
Perovskite nanocrystals passivated by aromatic phosphonic acid for high-performance light-emitting diodes†
Muhammad Imran
Saleem‡
a,
Siwei
He‡
b,
Seung Hyun
Kim
ac,
Jae-Wook
Kang
 *b and
Jeong-Hwan
Lee
*ac
*b and
Jeong-Hwan
Lee
*ac
aDepartment of Materials Science and Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: Jeong-hwan.lee@inha.ac.kr
bDepartment of Flexible and Printable Electronics, LANL-JBNU Engineering Institute-Korea, Jeonbuk National University, Jeonju 54896, Republic of Korea. E-mail: jwkang@jbnu.ac.kr
cProgram in Semiconductor Convergence, Inha University, Incheon 22212, Republic of Korea
First published on 12th September 2024
Abstract
Optimizing the content of inherent capping ligands in perovskite nanocrystals (PeNCs) is a crucial strategy for enhancing the performance and stability of perovskite light-emitting diodes (PeLEDs). However, the highly dynamic adsorption–desorption state of capping ligands and their detachment from the surface of PeNCs often result in degraded optical properties and the emergence of surface defects. In this study, we investigated the use of benzylphosphonic acid (BPA), an aromatic conductive molecule, for passivation to replace the native capping ligands of FAPbBr3 NCs. Comprehensive characterization results validate that the original long insulative ligands were partially substituted with short conductive phosphine oxide functional groups, effectively passivating the nonradiative defects of FAPbBr3 while enhancing electron injection into perovskite emitting layers. Moreover, this proposed ligand-exchange strategy significantly improved the photoluminescence quantum yield of FAPbBr3 NCs. Consequently, we achieved a high-performance PeLED with an external quantum efficiency of 12.9% at 4660 cd m−2 using BPA-passivated FAPbBr3 NCs. These findings underscore the potential of post-passivated PeNCs via aromatic acid in fabricating high-performance optoelectronic devices.
1. Introduction
Perovskite nanocrystals (PeNCs) have emerged as a groundbreaking class of semiconducting materials for optoelectronic applications. Their narrow emission spectrum, characterized by a full-width at half maximum (FWHM) of less than 20 nm, facilitates superior color gamut coverage. Moreover, their high photoluminescence quantum yield (PLQY) and spectral adjustability render them highly promising for next-generation lighting and display applications.1–7 In recent years, significant improvements in the performance of perovskite light-emitting diodes (PeLEDs) have been noted, attributed to the exceptional characteristics of nanocrystals as emitters.8–10 Despite notable strides in the performance of NC-based PeLEDs, exposure of PeNCs to harsh environmental conditions, such as polar molecules, extended heat exposure, or intense light illumination, can detrimentally impact their optical properties. Importantly, the highly dynamic state of capping ligand adsorption–desorption, the high surface-to-volume ratio of PeNCs, and surface defects at grain boundaries in thin films collectively contribute to an adverse effect on halide ion migration.11 Furthermore, native ligands have a tendency to de-coordinate from the surface of perovskite NCs over time due to their highly ionic nature and intrinsic instability.12,13 This phenomenon leads to the degradation of optical properties and fosters the formation of surface defects, aggregation, and phase transitions.14–16 Consequently, halide/Pb ion vacancies and Pb ion gaps, acting as non-radiative recombination centers, diminish the luminescence efficiency and operational stability of LEDs by quenching photogenerated excitons and carriers.17–20Modulating optical properties and mitigating surface defects have proven effective in enhancing the operational efficiency and durability of NC-based PeLEDs. Over the years, significant efforts have been directed towards enhancing bimolecular radiative recombination rates by addressing the inherently weak exciton binding energy and thermal instability of PeNCs. Strategies such as Cs/Pb site doping12,21–24 and surface engineering25–28 have been extensively explored for this purpose.
Ligand engineering is also an essential strategy for firmly anchoring onto PeNCs to eliminate structural defects. This approach effectively counters the thermodynamic instability associated with native-capped ligands like oleic acid, octylamine, or oleylamine while boosting the colloidal stability of PeNCs and increasing their PLQYs.16,29 For example, Zhang et al. reported PeNCs capped with aromatic amino acid ligands featuring cation–π interactions, enabling the creation of Rec. 2020 pure-red LEDs. The strong cation–π interactions significantly improved the optical properties and spectral stability of LEDs under continuous operation.30 Duan et al. introduced biogenic and amphiphilic capping agents, such as phosphatidyl-L-serine, which contain multi-binding motifs to firmly attach to PeNC surfaces and detach undercoordinated ions without causing additional crystal deformation. This ligand-exchange strategy enhanced the stability of colloidal NCs against diverse external stimuli like heat, water, and light, maintaining a persistent PLQY of over 80% for an extended 180-day period, demonstrating the robustness of these colloidal NCs capped with biogenic and amphiphilic agents. PeNC LEDs based on biogenic and amphiphilic passivated NCs exhibit remarkable stability, this compelling longevity suggests that NCs capped with phosphatidyl-L-serine stand out as promising candidates.16 Furthermore, the use of n-butylamine as a capping ligand is suggested to enhance ambient stability by establishing condensed hydrophobic protective layers, thereby impeding moisture infiltration and effectively passivating external defects. This contributes to the augmentation of PLQY by proficiently addressing surface defects in colloidal perovskite NCs.31 Thus, achieving highly stable colloidal NCs necessitates manipulating surface ligands that strongly anchor the PeNCs, enabling defect elimination while preserving colloidal stability and enhancing optical properties over time.
Here, we present benzylphosphonic acid (BPA)-passivated FAPbBr3 NCs. In earlier studies, BPA was used to modify the work function of solution-processed ZnO.32 In addition, BPA derivatives with various fluorine substitutions were used to adjust the work function of indium tin oxide (ITO).33 In contrast, in this study, BPA is used as a ligand to substitute the native capping ligands from the surface of FAPbBr3 NCs. This approach is designed to mitigate aggregation tendencies, enhance PLQY in thin films, and improve the optoelectrical performance of the LEDs. The incorporation of –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the phosphonic acid confers robust coordination capabilities and amphipathic properties. Unlike the commonly used carboxyl and sulfinyl groups, the electron-donating nature of –PO groups plays a crucial role in preventing superoxide formation. This is attributed to the significant electronegativity difference between P and O, leading to increased electron-cloud density for –PO,34 thereby enhancing both electron-donating and coordination abilities. Detailed characterization results confirm the replacement of native capped ligands (oleic acid and octylamine), as evidenced by the substantial decrease in the intensity of –CH3 and –CH2 stretching vibrations at 2926 cm−1 and 2854 cm−1 associated with carboxylate ligands. Meanwhile, the appearance of CC stretches at 1576 cm−1 and 1507 cm−1 exclusively in BPA-passivated FAPbBr3 NCs indicates successful exchange with the newly introduced aromatic phosphonic acid. The proposed ligand-exchange strategy significantly improves the PLQY from 77% (pristine NCs) to 90% (BPA-NCs). Furthermore, the stability of the colloidal dispersion of BPA-passivated NCs was enhanced compared to that of pristine NCs, improving oxidation and moisture durability. The enhancement in optical properties is attributed to BPA binding to the perovskite surface, forming a novel Pb–O–P covalent bond and displacing the bromide vacancy site. Finally, we fabricated PeLEDs and achieved a maximum external quantum efficiency (EQE) of 12.90% at 4660 cd m−2 and current efficiency (CE) of 55.83 cd A−1, representing an increase of ∼53% and ∼37% compared to control devices, respectively. These results underscore the potential of post-passivated perovskite NCs via aromatic phosphonic acid in fabricating high-performance devices.
O groups in the phosphonic acid confers robust coordination capabilities and amphipathic properties. Unlike the commonly used carboxyl and sulfinyl groups, the electron-donating nature of –PO groups plays a crucial role in preventing superoxide formation. This is attributed to the significant electronegativity difference between P and O, leading to increased electron-cloud density for –PO,34 thereby enhancing both electron-donating and coordination abilities. Detailed characterization results confirm the replacement of native capped ligands (oleic acid and octylamine), as evidenced by the substantial decrease in the intensity of –CH3 and –CH2 stretching vibrations at 2926 cm−1 and 2854 cm−1 associated with carboxylate ligands. Meanwhile, the appearance of CC stretches at 1576 cm−1 and 1507 cm−1 exclusively in BPA-passivated FAPbBr3 NCs indicates successful exchange with the newly introduced aromatic phosphonic acid. The proposed ligand-exchange strategy significantly improves the PLQY from 77% (pristine NCs) to 90% (BPA-NCs). Furthermore, the stability of the colloidal dispersion of BPA-passivated NCs was enhanced compared to that of pristine NCs, improving oxidation and moisture durability. The enhancement in optical properties is attributed to BPA binding to the perovskite surface, forming a novel Pb–O–P covalent bond and displacing the bromide vacancy site. Finally, we fabricated PeLEDs and achieved a maximum external quantum efficiency (EQE) of 12.90% at 4660 cd m−2 and current efficiency (CE) of 55.83 cd A−1, representing an increase of ∼53% and ∼37% compared to control devices, respectively. These results underscore the potential of post-passivated perovskite NCs via aromatic phosphonic acid in fabricating high-performance devices.
2. Materials and methods
Lead bromide (PbBr2, 99%), formamidinium bromide (FABr, >99.99%), octylamine (OTAm, 99%), oleic acid (OA, 90%), poly(9-vinylcarbazole) (PVK, Mn = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000), N,N-dimethylformamide (DMF, anhydrous, 99.8%), chloroform (anhydrous, 99%), acetonitrile (anhydrous, 99.8%), and hexane (anhydrous, 95%) were procured from Sigma-Aldrich. Benzylphosphonic acid (97%) was obtained from Thermo Fisher Scientific, France. All chemicals were utilized as received without any further purification.
000–50000), N,N-dimethylformamide (DMF, anhydrous, 99.8%), chloroform (anhydrous, 99%), acetonitrile (anhydrous, 99.8%), and hexane (anhydrous, 95%) were procured from Sigma-Aldrich. Benzylphosphonic acid (97%) was obtained from Thermo Fisher Scientific, France. All chemicals were utilized as received without any further purification.
2.1 Perovskite NC synthesis
The PeNCs were synthesized using the ligand-assisted reprecipitation (LARP) method. The precursor solution was prepared by dissolving 0.0367 g of PbBr2 (0.1 mmol) and 0.025 g of FABr (0.2 mmol) separately in 0.5 mL of DMF. The fully dissolved precursor solution (250 μL of PbBr2 and FABr) was added to a separate vial containing 250 μL of OA and 25 μL of OTAm. After stirring for 15 min, this solution was slowly added dropwise into 8 mL of chloroform, which acted as the solvent for inducing crystallization. The mixture was then vigorously stirred for 35 seconds. Purification was done by adding 3 mL of acetonitrile, followed by centrifugation at 10000 rpm for 5 min. The resulting supernatant, containing unreacted materials, was discarded, and the remaining precipitate underwent further purification by adding 2 mL of hexane. A subsequent centrifugation at 6000 rpm for 3 min allowed the removal of larger FAPbBr3 particles. Finally, the supernatant was collected and stored in darkness in a refrigerator at 6–8 °C for subsequent characterization and device fabrication.
The ligand solution was prepared by adding the appropriate amount of BPA (0.5, 1, and 2 mg) into 3 mL of acetonitrile. Continuous stirring was then carried out for 3 h at room temperature to ensure complete dissolution of BPA in the acetonitrile solution. This acetonitrile solution containing varying amounts of BPA was subsequently mixed with the perovskite solution to initiate the ligand exchange process.
2.2 LED fabrication method
Indium tin oxide (ITO) patterned glass substrates were first sonicated in acetone and then in isopropyl alcohol. The substrates were then dried by placing them in an oven at 60 °C for 15 min to remove any residual solvents. Prior to device fabrication, the cleaned substrates underwent treatment using a plasma cleaner. Subsequently, poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) solutions (PVP Al 4083, filtered through a 0.45μm polypropylene filter) were spin-coated onto the ITO-coated glass substrates at 2000rpm for 60 s and baked at 150 °C for 30min. The PEDOT:PSS-coated substrates were then transferred into a nitrogen-filled glove box. PVK (6 mg mL−1 in chlorobenzene) was spin-coated onto the substrates at 4000 rpm for 50 s and heated at 170 °C for 30 min to remove the organic solvent. Subsequently, the perovskite emissive layer was formed by spin-coating PeNCs at 1000 rpm for 50 s and then baked at 70 °C for 10 min. The 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi, 35 nm) and LiF/Al (1/100 nm) layers were evaporated using a thermal evaporator (located at the 3D Convergence Center of Inha University) under a base vacuum pressure of 10−7 torr at a rate of 1 Å s−1 and 0.1/4Å s−1, respectively. Finally, before measurement, all the devices were encapsulated with a glass lid using ultraviolet-curing epoxy resin in a glovebox filled with N2 gas.
2.3 Characterization
The absorbance analysis was conducted using a UV-visible-IR spectrophotometer, specifically the PerkinElmer LAMBDA-900 model. Photoluminescence quantum yield (PLQY) measurements were carried out using a Quantaurus-QY Absolute PL quantum yield spectrometer manufactured by Hamamatsu (model C11347-11).Fourier-transform infrared (FT-IR) measurements of the thin film prepared on a quartz substrate were conducted using a Bruker VERTEX 80V spectrometer. The spectral range analyzed was 400–4000 nm, and the measurements were performed in the attenuated total reflection (ATR) mode.
X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific Al Kα source. The PeNC films were deposited onto Si substrates. Scans were carried out at 0.1 eV intervals with a 50-eV pass energy.
The crystal structure of the PeNCs was confirmed using a grazing incidence X-ray diffraction (GI-XRD) diffractometer, specifically the X’Pert-PRO MRD model by Phillips. The perovskite NCs were spin-coated onto a Si substrate for analysis.
The nanocrystal shapes of FAPbBr3 NCs were confirmed using a field-emission transmission electron microscope (FE-TEM), specifically the JEM-2100F model.
3. Results and discussion
The FAPbBr3 NCs are synthesized under ambient environmental conditions using the previously demonstrated ligand-assisted reprecipitation (LARP) method.1,35 Carboxylic OA and OTAm serve as capping ligands during the synthesis of perovskite NCs. These capping ligands (OA, OTAm) are then partially removed from the surface of NCs by introducing stoichiometric amounts of benzylphosphonic acid (BPA-passivation) at 0.5, 1, and 2 mg concentrations. The BPA-passivation process initiates during the washing step in an acetonitrile solution. Detailed information about this ligand exchange process is provided in the experimental section. The morphology and crystal structure characteristics of pristine and BPA-passivated perovskite NCs were evaluated using high-resolution transmission electron microscopy (HR-TEM) and X-ray diffraction (XRD). TEM images (Fig. 1a–d) show that both pristine and BPA-passivated FAPbBr3 NCs have a similar distorted rectangular shape morphology with an average size in the range of 8–10 nm. We observed pronounced aggregation tendencies within the pristine FAPbBr3 NCs, while the BPA-passivated counterparts (0.5, 1, and 2 mg) rarely showed the phenomena. The histogram (Fig. 1e–h) further confirms that the dimensions and morphology of the NCs remained unchanged in the BPA-passivated NCs, indicating a reduction of their aggregation tendency compared to the control NCs. The current morphology of FAPbBr3 exhibits characteristics that are intermediate between NC-like and platelet-like structures (Fig. 1 and Fig. S1, ESI†), consistent with the reported literature.36,37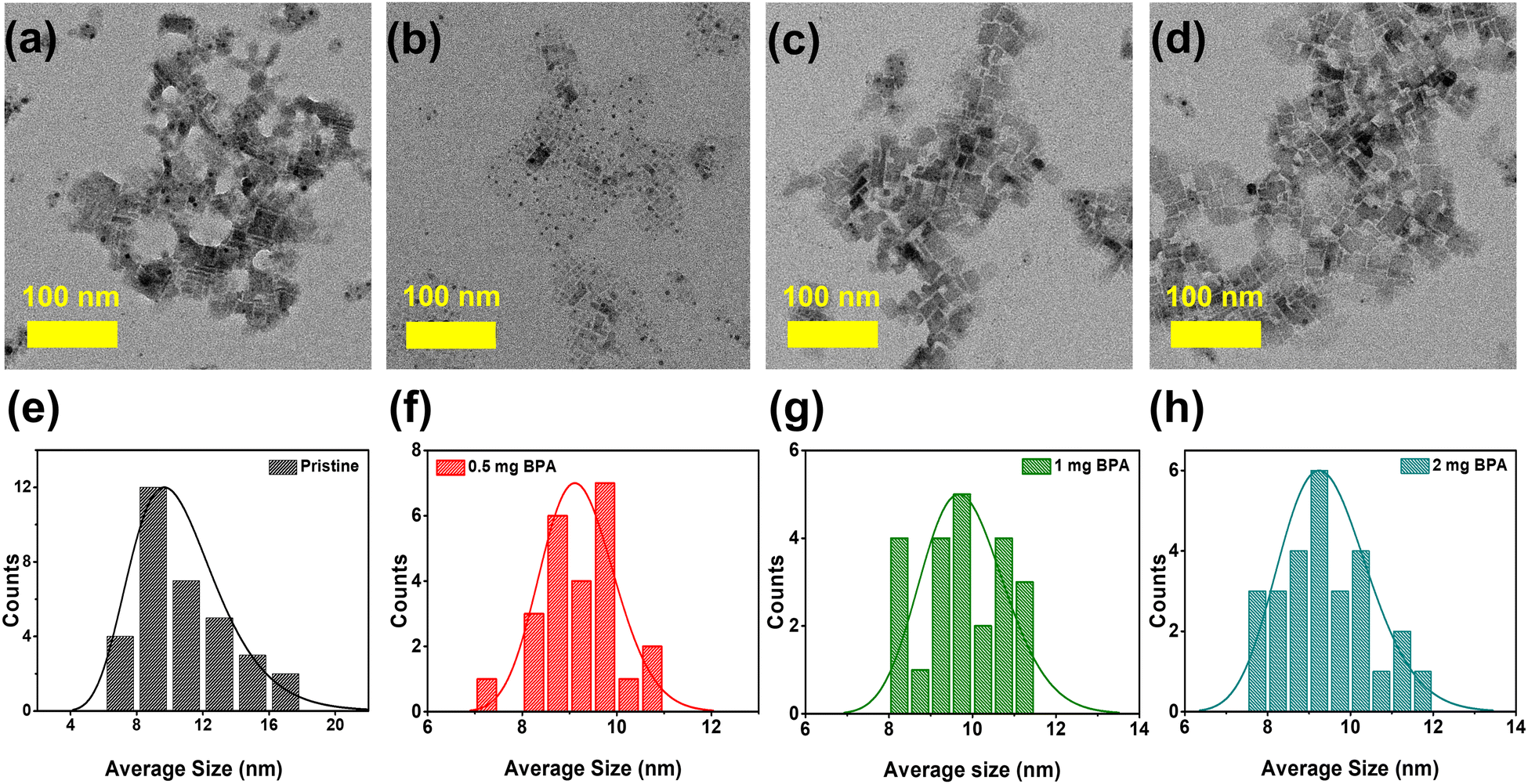 | ||
| Fig. 1 (a)–(d) Transmission electron microscope (TEM) images of pristine and (0.5, 1, and 2 mg) BPA-passivated FAPbBr3 NCs; (e)–(h) Histograms of the average sizes of pristine and BPA-passivated NCs. | ||
Furthermore, XRD data for pristine and BPA-passivated NCs matched well with those of the FAPbBr3 NC space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, no. 221 (JCD 87-0158).38 The peaks at 14.79°, 29.69°, and 33.34° are assigned to the (100), (200), and (210) planes of perovskite NCs, respectively. The absence of discernible alterations in the crystal structure of FAPbBr3 NCs following BPA passivation (Fig. 2a) suggests that BPA is not incorporated into the perovskite crystal structure. It is noted that BPA-passivation reduces the tendency for aggregation without causing obvious changes in crystal structure or morphology. The surface–ligand interactions between aromatic acids and NCs were analyzed using Fourier-transform infrared (FT-IR) (Fig. S2, ESI†) spectroscopy. The FT-IR spectra exhibit strong –CH3 and –CH2 stretching vibrations at 2926 cm−1 and 2854 cm−1, respectively (Fig. 2b), attributed to the native capping ligands (OA, OTAm).39 The peak at 1463 cm−1 in Fig. 2c corresponds to bending vibrations of –CH3 and –CH2. The decreased signal intensities observed at 2926, 2854, and 1463 cm−1 suggest partial removal of native carboxylate ligands (OA and OTAm). The appearance of CC stretching at 1576 cm−1 and 1507 cm−1 (Fig. 2c) exclusively in BPA-passivated FAPbBr3 NCs further indicates the successful exchange of carboxylate ligands with the newly introduced aromatic phosphonic acid (BPA-passivation).40 The stretching vibration of CC is ascribed to the aromatic ring that appears in the FT-IR curve of BPA-passivated FAPbBr3 NCs. Thus, in comparison to linear phase ligands, the incorporation of BPA introduces increased steric hindrance, thereby more effectively enhancing the thermal and photostability of BPA-passivated FAPbBr3 NC films. The PL spectra of pristine and BPA-passivated FAPbBr3 NCs are illustrated in Fig. 2d. The PL peaks of all the samples are located at 531 nm, and there is no significant change in the emission of FAPbBr3 NCs before and after passivation. The inset in Fig. 2d provides a focused image of the PL profiles of pristine and BPA-passivated NCs, where the absence of spectral broadening in the FAPbBr3 NCs subjected to BPA passivation can be clearly observed. Remarkably, the absolute PLQY values increased from 77% of pristine NCs to 90% of 1 mg BPA-passivated FAPbBr3 NCs (Fig. 2e). It can be inferred from the results that the optimized amount of 1 mg of BPA induces passivation of bromide side vacancies, consequently hindering nonradiative recombination processes.41
m, no. 221 (JCD 87-0158).38 The peaks at 14.79°, 29.69°, and 33.34° are assigned to the (100), (200), and (210) planes of perovskite NCs, respectively. The absence of discernible alterations in the crystal structure of FAPbBr3 NCs following BPA passivation (Fig. 2a) suggests that BPA is not incorporated into the perovskite crystal structure. It is noted that BPA-passivation reduces the tendency for aggregation without causing obvious changes in crystal structure or morphology. The surface–ligand interactions between aromatic acids and NCs were analyzed using Fourier-transform infrared (FT-IR) (Fig. S2, ESI†) spectroscopy. The FT-IR spectra exhibit strong –CH3 and –CH2 stretching vibrations at 2926 cm−1 and 2854 cm−1, respectively (Fig. 2b), attributed to the native capping ligands (OA, OTAm).39 The peak at 1463 cm−1 in Fig. 2c corresponds to bending vibrations of –CH3 and –CH2. The decreased signal intensities observed at 2926, 2854, and 1463 cm−1 suggest partial removal of native carboxylate ligands (OA and OTAm). The appearance of CC stretching at 1576 cm−1 and 1507 cm−1 (Fig. 2c) exclusively in BPA-passivated FAPbBr3 NCs further indicates the successful exchange of carboxylate ligands with the newly introduced aromatic phosphonic acid (BPA-passivation).40 The stretching vibration of CC is ascribed to the aromatic ring that appears in the FT-IR curve of BPA-passivated FAPbBr3 NCs. Thus, in comparison to linear phase ligands, the incorporation of BPA introduces increased steric hindrance, thereby more effectively enhancing the thermal and photostability of BPA-passivated FAPbBr3 NC films. The PL spectra of pristine and BPA-passivated FAPbBr3 NCs are illustrated in Fig. 2d. The PL peaks of all the samples are located at 531 nm, and there is no significant change in the emission of FAPbBr3 NCs before and after passivation. The inset in Fig. 2d provides a focused image of the PL profiles of pristine and BPA-passivated NCs, where the absence of spectral broadening in the FAPbBr3 NCs subjected to BPA passivation can be clearly observed. Remarkably, the absolute PLQY values increased from 77% of pristine NCs to 90% of 1 mg BPA-passivated FAPbBr3 NCs (Fig. 2e). It can be inferred from the results that the optimized amount of 1 mg of BPA induces passivation of bromide side vacancies, consequently hindering nonradiative recombination processes.41
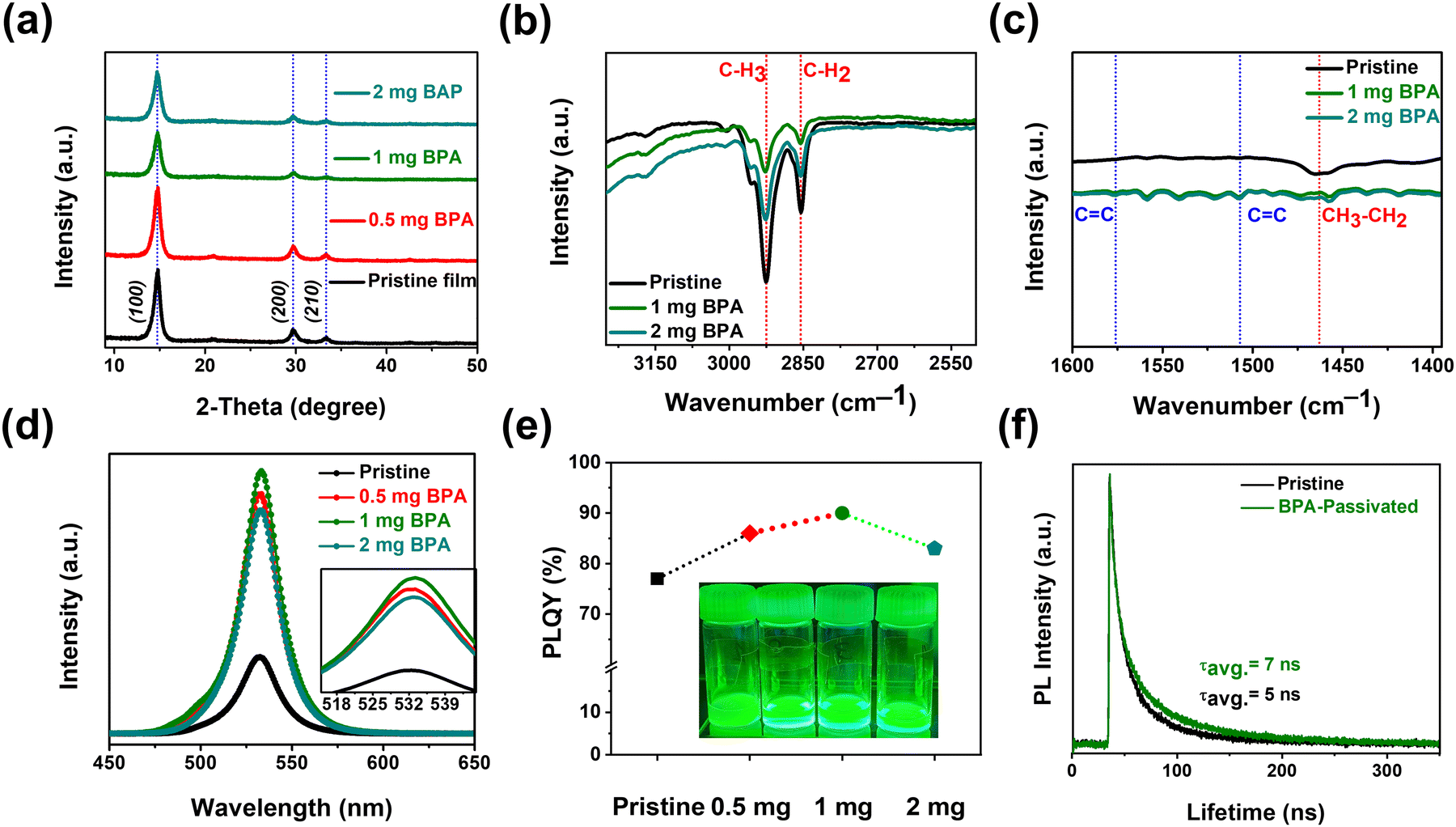 | ||
| Fig. 2 (a) X-ray diffraction (XRD) patterns of pristine and BPA-passivated FAPbBr3 NC films on a silicon substrate. (b) and (c) Fourier-transform infrared spectroscopy (FT-IR) spectra of pristine and BPA-passivate FAPbBr3 NC films on a quartz substrate. FT-IR measurements were conducted in the attenuated total reflection (ATR) mode. The red-dotted lines indicate the stretching associated with native alkylamine ligands, and the blue-dotted lines denote the CC stretching linked with aromatic BPA acid. (d) and (e) Photoluminescence (PL) spectra and corresponding PL quantum yield (PLQY) of pristine and BPA-passivated FAPbBr3 NCs. The inset of Fig. 2e demonstrates photos of pristine and BPA-passivated (0.5, 1, and 2 mg) NC solutions. (f) Time-resolved photoluminescence (TRPL) decay curves of pristine and BPA-passivated FAPbBr3 NC films. | ||
Time-resolved photoluminescence (TRPL) measurements were conducted to determine the carrier lifetime (Fig. 2f). The average lifetimes (τavg) were calculated by fitting the TRPL curves with a bi-exponential decay function. BPA-passivated films displayed a longer approximate PL lifetime of τavg = 7 ns compared to pristine perovskite films with an approximate time of τavg = 5 ns, as summarized in Table S1 (ESI†). The increased non-radiative recombination within the pristine perovskite films leads to rapid PL decay. Thus, the prolonged lifetime observed in films passivated with BPA indicates that the passivation layer mitigates non-radiative recombination by reducing the defect on the surface.
Fig. S3 (ESI†) illustrates the UV-vis absorption spectra of pristine and BPA-passivated FAPbBr3 NCs. The analysis indicates that the aromatic ligand exchange has no significant effect on the peak position, consistent with PL results. Furthermore, the superior stability of BPA-NCs was monitored by dispersing both pristine and BPA-NCs into DI water for 10 days, as shown in Fig. S4 (ESI†). The solution containing pristine FAPbBr3 NCs gradually became opaque, exhibiting agglomeration and forming a yellow bulk within 10 days, indicative of the inherent instability of perovskites due to their ionic nature.42 In contrast, the BPA-passivated FAPbBr3 NCs maintained their green color without agglomeration, demonstrating higher stability than pristine NCs in a colloidal state. Similar trends were observed in pristine (PLQY = 0.55) and BPA-passivated FAPbBr3 NCs (PLQY = 0.80) after two weeks of storage under refrigeration, with a retention factor of more than 88.9% (Fig. S5 and Table S2, ESI†). To further confirm whether superoxide formation played a significant role in BPA-passivated FAPbBr3 NC films under UV lamp photoexcitation, we monitored the photostability of perovskite films under continuous illumination. Upon photoexcitation, excitons on the perovskite surface can migrate toward adsorbed oxygen molecules, converting them into superoxide ions (O2−). This interaction could initiate the oxidation and subsequent decomposition of the perovskite NCs.43 As shown in Fig. S6 (ESI†), the pristine film degraded quickly, whereas the BPA-passivated FAPbBr3 NC films demonstrated significantly enhanced stability under the same conditions, suggesting a protective effect imparted by the BPA passivation against photoinduced degradation. These findings suggest that aromatic ligand exchange provides superior stability under various challenging conditions, effectively mitigating the rapid degradation of the optical properties of perovskite NCs.44,45
XPS was conducted to explore the surface chemical binding of pristine and BPA-passivated FAPbBr3 NCs. The XPS results of pristine and BPA-passivated FAPbBr3 NC structures are shown in Fig. S7 (ESI†). A new peak at 532.42 eV emerged exclusively in the O 1s spectra of BPA-passivated FAPbBr3 NCs, indicating the incorporation of aromatic benzylphosphonic acid within the perovskite structures (Fig. 3a). The presence of oxygen peak indicates the high electron density of BPA-passivated NCs as an electron donor to coordinate with unsaturated Pb2+. Furthermore, the pristine perovskite NCs (atomic ratio of approximately 0.1%) exhibited an almost negligible atomic ratio of P 2p. However, this ratio substantially increased in the case of BPA-passivated perovskite (atomic ratio of 0.26%). Moreover, the P 2p peak of BPA-passivated FAPbBr3 NCs exhibits a shift toward a higher binding energy of 127.88 eV compared to the 127.28 of the pristine NCs sample. This change in binding energy (0.6 eV) is attributed to the formation of covalent bonds during the adsorption of phosphonate onto a perovskite surface (Fig. S8, ESI†). The O 1s spectrum of the pristine BPA revealed predominant peaks owing to the oxygen moieties associated with the P–OH and PO groups. The results are consistent with previously reported O 1s spectra of phosphonic acid derivatives.46,47 Therefore, it can be concluded that the distinctive appearance of O 1s spectra solely in BPA-passivated perovskite structures results from the formation of covalent bonds during phosphonate adsorption onto the metallic surface.48 Importantly, BPA binds to the perovskite surface by forming a novel Pb–O–P covalent bond (Fig. S9, ESI†), displacing the bromide vacancy site. The Pb 4f and Br 3d peaks in BPA-passivated FAPbBr3 NCs exhibit higher binding energy values than pristine NCs (Fig. 3b–d). The high-resolution XPS reveals the presence of a peak at 533.10 eV in pristine NCs. Additionally, two smaller peaks are observed at 533.12 and 528.98 eV, corresponding to the chemical states associated with P–OH and Pb–O–P bonds, respectively (Fig. S9, ESI†). This is attributed to the greater electronegativity of oxygen atoms relative to bromine atoms, thereby influencing the Fermi level.4 Based on the abovementioned results and discussion, the mechanism of ligand exchange for pristine and BPA-passivated FAPbBr3 NCs is proposed and shown in Fig. 4. After the purification of FAPbBr3, halogen vacancies are generated on the surface of perovskite NCs. However, after ligand exchange with BPA, the BPA aromatic short ligands bind to the perovskite surface and replace the Br vacancies.
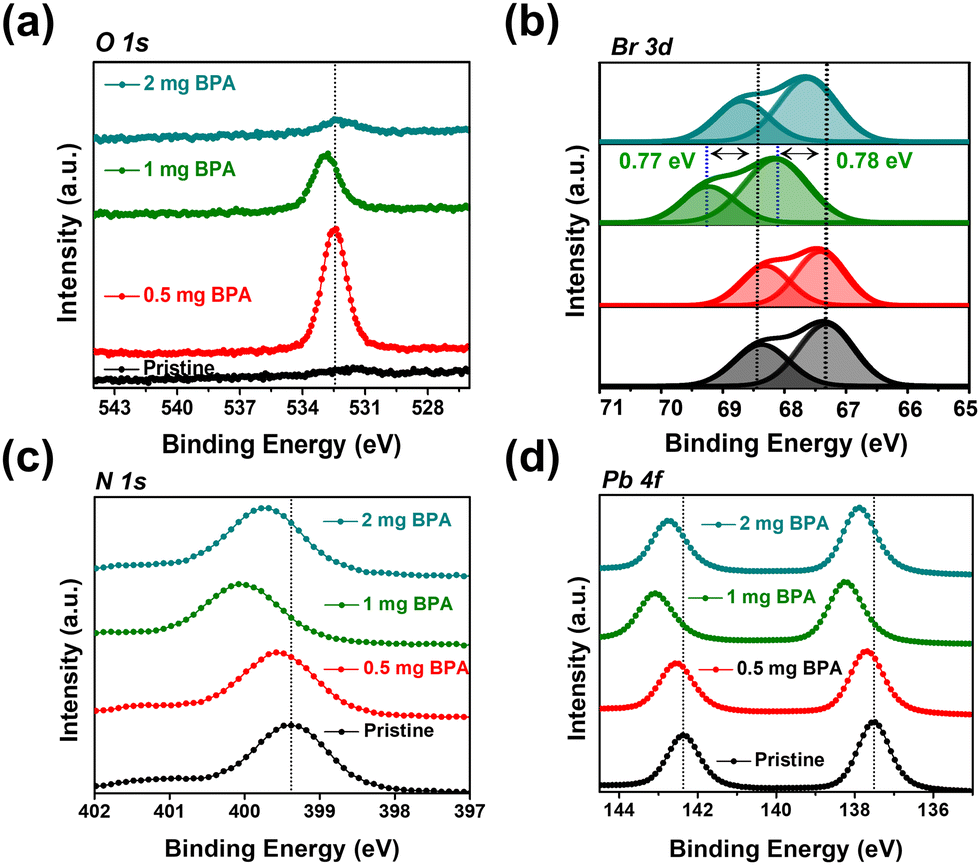 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) spectra of pristine and BPA-passivated FAPbBr3 NCs. (a) High-resolution spectra of O 1s, (b) Br 3d, (c) N 1s, and (d) Pb 4f of pristine and BPA-passivated FAPbBr3 NCs. | ||
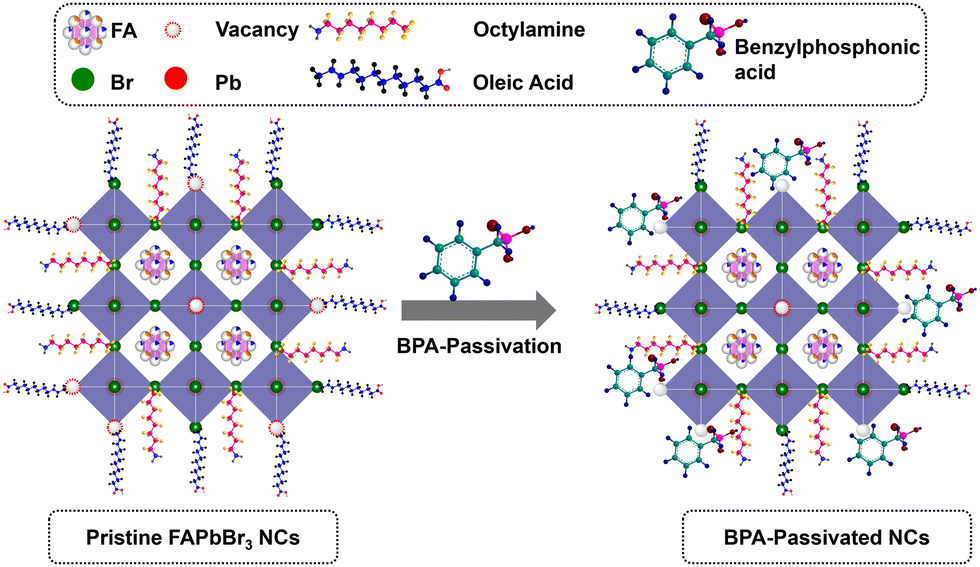 | ||
| Fig. 4 Schematic illustration of BPA-passivation in the FAPbBr3 NCs. | ||
To further evaluate the impact of aromatic phosphonic acid on the electroluminescent (EL) performance, PeLEDs were fabricated having a configuration of ITO/PEDOT: PSS/PVK/PeNCs/TPBi/LiF/Al (Fig. 5a). The PEDOT:PSS (60 nm)/PVK (10 nm) served as a hole-transporting/injecting layer. The TPBi layer is used as an electron-transporting layer. The FAPbBr3 NC thin film with a thickness of 35 nm (pristine or BPA-passivated) served as an emissive layer (EML), and ITO (70 nm) and LiF (1 nm)/Al (100 nm) were used as the anode and cathode, respectively. The current density–voltage–luminance (J–V–L) curves of both pristine and BPA-passivated FAPbBr3 NCs PeLEDs are illustrated in Fig. 5b. After BPA-passivation, the brightness of the PeLEDs improved from 27509 to 29280 cd m−2. It was observed that the turn-voltage (Von) of BPA-passivated PeLEDs decreased from 3.8 V (pristine PeLEDs) to 3.5 V. The low Von observed in BPA-passivated FAPbBr3 NCs LED compared to the control can be attributed to the modifications in the Fermi level, as supported by XPS analysis. Furthermore, the control (pristine) PeLEDs exhibited a maximum luminance (Lmax) of 27509 cd m−2 at a current density of 161.71 mA cm−2. In contrast, the BPA-passivated PeLEDs achieved a superior Lmax of 29280 cd m−2 at a significantly lower current density of 121.32 mA cm−2. These results suggest enhanced radiative recombination of carriers at low injected carrier concentrations, attributed to the BPA-passivated perovskite NC domains with increased quantum confinement.49,50 The current efficiency (CE) and power efficiency (PE) versus current density curves for pristine and BPA-passivated PeLEDs reveal remarkable differences. Pristine PeLEDs exhibited a peak CE of 37.08 cd A−1 and a corresponding power efficiency of 20.80 lm W−1. In contrast, BPA-passivated PeLEDs demonstrated superior performance, achieving a maximum CE and power efficiency of 55.83 cd A−1 and 34.39 lm W−1, respectively (Fig. 5c and d). Notably, the BPA-passivated PeLEDs displayed a substantially higher peak EQE of 12.90%, compared to the 8.44% observed in pristine PeLEDs (Fig. 5e). This increase in EQE, approximately 53% in BPA-passivated PeLEDs compared to that in pristine devices, is attributed to the efficient mitigation of halogen vacancies, consequently impeding nonradiative recombination processes.
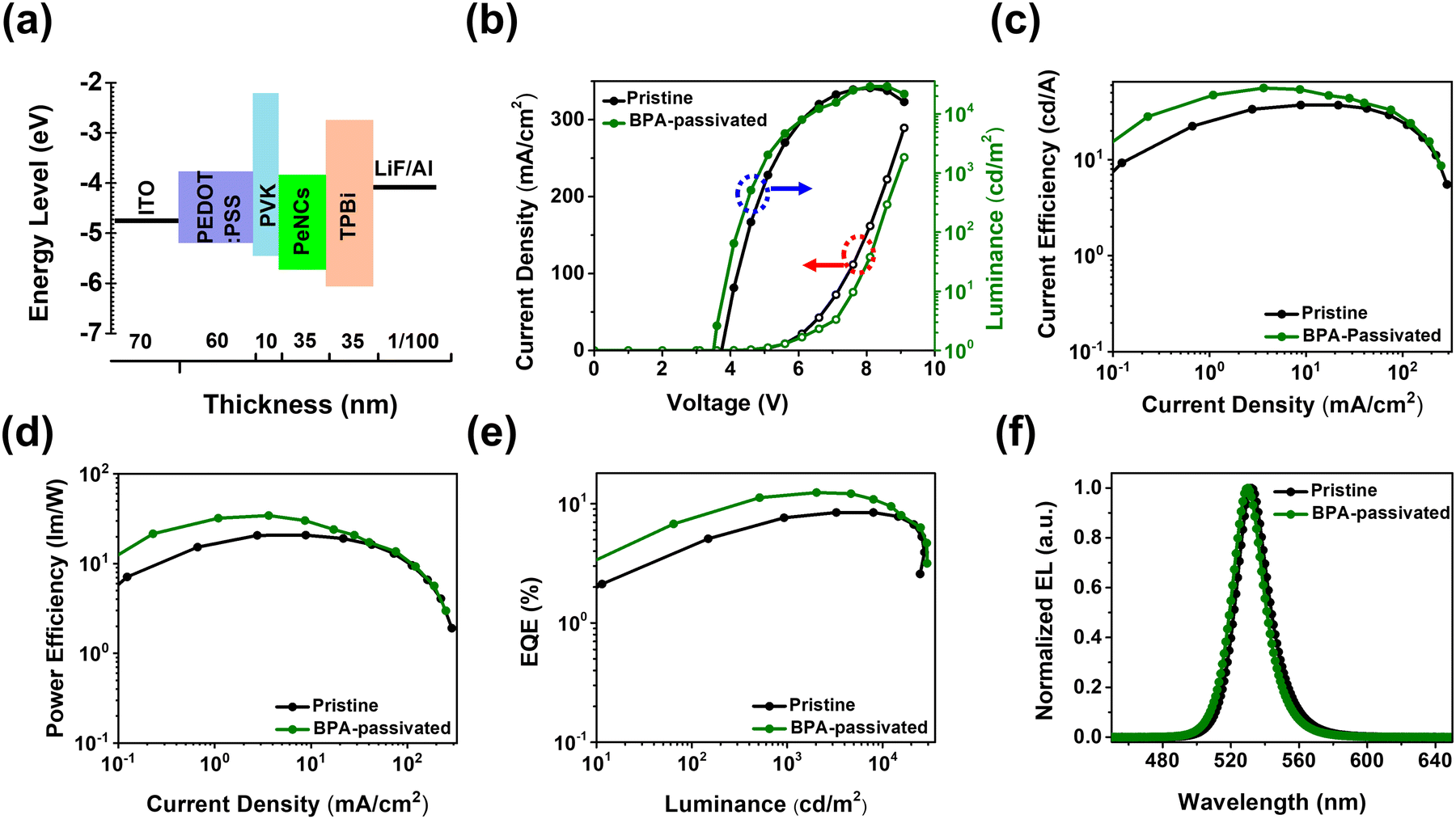 | ||
| Fig. 5 Optoelectrical performance of Perovskite LEDs (PeLEDs). (a) Schematic illustration of the energy level diagram of a PeLED. (b) Current density versus voltage curves (dotted line with red arrows) and luminance versus voltage curves (solid line with blue arrows). (c) Current efficiency versus current density curves. (d) Power efficiency versus current density curves. (e) External quantum efficiency (EQE) versus luminance curves, and (f) normalized electroluminescent (EL) spectra of pristine and BPA-passivated perovskite NC LEDs. | ||
The normalized EL spectra in Fig. 5f show a slight blue shift in the BPA-passivated LEDs compared to the pristine counterparts. The FWHM for BPA-passivated PeLEDs was approximately 21 nm, meeting the requirement of vivid displays, while it measured 22 nm for pristine PeLEDs. This discrepancy underscores the excellent color purity achieved by BPA-passivated PeLEDs. The Commission Internationale de L’Eclairage (CIE) chromaticity coordinates of the EL spectra for BPA-passivated NCs PeLEDs are illustrated in Fig. S10 (ESI†); the BPA-passivated NCs PeLEDs are a pure-green, with CIE 1931 coordinates of (0.19, 0.76). These coordinates closely approach the Rec. 2020 specification (0.098, 0.779), indicating the potential suitability of these devices for practical applications.35 Importantly, the EL peak demonstrates remarkable spectral stability, remaining unaltered even with a rise in the luminance of over 25000 cd m−2 and operational voltage (∼7.6 V), highlighting the excellent durability (Fig. S11, ESI†). All the key performance parameters are summarized in Table 1 and Table S3 (ESI†). We noticed that the BPA-passivated LED exhibited improved optoelectrical performance, as shown in Fig. 5. At low voltages, the BPA-passivated LED exhibited lower current but higher luminance compared to the control (pristine) devices, with a slightly different EL spectrum, indicating that the BPA-LED has better charge balance than the control. Although the strategy can enhance the EQE and luminance, the efficiency roll-off characteristics are insignificant after the modification. Engineering the interfacial barrier between the EML and the electron transporting layer to get a low energy barrier would help to further improve the efficiency roll-off.51
| Perovskite LED | V on | EL peak (nm) | FWHM (nm) | CE (cd A−1) | PE (lm W−1) | EQE (%) | L max (cd m−2) |
|---|---|---|---|---|---|---|---|
| Pristine | 3.8 | 533 | 22 | 37.08 (34.80 ± 1.68) | 20.80 (19.42 ± 0.98) | 8.44 (7.90 ± 0.39) | 27509 (26537 ± 1926) |
| BPA-passivated | 3.5 | 531 | 21 | 55.83 (50.69 ± 7.33) | 34.39 (31.28 ± 4.55) | 12.90 (11.66 ± 1.72) | 29280 (25204 ± 4207) |
4. Conclusions
In summary, an effective strategy has been implemented to enhance the colloidal stability of FAPbBr3 NCs and improve the performance of PeLEDs by partially replacing the native ligands on the surface of FAPbBr3 NCs with aromatic benzylphosphonic acid (BPA-passivation). By optimizing the content of aromatic phosphonic acid on the surface of NCs, a significant reduction in non-radiative recombination rates was observed, leading to a notable improvement in the PLQY from 77% for pristine NCs to 90% for BPA-passivated NCs. The exchange of native capped ligands (oleic acid and octylamine) with newly introduced aromatic phosphonic acid was successfully achieved. Furthermore, the proposed aromatic ligand-exchange strategy offers superior stability under diverse challenging conditions, effectively mitigating the rapid degradation of optical properties. Finally, PeLEDs were fabricated using both pristine and BPA-passivated FAPbBr3 NCs as emitters, achieving a maximum EQE of 12.90% for BPA-passivated devices, which is approximately 53% higher compared to pristine devices (EQE of 8.44%). This work provides an effective approach to fine-tuning the ligand chemistry of perovskite LEDs for high efficiency and excellent stability of perovskite NCs against various harsh environmental conditions.Author contributions
M. I. Saleem: conceptualization, methodology, data analysis, data curation, investigation, and writing – original draft. S. He: LED fabrication and TRPL measurement. S. H. Kim: investigation and validation. J.-W. Kang: funding acquisition, supervision, and review & editing. J.-H. Lee: funding acquisition, supervision, data validation, visualization, and review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science, ICT & Future Planning, and Technology Innovation Program (20014528, 20016283), funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). It was also supported by the Digital Research Innovation Institution Program Through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (RS-2023-00283597). This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2022R1A6A1A03051705).References
- H. Zhao, H. Chen, S. Bai, C. Kuang, X. Luo, P. Teng, Y. Yang, L. Duan, F. Gao and M. Liu, ACS Energy Lett., 2021, 6, 2395–2403 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- M. I. Saleem, R. Choi and J.-H. Lee, Mater. Chem. Front., 2023, 7, 2316–2336 RSC.
- J. S. Kim, J.-M. Heo, G.-S. Park, S.-J. Woo, C. Cho, H. J. Yun, D.-H. Kim, J. Park, S.-C. Lee and S.-H. Park, Nature, 2022, 611, 688–694 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang and C. Yan, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Z. Chu, Q. Ye, Y. Zhao, F. Ma, Z. Yin, X. Zhang and J. You, Adv. Mater., 2021, 33, 2007169 CrossRef CAS PubMed.
- S. Jeon, L. Zhao, Y.-J. Jung, J. W. Kim, S.-Y. Kim, H. Kang, J.-H. Jeong, B. P. Rand and J.-H. Lee, Small, 2019, 15, 1900135 CrossRef PubMed.
- Q. Wan, W. Zheng, C. Zou, F. Carulli, C. Zhang, H. Song, M. Liu, Q. Zhang, L. Y. Lin and L. Kong, ACS Energy Lett., 2023, 8, 927–934 CrossRef CAS.
- H.-D. Lee, S.-J. Woo, S. Kim, J. Kim, H. Zhou, S. J. Han, K. Y. Jang, D.-H. Kim, J. Park, S. Yoo and T.-W. Lee, Nat. Nanotechnol., 2024, 19, 624–631 CrossRef CAS PubMed.
- Y.-H. Kim, J. Park, S. Kim, J. S. Kim, H. Xu, S.-H. Jeong, B. Hu and T.-W. Lee, Nat. Nanotechnol., 2022, 17, 590–597 CrossRef CAS PubMed.
- M. I. Saleem, A. Katware, A. Amin, S.-H. Jung and J.-H. Lee, Nanomaterials, 2023, 13, 1366 CrossRef CAS PubMed.
- L. Zhang, W. Liang, L. Xu, M. Zhu, X. Wang, J. Su, L. Li, N. Liu, Z. Zhang and Y. Gao, Chem. Eng. J., 2021, 417, 129349 CrossRef CAS.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- Y. Shin, H. Suh, B.-H. Kwon, J. Park, H. Cho, C. W. Joo, S. Choi, J.-W. Shin, C.-m Kang, D. H. Ahn, W. J. Lee, Y.-H. Kim, J. Jang and B.-S. Bae, Chem. Eng. J., 2023, 474, 145889 CrossRef CAS.
- Y. Duan, K. Chordiya, M. U. Kahaly, F. E. Oropeza, V. A. de la Peña O'Shea, D.-Y. Wang and R. D. Costa, Adv. Opt. Mater., 2022, 10, 2201176 CrossRef CAS.
- J.-N. Yang, Z.-Y. Ma, J.-D. Luo, J.-J. Wang, C. Ye, Y. Zhou, Y.-C. Yin, X.-C. Ru, T. Chen and L.-Y. Li, Nano Lett., 2023, 23, 3385–3393 CrossRef CAS PubMed.
- J.-N. Yang, Y. Song, J.-S. Yao, K.-H. Wang, J.-J. Wang, B.-S. Zhu, M.-M. Yao, S. U. Rahman, Y.-F. Lan and F.-J. Fan, J. Am. Chem. Soc., 2020, 142, 2956–2967 CrossRef CAS PubMed.
- Y.-K. Wang, K. Singh, J.-Y. Li, Y. Dong, X.-Q. Wang, J. M. Pina, Y.-J. Yu, R. Sabatini, Y. Liu and D. Ma, Adv. Mater., 2022, 34, 2200854 CrossRef CAS PubMed.
- M. I. Saleem and J.-H. Lee, J. Inf. Disp., 2024, 25, 157–168 CrossRef CAS.
- H. Lv, X. Tang and M. Chen, ACS Appl. Nano Mater., 2023, 6(20), 18918–18925 CrossRef CAS.
- J. Pan, X. Li, X. Gong, J. Yin, D. Zhou, L. Sinatra, R. Huang, J. Liu, J. Chen, I. Dursun, A. M. El-Zohry, M. I. Saidaminov, H.-T. Sun, O. F. Mohammed, C. Ye, E. H. Sargent and O. M. Bakr, Angew. Chem., Int. Ed., 2019, 58, 16077–16081 CrossRef CAS PubMed.
- D. Ghosh, M. Y. Ali, A. Ghosh, A. Mandal and S. Bhattacharyya, J. Phys. Chem. C, 2021, 125, 5485–5493 CrossRef CAS.
- S. Huang, P. Huang, L. Wang, J. Han, Y. Chen and H. Zhong, Adv. Mater., 2019, 31, 1903830 CrossRef CAS PubMed.
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Adv. Mater., 2022, 34, 2105958 CrossRef CAS PubMed.
- D. Yan, T. Shi, Z. Zang, T. Zhou, Z. Liu, Z. Zhan, J. Du, Y. Leng and X. Tang, Small, 2019, 15, 1901173 CrossRef PubMed.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami and J. Liu, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- K. Yang, F. Li, H. Hu, T. Guo and T. W. Kim, Nano Energy, 2019, 65, 104029 CrossRef CAS.
- B. Lyu, X. Bao, D. Gao, X. Guo, X. Lu and J. Ma, ACS Sustainable Chem. Eng., 2022, 10, 5084–5091 CrossRef.
- J. Zhang, B. Cai, X. Zhou, F. Yuan, C. Yin, H. Wang, H. Chen, X. Ji, X. Liang and C. Shen, Adv. Mater., 2023, 35, 2303938 CrossRef CAS PubMed.
- F. Chun, B. Zhang, Y. Li, W. Li, M. Xie, X. Peng, C. Yan, Z. Chen, H. Zhang and W. Yang, Chem. Eng. J., 2020, 399, 125715 CrossRef CAS.
- L. Ilja, R. Sina, K. Juliane, P. Fortunato, P. Michael, H. Jana, B. Thomas, H. Stefan and N. Dieter, Appl. Phys. Lett., 2015, 106, 113302 CrossRef.
- J. H. Peter, L. Hong, B. P. Pavel, A. P. Sergio, C. J. Simon, R. A. Neal, L. B. Jean and R. M. Seth, Adv. Mater., 2009, 21, 4496–4501 CrossRef.
- H. Li, J. Shi, J. Deng, Z. Chen, Y. Li, W. Zhao, J. Wu, H. Wu, Y. Luo and D. Li, Adv. Mater., 2020, 32, 1907396 CrossRef CAS PubMed.
- S. He, H. B. Lee, K.-J. Ko, N. Kumar, J.-H. Jang, S.-O. Cho, M. Song, W. Zhou, B.-J. Lee, J.-H. Lee and J.-W. Kang, Adv. Opt. Mater., 2023, 11, 2300486 CrossRef CAS.
- Y. Zu, J. Xi, L. Li, J. Dai, S. Wang, F. Yun, B. Jiao, H. Dong, X. Hou and Z. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 2835 CrossRef CAS PubMed.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765 CrossRef CAS PubMed.
- T. Zhang, H. Li, P. Yang, J. Wei, F. Wang, H. Shen, D. Li and F. Li, Org. Electron., 2019, 68, 76–84 CrossRef CAS.
- L. Gao, T. Cheng, L. Gou, Y. Zhang, Y. Liu, L. Yuan, X. Zhang, Y. Wang, F. Men and J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 18125–18133 CrossRef CAS PubMed.
- X. Lv, Y.-L. Xu, S.-Q. Sun, Q. Sun, Y.-M. Xie and M.-K. Fung, J. Alloys Compd., 2023, 969, 172459 CrossRef CAS.
- L. Gao, T. Cheng, Q. Wang, L. Gou, Y. Zhang, X. Zhang, C.-H. Tan, W. Zheng, M. A. McLachlan and J. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 55380–55390 CrossRef CAS PubMed.
- S. Jeon, J. Ahn, M.-C. Jung, H. K. Woo, J. Bang, B. K. Jung, S. Oh, S. Y. Lee, K. J. Lee, T. Paik, D.-H. Ha, J.-P. Ahn, S. Jeong, D. H. Kim, J. H. Noh, H. S. Jang, M. J. Han and S. J. Oh, Small, 2023, 20, 2307032 CrossRef PubMed.
- L. Quan, D. Ma, Y. Zhao, O. Voznyy, H. Yuan, E. Bladt, J. Pan, F. Arquer, R. Sabatini, Z. Piontkowski, A. Emwas, P. Todorović, R. Quintero-Bermudez, G. Walters, J. Z. Fan, M. Liu, H. Tan, M. I. Saidaminov, L. Gao, Y. Li, D. H. Anjum, N. Wei, J. Tang, D. W. McCamant, M. B. J. Roeffaers, S. Bals, J. Hofkens, O. M. Bakr, Z. Lu and E. H. Sargent, Nat. Commun., 2020, 11, 170 CrossRef PubMed.
- F. Zaccaria, B. Zhang, L. Goldoni, M. Imran, J. Zito, B. van Beek, S. Lauciello, L. De Trizio, L. Manna and I. Infante, ACS Nano, 2022, 16, 1444–1455 CrossRef CAS PubMed.
- Y. Shynkarenko, M. I. Bodnarchuk, C. Bernasconi, Y. Berezovska, V. Verteletskyi, S. T. Ochsenbein and M. V. Kovalenko, ACS Energy Lett., 2019, 4, 2703–2711 CrossRef CAS PubMed.
- M. Wagstaffe, A. G. Thomas, M. J. Jackman, M. Torres-Molina, K. L. Syres and K. Handrup, J. Phys. Chem. C, 2016, 120, 1693–1700 CrossRef CAS.
- F. Li, H. Zhong, G. Zhao, S. Wang and G. Liu, Colloids Surf., A, 2016, 490, 67–73 CrossRef CAS.
- T. Xuan, X. Yang, S. Lou, J. Huang, Y. Liu, J. Yu, H. Li, K.-L. Wong, C. Wang and J. Wang, Nanoscale, 2017, 9, 15286–15290 RSC.
- Z. Chen, Z. Li, C. Zhang, X.-F. Jiang, D. Chen, Q. Xue, M. Liu, S. Su, H.-L. Yip and Y. Cao, Adv. Mater., 2018, 30, 1801370 CrossRef PubMed.
- C. Sun, Y. Jiang, M. Cui, L. Qiao, J. Wei, Y. Huang, L. Zhang, T. He, S. Li and H.-Y. Hsu, Nat. Commun., 2021, 12, 2207 CrossRef CAS PubMed.
- K. Elkhouly, R. Gehlhaar, J. Genoe, P. Heremans and W. Qiu, Adv. Opt. Mater., 2020, 8, 2000941 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02419b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |