
Boosting solar cell performance during highly thermo- and photo-stable asymmetric perylene diimide dimeric acceptors by selenium-annulation at the outside bay position†
Junfeng
Tong‡
 *a,
Jiayu
Fang‡
a,
Lili
An
b,
Youzhi
Huo
c,
Fushui
Di
a,
Pengzhi
Guo
a,
Chunyan
Yang
a,
Zezhou
Liang
d,
Jianfeng
Li
a and
Yangjun
Xia
*a
*a,
Jiayu
Fang‡
a,
Lili
An
b,
Youzhi
Huo
c,
Fushui
Di
a,
Pengzhi
Guo
a,
Chunyan
Yang
a,
Zezhou
Liang
d,
Jianfeng
Li
a and
Yangjun
Xia
*a
aGansu Provincial Engineering Research Center for Organic Semiconductor Materials and Application Technology, School of Materials Science and Engineering, Lanzhou Jiaotong University, Lanzhou 730070, China. E-mail: tongjunfeng139@163.com; xiayangjun2015@126.com
bSchool of Chemical Engineering, Northwest Minzu University, Lanzhou 730030, China
cHarbin Station, China Railway Harbin Group Co., Ltd, Harbin 150010, China
dKey Laboratory for Physical Electronics and Devices of the Ministry of Education & Shaanxi Key Lab of Information Photonic Technique, School of Electronics and Information Engineering, Xi′an Jiaotong University, Xi'an 710049, China
First published on 15th July 2024
Abstract
Asymmetric perylene diimide (PDI)-based dimeric electron acceptors have received relatively little attention. Herein, two asymmetric fused PDI dimers, namely PDI-TPDI and PDI-SePDI, linked through one single bond (σ bond), were synthesized by S/Se-annulation at the outside bay position. Both of them exhibited outstanding thermal-stability with a 5% decomposition temperature of higher than 400 °C and good photostability. After replacing S-annulation with Se-annulation, a blue-shifted absorption peak, weakened aggregation, deepened ELUMO, but almost unchanged molecular twisting angle between two aromatic rings were observed. Accordingly, the Se-annulated PDI-SePDI-based device achieved a 56.64% elevated PCE as high as 5.31% with a slightly decreased VOC of 0.70 V, a 55.91% increased JSC of 14.64 mA cm−2, a 3.45% enhanced FF of 51.84%, and the S-annulated PDI-TPDI-based device obtained the PCE of 3.39%. The PCE enhancement was mainly due to the enhanced exciton dissociation, suppressed charge recombination, and increased charge mobility, benefiting from the beneficial microstructural morphology as the result of larger heteroatom Se-annulation. This work revealed the regulating mechanism of Se-annulation of PDI at the outside position on molecular distortion, morphology and photovoltaic performance.
Introduction
Recently, non-fullerene electron acceptors (NFEAs) have received increasing attention due to unparalleled advantages including synthetic flexibility, no batch-to-batch difference, broad and strong absorption ranging from ultraviolet-visible (UV-vis) to near-infrared (NIR) area, tuneable energy levels, improved morphological stability, and small driving force to guarantee efficient exciton dissociation.1–7 Furthermore, the power conversion efficiencies (PCEs) of NFEAs-based organic solar cells (OSCs) have increased sharply8–10 and surpassed those of fullerene-based OSCs.11 Commonly, solution processed NFEAs were classified into two categories on the basis of chemical structure, including the fused aromatic ITIC derivatives featuring the acceptor–donor–acceptor (A–D–A) framework12,13 and/or ladder type Y series acceptors14 and rylene imide derivatives, especially perylene diimide (PDI) derivatives with an A–DA′D–A structure.15–18 Although the development of PDI-based acceptors has lagged behind that of A–D–A type acceptors, they are considered to be very promising building blocks for constructing efficient photovoltaic (PV) semiconductors, owing to their attractive characteristics including the excellent light-absorbing (400–600 nm) and electron-withdrawing ability, outstanding thermal- and photo-robustness, high electron mobility up to 10−4–10−3 cm2 V−1 s−1, and facile chemical modifications at multiple positions.1,6,19–25Apart from the above merits, PDI derivatives with low synthetic complexity and cheap production costs also possess excellent photochemical stability and have been widely applied in car paint, which remains stable for many years under the conditions of sunlight.1,26 Despite these advantages, the anisotropic characteristics made PDI molecules tend to form microscale or sub-microscale aggregates along the direction of a planar perylene backbone, resulting in an oversized phase separation in the bulk-heterojunction (BHJ) blend film.27–29 Additionally, the large aggregation would precipitate the formation of an excimer, leading to an irreversible loss of photo-induced excitons, which trapped excitons by shortening the diffusion length.30 Although charge mobility can be enhanced to a certain degree, owing to the 5–20 nm excitation diffusion length, it will undoubtedly impede the excitation diffusion. Additionally, it will reduce the exciton dissociation because of the slashed D/A interfaces and eventually hurt the device performance. For overcoming this issue and alleviating the strong aggregation, several productive strategies have been put forward and many efficient twisted PDI acceptors have been designed. Constructing a PDI-based monomer PDFC with an A–DA′D–A motif,31 dimers SF-PDI215 and SF-iPDI232 with a spirofluorene (SF) linker and SdiPBI-Se with a single bond linker,16 trimers Ta-PDI with an electron-deficient 1,3,5-triazine (Ta) linker1 and TPO-PDI with a triphenylphosphine monoxide (TPO) linker,17 tetramers FTTB-PDI4 with a tetrathienylbenzene (TTB) linker18 and SF-iPDI432 and SF-PDI433 with an SF linker and BPT-Se1 with an i-BDT-Th linker,3 multimer CRP-1 with a corannurylene pentapetalae (CRP) linker34etc. achieved a balance between high exciton dissociation and efficient charge transfer and thereby acquired an inspirational PV performance with a PCE ranging from 5.31% to 12.56%. Early in 2014, the dovetail shape side chain-containing PDI dimer (s-diPBI) with a single linker exhibited the 3.63% PCE, which was significantly higher than those (1.54% and 1.36%) of (d-diPBI) with double linkage and (t-diPBI) with triple linkage when paired with PBDTTT-C-T.35 Meanwhile, Jen et al. extended the dovetail side chain from heptane-4-yl to undecane-6-yl and obtained di-PBI, the PCE of the PBDTT-F-TT:di-PBI-based device was significantly increased to 5.90% after optimization by the inverted device and the modified zinc oxide interlayer.36 Afterwards, they continued to change the linking modes and developed two dimers H-di-PDI and B-di-PDI, with the torsion angle between two PDI subunits of 70° for bay-linking B-di-PDI and 90° for imide-linking H-di-PDI. Hence, the PTB7-Th:H-di-PDI-based device afforded a higher PCE of 6.41% due to the high-efficient exciton dissociation and charge percolation pathways as the result of better miscibility, predominate face-on orientation and suitable aggregation domains.37 In 2017, three PDI diploids with a shorter pentan-3-yl side chain, namely, bb-2PDI (bay-/bay-), oo-2PDI (ortho-/ortho-) and bo-2PDI (bay-/ortho-), connected at different linking positions were developed via the Ullmann coupling reaction, with the dihedral angles of 70°, 87° and 67°, respectively. Dimer oo-2PDI exhibited the highest ELUMO of −3.90 eV, resulting in the relatively higher open-circuit voltage (VOC) of 0.799 V, and favourable morphology, relatively higher and balanced charge mobility made PTB7-Th:oo-2PDI-based device afford the excellent short-circuit current density (JSC) up to 18.79 mA cm−2 and markedly outperformed PCE of 8.30%.38 Apparently, constructing PDI dimers with appropriate twisted architecture and fewer synthesis steps was an effective method for exploring high-performance NFEAs.
Recently, heteroatom annulation was demonstrated to be one effective and feasible strategy for fine-tuning the molecular structure and electronic density due to van der Waals and heteroatom–heteroatom interaction.16,39–41 In 2015, Wang et al. modulated the molecule via the S-annulation strategy and obtained the dimer SdiPBI-S with a dihedral angle of 80°, a higher absorption coefficient (ε) up to 1.4 × 105 M−1 cm−1 at 504 nm, and an elevated ELUMO of −3.85 eV. Thus, an excellent PCE of 7.16% was found when blending with PDBT-T1, exhibiting a high VOC of 0.90 V, a JSC of 11.98 mA cm−2, and an FF of 66.1% after applying 0.75% DIO additive.39 As an alternative to the sulfur atom, the more easily polarized selenium atom possesses a larger and looser outermost electron cloud, effectively enhancing the orbital overlap and improving the charge transfer capability. Inspired by this, the Se-annulation analogue SdiPBI-Se, exhibiting the slightly reduced dihedral angle of 77°, the comparable ε of 1.04 × 105 M−1 cm−1, and the slightly down-shifted ELUMO of −3.87 eV, was developed. The better photon absorption, charge transfer and ultrafast charge generation promoted the PDBT-T1:SdiPBI-Se-based device to deliver the synergistically increased VOC of 0.96 V, JSC of 12.49 mA cm−2, and FF of 70.2% and thus elevated the PCE up to 8.4%.16 In 2020, Zhou et al. chose the planar vinylene as a central linker and synthesized a series of fused PDI dimers, V-TDI2 and V-FDI2 fused with thiophene and furan at the inside-bay position and V-PDIS2 and V-PDISe2 fused with the S atom and the Se atom with a larger radius at the outside-bay position, utilizing fusion-free V-PDI2 as the reference. It was exhibited that V-TDI2 and V-FDI2 afforded a planar molecular structure and raised the ELUMO, giving a higher VOC of 0.97 and 1.00 V, while V-PDIS2 and V-PDISe2 adopted the twisted molecular configuration with a dihedral angle of 81–82°, delivering the larger JSC of 9.41, 10.00 mA cm−2 and a lower ELUMO. Accordingly, the external-annulation PBDB-T:V-PDIS2- and PBDB-T:V-PDISe2-based devices got the winning PCE of 5.76% and 6.51%.40 Meanwhile, Bo et al. synthesized the two Se-annulation PDI acceptors VDP-Se and NDP-Se at the outside-bay position and found that VDP-Se had a slightly twisted configuration (11.7°), while NDP-Se exhibited a distorted naphthalene core and X-shaped skeleton with a dihedral angle of 34.9°. Two planar PDI arms not only guaranteed enough solubility but also suppressed the oversized aggregation. Finally, the PBDB-T:NDP-Se-based device yielded the 23.29% increased PCE of 7.41%.41 It should be noted that constructing the asymmetric molecules was also a powerful method for developing efficient PV materials due to the higher dipole moment and larger intermolecular binding energy than their symmetric counterparts.42,43 In 2018, the asymmetric linker 6-(thien-2-yl)-benzo[b]thiophene (T-BTh) and the two dimers unfused A101 and fused A102 were developed. The increased effective π-conjugation and decreased torsion angle produced a favourable morphology and enhanced π–π stacking, resulting in better charge mobility. Thus, a 57.82% increased PCE of 5.65% was achieved in the PBDB-T:A102-based device.44 Meanwhile, Zhang et al. developed two Se-annulation PDI dimers FPDI-Se with one selenophene heterocycle and FPDI-2Se with two selenophene heterocycles and found that the facilitated molecular stacking and intermolecular charge transport made the asymmetric PTB7-Th:FPDI-Se-based device obtain the 48.54% elevated PCE of 6.61%.45 Assisted by Se-annulation and fused by a thiophene at the outside-bay position, three asymmetric fused dual-PDI acceptors with the dovetail shape tridecane-7-yl side chain, termed as PDI2-Se fused with the Se atom, PDI2-FT fused with the thiophene ring and PDI2Se-FT both fused with Se atom and thiophene were developed by Xia's group, exhibiting the X-shaped configuration with similar dihedral angles of 24.2°–25.2° and close ELUMO of −3.76 to −3.71 eV. PDI2Se-FT afforded the highest PCE of 6.96% when paired with PBDB-T.46 However, the correlation between the molecular structure and PV performance during these hetero-atom annulation PDI dimers is still unclear. It is not definite that the twisted molecular structure was most favourable for enhancing the device performance. Accordingly, it was of significant importance and highly urgent to explore the new asymmetric PDI dimers and further elaborate the interplay of structure–property–performance.
In consideration of the abovementioned merits of S/Se-annulation and the advantages of asymmetric PDI acceptors, herein, two asymmetric S/Se-annulation PDI dimeric electron acceptors, named PDI-TPDI and PDI-SePDI (Chart 1), were synthesized via the tetrakis(triphenylphosphine)platinum(0)-catalyzed Suzuki coupling reaction. The influence of the different annulation types on the molecular twisting configuration, thermo-/photo-stability, absorption, energy level, molecular stacking and aggregation, charge transfer, miscibility and morphology of the blend film together with the PV performance were systematically investigated. Both asymmetric acceptors possessed excellent thermo-/photo-stability and a similar dihedral angle between two PDI sub-planes of approximately 67°. Furthermore, replacing S-annulation with Se-annulation weakened the molecular aggregation, making the PTB7-Th:PDI-SePDI-based device achieve a PCE up to 5.31%.
 | ||
| Chart 1 Chemical structure of the studied asymmetric S/Se-annulated dimers PDI-TPDI and PDI-SePDI and donor PTB7-Th in this study. | ||
Results and discussion
Synthesis and characterization
As elucidated in Scheme 1, the monobromides TPDIBr and SePDIBr were synthesized via two synthetic steps including S/Se-annulation and mono-bromination reactions. The starting material mononitrated PDI-HD-NO2 was synthesized according to a reported method.21 Annulation with S or Se powder in the solvent of N-methylpyrrolidone (NMP) at a high temperature of 190 °C produced the fused PDI moieties TPDI and SePDI with yields of 67% and 65%, respectively. Next, monobrominating TPDI and SePDI with liquid bromine (Br2) utilizing ferric chloride (FeCl3) as a catalyst gave rise to monobromide TPDIBr and SePDIBr in yields of 63% and 61%, respectively. Meanwhile, the PDI-HDBr reacted with bis(pinacolato)diboron, utilizing the Pd(dppf)Cl2 as a catalyst in the presence of K2HPO4, forming the monoborate ester PDI-Bpin in a yield of 42%.47 The targeted asymmetric S/Se-annulation PDI dimeric acceptors PDI-TPDI and PDI-SePDI were prepared via a classical Suzuki coupling reaction with yields of 25% and 23% (Table S1, ESI†), respectively.48 Chemical structures of the S-annulation intermediates TPDI and Se-annulation one SePDI, mono-bromides TPDIBr and SePDIBr, monoboric ester PDI-Bpin, and targeted asymmetric dimeric acceptors PDI-TPDI and PDI-SePDI were determined by 1H NMR (Fig. S1–S6 and S8, ESI†). It was noted that, S-annulation made the chemical shifts of the aromatic hydrogen increase during the 1H NMR spectra of TPDI and TPDIBr (Fig. S1 and S2, ESI†) compared with those of the unfused PDI-HD and its monobromide PDI-HDBr.49 Furthermore, the corresponding chemical shifts of the aromatic hydrogens of SePDI and mono-brominated SePDIBr were slightly increased (Fig. S3 and S4, ESI†). The reason was likely due to an enhanced deshielding effect (triggered by the anisotropy of the enlarged conjugated aromatic rings) as a result of introducing the Se atom with its bigger and looser outermost electron cloud compared to that of the S atom, and the latter DFT calculation excluded the steric effect. For S-annulation PDI-TPDI, chemical shifts ranging from 9.52 to 7.84 ppm belonged to the hydrogens of unfused PDI and S-annulated TPDI aromatic rings, one in the range of 4.13–3.98 ppm was attributed to hydrogens of methylene directly linked to imide N, one ranging from 1.98 to 1.87 ppm was ascribed to the hydrogens of methane, and the remainder were assigned to hydrogens of –CH2– and –CH3 groups (Fig. S6, ESI†). Because of the stronger deshielding effect of the newly formed selenophene ring, the chemical shifts were all slightly increased and moved forward the low field (Fig. S8, ESI†). The structures of the designed asymmetric dimeric acceptors S-annulation PDI-TPDI and Se-annulation PDI-SePDI were further characterized by the 13C NMR spectra (Fig. S7 and S9, ESI†). | ||
| Scheme 1 Synthesis of monobromides TPDIBr and SePDIBr, monoboric ester PDI-Bpin and asymmetric PDI dimeric acceptors PDI-TPDI and PDI-SePDI. | ||
Both asymmetric acceptors exhibited excellent solubility in chlorobenzene (CB) and chloroform (CF) solvents. As shown in Fig. 1a, the thermal decomposition temperatures (Td, 5% weight-loss) were observed to be 426 °C for PDI-TPDI and 409 °C for PDI-SePDI, determined by thermogravimetric analysis (TGA) in the nitrogen atmosphere. The observed 17 °C decreased Td could be interpreted by the weakened solid molecular stacking interaction found in the later XRD measurement. Overall, the exhibited Td higher than 400 °C indicated that both asymmetric PDI-TPDI and PDI-SePDI possessed excellent thermo-stability during the fabrication procedure of OSCs.
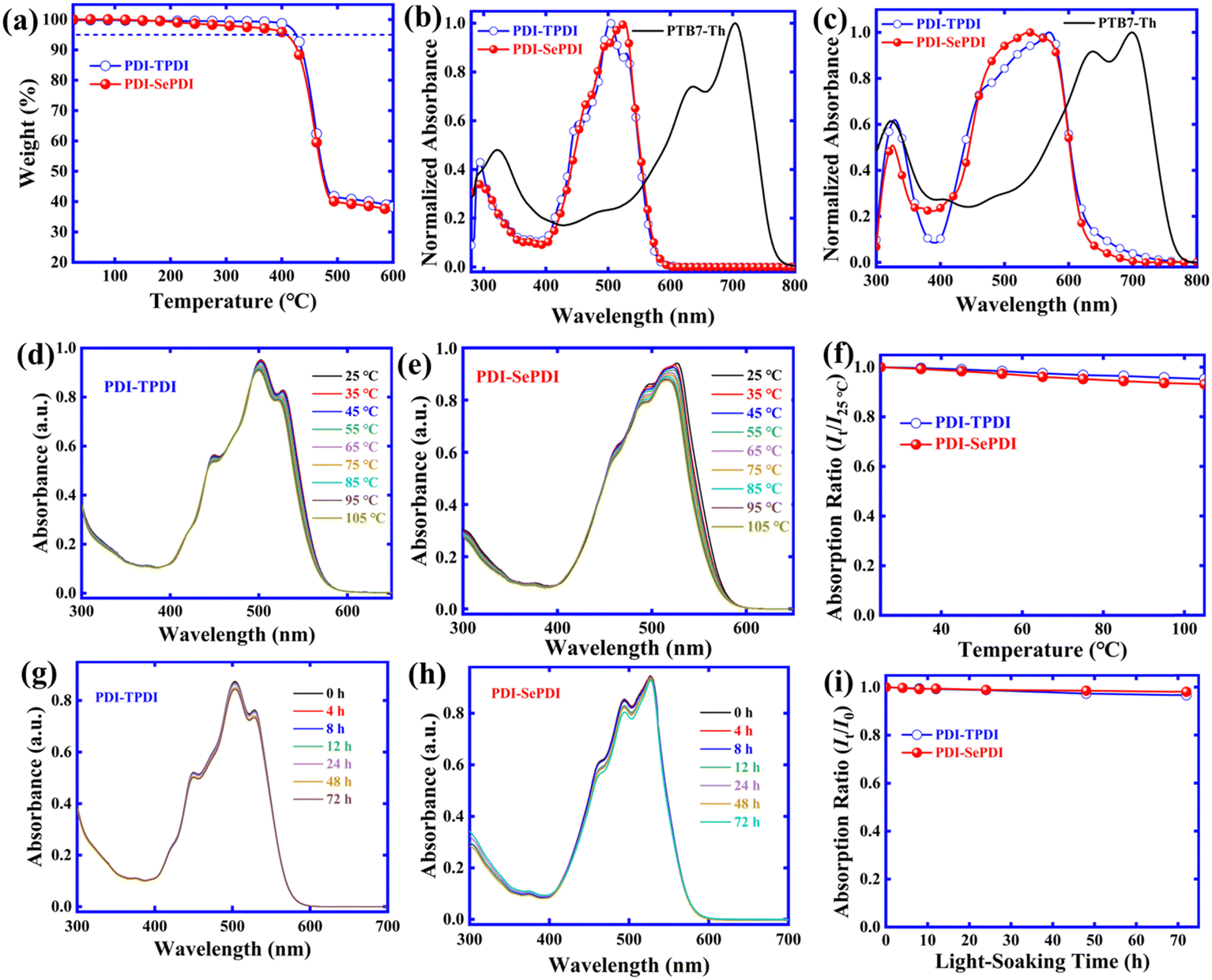 | ||
| Fig. 1 TG curve for asymmetric acceptors PDI-TPDI and PDI-SePDI (a), absorption in dilute CH2Cl2 (b) and as neat films (c), temperature-dependent absorption for PDI-TPDI (d) and PDI-SePDI (e) as well as the curve of maximum absorption intensity relative to temperature (f), changes for UV-vis absorption for PDI-TPDI (g) and PDI-SePDI (h) in chlorobenzene after illumination, and absorption intensity variation trend relative to light-soaking time (i). | ||
Optical properties, photostability, aggregation and energy levels
The normalized UV-Vis absorption spectra for PDI-TPDI and PDI-SePDI both in diluted CH2Cl2 solution and film were implemented in order to investigate the light absorption properties during these asymmetric NFEAs. The obtained results are shown in Fig. 1b, c and Table 1. Two asymmetric acceptors PDI-TPDI and PDI-SePDI possessed a wide absorption in the wavelength range of 400–600 nm, which was similar to the absorption profile with the symmetric analogues di-PBI36 and SdiPBI-S.39 As for S-annulated PDI-TPDI, absorption peaks at 293 and 503 nm together with one high energy shoulder peak at 448 nm and one low energy shoulder peak at 529 nm in diluted CH2Cl2 solution were found, whilst absorption peaks at 288 and 526 nm and two high-energy shoulder peaks at 464 and 498 nm in PDI-SePDI were found. The chlorobenzene solution state molar extinction coefficient (ε) for these studied asymmetric acceptors was further measured, as exhibited in Fig. S10 (ESI†). It was elucidated that the values of ε were in turn calculated to be 6.71 × 104 M−1 cm−1 at 503 nm for PDI-TPDI and 5.52 × 104 M−1 cm−1 at 526 nm for PDI-SePDI (Table S1, ESI†), indicating a slightly decreased ε in Se-annulation PDI-SePDI. What is more, moving from solution to film, the large red-shift of approximately 36 nm from 293 nm to 329 nm and 67 nm from 503 nm to 570 nm together with a high-energy shoulder peak at 471 nm were found in S-annulation PDI-TPDI, while after applying the larger atomic radius Se-annulation PDI-SePDI, the red-shifted value of 37 nm from 288 nm to 325 nm and 12 nm from 526 nm to 538 nm as well as the low-energy shoulder peak at 570 nm were found. The close red-shift value of high energy peak but significantly small red-shift value of the low energy peak indicated that the Se-annulation PDI-SePDI possessed the weak aggregation.50 Interestingly, the PDI-SePDI had a stronger absorption ranging from 450–580 nm than that of PDI-TPDI. Moreover, PDI-TPDI- and PDI-SePDI-based films exhibited absorption band edges (λfilmonset) of 633 and 625 nm, corresponding to the optical bandgaps (Eoptg) of 1.96 and 1.98 eV, respectively, which were slightly smaller than their symmetric analogues.36,39| Material | Solution | Film |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
φ onsetox (V) | φ onsetred (V) | E HOMO (eV) | E LUMO (eV) |
E
ecg
(eV) |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) | λ sh (nm) | λ max (nm) | λ sh (nm) | λ onset (nm) | |||||||
| a Estimated from the absorption onset of the asymmetric acceptors in the film (Eoptg = 1240/λfilmonset). b Obtained from the oxidation potential for asymmetric acceptors (EHOMO = −e(φonsetox − φFcox + 4.80) (eV)). c Calculated from the reduction potential for asymmetric acceptors (ELUMO = −e(φonsetred − φFcox + 4.80) (eV)). d Calculated from Eecg = −(EHOMO − ELUMO) (eV). | |||||||||||
| PDI-TPDI | 293, 503 | 448, 529 | 329, 570 | 471 | 633 | 1.96 | 1.68 | –0.86 | –6.38 | –3.84 | 2.54 |
| PDI-SePDI | 288, 526 | 464, 498 | 325, 538 | 570 | 625 | 1.98 | 1.58 | –0.81 | –6.28 | –3.89 | 2.39 |
| PTB7-Th | 321, 705 | 636 | 323, 698 | 636 | 761 | 1.63 | 0.53 | –1.13 | –5.23 | –3.57 | 1.66 |
The fluorescence spectra of the two asymmetric acceptors PDI-TPDI and PDI-SePDI in diluted dichloromethane (DCM) solution and film states were also measured in Fig. S11 (ESI†). The excitation wavelength (λex) was situated at 503 nm in DCM and 480 nm in the film for PDI-TPDI, whereas the λex was placed at 480 nm in DCM and 538 nm in film for PDI-SePDI. The S-annulated PDI-TPDI exhibited the emission peak at 584 nm in DCM solution and 628 nm in film, in contrast, Se-annulated PDI-SePDI showed the distinctive dual peaks at 532 nm and 575 nm as well as 634 nm in film (Table S1, ESI†).
The solution aggregation behaviour was estimated by applying the temperature-dependent absorption (TD-Abs)45,51 and the related results are shown in Fig. 1. We can see that when the temperature is heated from 25 °C to 105 °C with 10 °C as an interval in CB solution, a 3-nm blue-shift from 503 nm to 500 nm and a 4.64% decreased absorption intensity from 0.9528 to 0.9086 of the maximum peak, as well as a 7-nm blue-shift from 527 nm to 520 nm and 5.07% decreased intensity from 0.8287 to 0.7867 of the low-energy shoulder peak are observed in PDI-TPDI (Fig. 1d). Meanwhile, the 11-nm blue-shift from 526 nm to 515 nm and 7.07% decreased absorption intensity from 0.9405 to 0.8740 of the maximum peak, as well as the 5-nm blue-shift from 498 nm to 493 nm and 8.52% decreased intensity from 0.8620 to 0.7886 of the high-energy shoulder peak are observed in PDI-SePDI (Fig. 1e). The observed larger blue-shifted value for the maximum absorption peak and more significant decreased value of intensity suggested that Se-annulated PDI-SePDI possessed relatively weaker solution aggregation (Fig. 1f).22,52
The photostability of these resultant asymmetric small molecular acceptors PDI-TPDI and PDI-SePDI and the influence of the different annulation method at the outer bay positions were evaluated in Fig. 1. As for PDI-TPDI, the maximum peak slightly changed and the intensity was decreased by 96.51%, and the bilateral shoulder peaks exhibited 96.26% and 96.16% decreased intensity, while PDI-SePDI exhibited a relatively smaller intensity decrease (98.31%) and the shoulder peaks exhibited 92.16% and 94.76% decreased intensity, after exposing the light for 72 h in CB solution. These observations demonstrated that these asymmetric acceptors PDI-TPDI and PDI-SePDI both had high photostability in solution state (Fig. 1i). This attractive photostability was also observed in these PDI dimeric molecules 2T-(PDI-HD)2 with bithiophene (2T) as linker and F2T-(PDI-HD)2 with fluorinated 2T (F2T) as linker,41 and outer bay N-annulated dimers with high charge mobility TVT and TYT as linkers,21 as well as A–A type PDI-containing polymer acceptors (PAs).23
The molecular stacking interaction and solid aggregation as well as the influence of different annulation types were estimated by the X-ray diffraction (XRD) analyses, as indicated in Fig. S12 (ESI†). The PDI-TPDI-based film exhibited a sharp and strong peak at 2θ = 3.99°, while the PDI-SePDI-based film exhibited a very weak peak at 2θ = 3.77°, corresponding to the inter-chain stacking distance of 22.12 Å and 23.41 Å, respectively. Unfortunately, no obvious diffraction peaks in the large angle region were found in the resultant asymmetric PDI small molecular acceptors.53 The enlarged inter-chain stacking distance in Se-annulated PDI-SePDI suggested the presence of degraded solid stacking interactions, which is in agreement with the smaller red-shift value from solution to film state and relatively weaker solution aggregation from TD-Abs analysis.
Electrochemical properties
To know the energy level information of the asymmetric dimeric acceptors and the impact of different annulation methods, the cyclic voltammetry (CV) method was applied to measure the electrochemical properties of these resulting asymmetric PDI acceptors. Note that the redox couple (φ1/2) for Fc/Fc+ was 0.10 V relative to the Ag/AgNO3 reference electrode.54 As can be seen from Fig. 2a and Table 1, the oxidation onset potential (φonsetox) and reduction onset potential (φonsetred) in the film state were separately found to be 1.68 V and −0.86 V for S-annulated PDI-TPDI, together with 1.58 V and −0.81 V for Se-annulated PDI-SePDI. Thereupon, the corresponding EHOMO, ELUMO and electrochemical band gap (Eecg) were in sequence estimated to be −6.38, −3.84 and 2.54 eV for S-annulated PDI-TPDI and −6.28, −3.89 and 2.39 eV for Se-annulated PDI-SePDI.51 The LUMO values of these asymmetric PDI-TPDI and PDI-SePDI were close to those of the symmetric analogues (−3.85 eV for SdiPBI-S and −3.87 eV SdiPBI-Se).16,39 Clearly, after applying Se-annulation, the 0.05 eV down-shifted ELUMO from −3.84 to −3.89 eV was observed. Furthermore, we found that there existed the 0.27–0.32 eV LUMO energy offset between PTB7-Th and these studied asymmetric acceptors PDI-TPDI and PDI-SePDI, which could guarantee the high-performance exciton dissociation.41,55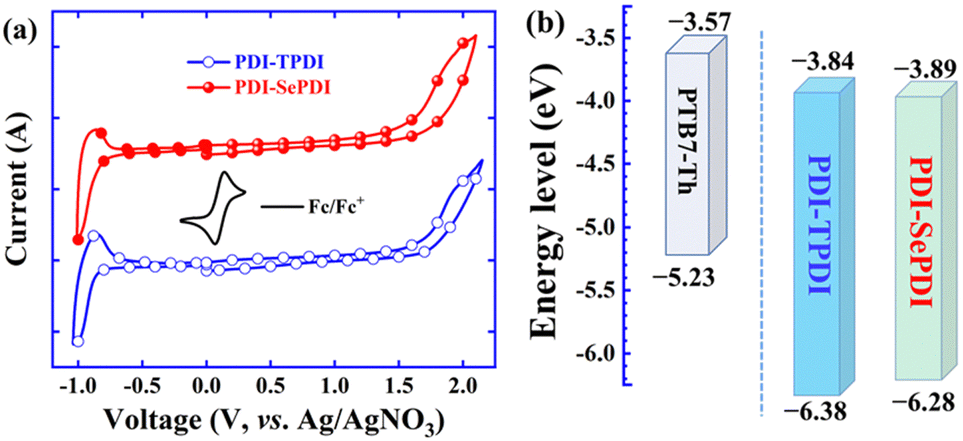 | ||
| Fig. 2 CV curves of asymmetric acceptors PDI-TPDI and PDI-SePDI (a) and energy level alignment for the used donor and acceptors (b). | ||
Theoretical calculations
In order to clarify the internal configuration and the influence of modulation of different annulation on the molecular twisting degree, theoretical calculations were carried out using density-functional theory (DFT) at the B3LYP/6-311G(d,p) level (Gaussian 09).56 For predigesting the calculations and saving time, considering that the flexible 2-hexyldecyl (HD) side chain played a negligible effect on electron distribution and energy level, the HD group of these resulting asymmetric acceptors PDI-TPDI and PDI-SePDI at the imide position was replaced with the iso-butyl (iBu) group. As presented in Fig. 3, during these PDI-TPDI and PDI-SePDI model molecules, the electron clouds of HOMO and LUMO orbitals were distributed across the whole aromatic skeleton, leading to the large band gap of 2.40–2.42 eV. Also, the EHOMO and ELUMO values were −6.30 eV and −3.88 eV for PDI-TPDI, respectively, after Se-annulation, the corresponding values were in turn moved to −6.27 eV and −3.87 eV. After replacing S-annulation with Se-annulation, the EHOMO was increased by 0.30 eV, the ELUMO was up-shifted by 0.01 eV, and the bandgap was reduced by 0.02 eV, which was similar to the results obtained in the CV test. Interestingly, the dihedral angle between the PDI ring and the heteroatom S/Se-annulation aromatic ring were in turn calculated to be 67.23° for PDI-TPDI and 67.06° for PDI-SePDI (Fig. 3). Moreover, the values of the torsion angle (θ) between the PDI ring and S/Se-annulated PDI rings were found to be 107.98° for PDI-TPDI and 108.19° for PDI-SePDI (Table S2, ESI†). In addition, the dihedral angle between the PDI ring and heterocyclic ring during the heteroatom-annulated PDI subunit was found to be 1.59° for PDI-TPDI and 1.57° for PDI-SePDI, respectively (Fig. S13, ESI†), indicating the presence of similar steric hindrance. These close dihedral angles and torsion angles suggested the different annulation methods played a negligible effect on the molecular twisting degree. The dipole moment of these asymmetric acceptors was found to be 0.8891 Debye for PDI-TPDI and 0.3827 Debye for PDI-SePDI, respectively.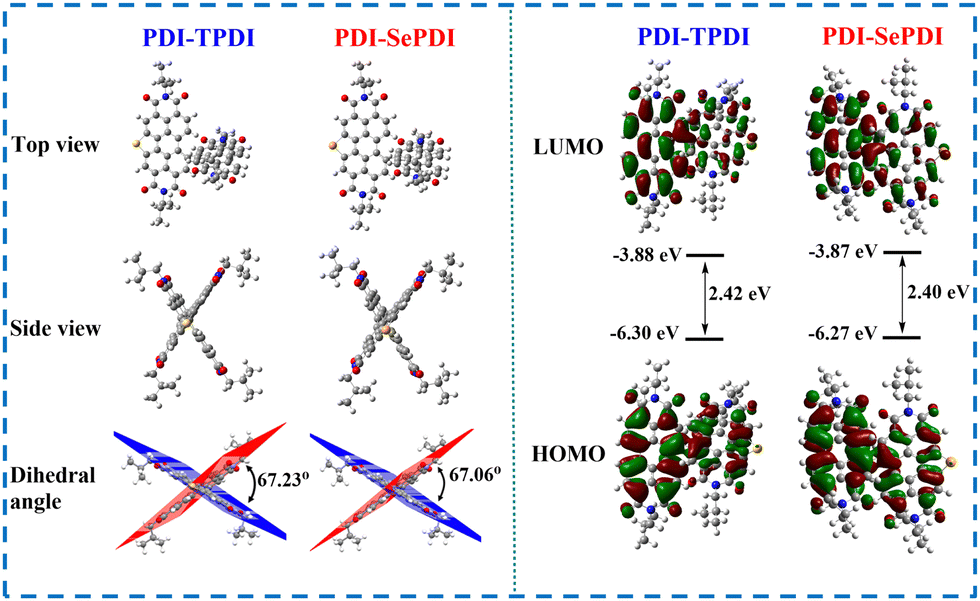 | ||
| Fig. 3 Top-/side-view of the optimized molecular structures, HOMO/LUMO orbital distributions and the calculated energy levels for model compounds PDI-TPDI and PDI-SePDI. | ||
To gain further insight into the impact of different annulation types on the molecular surface electrostatic potential (ESP) and ESP area distributions, the corresponding theoretical calculations were performed.57,58 As can be seen from Fig. S14 (ESI†), the negative region was principally distributed on the oxygen of the imide from fused TPDI/SePDI and unfused PDI aromatic rings. After replacing S-annulation with Se-annulation, the molecular polarity index (MPI) was decreased from 8.96 kcal mol−1 to 8.90 kcal mol−1, while the overall average ESP was also descended from 2.85 kcal mol−1 to 2.71 kcal mol−1. The difference of the overall average ESP between donor and acceptor was reduced from 4.46 kcal mol−1 of the (TB7-Th)2:PDI-TPDI system to 4.32 kcal mol−1 of the (TB7-Th)2:PDI-SePDI system (Table S3, ESI†), indicating the presence of reduced intermolecular interaction.59 Fig. S14b (ESI†) exhibited that PDI-TPDI possessed a wider ESP distribution range. The Se-annulation made the overall surface area increase from 927.35 Å2 to 931.09 Å2, which benefited from annulation of the larger Se atom in PDI-SePDI.
Photovoltaic properties
In order to understand the solar cell performance of these studied asymmetric acceptors and clarify the influence of different annulation methods in PDI-TPDI and PDI-SePDI, devices adopting the traditional structure (ITO/PEDOT:PSS/PTB7-Th:asymmetric acceptors/PDINO/Al) were fabricated. The detailed testing process embodied screening the weight ratio of D/A, utilizing the high boiling point 1,8-diiodooctane (DIO) solvent additive and thermal annealing (TA) at 110 °C for 10 min and so on. As for S-annulation PDI-TPDI, the D:A ratios (from 1:1.2 to 1:1 then to 1.2:1) were tested, and it was found that the best weight ratio was 1:1. Meanwhile, for the Se-annulation PDI-SePDI, the weight ratio was varied from 1:1.5 to 1:1.8 then to 1:2, and the optimal weight ratio was 1:1.8 (Fig. S15 and Table S4, ESI†). Under the conditions of each best D:A ratio, PDI-TPDI exhibited the VOC of 0.72 V, a JSC of 7.02 mA cm−2 and a high FF of 61.34%, leading to the PCE of 3.10%. After utilizing Se-annulation, the PDI-SePDI-based device afforded the VOC of 0.71 V, the 52.85% increased JSC of 10.73 mA cm−2, and the decreased FF of 45.83%, resulting in the 12.90% enhanced PCE of 3.50%. Since the DIO could tune the morphology of the photoactive layer in the OSCs and form the interpenetrating network BHJ structure with nano-sized phase separation, 3% DIO was applied aiming to boost the PV performance.23,51 After the 3% DIO was applied, the PDI-TPDI-based device afforded the unchanged VOC of 0.72 V, the 39.17% decreased JSC of 4.27 mA cm−2, and the 24.73% down-shifted FF of 46.17%, leading to the 53.87% dropped PCE of 1.43%. Meanwhile, the PDI-SePDI-based device yielded the slightly decreased VOC of 0.70 V, 51.54% reduced JSC of 5.20 mA cm−2, and 23.78% upshifted FF of 56.73%, producing the 40.86% dropped PCE of 2.07% (Fig. S16 and Table S4, ESI†). Unfortunately, the DIO solvent additive had a negative impact on solar cell performance and never elevated the photovoltaic performance. Considering the optimization of the morphology and thus promotion of the PV performance of thermal annealing, the TA at 110 °C for about 10 min was further applied.21,60 When using TA treatment, the S-annulation PDI-TPDI-based device gained the stable VOC of 0.72 V, the 33.76% increased JSC of 9.39 mA cm−2, and the 18.31% decreased FF of 50.11%, contributing to the 9.4% elevated PCE of 3.39%. In the meantime, the Se-annulation PDI-SePDI gained the slightly decreased VOC of 0.70 V, the 36.44% elevated JSC up to 14.64 mA cm−2, and the 13.11% enhanced FF of 51.84%, giving rise to the 51.71% up-shifted PCE as high as 5.31% (Fig. S16 and Table S4, ESI†).
Going through a series of device optimizations, the best current density relative to voltage curves and corresponding EQE spectra are exhibited in Fig. 4. It was found that, under optimal conditions, S-annulation PDI-TPDI-based device exhibited a VOC of 0.72 V, a JSC of 9.39 mA cm−2, an FF of 50.11% and thus a PCE of 3.39%, after Se-annulation, PDI-SePDI achieved the slightly decreased VOC of 0.70 V, the 55.91% increased JSC up to 14.64 mA cm−2, the 3.45% enhanced FF of 51.84% and thus the 56.64% elevated PCE as high as 5.31%. The marked increase in JSC was verified by the enhancement of EQE ranging from 400 nm to 750 nm (Fig. 7b) and slightly enhanced FF was explained by the downgraded series resistance (RS) from 11.14 Ω m2 to 6.47 Ω m2 (Table 2). Additionally, based on the EQE spectra, the integrated JSC was in sequence calculated to be 9.30 and 14.52 mA cm−2, exhibiting a tolerable error of less than 5%, indicative of the high creditability of photocurrent measured from the J–V test. Additionally, the corresponding device stability was estimated in Fig. S17 (ESI†).23 When the light exposure lasted for 72 h, the devices parameters VOC, JSC, FF and PCE remained approximately 98%, 88%, 91% and 79% of their initial values, suggesting that these asymmetric PDI dimers exhibited relatively higher stability.
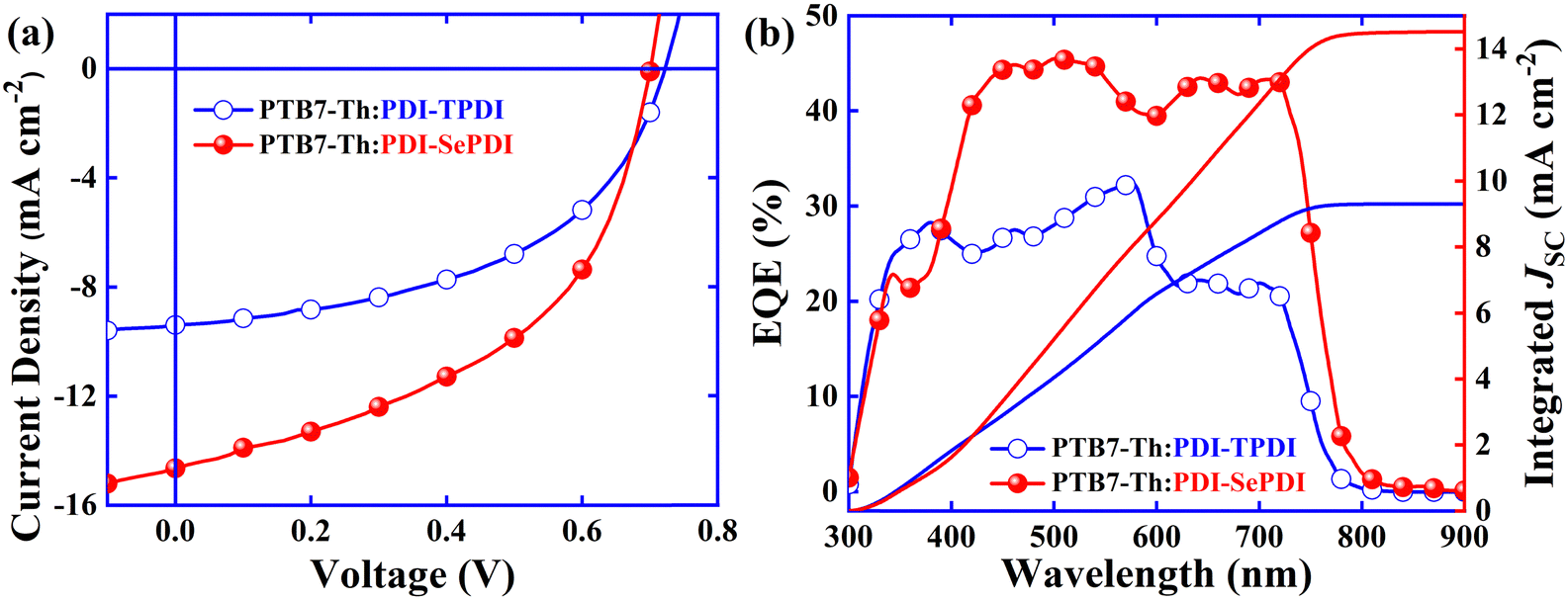 | ||
| Fig. 4 Best J–V (a) and EQE (b) curves for PDI-TPDI and PDI-SePDI-based devices. | ||
| Acceptor | Conditions | V OC (V) | J SC (mA cm−2) | FF (%) | PCEb (%) | R SH (Ωm2) | R S (Ωm2) |
|---|---|---|---|---|---|---|---|
| a Integrated JSC calculated from EQE. b Statistics in parentheses from 10 independent cells. | |||||||
| PDI-TPDI | 1:1/TA |
0.72 ± 0.01 | 9.39 ± 0.08 (9.30) | 50.11 ± 0.43 | 3.39 ± 0.13 | 435.97 | 11.14 |
| PDI-SePDI | 1:1.8/TA |
0.70 ± 0.01 | 14.64 ± 0.22 (14.52) | 51.84 ± 0.38 | 5.31 ± 0.24 | 137.02 | 6.47 |
Exciton dissociation, recombination and charge transport properties
To deeply understand the reason for the Se-annulated PDI-SePDI-based device obtaining the higher JSC and determine the impact of different annulation methods on dissociation and collection procedure of excitons, photocurrent (Jph) versus effective voltage (Veff) was measured in Fig. 5a and b. The Jph reached the saturated value (Jsat) of 9.32 mA cm−2 for PDI-TPDI and 14.56 mA cm−2 for PDI-SePDI when Veff was larger than 2.50 V. The exciton dissociation probability (ηdiss) was calculated to be 81.9% for PDI-TPDI and 89.7% for PDI-SePDI, meanwhile, the charge collection probability (ηcoll) was in turn found to be 71.4% for PDI-TPDI and 70.6% for PDI-SePDI (Table 3). Obviously, the increased ηdiss and close ηcoll could partially account for the 55.91% increased JSC and slightly elevated FF.61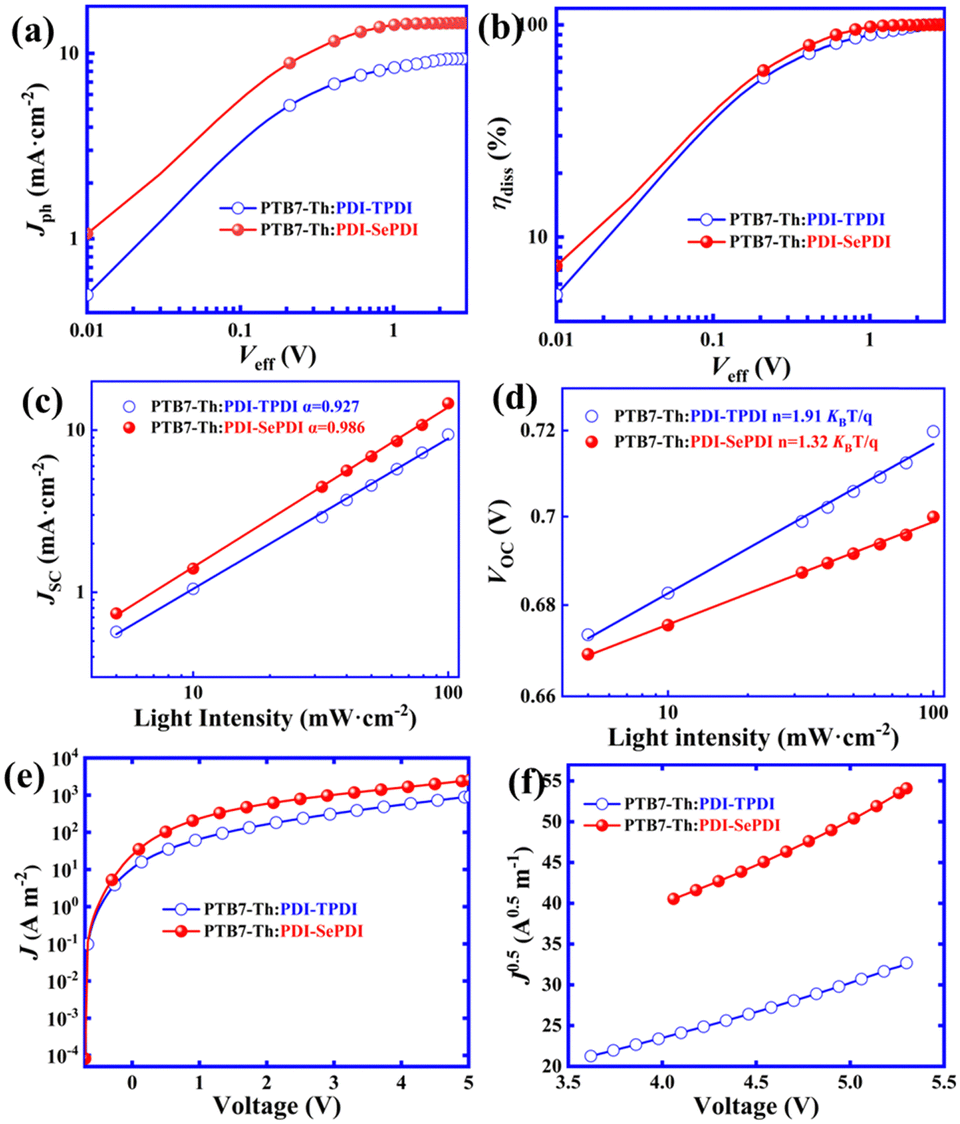 | ||
| Fig. 5 Plots of Jphvs. effective voltage (Veff) (a), ηdissvs. Veff (b), JSCvs. light intensity (c), VOCvs. light intensity (d), J–V curve (e) and fitting J0.5–V curve (f) for PDI-TPDI and PDI-SePDI-based devices. | ||
| Acceptor | J sat (mA cm−2) |
J
ph
s
(mA cm−2) |
J
ph
m
(mA cm−2) |
η diss (%) | η coll (%) | μ e (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| a Saturated photocurrent density. b Photocurrent density at short-circuit condition. c Photocurrent density at maximal power output. d η diss = Jphs/Jsat. e η coll = Jphm/Jsat. | ||||||
| PDI-TPDI | 9.32 | 7.63 | 6.65 | 81.9 | 71.4 | 1.24 × 10−5 |
| PDI-SePDI | 14.56 | 13.06 | 10.28 | 89.7 | 70.6 | 3.98 × 10−5 |
J
SC relative to light intensity (Plight) tests were conducted to investigate the biomolecular recombination behaviors of these resultant asymmetric acceptors (Fig. 5c and d). The JSC and Plight obeyed the power law equation of JSC ∝ (Plight)α, where the value of α (slope of the fitting line) approaching to 1 implied a low bimolecular recombination.23,61,62 The α values of 0.927 for PDI-TPDI and 0.986 for PDI-SePDI were observed in short-circuit conditions, which is indicative of the suppressed bimolecular recombination after Se-annulation (Fig. 5c). Furthermore, the monomolecular or trap-assisted recombination behavior was further evaluated via testing the variation of VOC with respect to Plight, as depicted in Fig. 5d. The VOC relative to Plight relationship was described as VOC ∝ (nKBT/q)ln(Plight), in which KB, T and q stand for Boltzmann constant, absolute temperature and elementary charge, respectively. Note that the slope of nKBT/q was equal to kBT/q, indicating that bimolecular recombination was dominant, while nKBT/q was close to 2kBT/q, which implied that monomolecular or trap-assisted recombination took place. It was exhibited that the n values were separately found to be 1.91 for PDI-TPDI and 1.32 for PDI-SePDI, suggesting that the suppressed monomolecular or trap-assisted recombination mechanism played a dominant role in the PDI-SePDI-based device.63 Apparently, the suppressed bimolecular- and monomolecular- and/or trap-assisted recombination could in part explain why the PDI-SePDI-based device acquired the increased JSC and thus enhanced PCE.
To find out the origin why PDI-SePDI-containing devices obtained a significantly enhanced JSC and slightly elevated FF and to determine the impact of different annulation methods at the bay position of PDI, the space-charge-limited-current (SCLC) electron mobility (μe) was measured applying the vertical electron-only devices in the structure of ITO/ZnO/PTB7-Th: PDI-TPDI or PDI-SePDI/PDINO/Al (Section 1.3 in ESI†).51,64 Note that the thickness values of these resultant asymmetric acceptors PDI-TPDI and PDI-SePDI based films were found to be 100 nm and 105 nm, respectively. Obviously, after utilizing the Se-annulation to replace S-annulation, the μe value was increased from 1.24 × 10−5 cm2 V−1 s−1 to 3.98 × 10−5 cm2 V−1 s−1 (Fig. 5e, f and Table 3). The 3.21-fold elevated electron mobility could in part explain the origin of the 55.91% elevated JSC and 3.45% elevated FF in the PDI-SePDI-based device.
Morphology characteristics of the active layers
To further understand the influence of different annulation methods on miscibility and interaction between PTB7-Th and PDI-TPDI/PDI-SePDI, we measured the corresponding contact angle (CA), and the related parameters including contact angles (θ), surface tension (γ), and surface energy parameters (χ), which are listed in Table S6 (ESI†). As can be seen from Fig. 6, the γ values of the used electron donor PTB7-Th, and the resultant asymmetric acceptors PDI-TPDI and PDI-SePDI in the pristine films were separately estimated to be 29.65, 20.53 and 23.47 mN m−1. Then, the Flory–Huggins interaction parameter χD–A was calculated from the formula,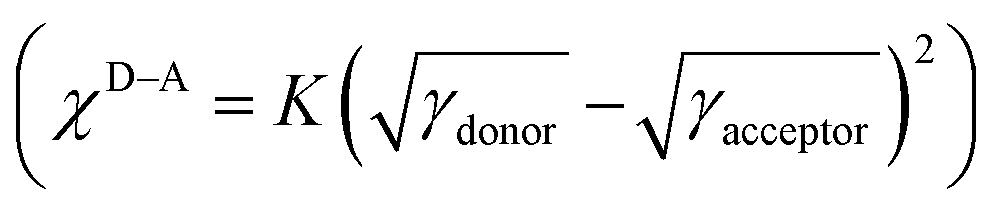 to be 0.863K for PTB7-Th:PDI-TPDI combination and 0.361K for PTB7-Th:PDI-SePDI system.49,65 Apparently, replacing S-annulation with Se-annulation in the PTB7-Th:PDI-SePDI system resulted in the decreased χD–A, suggesting that PDI-SePDI showed the enhanced miscibility with donor PTB7-Th, which was helpful to form the advantageous morphology. This correlated well with the following observed relatively smoother surface after Se-annulation.
to be 0.863K for PTB7-Th:PDI-TPDI combination and 0.361K for PTB7-Th:PDI-SePDI system.49,65 Apparently, replacing S-annulation with Se-annulation in the PTB7-Th:PDI-SePDI system resulted in the decreased χD–A, suggesting that PDI-SePDI showed the enhanced miscibility with donor PTB7-Th, which was helpful to form the advantageous morphology. This correlated well with the following observed relatively smoother surface after Se-annulation.
 | ||
| Fig. 6 Contact angle on PDI-TPDI, PDI-SePDI and PTB7-Th films. | ||
To deeply figure out the reason why the Se-annulation PDI-SePDI-based device obtained the significantly increased JSC and slightly enhanced FF and the influence of different annulation on surface morphology, the tapping-model AFM was applied, as depicted in Fig. 7a–d. After utilizing Se-annulation PDI-SePD instead of S-annulation PDI-TPDI, the values of the root-mean-square (RMS) was decreased from 2.452 nm to 2.293 nm. Furthermore, we further measured the scanning electron microscopy (SEM) images in Fig. 7e and f. A smoother surface was found in the Se-annulated PTB7-Th:PDI-SePDI-based blend film. The observed reduced surface roughness and flattened blend film was in accordance with the weakened aggregation both in the CB solution and film states and the improved miscibility with PTB7-Th obtained from the CA measurement. The improved morphology could in part account for the 3.21-times increased electron mobility, the 55.91% elevated photocurrent and thus the 56.64% enhanced photovoltaic efficiency.66,67 We also observed the presence of some bulges during these resultant asymmetric PDI-TPDI and PDI-SePDI-based photoactive layers which did not favour formation of the desired microstructural morphology and thus restricted the device performance. There was still some room for the morphology optimization of the photoactive layer, so more solvent additives and post-treatment could be adopted in future work.
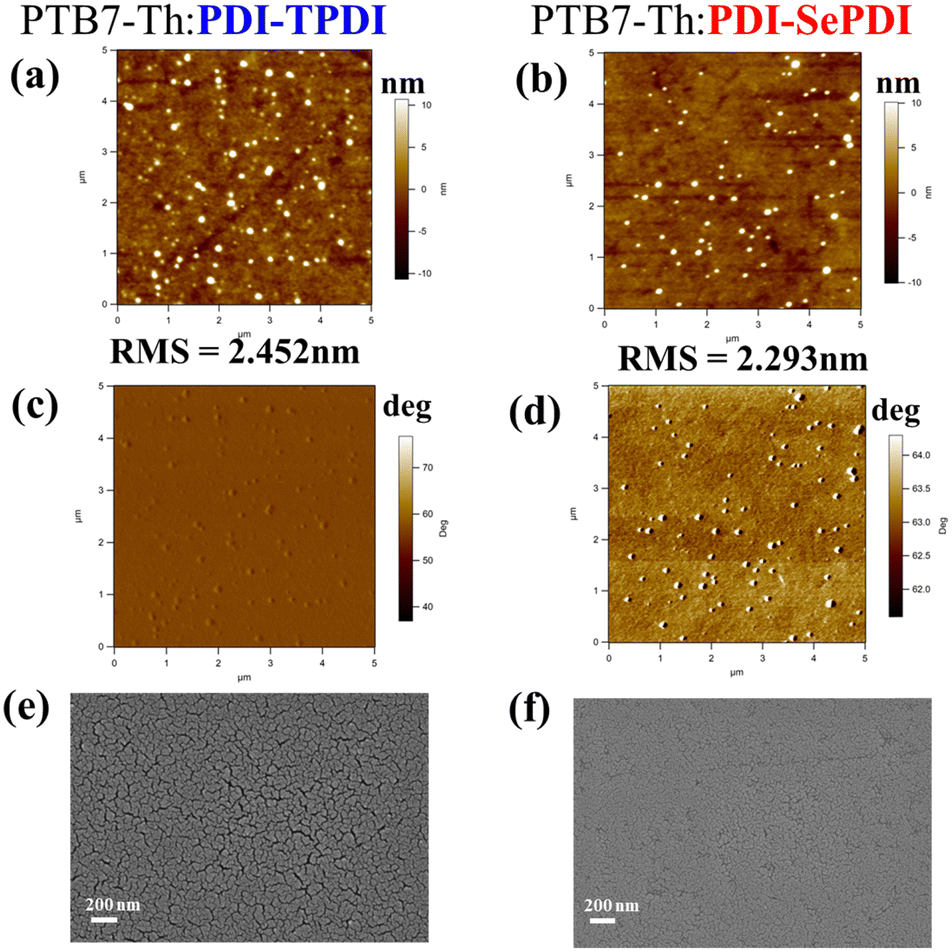 | ||
| Fig. 7 AFM height images with (a) and (b), phase images with (c) and (d) and SEM images (e) and (f) for PDI-TPDI- (a), (c) and (e) and PDI-SePDI-containing (b), (d) and (f) blend films. | ||
Conclusions
To sum up, two asymmetric PDI-based dimeric acceptors, named PDI-TPDI and PDI-SePDI, were developed by heteroatom S/Se-annulation at the outside position of a PDI aromatic ring. The resultant asymmetric acceptors exhibited outstanding thermo- and photo-stability as well as a similar dihedral angle of 67° between two PDI planes. Replacing S-annulation with Se-annulation led to a blue-shifted maximal absorption peak, weakened aggregation both in solution and solid film states, and deepening of the ELUMO energy level, as well as having an inconsequential effect on the twisting angle between the PDI and S/Se-annulation aromatic rings. Thereupon, the S-annulated PDI-TPDI-based device obtained the VOC of 0.72 V, JSC of 9.39 mA cm−2, and FF of 50.11%, giving rise to the PCE of 3.39%, while Se-annulated PDI-SePDI achieved the slightly decreased VOC of 0.70 V, the increased JSC of 14.64 mA cm−2 and FF of 51.84%, contributing to a 56.64% elevated PCE up to 5.31%. The increase of PCE in the PDI-SePDI-containing device principally benefited from the enhanced exciton dissociation, suppressed charge recombination, and increased charge mobility originating from the advantageous microstructural morphology generated by larger heteroatom Se-annulation. The structure–property–performance relationship study proved that Se-annulation of PDI at the outside position was an effective strategy for tuning the molecular aggregation, morphology and further boosting the solar cell performance.Experimental section
Materials
Chemical reagents used in this work were obtained from commercial companies. Starting material mononitrated PDI-HD-NO221 and monobromide PDI-HDBr49 were synthesized according to the corresponding method. The synthetic details for the used key monobromides TPDIBr, SePDIBr and monoborate ester PDI-Bpin can be found in the ESI.†:CH2Cl2 = 1:1) as the eluent to acquire 34 mg PDI-TPDI as a red solid (yield = 25%). M.p. > 280 °C. 1H NMR (500 MHz, CDCl3), δ (ppm): 9.51 (s, 1H, H of PDI), 9.43 (s, 1H, H of PDI), 8.89–8.83 (m, 2H, H of PDI), 8.76 (s, 2H, H of PDI), 8.59 (d, 3J = 8.5 Hz, 1H, H of PDI), 8.53 (s, 1H, H of PDI), 8.48 (s, 1H, H of PDI), 8.44 (d, 3J = 8.5 Hz, 1H, H of PDI), 8.12 (d, 3J = 8.5 Hz, 1H, H of PDI), 7.84 (d, 3J = 8.5 Hz, 1H, H of PDI), 4.13 (d, 3J = 6.5 Hz, 4H, >N–CH2–), 4.06 (d, 3J = 6.5 Hz, 2H, >N–CH2–), 3.98 (br, 2H, >N–CH2–), 1.98 (br, 2H, >CH–), 1.87 (br, 2H, >CH–), 1.42–1.15 (m, 96H, –CH2–), 0.83–0.76 (m, 24H, –CH3). 13C NMR (125 MHz, CDCl3), δ (ppm): 163.52, 163.31, 163.13, 142.28, 140.19, 138.93, 135.04, 134.12, 133.30, 131.92, 131.57, 131.12, 129.46, 129.04, 128.70, 128.48, 127.70, 127.29, 126.67, 125.78, 124.15, 123.43, 44.75, 31.86, 31.81, 31.57, 30.00, 29.66, 29.55, 29.26, 22.63, 14.06. Elemental anal. calcd for C112H144N4O8S (%): C, 78.83; H, 8.51; N, 3.28. Found (%), C, 78.76; H, 8.44; N, 3.39.
Author contributions
Junfeng Tong: conceptualization, investigation, writing – original draft. Jiayu Fang: writing & investigation. Lili An: investigation. Youzhi Huo: investigation. Fushui Di: investigation. Pengzhi Guo: calidation. Chunyan Yang: supervision. Zezhou Liang: supervision. Jianfeng Li: supervision. Yangjun Xia: review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Raw data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (62064006 and 52262026), Industrial support plan project of colleges and universities in Gansu Province (No.: 2021CYZC-19 and 2020C-07), West Light Foundation of the Chinese Academy of Sciences (No. 22JR9KA035), and the Natural Science Foundation of Gansu Province (23JRRA878).References
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef PubMed.
- G. Han and Y. Yi, Acc. Chem. Res., 2022, 55, 869–877 CrossRef CAS PubMed.
- Z. Luo, T. Liu, Z. Chen, Y. Xiao, G. Zhang, L. Huo, C. Zhong, X. Lu, H. Yan, Y. Sun and C. Yang, Adv. Sci., 2019, 6, 1802065 CrossRef PubMed.
- L. Ye, W. Ye and S. Zhang, J. Semicond., 2021, 42, 101607 CrossRef CAS.
- X. Wang, Z. Li, X. Zheng, C. Xiao, T. Hu, Y. Liao and R. Yang, Adv. Funct. Mater., 2023, 33, 2300323 CrossRef CAS.
- L. Zhang, Z. Chen, F. Sun, Y. Wang, H. Bao, X. Gao and Z. Liu, J. Electron. Mater., 2022, 51, 4224–4237 CrossRef CAS.
- D. Zhou, J. Wang, Z. Xu, H. Xu, J. Quan, J. Deng, Y. Li, Y. Tong, B. Hu and L. Chen, Nano Energy, 2022, 103, 107802 CrossRef CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, H. Xu, J. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- G. Zhang, J. Feng, X. Xu, W. Ma, Y. Li and Q. Peng, Adv. Funct. Mater., 2019, 9, 1906587 CrossRef.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef CAS PubMed.
- K. Bhardwaj, S. Naqvi, S. K. Saini, M. Kumar and R. Kumar, Sol. Energy, 2022, 246, 320–330 CrossRef CAS.
- P. Cheng, X. Zhao and X. Zhan, Acc. Mater. Res., 2022, 3, 309–318 CrossRef CAS.
- W. Liu, J. Fang, J. Tong, J. Qin, L. An, P. Guo, C. Yang, Z. Liang, J. Li and Y. Xia, Dyes Pigm., 2024, 224, 112038 CrossRef CAS.
- J. Tong, X. Jiang, Z. Dong, L. An, P. Guo, C. Yang, Z. Liang, J. Li and Y. Xia, Dyes Pigm., 2022, 208, 110816 CrossRef CAS.
- J. Tong, W. Liu, Y. Huang, Z. Li, Y. Wang, S. Bai, Z. Liang, L. Yan, J. Li and Y. Xia, J. Mater. Chem. C, 2023, 11, 6951–6962 RSC.
- M. Zhang, Y. Bai, C. Sun, L. Xue, H. Wang and Z.-G. Zhang, Sci. China: Chem., 2022, 65, 462–485 CrossRef CAS.
- R. Singh, E. Giussani, M. M. Mróz, F. D. Fonzo, D. Fazzi, J. Cabanillas-González, L. Oldridge, N. Vaenas, A. G. Kontos, P. Falaras, A. C. Grimsdale, J. Jacob, K. Müllen and P. E. Keivanidis, Org. Electron., 2014, 15, 1347–1361 CrossRef CAS.
- R. Singh, E. Aluicio-Sarduy, Z. Kan, T. Ye, R. C. I. MacKenzie and P. E. Keivanidis, J. Mater. Chem. A, 2014, 2, 14348–14353 RSC.
- J. Xiao, H.-J. Yu, D.-X. Xie, A. Shinohara, T. Fan, J. Yi, J. Y. L. Lai, G. Shao and H. Yan, Sustainable Energy Fuels, 2023, 7, 294–299 RSC.
- B. Carlotti, Z. Cai, H. Kim, V. Sharapov, I. K. Madu, D. Zhao, W. Chen, P. M. Zimmerman, L. Yu and T. Goodson, III, Chem. Mater., 2018, 30, 4263–4276 CrossRef CAS.
- P. Murugan, E. Ravindran, V. Sangeetha, S.-Y. Liu and J. W. Jung, J. Mater. Chem. A, 2023, 11, 26393–26425 RSC.
- S. Amiralaei, D. Uzun and H. Icil, Photochem. Photobiol. Sci., 2008, 7, 936–947 CrossRef CAS PubMed.
- K. Ding, T. Shan, J. Xu, M. Li, Y. Wang, Y. Zhang, Z. Xie, Z. Ma, F. Liu and H. Zhong, Chem. Commun., 2020, 56, 11433–11436 RSC.
- K. C. Song, R. Singh, J. Lee, D. H. Sin, H. Lee and K. Cho, J. Mater. Chem. C, 2016, 4, 10610–10615 RSC.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76 CrossRef CAS PubMed.
- S. Chen, D. Meng, J. Huang, N. Liang, Y. Li, F. Liu, H. Yan and Z. Wang, CCS Chem., 2021, 3, 78–84 CrossRef CAS.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K.-Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- C.-H. Wu, C.-C. Chueh, Y.-Y. Xi, H.-L. Zhong, G.-P. Gao, Z.-H. Wang, L. D. Pozzo, T.-C. Wen and A. K.-Y. Jen, Adv. Funct. Mater., 2015, 25, 5326–5332 CrossRef CAS.
- H. Wang, L. Chen and Y. Xiao, J. Mater. Chem. A, 2017, 5, 22288–22296 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- J. Yang, F. Chen, P. Cong, H. Xiao, Y. Geng, Z. Liao, L. Chen, B. Zhang and E. Zhou, J. Energy Chem., 2020, 40, 112–119 CrossRef.
- M. Li, H. Wang, Y. Liu, Y. Zhou, H. Lu, J. Song and Z. Bo, Dyes Pigm., 2020, 175, 108186 CrossRef CAS.
- H. Lu, H. Jin, H. Huang, W. Liu, Z. Tang, J. Zhang and Z. Bo, Adv. Funct. Mater., 2021, 31, 2103445 CrossRef CAS.
- C. Li, H. Fu, T. Xia and Y. Sun, Adv. Energy Mater., 2019, 9, 1900999 CrossRef.
- F. Chen, G. Ding, A. Tang, B. Xiao, J. Li and E. Zhou, J. Mater. Chem. C, 2018, 6, 2580–2587 RSC.
- Y. Yin, J. Song, F. Guo, Y. Sun, L. Zhao and Y. Zhang, ACS Appl. Energy Mater., 2018, 1, 6577–6585 CrossRef CAS.
- L. Wang, H. Shen, X. You, D. Wu and J. Xia, Eur. J. Org. Chem., 2022, e202101554 CrossRef CAS.
- G. Liu, T. Koch, Y. Li, N. L. Doltsinis and Z. Wang, Angew. Chem., 2019, 131, 184–189 CrossRef.
- S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- J. Tong, X. Jiang, H. Li, L. An, C. Yang, Y. Huang, P. Guo, Z. Liang, C. Yang, J. Li and Y. Xia, Opt. Mater., 2021, 121, 111593 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- J. Tong, J. Li, P. Zhang, X. Ma, M. Wang, L. An, J. Sun, P. Guo, C. Yang and Y. Xia, Polymer, 2017, 121, 183–195 CrossRef CAS.
- Z. Chen, W. Zhang, J. Huang, D. Gao, C. Wei, Z. Lin, L. Wang and G. Yu, Macromolecules, 2017, 50, 6098 CrossRef CAS.
- J. Tong, L. An, J. Li, J. Lv, P. Guo, L. Li, P. Zhang, C. Yang, Y. Xia and C. Wang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2059–2071 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mark, H. Bässles, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- H. Yao, D. Qian, H. Zhang, Y. Qin, B. Xu, Y. Cui, R. Yu, F. Gao and J. Hou, Chin. J. Chem., 2018, 36, 491–494 CrossRef CAS.
- Q. Xie, Y. Liu, X. Liao, Y. Cui, S. Huang, L. Hu, Q. He, L. Chen and Y. Chen, Macromol. Rapid Commun., 2020, 41, 2000454 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, Jr., C. S. Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- T. Ye, R. Singh, H.-J. Butt, G. Floudas and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2013, 5, 11844–11857 CrossRef CAS PubMed.
- C. Han, H. Gao, Y. Sun, Y. Kan, Z. Bi, W. Ma, Y. Zhang, J. A. Zapien, Y. Yang and K. Gao, J. Energy Chem., 2024, 93, 601–608 CrossRef CAS.
- Z. Liang, J. Tong, H. Li, Y. Wang, N. Wang, J. Li, C. Yang and Y. Xia, J. Mater. Chem. A, 2019, 7, 15841–15850 RSC.
- S. Rasool, Q. V. Hoang, D. V. Vu, C. E. Song, H. K. Lee, S. K. Lee, J.-C. Lee, S.-J. Moon and W. S. Shin, J. Energy Chem., 2022, 64, 236–245 CrossRef CAS.
- J. Li, Y. Wang, Z. Liang, N. Wang, J. Tong, C. Yang, X. Bao and Y. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 7022–7029 CrossRef CAS PubMed.
- C. Gu, Y. Zhao, X. Kang, X. Zhou, F. Bi, Y. Tian, Y. Li, J. Ma and X. Bao, Polymer, 2024, 299, 126926 CrossRef CAS.
- L. Ye, Y. Xie, K. Weng, H. S. Ryu, C. Li, Y. Cai, H. Fu, D. Wei, H. Y. Woo, S. Tan and Y. Sun, Nano Energy, 2019, 58, 220–226 CrossRef CAS.
- X. Liu, Z. Liang, S. Du, X. Niu, J. Tong, C. Yang, X. Lu, X. Bao, L. Yan, J. Li and Y. Xia, ACS Appl. Mater. Interfaces, 2022, 14, 9386–9397 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc01986e |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |