projects.16,17While plasmonics mostly use metallic materials, several materials with free charge-carriers are capable of supporting SPR. Hence, LSPR has also been observed in other materials such as inorganic semiconductors,18,19 2D materials such as graphene20 and organic materials.21,22 Regarding the latter class, it has been demonstrated that the two key attributes of plasmonics, field enhancement and subwavelength field confinement, may be achieved following two different strategies, using either free charge-carriers as in doped conducting polymers,21–27 or by exploitation of surface polaritons, arising from organic excitonic materials exhibiting strong absorption band resonance.28–31 Hence, organic plasmonic materials offer an innovative alternative to control light at the nanoscale and present the advantages of being cheap and easily tunable in terms of optoelectronic and structural properties. The specific properties of organic plasmonic materials could also potentially be used to create tunable metamaterials,23,24,30,32–34 or even new spectroscopic enhancement35 or detection schemes36 thanks to specific chemical bonds between a plasmonic organic material and a chemical target.
In this work, we use a conductive polymer (CP) as a plasmonic material supporting LSPR by creating arrays of well-organized CP NPs of various sizes. To do so, we used e-beam lithography combined with electropolymerization. We demonstrate that the LSPR of the CP NPs can be tuned and be pushed towards the visible range by manipulating their properties.
Results and discussion
We have used poly(3,4-ethylenedioxythiophene) (PEDOT) as a conjugated polymer, as it can be easily p-doped in air, to achieve high conductivities,37 and it has already been used to evidence LSPR in CPs.22–24The real part of the dielectric constant of PEDOT is known to be negative (which is a requirement for supporting LSPR38) for light irradiation around or above 1 eV.22,39–42 We have nanostructured this material into NPs of various sizes in order to confine the oscillations of the quasi-free carriers. To do so, we used e-beam lithography combined with electropolymerization.
Lithography was used to create nanoholes of various diameters in an electron-sensitive polymethacrylate (PMMA) layer (100 nm-thick) deposited on an ITO substrate. These steps were fully described in previous papers.11,43 Characterization of the PMMA holes by SEM and AFM is shown in Fig. S1 (ESI†). We then deposited PEDOT in the nanoholes by electropolymerization in aqueous sodium dodecylsulfate (SDS) solutions at low potential,44 in order to control PEDOT growth, and to avoid filling the holes and losing the patterns of the template. DS counter-ions are similar to the commonly used polystyrene sulfonate (PSS) ions but several advantages of using DS have been pointed out.44,45 PEDOT/DS is more conductive and more stable than PEDOT/PSS.
Electrodeposition of PEDOT/DS using cyclic voltammetry is shown in Fig. S2 (ESI†) where experimental conditions are fully explained (aqueous solution of 0.1 M LiClO4, 5 mM EDOT and 0.07 M SDS). The onset of EDOT oxidation occurs around 0.9 V versus SCE. It can be seen that the CVs do not show ultramicroelectrode behavior.46–48 In fact, PMMA was not deposited on the entire ITO surface, so the current recorded was not that from the PMMA holes only but also comes from the uncovered part of the ITO electrode. After PEDOT/DS electrodeposition in the nanoholes, PMMA was removed from the surface by soaking in a 40 °C acetone bath for 30 min.
Fig. 1 and Fig. S3 (ESI†) show the SEM images of an array of PEDOT NPs obtained using this process. Various sizes and shapes of PEDOT/DS NPs are obtained. In Fig. 1a, nanodisks as small as 90 nm in diameter are obtained. They are organized in a regular square array with a center-to-center distance of 360 nm. Fig. 1b also shows the Raman spectra of the various PEDOT arrays. They are very close to those observed for a thin film of a similar thickness and those published in the literature.49 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch vibration signal at 1422 cm−1 does not shift upon nanostructuration which clearly indicates that the oxidation level of the PEDOT/SDS NPs does not change with the NP size.49 Another interesting observation is a small exaltation of the CC stretch vibration signals between 1400 and 1600 cm−1 as a consequence of the nanostructuration. The cylindrical PEDOT NP arrays have also been characterized by AFM. Fig. 1c shows the topography obtained in the tapping mode while Fig. 1d shows a profile of the surface above 5 adjacent NPs. The NPs have almost the same diameter (here 150 nm) and their heights are between 60 and 70 nm (average value 63 nm). AFM reveals that the NPs are not perfect disks but have a volcano-shape (Fig. S4, ESI†). The growth of PEDOT/DS seems to be slightly favored near the PMMA/solution interface. Such a behavior has already been observed using PS beads in experiments combining nanosphere lithography and PEDOT electropolymerization.50,51
C stretch vibration signal at 1422 cm−1 does not shift upon nanostructuration which clearly indicates that the oxidation level of the PEDOT/SDS NPs does not change with the NP size.49 Another interesting observation is a small exaltation of the CC stretch vibration signals between 1400 and 1600 cm−1 as a consequence of the nanostructuration. The cylindrical PEDOT NP arrays have also been characterized by AFM. Fig. 1c shows the topography obtained in the tapping mode while Fig. 1d shows a profile of the surface above 5 adjacent NPs. The NPs have almost the same diameter (here 150 nm) and their heights are between 60 and 70 nm (average value 63 nm). AFM reveals that the NPs are not perfect disks but have a volcano-shape (Fig. S4, ESI†). The growth of PEDOT/DS seems to be slightly favored near the PMMA/solution interface. Such a behavior has already been observed using PS beads in experiments combining nanosphere lithography and PEDOT electropolymerization.50,51
 | ||
| Fig. 1 (a) SEM of PEDOT/DS NP disks with an average diameter of 90 nm. (b) Raman spectra of PEDOT/DS NPs and thin-film PEDOT/DS obtained with laser irradiation of 633 nm and an acquisition time of 3 × 10 s. The baseline was corrected using a polynomial fit. (c) AFM image of PEDOT/DS NP disks with an average diameter of 150 nm. (d) Height profile of PEDOT NPs. | ||
These results demonstrate that the shapes of the PEDOT NPs and their organization can be finely tuned thanks to the combined use of e-beam lithography and electropolymerization.
Fig. 2 and Fig. S5 (ESI†) show the visible-near infrared spectra of the nano-disks with different diameters (90, 120 and 150 nm) and a comparison with those of a thin film of PEDOT/DS deposited under the same electrochemical conditions. The spectrum of the thin PEDOT film does not present a maximum and is similar to those reported in the literature with a broad band, attributed to a bipolaron, starting at 800 nm and covering the whole wavelength range up to 1650 nm. The clear shoulder at 800 nm has been attributed to a polaronic band.52–57
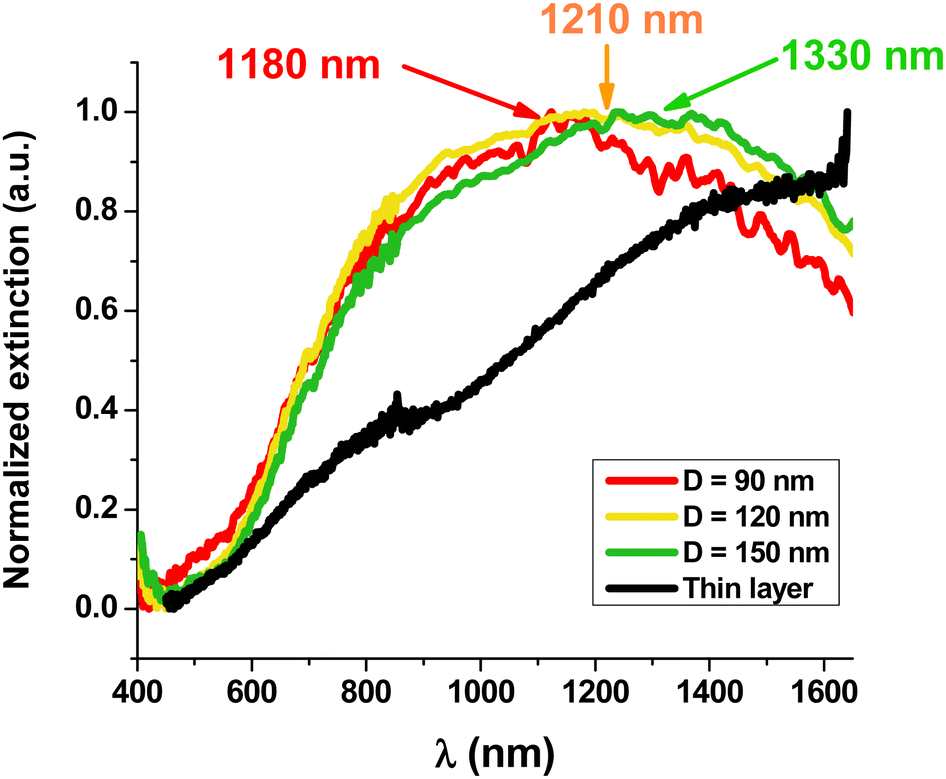 | ||
| Fig. 2 Normalized extinction spectra of nano-disks of PEDOT/DS for different diameters (90 nm-red curve, 120 nm-yellow curve and 150 nm-green curve) and of a thin film (black curve) of electropolymerized PEDOT/DS under the same conditions. | ||
In contrast, the three spectra of the PEDOT arrays bearing NPs with diameters of 150, 120 and 90 nm show clear maxima at 1330, 1210 and 1180 nm, respectively. Despite a quite large full-width at half-maximum (FWHM), we clearly observe that decreasing the diameter of the PEDOT NP leads to a blue shift of the absorption wavelength. This effect is widely known with metallic NPs for which a strong blue shift is observed when their size decreases.38,58 Moreover, the blue shift observed with PEDOT NPs is also in accordance with previous experimental results and theoretical calculations.22,24Table 1 compares the maximum wavelengths for PEDOT NPs reported in this work with those observed by Jonsson for PEDOT nano-disks of similar or larger thicknesses deposited by vapor phase deposition and nanostructured by colloidal lithography.24
The maximum wavelength of PEDOT/DS NPs with 150 nm diameter is closer to the visible range than that of PEDOT/sulf NPs with a similar diameter and height.24 This difference demonstrates that the counter-anion (sulf versus DS) and the differences in the fabrication procedure (vapor phase deposition versus electropolymerization) have a great effect on the PEDOT NP LSPR wavelength. Indeed, counter-ions are known to change the conductive properties of PEDOT.45 Moreover, PEDOT/DS deposited by electropolymerization is most probably better organized, with DS micelles guiding the growth of PEDOT wires.44
The FWHM has also been used to characterize LSPR peaks. It is around 0.75 eV (1000 nm) for PEDOT/DS NPs (Fig. 2). This value can be compared to that of gold NPs absorbing at high wavelength centered at 725 nm or 745 nm (Fig. S6, ESI†) (i.e. gold NPs with diameters of 210 and 240 nm) for which the FWHM is between 0.35 eV (140 nm) and 0.45 eV (200 nm). The larger FWHM of PEDOT is not due to the lack of control of the sizes but suggests that the plasmons in PEDOT have a shorter lifetime. The lifetime of plasmons is related to the electrical conduction relaxation time called 〈τ〉 in this article. τ of PEDOT/PSS was reported to be 7 fs39 while that of copper is 27 fs and that of gold is 9.2 fs.59 The lower electrical conduction relaxation time of PEDOT/DS compared to that of gold results in more losses in the plasmonic NPs, so the lifetime of LSPR is shortened and the FWHM enlarged.60,61
It is well known that the removal of PSS from PEDOT/PSS chains and their replacement by smaller anions lead to an enhancement of the conductivity. Many methods, called secondary doping, have been used for this purpose.56 We have thus used similar methods to enhance PEDOT/DS conductivity. Unfortunately, the adhesion of electropolymerized PEDOT to ITO is poor and, consequently, any attempt to perform a secondary doping of the pristine PEDOT CP arrays, generated by simple PEDOT electropolymerization in aqueous SDS, results in the destruction of the arrays. A small layer of oligothiophenes, 5 nm-thick, was therefore first grafted by diazonium electroreduction in the nanoholes, and PEDOT was then electropolymerized following an already published procedure.62–66 This two-step process considerably increases PEDOT adhesion to the substrate, and secondary doping proved then to be possible without damaging the PEDOT NP arrays (see Fig. S7, ESI†). Among the many available secondary doping processes, we have used acid treatment to remove DS in order to improve the conductivity of the material (this method is known to improve the conductivity by a factor of 1000).67 A droplet of 10 M acetic acid was placed on the NPs and heated at 180 °C for 5 min. Thermal activation facilitates the exchange of acetate and DS anions. These smaller anions replace DS and change the supramolecular structures of the polymer, leading to better inter-chain charge transfer, by analogy with the process already described for PEDOT/PSS.68
The SEM images of the NPs before and after acid treatment are shown in Fig. S7 (ESI†). The NPs did not show changes of geometry and their diameters remain the same. The extinction spectrum, however, showed a tremendous blue shift of almost 300 nm (Fig. 3a). A new maximum peak at 960 nm was recorded and is associated with a FWHM of 0.6 eV (i.e. closer to that of gold NPs with LSPR at the same wavelength). The blue-shift of the plasmonic band maximum was reproducibly observed using arrays of NPs of different sizes (diameters of 120 nm and 240 nm and a height of 40 nm) as shown in Fig. S8 (ESI†). These giant shifts can be attributed to the change of the material properties with permittivity and conductivity changes directly impacting the plasmon resonance.38,59 Note that the Raman spectra did not change upon secondary doping (Fig. S9, ESI†) which indicates that the oxidation level of PEDOT does not change significantly. An important result is that secondary doping of electropolymerized PEDOT films is demonstrated to be a novel approach to generate redox-tunable conducting polymer nanoantennas for visible light.
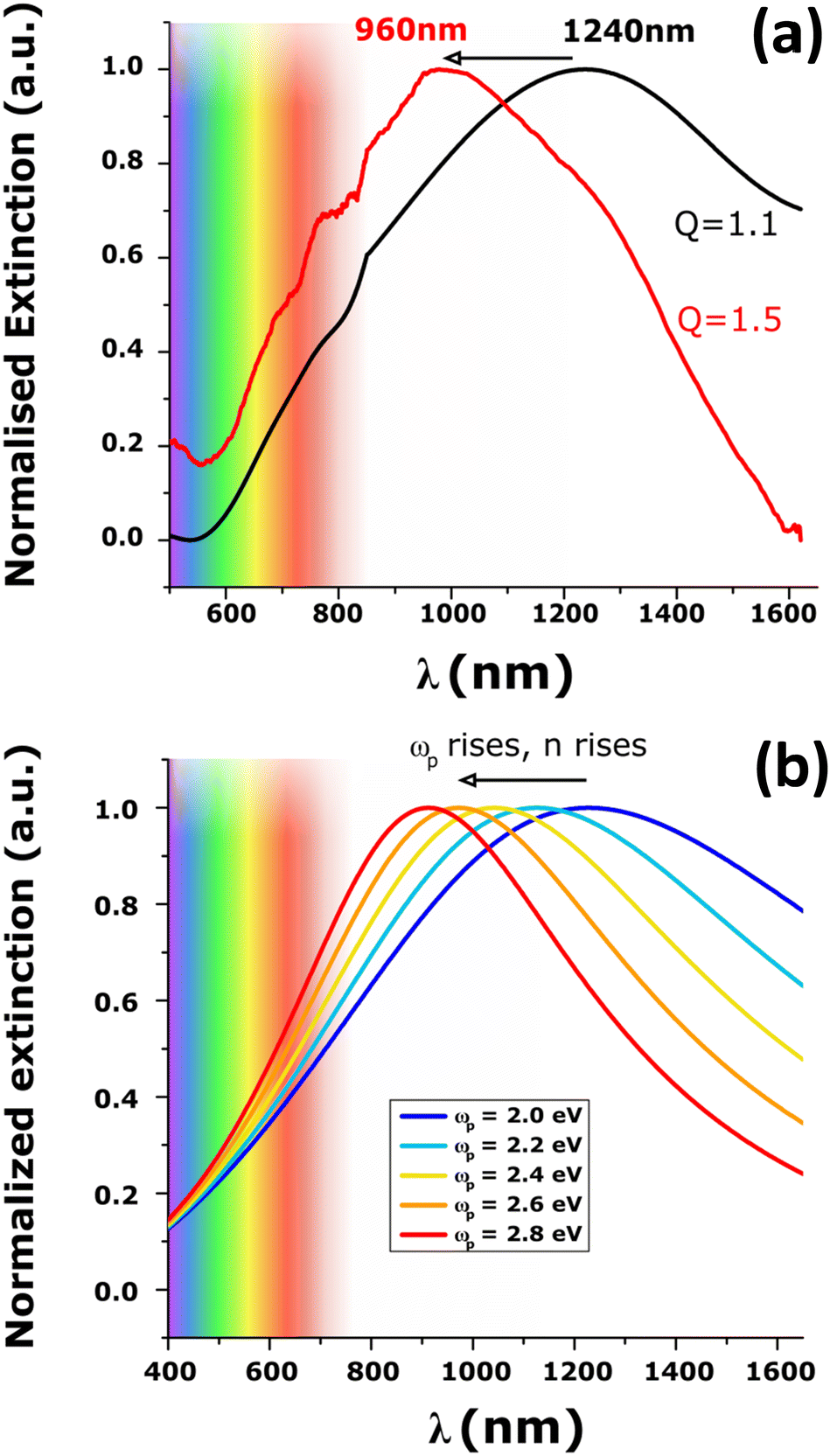 | ||
| Fig. 3 (a) Normalized extinction spectra of PEDOT NPs (diameter 90 nm, height 65 nm) before (black curve) and after (red curve) acid treatment. (b) Simulation of the effect of plasma frequency, i.e. the density of charge-carriers, on the LSPR wavelength calculated using the Drude model and using ωp values from 2 eV to 2.8 eV. | ||
Previous studies demonstrated that optical constants and charge transport properties of CP, including PEDOT can be approximately described, at high frequencies, by the Drude model whereas the localized modified Drude model or other corrected Drude type models need to be used in the low frequency regime.22,24,39–41,69–76
Within the Drude model, the plasma frequency ωp is given by:
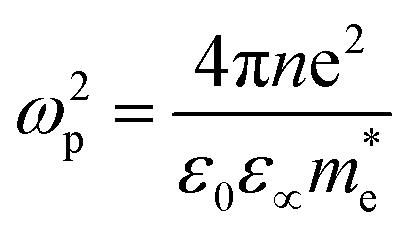
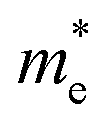 is the effective mass of charge carriers, and ε∝ and ε0 represent the dielectric constants of the medium and free space, while the electrical conductivity in this model is given by:
is the effective mass of charge carriers, and ε∝ and ε0 represent the dielectric constants of the medium and free space, while the electrical conductivity in this model is given by:where 〈τ〉 is the mean value of the electrical conduction relaxation time, i.e. the mean value of the time between two successive collisions, and μ is the charge-carrier mobility. Hence from these two equations we have:
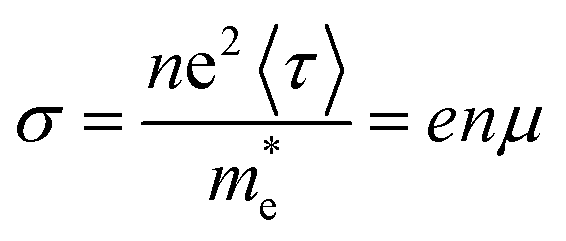
and one can note that, within this simplified model ωp is proportional to
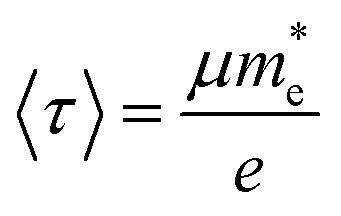
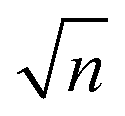 , with n being the charge density and τ being proportional to μ, the charge mobility (in doing so we keep constant the effective mass of charge carriers
, with n being the charge density and τ being proportional to μ, the charge mobility (in doing so we keep constant the effective mass of charge carriers 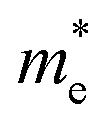 which could also vary with PEDOT wire orientation).41
which could also vary with PEDOT wire orientation).41
The plasmonic peaks for a cylindrical particle of radius r and height h can then be simulated using the following equations:22,38,77
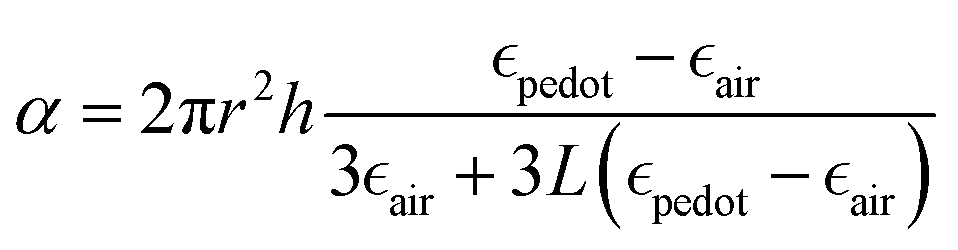
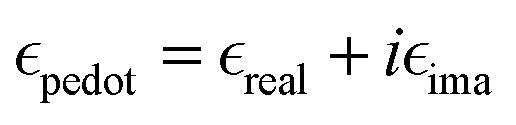 given by the plasma equations:
given by the plasma equations:where the form factor L depends on the NP diameter over the height ratio (the calculation of L is shown in the ESI files and its variation with d/h is depicted in Fig. S10, ESI†).
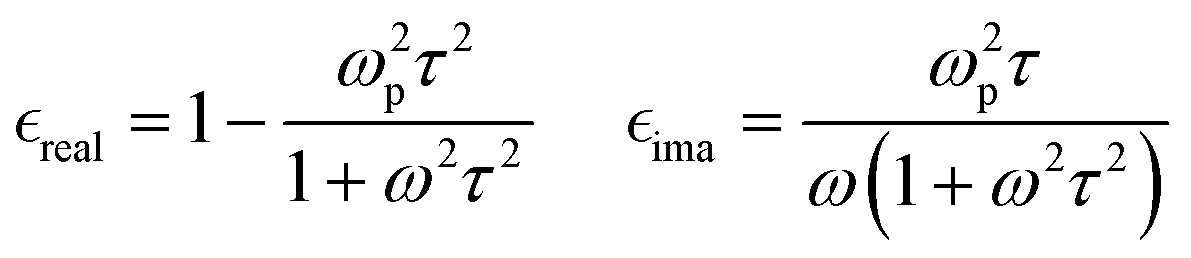
The coefficients of absorbance, diffusion and extinction can then be calculated using:
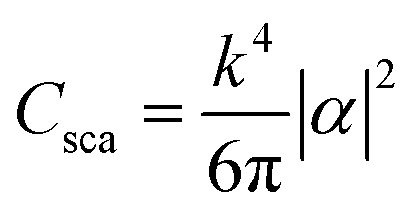
| Cabs = kIm(α) |
| Cext = Csca + Cabs |
With these assumptions, we can simulate the extinction spectra of cylindrical PEDOT NPs and tune r, h, ωp and τ to simulate a material with more or less conductivity.
The effects of r and h on the LSPR are shown in Fig. S11 and S12 (ESI†), where a blueshift of the plasmonic peak maximum with decreasing NP radius (at constant height) or with increasing NP height (at constant radius) can be seen because of the change of the form factor L. These observations are in good agreement with previous studies and with the results shown in Fig. 2 and Fig. S8 (ESI†).
Simulation of the LSPR peak shown in Fig. 3 at 1240 nm leads to ωp = 1.95 eV and τ = 0.75 fs. These values can be compared to those reported for PEDOT films by Girtan et al.: ωp = 1.8 eV and τ = 7.9 fs39 and are consistent with values already published for PEDOT using ellipsometry or charge transport measurements.40,71 One must note that even if the simple Drude model, used in this study, can describe well the spectroscopic optical data, the interpretation of the model parameters cannot be rigorous but phenomenological. In other words, due to the variations in the model parameters in a series of samples, it is meaningful to monitor relative changes but their absolute values are less significant. The high ωp and low τ values obtained here may thus indicate that the simple Drude model over-estimates ωp and under-estimates τ.40,71 However, the small discrepancy for ωp and τ can also be due to the underlying oligothiophene BTB adhesion layer, covalently bonded to the electrode or to some disparity in the physical properties of each PEDOT NP. Indeed, the SEM image (Fig. 1a) shows that some NPs appear lighter than others. Such contrast on the SEM image can be attributed to variations of conductivity of PEDOT NPs leading to higher FWHM and thus to an underestimation of τ and an overestimation of ωp.
Fig. S13 (ESI†) shows the effect of changing τ (at constant ωp). Increasing τ leads to a strong decrease in the FWHM associated with a minimal blue shift and cannot explain the modification of the spectra upon secondary doping. In contrast, the effect of an increase in plasma frequency on the LSPR (at constant τ), depicted in Fig. S14 (ESI†) and in Fig. 3b, leads to a large blue shift and is very similar to the change in LSPR observed experimentally.
These simulations thus provide precious insights into the changes in the shape of the spectra of PEDOT NPs upon secondary doping. Overall, the blue shift of the LSPR band and the lower FWHM are coherent, and indicate, a higher charge density and a small increase of the mobility of the charge after secondary doping. With this Drude model, a blue shift of 300 nm starting at 1200 nm is observed when ωp is increased by a factor of 1.4, meaning that n is increased by a factor of 1.96. This value has to be compared with those reported in the literature.68 Secondary doping of PEDOT has been associated with better mobility but also with higher charge-carrier density68,78–80 by a factor of 5 in the best-case scenario. We can conclude that the 300 nm blue shift observed is fully coherent with previous publications and that measuring and understanding the LSPR spectra of PEDOT NPs is a way of obtaining new information about the density and the mobility of the charge carriers and the conductivity of PEDOT NPs after secondary doping.
Conclusions
This work highlights the emerging field of organic plasmonics using conductive polymers. We show that, like metallic plasmonic NPs, the confinement of charges in PEDOT NPs leads to a strong modification of their extinction spectra with a LSPR in the near infrared region. We also show that the LSPR maximum wavelength shifts according to the diameter of the NPs. It is also closely related to the charge-carrier density while its FWHM is a measure of the charge-carrier lifetime. Consequently, this resonance can be tuned by manipulating the shapes of the NPs with lithography and the deposition method. The important finding of this work is that the LSPR can also be tuned by an acid treatment which leads to an impressive blue shift of 300 nm and brings PEDOT LSPR closer to the visible range. From a more general point of view, measuring the LSPR properties of PEDOT NPs is a new means of studying the secondary doping of conducting polymers and can help to better understand such phenomena which are widely used in the quest for replacing ITO with organic electrodes in many applications.Organic plasmonics is still in its infancy and can potentially offer brand new properties thanks to the various conductive polymers available. It opens the gate for new sensors with more specificity and possibly new detection schemes, new active plasmonic devices with tunable metasurfaces, and challenges different concepts in plasmonics and conductive polymers.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availabilibity
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Stephanie Lau for the Raman spectra and Dr John S. Lomas for editing our manuscript. We are grateful to the Ile-de-France region and IDEX for financial support of the AFM-Beam-Rex platform. P. B. thanks Ecole Normale Supérieure Paris-Saclay for his PhD scholarship. The Agence Nationale de la Recherche (ANR, France) is gratefully acknowledged for its financial support of this work (ANR-19-CE09-APMJ).Notes and references
- H. Xu, E. J. Bjerneld, M. Käll and L. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nanosci. Technol., 2009, 7, 308–319 Search PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- N. T. Fofang, T.-H. Park, O. Neumann, N. A. Mirin, P. Nordlander and N. J. Halas, Nano Lett., 2008, 8, 3481–3487 CrossRef CAS PubMed.
- A. J. Wilson and K. A. Willets, Annu. Rev. Anal. Chem., 2016, 9, 27–43 CrossRef PubMed.
- J.-C. Lacroix, Q. van Nguyen, Y. Ai, Q. van Nguyen, P. Martin and P.-C. Lacaze, Polym. Int., 2019, 68, 607–619 CrossRef CAS.
- V. Stockhausen, P. Martin, J. Ghilane, Y. Leroux, H. Randriamahazaka, J. Grand, N. Felidj and J. C. Lacroix, J. Am. Chem. Soc., 2010, 132, 10224–10226 CrossRef CAS PubMed.
- D. Schaming, V.-Q. Nguyen, P. Martin and J.-C. Lacroix, J. Phys. Chem. C, 2014, 118, 25158–25166 CrossRef CAS.
- Y. Lu, S. H. Lam, W. Lu, L. Shao, T. H. Chow and J. Wang, Nano Lett., 2022, 22, 1406–1414 CrossRef CAS PubMed.
- P. Bléteau, M. Bastide, S. Gam-Derouich, P. Martin, R. Bonnet and J.-C. Lacroix, ACS Appl. Nano Mater., 2020, 3, 7789–7794 CrossRef.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- S. Kawata, Y. Inouye and P. Verma, Nat. Photonics, 2009, 3, 388–394 CrossRef CAS.
- A. Horrer, Y. Zhang, D. Gérard, J. Béal, M. Kociak, J. Plain and R. Bachelot, Nano Lett., 2020, 20, 509–516 CrossRef CAS PubMed.
- D. Gryb, F. J. Wendisch, A. Aigner, T. Gölz, A. Tittl, L. de, S. Menezes and S. A. Maier, Nano Lett., 2023, 23, 8891–8897 CrossRef CAS PubMed.
- J. B. Pendry, D. Schurig and D. R. Smith, Sciencexpress, 2006, 312, 1780–1782 CAS.
- U. Leonhardt and T. G. Philbin, New J. Phys., 2006, 8, 247 CrossRef.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- G. Garcia, R. Buonsanti, E. L. Runnerstrom, R. J. Mendelsberg, A. Llordes, A. Anders, T. J. Richardson and D. J. Milliron, Nano Lett., 2011, 11, 4415–4420 CrossRef CAS PubMed.
- A. N. Grigorenko, M. Polini and K. S. Novoselov, Nat. Photonics, 2012, 6, 749–758 CrossRef CAS.
- M. Yilmaz, E. Babur, M. Ozdemir, R. L. Gieseking, Y. Dede, U. Tamer, G. C. Schatz, A. Facchetti, H. Usta and G. Demirel, Nat. Mater., 2017, 16, 918–924 CrossRef CAS PubMed.
- S. Chen, E. S. H. Kang, M. Shiran Chaharsoughi, V. Stanishev, P. Kühne, H. Sun, C. Wang, M. Fahlman, S. Fabiano, V. Darakchieva and M. P. Jonsson, Nat. Nanotechnol., 2020, 15, 35–40 CrossRef CAS PubMed.
- J. Karst, M. Floess, M. Ubl, C. Dingler, C. Malacrida, T. Steinle, S. Ludwigs, M. Hentschel and H. Giessen, Science, 2021, 374, 612–616 CrossRef CAS PubMed.
- A. Karki, G. Cincotti, S. Chen, V. Stanishev, V. Darakchieva, C. Wang, M. Fahlman and M. P. Jonsson, Adv. Mater., 2022, 34, 2107172 CrossRef CAS PubMed.
- G. Demirel, H. Usta, M. Yilmaz, M. Celik, H. A. Alidagi and F. Buyukserin, J. Mater. Chem. C, 2018, 6, 5314–5335 RSC.
- A. Karki, Y. Yamashita, S. Chen, T. Kurosawa, J. Takeya, V. Stanishev, V. Darakchieva, S. Watanabe and M. P. Jonsson, Commun. Mater., 2022, 3, 48 CrossRef CAS.
- Y. Lu, T. H. Chow, K. K. Chui and J. Wang, J. Phys. Chem. C, 2023, 127, 5643–5652 CrossRef CAS.
- M. J. Gentile, S. Núñez-Sánchez and W. L. Barnes, Nano Lett., 2014, 14, 2339–2344 CrossRef CAS PubMed.
- J. Lagois and B. Fischer, Phys. Rev. Lett., 1976, 36, 680–683 CrossRef CAS.
- J. Karst, Y. Lee, M. Floess, M. Ubl, S. Ludwigs, M. Hentschel and H. Giessen, Nat. Commun., 2022, 13, 7183 CrossRef CAS PubMed.
- E. S. H. Kang, S. Kk, I. Jeon, J. Kim, S. Chen, K. Kim, K. Kim, H. S. Lee, F. Westerlund and M. P. Jonsson, Adv. Sci., 2022, 9, 2201907 CrossRef CAS PubMed.
- S. Chen and M. P. Jonsson, ACS Photonics, 2023, 10, 571–581 CrossRef CAS.
- D. de Jong, J. Karst, D. Ludescher, M. Floess, S. Moell, K. Dirnberger, M. Hentschel, S. Ludwigs, P. V. Braun and H. Giessen, Nanophotonics, 2023, 12, 1397–1404 CrossRef CAS PubMed.
- Y. Lee, J. Karst, M. Ubl, M. Hentschel and H. Giessen, Nanophotonics, 2023, 12, 2865–2871 CrossRef CAS.
- X. Li, S. Zhu, G. Zhu, J. Wang, Y. Ding, W. Du and T. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 14357–14363 CrossRef CAS PubMed.
- G. Demirel, R. L. M. Gieseking, R. Ozdemir, S. Kahmann, M. A. Loi, G. C. Schatz, A. Facchetti and H. Usta, Nat. Commun., 2019, 10, 5502 CrossRef CAS PubMed.
- B. Cho, K. S. Park, J. Baek, H. S. Oh, Y. E. Koo Lee and M. M. Sung, Nano Lett., 2014, 14, 3321–3327 CrossRef CAS PubMed.
- S. A. Maier, Plasmonics: Fundamental and Applications, Springer New York, NY, New York, 1st edn, 2007 Search PubMed.
- M. Girtan, R. Mallet, M. Socol and A. Stanculescu, Mater. Today Commun., 2020, 22, 100735 CrossRef CAS.
- Y. Chang, K. Lee, R. Kiebooms, A. Aleshin and A. J. Heeger, Synth. Met., 1999, 105, 203–206 CrossRef CAS.
- Y. Duan, A. Rahmanudin, S. Chen, N. Kim, M. Mohammadi, K. Tybrandt and M. P. Jonsson, Adv. Mater., 2023, 35, 2303949 CrossRef CAS PubMed.
- M. Kong, M. Garriga, J. S. Reparaz and M. I. Alonso, ACS Omega, 2022, 7, 39429–39436 CrossRef CAS PubMed.
- M. Bastide, S. Gam-Derouich and J.-C. Lacroix, Nano Lett., 2022, 22, 4253–4259 CrossRef CAS PubMed.
- N. Sakmeche, S. Aeiyach, J. Aaron, M. Jouini, J. C. Lacroix and P. Lacaze, Langmuir, 1999, 15, 2566–2574 CrossRef CAS.
- X. Wu, W. Pei, H. Zhang, Y. Chen, X. Guo, H. Chen and S. Wang, J. Electroanal. Chem., 2015, 758, 26–32 CrossRef CAS.
- K. Stulík, C. Amatore, K. Holub, V. Marecek and W. Kutner, Pure Appl. Chem., 2000, 72, 1483–1492 CrossRef.
- C. Amatore, J. M. Savéant and D. Tessier, J. Electroanal. Chem. Interfacial Electrochem., 1983, 147, 39–51 CrossRef CAS.
- V.-Q. Nguyen, D. Schaming, D. L. Tran and J.-C. Lacroix, ChemElectroChem, 2016, 3, 2264–2269 CrossRef CAS.
- M. Stavytska-Barba and A. M. Kelley, J. Phys. Chem. C, 2010, 114, 6822–6830 CrossRef CAS.
- V. Q. Nguyen, D. Schaming, P. Martin and J.-C. Lacroix, ACS Appl. Mater. Interfaces, 2015, 7, 21673–21681 CrossRef CAS PubMed.
- L. Santos, P. Martin, J. Ghilane, P.-C. Lacaze, H. Randriamahazaka, L. M. Abrantes and J.-C. Lacroix, Electrochem. Commun., 2010, 12, 872–875 CrossRef CAS.
- N. Massonnet, A. Carella, O. Jaudouin, P. Rannou, G. Laval, C. Celle and J. P. Simonato, J. Mater. Chem. C, 2014, 2, 1278–1283 RSC.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, ACS Appl. Polym. Mater., 2019, 1, 83–94 CrossRef CAS.
- G. Rebetez, O. Bardagot, J. Affolter, J. Réhault and N. Banerji, Adv. Funct. Mater., 2022, 32, 2105821 CrossRef CAS.
- S. S. Kalagi and P. S. Patil, Synth. Met., 2016, 220, 661–666 CrossRef CAS.
- J. Ouyang, Displays, 2013, 34, 423–436 CrossRef CAS.
- J. Ouyang, ACS Appl. Mater. Interfaces, 2013, 5, 13082–13088 CrossRef CAS PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- A. Boltasseva and J. B. Khurgin, MRS Bull., 2012, 37, 768–779 CrossRef.
- J. B. Khurgin, Nat. Nanotechnol., 2015, 10, 2–6 CrossRef CAS PubMed.
- L. M. Santos, J. Ghilane, C. Fave, P.-C. Lacaze, H. Randriamahazaka, L. M. Abrantes and J.-C. Lacroix, J. Phys. Chem. C, 2008, 112, 16103–16109 CrossRef CAS.
- E. Villemin, B. Lemarque, T. T. Vũ, V. Q. Nguyen, G. Trippé-Allard, P. Martin, P.-C. Lacaze and J.-C. Lacroix, Synth. Met., 2019, 248, 45–52 CrossRef CAS.
- V. Stockhausen, G. Trippé-Allard, V. Q. Nguyen, J. Ghilane and J. C. Lacroix, J. Phys. Chem. C, 2015, 119, 19218–19227 CrossRef CAS.
- V. Stockhausen, V. Q. Nguyen, P. Martin and J. C. Lacroix, ACS Appl. Mater. Interfaces, 2017, 9, 610–617 CrossRef CAS PubMed.
- L. Santos, J. Ghilane and J.-C. Lacroix, J. Am. Chem. Soc., 2012, 134, 5476–5479 CrossRef CAS PubMed.
- Y. Xia and J. Ouyang, ACS Appl. Mater. Interfaces, 2010, 2, 474–483 CrossRef CAS PubMed.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J. P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- V. N. Prigodin and A. J. Epstein, Synth. Met., 2001, 125, 43–53 CrossRef.
- A. Aleshin, R. Kiebooms, R. Menon, F. Wudl and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 3659–3663 CrossRef CAS.
- K. Lee and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 35201 CrossRef.
- N. Kim, B. H. Lee, D. Choi, G. Kim, H. Kim, J.-R. Kim, J. Lee, Y. H. Kahng and K. Lee, Phys. Rev. Lett., 2012, 109, 106405 CrossRef PubMed.
- F. Yan, E. P. J. Parrott, B. S.-Y. Ung and E. Pickwell-MacPherson, J. Phys. Chem. C, 2015, 119, 6813–6818 CrossRef CAS.
- Z. Guo, T. Sato, Y. Han, N. Takamura, R. Ikeda, T. Miyamoto, N. Kida, M. Ogino, Y. Takahashi and N. Kasuya, Commun. Mater., 2024, 5, 26 CrossRef CAS.
- C. Dingler, R. Walter, B. Gompf and S. Ludwigs, Macromolecules, 2022, 55, 1600–1608 CrossRef CAS.
- S. Chen, P. Kühne, V. Stanishev, S. Knight, R. Brooke, I. Petsagkourakis, X. Crispin, M. Schubert, V. Darakchieva and M. P. Jonsson, J. Mater. Chem. C, 2019, 7, 4350–4362 RSC.
- C. Langhammer, M. Schwind, B. Kasemo and I. Zoric, Nano Lett., 2008, 8, 1461–1471 CrossRef CAS PubMed.
- W. Shi, T. Zhao, J. Xi, D. Wang and Z. Shuai, J. Am. Chem. Soc., 2015, 137, 12929–12938 CrossRef CAS PubMed.
- Q. Wei, M. Mukaida, K. Kirihara, Y. Naitoh and T. Ishida, Materials, 2015, 8, 732–750 CrossRef CAS PubMed.
- E. Jin Bae, Y. Hun Kang, K. S. Jang and S. Yun Cho, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00950a |
| This journal is © The Royal Society of Chemistry 2024 |