
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of degradable networked-organic semiconductors and effects on charge carrier mobility in organic thin-film transistors†
Hyunjung
Jin‡
 ab,
Kyeongmin
Kim‡
ab,
Kyuyeon
Kim‡
ac,
Sungmin
Park
d,
Eul-Yong
Shin
a,
Jae Won
Heo
efg,
Hyunjoo
Lee
hi,
Se-Woong
Baek
c,
In Soo
Kim
efg,
Hyungju
Ahn
*j and
Hae Jung
Son
*ab
ab,
Kyeongmin
Kim‡
ab,
Kyuyeon
Kim‡
ac,
Sungmin
Park
d,
Eul-Yong
Shin
a,
Jae Won
Heo
efg,
Hyunjoo
Lee
hi,
Se-Woong
Baek
c,
In Soo
Kim
efg,
Hyungju
Ahn
*j and
Hae Jung
Son
*ab
aAdvanced Photovoltaics Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: hjson@kist.re.kr
bGraduate School of Energy and Environment (KU-KIST Green School), Korea University, Seoul 02841, Republic of Korea
cDepartment of Chemical and Biological Engineering, Korea University, Seoul, 02841, Republic of Korea
dPhotonics Convergence Display Research Center, ICT Convergence Research Division, Gumi Electronics and Information Technology Research Institute (GERI), Gumi, Gyeongbuk 39253, Republic of Korea
eNanophotonics Research Center, Korea Institute of Science and Technology (KIST), Seoul, 02792, Republic of Korea
fSchool of Advanced Materials Science and Engineering, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
gKIST-SKKU Carbon-Neutral Research Center, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
hClean Energy Research Center, Korea Institute of Science and Technology (KIST), 02792, Seoul, Republic of Korea
iDivision of Energy & Environment Technology, KIST School, Korea University of Science and Technology, Seoul 02792, Republic of Korea
jPohang Accelerator Laboratory, POSTEHC, Pohang, Gyungbuk 37673, Republic of Korea. E-mail: hyungju@postech.ac.kr
First published on 1st May 2024
Abstract
As a promising solution to address the growing concern of electronic waste, transient electronics, such as biomedical implants, environmental sensors, and hardware-secured devices, have garnered considerable attention. These devices can be engineered to exhibit partial solubility, degradability, or physical dissolution. For the development of degradable transient organic electronics, it is important to develop degradable polymeric organic semiconductors. However, few studies have focused on controlling the degradation rates of polymers in films. In this study, we designed and synthesized three different degradable polymers, namely, PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO, through in situ polymerization in the film state. Notably, PY-t-TIPO exhibited a significantly higher charge mobility of 1.67 × 10−3 cm2 V−1 s−1 in organic thin-film transistors (OTFTs) than PY-m-TIPO and PY-p-TIPO although all the degradable polymer films exhibited similar amorphous properties. We demonstrated that the number of bonding arms affects not only the degradation rate of the polymer films but also the charge mobility of the corresponding OTFTs. Our study provides design guidelines for developing degradable polymers with controllable degradation rates for high charge mobility in transient OTFTs.
Introduction
Transient electronics, an emerging class of functional electronics, has attracted considerable attention in the field of materials science.1,2 Unlike conventional electronic devices, which are designed to remain functional and stable over an extended period, transient electronic devices are typically designed to operate over a short and predefined duration and disintegrate completely or partially upon fulfilling their intended purposes.3 Examples of transient electronic devices include electrophysiological sensors,4–6 diagnostic brain implants,7 therapeutic implants8–11 for therapy and biosensing, degradable microwave electronic devices12,13 for temporal medical implant applications,14 disposable sensors,15,16 data-securing hardware systems, and environmentally friendly degradable devices.17,18The early prototypes of transient electronics were reported to be silicon-based devices.2,19–22 However, inorganic semiconductors, such as silicon, have drawbacks such as mechanical brittleness, toxicity, and high manufacturing costs. Furthermore, elements such as gallium and indium have limited global reserves. Organic semiconductors have attracted significant attention as an alternative to silicon-based devices. These materials are known for their ease of fabrication, mechanical flexibility,23 and tunable properties. Organic semiconductors can be applied in various systems such as organic light-emitting diodes,24–28 organic photovoltaics,29–33 and organic sensors.34–36 Compared to inorganic active materials, polymers are a promising class of materials that exhibit the desired combination of good electrical properties and degradability, owing to the vast chemical design space enabled by synthetic methodologies. To facilitate their application in transient electronics, developing organic semiconductors that simultaneously exhibit excellent electrical properties and degradability to promote their application in transient electronics is a challenging research task. In particular, it is important to develop organic semiconductors that can control the degradation rate of thin films,37 which may depend on the chemical structure of the organic semiconductor38 and the morphology of the thin film.39
In this study, we developed various degradable polymers through the reaction between an aldehyde-attached small molecule, Y5-TA-Cl, and the geometrical isomers of m-PO-N, p-PO-N, and t-PO-N, which yielded PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO, respectively, as shown in Fig. 1a. We elucidated the effects of the number of bonding arms in the network core and the geometrical-isomeric structures of the degradable polymers on the charge mobilities of the polymer-film-based organic thin-film transistors (OTFTs) and degradation behavior under acidic conditions. Furthermore, the synthesized degradable polymers were used in OTFTs to investigate their electronic properties and applicability in the development of degradation-controllable transient electronics.
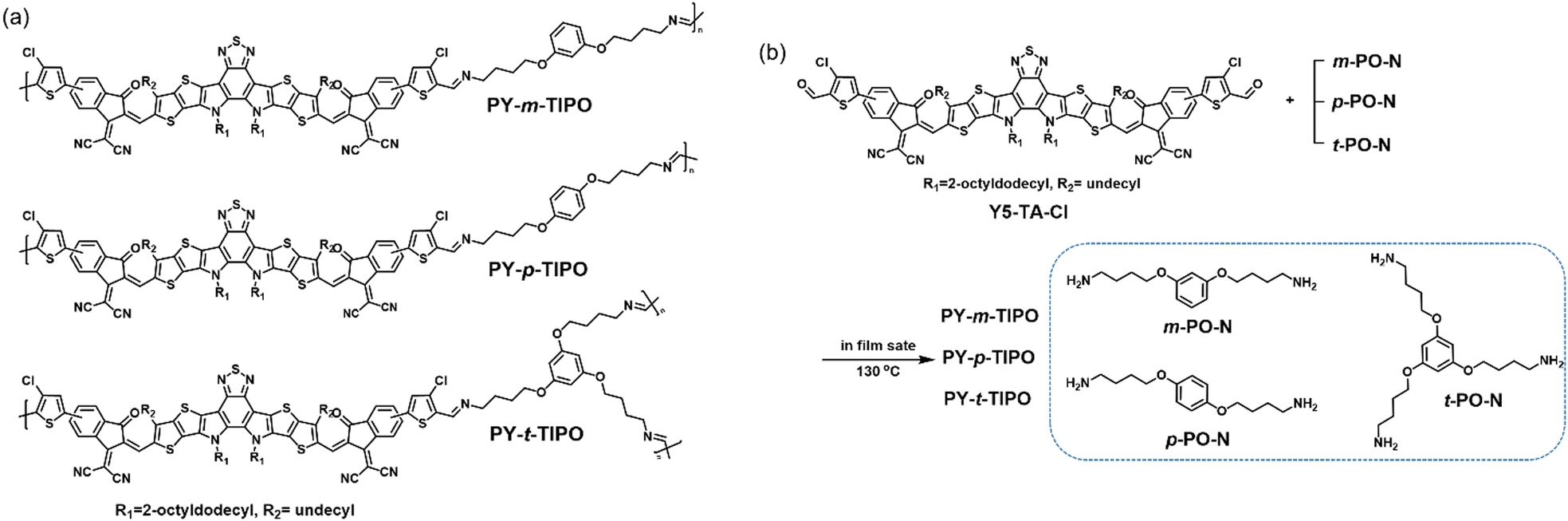 | ||
| Fig. 1 (a) Chemical structures and (b) synthetic route of the degradable polymers PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO. | ||
Results and discussion
Design and synthesis of degradable polymers
We prepared a series of degradable polymers, namely, PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO, via the reaction between Y5-TA-Cl and amine-functionalized crosslinkers m-PO-N, p-PO-N, and t-PO-N, as shown in Fig. 1b. Y5-TA-Cl is a chlorinated acceptor Y5-derivative40 functionalized with the aldehyde groups in indanone. Details of the synthesis and characterization of each monomer and degradable polymer are presented in Schemes S1–S3 and Fig. S1–S6 (ESI†).41The key strategy for controlling the degradation rate of degradable polymers is to vary the number and positions of amines in the crosslinker, which may also affect the molecular packing structures and morphology of the thin films. In this study, degradable polymers were synthesized in the film state through in situ one-shot preparations using the Schiff reaction.41–43 To confirm the occurrence of in situ one-shot polymerization in the film state, we conducted a washing test by spin-coating the films with dichloromethane. The films with added PO-N retained their absorption peaks even after washing. Additionally, the films with added t-PO-N, which had the highest amine content, exhibited the highest amount of residual films.
We performed Fourier transform infrared (FT-IR) and ultraviolet-visible (UV-Vis) spectroscopy measurements to investigate the reaction between Y5-TA-Cl and the amine-functionalized crosslinkers in the film. The changes in the FT-IR peaks at 1640–1720 cm−1, which include the peaks assigned to ketone (1694 cm−1) and aldehyde (1679 cm−1) groups, were monitored via deconvolution and curve-fitting for diverse stoichiometric ratios of PO-N (Y5-TA-Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PO-N = 1:0, 1:0.25, 1:0.5, 1:0.75, 1:1). Fig. 2 and Fig. S7 (ESI†) present the summary of the results. Irrespective of the PO-N types, the intensities of the aldehyde peak at 1679 cm−1 and the imine peak at 1630 cm−1 simultaneously decreased and increased, respectively, with increasing PO-N concentration. We estimated the polymerization yield by comparing the decreased peak area for the aldehyde group with that for the ketone group at 1694 cm−1, as shown in Table 1. After the reaction, some aldehyde monomers were retained in the p-PO-N film even when the Y5-TA-Cl:p-PO-N stoichiometric ratio was 1:1. This resulted in a lower polymerization yield of 89% than the 95% yield of the m-PO-N film. This is probably because the linear polymers prepared using p-PO-N had a relatively low degree of molecular movement in the film, which decreased the reactivity of the Schiff reaction. The polymerization yield of the resulting polymer films was confirmed again at around 88% by performing the gel-permission chromatography (GPC) measurements and the GPC data are shown in Fig. S8 (ESI†).
:PO-N = 1:0, 1:0.25, 1:0.5, 1:0.75, 1:1). Fig. 2 and Fig. S7 (ESI†) present the summary of the results. Irrespective of the PO-N types, the intensities of the aldehyde peak at 1679 cm−1 and the imine peak at 1630 cm−1 simultaneously decreased and increased, respectively, with increasing PO-N concentration. We estimated the polymerization yield by comparing the decreased peak area for the aldehyde group with that for the ketone group at 1694 cm−1, as shown in Table 1. After the reaction, some aldehyde monomers were retained in the p-PO-N film even when the Y5-TA-Cl:p-PO-N stoichiometric ratio was 1:1. This resulted in a lower polymerization yield of 89% than the 95% yield of the m-PO-N film. This is probably because the linear polymers prepared using p-PO-N had a relatively low degree of molecular movement in the film, which decreased the reactivity of the Schiff reaction. The polymerization yield of the resulting polymer films was confirmed again at around 88% by performing the gel-permission chromatography (GPC) measurements and the GPC data are shown in Fig. S8 (ESI†).
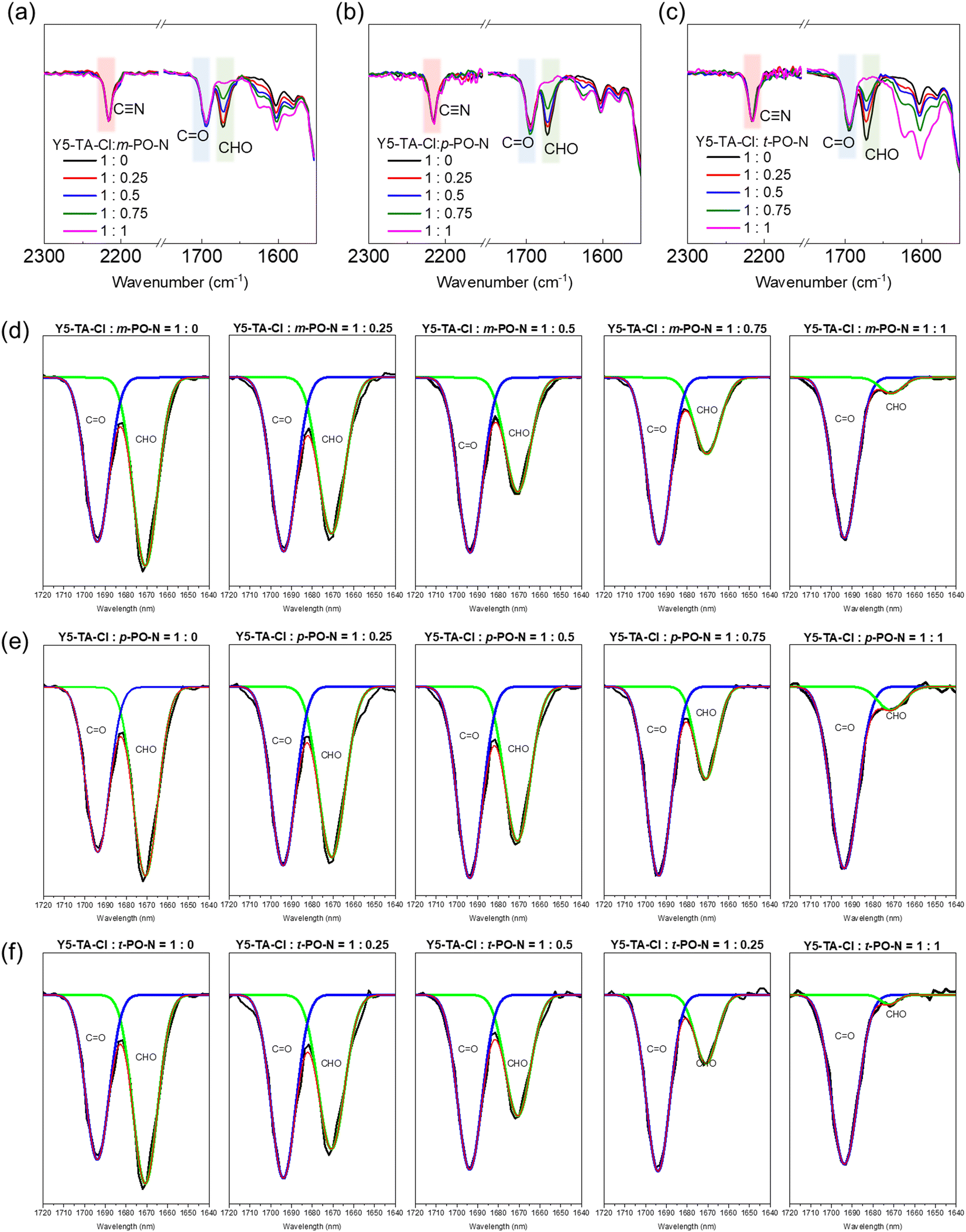 | ||
| Fig. 2 The normalized FT-IR spectra of Y5-TA-Cl with (a) m-PO-N, (b) p-PO-N, and (c) t-PO-N annealed films in various stoichiometric ratios (Y5-TA-Cl:PO-N = 1:0, 1:0.25, 1:0.5, 1:0.75, 1:1). The deconvoluted FT-IR spectra by Gaussian fitting of Y5-TA-Cl with (d) m-PO-N, (e) p-PO-N, and (f) t-PO-N. | ||
| Crosslinker | Stoichiometric ratio of Y5-TA-Cl:PO-N |
Normalized value of integrated area
|
Integration area ratio | Polymerization yield [%] |
|---|---|---|---|---|
| m-PO-N | 1:0 |
1.134 | 1 | — |
| 1:0.25 |
0.953 | 0.84 | 16.0 | |
| 1:1.5 |
0.675 | 0.59 | 40.5 | |
| 1:0.75 |
0.496 | 0.34 | 56.3 | |
| 1:1 |
0.091 | 0.08 | 92.0 | |
| p-PO-N | 1:0 |
1.170 | 1 | — |
| 1:0.25 |
1.030 | 0.88 | 12.0 | |
| 1:1.5 |
0.826 | 0.71 | 29.4 | |
| 1:0.75 |
0.421 | 0.36 | 64.1 | |
| 1:1 |
0.124 | 0.11 | 89.4 | |
| t-PO-N | 1:0 |
1.112 | 1 | — |
| 1:0.25 |
0.841 | 0.76 | 24.4 | |
| 1:1.5 |
0.693 | 0.62 | 37.7 | |
| 1:0.75 |
0.389 | 0.35 | 65.0 | |
| 1:1 |
0.056 | 0.05 | 95.0 |
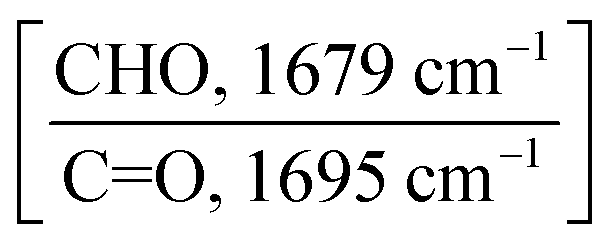
The UV-Vis absorption spectra of the PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO degradable polymers and Y5-TA-Cl films are shown in Fig. 3a and summarized in Table S1 (ESI†). After polymerization, the absorption spectra of all the films were blue-shifted compared with the Y5-TA-Cl spectrum. Therefore, the maximum absorption peaks of PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO were observed at 788, 790, and 792 nm, respectively. Additionally, the 0–0 peaks of the polymers were less intense than those of Y5-TA-Cl, and the absorption onsets of the polymers were blue-shifted. These results are because polymerization disturbed the packing structure of Y5-TA-Cl in the film, increasing the amorphous properties of the polymers. The absorption spectra of PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO exhibited similar features to each other.
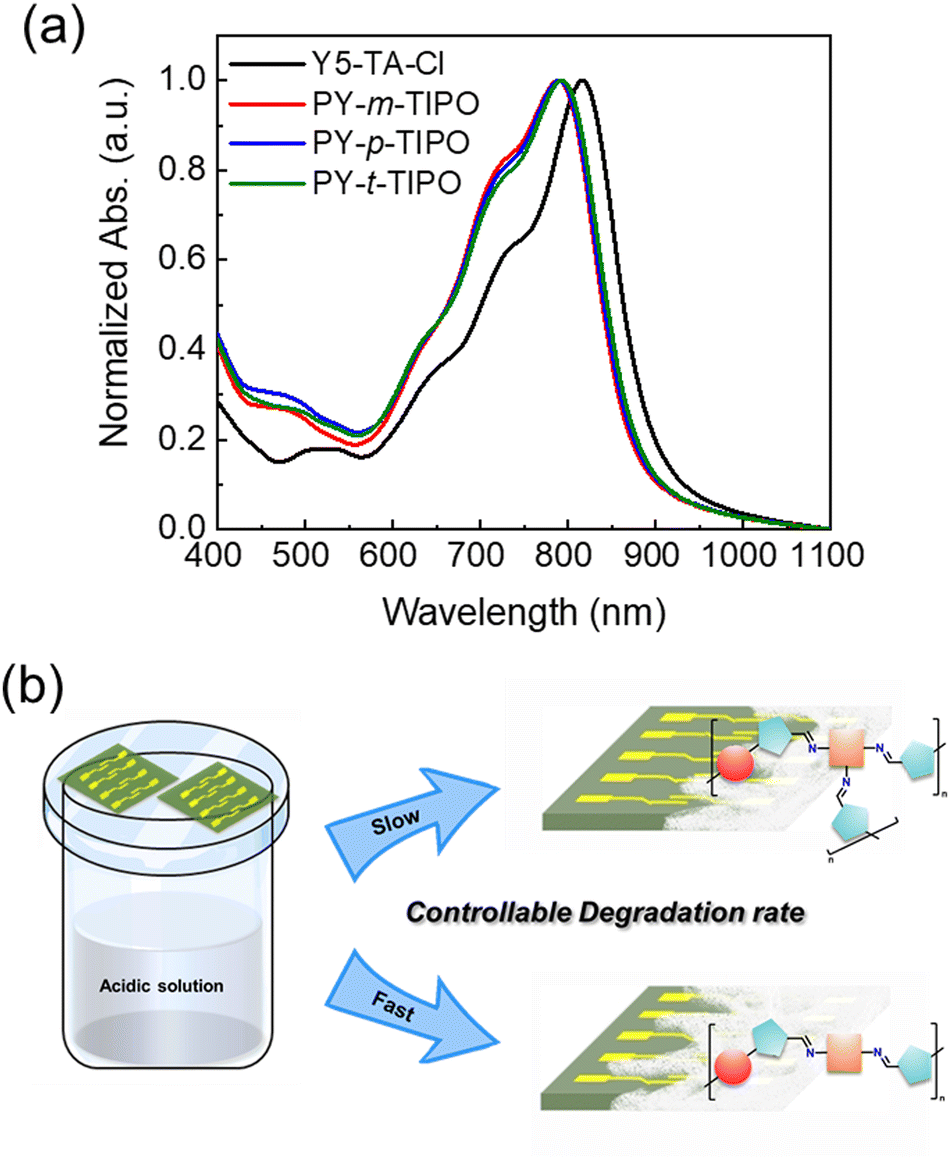 | ||
| Fig. 3 (a) The normalized UV-Vis absorption spectra of Y5-TA-Cl and degradable polymers in film. (b) Illustration of degradable polymers in film during the degradation procedure with controllable degradation time. | ||
Degradation studies
To investigate the degradation characteristics of the PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO degradable polymer films, we conducted acid exposure time-dependent UV-Vis and FT-IR measurements. We observed the variations in the optical properties of the polymer films and the chemical degradation of the imine (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) bond. Additionally, we tracked the formation of aldehydes (CHO). Each polymer film underwent degradation upon their exposure to vapors from 0.1 M trifluoroacetic acid (TFA) solution (Fig. 3b).
N–) bond. Additionally, we tracked the formation of aldehydes (CHO). Each polymer film underwent degradation upon their exposure to vapors from 0.1 M trifluoroacetic acid (TFA) solution (Fig. 3b).
The UV-Vis spectroscopy was performed to analyze the absorption profiles of the polymer films during their degradation process (Fig. S9, ESI†). Prior to degradation, the polymer films exhibited two primary absorption peaks, denoted as 0–0 and 0–1 transition peaks at 792 and 723 nm, respectively. The 0–0 peak-normalized data revealed that the intensity of the 0–1 peak gradually increased as the degradation time proceeded and that the peak was slightly redshifted. This shift is attributed to the depolymerization of the imine-based polymers to aldehyde monomers.44 Notably, under acidic conditions, PY-t-TIPO, which features three bonding arms, exhibited higher stability than PY-m-TIPO and PY-p-TIPO, each with two bonding arms. This observation suggests that the polymer degradation accelerates with decreasing number of bonding arms. The structural differences in the positions of the bonding arms also influence the degradation rate of the polymers.
To quantitatively analyze the degradation behavior, we systematically monitored and compared the degradable polymers in the films through FT-IR spectroscopy. The spectra were normalized in relation to the peak ascribed to the unaltered cyanide (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) stretching vibration45,46 at 2216 cm−1, as shown in Fig. 4a–c. Gaussian fitting was applied to deconvolute the peaks, as shown in Fig. S10–S12 (ESI†). The intensity of the aldehyde vibration peak47 at 1678 cm−1 progressively increased, while that of the imine stretching vibration peak48 at 1630 cm−1 decreased during the polymer degradation process. A comparison between the polymer films revealed that the degradation rate of the polymer films decreased in the order of PY-p-TIPO, PY-m-TIPO, and PY-t-TIPO, as shown in Fig. 4d and Tables S2–S4 (ESI†). Although the slowest degradation rate of PY-t-TIPO is due to three bonding arms, the slightly different degradation between PY-p-TIPO and PY-m-TIPO may be affected by different molecular conformations of the polymers; the linear shape of PY-p-TIPO would be more favorable for exposure to acid vapors. These findings highlighted the significance of the number of bonding arms and their positions as design parameters for controlling the degradation rate of organic transient electronic devices.
N) stretching vibration45,46 at 2216 cm−1, as shown in Fig. 4a–c. Gaussian fitting was applied to deconvolute the peaks, as shown in Fig. S10–S12 (ESI†). The intensity of the aldehyde vibration peak47 at 1678 cm−1 progressively increased, while that of the imine stretching vibration peak48 at 1630 cm−1 decreased during the polymer degradation process. A comparison between the polymer films revealed that the degradation rate of the polymer films decreased in the order of PY-p-TIPO, PY-m-TIPO, and PY-t-TIPO, as shown in Fig. 4d and Tables S2–S4 (ESI†). Although the slowest degradation rate of PY-t-TIPO is due to three bonding arms, the slightly different degradation between PY-p-TIPO and PY-m-TIPO may be affected by different molecular conformations of the polymers; the linear shape of PY-p-TIPO would be more favorable for exposure to acid vapors. These findings highlighted the significance of the number of bonding arms and their positions as design parameters for controlling the degradation rate of organic transient electronic devices.
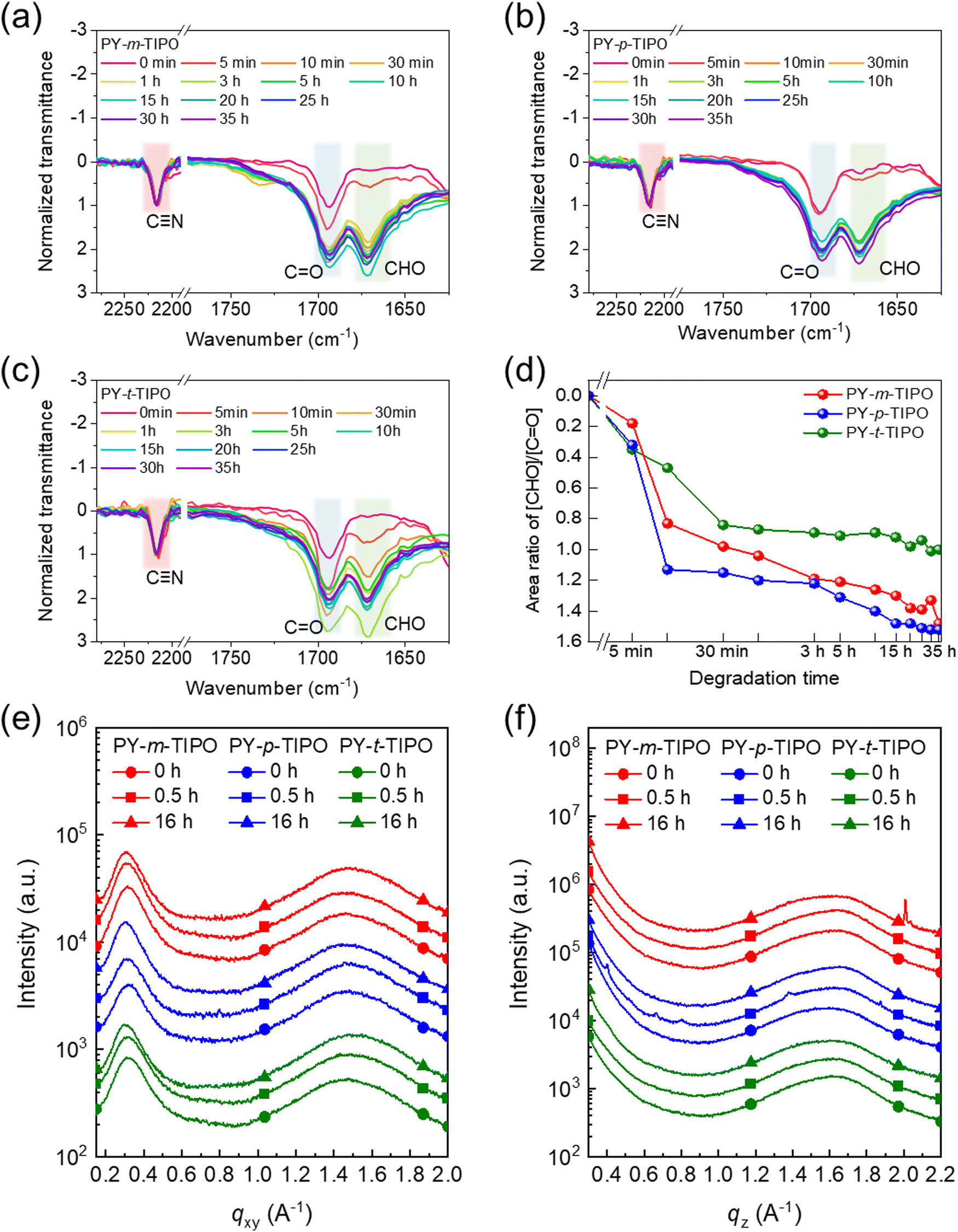 | ||
| Fig. 4 The normalized FT-IR spectra at cyanide transmittance (2216 cm−1) of films based on (a) PY-m-TIPO, (b) PY-p-TIPO and (c) PY-t-TIPO in 0.1 M TFA during the degradation procedure. (d) The degradation rate defined by the ratio of integrated areas of aldehyde and ketone peaks. (e) In-plane and (f) out-of-plane line-cut profiles from 2D-GIWAXS patterns. | ||
Molecular packing properties and morphology of films
To compare the morphological characteristics of the Y5-TA-Cl and degradable polymers PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO in film, we performed two-dimensional grazing-incidence wide-angle X-ray scattering (2D-GIWAXS). The 2D-GIWAXS images and line-cut profiles of the polymer and monomer films are shown in Fig. S13 and S14 (ESI†) and Fig. 4e and f, and their crystallographic parameters are summarized in Table S5 (ESI†). The 2D-GIWAXS patterns of degradable polymers exhibited circular shape of peaks, which indicates random orientation unlike that of Y5-TA-Cl. In the line profile of out-of-plane direction (OOP), the Y5-TA-Cl, PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO films showed (010) peaks at qz = 1.66, 1.58, 1.56 and 1.58 Å−1 which are correlated to the π–π stacking of the them. The coherence lengths (LC) of the (010) peaks, as calculated using the simple Scherrer equation, for the Y5-TA-Cl and degradable polymer films were 49.38, 12.21, 11.83, and 12.52 Å, respectively. Notably, the films with crosslinkers m-PO-N, p-PO-N, and t-PO-N showed a lot of changes in the GIWAXS images and line-cut profiles after they underwent polymerization and commonly exhibited amorphous properties. Probably, the packing structure of Y5-TA-Cl in the film was destructed during heating and polymerization. We also investigated the variations in the molecular packing of the polymer films during exposure to the vapors of 0.1 M TFA solution. From the line-cut profiles after 0.5 and 16 h (Fig. 4e and f), it is revealed that the amorphous properties of the films were maintained during degradation, and the films were expected to remain in an amorphous state even after the polymers were completely degraded to individual monomers. Moreover, the AFM images of degradable polymers and Y5-TA-Cl films were obtained and morphologies before and after acidic vapor treatments were compared as shown in Fig. S15 (ESI†). The Y5-TA-Cl film shows a smooth surface with an RMS of 0.81 nm and the degradable polymers also show comparable roughness. Notably, the RMS value for PY-t-TIPO was slightly increased due to the relatively large morphological change during the polymerization. However, after acid vapor treatment, the smooth surface of degradable polymer films was completely changed to a largely agglomerated morphology in an amorphous state.OTFT performance
The electrical performance of the degradable polymer films was evaluated using top-gate bottom-contact (TGBC) OTFTs.49 The Y5-TA-Cl solution was spin-cast onto a glass substrate with a patterned source–drain, and the polymer layer was formed through annealing at 130 °C for 20 min. During thermal annealing, in situ polymerization of Y5-TA-Cl occurred in the film state.41 The structure of the OTFT device is shown in Fig. 5a and the detailed fabrication procedures for the device are described in the experimental section (ESI†). The transfer characteristics of the Y5-TA-Cl and degradable polymers are shown in Fig. 5b–e and Fig. S16 (ESI†).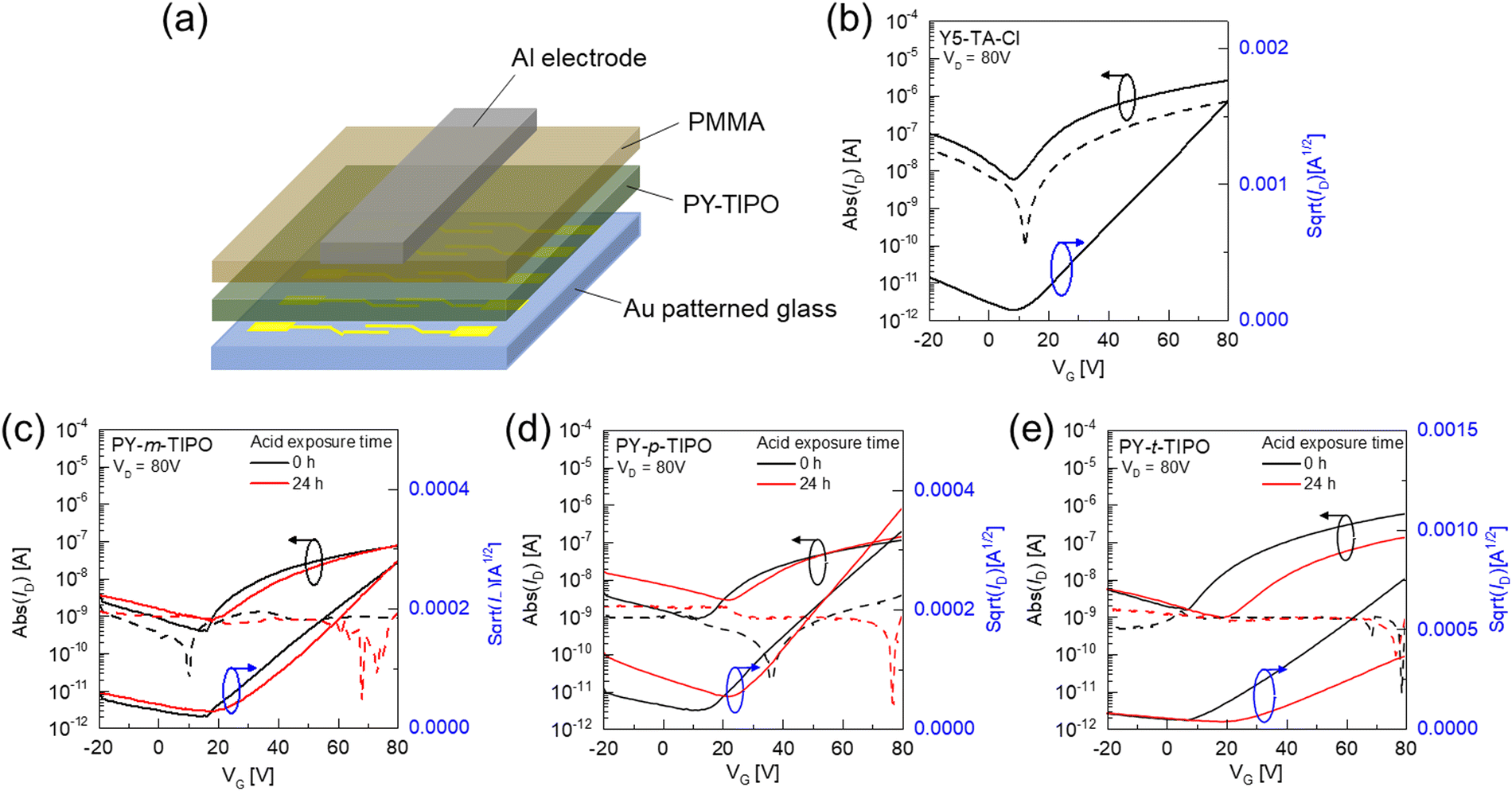 | ||
| Fig. 5 (a) Top-gate bottom-contact (TGBC) OTFT device structure. Transfer characteristics of (b) Y5-TA-Cl, (c) PY-m-TIPO, (d) PY-p-TIPO and (e) PY-t-TIPO-based OTFTs before and after exposure to acid vapor for 24 h (VD: drain–source voltage = 80 V, ID: drain–source current; VG: gate–source voltage). | ||
We investigated the effect of degradable polymers with different geometrical crosslinkers on the electrical properties of the OTFTs. For all of the OTFT devices with degradable polymers, the electron mobility (μ) and threshold voltage (Vth) were calculated using the conventional gradual channel approximation equations at VD = 80 V. The detailed electrical parameters of the OTFT devices are summarized in Table 2. PY-t-TIPO showed an electron mobility of 1.67 × 10−3 cm2 V−1 s−1. This is a decreased value compared to 6.87 × 10−3 cm2 V−1 s−1 of Y5-TA-Cl. Nevertheless, it is a remarkable result because the GIWAXS results revealed that the PY-t-TIPO film showed unfavorable amorphous properties. This demonstrates that the degradable polymer is favorable for formation of an interconnected pathway for charge transport in OTFTs. Although the PY-m-TIPO and PY-p-TIPO films exhibited amorphous properties similar to those of PY-t-TIPO, the electron mobilities of the OTFTs prepared using PY-m-TIPO and PY-p-TIPO decreased to 0.18 and 0.24 × 10−3 cm2 V−1 s−1, respectively. It is expected that the chemical structure of the three bonding arms is important to form the networked structure of the polymer for it promotes efficient charge transport. Additionally, geometrical isomerism of m-PO-N and p-PO-N is not significantly important for determining the charge carrier mobility of the polymer.
| Material | Acid vapor exposure time | Electron mobility [cm2 V−1 s−1] | V th [V] | I on/Ioff |
|---|---|---|---|---|
| Y5-TA-Cl | 0 h | 6.87 × 10−3 | 10.0 | 4.2 × 102 |
| PY-m-TIPO | 0 h | 0.18 × 10−3 | 6.41 | 1.9 × 102 |
| 24 h | 0.31 × 10−3 | 22.9 | 0.9 × 102 | |
| PY-p-TIPO | 0 h | 0.24 × 10−3 | 4.58 | 1.2 × 102 |
| 24 h | 0.45 × 10−3 | 18.2 | 0.5 × 102 | |
| PY-t-TIPO | 0 h | 1.67 × 10−3 | 13.8 | 3.8 × 102 |
| 24 h | 0.53 × 10−3 | 23.5 | 1.4 × 102 |
Conclusions
We developed a series of degradable polymers, namely, PY-m-TIPO, PY-p-TIPO, and PY-t-TIPO, via in situ polymerization. The degree of molecular degradation of the polymers could be controlled by varying the number of bonding arms in the network structure. Such different networked-bonding numbers affect the stability of the polymer films under exposure to acidic conditions. More importantly, the PY-t-TIPO-based OTFT shows one-order of magnitude higher charge mobility of 1.67 × 10−3 cm2 V−1 s−1 in OTFT compared with PY-m-TIPO and PY-p-TIPO although all degradable polymer films exhibited similar amorphous properties. Therefore, the presence of three bonding arms in the network core is important not only for retarding the degradation rate of the polymer film but also for achieving high charge mobility by providing an effective interconnected pathway for charge carriers. This research result provides a design guideline to develop degradable polymers with a controllable degradation rate toward the development of high charge mobility transient OTFTs.Author contributions
Hyunjung Jin: conceptualization, investigation, formal analysis, data curation, writing – original draft. Kyeongmin Kim: conceptualization, formal analysis, data curation. Kyuyeon Kim: investigation, formal analysis, data curation. Sungmin Park: conceptualization, investigation, formal analysis, data curation, writing – original draft. Eul-Yong Shin: investigation, formal analysis, data curation. Jae Won Heo: investigation. Hyunjoo Lee: investigation. Se-Woong Baek: investigation. In Soo Kim: investigation, data curation. Hyungju Ahn: supervision, methodology, formal analysis, writing – review & editing. Hae Jung Son: supervision, conceptualization, methodology, formal analysis, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00279418).References
- R. Jamshidi, M. Taghavimehr, Y. Chen, N. Hashemi and R. Montazami, Adv. Sustainable Syst., 2022, 6, 2100057 CrossRef.
- S.-W. Hwang, H. Tao, D.-H. Kim, H. Cheng, J.-K. Song, E. Rill, M. A. Brenckle, B. Panilaitis, S. M. Won, Y.-S. Kim, Y. M. Song, K. J. Yu, A. Ameen, R. Li, Y. Su, M. Yang, D. L. Kaplan, M. R. Zakin, M. J. Slepian, Y. Huang, F. G. Omenetto and J. A. Rogers, Science, 2012, 337, 1640–1644 CrossRef CAS PubMed.
- W. B. Han, G.-J. Ko, K.-G. Lee, D. Kim, J. H. Lee, S. M. Yang, D.-J. Kim, J.-W. Shin, T.-M. Jang, S. Han, H. Zhou, H. Kang, J. H. Lim, K. Rajaram, H. Cheng, Y.-D. Park, S. H. Kim and S.-W. Hwang, Nat. Commun., 2023, 14, 2263 CrossRef CAS PubMed.
- G. Ye, D. Song, J. Song, Y. Zhao and N. Liu, Adv. Funct. Mater., 2023, 33, 2303990 CrossRef CAS.
- H. Ullah, M. A. Wahab, G. Will, M. R. Karim, T. Pan, M. Gao, D. Lai, Y. Lin and M. H. Miraz, Biosensors, 2022, 12, 630 CrossRef CAS PubMed.
- S. Imani, A. J. Bandodkar, A. V. Mohan, R. Kumar, S. Yu, J. Wang and P. P. Mercier, Nat. Commun., 2016, 7, 11650 CrossRef CAS PubMed.
- W. H. Lee, G. D. Cha and D.-H. Kim, Curr. Opin. Biotechnol, 2021, 72, 13–21 CrossRef CAS PubMed.
- X. Peng, K. Dong, Z. Wu, J. Wang and Z. L. Wang, J. Mater. Sci., 2021, 56, 16765–16789 CrossRef CAS.
- X. Chen, Y. J. Park, M. Kang, S.-K. Kang, J. Koo, S. M. Shinde, J. Shin, S. Jeon, G. Park and Y. Yan, Nat. Commun., 2018, 9, 1690 CrossRef PubMed.
- Y. S. Rim, H. Chen, B. Zhu, S. H. Bae, S. Zhu, P. J. Li, I. C. Wang and Y. Yang, Adv. Mater. Interfaces, 2017, 4, 1700020 CrossRef.
- S.-W. Hwang, C. H. Lee, H. Cheng, J.-W. Jeong, S.-K. Kang, J.-H. Kim, J. Shin, J. Yang, Z. Liu and G. A. Ameer, Nano Lett., 2015, 15, 2801–2808 CrossRef CAS PubMed.
- V. Naresh and N. Lee, Sensors, 2021, 21, 1109 CrossRef CAS PubMed.
- H. Tran, V. R. Feig, K. Liu, H.-C. Wu, R. Chen, J. Xu, K. Deisseroth and Z. Bao, ACS Cent. Sci., 2019, 5, 1884–1891 CrossRef CAS PubMed.
- Y. H. Jung, T.-H. Chang, H. Zhang, C. Yao, Q. Zheng, V. W. Yang, H. Mi, M. Kim, S. J. Cho and D.-W. Park, Nat. Commun., 2015, 6, 7170 CrossRef PubMed.
- N. Thiyagarajan, J.-L. Chang, K. Senthilkumar and J.-M. Zen, Electrochem. Commun., 2014, 38, 86–90 CrossRef CAS.
- Z. Zou, A. Jang, E. MacKnight, P.-M. Wu, J. Do, P. L. Bishop and C. H. Ahn, Sens. Actuators, B, 2008, 134, 18–24 CrossRef CAS.
- X. Liu, M. Shi, Y. Luo, L. Zhou, Z. R. Loh, Z. J. Oon, X. Lian, X. Wan, F. B. L. Chong and Y. Tong, Appl. Sci., 2020, 10, 1320 CrossRef CAS.
- X. Huang, D. Wang, Z. Yuan, W. Xie, Y. Wu, R. Li, Y. Zhao, D. Luo, L. Cen and B. Chen, Small, 2018, 14, 1800994 CrossRef PubMed.
- M. Mooney, A. Nyayachavadi and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 14645–14664 RSC.
- S.-K. Kang, G. Park, K. Kim, S.-W. Hwang, H. Cheng, J. Shin, S. Chung, M. Kim, L. Yin and J. C. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 9297–9305 CrossRef CAS PubMed.
- S. K. Kang, S. W. Hwang, H. Cheng, S. Yu, B. H. Kim, J. H. Kim, Y. Huang and J. A. Rogers, Adv. Funct. Mater., 2014, 24, 4427–4434 CrossRef CAS.
- S.-K. Kang, R. K. J. Murphy, S.-W. Hwang, S. M. Lee, D. V. Harburg, N. A. Krueger, J. Shin, P. Gamble, H. Cheng, S. Yu, Z. Liu, J. G. McCall, M. Stephen, H. Ying, J. Kim, G. Park, R. C. Webb, C. H. Lee, S. Chung, D. S. Wie, A. D. Gujar, B. Vemulapalli, A. H. Kim, K.-M. Lee, J. Cheng, Y. Huang, S. H. Lee, P. V. Braun, W. Z. Ray and J. A. Rogers, Nature, 2016, 530, 71–76 CrossRef CAS PubMed.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- V. N. Hamanaka, E. Salsberg, F. J. Fonseca and H. Aziz, Org. Electron., 2020, 78, 105509 CrossRef CAS.
- J.-K. Chang, H. Fang, C. A. Bower, E. Song, X. Yu and J. A. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5522–E5529 CAS.
- J. K. Salunke, F. Wong, K. Feron, S. Manzhos, M. F. Lo, D. Shinde, A. Patil, C. Lee, V. Roy and P. Sonar, J. Mater. Chem. C, 2016, 4, 1009–1018 RSC.
- M. Pfeiffer, K. Leo, X. Zhou, J. Huang, M. Hofmann, A. Werner and J. Blochwitz-Nimoth, Org. Electron., 2003, 4, 89–103 CrossRef CAS.
- Y. Chen and D. Ma, J. Mater. Chem., 2012, 22, 18718–18734 RSC.
- S. Yoon, S. Park, S. H. Park, S. Nah, S. Lee, J.-W. Lee, H. Ahn, H. Yu, E.-Y. Shin, B. J. Kim, B. K. Min, J. H. Noh and H. J. Son, Joule, 2022, 6, 2406–2422 CrossRef CAS.
- M. Shibata, Y. Sakai and D. Yokoyama, J. Mater. Chem. C, 2015, 3, 11178–11191 RSC.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 RSC.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- J. D. Myers and J. Xue, Polym. Rev., 2012, 52, 1–37 CrossRef CAS.
- J. Borges-González, C. J. Kousseff and C. B. Nielsen, J. Mater. Chem. C, 2019, 7, 1111–1130 RSC.
- Y.-W. Su, S.-C. Lan and K.-H. Wei, Mater. Today, 2012, 15, 554–562 CrossRef CAS.
- D. A. Bernards, R. M. Owens and G. G. Malliaras, 2008.
- J. A. Chiong, Y. Zheng, S. Zhang, G. Ma, Y. Wu, G. Ngaruka, Y. Lin, X. Gu and Z. Bao, J. Am. Chem. Soc., 2022, 144, 3717–3726 CrossRef CAS PubMed.
- J. A. Chiong, L. Michalek, A. E. Peña-Alcántara, X. Ji, N. J. Schuster and Z. Bao, J. Mater. Chem. C, 2023, 11, 15205–15214 RSC.
- S. Yoon, E.-Y. Shin, N.-K. Cho, S. Park, H. Y. Woo and H. J. Son, J. Mater. Chem. A, 2021, 9, 24729–24758 RSC.
- S. Park, S. H. Park, H. Jin, S. Yoon, H. Ahn, S. Shin, K. Kwak, S. Nah, E.-Y. Shin, J. H. Noh, B. K. Min and H. J. Son, Nano Energy, 2022, 98, 107187 CrossRef CAS.
- H. Jin, K. Kim, S. Park, J. Rhee, H. Ahn, D. J. Kim, K. Kim, J. H. Noh, T. S. Kim, E. Y. Shin and H. J. Son, Adv. Funct. Mater., 2023, 33, 2304930 CrossRef CAS.
- A. M. Cantón-Díaz, B. M. Munoz-Flores, I. Moggio, E. Arias, A. De León, M. C. García-López, R. Santillán, M. E. Ochoa and V. M. Jiménez-Pérez, New J. Chem., 2018, 42, 14586–14596 RSC.
- A. W. Jeevadason, K. K. Murugavel and M. Neelakantan, Renewable Sustainable Energy Rev., 2014, 36, 220–227 CrossRef.
- N. Nozaki, A. Uva, H. Matsumoto, H. Tran and M. Ashizawa, RSC Appl. Polym., 2024, 2, 163–171 RSC.
- J.-Y. Liang, S.-R. Shin, S.-H. Lee and D.-S. Lee, Polymers, 2019, 11, 1674 CrossRef CAS PubMed.
- L. Liu, Y. Gao, J. Zhao, L. Yuan, C. Li, Z. Liu and Z. Hou, Polymers, 2018, 10, 1125 CrossRef PubMed.
- J. Shi, D. Xing and J. Lia, Energy Procedia, 2012, 16, 758–762 CrossRef CAS.
- S.-H. Lee, S.-R. Shin and D.-S. Lee, Mater. Des., 2019, 172, 107774 CrossRef CAS.
- W. H. Lee and Y. D. Park, Polymers, 2014, 6, 1057–1073 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthesis of monomers and polymers, additional FT-IR results, and NMR results. See DOI: https://doi.org/10.1039/d4tc01377h |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |