
Conjugated polymers with near-infrared (NIR) optical absorption: structural design considerations and applications in organic electronics
Tanzida
Zubair†
,
Md Mahmudul
Hasan†
,
Raul S.
Ramos†
and
Robert M.
Pankow
 *
*
Department of Chemistry and Biochemistry, University of Texas at El Paso, El Paso, Texas 79968, USA. E-mail: rmpankow@utep.edu
First published on 14th May 2024
Abstract
Conjugated polymers (CPs) have revolutionized soft-matter electronics by enabling the manufacture of non-toxic, low-cost, and mechanically robust materials and devices for various technologies, including organic photovoltaics (OPV), electrochromic devices (ECDs), organic field effect transistors (OFET), and organic bioelectronics. Coupled with breakthroughs in synthetic chemistry and structure-function properties, the advancements in organic electronics have been firmly intertwined with CP structural developments. Recent research efforts have focused on the molecular engineering of CPs to broaden the optical absorption to the near-infrared (NIR) region (>750 nm) to enable new capabilities in photon harvesting for photovoltaics, optical switching and detection, thermal energy management, active camouflage, and to tune energy levels to achieve n-type and ambipolar charge transport. This review presents a comprehensive overview of the design and synthesis of CPs with NIR optical absorption, and it describes their incorporation in established and emerging device applications. A central focus throughout is how the polymer structures can be tailored specifically for each application with included strengths and shortcomings. An emphasis is placed on recent and emerging polymer structures with an outlook on areas for future work.
 Robert M. Pankow | Robert M. Pankow received his BA in Chemistry from the University of California, Santa Barbara (2012) and his MS in Chemistry from California State University, Northridge (2015). He then earned his PhD in Chemistry under the guidance of Prof. Barry C. Thompson at the University of Southern California (2020). Robert then moved Northwestern University where he was an Intelligence Community Postdoctoral Fellow in the lab of Prof. Tobin J. Marks. Robert then began his independent career as an Assistant Professor in the Department of Chemistry and Biochemistry at The University of Texas at El Paso in August 2023. |
1. Introduction
The prodigious advancements in conjugated polymer (CP) structural engineering over the last few decades have been directly coupled with the development of new synthetic methodologies for monomer functionalization, polymerization, and an improved understanding of the structure–function relationships that enable state-of-the-art performance metrics in electrochromic devices (ECDs),1–4 organic photovoltaics (OPV),5–8 organic photodiodes and phototransistors (OPD and OPTs),9–12 organic field effect transistors (OFET),13–16 and organic electrochemical transistors (OECTs).17–20 Recently, significant research effort has been focused on the development of CPs with near-infrared (NIR) optical absorption (750–2500 nm) to increase light harvesting capabilities in OPV,21,22 provide optical transparency for smart-windows and photovoltaic green houses,23,24 enable new capabilities in active camouflage,25,26 provide discrete, transparent electronics,27,28 and narrow bandgaps to enable ambipolar charge transport.14,29,30 Compared to their inorganic counterparts possessing NIR-IR optical absorption, e.g. bulk III–V semiconductors,31,32 quantum dots,33 and 2D materials,34,35 NIR-CPs are overall more sustainable and relatively non-toxic, are mechanically robust (flexible and stretchable), and provide ease of tuning the optical, electronic, and physicochemical properties.36In previous decades, significant research effort in NIR-CPs was focused on developing new light harvesting polymers for applications in polymer-fullerene organic solar cells, where the development of a polymer spectral response complimentary to fullerene and covering the visible wavelength range (380–700 nm) was primarily targeted.37,38 Additionally, for ECDs CPs with optical switching and coloration within the visible wavelength range were heavily pursued, since this boasted the greatest benefit for smart-window and display technologies.1,39 A characteristic of narrow bandgap CPs is ambipolar and n-type charge transport, which has been of great interest in OFET and OECT technologies, and the advent of electronic skin (e-skin) has stimulated renewed interest for NIR-CPs.40,41
In this review, the relevant aspects of NIR-CP design and their impact on the optical, electronic, and morphological/microstructural properties are first introduced. This includes ECDs, OPVs, OPDs, OPTs, OFETs, and OECTs. Next, different classes of NIR-CPs organized by repeat unit are provided with their distinct structural, optical and electronic characteristics, and critical synthetic steps, and aspects of sustainability indicated. While λmax is used as a point of comparison for detailing the relative region of the polymer optical absorption profile, it is important to note that many of the NIR-CPs have optical absorption extending further beyond this value, so CPs with an optical bandgap (Eg) less than 1.65 eV where λonset = 750 nm are considered. Notable organic electronic device applications and relevant performance metrics are also indicated. An emphasis is placed on various electron acceptor repeat units in alternating donor–acceptor copolymers, since many of the unique developments and advancements in CP structure can be ascribed to this unit. Lastly, an outlook and concluding remarks are provided.
2. General considerations for NIR-CP design
The structural diversity present in NIR-CPs is the result of extensive materials evaluation and the establishment of structure–function relationships provided from evaluating device performance trends. In this section, we present key structural engineering strategies for the design and synthesis of NIR-CPs and provide an overview of factors influencing CP self-assembly, morphology and microstructure, and the specificity of these for different organic electronic device applications.2.1 Donor–acceptor copolymers
The discovery of the donor–acceptor (D–A) copolymer by Havinga et al. ushered in a new era in CP structural design (see Fig. 1A).42,43 Briefly, an alternating donor–acceptor conjugated copolymer consists of an electron donating unit and an electron accepting unit forming the π-conjugated backbone. The linkages and connectivity between these units are carefully controlled so that donor–donor (D–D) or acceptor–acceptor (A–A) homocouplings are minimized, which can be detrimental to achieving the desired optical and electronic properties.44,45 This polymer design strategy can be further adjusted by incorporation of a second electron donor (yielding D1–A–D2–A) or electron acceptor unit (yielding D–A1–D–A2) to yield a semi-random terpolymer (Fig. 1A).46 Following the principles of perturbation theory, by pairing an electron donor with a high lying highest occupied molecular orbital (HOMO) with an electron acceptor possessing a relatively low-lying lowest unoccupied molecular orbital (LUMO), the energy levels of the CP, e.g. the valence band and conduction band, can be dramatically altered to afford a narrow bandgap (Fig. 1A). Examples of donor (1–7) and acceptor (8–12) repeat units are provided in Fig. 1B, where it is shown that electron donating, e.g. electron rich aromatic heterocycles (1–4) or fused-ring aromatics (5–7), and electron accepting functionalities, e.g. electronegative heteroatoms and electron withdrawing substituents (8–12), are the defining structural features, respectively. Note, the utility of D–A copolymers was enabled by advancements in CP polymerization methods, such as the optimization of Suzuki–Miyaura and Stille polymerizations. These methodologies provide a high functional group tolerance thereby facilitating the rapid development of large libraries of polymer structures, due to the combinatorial screening and syntheses that can be easily accomplished when various donor comonomers are paired with various acceptors.36,47,48 Thus, D–A copolymers with tailored bandgaps can be easily obtained providing either a wide optical bandgap (Eg > 1.8 eV), a medium bandgap (1.60 eV < Eg < 1.75 eV), or a narrow bandgap (Eg < 1.60 eV).7,49 | ||
| Fig. 1 (A) Overview of the donor–acceptor (D–A) conjugated copolymer and terpolymer design strategy towards the preparation of narrow bandgap conjugated polymers, and the mixing between donor monomer/acceptor monomer HOMOs (HOMOD/HOMOA) and LUMOs (LUMOD/LUMOA) to achieve a reduced bandgap (Eg = LUMO–HOMO). (B) Select examples of donor repeat units and acceptor repeat units to highlight the structural diversity and distinguishing functional groups that define these classes of monomers. (C) Examples of heteroatom modification, ring-fusion, and π-extension using benzothiadiazole (BT) as the example. (D) Examples of sidechain engineering including the design of linear alkyl, branched alkyl, and the incorporation of heteroatoms to afford oligo(ethylene glycol) (EGn) and siloxane based sidechains. | ||
2.2 Heteroatom substitution, ring fusion, and π-extension
The strategic incorporation of halogens (e.g., F or Cl), chalcogens (e.g., Se or Te), or pnictogens (e.g., N), is a simple yet powerful tool for narrowing the optical bandgap and tuning energetics to increase electron affinity and facilitate electron transport.50–53 Examples of this are shown in Fig. 1C (11–20) starting from benzothiadiazole (BT). The incorporation of heteroatoms, such as fluorine and other halogens, can have profound effects on controlling the polymer morphology and self-assembly.54 Notably, fluorination (13) and cyanation (14) have been shown to increase the electron affinity (EA), and halogenation can further improve the polymer conformational coplanarity via halogen-chalcogen and halogen-hydrogen non-covalent interactions.55–58 The incorporation of other chalcogens in place of sulphur to afford other chalcogenophenes (16), such as selenophene and tellurophene, is an additional strategy for tuning the polymer optical/electronic and physicochemical properties.53,59–61 Relative to thiophene, the inter-ring bond distances of selenophene and tellurophene are shorter indicating greater quinoidal character and improved π-orbital overlap within the π-conjugated system.53,62,63 Thus, a reduction in the optical bandgap is observed. Additionally, strong Se–Se and Te–Te non-covalent interactions drive polymer self-assembly and can increase polymer semicrystallinity providing extended crystalline domains to accommodate charge transport.63,64Next, ring fusion and π-extension (17–20) are additional strategies for rigidifying or increasing the extent of π-orbital overlap within the π-conjugated system of the CP, which in turn narrows the bandgap and facilitates charge transport.65–67 In the case of ring-fusion for 11 this often invokes the functionalization of naphthalene and anthracene analogues and derivatives. However, extended fused ring monomers can also be synthesized from simpler molecular building blocks (6, 7, and 12) via linear or convergent synthetic pathways.68–71
2.3 Sidechain engineering
The CP sidechains facilitate solution processing and dictate the identity of processing solvent, e.g., halogenated solvents (chloroform, chlorobenzene, or dichlorobenzene) or non-halogenated solvents (e.g., toluene, xylenes, alcohols, ethers, and water).72 In addition, they also direct the polymer self-assembly with direct influence over the polymer crystallization, aggregation, polymer chain entanglement, sidechain interdigitation, and polymer orientation relative to the substrate.73–77 Thus, proper tailoring of the CP sidechains is imperative for achieving desired optical/electronic properties and device performance metrics.72,78–81 Briefly, CP sidechains are often linear (21–23) or branched alkyl substituents (24–26) where linear sidechains provide increased sidechain interdigitation thereby affording CP crystalline domains with a high-degree of structural order (Fig. 1D).82 Relative to their linear counterparts, branched alkyl sidechains provide improved polymer solubility in organic processing solvents, regulate polymer aggregation, and increase polymer chain entanglement.83–85Lastly, the incorporation of heteroatoms within sidechains, such as oxygen and silicon, to yield oligo(ethylene glycol) (EGn) or siloxane terminated sidechains (27–29) can impart solution processing with more polar solvents, such as alcohols and water, or more sustainable non-halogenated solvents (Fig. 1D), respectively.86–89 Additionally, the incorporation of EGn sidechains can provide improved electrolyte penetration and exchange, which is essential for ECDs and OECTs.90–94
2.4 Conjugated polymer morphology and microstructure
Precise control over the conjugated polymer morphology and microstructure is imperative for achieving desirable optoelectronic and charge transport properties and ultimately state-of-the-art performance metrics.95–97 As mentioned before, CPs can self-assemble via non-covalent interactions directed by the heteroatoms present within the conjugated backbone and via sidechain interdigitation to form well organized lamellae. Detailed in Fig. 2A, CP thin films can be comprised of a mixture of crystalline and amorphous domains where the crystalline domains are distinguished by improved polymer self-assembly and highly ordered nanostructuring. In contrast, the amorphous domains contain polymer chains with a random orientation and lack a well-defined microstructure. In a fully amorphous polymer, there is an absence of observable crystalline domains, and the polymer chains assume a random orientation (Fig. 2A). It is important to note that many semicrystalline CPs afford excellent charge transport properties, since the intermolecular and π–π interactions that facilitate charge delocalization and transport between polymer chains have an increased presence in the crystalline domain.98 Note, amorphous polymers with well-defined electronic coupling between polymer chains are still capable of possessing efficient charge transport.99 | ||
| Fig. 2 (A) Depiction of the different semicrystalline and amorphous morphologies possible with conjugated polymers, as well as a depiction of a polymer blend composed of two semicrystalline polymers (polymer A and polymer B). (B) Depiction of the different polymer orientations relative to the substrate face, including: π-face on, bimodal, edge on, and random. | ||
Additionally, different organic electronic device applications require specific polymer morphologies and microstructures to achieve desirable performance. Specifically, for ECDs and OECTs, where device operation is tethered to the ability of the electrolyte to penetrate and exchange within the polymer film, having a CP with large crystalline domains can lead to electrolyte ion trapping which is detrimental to device performance.92,100,101 Conversely, in OFETs having a large crystalline domain size can be beneficial to ensuring efficient charge transport and high hole and electron mobilities.98,102,103 Larger crystalline domain sizes are also important in OPV, but in this setting the domain sizes must be carefully controlled to minimize extensive aggregation within the active layer blend to provide efficient photocurrent generation and charge extraction.104,105 This concept of controlling the polymer blend morphology is detailed in Fig. 2A, where the different polymer constituents (polymer A and polymer B) can form separated domains and interpenetrating networks of crystalline and amorphous domains.106,107 Lastly, the parameters influencing polymer self-assembly can dictate the orientation of the polymer chain, e.g., whether the CP π-conjugated backbone is parallel or perpendicular to the face of the substrate (Fig. 2B). This orientational preference and the microstructural characterization described in Section 2.4 can be probed using grazing incidence wide-angle X-ray scattering (GIWAXS).108,109 The polymer orientation is of critical importance to the device performance metrics for OFET and OPV, since the direction of polymer chain alignment indicates the preferred direction of charge transport. Specifically, a preferential π-face-on orientation facilitates charge transport in the vertical direction relative to the substrate face, which is beneficial for OPV. In contrast, an edge-on orientation facilitates charge transport in the horizontal direction, which is of greater utility to OFETs/OECTs. It is possible that preferential alignment is not observed in which case a bimodal or random orientation is obtained.110
To summarize, the D–A copolymer structure has enabled the rapid development of state-of-the-art NIR-CPs for a variety of organic electronic device technologies. Key functionalization strategies for tuning the polymer optical/electronic, physicochemical, and morphology and microstructure include heteroatom incorporation, ring fusion, π-extension, and sidechain engineering. Lastly, it is important to note that the different organic electronic devices each require a specific polymer morphology (semicrystalline or amorphous), microstructure, and chain orientation (π-face on, edge on, bimodal, or random), which is further elaborated upon in the following section.
3. Organic electronic device applications for NIR-CPs
In this section the basic overview of device operation and key performance metrics for ECDs, OPV, OPDs, OFETs, and OECTs are briefly described to establish the broad utility of NIR-CPs and highlight the structural characteristics specific to each technology. Since the focus of this review is not on device physics or optimization of the device architecture, relevant reviews and articles detailing these topics are provided.3.1 Electrochromic devices (ECD)
ECD technologies have traversed into new applications, including thermal energy management, energy storage and active camouflage, which traverse beyond the smart-window and display technology applications originally envisioned.3,111,112 Central to these new capabilities for ECDs, are the advent of electrochemically robust NIR-CPs and advancements in ECD device architecture and engineering (Fig. 3).111–113 For the vertical ECD architecture, a CP film is coated onto an optically transparent electrode and coated with an electrolyte. A second electrode coated with an ion-storage layer (ISL) covers the polymer CP film. For the lateral ECD architecture, the NIR-CP and the ISL are adjacent (in the same lateral plane), and each is coated with the electrolyte. Briefly, starting with a neutral CP a positive or negative bias is applied to electrochemically oxidize or reduce the CP layer, which in turn generates a p/n-polaron, respectively.2,92,114 This leads to bleaching of the ground state optical absorption band and emergence of a polaron/bipolaron absorption band. Note, since this is an electrolyte coupled process, efficient and reversible electrochemical oxidation or reduction and electrolyte exchange within the CP layer are of critical importance. For NIR-IR optical switching, it is also important to consider the optical transparency of the electrode and substrate.115 Key performance metrics for ECDs include the optical contrast (ΔT%), the switching time (t95), coloration efficiency (CE), cycling stability, and optical memory.2,111,114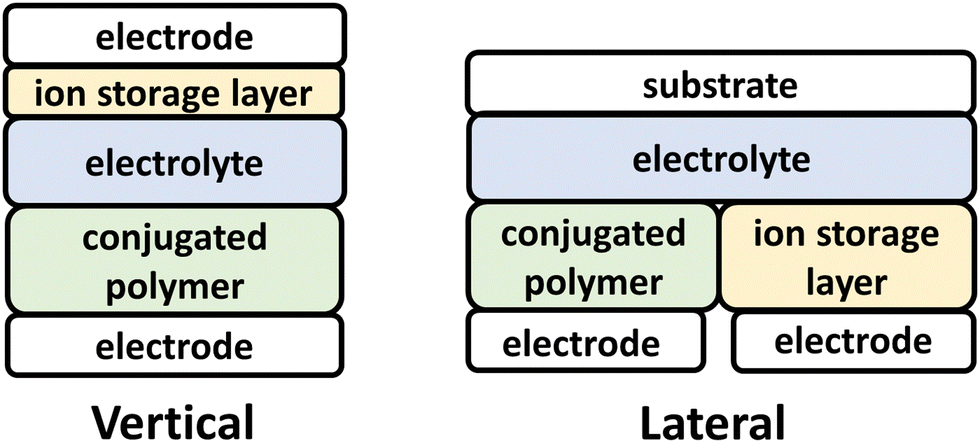 | ||
| Fig. 3 Vertical (left) and lateral (right) ECD architectures. | ||
Specific for considerations for conjugated polymer design include incorporation of functionalities and repeat units that impart electrochemical robustness, such as 3,4-ethylenedioxythophene (EDOT), and oligo(ethylene glycol) (EGn) sidechains to facilitate electrolyte penetration and exchange.114 NIR-CPs for ECDs have been synthesized primarily via Stille polymerization, but it is notable that Reynolds et al. have demonstrated that CPs for ECDs can be prepared using direct arylation polymerization (DArP), which is more sustainable since it proceeds via C–H functionalization and avoids the use of toxic trialkylstannane residues.116–119 In this review, NIR-CPs employed for ECDs include isoindigo (Section 4.1) and diketopyrrolopyrrole analogues (Section 4.2).
3.2 Organic photovoltaics (OPV)
OPV seeks to provide a sustainable energy capture resource to combat fossil fuel dependence, and broadening CP optical absorption into the NIR wavelength region has been a longstanding goal to enable efficient solar energy harvesting.8,37,120 OPV device architectures are shown in Fig. 4 where the NIR-CP resides in the active layer. The active layer is a blend of an electron donor polymer (Fig. 5) and an electron acceptor small-molecule or an electron acceptor polymer (Fig. 8–12). This blend forms a bulk-heterojunction, which is a phase-separated interpenetrating bicontinuous network composed of the donor and acceptor domains or phases (see Fig. 2A).121 Note, the active layer can also be composed of a single-component where the donor and acceptor are covalently linked, e.g., single-component solar cells and double cable polymers.122 The active layer is then interfaced with a hole and an electron transport layer, which are in contact with the electrodes used for charge collection. The nomenclature used (conventional or inverted), simply refers to whether the bottom electrode is in contact with the hole transport layer (conventional) or the electron transport layer (inverted). Briefly, device operation initiates when the donor polymer absorbs sunlight generating an exciton that travels to the donor–acceptor interface forming the charge-transfer (CT) state. Charge separation can then occur forming a hole and an electron in the donor and acceptor phases, respectively. These charges can then travel through the donor/acceptor domains until collected by the electrodes yielding photocurrent. Key performance metrics used for evaluating OPV device performance include the short-circuit current (Jsc), the open-circuit voltage (Voc), fill-factor (FF), and the power conversion efficiency (PCE), which have been described in detail in relevant reviews.123–126 | ||
| Fig. 4 Conventional (left) and inverted (right) OPV device architectures. | ||
 | ||
| Fig. 5 Examples of donor polymers used in OPV, OPDs, and OPTs. | ||
The design of donor and acceptor polymers for OPV is highly complex and requires the consideration various facets including: (i) the ionization potential (IP) and electron affinity (EA) of the donor polymer and acceptor polymer or small-molecule for efficient charge transfer and limiting charge recombination, (ii) ensuring complimentary optical absorption of the donor and acceptor to maximize spectral coverage, (iii) balanced and maximized hole and electron mobilities of the donor and acceptor, respectively, for efficient charge transport and collection, and (iv) optimal morphology/microstructure of the donor and acceptor phases to limit charge recombination/traps and maximize hole and electron charge transport to their respective electrodes.8,38,105 NIR-IR optical absorption has been targeted for both the donor and acceptor components, but recent state-of-the-art PCEs have been achieved by employing new polymer acceptors with NIR optical absorption and pairing with wide to mid-bandgap polymer donors.5,6
NIR-CPs for OPV are most often synthesized using Stille polymerization, since Stille affords broad functional group tolerance, high molecular weights (Mn), and polymer products with excellent structural fidelity. To avoid the generation of toxic tin waste and increase the sustainable aspects of OPV, there have been significant developments in the area of DArP and Zn-anion radical polymerizations to afford NIR-CPs that provide OPV performance metrics that converge or surpass their Stille analogues.127–130 In this review, NIR-CPs incorporated into OPV devices include diketopyrrolopyrrole analogues (Section 4.2), naphthalene diimide (Section 4.3), benzothiadiazole (Section 4.4), polymerized small-molecule acceptors (Section 4.5), and cyclopentadithiophene analogues (Section 4.6).
3.3 Organic photodiodes (OPD)
OPDs seek to convert optical illumination into an electrical signal, and the incorporation of NIR-CPs has enabled new capabilities in optical sensing, biomedical imaging, and optical communication.11,12 Note, organic photodetectors have various architectures, such as phototransistors (OPTs) and photoconductors, but only the photodiode architecture, which is analogous to the conventional and inverted OPV device architectures, is considered in this section (Fig. 4).11 OPDs consist of an active layer analogous to OPV (blend of polymer donor and polymer or small-molecule acceptor), which has electron or hole transport layers above and below to ensure efficient charge transport and collection. The operation mechanism of an OPD is like that of OPV, where incident light is absorbed by the active layer leading to the formation of excitons, which in turn undergo charge separation and transport to the electrodes yielding photocurrent. Key figures of merit include the external quantum efficiency (EQE) at a given wavelength, the photocurrent/dark current ratio (P), the responsivity (R), and the detectivity (D), and the considerations for NIR-CP design are similar to that of OPV. Namely, the optical absorption of the active layer should overlap with the targeted wavelengths or ranges, and the energy levels of the donor polymer and acceptor polymer should be sufficiently offset to ensure efficient hole and electron charge separation. In this review, NIR-CPs incorporated into OPDs include benzothiadiazole (Section 4.4), polymerized small-molecule acceptors (Section 4.5), and cyclopentadithiophene analogues (Section 4.6).3.4 Organic field effect transistors (OFET) and organic phototransistors (OPTs)
OFETs have enabled new capabilities in flexible and stretchable electronics, chemical/strain sensors, complimentary circuits, and electronic skin.41,131–133 Briefly, the device architecture consists of a source and drain electrodes, the CP, which serves as the channel material, a dielectric layer, and a gate electrode (Fig. 6). These components can be arranged to afford a variety of different architectures, such as top gate bottom contact (TGBC), top gate top contact (TGTC), bottom gate bottom contact (BGBC), and bottom gate top contact (BGTC).14,103 Device operation proceeds with application of positive or negative bias between the gate and source electrode (VGS), which in turn polarizes the channel material and renders the OFET to its “ON” state. Application of a bias between the source and drain electrodes (VDS) induces current flow through the channel and the drain current (ID) is then measured.103,134 Key performance metrics include the saturated current charge mobility (μsat), on/off current ratio (Ion/Ioff), threshold voltage (Vth), and cycling stability. The operation of OPTs invokes a similar mechanism to OFETs, but optical absorption by the channel material (NIR-CP) modulates the charge carrier and transport properties leading to distinct metrics for channel current and threshold voltages between the illuminated and dark states.135 As with OPDs, the key figures of merit for OPTs include P, R, and D.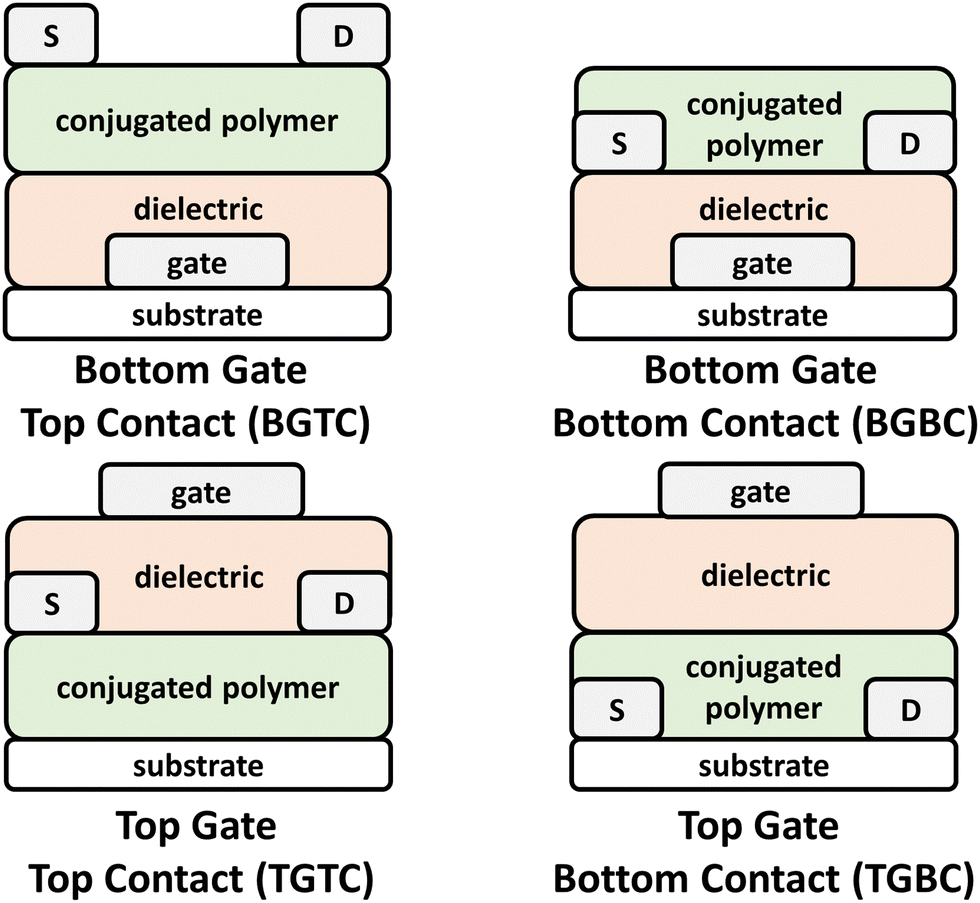 | ||
| Fig. 6 Bottom gate top contact (BGTC), bottom gate bottom contact (BGBC), top gate top contact (TGTC), and top gate bottom contact (TGTC) OFET device architectures. | ||
Developments in transparent OFET technologies, NIR OPTs, and interest in high mobility n-type and ambipolar charge transport, which requires an increased EA, decreased IP, and therefore narrower bandgap, necessitates the pursuit of CPs with NIR optical absorption. However, unlike the previously described NIR-CP device applications, such as ECD, OPV, and OPD, the OFET operation mechanism does not directly depend on the optical absorption of the polymer. Additionally, the potential for these polymers to be employed in other organic electronic device applications outside of OFETs is significant, and so their inclusion within this review will hopefully catalyse these efforts.
As with OPV, there are numerous considerations regarding the overall structural design of CPs to achieve state-of-the-art performance metrics in OFET, e.g., polymer morphology/microstructure and energy levels for charge transport. In general, highly crystalline polymers with short π–π distances, and a π-edge on orientation relative to the substrate are desired to achieve efficient charge transport. A notable structural optimization strategy for OFETs is sidechain engineering, where the inclusion of heteroatoms, such as silanes, and modification of the branching point in long chain branched alkyls provides efficient polymer packing and improved intermolecular π–π overlap.
NIR-CPs for OFETs are commonly synthesized via Stille polymerization, but more recently DArP as well as acid-catalysed aldol condensation-elimination and imine condensation polymerizations, which are tin and transition metal catalyst free, have afforded NIR-CPs with desirable performance metrics.136,137 In this review, NIR-CPs incorporated into OFET and OPTs include diketopyrrolopyrrole (Section 4.2), naphthalene diimide (Section 4.3), and benzothiadiazole analogues (Section 4.4).
3.5 Organic electrochemical transistors (OECT)
Recently, OECTs have generated a resurgence of interest and have uncovered new frontiers in organic electronics and CP structural design. OECTs have numerous applications in biological sensors,138 bioelectronics,132,139 artificial synapses,140 neuromorphic devices,141,142 and complimentary circuits at the machine and biological tissue interface.17,93,94 As with OFETs, the n-type and ambipolar charge transport imparted by narrow bandgap NIR-CPs have made these targeted polymers for OECTs. The OECT device architecture consists of source/drain electrodes, a gate electrode, and a conjugated polymer layer, but in place of the dielectric layer a liquid electrolyte (typically aqueous) is used (Fig. 7). Additionally, a polymer insulator, such as parylene, can be coated over the source/drain and the CP layer and the channel patterned using lithography. Common architectures include top contact, bottom contact, coplanar, and vertical. For OECTs operating in accumulation mode, application of a gate voltage (VGS) is applied that induces the injection of the electrolyte ions and the electrochemical doping the channel material rendering the device to its “ON” state.143 Key performance metrics for OECTs include the transconductance (gm), voltage hysteresis (ΔV), on/off current ratio (Ion/Ioff), and cycling stability. | ||
| Fig. 7 OECT device architectures including top contact, bottom contact, coplanar, and vertical. | ||
For efficient operation of OECTs, it is imperative that the channel material provides excellent charge/electronic transport while facilitating the penetration and intercalation of electrolyte ions. The simultaneous transport of electronic/ionic charge is referred to as organic mixed ionic-electronic conduction (OMIEC), and the optimization of this parameter has generated new avenues in the structural engineering of CPs. In general, the structures of CPs in OECTs require similar characteristics as that of OFETs, but a key difference is the inclusion of EGn sidechains to increase the hydrophilicity of the polymer (essential when aqueous electrolytes are used) and to support electrolyte cation transport. Additionally, as with OFETs, the device operational mechanism of OECTs is not directly dependent on the polymer optical absorption; however, many state-of-the-art polymers demonstrate NIR optical absorption due to the preferential arrangement of energy levels, e.g. the polymer IP/EA and corresponding optical bandgap. These polymers are most often synthesized via Stille polymerization, indicating a need for the introduction of more sustainable polymerization methods. Although Mei et al. and Thelakkat et al. have demonstrated synthesis of DPP NIR-CPs via DArP and their incorporation into OECTs.144,145 In this review, NIR-CPs incorporated into OECTs include isoindigo (Section 4.1) and diketopyrrolopyrrole analogues (Section 4.2).
4. Donor–acceptor conjugated copolymers with NIR-optical absorption
In this section, copolymers containing isoindigo (IID), diketopyrrolopyrrole (DPP), naphthalene diimide (NDI), fused-ring electron acceptors (FREAs), and cyclopentadithiophene (CDT) and analogues are described. The polymer λmax/Eg, EA/IP, and notable organic electronic device performance are tabulated, and key synthetic steps, functionalization strategies, and aspects synthetic sustainability are indicated when relevant.4.1 Isoindigo (IID) analogues
IID has remained a prominent component of CPs since its original disclosure by Reynolds et al.146,147 The IID core is highly modular, providing facile access to a variety of structural analogues, and can be prepared in a few number of scalable synthetic steps.147,148 It has demonstrated utility in a variety of organic electronic applications notably ECDs, OPV, OFETs, and OECTs. IID monomers have also been employed in DArP including flow-synthesis, which avoids polymer batch-to-batch variation and provides large-scale continuous polymer synthesis.127,149 Polymer structures are provided in Fig. 8 with optical absorption (λmax), optical bandgap (Eg), IP/EA, and device performance metrics tabulated in Table 1. Through extensive structural optimization, such as sidechain engineering and proper pairing of the electron donor repeat unit, excellent charge motilities in OFETs have been achieved (μh = 3.62 cm2 V−1 s−1) with P1 (IIDDT-C3), which possesses optical absorption at the edge of the NIR wavelength region (λmax = 719 nm and Eg = 1.58 eV).150 The elegantly designed sidechain of P1 (IIDDT-C3) provides a branching site further away from the conjugated polymer backbone allowing for more efficient polymer packing and shorter π–π distances (3.57 Å). Changing the identity of the donor repeat unit to one that is more electron donating, e.g, bis-EDOT, can narrow the bandgap and shift the optical absorption further into the NIR region, such as with P2 [p(IID-2EDOT)] which provides a red shift (λmax = 740 nm and Eg = 1.32 eV) and enables NIR optical switching in ECDs [CE (1040 nm) = 433 cm2 C−1].151P2 [p(IID-2EDOT)] also provided the shortest bleaching times (tb) when compared to its bithiophene analogue (8.7 s versus 30.5 s, respectively). The Incorporation of EGn sidechains, such as with P3 (IG-T) imparts hydrophilicity essential for OECT operation affording gm = 0.023 S cm−1, and thiophene is used in this case leading to blue shift in optical absorption relative to P1 (λmax = 712 nm and Eg = 1.57 eV).152 Inclusion of a vinylene or thienothiophene π-spacer between IID units and the strategic incorporation of heteroatoms enables ambipolar charge transport in OFETs for P4 (PF4IVI2T) and P5 (PITTI-BT) (μh/μe = 1.03/1.82 cm2 V−1 s−1 and 3.06/2.81 cm2 V−1 s−1) allowing optical absorption to extend further into the NIR wavelength range (Eg = 1.57 and 1.52 eV), respectively.153,154 For P4 (PF4IVI2T) and P5 (PITTI-BT) incorporation of a π-spacer and fluorine substituents provided stronger intermolecular interactions and improved polymer packing, including a preferential edge-on orientation. Additionally, P5 (PITTI-BT) was processed using p-xylene, which is a more sustainable processing solvent, in place of halogenated solvents.154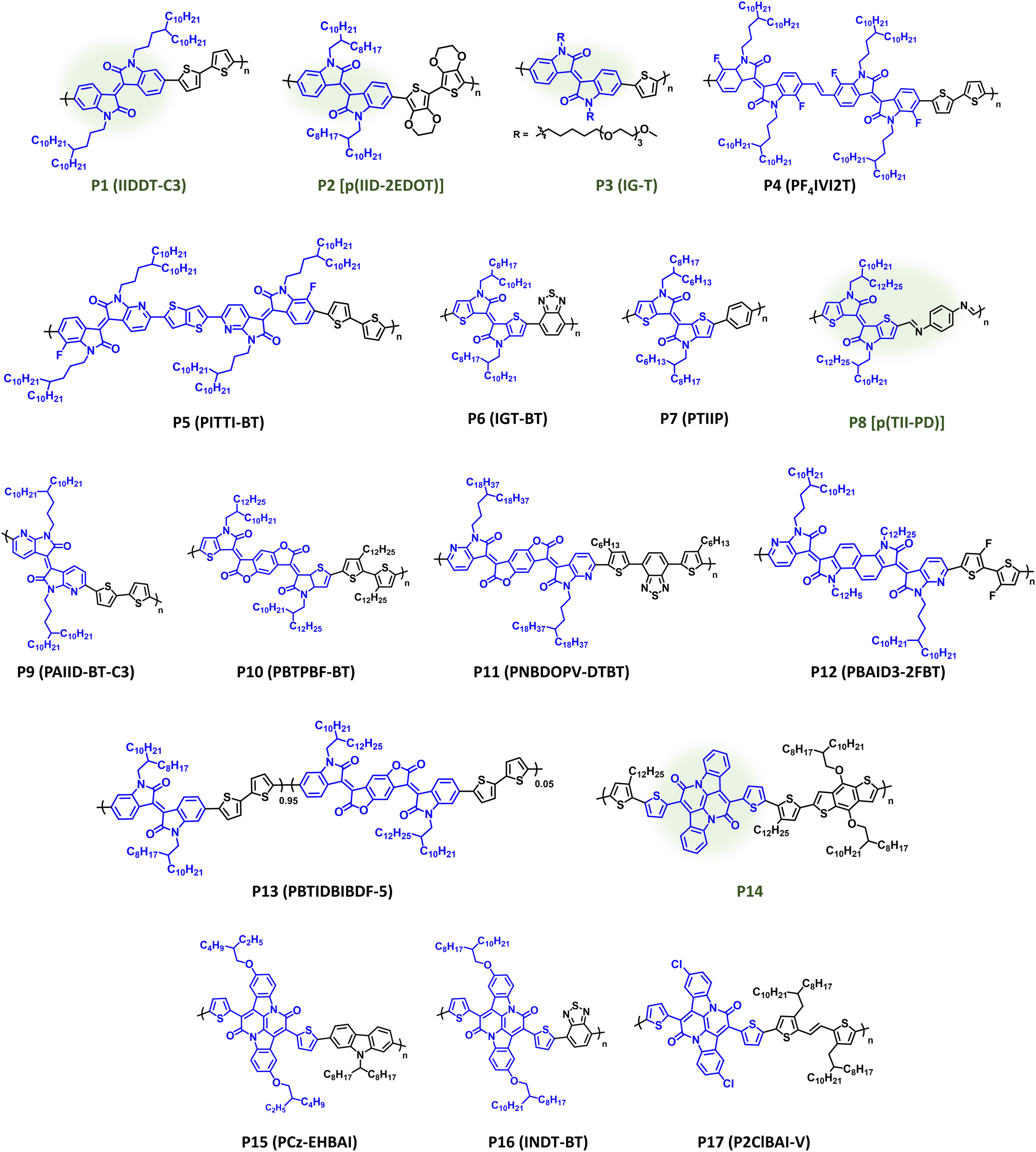 | ||
| Fig. 8 Select examples of NIR-CPs containing IID, IID analogues, and BAI repeat units. IID analogues repeat units and NIR-CPs with relatively sustainable syntheses are highlighted in green. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P1 (IIDDT-C3) | 719; 1.58 | −5.52/−3.74 | OFET (BGTC) | μ h = 3.62 cm2 V−1 s−1 | 150 |
| P2 [p(IID-2EDOT)] | 740; 1.32 | −5.06/−4.05 | ECD (vertical) | CE (1040 nm) = 433 cm2 C−1 | 151 |
| P3 (IG-T) | 712; 1.57 | −5.3/−3.8 | OECT (top contact) | g m = 0.023 S cm−1 | 152 |
| P4 (PF4IVI2T) | 728; 1.57 | −5.74/−4.17 | OFET (TGBC) | μ h/μe = 1.03/1.82 cm2 V−1 s−1 | 153 |
| P5 (PITTI-BT) | 752; 1.52 | −5.71/−4.19 | OFET (TGBC) | μ h/μe = 3.06/2.81 cm2 V−1 s−1 | 154 |
| P6 (IGT-BT) | 1035; 0.92 | −4.86/−3.73 | OFET (TGBC) | μ h/μe = 0.10/0.16 cm2 V−1 s−1 | 155 |
| P7 (PTIIP) | 872; 1.18 | −4.99/−3.63 | OFET (BGBC) | μ h = 5 × 10−3 cm2 V−1 s−1 | 156 |
| P8 [p(TII-PD)] | 770; 1.32 | −5.21/−3.89 | OFET (BGTC) | μ h = 1.83 × 10−2 cm2 V−1 s−1 | 157 |
| P9 (PAIID-BT-C3) | 756; 1.50 | −5.67/−3.64 | OFET (BGBC) | μ h/μe = 0.48/0.78 cm2 V−1 s−1 | 158 |
| P10 (PBTPBF-BT) | 1107; 0.43 | −5.18/−3.94 | OFET (BGTC) | μ h/μe = 0.34/0.13 cm2 V−1 s−1 | 159 |
| P11 (PNBDOPV-DTBT) | 876; 1.26 | −5.64/−4.38 | OFET (TGBC) | μ h/μe = 5.97/7.07 cm2 V−1 s−1 | 160 |
| P12 (PBAID3-2FBT) | 797; 1.27 | −5.17/−3.90 | OFET (BGTC) | μ h/μe = 1.68/1.37 cm2 V−1 s−1 | 161 |
| P13 (PBTIDBIBDF-5) | n.r./1.51 | −5.58/−3.74 | OPT (BGTC) | R (650 nm) = 128 A W−1 | 162 |
| P14 | 783; 1.24 | −4.91/−3.63 | OFET (BGBC) | μ h/μe = 1.5/0.41 cm2 V−1 s−1 | 163 |
| P15 (PCz-EHBAI) | 692; 1.52 | −5.21/−3.57 | ECD (vertical) | CE (1100 nm) = 451 cm2 C−1 | 164 |
| P16 (INDT-BT) | 820; 1.23 | −4.97/−3.74 | OFET (TGBC) | μ h/μe = 0.52/3.11 cm2 V−1 s−1 | 165 |
| P17(P2ClBAI-V) | 897; 1.04 | −5.32/−3.70 | OFET (TGBC) | μ h/μe = 4.04/1.06 cm2 V−1 s−1 | 166 |
To alleviate steric hindrance between the phenyl C–H and the lactone carbonyl C–O in the IID core, McCulloch et al. developed thienoisoindigo (TIID), which has favourable S–O chalcogen interactions and reduced steric hindrance thereby affording a more conformationally coplanar structure with improved π–π orbital overlap.155 The polymer P6 (IGT-BT) possesses strong NIR optical absorption (λmax = 1035 nm), a very narrow optical bandgap (Eg = 0.92 eV), and balanced ambipolar charge transport properties (μh/μe = 0.10/0.16 cm2 V−1 s−1).155 Even with phenylene as a repeat unit [P7 (PTIIP)], TIID affords desirable NIR optical absorption (λmax = 872 nm) and narrow optical bandgap (Eg = 1.18 eV), albeit with sacrificed charge transport properties (μh = 5 × 10−3 cm2 V−1 s−1).156 A unique approach to improve the sustainability of CP synthesis includes the preparation of P8 [p(TII-PD)] by Nozaki et al., which can be prepared via acid catalysed condensation between the respective aldehyde and amine functionalized monomers.157P8 [p(TII-PD)] provides good NIR optical absorption (λmax = 770 nm and Eg = 1.32 eV) with respectable OFET charge mobility (μh = 1.83 × 10−2 cm2 V−1 s−1). Characterization of the morphology of microstructure of P8 [p(TII-PD)] indicate a fibrous, bundled polymer aggregates with large voids and a bimodal orientation. Notably, P8 [p(TII-PD)] can be recycled to its monomers via acid catalysed hydrolysis and repolymerized. This demonstrates a unique capability for this polymer, and the imperative task of generating more sustainable polymerization methods should generate renewed interest in poly(azomethines).167–170
Another variation of isoindigo includes diazaisoindigo, which incorporates pyridine to alleviate steric hindrance and provide a more coplanar conformation to increase π-conjugation throughout the π-system.158,161,171 Towards the synthesis of P9 (PAIID-BT-C3), Huang et al. demonstrated a targeted approach towards the development of new polymer structures by first using computation to model monomers to ensure desirable torsion angles and coplanarity.158P9 (PAIID-BT-C3) then afforded broad optical absorption throughout the visible wavelength range and into the NIR region (λmax = 756 nm and Eg = 1.50 eV) with ambipolar charge transport in OFETs (μh/μe = 0.10/0.16 cm2 V−1 s−1).
Incorporation of the highly electron deficient benzodifurandione (BDO) between lactone units in IID has proven to be an effective strategy to improve charge transport and significantly red shift optical absorption of IID analogues.136,159,172–175 The TIID analogue P10 (PBTPBF-BT) affords strong NIR optical absorption (λmax = 1107 nm and Eg = 0.43 eV), while achieving ambipolar charge transport in OFETs (μh/μe = 0.34/0.13 cm2 V−1 s−1).159 The polymer demonstrated robust ambient stability, since it was found that the OFET charge mobilities closely maintained their original values (μh/μe = 0.34/0.13 cm2 V−1 s−1) when tested under ambient conditions. P11 (PNBDOPV-DTBT), which is an azaisoindigo analogue, provides desirable NIR optical absorption (λmax = 797 nm and Eg = 1.27 eV) with superior OFET charge mobilities (μh/μe = 5.97/7.07 cm2 V−1 s−1) under ambient testing conditions.160P11 (PNBDOPV-DTBT) was then incorporated into a complementary inverter on a flexible PET substrate yielding a gain of 148. Lastly, P12 (PBAID3-2FBT), which incorporates a naphthalene bis-isatin flanked by azaisoindigo analogues additionally affords ambipolar charge transport in OFETs (μh/μe = 1.68/1.37 cm2 V−1 s−1) with a slight blue shift relative to P10/P11 (λmax = 876 nm and Eg = 1.26 eV).161 Following storage of P12 OFET devices for 173 days, the charge mobilities were maintained relatively close to their initial values (μh/μe = 0.428/0.276 cm2 V−1 s−1) indicating excellent stability. P13 (PBTIDBIBDF-5), is a semi-random copolymer incorporating isoindigo and BDO acceptor repeat units where the composition and percentage of BDO is finely tuned to increase NIR optical absorption (Eg = 1.51 eV) while maintaining suitable charge transport properties for use in OPTs.162 A responsivity (650 nm) of 128 A W−1 was obtained from OPTs incorporating P13 blended with 1% PBA. Flexible OPTs using PET as the substrate were found to provide R (650 nm) of 180 mA W−1. As detailed here, the incorporation of benzodifuradione and its structural analogues is a promising strategy to impart excellent hole and electron charge transport properties with ambient stability and strong optical absorption in the NIR. Additionally, McCulloch et al. have demonstrated naphthalene bis-isatin containing polymers can be prepared via aldol condensation polymerization, which provides a more sustainable polymerization methodology.136
Bay annulated indigo (BAI) or indolonapthyridine can be synthesized directly from the naturally occurring dye, indigo, via a single, scalable condensation step to afford the π-extended fused ring structure.163,165,176 The fused ring structure and structural tunability of BAI impart excellent optical absorption and charge transport properties in the corresponding polymers (P14–P17). Lie et al. disclosed the initial synthesis of BAI towards the preparation of P14, which provided optical absorption on the edge of the NIR wavelength range (λmax = 783 nm and Eg = 1.24 eV) and desirable ambipolar charge transport in OFETs (μh/μe = 1.5/0.41 cm2 V−1 s−1).163 This work was followed up with the introduction of sidechains onto the BAI core and copolymerization with carbazole to yield P15 (PCz-EHBAI), which provided ECDs with remarkably stable optical switching (>7500 cycles) at NIR wavelengths [CE (1100 nm) = 451 cm2 C−1].164 Suzuki–Miyaura polymerization of alkylated BAI with benzothiadiazole afforded P16 (INDT-BT), which provides strong NIR optical absorption (λmax = 820 nm and Eg = 1.23 eV) and notable ambipolar charge transport in OFETs (μh/μe = 0.52/3.11 cm2 V−1 s−1).165 The strategic incorporation of heteroatoms on BAI and an alkylated TVT comonomer affords P17 (P2CIBAI-V), which provided a significant red shift in the optical absorption (λmax = 897 nm and Eg = 1.04 eV) with excellent OFET charge mobilities (μh/μe = 4.04/1.06 cm2 V−1 s−1).166 Microstructural characterization of P17 (P2CIBAI-V) using GIWAXS indicated a bimodal orientation and excellent π–π stacking with distances of 3.54 Å. Overall, BAI is a promising candidate for numerous device applications in organic electronics, and its inherent sustainability, due to indigo serving as a naturally sourced monomer feedstock, and ease of synthesis relative to other acceptor repeat units makes it a valuable target for future work.
4.2 Diketopyrrolopyrrole (DPP) analogues
DPP is one of the most prevalent acceptor repeat units in CPs given its notable utility in OPV, OFETs, and OECTs following disclosure by Janssen et al. of high-performance DPP donor polymers for fullerene solar cells.177–181 As with IID, DPP possesses high structural tunability via facile modification of the pendant aryl groups and alkyl substituents, relative ease of synthesis, and desirable optical and electronic properties suitable for a wide range of organic electronic device applications.182 It is notable that many DPP containing CPs have been synthesized via DArP, which significantly improves the overall sustainability of the CP synthesis.129,130,144,145,183–186 DPP containing NIR-CPs and analogues described here are provided in Fig. 9 with optical absorption (λmax) and bandgap (Eg), IP/EA, and device performance metrics tabulated in Table 2. Pairing DPP with the π-extended donor TVT affords P18 (PDVT-10), which is a well-studied NIR-CP (λmax = 783 nm and Eg = 1.40 eV) exhibiting state-of-the-art hole mobilities under ambient testing conditions (μh = 11.0 cm2 V−1 s−1).187,188P18 (PDVT-10) has also been incorporated into flexible and stretchable OFETs, electronic skin, and photodetectors demonstrating the broad utility of this NIR-CP.187–190 Incorporation of a more electron donating repeat unit, EDOT, provides P19 (DPP-E), which demonstrates a significant red shift in the optical absorption relative to P18 (PDVT-10) (λmax = 914 nm and Eg = 1.19 eV) and imparts electrochemical stability rendering the polymer suitable for use in NIR-ECDs [CE (1180 nm) = 901 cm2 C−1].191–193 Replacement of the alkyl substituents on P19 (DPP-E) with EGn sidechains to improve electrolyte uptake provides P20 (gDPP-E), which affords improved ΔT% metrics in NIR-ECDs relative to P19 (24.2% versus 18.8%, respectively) and has demonstrated utility in OECTs.92,194 Copolymerization of glycolated DPP with EGn functionalized bithiophene yields P21 (gDPP-g2T),which further red shifts the optical absorption (λmax = 834 nm and Eg = 1.08 eV), and it affords notable performance metrics in OECTs (gm,A = 226.1 μS μm2) where the incorporation of EGn sidechains increases hydrophilicity and electrolyte exchange.93P22 (DPP-DTT) incorporates the fused thiophene donor unit thieno[3,2-b]thiophene (TT) providing broad NIR optical absorption with λmax/Eg of 818 nm/1.36 eV.195,196 Blends of P22:polystyrene yielded nanowire networks for efficient charge transport, and coupled with the strong NIR optical absorption an R (850 nm) = 246 A W−1 was achieved.196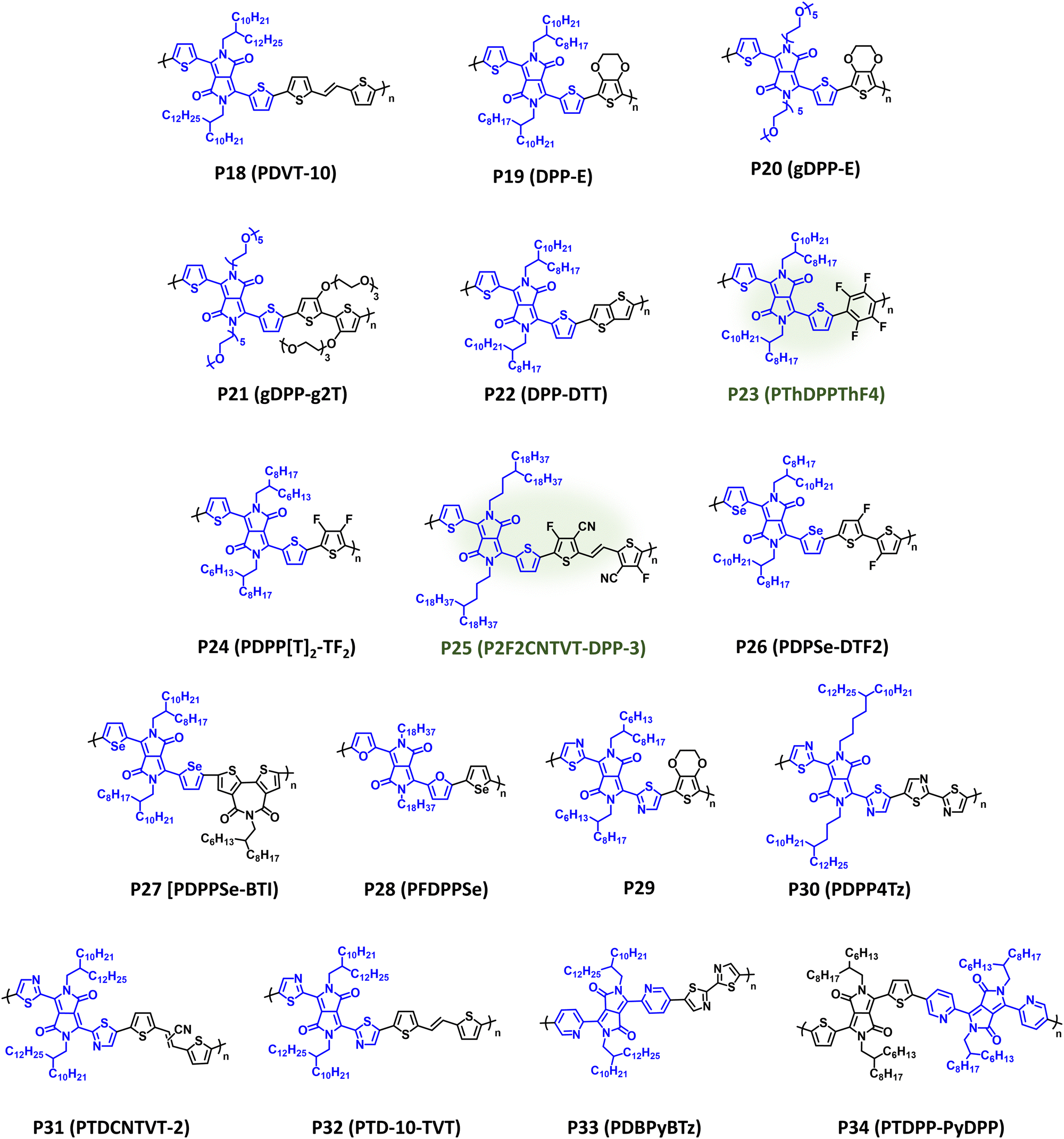 | ||
| Fig. 9 Select examples of NIR-CPs containing DPP analogues. DPP NIR-CPs with relatively sustainable syntheses are highlighted in green. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P18 (PDVT-10) | 783; 1.40 | −5.28; −3.60 | OFET (BGTC) | μ h = 11.0 cm2 V−1 s−1 | 187 |
| P19 (DPP-E) | 834; 1.19 | −5.04/−3.85 | ECD (vertical) | CE (1180 nm) = 901 cm2 C−1 | 192 |
| P20 (gDPP-E) | 853; 1.12 | −4.60/n.r. | ECD (vertical) | ΔT% (853 nm) = 24.2% | 92 |
| P21 (gDPP-g2T) | 914; 1.08 | n.r. | OECT (vertical) | g m,A = 226.1 μS μm2 | 93 |
| P22 (DPP-DTT) | 818; 1.36 | −5.22/−3.86 | OPT (BGTC) | R (850 nm) = 246 A W−1 | 196 |
| P23 (PThDPPThF4) | 775; 1.45 | −4.65/−3.20 | OFET (TGBC) | μ e = 0.44 cm2 V−1 s−1 | 183 |
| P24 (PDPP[T]2-TF2) | 836; 1.3 | −5.91/−3.70 | OFET (BGBC) | μ h/μe = 0.22/0.19 cm2 V−1 s−1 | 197 |
| P25 (P2F2CNTVT-DPP-3) | 864; 1.32 | −5.43/−3.67 | OFET (TGBC) | μ e = 1.61 cm2 V−1 s−1 | 184 |
| P26 (PDPSe-DTF2) | 836; 1.33 | −5.30/−3.97 | OFET (BGTC) | μ h/μe = 0.16/0.006 cm2 V−1 s−1 | 198 |
| P27 (PDPPSe-BTI) | 804; 1.32 | −5.38/−3.51 | OFET (BGTC) | μ h/μe = 0.025/0.154 cm2 V−1 s−1 | 199 |
| P28 (PFDPPSe) | 832, 1.34 | −5.5/−3.8 | OPV (inverted) | PCE = 6.16% | 200 |
| P29 | 820; 1.30 | −5.10/−3.61 | ECD (vertical) | t b/tc (760 nm) = 6.9/0.5 s | 191 |
| P30 (PDPP4Tz) | 715; 1.34 | −5.71/−3.9 | OFET (BGBC) | μ e = 0.067 cm2 V−1 s−1 | 201 |
| P31 (PTDCNTVT-2) | 776; 1.39 | −5.80/−3.72 | OFET (TGBC) | μ h/μe = 1.28/0.97 cm2 V−1 s−1 | 202 |
| P32 (PTD-10-TVT) | 770; 1.47 | −5.40/−3.64 | OFET (BGBC) | μ h/μe = 0.17/0.0097 cm2 V−1 s−1 | 203 |
| P33 (PDBPyBTz) | 656; 1.47 | −5.45/−3.84 | OFET (BGBC) | μ e = 0.023 cm2 V−1 s−1 | 204 |
| P34 (PTDPP-PyDPP) | 792; 1.43 | −5.96/−4.00 | OPV (inverted) | PCE = 0.2% | 205 |
While DPP is typically used for hole transport materials or p-type CPs, copolymerization with fluorinated donor units tunes the EA providing CPs with energy levels suitable for electron transport, while retaining NIR optical absorption with P23–P26 as examples. P23 (PThDPPThF4), which was synthesized via DArP and affords desirable electron mobilities in OFETs (μe = 0.44 cm2 V−1 s−1) and broad optical absorption extending into the NIR (λmax = 775 nm and Eg = 1.45 eV).183 Incorporation of a fluorinated thiophene donor provides a substantial red shift in the optical absorption for P25 (PDPP[T]2-TF2) (λmax = 836 nm and Eg = 1.3 eV) and imparts ambipolar charge transport characteristics in OFETs (μh/μe = 0.22/0.19 cm2 V−1 s−1).197 These excellent charge transport properties were attributed to the improved crystallinity and intermolecular π–π interactions imparted through incorporation of the perfluorothiophene comonomer. P25 (P2F2CNTVT-DPP-3), which was synthesized via DArP, demonstrated strategic sidechain engineering and functionalization of the TVT donor with nitrile and fluorine substituents affording excellent OFET electron mobility (μe = 1.61 cm2 V−1 s−1) with a λmax/Eg of 864 nm/1.32 eV.184 The extensive functionalization of P25 (P2F2CNTVT-DPP-3) is a testament to the functional group tolerance of DArP, and a masterful demonstration of structural engineering to maximize crystalline domains, control polymer chain orientation, and afford a well-ordered fibril morphology.
As previously noted, the incorporation of selenophene and other chalcogenophenes provides another handle for tailoring the optical and electronic properties of DPP based NIR-CPs via modification of the orbital overlap in the π-system and tuning the intermolecular interactions and self-assembly between polymers and chalcogen centers.60,200,206–209P26 (PDPSe-DTF2) and P27 (PDPPSe-BTI) incorporate the selenophenyl aryl substituent in place of the 2-thienyl, and both possess similar optical bandgaps (λmax = 836 nm and Eg = 1.33 eV and λmax = 804 nm and Eg = 1.32 eV) with desirable OFET charge transport characteristics (μh/μe = 0.16/0.006 cm2 V−1 s−1 and 0.025/0.154 cm2 V−1 s−1), respectively.198,199P28 (PFDPPSe) provides strong NIR-optical absorption (λmax = 832 nm and Eg = 1.34 eV) suitable for harvesting NIR wavelengths in OPV applications, and it afforded a champion PCE of 6.16% when blended with PC71BM and with a notable active layer thickness of 210 nm.200
As with IID, the incorporation of azaheterocycles, such as 2-thiazolyl,183,191,201,204,210 2-pyridiyl,197,198,204,205 and 2-pyrazinyl,211,212 into the DPP scaffold can be readily accomplished with P29–P34 provided as examples. P29, which is the 2-thiazolyl analogue of P29, demonstrates a blue shift in optical absorption relative to P29 (λmax = 820 nm and Eg = 1.30 eV) and NIR optical switching when incorporated into ECDs [tb/tc (760 nm) = 6.9/0.5 s].191 Sidechain engineering and strategic pairing with a bithiazole comonomer afforded the n-channel NIR-CP P30 (PDPP4Tz), which possesses broad optical absorption extending throughout the visible wavelength range and into the NIR (λmax = 715 nm and Eg = 1.34 eV) and efficient OFET electron mobilities (μe = 0.067 cm2 V−1 s−1).201 It is worth noting P30 (PDPP4Tz) was processed using blade coating using the less toxic and more sustainable solvent p-xylene, and the devices demonstrated excellent ambient stability for over 100 days. Comparing polymers P31 (PTDCNTVT-2) and P32 (PTD-10-TVT), which contain a nitrile functionalized TVT and unfunctionalized TVT, incorporation of the nitrile provides a slight red shift in the optical absorption (λmax = 776 nm and Eg = 1.39 eV versus λmax = 770 nm and Eg = 1.47 eV) and a significant increase in OFET charge mobilities (μh/μe = 1.28/0.97 cm2 V−1 s−1versus μh/μe = 0.17/0.0097 cm2 V−1 s−1).202 Notably, OFET characterization for P31 (PTDCNTVT-2) occurred under ambient conditions, and inverters incorporating P31 (PTDCNTVT-2) provided an impressive gain of 168.202,203 Lastly, incorporation of the 2-pyridyl aryl group in DPP is depicted with the polymers P33 (PDBPyBTz) and P34 (PTDPP-PyDPP), which show a strong dependence of the comonomer identity on the polymer optical and electronic properties.204 Specifically, P33 (PDBPyBTz) provides optical absorption extending throughout the visible spectrum and into the NIR wavelength region (λmax = 656 nm and Eg = 1.47 eV) and achieves good OFET electron mobility (μe = 0.023 cm2 V−1 s−1).204,205P34 (PDTDPP-PyDPP) provides strong NIR optical absorption (λmax = 792 nm and Eg = 1.43 eV) but affords a only modest PCE of 0.2% in all-polymer solar cells (APSCs) when paired with donor polymer PTB7-Th (Fig. 5), due to an unfavourable energy level alignment, which indicates a potential area for future structural optimization.205
Overall, DPP and its analogues afford NIR-CPs with numerous handles for structural modification and functionalization, compatibility with more sustainable polymerization methods, such as DArP, and processing conditions, such as the use non-halogenated solvents, excellent overall stability, and robust mechanical properties. Thus, it is foreseeable that DPP based polymers will continue to provide a substantial contribution towards the further advancement of NIR-CPs in organic electronics.
4.3 Naphthalene diimide (NDI) analogues
Following independent disclosure by Facchetti et al. and Watson et al., NDI CPs have provided exceptional electron transport properties with robust ambient stability and extensive use in OPV, OFETs, and OECTs.182,213–215 Although not as modular or structurally tuneable as IID and DPP, numerous NIR-CPs containing NDI have been reported with select examples provided in Fig. 10 and λmax/Eg, IP/EA, and device performance metrics tabulated in Table 3. The polymer P35 (N2200) achieved remarkable initial OFET electron mobilities of 0.85 cm2 V−1 s−1 when processed from dichlorobenzene and 0.45 cm2 V−1 s−1 when processed from xylenes.216 By controlling the polymer chain orientational alignment and self-assembly the electron mobility of P35 (N2200) was increased to 6.4 cm2 V−1 s−1 when processed from mesitylene solutions.217 In APSCs, P35 (N2200) provides complimentary NIR optical absorption (λmax = 700 nm and Eg = 1.50 eV) to many medium or wide bandgap donor polymers, and it has afforded PCEs of 11% after being blended with the donor polymer PTzBI-Si (Fig. 5) and processed from the sustainable solvent cyclopentyl methyl ether (CPME).88 Additionally, P35 (N2200) has been synthesized via DArP with extensive optimization of the polymerization conditions and defect pathways provided by Sommer et al.218,219 Thus, P35 (N2200) is exemplary n-type polymer capable of being processed using sustainable solvents and synthesized using more sustainable polymerization conditions; however, due to the lack of structural tunability and steric hindrance imparted by the naphthalene core, structural engineering is often limited to sidechain modification or pairing with a different comonomer. Examples of NDI NIR-CPs with different donor repeat units other than bithiophene are provided with polymers P36–P42. P36 [P(NDI2OD-FT2)] includes fluorine substitution on the bithiophene donor unit, which slightly widened the bandgap and provided a pronounced blue shift in optical absorption (λmax = 630 nm and Eg = 1.59 eV).220 Blending with PTB7-Th afforded a champion PCE of 6.71% in APSCs. P37 (PNDI-T10) is a semi-random copolymer where two different donor comonomers (thiophene and bithiophene) were copolymerized with the NDI acceptor. 10% incorporation of thiophene afforded the optimal polymer composition by finely tuning the polymer microstructure and facilitating improved donor–acceptor polymer miscibility in APSCs.221 Thus, a PCE of 9.0% was achieved in ternary APSCs when P37 (PNDI-T10) was blended with the donor polymers PTB7-Th and PBDTTS-FTAZ (Fig. 5).222 As with DPP, incorporation of different chalcogenophenes, such as selenophene, is another common strategy for structurally engineering NDI based NIR-CPs. Examples of this include P38–P40, which have been incorporated into OFETs and APSCs and provide comparable optical absorption to P35 (N2200).128,223,224 Notably, P38 (PNDIThSe) was synthesized using the more sustainable Zn-anionic radical polymerization, which was first reported by Kiriy et al. for the synthesis of P35 (N2200).128,225,226 As with P37 (PNDI-T10), the asymmetric structure of P38 (PNDIThSe) helps suppress crystallization and improves miscibility between the donor and acceptor polymers in APSCs.128 This provides a PCE of 6.41% when blended with PBDB-T (Fig. 5). P39 (PNDI-SVS), which incorporates a selenophene TVT analogue as the donor unit, provides a red shift in optical absorption and narrower bandgap (λmax = 748 nm and Eg = 1.31 eV) relative to P35 (N2200).223P39 (PNDI-SVS) possesses an EA of −3.98 eV, which affords an impressive OFET electron mobility of 2.4 cm2 V−1 s−1, and demonstrates excellent stability when subjected to constant biasing for 3 h under ambient conditions with only a 3% loss in performance metrics. P40 (PNDI-VSV) shows a unique functionalization strategy with the incorporation of two vinyl π-spacers attached to a selenophene donor unit.224 Relative to P39 (PNDI-SVS), P40 (PNDI-VSV) shows a reduction in crystal coherence lengths and a preferential face-on orientation via GIWAXS measurements, where P39 (PNDI-SVS) provides a bimodal orientation containing crystalline domains with face-on and edge-on orientation, which may contribute to the lower electron mobility (μe = 0.7 cm2 V−1 s−1) of P40 (PNDI-VSV). Notably, P40 (PNDI-VSV) was synthesized via Suzuki–Miyaura polymerization. | ||
| Fig. 10 Select examples of NIR-CPs containing repeat units of NDI and analogues. NDI NIR-CPs with relatively sustainable syntheses are highlighted in green. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P35 (N2200) | 700; 1.50 | −5.94/−3.80 | OPV (conventional) | PCE = 11.0% | 88 |
| P36 [P(NDI2OD-FT2)] | 630; 1.59 | −5.50/−3.91 | OPV (inverted) | PCE = 6.71% | 220 |
| P37 (PNDI-T10) | 694; 1.55 | −6.36/−4.05 | OPV (conventional) | PCE = 9.0% | 222 |
| P38 (PNDIThSe) | 697; 1.50 | −5.85/−3.81 | OPV (conventional) | PCE = 6.41% | 128 |
| P39 (PNDI-SVS) | 748; 1.31 | −5.29/−3.98 | OFET (TGBC) | μ e = 2.4 cm2 V−1 s−1 | 223 |
| P40 (PNDI-VSV) | 714; 1.49 | −5.46/−4.06 | OFET (TGBC) | μ e = 0.70 cm2 V−1 s−1 | 224 |
| P41 [P(NDI-TCPDTT)] | 830; 1.25 | −5.35/−4.15 | OPV (conventional) | PCE = 1.1% | 227 |
| P42 [P(IDT-NDI)] | 730; 1.51 | −5.75/−3.84 | OPV (conventional) | PCE = 5.33% | 228 |
| P43 (PIDTT-NDI) | n.r.; 1.48 | −5.69/−4.21 | OPT (BGTC) | R (754 nm) = 8.42 mA W−1 | 229 |
| P44 (PDPP-NDI) | 910; 1.03 | −5.46/−3.97 | OFET (BGTC) | μ e = 0.0378 cm2 V−1 s−1 | 230 |
| P45 (PNDIBTOC8) | 680; 1.46 | −5.82/−3.72 | OPV (conventional) | PCE = 3.14% | 231 |
| P46 (PNIT) | 697; 1.59 | −5.94/−3.99 | OPV (conventional) | PCE = 5.32% | 127 |
| P47 (PNDTI-BT) | 800; 1.2 | −5.6/−4.4 | OFET (BGTC) | μ h/μe = 0.10/0.27 cm2 V−1 s−1 | 232 |
| P48 (PNDTI-TT) | n.r.; 1.3 | −5.7/−4.1 | OFET (BGTC) | μ h/μe = 0.046/0.26 cm2 V−1 s−1 | 232 |
| P49 (PNDTI-DTT) | 833; 1.3 | −5.6/−4.0 | OPV (conventional) | PCE = 3.2% | 233 |
| P50 [P(NDI2DT-DTYA2-1T)] | 918; 1.25 | −5.82/−4.25 | OFET (BGBC) | μ e = 0.38 cm2 V−1 s−1 | 234 |
The polymers P41 [P(NDI-TCPDTT)] and P42 [P(IDT-NDI)] provide examples of NDI based NIR-CPs incorporating fused ring donors, such as CDT and indacenodithiophene (IDT), respectively.227,228P41 [P(NDI-TCPDTT)] possesses strong NIR optical absorption (λmax = 830 nm and Eg = 1.25 eV) and when blended with the donor polymer P3HT (Fig. 5) affords a modest PCE of 1.1%. However, it should be noted the APSC was fabricated using tetralin as the processing solvent with an active layer thickness of 410 nm. P42 [P(IDT-NDI)] provides a red shift relative P35 (N2200) (λmax = 730 nm and Eg = 1.51 eV), and when blended with donor polymer J51 (Fig. 5) in APSCs a champion PCE of 5.33% is obtained.228 This improved PCE compared to P42 [P(IDT-NDI)] with J50 (PCE = 4.12%) or PTB7-Th (PCE =3.63%) was ascribed to a more complimentary optical absorption between the donor/acceptor polymers and a suitable offset in energy levels (IP/EA) to facilitate charge transfer. P43 (PIDTT-NDI) contains a π-extended indacenodithienothiophene donor unit affording a narrower bandgap compared to P42 [P(IDT-NDI)] (Eg = 1.48 eV).229P43 (PIDTT-NDI) was then incorporated into OPTs providing an R (754 nm) = 8.42 mA W−1, and it retained suitable responsivity metrics in flexible OPTs utilizing PEN substrates (R = 3.75 mA W−1).
Copolymerization with another acceptor unit is also an effective strategy to tune the optical and electronic properties of NDI containing NIR-CPs with polymers P44–P46 as examples.127,230,231 Notably, P45 (PNDIBTOC8) and P46 (PNIT),which was synthesized using DArP, were both used as acceptor polymers in APSCs with P46 (PNIT) providing a red shift in optical absorption (λmax = 697 nm) but wider optical bandgap (Eg = 1.59 eV) relative to P45 (PNDIBTOC8) (λmax = 680 nm and Eg = 1.46 eV). When blended with the donor polymer PBDB-T, P46 (PNIT) affords a maximum PCE of 5.32%.127P45 (PNDIBTOC8) afforded a comparable PCE of 3.14% when blended with the donor polymer PBDTTT-C-T (Fig. 5) in APSCs.231
Although there are limited strategies for further functionalization of NDI to modify the optical and electronic properties, Takimiya et al. discovered an elegant approach for extension of the π-system yielding naphthodithiophene diimide (NDTI), which is depicted with polymers P47–P49.232,235 NDTI can be synthesized in only two steps from the dibrominated NDI precursor. P47 (PNDTI-BT) provides broad optical absorption extending throughout the visible wavelength range deep into the NIR (λmax = 800 nm and Eg = 1.2 eV). Notably, the polymer also affords ambipolar charge transport in OFETs μh/μe = 0.10/0.27 cm2 V−1 s−1, and shows good ambient stability with the device measurements proceeding in air.232P49 (PNDTI-DTT) provides a red shift in optical absorption relative to P47 (PNDTI-BT) (λmax = 833 nm) with a slight increase in optical bandgap (Eg = 1.3 eV), and provided a respectable PCE of 3.2% in APSCs when blended with PTB7.233 Another example of ring extension with NDI is shown with the polymer P50 [P(NDI2DT-DTYA2-1T)].234P50 [P(NDI2DT-DTYA2-1T)] provides a narrow optical absorption band almost exclusively in the NIR wavelength region (λmax = 918 nm and Eg = 1.25 eV). Additionally, despite having an amorphous morphology P50 [P(NDI2DT-DTYA2-1T)] provides an impressive electron mobility (μe = 0.38 cm2 V−1 s−1).
Overall, NIR-CPs incorporating NDI and its analogues afford superb electron mobilities, ambient stability, and broad utility across the field of organic electronics. While there is limited capability with altering the structure of NDI relative to IID or DPP, creative strategies, such as the π-extension and ring fusion with PNDTI, indicate new opportunities for further development and functionalization.
4.4 Benzothiadiazole (BT) analogues
Since disclosure by Meijer et al., benzothiadiazole has afforded numerous state-of-the-art p-type and n-type CPs with prominent utility in OPV and OFET devices.236–238 As previously discussed, the BT core offers a variety of options for functionalization and derivatization via heteroatom substitution, π-extension, and fused ring systems with relevant NIR-CPs (P51–P67) provided in Fig. 11 and λmax/Eg, IP/EA, and device performance metrics tabulated in Table 4. | ||
| Fig. 11 Select examples of NIR-CPs containing BT analogues. BT NIR-CPs with relatively sustainable syntheses are highlighted in green. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P51 (PDPP-TBT) | 915; 1.2 | −5.2/−4.0 | OFET (BGTC) | μ h/μe = 0.35/0.40 cm2 V−1 s−1 | 239 |
| P52(PDPP-FBT) | 816; 1.20 | −5.22/−3.49 | OFET (BGTC) | μ h/μe = 0.21/0.42 cm2 V−1 s−1 | 240 |
| P53 (PDPP-VBBT) | 664; 1.35 | −5.34/−3.43 | OFET (BGTC) | μ h/μe = 0.20/0.15 cm2 V−1 s−1 | 241 |
| P54 | 829; 1.27 | −5.19/−3.42 | OPT (BGBC) | R (850 nm) = 118 A W−1 | 242 |
| P55 [P(Ge-DTDCNBT)] | 754; 1.27 | −5.14/−3.77 | OFET (TGBC) | μ e = 2.8 × 10−3 cm2 V−1 s−1 | 58 |
| P56 (DCNBT-TPC) | 841; 1.38 | −5.51/−3.87 | OPV (conventional) | PCE = 10.2% | 243 |
| P57 (PDPP-BO) | 964; 1.17 | −5.4/−4.2 | OPT (TGBC) | D* (940 nm) = 1 × 1012 Jones | 244 |
| P58 | 635; 1.58 | −5.15/−3.55 | OFET (BGTC) | μ h = 4.65 × 10−4 cm2 V−1 s−1 | 245 |
| P59 (pN-BBSeE) | 848; 1.36 | −5.53/−4.05 | OFET (TGBC) | μ h/μe = 10.65/10.72 cm2 V−1 s−1 | 29 |
| P60 (PBBT12DPP) | 1280; 0.65 | −4.55/−3.9 | OFET (BGBC) | μ h/μe = 1.17/1.32 cm2 V−1 s−11 | 246 |
| P61 (PNDTI-BBT-DP) | 914; 0.96 | −5.5/−4.4 | OFET (BGTC) | μ h/μe = 0.31 cm2 V−1 s−1 | 247 |
| P62 | 1106; 1.37 | −5.31/−3.94 | OFET (BGBC) | μ h = 0.11 cm2 V−1 s−1 | 248 |
| P63 (PBTTQCN-TT) | 1270; 0.66 | −5.20/−4.10 | OFET (TGBC) | μ h/μe = 1.3 × 10−3/2.0 × 10−3 cm2 V−1 s−1 | 249 |
| P64 (PBDTTQ-2) | 978; 1.03 | −5.48/−4.01 | OFET (BGBC) | μ h/μe = 1.2 × 10−3/6.0 × 10−4 cm2 V−1 s−1 | 250 |
| P65 (PCZ-TZBTTT) | 785; 1.35 | −5.18/−3.83 | OPV (conventional) | PCE = 3.17% | 251 |
| P66 (PTZBTTT-BDT) | 810; 1.1 | −5.14/−4.04 | OPD | D* (800 nm) = 1.75 × 1013 Jones | 252 |
| P67 (PDTTZ-TT) | 865; 0.98 | −4.96/−3.98 | OPD | D* (1160 nm) = 7.3 × 1012 Jones | 10 |
DPP and BT copolymers (P51–P54 in Table 4) have been shown to provide broad NIR optical absorption and ambipolar charge transport characteristics in OFETs. Specifically, P51 (PDPP-TBT) affords strong NIR optical absorption (λmax = 915 nm) with a narrow optical bandgap (Eg = 1.2 eV), and provides balanced ambipolar charge transport (μh/μe = 0.35/0.40 cm2 V−1 s−1) and a gain of 35 when incorporated into inverters.239,253P51 (PDPP-TBT) was synthesized using Suzuki–Miyaura polymerization avoiding the toxicity associated with the alkyl-tin residues of Stille polymerization. Additionally, the excellent NIR optical absorption of P51 makes it suitable for OPTs affording R (935 nm) = 34.78 mA W−1.254P52 (PDPP-FBT) and P53 (PDPP-VBBT) provide examples of heteroatom substitution and π-extension using the P51 (PDPP-TBT) scaffold, respectively. P52 (PDPP-FBT) demonstrates a blue shift in optical absorption (λmax = 816 nm) relative to P51 (PDPP-TBT) and comparable charge mobilities (μh/μe = 0.21/0.42 cm2 V−1 s−1), albeit with an improved electron mobility.240 Additionally, P52 (PDPP-FBT) adopts a preferential edge-on orientation with π–π distances (3.8 Å) suitable for efficient charge transport in OFETs. In comparison, P53 (PDPP-VBBT), which incorporates a vinylene π-spacer, demonstrates a blue shift relative to P51 and P52 (λmax = 918 nm and Eg = 1.25 eV) and provides comparable ambipolar charge transport mobilities (μh/μe = 0.20/0.15 cm2 V−1 s−1).241P54 incorporates BT with a π-extended fused ring DPP derivative as a second acceptor repeat unit affording strong NIR optical absorption (λmax/Eg = 829 nm/1.27 eV).242 When blended with PC71BM in OPTs, P54 provides an R (850 nm) = 118 A W−1 with improved photoresponsivity at lower light intensities (1.8 μW cm−2). This trend in responsivity is attributed to a desirable morphology/microstructure for the P54/PC71BM blends.
In addition to fluorination, cyanation is another effective strategy to tune the EA of benzothiadiazole CPs to facilitate electron transport with polymers P55 [P(Ge-DTDCNBT)] and P56 (DCNBT-TPC) as examples.58,243P55 [P(Ge-DTDCNBT)] affords broad NIR optical absorption (λmax = 754 nm and Eg = 1.27 eV) and demonstrates good electron transport (μe = 2.8 × 10−3 cm2 V−1 s−1).58 By pairing with a strategically designed fused-ring donor unit, P56 (DCNBT-TPC) affords a substantial red shift in optical absorption (λmax = 841 nm and Eg = 1.38 eV) relative to P55 [P(Ge-DTDCNBT)]. This provides capabilities for harvesting NIR wavelengths allowing it to achieve a remarkable PCE of 10.2% in ternary APSCs.243
Chalcogen substitution of benzothiadiazole can also be performed yielding benzooxadiazole or benzoselenodiazole, which is shown with polymers P57–P59.29,245 As described above, chalcogen substitution is an effective way to modifying the energetics and non-covalent interactions of the polymer. Notably, P57 which substitutes the sulphur in BT for the more electronegative oxygen, provides a significant redshift in optical absorption and narrower bandgap compared to P51 (λmax = 964 nm and Eg = 1.17 eV).244 This strategic heteroatom modification enables ambipolar OPTs with D* = 1 × 1012 and 7 × 1011 Jones for n-type and p-type operation, respectively, at a NIR wavelength (940 nm). P58 was synthesized via DArP and provides broad optical absorption throughout the visible wavelength region and into the NIR.245 When incorporated into an OFET, P58 affords a modest hole mobility (μh = 4.65 × 10−4 cm2 V−1 s−1). P59 (pN-BBSeE) incorporates chalcogen substitution and π-extension, as well as NDTI as an additional acceptor unit. This judiciously designed polymer provides strong NIR optical absorption (λmax = 841 nm and Eg = 1.38 eV) with an additional π–π* absorption band present at 515 nm. Additionally, it affords exceptionally high and well balanced ambipolar charge mobilities (μh/μe = 10.65/10.72 cm2 V−1 s−1).29
A further reduction in bandgap and increase in the EA can be accomplished through the addition of a second thiadiazole functionality on BT to yield benzobisthiadiazole (BBT), which is shown with polymers P60–P62. Compared to BT polymers P51–P59, P60 (PBBT12DPP), which is a dual-acceptor alternating copolymer containing DPP and BBT repeat units, displays a significant red shift in optical absorption (λmax = 1280 nm) with an ultra-narrow optical bandgap of 0.65 eV. P60 (PBBT12DPP) was also found to provide desirable ambipolar charge transport in OFETs (μh/μe = 1.17/1.32 cm2 V−1 s−1) facilitated by the narrow bandgap of the NIR-CP.246P61 (PNDTI-BBT-DP) also possesses a dual-acceptor alternating copolymer structure incorporating NDTI and BBT. This polymer also affords a very narrow optical band gap of 0.96 eV, and excellent electron mobility (μe = 0.31 cm2 V−1 s−1).247 Lastly, P62 incorporates bithiazole and BBT providing optical absorption across the visible wavelength range and into the NIR (λmax = 1106 nm and Eg = 1.37 eV). Instead of ambipolar charge transport, only unipolar hole transport (μh = 0.11 cm2 V−1 s−1) was observed with P62.248 Employing computational tools to characterize the electronic structure, this was ascribed to a highly localized LUMO on BBT, which was posited as being unfavourable for electron transport. Additional functionalization strategies to alter the optical and electronic properties of BT based polymers are shown with P63–P67. P63 (PBTTQCN-TT) and P64 (PBDTTQ-2) each contain a BT unit with π-extended fused ring system.249,250 For P63 (PBTTQCN-TT) the indanone functionalized thiadazolo[3,4-g]quinoxaline unite imparts an ultra-narrow bandgap of 0.66 eV with strong NIR optical absorption (λmax = 1270 nm), and with P64 (PBDTTQ-2) an optical bandgap of 1.03 eV is achieved with λmax = 978 nm. P63 (PBTTQCN-TT) and P64 (PBDTTQ-2) afford ambipolar charge transport, albeit with relatively low charge mobilities (μh/μe = 1.3 × 10−3/2.0 × 10−3 cm2 V−1 s−1 and 1.2 × 10−3/6.0 × 10−4 cm2 V−1 s−1, respectively). However, P63 (PBTTQCN-TT) has also been incorporated into organic phototransistors providing detection up to 1550 nm.255
Introduction of a triazole unit to yield [1,2,5]thiadiazolo[3,4-f]benzotriazole is an efficient strategy for facilitating electron transport via improved EA with an additional handle for tuning the morphology/microstructure via sidechain engineering on the triazole unit with polymers P65 (PCZ-TZBTTT) and P66 (PTZBTTT-BDT) as examples.251,252λmax/Eg for P65 (PCZ-TZBTTT) and P66 (PTZBTTT-BDT) were measured to be 785 nm/1.35 eV and 810 nm/1.1 eV, respectively. Notably, P65 (PCZ-TZBTTT) was blended with PC61BM in OPV devices affording a PCE of 3.17%, and P66 (PTZBTTT-BDT) was incorporated into OPDs with PC61BM as the acceptor providing a specific detectivity (D*) of 1.75 × 1013 Jones at 800 nm. Lastly, the polymer P67 (PDTTZ-TT) incorporates thieno[3,4-c]thiadiazole as an acceptor with a simplified structure in place of BT affording a measured λmax/Eg of 865 nm/0.98 eV.10 When blended with PC71BM in OPDs a detectivity of 7.3 × 1012 Jones is achieved at 1160 nm. This is particularly unique D–A polymer structure, since P67 (PDTTZ-TT) is composed entirely of functionalized thiophenes with the thieno[3,4-c]thiadiazole acceptor unit synthesized within only a few steps, and the polymer shows optical absorption extending from 400–1200 nm.10
Overall, BT provides a relative simple structure with a high degree of structural tunability including heteroatom substitution, π-extension, and ring fusion. It has also been polymerized using Suzuki–Miyaura and DArP, which demonstrates improved sustainability for this class of NIR-CPs. In many cases, NIR-CPs incorporating BT provide highly crystalline morphologies with a polymer orientation that is preferentially edge-on, which is suitable for OFETs. However, as detailed here, there are examples of BT containing NIR-CPs with utility in OPV/OPDs indicating the morphology can be tailored for the desired device application.
4.5 Fused ring electron acceptors (FREAs) and polymerized small-molecule acceptors (PSMAs)
Fused ring electron acceptors (FREAs) have reignited the field of organic small-molecule and polymer solar cells, due to their strong NIR optical absorption that is complimentary to many state-of-the-art donor polymers, excellent electron transport, and morphological stability relative to fullerenes.5,6,8,256–260 Their characteristic structural features include an expansive π-conjugated network partitioned into different sections of electron donor (D) or acceptor (A) character to yield A–D–A or A–D–A–D–A structures, which have been the topic of many reviews.5,6,8,256,257 FREAs are typically comprised of an IDT or thieno[2′,3′:4,5]pyrrolo[3,2-e:2′,3′-g][2,1,3]benzothiadiazole (TPB) core flanked by functionalized indanones, and following polymerization yield a class of polymers referred to as polymerized small-molecule acceptors (PSMAs) with examples provided in Fig. 12 (P68–P82) and the optical absorption (λmax), optical bandgap (Eg), IP/EA, and device performance metrics tabulated in Table 5. | ||
| Fig. 12 Select examples of NIR-CPs containing FREAs. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P68 (PZ1) | 704; 1.55 | −5.74/−3.86 | OPV (conventional) | PCE = 11.2% | 261 |
| P69 (PJ1) | 798; 1.41 | −5.64/−3.82 | OPV (conventional) | PCE = 14.4% | 262 |
| P70 (PY3Se-1V) | 850; 1.25 | −5.51/−3.90 | OPD | R (960 nm) = 28 A W−1 | 263 |
| P71 (PATIC-HD-Th) | 803; n.r. | −5.31/−3.58 | OPV (conventional) | PCE = 12.47% | 264 |
| P72 (PY-IT) | 808; 1.39 | −5.68/−3.94 | OPV (conventional) | PCE = 15.05% | 265 |
| P73 (PYF-T-o) | 824; 1.38 | −5.73/−3.81 | OPV (conventional) | PCE = 15.2% | 266 |
| P74 (PYN-BDTF) | 820; 1.38 | −5.67/−3.77 | OPV (conventional) | PCE = 13.22% | 21 |
| P75 (PYTT-2) | 832; 1.49 | −5.73/−3.83 | OPV (conventional) | PCE = 14.32% | 267 |
| P76 (PY-SSe-V) | 912; 1.36 | −5.65/−3.78 | OPV (conventional) | PCE = 18.14% | 268 |
| P77 (PFY-1Se) | 800; 1.79 | −5.68/−3.89 | OPV (conventional) | PCE = 13.8% | 269 |
| P78 (PFY-2Se) | 825; 1.73 | −5.64/−3.91 | OPV (conventional) | PCE = 14.7% | 269 |
| P79 (PZT-γ) | 838; 1.36 | −5.57/−3.78 | OPV (conventional) | PCE = 15.8% | 270 |
| P80 (PYFTET) | 866; 1.43 | −5.76/−4.08 | OPV (conventional) | PCE = 9.53% | 271 |
| P81 (PFY-DTC) | n.r.; 1.40 | −5.53/−3.87 | OPV (conventional) | PCE = 11.08% | 272 |
| P82 (PY5-BSeI) | 812; 1.35 | −5.76/−4.00 | OPV (conventional) | PCE = 17.77% | 273 |
Polymers P68–P71 in Fig. 12 provide a general overview of structural modifications to the central IDT or TPB core observed with FREAs/PSMAs.261–264 Remarkable levels of structural engineering are observed with these polymers, including sidechain engineering, π-extension, ring fusion, heteroatom substitution, and controlling the polymer regioregularity. Possessing an IDT core, P68 (PZ1) was the first demonstration of a PSMA and their utility in APSCs providing complimentary optical absorption (λmax = 704 nm and Eg = 1.55 eV) to the donor polymer PM6 (Fig. 5).261 Notably, APSCs with an active layer P68 (PZ1):PM6 provided a PCE of 11.2%, with exceptional morphological stability (90% retention of the initial PCE after 80 d of storage), and scalability to larger-area devices (1.1 cm2). P69 (PJ1) substitutes the IDT core for TPB providing a significant red shift in the optical absorption (λmax = 798 nm) and reduction in the optical bandgap (Eg = 1.41 eV) relative to P68 (PZ1). When blended with the donor polymer PBDB-T a PCE of 14.4% was achieved, and excellent thermal stability of the active layer was observed following annealing at 150 °C for 180 m (PCE = 12.5%).262 Additionally, with thick (305 nm) active layers a PCE of 12.1% is obtained, indicating potential viability with large scale, foundry-compatible device fabrication. P70 (PY3Se-1V) provides an example of π-extension via a vinylene spacer and heteroatom substitution via selenophene, which is described further with polymers P76–P79.263 The incorporation of the vinylene spacer was found to extend the optical absorption of P70 (PY3Se-1V) deeper into the NIR wavelength region providing a λmax/Eg of 850 nm/1.25 eV.263P70 (PY3Se-1V) was blended with PBDB-T in APSCs (PCE = 13.2%) and P3HT in OPDs [R (960 nm) = 28 A W−1]. Lastly, P71 (PATIC-HD-Th) provides an example of modification of the TPB central core via incorporation of quinoxaline analogue derived from phenanthraquinone.264 A blue shift in optical absorption (λmax = 803 nm) relative to P68–P70 was observed, and P71 (PATIC-HD-Th) provided a PCE of 12.47% in APSCs when blended with PBDB-T.
Structural modification of the indanone linker in PSMAs can also be accomplished, which is highlighted with polymers P72–P75. This includes synthesis of single structural isomers to control regioregularity [P72 (PY-IT)], heteroatom modification [P73 (PYF-T-o)], π-extension [P74 (PYN-BDTF)], and modification of indanone to incorporate other aromatic heterocycles [P75 (PYTT-2)].21,265–267,274 Controlling the regioregularity of the CP structure from the regio-random P69 (PJ1) to the regio-regular P72 (PY-IT) yields a red shift in the optical absorption (λmax = 808 nm) and boosts the APSC PCE to 15.05% when blended with PM6.265 Additionally, diligent optimization of the active layer morphology via the introduction of solid additives into PM6:P72 (PY-IT) blends affords a landmark APSC PCE of 18.3%.274 Incorporating both strategies of controlling regio-selectivity and heteroatom incorporation, P73 (PYF-T-o) employs a fluorination strategy to modify the optical properties and polymer self-assembly.266P73 (PYF-T-o) provides a red shift in optical absorption (λmax = 824 nm) relative to P72 (PY-IT) indicating increased π-orbital overlap and intermolecular interactions, and PM6:P73 (PYF-T-o) blends provide a boost in APSC PCE to 15.2%. P74 (PYN-BDTF) introduces a π-extended indanone group providing a redshift in the optical absorption (λmax = 820 nm) relative to P72 (PY-IT).21 Incorporation of the fluorinated donor comonomer with P74 (PYN-BDTF) in blends with PM6 provided a preferential face on orientation and improved crystallinity relative to the non-fluorinated analogue affording a PCE of 13.22%. P75 (PYTT-2) incorporates a thiophene analogue of indanone, which provides a regio-regular configuration compared to the regio-random configuration with P69 (PJ1). The thiophene functionalized end-group provides desirable optical properties (λmax = 832 nm and Eg = 1.49 eV) and a PCE of 14.32% when blended with PBDB-T. Additionally, it provides a robust morphology with 80% PCE retention after annealing at 120 °C for 100 h.
As shown with previous NIR-CPs, chalcogen substitution from the commonly employed sulphur/thiophene to its selenium/selenophene analogues or to pnictogens, such as nitrogen, is an effective strategy for tuning the optical properties, polymer energetics, and self-assembly with P76–P79 provided as examples for FREAs/PSMAs.268–270 Of particular note is P76 (PY-SSe-V), which possesses a strategic asymmetric substitution of the TPB core to incorporate a single selenophene unit and was polymerized with vinylene to yield a regio-regular NIR-CP suitable for APSCs.268P76 (PY-SSe-V) afforded a substantial red shift relative to the other PSMAs (λmax = 912 nm) and a PCE of 17.03% when blended with PM6 in binary devices and 18.14% in ternary solar cells. Jen et al. investigated the systematic incorporation of selenium within the PJ1 framework by preparing polymers P77 (PFY-1Se) and P78 (PFY-2Se).269 Increasing selenium content affords a redshift in optical absorption [λmax = 800 nm/825 nm for P77 (PFY-1Se)/P78 (PFY-2Se)] and an increase in PCE [PCE = 13.8%/14.7% for P77 (PFY-1Se)/P78 (PFY-2Se)] with PBDB-T as the donor polymer. The improved APSC performance metrics were attributed to increased crystallinity and shorter π–π distances, which are a characteristic of the heavier chalcogens. Next, P79 (PZT-γ) is functionalized with a central triazole core rather than BT and incorporates the regio-regular polymerization strategy to achieve broader optical absorption with a λmax/Eg of 838 nm/1.36 eV.270 It was found the regio-regular structure and incorporation of triazole with P79 (PZT-γ) provides ideal phase separation and increased crystalline domains when blended with the donor polymer PBDB-T, which affords a PCE of 15.8%.
Lastly, variation of the donor comonomer is another effective strategy for tuning the optical, electronic, morphological/microstructural, and mechanical properties of PSMAs with P80–P82 as examples.271–273,275 Notably, P80 (PYFTET) incorporates a thienylene-ethylene-thienylene (TET) donor unit, which is the non-conjugated form of TVT.271,276 This serves as a conjugation break spacer (CBS), which imparts mechanical flexibility and stretchability within the polymer relative to its conjugated counterpart.277,278P80 (PYFTET) provides a λmax/Eg of 866 nm/1.43 eV with a PCE of 9.53% when blended with PM6, and when incorporated into ultra-flexible APSCs using parylene substrates P80 (PYFTET) demonstrates 90% PCE retention after 5000 bending cycles at a radius of 14 μm.271P81 (PFY-DTC) incorporates the strong electron donor unit CDT, which is discussed further in Section 4.6.272P81 (PFY-DTC) provides extensive coverage of the visible wavelength range into the NIR region with an optical bandgap of 1.40 eV. Blends of PBDB-T:P81 (PFY-DTC) displayed strong aggregation and large crystalline domain sizes via GIWAXS and provided PCEs of 11.08% in APSCs. Finally, P82 (PY5-BSeI) is unique within this class of polymers since it possesses a dual-acceptor alternating copolymer structure with the strategically designed biselenophene imide comonomer.273P82 (PY5-BSeI) provides strong optical absorption extending from 400–900 nm with a λmax/Eg of 812 nm/1.35 eV. APSCs incorporating PM6: P82 (PY5-BSeI) afforded an exceptional PCE of 17.77%, which was attributed to optimal donor–acceptor phase separation, ideal polymer packing and self-assembly, and high crystallinity.
To summarize, FREAs are a class of repeat units that demonstrate the highest levels of structural engineering in NIR-CPs. They afford state-of-the-art performance metrics in OPV devices and desirable performance in OPDs surpassing fullerene and other acceptor polymers in their ability to harvest solar illumination and generate photocurrent. With that taken into account it is worth noting that the syntheses often invoke arduous synthetic pathways, which can increase costs and tarnish the sustainable aspects OPV seeks to provide.279 Additionally, PSMAs are typically synthesized via Stille polymerization so there are many opportunities for future work to address these deficiencies and develop more sustainable synthetic pathways and polymerization procedures suitable for high-performance PSMAs.
4.6 Cyclopentadithiophene (CDT) and analogues
Although differences in the structure and functionalization of acceptor units have been a primary focus of this review, it is important to highlight the CDT donor unit and its analogues, which have afforded numerous NIR-CPs, due to the strong electron donating capabilities imparted by the fused bithiophene framework, with select examples (P83–P90) provided in Fig. 13 and the optical absorption (λmax), optical bandgap (Eg), IP/EA, and device performance metrics tabulated in Table 6. P83–P86 all possess the CDT donor and various functionalized BT acceptor units with λmax/Eg ranging from 702–920 nm/1.20–1.54 eV.280–288 | ||
| Fig. 13 Select examples of NIR-CPs containing CDT repeat units and analogues. | ||
| Polymera | λ max (nm); Egb (eV) | IP/EA (eV) | Device applicationc | Notable performance metric | Ref. |
|---|---|---|---|---|---|
| a Abbreviation in parentheses indicates common name. b Estimated in all cases using λonset where optical Eg = 1240/λonset. c Device architecture is indicated in parentheses (see Section 3). n.r. = not reported. | |||||
| P83 | 776; 1.54 | −5.05/−3.30 | OFET (BGBC) | μ h = 0.56 cm2 V−1 s−1 | 280 |
| P84 | 702; 1.46 | −5.21/−2.85 | OECT (planar) | g m,norm = 35.8 S cm−1 | 281 |
| P85 | 920; 1.37 | −5.07/−3.70 | OFET (BGTC) | μ h = 0.4 cm2 V−1 s−1 | 282 |
| P86 (PCPPC) | 784; 1.20 | −5.28/−3.41 | OFET | μ h/μe = 1.5/0.41 cm2 V−1 s−1 | 283 |
| P87 | 1270; <0.5 | −5.15/−4.44 | n.r. | n.r. | 284 |
| P88 | 1080; 0.85 | −4.80/−3.66 | OPD | D* (1200 nm) = 3 × 1011 Jones | 285 |
| P89 (PDPP2TzDTP) | n.r.; 1.28 | −5.61/−3.94 | OPV | PCE = 5.6% | 286 |
| P90 (PSBTBT) | n.r.; 1.45 | −5.05/−3.27 | OPV | PCE = 17.35% | 288 |
P83 and P84 incorporate a mono-fluorinated and di-fluorinated BT acceptor unit copolymerized with CDT.280,281P83 provides a redshift in optical absorption relative to P84 (λmax = 776 nm versus 702 nm, respectively), and P83 demonstrated excellent ambient stability when incorporated into OFETs with an increase in the initial mobility (μh = 0.56 cm2 V−1 s−1) by 4.8% after ambient storage for 12 d. P84 was incorporated into OECTs using the ionic-liquid [EMIM][TFSI] as the electrolyte providing a normalized transconductance (gm,norm) of 35.8 S cm−1. Use of an ionic liquid is likely critical here, given the absence of hydrophilic EGn sidechains to facilitate aqueous electrolyte penetration and uptake.
P85 and P86 (PCPPC) each incorporate pyridal[2,1,3]-thiadiazole acceptor units. P85 was strategically synthesized to ensure a regio-regular structure rather than a random orientation of the pyridal[2,1,3]thiadiazole acceptor. This careful control affords strong NIR optical absorption (λmax = 920 nm and Eg = 1.37 eV) and good hole mobilities (μh = 0.4 cm2 V−1 s−1). P86 (PCPPC) possesses a narrower bandgap (Eg = 1.20 eV) and an elevated conduction band relative to P85 (−3.41 versus −3.70 eV, respectively). Yet, P86 demonstrated ambipolar charge transport in OFETs (μh/μe = 1.50/0.41 cm2 V−1 s−1), and inverters containing P86 (PCPPC) were found to provide a gain of 165.283
P87 and P88, which possess a bridgehead imine-substituted CDT and an exocyclic olefin substituted CDT as the donor units, respectively, provide broad optical absorption extending far into the NIR (λmax/Eg = 1270 nm/<0.5 eV (P87) and 1080 nm/0.85 eV (P88)). This class of donor unit and the corresponding NIR-CPs were pioneered by Azoulay et al. and have demonstrated utility in OPDs with a D* (1200 nm) of 3 × 1011 Jones provided by P88 and PC70BM blends.285,289–291 Lastly, the polymers P89 (PDPP2TzDTP) and P90 (PSBTBT), which contain the silole and pyrrole analogues of CDT, each provide narrow bandgaps (Eg = 1.28 and 1.45 eV, respectively).286,287P89 (PDPP2TzDTP) and P90 (PSBTBT) served as a donor polymers in OPV providing PCEs of 5.6% and 4.7% when blended with PC70BM. More recently, P90 (PSBTBT) was incorporated into ternary solar cells with the donor polymer D18-Cl and small-molecule acceptor BTP-eC9.288 In this setting, P90 (PSBTBT) provided complementary energy levels to the other components enabling efficient charge transfer and more complimentary spectral coverage across the visible wavelength range and into the NIR. Thus, an excellent PCE of 17.35% was obtained. The application of these structurally simple yet powerful electron donating units into NIR-CPs for ternary solar cells should generate increased research interest for these polymers.
5. Summary and perspective
In summary, an overview of NIR-CPs containing IID, DPP, NDI, BT, FREAs, CDT and analogues with applications in ECDs, OPV, OPDs, OPTs, OFETS, and OECTs was presented. The defining structural characteristics for each polymer class, and strategies for structural engineering to achieve NIR optical absorption and desired performance metrics were also discussed. Common to each of these polymer classes is the alternating D–A copolymer structure, which affords narrow bandgap polymers, tuneable energy levels, and allows for the generation of large libraries of polymers structures.Of the NIR-CPs presented, it is clear that specific polymer repeat units are best suited for certain organic electronic device applications. Specifically, IID, DPP, NDI, and BT are particularly suited for OFETs and OPTs while FREAs are the optimal choice for achieving state-of-the-art performance metrics in OPVs. Given that all the polymers possess suitable NIR optical absorption, this pairing between polymer and application indicates additional preferred material characteristics, such as energetics and energy level alignment and polymer morphology and microstructure. Thus, even though a polymer may have suitable NIR optical absorption for use in APSCs, the polymer may not have a suitable IP/EA energy offset with the donor polymer to facilitate charge separation. Additionally, the morphology and polymer orientation (e.g., π-face on or edge on) must be carefully considered, since large crystalline domain sizes and polymer aggregation may be detrimental rather than beneficial, and desirable performance metrics are more probable when the orientation of the polymer π–π stacking directions aligns with the direction of charge transport, e.g. vertical direction for OPV or horizontal direction for OFET.
Currently, there are limited examples of NIR-CPs in ECDs that can match the performance metrics of CPs with optical switching in the visible wavelength range (ΔT% = 60–80%, t95 < 1.0 s, and cycling stability >3000 cycles).2,292 Thus, there is significant opportunity here for the development of more electrochemically robust NIR-CPs. Additionally, the development of ultra-flexible and stretchable organic NIR-ECDs lags behind OFET and OPV.41,293 With increasing research interest in wearable electronics, e-skin, soft robotics, and IoT applications, the potential for NIR-ECDs to advance capabilities in thermal energy management, energy storage, and active camouflage is significant.294
For OPV, there has been rapid development of APSC performance metrics since the discovery of PSMAs. However, the PCE for APSCs still falls behind donor polymer:acceptor small-molecule blends, which have certified PCEs approaching 20%.295 As with ECDs, the development of flexible and stretchable OPVs with uncompromised performance metrics remains a continued challenge. It is foreseeable that the continued exploration and incorporation of new functionalities, such as hydrogen bonding groups or moieties capable of cross-linking, will assist in the development of more mechanically robust NIR-CPs.131,296–298
Aside from developing more mechanically robust materials, another aspect to consider is the advancement of materials operation in extreme environments. Recently, there have been developments in the fabrication of polymer composite materials for thermally robust OFETs and gas sensors where temperatures were set to 220 °C while maintaining hole mobilities >2.0 cm2 V−1 s−1.299,300 These CP and non-conjugated polymer composite materials enable operation at extreme temperatures, which opens new opportunities for organic electronics where previously only inorganics offered viable technologies. This research area could be expanded to include NIR-CPs in OPV, where thermal stability test are often <100 °C, and ECDs where device operational stability at temperatures of 180 °C has been demonstrated.301
Opportunities to further increase sustainability in polymer synthesis and processing should also not be ignored. Currently, DArP has demonstrated utility for multiple classes of CPs; however, the successful application of this polymerization methodology towards PSMAs to achieve convergence in performance metrics with polymers synthesized via Stille polymerization has yet to be disclosed, to our knowledge. Additionally, the FREA repeat units incorporated into PSMAs often invoke arduous synthetic pathways that diminish the overall sustainability of OPV. Thus, the grand challenge of identifying structurally simple acceptor polymers while still maintaining high PCEs (>15%) remains an outstanding challenge. Additionally, the solution processing of NIR-CPs often requires toxic halogenated solvents, and so identifying a suitable replacement that can be broadly applied across the field of organic electronics is imperative.
It is envisioned that the broad scope presentation of NIR-CPs provided here will inspire that next generation of structural developments to achieve improved performance metrics or unlock new technological capabilities. It is important to note that the structural advancements are directly tied to an improved understanding of the device operation and underlying physics, and in many instances the performance metrics obtained from device characterization serve as a feedback loop for tailoring polymer design. Thus, the continuity of organic electronics and the continued advancement of NIR-CP structural development is reliant on furthering interdisciplinary interactions between synthetic chemists, materials scientists, engineers, and physicists.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the UT System STARS Program and UTEP Startup Awards.References
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
- X. Li, K. Perera, J. He, A. Gumyusenge and J. Mei, J. Mater. Chem. C, 2019, 7, 12761–12789 RSC.
- H. Fu, L. Zhang, Y. Dong, C. Zhang and W. Li, Mater. Chem. Front., 2023, 7, 2337–2358 RSC.
- C. Gu, A.-B. Jia, Y.-M. Zhang and S. X.-A. Zhang, Chem. Rev., 2022, 122, 14679–14721 CrossRef CAS PubMed.
- L. Zhang, Z. Yao, H. Wang, J. Zhang, X. Ma and F. Zhang, Sol. RRL, 2023, 7, 2300219 CrossRef CAS.
- T. Wang, M. Chen, R. Sun and J. Min, Chem, 2023, 9, 1702–1767 CAS.
- Y. Cai, L. Huo and Y. Sun, Adv. Mater., 2017, 1605437 CrossRef PubMed.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- C. Liu, L. Shao, S. Chen, Z. Hu, H. Cai and F. Huang, Prog. Polym. Sci., 2023, 143, 101711 CrossRef CAS.
- X. Cao, J. Tong, Z. He, M. Zhang, X. Zhang, J. Ma, P. Gao, J. Li, P. Zhang, C. Wang, Y. Xia and H. Wu, Dyes Pigm., 2018, 158, 319–325 CrossRef CAS.
- H. Ren, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Adv. Sci., 2021, 8, 2002418 CrossRef CAS PubMed.
- Q. Li, Y. Guo and Y. Liu, Chem. Mater., 2019, 31, 6359–6379 CrossRef CAS.
- P. Hu, X. He and H. Jiang, InfoMat, 2021, 3, 613–630 CrossRef CAS.
- S. Griggs, A. Marks, H. Bristow and I. McCulloch, J. Mater. Chem. C, 2021, 9, 8099–8128 RSC.
- T. Leydecker, Z. M. Wang, F. Torricelli and E. Orgiu, Chem. Soc. Rev., 2020, 49, 7627–7670 RSC.
- S. Park, S. H. Kim, H. H. Choi, B. Kang and K. Cho, Adv. Funct. Mater., 2020, 30, 1904590 CrossRef CAS.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- S. T. M. Tan, A. Gumyusenge, T. J. Quill, G. S. LeCroy, G. E. Bonacchini, I. Denti and A. Salleo, Adv. Mater., 2022, 2110406 CrossRef CAS PubMed.
- M. Moser, J. F. Ponder Jr., A. Wadsworth, A. Giovannitti and I. McCulloch, Adv. Funct. Mater., 2019, 29, 1807033 CrossRef.
- Y. Wang, S. Wustoni, J. Surgailis, Y. Zhong, A. Koklu and S. Inal, Nat. Rev. Mater., 2024, 9, 249–265 CrossRef CAS.
- N. Su, R. Ma, G. Li, T. Liu, L.-W. Feng, C. Lin, J. Chen, J. Song, Y. Xiao, J. Qu, X. Lu, V. K. Sangwan, M. C. Hersam, H. Yan, A. Facchetti and T. J. Marks, ACS Energy Lett., 2021, 6, 728–738 CrossRef CAS.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- C. J. M. Emmott, J. A. Röhr, M. Campoy-Quiles, T. Kirchartz, A. Urbina, N. J. Ekins-Daukes and J. Nelson, Energy Environ. Sci., 2015, 8, 1317–1328 RSC.
- E. Pascual-San-José, G. Sadoughi, L. Lucera, M. Stella, E. Martínez-Ferrero, G. E. Morse, M. Campoy-Quiles and I. Burgués-Ceballos, J. Mater. Chem. A, 2020, 8, 9882–9895 RSC.
- J. Niu, Y. Wang, X. Zou, Y. Tan, C. Jia, X. Weng and L. Deng, Appl. Mater. Today, 2021, 24, 101073 CrossRef.
- Z. Wu, Q. Zhao, X. Luo, H. Ma, W. Zheng, J. Yu, Z. Zhang, K. Zhang, K. Qu, R. Yang, N. Jian, J. Hou, X. Liu, J. Xu and B. Lu, Chem. Mater., 2022, 34, 9923–9933 CrossRef CAS.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. B. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 3005 CrossRef PubMed.
- B. Park, H. Kang, Y. H. Ha, J. Kim, J.-H. Lee, K. Yu, S. Kwon, S.-Y. Jang, S. Kim, S. Jeong, S. Hong, S. Byun, S.-K. Kwon, Y.-H. Kim and K. Lee, Adv. Sci., 2021, 8, 2100332 CrossRef CAS PubMed.
- D. Liu, Y. Zhao, J. Zhang, Z. Wei, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2024, e202400061 CAS.
- J. Han, X. Rong, C. Xu, Y. Deng, Y. Geng, G. Dong and L. Duan, Adv. Electron. Mater., 2023, 9, 2201288 CrossRef CAS.
- F. P. García de Arquer, A. Armin, P. Meredith and E. H. Sargent, Nat. Rev. Mater., 2017, 2, 16100 CrossRef.
- C. Xie, X.-T. Lu, X.-W. Tong, Z.-X. Zhang, F.-X. Liang, L. Liang, L.-B. Luo and Y.-C. Wu, Adv. Funct. Mater., 2019, 29, 1806006 CrossRef.
- Y. Ma, Y. Zhang and W. W. Yu, J. Mater. Chem. C, 2019, 7, 13662–13679 RSC.
- C. Liu, J. Guo, L. Yu, J. Li, M. Zhang, H. Li, Y. Shi and D. Dai, Light: Sci. Appl., 2021, 10, 123 CrossRef CAS PubMed.
- N. Hiremath, R. Kumar, K. C. Hwang, I. Banerjee, S. Thangudu and R. Vankayala, ACS Appl. Nano Mater., 2022, 5, 1719–1733 CrossRef CAS.
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299–306 CrossRef CAS.
- J. Kuwabara, Y. Fujie, K. Maruyama, T. Yasuda and T. Kanbara, Macromolecules, 2016, 49, 9388–9395 CrossRef CAS.
- M. Streiter, D. Beer, F. Meier, C. Göhler, C. Lienert, F. Lombeck, M. Sommer and C. Deibel, Adv. Funct. Mater., 2019, 29, 1903936 CrossRef CAS.
- J. B. Howard and B. C. Thompson, Macromol. Chem. Phys., 2017, 218, 1700255 CrossRef.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- Z. Bao, W. Chan and L. Yu, Chem. Mater., 1993, 5, 2–3 CrossRef CAS.
- L. Sun, X. Xu, S. Song, Y. Zhang, C. Miao, X. Liu, G. Xing and S. Zhang, Macromol. Rapid Commun., 2019, 40, 1900074 CrossRef PubMed.
- G. Li, X. Zhang, L. O. Jones, J. M. Alzola, S. Mukherjee, L. Feng, W. Zhu, C. L. Stern, W. Huang, J. Yu, V. K. Sangwan, D. M. DeLongchamp, K. L. Kohlstedt, M. R. Wasielewski, M. C. Hersam, G. C. Schatz, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2021, 143, 6123–6139 CrossRef CAS PubMed.
- B. Shahid, D. Zhu, Q. Wang, X. Yuan, I. Ismail, Y. Wu, Z. Du and R. Yang, Polym. Int., 2020, 69, 564–570 CrossRef CAS.
- M. Li, C. An, W. Pisula and K. Müllen, Acc. Chem. Res., 2018, 51, 1196–1205 CrossRef CAS PubMed.
- M. Planells, B. C. Schroeder and I. McCulloch, Macromolecules, 2014, 47, 5889–5894 CrossRef CAS.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- Y.-S. Lee, J. Y. Lee, S.-M. Bang, B. Lim, J. Lee and S.-I. Na, J. Mater. Chem. A, 2016, 4, 11439–11445 RSC.
- H. Yu, Z. Qi, J. Yu, Y. Xiao, R. Sun, Z. Luo, A. M. H. Cheung, J. Zhang, H. Sun, W. Zhou, S. Chen, X. Guo, X. Lu, F. Gao, J. Min and H. Yan, Adv. Energy Mater., 2020, 11, 2003171 CrossRef.
- W. Zhong, J. Xiao, S. Sun, X.-F. Jiang, L. Lan, L. Ying, W. Yang, H.-L. Yip, F. Huang and Y. Cao, J. Mater. Chem. C, 2016, 4, 4719–4727 RSC.
- A. Casey, Y. Han, Z. Fei, A. J. P. White, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. C, 2015, 3, 265–275 RSC.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- S. Shi, H. Wang, P. Chen, M. A. Uddin, Y. Wang, Y. Tang, H. Guo, X. Cheng, S. Zhang, H. Y. Woo and X. Guo, Polym. Chem., 2018, 9, 3873–3884 RSC.
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422–433 RSC.
- E. I. Carrera and D. S. Seferos, Macromolecules, 2014, 48, 297–308 CrossRef.
- A. V. Marsh and M. Heeney, Polym. J., 2023, 55, 375–385 CrossRef CAS.
- J.-L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5003 CrossRef CAS PubMed.
- H. Mori, S. Nishinaga, R. Takahashi and Y. Nishihara, Macromolecules, 2018, 51, 5473–5484 CrossRef CAS.
- H. Mori, R. Hosogi, Y. Minagawa, H. Yamane and Y. Nishihara, ACS Appl. Polym. Mater., 2024, 6, 3883–3893 CrossRef CAS.
- G. Bianchi, C. Carbonera, L. Ciammaruchi, N. Camaioni, N. Negarville, F. Tinti, G. Forti, A. Nitti, D. Pasini, A. Facchetti, R. M. Pankow, T. J. Marks and R. Po, Sol. RRL, 2022, 6, 2200643 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y.-J. Cheng, Y.-J. Ho, C.-H. Chen, W.-S. Kao, C.-E. Wu, S.-L. Hsu and C.-S. Hsu, Macromolecules, 2012, 45, 2690–2698 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. DeLongchamp, R. J. Kline, M. Cölle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091–1109 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- Y. Wu, S. Schneider, C. Walter, A. H. Chowdhury, B. Bahrami, H.-C. Wu, Q. Qiao, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2020, 142, 392–406 CrossRef CAS PubMed.
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017 CrossRef CAS PubMed.
- Y. Kim, J. Choi, C. Lee, Y. Kim, C. Kim, T. L. Nguyen, B. Gautam, K. Gundogdu, H. Y. Woo and B. J. Kim, Chem. Mater., 2018, 30, 5663–5672 CrossRef CAS.
- C. Lee, H. R. Lee, J. Choi, Y. Kim, T. L. Nguyen, W. Lee, B. Gautam, X. Liu, K. Zhang, F. Huang, J. H. Oh, H. Y. Woo and B. J. Kim, Adv. Energy Mater., 2018, 8, 1802674 CrossRef.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- S. Moro, N. Siemons, O. Drury, D. A. Warr, T. A. Moriarty, L. M. A. Perdigão, D. Pearce, M. Moser, R. K. Hallani, J. Parker, I. McCulloch, J. M. Frost, J. Nelson and G. Costantini, ACS Nano, 2022, 16, 21303–21314 CrossRef CAS PubMed.
- C. Liu, C. Xiao, J. Wang, B. Liu, Y. Hao, J. Guo, J. Song, Z. Tang, Y. Sun and W. Li, Macromolecules, 2022, 55, 5964–5974 CrossRef CAS.
- L. Sun, N. Sun, L. Bai, X. An, B. Liu, C. Sun, L. Fan, C. Wei, Y. Han, M. Yu, J. Lin, D. Lu, N. Wang, L. Xie, K. Shen, X. Zhang, Y. Xu, J. Cabanillas-Gonzalez and W. Huang, Chin. Chem. Lett., 2019, 30, 1959–1964 CrossRef CAS.
- C. Pan, Z. Guo and J. Xu, ACS Appl. Eng. Mater., 2024, 2, 49–55 CrossRef CAS.
- J. Neu, S. Samson, K. Ding, J. J. Rech, H. Ade and W. You, Macromolecules, 2023, 56, 2092–2103 CrossRef CAS.
- Z. Chen, L. Yan, J. J. Rech, J. Hu, Q. Zhang and W. You, ACS Appl. Polym. Mater., 2019, 1, 804–814 CrossRef CAS.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- M. Moser, L. R. Savagian, A. Savva, M. Matta, J. F. Ponder, T. C. Hidalgo, D. Ohayon, R. Hallani, M. Reisjalali, A. Troisi, A. Wadsworth, J. R. Reynolds, S. Inal and I. McCulloch, Chem. Mater., 2020, 32, 6618–6628 CrossRef CAS.
- A. A. Advincula, A. L. Jones, K. J. Thorley, A. M. Österholm, J. F. Ponder and J. R. Reynolds, Chem. Mater., 2022, 34, 4633–4645 CrossRef CAS.
- R. M. Pankow, B. Kerwin, Y. Cho, S. Jeong, G. Forti, B. Musolino, C. Yang, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2023, 34, 2309428 CrossRef.
- W. Huang, J. Chen, Y. Yao, D. Zheng, X. Ji, L.-W. Feng, D. Moore, N. R. Glavin, M. Xie, Y. Chen, R. M. Pankow, A. Surendran, Z. Wang, Y. Xia, L. Bai, J. Rivnay, J. Ping, X. Guo, Y. Cheng, T. J. Marks and A. Facchetti, Nature, 2023, 613, 496–502 CrossRef CAS PubMed.
- J. Kim, R. M. Pankow, Y. Cho, I. D. Duplessis, F. Qin, D. Meli, R. Daso, D. Zheng, W. Huang, J. Rivnay, T. J. Marks and A. Facchetti, Nat. Electron., 2024, 7, 234–243 CrossRef CAS.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Prog. Polym. Sci., 2023, 136, 101626 CrossRef CAS.
- N. A. Kukhta and C. K. Luscombe, Chem. Commun., 2022, 58, 6982–6997 RSC.
- W. J. Mullin, S. A. Sharber and S. W. Thomas III, J. Polym. Sci., 2021, 59, 1643–1663 CrossRef CAS.
- A. Salleo, Mater. Today, 2007, 10, 38–45 CrossRef CAS.
- R. Noruzi, E. Lim, B. S. S. Pokuri, M. L. Chabinyc and B. Ganapathysubramanian, npj Comput. Mater., 2022, 8, 38 CrossRef.
- S. E. Chen, L. Q. Flagg, J. W. Onorato, L. J. Richter, J. Guo, C. K. Luscombe and D. S. Ginger, J. Mater. Chem. A, 2022, 10, 10738–10749 RSC.
- C. G. Bischak, L. Q. Flagg, K. Yan, T. Rehman, D. W. Davies, R. J. Quezada, J. W. Onorato, C. K. Luscombe, Y. Diao, C.-Z. Li and D. S. Ginger, J. Am. Chem. Soc., 2020, 142, 7434–7442 CrossRef CAS PubMed.
- H. Sirringhaus, M. Bird and N. Zhao, Adv. Mater., 2010, 22, 3893–3898 CrossRef CAS PubMed.
- G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS.
- S. Holliday, Y. Li and C. K. Luscombe, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- N. S. Gobalasingham, S. Noh, J. B. Howard and B. C. Thompson, ACS Appl. Mater. Interfaces, 2016, 8, 27931–27941 CrossRef CAS.
- G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, NJ, 4th edn, 2004 Search PubMed.
- A. Mahmood and J.-L. Wang, Sol. RRL, 2020, 4, 2000337 CrossRef CAS.
- P. Müller-Buschbaum, Adv. Mater., 2014, 26, 7692–7709 CrossRef PubMed.
- M. Pandey, N. Kumari, S. Nagamatsu and S. S. Pandey, J. Mater. Chem. C, 2019, 7, 13323–13351 RSC.
- G. Cai, J. Wang and P. S. Lee, Acc. Chem. Res., 2016, 49, 1469–1476 CrossRef CAS PubMed.
- D. K. Pathak and H. C. Moon, Mater. Horiz., 2022, 9, 2949–2975 RSC.
- A. L.-S. Eh, A. W. M. Tan, X. Cheng, S. Magdassi and P. S. Lee, Energy Technol., 2018, 6, 33–45 CrossRef.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- M. Nikolou, A. L. Dyer, T. T. Steckler, E. P. Donoghue, Z. Wu, N. C. Heston, A. G. Rinzler, D. B. Tanner and J. R. Reynolds, Chem. Mater., 2009, 21, 5539–5547 CrossRef CAS.
- X. Cheng, Y. Ma, X. Ju, W. Zhao, J. Zhao, Q. Li, Z. Sang, H. Du and Y. Zhang, Synth. Met., 2020, 270, 116589 CrossRef CAS.
- Y. Zhang, L. Kong, Y. Zhang, H. Du, J. Zhao, S. Chen, Y. Xie and Y. Wang, Org. Electron., 2020, 81, 105685 CrossRef CAS.
- J. A. Kerszulis, R. H. Bulloch, N. B. Teran, R. M. W. Wolfe and J. R. Reynolds, Macromolecules, 2016, 49, 6350–6359 CrossRef CAS.
- L. A. Estrada, J. J. Deininger, G. D. Kamenov and J. R. Reynolds, ACS Macro Lett., 2013, 2, 869–873 CrossRef CAS PubMed.
- C. J. Brabec, A. Distler, X. Du, H.-J. Egelhaaf, J. Hauch, T. Heumueller and N. Li, Adv. Energy Mater., 2020, 10, 2001864 CrossRef CAS.
- Z. Wang, K. Gao, Y. Kan, M. Zhang, C. Qiu, L. Zhu, Z. Zhao, X. Peng, W. Feng, Z. Qian, X. Gu, A. K.-Y. Jen, B. Z. Tang, Y. Cao, Y. Zhang and F. Liu, Nat. Commun., 2021, 12, 332 CrossRef CAS PubMed.
- G. Feng, J. Li, F. J. M. Colberts, M. Li, J. Zhang, F. Yang, Y. Jin, F. Zhang, R. A. J. Janssen, C. Li and W. Li, J. Am. Chem. Soc., 2017, 139, 18647–18656 CrossRef CAS.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2018, 58, 4129 CrossRef.
- N. K. Elumalai and A. Uddin, Energy Environ. Sci., 2016, 9, 391–410 RSC.
- D. Gupta, S. Mukhopadhyay and K. S. Narayan, Sol. Energy Mater. Sol. Cells, 2010, 94, 1309–1313 CrossRef CAS.
- M.-H. Jao, H.-C. Liao and W.-F. Su, J. Mater. Chem. A, 2016, 4, 5784–5801 RSC.
- R. M. Pankow, J. Wu, A. Harbuzaru, B. Kerwin, Y. Chen, R. P. Ortiz, A. Facchetti and T. J. Marks, Chem. Mater., 2022, 34, 3267–3279 CrossRef CAS.
- J. Wu, C. Fu, R. M. Pankow, Y. Chen, D. Zheng, Z. Lu, Y. Huang, T. J. Marks and A. Facchetti, Chem. Mater., 2023, 35, 10106–10118 CrossRef CAS.
- T. J. Aldrich, A. S. Dudnik, N. D. Eastham, E. F. Manley, L. X. Chen, R. P. H. Chang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Macromolecules, 2018, 51, 9140–9155 CrossRef CAS.
- N. S. Gobalasingham, R. M. Pankow, S. Ekiz and B. C. Thompson, J. Mater. Chem. A, 2017, 5, 14101–14113 RSC.
- J. Y. Oh, S. Rondeau-Gagné, Y.-C. Chiu, A. Chortos, F. Lissel, G.-J. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W.-G. Bae, Y. Kim, L. Jin, J. W. Chung, J. B.-H. Tok and Z. Bao, Nature, 2016, 539, 411–415 CrossRef CAS PubMed.
- G. Dufil, I. Bernacka-Wojcik, A. Armada-Moreira and E. Stavrinidou, Chem. Rev., 2022, 122, 4847–4883 CrossRef CAS PubMed.
- R. Kubota, Y. Sasaki, T. Minamiki and T. Minami, ACS Sens., 2019, 4, 2571–2587 CrossRef CAS PubMed.
- Z. A. Lamport, H. F. Haneef, S. Anand, M. Waldrip and O. D. Jurchescu, J. Appl. Phys., 2018, 124, 071101 CrossRef.
- Y.-C. Lin, W.-C. Yang, Y.-C. Chiang and W.-C. Chen, Small Sci., 2022, 2, 2100109 CrossRef CAS.
- Y. Guo, X. Yang, L. Wang, J. Duan, Y. Zhou, C. B. Nielsen, Y. Yu, J. Yang, Y. Guo, Z. Li, W. Yue, Y. Liu and I. McCulloch, Macromolecules, 2021, 54, 10312–10320 CrossRef CAS.
- J. F. Ponder Jr, H. Chen, A. M. T. Luci, S. Moro, M. Turano, A. L. Hobson, G. S. Collier, L. M. A. Perdigão, M. Moser, W. Zhang, G. Costantini, J. R. Reynolds and I. McCulloch, ACS Mater. Lett., 2021, 3, 1503–1512 CrossRef CAS.
- J. Chen, W. Huang, D. Zheng, Z. Xie, X. Zhuang, D. Zhao, Y. Chen, N. Su, H. Chen, R. M. Pankow, Z. Gao, J. Yu, X. Guo, Y. Cheng, J. Strzalka, X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2022, 21, 564–571 CrossRef CAS PubMed.
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- S. G. Kim, J. S. Han, H. Kim, S. Y. Kim and H. W. Jang, Adv. Mater. Technol., 2018, 3, 1800457 CrossRef.
- Y. van de Burgt, A. Melianas, S. T. Keene, G. Malliaras and A. Salleo, Nat. Electron., 2018, 1, 386–397 CrossRef.
- M. Berggren, X. Crispin, S. Fabiano, M. P. Jonsson, D. T. Simon, E. Stavrinidou, K. Tybrandt and I. Zozoulenko, Adv. Mater., 2019, 31, 1805813 CrossRef PubMed.
- D. Ohayon, V. Druet and S. Inal, Chem. Soc. Rev., 2023, 52, 1001–1023 RSC.
- A. Erhardt, A. Hochgesang, C. R. McNeill and M. Thelakkat, Adv. Electron. Mater., 2023, 9, 2300026 CrossRef CAS.
- X. Luo, H. Shen, K. Perera, D. T. Tran, B. W. Boudouris and J. Mei, ACS Macro Lett., 2021, 10, 1061–1067 CrossRef CAS PubMed.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- Y. Wang, Y. Yu, H. Liao, Y. Zhou, I. McCulloch and W. Yue, Acc. Chem. Res., 2020, 53, 2855–2868 CrossRef CAS PubMed.
- F. Grenier, B. R. Aïch, Y.-Y. Lai, M. Guérette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137–2143 CrossRef CAS.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- M. H. Chua, S. H. G. Toh, P. J. Ong, Z. M. Png, Q. Zhu, S. Xiong and J. Xu, Polym. Chem., 2022, 13, 967–981 RSC.
- Z. S. Parr, J. Borges-González, R. B. Rashid, K. J. Thorley, D. Meli, B. D. Paulsen, J. Strzalka, J. Rivnay and C. B. Nielsen, Adv. Mater., 2022, 34, 2107829 CrossRef CAS PubMed.
- L. Xu, Z. Zhao, M. Xiao, J. Yang, J. Xiao, Z. Yi, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 40549–40555 CrossRef CAS PubMed.
- J. Yang, Y. Jiang, Z. Zhao, X. Yang, Z. Zhang, J. Chen, J. Li, W. Shi, S. Wang, Y. Guo and Y. Liu, Natl. Sci. Rev., 2022, 9, nwab145 CrossRef PubMed.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- G. W. P. Van Pruissen, F. Gholamrezaie, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2012, 22, 20387–20393 RSC.
- N. Nozaki, A. Uva, H. Matsumoto, H. Tran and M. Ashizawa, RSC Appl. Polym., 2024, 2, 163–171 RSC.
- J. Huang, Z. Mao, Z. Chen, D. Gao, C. Wei, W. Zhang and G. Yu, Chem. Mater., 2016, 28, 2209–2218 CrossRef CAS.
- G. Zhang, Z. Ye, P. Li, J. Guo, Q. Wang, L. Tang, H. Lu and L. Qiu, Polym. Chem., 2015, 6, 3970–3978 RSC.
- K. Shi, W. Zhang, D. Gao, S. Zhang, Z. Lin, Y. Zou, L. Wang and G. Yu, Adv. Mater., 2018, 30, 1705286 CrossRef PubMed.
- K. Huang, X. Zhao, Y. Du, S. Kim, X. Wang, H. Lu, K. Cho, G. Zhang and L. Qiu, J. Mater. Chem. C, 2019, 7, 7618–7626 RSC.
- X. Wang, F. Zhao, Z. Xue, Y. Yuan, M. Huang, G. Zhang, Y. Ding and L. Qiu, Adv. Electron. Mater., 2019, 5, 1900174 CrossRef.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS PubMed.
- B. He, W. T. Neo, T. L. Chen, L. M. Klivansky, H. Wang, T. Tan, S. J. Teat, J. Xu and Y. Liu, ACS Sustainable Chem. Eng., 2016, 4, 2797–2805 CrossRef CAS.
- K. J. Fallon, N. Wijeyasinghe, E. F. Manley, S. D. Dimitrov, S. A. Yousaf, R. S. Ashraf, W. Duffy, A. A. Y. Guilbert, D. M. E. Freeman, M. Al-Hashimi, J. Nelson, J. R. Durrant, L. X. Chen, I. McCulloch, T. J. Marks, T. M. Clarke, T. D. Anthopoulos and H. Bronstein, Chem. Mater., 2016, 28, 8366–8378 CrossRef CAS.
- J. Yang, Y. Jiang, Z. Tu, Z. Zhao, J. Chen, Z. Yi, Y. Li, S. Wang, Y. Yi, Y. Guo and Y. Liu, Adv. Funct. Mater., 2019, 29, 1804839 CrossRef.
- C.-J. Yang and S. A. Jenekhe, Macromolecules, 1995, 28, 1180–1196 CrossRef CAS.
- A. Bolduc, S. Barik, M. R. Lenze, K. Meerholz and W. G. Skene, J. Mater. Chem. A, 2014, 2, 15620–15626 RSC.
- S. Barik and W. G. Skene, Polym. Chem., 2011, 2, 1091–1097 RSC.
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao, K.-T. Cheng, J. B.-H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107 CrossRef CAS PubMed.
- Y. Ding, F. Zhao, S. Kim, X. Wang, H. Lu, G. Zhang, K. Cho and L. Qiu, ACS Appl. Mater. Interfaces, 2020, 12, 41832–41841 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed.
- G. Zhang, P. Li, L. Tang, J. Ma, X. Wang, H. Lu, B. Kang, K. Cho and L. Qiu, Chem. Commun., 2014, 50, 3180–3183 RSC.
- Z. Yan, B. Sun and Y. Li, Chem. Commun., 2013, 49, 3790–3792 RSC.
- H. Tang, Y. Liang, C. Liu, Z. Hu, Y. Deng, H. Guo, Z. Yu, A. Song, H. Zhao, D. Zhao, Y. Zhang, X. Guo, J. Pei, Y. Ma, Y. Cao and F. Huang, Nature, 2022, 611, 271–277 CrossRef CAS PubMed.
- K. J. Fallon and H. Bronstein, Acc. Chem. Res., 2021, 54, 182–193 CrossRef CAS PubMed.
- W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- Y. Patil and R. Misra, J. Mater. Chem. C, 2019, 7, 13020–13031 RSC.
- W. K. Chan, Y. Chen, Z. Peng and L. Yu, J. Am. Chem. Soc., 1993, 115, 11735–11743 CrossRef CAS.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 RSC.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Q. Wang, S. Böckmann, F. Günther, M. Streiter, M. Zerson, A. D. Scaccabarozzi, W. L. Tan, H. Komber, C. Deibel, R. Magerle, S. Gemming, C. R. McNeill, M. Caironi, M. R. Hansen and M. Sommer, Chem. Mater., 2021, 33, 2635–2645 CrossRef CAS.
- C. Zhang, W. L. Tan, Z. Liu, Q. He, Y. Li, J. Ma, A. S. R. Chesman, Y. Han, C. R. McNeill, M. Heeney and Z. Fei, Macromolecules, 2022, 55, 4429–4440 CrossRef CAS.
- J. Kuwabara, N. Takase, T. Yasuda and T. Kanbara, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2337–2345 CrossRef CAS.
- S. Broll, F. Nübling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481–7488 CrossRef CAS.
- X. Liu, Y. Guo, Y. Ma, H. Chen, Z. Mao, H. Wang, G. Yu and Y. Liu, Adv. Mater., 2014, 26, 3631–3636 CrossRef CAS PubMed.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- R. Gao, P. Yuan, X. Zhang, S. Sang, X.-C. Hang, H. Nan, C. Liu, C. Zhang, X. Gao, F. Chen, X. Guo and Z.-K. Chen, ACS Appl. Electron. Mater., 2019, 1, 1233–1242 CrossRef CAS.
- Y. Zheng, S. Zhang, J. B.-H. Tok and Z. Bao, J. Am. Chem. Soc., 2022, 144, 4699–4715 CrossRef CAS PubMed.
- R. M. Pankow, A. Harbuzaru, D. Zheng, B. Kerwin, G. Forti, I. D. Duplessis, B. Musolino, R. Ponce Ortiz, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2023, 145, 13411–13419 CrossRef CAS PubMed.
- J. Z. Low, W. T. Neo, Q. Ye, W. J. Ong, I. H. K. Wong, T. T. Lin and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1287–1295 CrossRef CAS.
- C. J. Mueller, C. R. Singh and M. Thelakkat, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 639–648 CrossRef CAS.
- G. Krauss, F. Meichsner, A. Hochgesang, J. Mohanraj, S. Salehi, P. Schmode and M. Thelakkat, Adv. Funct. Mater., 2021, 31, 2010048 CrossRef CAS.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Sol. Energy Mater. Sol. Cells, 2011, 95, 1168–1173 CrossRef CAS.
- Y. Lei, N. Li, W.-K. E. Chan, B. S. Ong and F. Zhu, Org. Electron., 2017, 48, 12–18 CrossRef CAS.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
- Q. Liu, Y. Wang, A. Kohara, H. Matsumoto, S. Manzhos, K. Feron, S. E. Bottle, J. Bell, T. Michinobu and P. Sonar, Adv. Funct. Mater., 2020, 30, 1907452 CrossRef CAS.
- Q. Liu, W. He, Y. Shi, S. Otep, W. L. Tan, S. Manzhos, C. R. McNeill, X. Guo, P. Sonar, T. Michinobu and A. K. K. Kyaw, Chem. Mater., 2022, 34, 3140–3151 CrossRef CAS.
- G. Oklem, X. Song, L. Toppare, D. Baran and G. Gunbas, J. Mater. Chem. C, 2018, 6, 2957–2961 RSC.
- Z. Yuan, B. Fu, S. Thomas, S. Zhang, G. DeLuca, R. Chang, L. Lopez, C. Fares, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2016, 28, 6045–6049 CrossRef CAS.
- Z. Chen, D. Gao, J. Huang, Z. Mao, W. Zhang and G. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 34725–34734 CrossRef CAS PubMed.
- D. Gao, Z. Chen, J. Huang, W. Zhang, C. Wei, Z. Lin, D. Li and G. Yu, J. Mater. Chem. C, 2017, 5, 3568–3578 RSC.
- C. Buckley, S. Thomas, M. McBride, Z. Yuan, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2019, 31, 3957–3966 CrossRef CAS.
- Z. Li, X. Xu, W. Zhang, Z. Genene, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Mater. Chem. A, 2017, 5, 11693–11700 RSC.
- H.-J. Yun, J. Cho, D. S. Chung, Y.-H. Kim and S.-K. Kwon, Macromolecules, 2014, 47, 7030–7035 CrossRef CAS.
- J. Lee, A.-R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed.
- S.-C. Chen, Q. Zheng, Q. Zhang, D. Cai, J. Wang, Z. Yin and C. Tang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1999–2005 CrossRef CAS.
- Z. Wang, Y. Shi, Y. Deng, Y. Han and Y. Geng, Adv. Funct. Mater., 2021, 31, 2104881 CrossRef CAS.
- D. Gao, Z. Chen, Z. Mao, J. Huang, W. Zhang, D. Li and G. Yu, RSC Adv., 2016, 6, 78008–78016 RSC.
- X. Yan, M. Xiong, X.-Y. Deng, K.-K. Liu, J.-T. Li, X.-Q. Wang, S. Zhang, N. Prine, Z. Zhang, W. Huang, Y. Wang, J.-Y. Wang, X. Gu, S. K. So, J. Zhu and T. Lei, Nat. Commun., 2021, 12, 5723 CrossRef CAS PubMed.
- X. Yan, M. Xiong, J.-T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z.-Y. Wang, Z.-F. Yao, J.-Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- N. Zhou and A. Facchetti, Mater. Today, 2018, 21, 377–390 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- S. G. Bucella, A. Luzio, E. Gann, L. Thomsen, C. R. McNeill, G. Pace, A. Perinot, Z. Chen, A. Facchetti and M. Caironi, Nat. Commun., 2015, 6, 8394 CrossRef CAS PubMed.
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705–6711 CrossRef CAS PubMed.
- R. Matsidik, H. Komber and M. Sommer, ACS Macro Lett., 2015, 4, 1346–1350 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K.-Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganäs, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935–10944 CrossRef CAS PubMed.
- Z. Li, X. Xu, W. Zhang, X. Meng, Z. Genene, W. Ma, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, Energy Environ. Sci., 2017, 10, 2212–2221 RSC.
- M. J. Sung, A. Luzio, W.-T. Park, R. Kim, E. Gann, F. Maddalena, G. Pace, Y. Xu, D. Natali, C. de Falco, L. Dang, C. R. McNeill, M. Caironi, Y.-Y. Noh and Y.-H. Kim, Adv. Funct. Mater., 2016, 26, 4984–4997 CrossRef CAS.
- K. Park, E.-Y. Shin, X. Jiao, C. R. McNeill, Y.-H. Kim, S.-K. Kwon and Y.-Y. Noh, ACS Appl. Mater. Interfaces, 2019, 11, 35185–35192 CrossRef CAS PubMed.
- V. Senkovskyy, R. Tkachov, H. Komber, M. Sommer, M. Heuken, B. Voit, W. T. S. Huck, V. Kataev, A. Petr and A. Kiriy, J. Am. Chem. Soc., 2011, 133, 19966–19970 CrossRef CAS PubMed.
- R. Tkachov, Y. Karpov, V. Senkovskyy, I. Raguzin, J. Zessin, A. Lederer, M. Stamm, B. Voit, T. Beryozkina, V. Bakulev, W. Zhao, A. Facchetti and A. Kiriy, Macromolecules, 2014, 47, 3845–3851 CrossRef CAS.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- L. Xue, Y. Yang, Z.-G. Zhang, X. Dong, L. Gao, H. Bin, J. Zhang, Y. Yang and Y. Li, J. Mater. Chem. A, 2016, 4, 5810–5816 RSC.
- S. Lee, C. Lee, H. Kim and Y. Kim, J. Mater. Chem. C, 2020, 8, 15778–15787 RSC.
- M.-K. Jeong, E. H. Suh, K. Lee, J. Jang and I. H. Jung, Org. Electron., 2020, 86, 105921 CrossRef CAS.
- F. Liu, H. Li, Y. Wu, C. Gu and H. Fu, RSC Adv., 2015, 5, 92151–92158 RSC.
- K. Nakano, M. Nakano, B. Xiao, E. Zhou, K. Suzuki, I. Osaka, K. Takimiya and K. Tajima, Macromolecules, 2016, 49, 1752–1760 CrossRef CAS.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. Tang and E. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- Z. Zhao, F. Zhang, Y. Hu, Z. Wang, B. Leng, X. Gao, C. Di and D. Zhu, ACS Macro Lett., 2014, 3, 1174–1177 CrossRef CAS PubMed.
- Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 CrossRef CAS PubMed.
- H. A. M. van Mullekom, J. A. J. M. Venkemans and E. W. Meijer, Chem. Commun., 1996, 2163–2164 RSC.
- P. Cong, Z. Wang, Y. Geng, Y. Meng, C. Meng, L. Chen, A. Tang and E. Zhou, Nano Energy, 2023, 105, 108017 CrossRef CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- P. Sonar, S. P. Singh, Y. Li, M. S. Soh and A. Dodabalapur, Adv. Mater., 2010, 22, 5409–5413 CrossRef CAS PubMed.
- J. Lee, M. Jang, S. M. Lee, D. Yoo, T. J. Shin, J. H. Oh and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 20390–20399 CrossRef CAS PubMed.
- J. Kim, A.-R. Han, J. Hong, G. Kim, J. Lee, T. J. Shin, J. H. Oh and C. Yang, Chem. Mater., 2014, 26, 4933–4942 CrossRef CAS.
- L. Ma, Z. Li, B. Chen, P. Xue, Z. Wang, Y. Wu, X. Zhan, Y. Liu and X. Chen, Adv. Electron. Mater., 2022, 8, 2101297 CrossRef CAS.
- K. Feng, Z. Wu, M. Su, S. Ma, Y. Shi, K. Yang, Y. Wang, Y. Zhang, W. Sun, X. Cheng, L. Huang, J. Min, H. Y. Woo and X. Guo, Adv. Funct. Mater., 2021, 31, 2008494 CrossRef CAS.
- D. Nodari, S. Sharma, W. Jia, A. V. Marsh, Y.-H. Lin, Y. Fu, X. Lu, A. Russkikh, G. T. Harrison, S. Fatayer, N. Gasparini, M. Heeney and J. Panidi, Adv. Mater., 2024, 2402568 CrossRef PubMed.
- C. N. Scott, M. D. Bisen, D. M. Stemer, S. McKinnon and C. K. Luscombe, Macromolecules, 2017, 50, 4623–4628 CrossRef CAS.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS PubMed.
- Y. Wang, M. Nakano, T. Michinobu, Y. Kiyota, T. Mori and K. Takimiya, Macromolecules, 2017, 50, 857–864 CrossRef CAS.
- C. Zhang, J. Zhang, W. Zeng, N. Zheng, W. Li, W. Gao, G. Yu and C. Yang, Polym. Chem., 2016, 7, 2808–2814 RSC.
- H. Chen, G. Cai, A. Guo, Z. Zhao, J. Kuang, L. Zheng, L. Zhao, J. Chen, Y. Guo and Y. Liu, Macromolecules, 2019, 52, 6149–6159 CrossRef CAS.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- Y. Dong, W. Cai, X. Hu, C. Zhong, F. Huang and Y. Cao, Polymer, 2012, 53, 1465–1472 CrossRef CAS.
- X. Hu, Y. Dong, F. Huang, X. Gong and Y. Cao, J. Phys. Chem. C, 2013, 117, 6537–6543 CrossRef CAS.
- S. Cho, J. Lee, M. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910–1916 CrossRef CAS.
- J. Park, C. Lee, T. Kim, H. Kim and Y. Kim, Adv. Electron. Mater., 2021, 7, 2000932 CrossRef CAS.
- Q. Li, Y. Ran, W. Shi, M. Qin, Y. Sun, J. Kuang, H. Wang, H. Chen, Y. Guo and Y. Liu, Appl. Mater. Today, 2021, 22, 100899 CrossRef.
- X. Liu, M. Sha, H. Yin and X. Hao, APL Energy, 2023, 1, 031501 CrossRef.
- Y. Zhang, Y. Ji, Y. Zhang, W. Zhang, H. Bai, M. Du, H. Wu, Q. Guo and E. Zhou, Adv. Funct. Mater., 2022, 32, 2205115 CrossRef CAS.
- P. Xue, P. Cheng, R. P. S. Han and X. Zhan, Mater. Horiz., 2021, 9, 194–219 RSC.
- F. Zhao, H. Zhang, R. Zhang, J. Yuan, D. He, Y. Zou and F. Gao, Adv. Energy Mater., 2020, 10, 2002746 CrossRef CAS.
- D. Li, X. Zhang, D. Liu and T. Wang, J. Mater. Chem. A, 2020, 8, 15607–15619 RSC.
- Y. Meng, J. Wu, X. Guo, W. Su, L. Zhu, J. Fang, Z.-G. Zhang, F. Liu, M. Zhang, T. P. Russell and Y. Li, Sci. China Chem., 2019, 62, 845–850 CrossRef CAS.
- T. Jia, J. Zhang, W. Zhong, Y. Liang, K. Zhang, S. Dong, L. Ying, F. Liu, X. Wang, F. Huang and Y. Cao, Nano Energy, 2020, 72, 104718 CrossRef CAS.
- Q. Fan, H. Fu, M. Liu, J. Oh, X. Ma, F. R. Lin, C. Yang, F. Zhang and A. K.-Y. Jen, ACS Appl. Mater. Interfaces, 2022, 14, 26970–26977 CrossRef CAS PubMed.
- H. Lai, H. Chen, Y. Zhu, H. Wang, Y. Li and F. He, Macromolecules, 2022, 55, 3353–3360 CrossRef CAS.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS PubMed.
- H. Yu, M. Pan, R. Sun, I. Agunawela, J. Zhang, Y. Li, Z. Qi, H. Han, X. Zou, W. Zhou, S. Chen, J. Y. L. Lai, S. Luo, Z. Luo, D. Zhao, X. Lu, H. Ade, F. Huang, J. Min and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 10137–10146 CrossRef CAS PubMed.
- T. Wang, R. Sun, W. Wang, H. Li, Y. Wu and J. Min, Chem. Mater., 2021, 33, 761–773 CrossRef CAS.
- X. Yang, R. Sun, Y. Wang, M. Chen, X. Xia, X. Lu, G. Lu and J. Min, Adv. Mater., 2023, 35, 2209350 CrossRef CAS PubMed.
- Q. Fan, H. Fu, Q. Wu, Z. Wu, F. Lin, Z. Zhu, J. Min, H. Y. Woo and A. K.-Y. Jen, Angew. Chem., Int. Ed., 2021, 60, 15935–15943 CrossRef CAS PubMed.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K.-Y. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- F. Qin, G. Li, Y. Liu, Y. Cho, R. M. Pankow, D. Zhang, L. Feng, Y. Wang, S. Jeong, G. Forti, D. Zheng, C. Yang, Y. Zhou, T. J. Marks and A. Facchetti, ACS Energy Lett., 2023, 8, 4733–4745 CrossRef CAS.
- Q. Fan, R. Ma, T. Liu, J. Yu, Y. Xiao, W. Su, G. Cai, Y. Li, W. Peng, T. Guo, Z. Luo, H. Sun, L. Hou, W. Zhu, X. Lu, F. Gao, E. Moons, D. Yu, H. Yan and E. Wang, Sci. China Chem., 2021, 64, 1380–1388 CrossRef CAS.
- S. Ma, B. Li, S. Gong, J. Wang, B. Liu, S. Young Jeong, X. Chen, H. Young Woo, K. Feng and X. Guo, Angew. Chem., Int. Ed., 2023, 62, e202308306 CrossRef CAS PubMed.
- W. Feng, T. Chen, Y. Li, T. Duan, X. Jiang, C. Zhong, Y. Zhang, J. Yu, G. Lu, X. Wan, B. Kan and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202316698 CrossRef CAS PubMed.
- G. Forti, R. M. Pankow, F. Qin, Y. Cho, B. Kerwin, I. Duplessis, A. Nitti, S. Jeong, C. Yang, A. Facchetti, D. Pasini and T. J. Marks, Chem. – Eur. J., 2023, 29, e202300653 CrossRef CAS PubMed.
- J. Zhang, C.-H. Tan, K. Zhang, T. Jia, Y. Cui, W. Deng, X. Liao, H. Wu, Q. Xu, F. Huang and Y. Cao, Adv. Energy Mater., 2021, 11, 2102559 CrossRef CAS.
- Y. Zhao, X. Zhao, Y. Zang, C. Di, Y. Diao and J. Mei, Macromolecules, 2015, 48, 2048–2053 CrossRef CAS.
- E. L. Melenbrink, K. M. Hilby, M. A. Alkhadra, S. Samal, D. J. Lipomi and B. C. Thompson, ACS Appl. Mater. Interfaces, 2018, 10, 32426–32434 CrossRef CAS PubMed.
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, Macromolecules, 2015, 48, 453–461 CrossRef CAS.
- S. Park, Y. Kim, C. Choi, H. Ahn, T. Park, S. H. Lee, Y. H. Jang and B. H. Lee, Macromol. Rapid Commun., 2022, 43, 2100709 CrossRef CAS PubMed.
- S. Heo, J. Kwon, M. Sung, S. Lee, Y. Cho, H. Jung, I. You, C. Yang, J. Lee and Y.-Y. Noh, ACS Appl. Mater. Interfaces, 2023, 15, 1629–1638 CrossRef CAS PubMed.
- L. Ying, B. B. Y. Hsu, H. Zhan, G. C. Welch, P. Zalar, L. A. Perez, E. J. Kramer, T.-Q. Nguyen, A. J. Heeger, W.-Y. Wong and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 18538–18541 CrossRef CAS PubMed.
- H. Yoo, M. Sung, H. Ahn, D. Yang, J. S. Yoo, J. Lee and B. H. Lee, Chem. Mater., 2023, 35, 9562–9571 CrossRef CAS.
- M. E. Foster, B. A. Zhang, D. Murtagh, Y. Liu, M. Y. Sfeir, B. M. Wong and J. D. Azoulay, Macromol. Rapid Commun., 2014, 35, 1516–1521 CrossRef CAS PubMed.
- A. E. London, L. Huang, B. A. Zhang, M. B. Oviedo, J. Tropp, W. Yao, Z. Wu, B. M. Wong, T. N. Ng and J. D. Azoulay, Polym. Chem., 2017, 8, 2922–2930 RSC.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed.
- Z. Yang, Z. Liu and L. Chen, ACS Appl. Polym. Mater., 2023, 5, 7658–7665 CrossRef CAS.
- W. Yao, Z. Wu, E. Huang, L. Huang, A. E. London, Z. Liu, J. D. Azoulay and T. N. Ng, ACS Appl. Electron. Mater., 2019, 1, 660–666 CrossRef CAS.
- Z. Wu, W. Yao, A. E. London, J. D. Azoulay and T. N. Ng, Adv. Funct. Mater., 2018, 28, 1800391 CrossRef.
- J. D. Azoulay, Z. A. Koretz, B. M. Wong and G. C. Bazan, Macromolecules, 2013, 46, 1337–1342 CrossRef CAS.
- C. Park, J. M. Kim, Y. Kim, S. Bae, M. Do, S. Im, S. Yoo and J. H. Kim, ACS Appl. Electron. Mater., 2021, 3, 4781–4792 CrossRef CAS.
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- S. Chang, J. H. Koo, J. Yoo, M. S. Kim, M. K. Choi, D.-H. Kim and Y. M. Song, Chem. Rev., 2024, 124, 768–859 CrossRef CAS PubMed.
- J. Fu, Q. Yang, P. Huang, S. Chung, K. Cho, Z. Kan, H. Liu, X. Lu, Y. Lang, H. Lai, F. He, P. W. K. Fong, S. Lu, Y. Yang, Z. Xiao and G. Li, Nat. Commun., 2024, 15, 1830 CrossRef CAS PubMed.
- Q. Wan, S. Seo, S.-W. Lee, J. Lee, H. Jeon, T.-S. Kim, B. J. Kim and B. C. Thompson, J. Am. Chem. Soc., 2023, 145, 11914–11920 CrossRef CAS PubMed.
- M. Y. Lee, S. Dharmapurikar, S. J. Lee, Y. Cho, C. Yang and J. H. Oh, Chem. Mater., 2020, 32, 1914–1924 CrossRef CAS.
- A. Nyayachavadi, A. K. Sur, P. Kulatunga, Y. Wang, T. C. Gomes, M. Mooney, G. T. Mason, A. Hu, X. Gu and S. Rondeau-Gagné, Chem. Mater., 2023, 35, 9682–9691 CrossRef CAS.
- G. G. Yang, D.-H. Kim, S. Samal, J. Choi, H. Roh, C. E. Cunin, H. M. Lee, S. O. Kim, M. Dincă and A. Gumyusenge, ACS Sens., 2023, 8, 3687–3692 CrossRef CAS PubMed.
- A. Gumyusenge, D. T. Tran, X. Luo, G. M. Pitch, Y. Zhao, K. A. Jenkins, T. J. Dunn, A. L. Ayzner, B. M. Savoie and J. Mei, Science, 2018, 362, 1131–1134 CrossRef CAS PubMed.
- R. Ji, Y.-Q.-Q. Yi, X. Wang, X. Wu, C. Huang, W. Su and Z. Cui, Thin Solid Films, 2024, 790, 140219 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |