
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrobially mediated synthesis of vivianite by Desulfosporosinus on the way to phosphorus recovery †
Yuriy V.
Knyazev
 *ab,
Mikhail S.
Platunov
*c,
Olga P.
Ikkert
b,
Sergey V.
Semenov
ae,
Oleg A.
Bayukov
a,
Anton D.
Nikolenko
c,
Vladimir P.
Nazmov
df,
Mikhail N.
Volochaev
a,
Andrey A.
Dubrovskiy
a,
Maksim S.
Molokeev
a,
Ekaterina D.
Smorodina
e,
Dmitry A.
Balaev
ae and
Olga V.
Karnachuk
b
*ab,
Mikhail S.
Platunov
*c,
Olga P.
Ikkert
b,
Sergey V.
Semenov
ae,
Oleg A.
Bayukov
a,
Anton D.
Nikolenko
c,
Vladimir P.
Nazmov
df,
Mikhail N.
Volochaev
a,
Andrey A.
Dubrovskiy
a,
Maksim S.
Molokeev
a,
Ekaterina D.
Smorodina
e,
Dmitry A.
Balaev
ae and
Olga V.
Karnachuk
b
aKirensky Institute of Physics, Federal Research Center KSC SB RAS, Krasnoyarsk, Russia. E-mail: yuk@iph.krasn.ru
bDepartment of Plant Physiology, Biotechnology, and Bioinformatics, Tomsk State University, Tomsk, Russia
cSynchrotron Radiation Facility SKIF, Boreskov Institute of Catalysis SB RAS, Kol'tsovo, Russia. E-mail: m.s.platunov@srf-skif.ru
dBudker Institute of Nuclear Physics, Novosibirsk, Russia
eSiberian Federal University, Krasnoyarsk, Russia
fInstitute of Solid State Chemistry and Mechanochemistry, Novosibirsk, Russia
First published on 30th April 2024
Abstract
We explored the role of biomineralization in industrial waste sludge formation, using the laboratory cultivation of Desulfovibrio sp. OL sulfate reducing species isolated from the Komsomolsky waste sludge (Russia). The most frequently reported sulfate-reducing bacteria (SRB) biomineralization products are various iron sulfides. Here we present first studies of the products of Desulfosporosinus metallidurans, acidophilic SRB from acid mine drainage. We analyzed the biomineralized sample using X-ray diffraction, electron microscopy, X-ray absorption and Mössbauer spectroscopies, and magnetization measurements via First-Order Reversal Curve (FORC) diagram analysis. Our findings show that the biomineralization occurring under pure culture conditions leads to the formation of greigite (Fe3S4) nanorods, along with larger microbially mediated crystals of vivianite (Fe3(PO4)2·8H2O) and siderite (FeCO3). Energy dispersive X-ray spectroscopy revealed that the crystal sizes of vivianite and siderite were comparatively larger than those of the nanorod-shaped greigite. Transmission electron microscopy and Mössbauer spectroscopy detected ultrafine ferrihydrite (Fe2O3·nH2O) superparamagnetic nanoparticles with an average size of 2.5 nm. FORC analysis showed significant magnetic interactions among these nanoparticles, suggesting their potential for magnetic separation applications. The current study demonstrates that ferrihydrite nanoparticles have a strong magnetic affinity for other crystal phases produced by Desulfosporosinus metallidurans. Therefore, we believe that the investigated bacterial species can be exploited in advanced magnetic separation techniques. This offers a cost-effective and environmentally friendly method for purifying sediments in industrial waste sludge.
Environmental significanceIndustrial waste, such as tailings, often contains valuable chemical elements essential for agriculture, including phosphorus and iron. However, these waste deposits pose a significant threat of soil pollution and contain large volumes of contaminated water. Additionally, the costly separation process presents significant challenges in recycling these elements. In this study, we explore biomineralization mechanisms that not only facilitate the extraction of phosphorus-rich inorganic compounds like vivianite but also support the purification of water within tailing ponds. Successfully implementing this approach could greatly reduce soil pollution, conserve water resources, and reclaim valuable elements from industrial waste, thereby promoting a more sustainable and circular economy. |
1. Introduction
The challenge of treating wastewater from industrial and domestic human activities is growing more urgent with each passing year. Over the past few decades, water resources have suffered increasing contamination from toxic chemicals,1 impacting both wildlife and human well-being. According to the World Health Organization data, in 2012, approximately one-sixth of the global population lacked access to safe drinking water. By 2020, this figure had risen to nearly 25%.Addressing this challenge requires significant resources and the development of cost-effective cleaning techniques. While water discharged during mining operations holds promise as a valuable resource, it remains susceptible to contamination from domestic and industrial wastewater.2 Conventional environmental engineering technologies, such as artificial wetland treatment systems,3–6 offer a solution. Constructed wetlands leverage natural microbial, biological, physical, and chemical processes to treat wastewater at minimal financial cost.
As previously noted,7,8 sewage sludge in wetlands harbors significant amounts of phosphorus, posing a considerable risk of eutrophication.7–9 Moreover, the production of in-sludge phosphorus through a sulfate-reducing process exacerbates this issue.8,10 Phosphorus, essential for primary producers in both aquatic and terrestrial ecosystems, can only be removed from the water column through sedimentation followed by disposal.11,12 However, efficient phosphorus separation technologies are lacking, particularly under non-sulfide reducing conditions where vivianite (Fe3(PO4)2·8H2O), the most wide-spread stable ferrous phosphate mineral, prevails.13 Recent breakthroughs have demonstrated that this mineral can be extracted through wet magnetic separation,7 offering prospects for cleaning wetlands, supplying clean water for human use, and facilitating phosphorus recovery, notably in agriculture.14,15
Vivianite offers significant potential as a raw material for phosphorus fertilizers, synthetic components for lithium batteries, and materials for photocatalytic CO2 reduction, among other applications.16–18 It also functions as a major phosphorus sink in both freshwater19 and marine environments.20–22 In anoxic environments rich in phosphate, the formation of vivianite competes with sulfidization for free Fe(II).20,21,23 Despite the existing uncertainties about the natural formation of vivianite, it is generally agreed that its genesis is influenced by the rate of sulfide production, which in turn depends on the availability of Fe(II).19,24
Studying the biochemical and biophysical processes in wastewater sludges is essential for advancing artificial treatment facilities.5,8,15 Moreover, the role of microorganisms in enhancing connectivity between aquatic habitats, especially in isolated wetlands, is not yet fully understood.25 There is clear evidence of a complex interplay between bacterial reduction – specifically the rate of Fe(II) release – and aqueous geochemical conditions, which include composition, pH, system kinetics, thermodynamics, and the surface chemistry of oxides.26 These interactions lead to the formation of either a dominant phase or a mixture of phases. The dynamics between biotic and abiotic factors involved in mineralization, as well as the conditions that affect the distribution of reduced phases such as magnetite and siderite during microbial Fe(III) reduction, remain poorly understood. It is well-known that the metabolism of anaerobic bacteria promotes the formation of magnetite and siderite, with their distribution being partially dependent on pH and reactivity.26,27 The similarity in mineralization between vivianite and siderite underscores the importance of biomineralization in the formation of wetland deposits, highlighting the need for comprehensive investigations.28,29 This emphasizes the role of biomineralization processes in the formation of wetland deposits and requires thorough investigations.
The resurgence of interest in biogenic vivianite (Fe3(PO4)2·8H2O), which contains bound phosphorus, stems from the role of iron in the long-term retention of phosphorus under anoxic conditions and its potential for phosphorus removal from wastewater.24,30 Recent research on biotic mineralization by sulfate-reducing bacteria (SRB)14,24,31–33 has demonstrated that Fe3+ is primarily reduced by biogenic S2−, which promotes the formation of vivianite associated with the SRB. Moreover, SRB are employed in various wastewater treatment technologies, including bioreactors and constructed wetlands, for metal removal.34 The purification process using SRB depends on the binding of metal ions to H2S produced during bacterial metabolism.34 In bioreactor technologies, Desulfobacterota SRB are particularly critical, while in wetlands, a range of Desulfosporosinus species are predominant. The capability of pure Desulfosporosinus cultures to produce sulfides is well-established, leading to intensive research into the effects of SRB on the formation of sedimentary iron sulfide minerals.35 Mackinawite and greigite are the most frequently reported biomineralized phases of SRB cultures.36–43 Recent studies have suggested that merging anaerobic digestion of waste activated sludge with in situ sulfate reduction by SRB could be a novel method for phosphate release.8,44 However, phosphorus-containing minerals in SRB species have not been thoroughly investigated.
Thus, this study focuses on the biomineralization processes during the lifecycle of Desulfosporosinus metallidurans, isolated from the Komsomolskoye tailing dump in Kemerovo oblast, Russia. The primary aim is to thoroughly investigate for the first time the phase composition and the magnetic behavior of the sample produced by Desulfosporosinus metallidurans under laboratory conditions to potentially recover phosphorus.
2. Materials and methods
2.1 Bacterial strain and biomineralization experiments
Desulfosporosinus metallidurans OLT was isolated from a microbial mat in a tailing dam at a gold ore mining site in Komsomolsk, Kemerovo oblast, Russia.45 The tailing dump is located on the territory of the former Komsomolsk gold recovery plant (Fig. 1). The plant processed gold-arsenopyrite quartz ores using cyanidation. Gold-containing waste was also processed at the plant. The tailing dump is a natural basin that has been filled with runoff from the gold recovery plant since 1964. The dump covers an area of 146 thousand m2 and has a volume of approximately 810 thousand m3. It contains approximately 1.1 million m3 of accumulated material The dump is surrounded by terrain on three sides and has an embankment dam on the fourth side. | ||
| Fig. 1 Location of the tailing dump in the Komsomolsk location, Kemerovo oblast, Russia. | ||
The OL strain has an acidic growth optimum of 5.5 and can tolerate elevated concentrations of metal ions, including Cu (500 mg L−1), Ni (700 mg L−1), and Co (250 mg L−1). For the biomineralization experiments, the culture was grown in serum bottles (500 mL) containing Widdel & Bak (WB) medium.46 The WB medium contained the following components per liter: Na2SO4 (4 g), KH2PO4 (0.2 g), NH4Cl (0.25 g), NaCl (1 g), MgCl2·6H2O (0.4 g), KCl (0.5 g), CaCl2 (0.113 g), vitamin solution (2 mL), micro element solution (1 mL), Na2SeO3 (final concentration of 23.6 μM), and Na2WO4 (final concentration of 24.2 μM) solutions (1 mL each). The pH of the medium was adjusted to 5.5 using 1 M H2SO4 solution. Na2S·9H2O (0.36 g L−1) was used as a reducing agent. Each bottle also contained an iron wire (100% Fe), as described previously.47,48 Lactate (18 mM) was used as an electron donor. Control bottles were not inoculated with D. metallidurans. The serum bottles were filled to the top, closed, and sealed with aluminum caps. The bottles were incubated in the dark at 28 °C for 14 days. The precipitates obtained in the experiment were harvested by centrifugation (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g, 15 min, 15 °C), washed with distilled water (10 min), dried, and ground manually.
100 g, 15 min, 15 °C), washed with distilled water (10 min), dried, and ground manually.
2.2 Measurement methods
X-ray powder diffraction data for the Rietveld analysis were collected using a Bruker D8 ADVANCE powder diffractometer with Cu-Kα radiation and a VANTEC linear detector at room temperature. The 2θ step was 0.016° and the counting time was 1 second per step.Electron microscopy and microdiffraction investigations were carried out on a Hitachi HT7700 transmission electron microscope with an accelerating voltage of 100 kV and an energy-dispersive X-ray spectroscopy (EDS) microanalysis system. Specimens were prepared by shaking the nanoparticle powder in alcohol in an ultrasonic bath and depositing the obtained suspension onto support meshes with a perforated carbon coating. The time of accumulation for the EDS analysis was determined by the spectrum assembly quality that allows for the quantitative processing and was no shorter than 10 minutes. The phase composition of the sample in a local field was determined by the selected-area electron diffraction (SAED) technique.
Mössbauer spectra of the sample were obtained on an MS-1104Em spectrometer (Research Institute of Physics, Southern Federal University) in the transmission geometry with a Co58(Rh) radioactive source. The measurements were performed in the temperature range of 4–300 K using a CFSG-311-MESS cryostat with a sample in the exchange gas based on a closed-cycle Gifford-McMahon cryocooler (Cryotrade Engineering, LLC). The spectra were processed by varying the entire set of hyperfine parameters using the least squares method in the linear approximation. The spectra were fitted using Lorentzian lines, taking into account the broadening caused by the magnetic and crystalline inhomogeneity of the sample.
X-ray absorption measurements were carried out at the Cosmos beamline of the VEPP-4M storage ring at the Budker Institute of Nuclear Physics SB RAS (Novosibirsk).49 The storage ring was operated at 2.5 GeV with an average current of 7 mA. The synchrotron radiation was monochromatized by a double crystal monochromator with two Si(111) crystals. Sulfur K-edge X-ray absorption near-edge spectra (XANES) were measured in the total fluorescence yield mode using a precision silicon photodiode, SPD.50 The samples were prepared by mixing 1 cm by 1 cm by ∼50 μm polymethyl methacrylate (PMMA51) with the sample powder. All X-ray absorption measurements were done at room temperature.
Magnetic measurements were performed using an original vibrating sample magnetometer (VSM)52 and a Quantum Design Physical Property Measurement System (PPMS-9). The temperature dependences of the magnetization were measured in the zero-field cooling (ZFC) and field cooling (FC) modes in the temperature range of 4–300 K. First-order reversal curve (FORC) measurements were performed on a LakeShore 8604 VSM at a temperature of 300 K in dc magnetic fields of 0–15000 Oe with a step of 50 Oe. The FORC diagrams were calculated using the open-source FORCsensei package,53 which is freely available from https://forcaist.github.io. The construction of FORC diagrams is described in detail, e.g., in ref. 54.
3. Results
3.1 X-ray diffraction
Fig. 2 shows the experimental XRD pattern. Due to the peak overlap and low peak/background ratio, atomic coordinates and thermal parameters were fixed. The refinement was stable and yielded low R factors (Table 1). The pattern exhibits low intensity and signal-to-noise ratio, which may be attributed to the low crystallinity of the sample. As shown below by the TEM, Mössbauer, and XPS measurements, the investigated biological sample contains a significant amount of ultrafine ferrihydrite nanoparticles.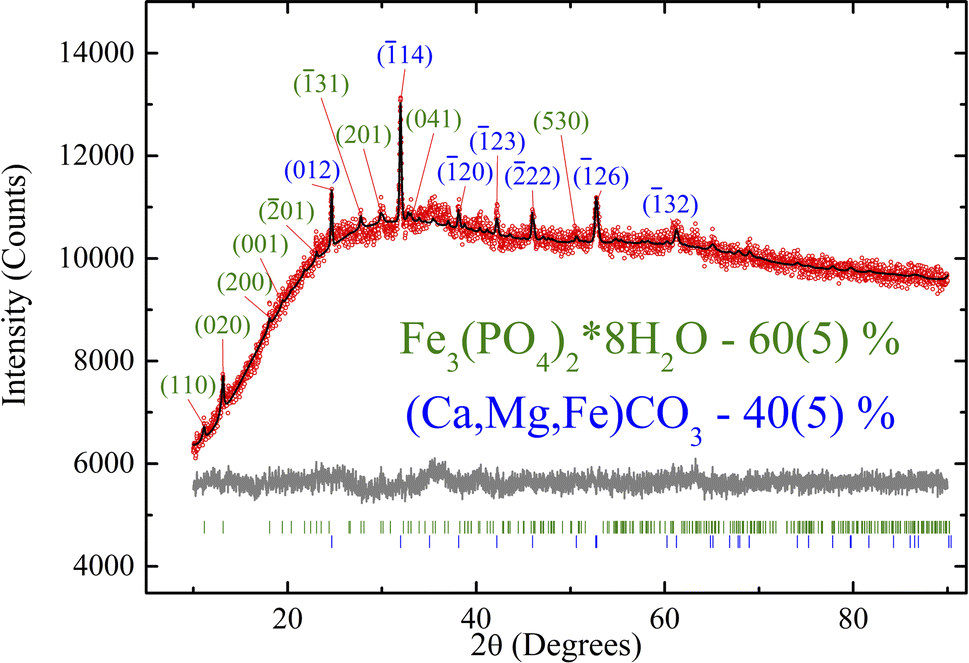 | ||
| Fig. 2 Rietveld-refined XRD patterns. Red dots show the experimental XRD data, and black ones show the theoretical fitting by the XRD data. The difference pattern between the experiment and theoretical fit is shown at the bottom. The characteristic Miller indices of vivianite and siderite are indicated. | ||
| Crystalline phase | Fe3(PO4)2·8H2O | Ca(1−x−y)Mg(x)Fe(y)CO3 |
|---|---|---|
| Weight, % | 60(5) | 40(5) |
| Sp. gr | C2/m |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
| a (Å) | 10.116(9) | 4.713(2) |
| b (Å) | 13.43(1) | 4.713(2) |
| c (Å) | 4.709(5) | 15.345(6) |
| α (°) | 90 | 90 |
| β (°) | 104.52(6) | 90 |
| γ (°) | 90 | 120 |
| V (Å3) | 619(1) | 295.2(2) |
| R B, % | 0.29 | 0.88 |
| 2θ-interval, ° | 10–90 | |
| R wp, % | 1.26 | |
| R p, % | 0.99 | |
| χ 2 | 1.25 | |
All the crystal peaks are indexed in Fig. 2. The XRD data show only the row crystallographic phases. The siderite crystalline phase (40 wt%) shows the indices typical of trigonal symmetry (Rc), and vivianite (60 wt%) refers to monoclinic C2/m sp. gr. Both phases exhibit specific parameters similar to those reported previously in ref. 55,56. Therefore, this structure was taken as a starting model for the Rietveld refinement in the TOPAS 4.2 software (Bruker AXS, Karlsruhe, Germany, 2008). The refinement was stable and yielded low R factors (see Table 1 and Fig. 2).
Unfortunately, small quantities and nanoscale crystallites are poorly detected using X-ray diffraction. Additionally, the broad background signal can be attributed to ferrihydrite nanoparticles. Furthermore, the investigated sample exhibits sulfur-containing crystal phases, as shown by other experimental techniques described below. XRD analysis is unfortunately unable to provide the exact atomic substitution degree in the obtained phases. To elucidate this, we measured the Néel temperature of these crystal phases using magnetometry, which provides at least qualitative analysis (Section 3.4).
3.2 Transmission electron microscopy
The morphology and electron diffraction of the sample were studied using TEM to analyze local fields. The microphotographs presented in Fig. 3 show the composition of vivianite and siderite microcrystals and Fe3S4 nanorod crystals surrounded by an ensemble of quasi-spherical ultrafine nanoparticles. In contrast, the reference sample contains no traces of iron sulfide rods or siderite and vivianite crystals (Fig. S1†). Despite the XRD data, our microscopic measurements unambiguously indicate the presence of an Fe3S4 nanorod phase, which can be determined from the interatomic spacings and microdiffraction pattern of the investigated local area (inset in Fig. 3). The obtained interatomic distance of 2.98 Å corresponds well to the greigite mineral. | ||
| Fig. 3 Microphotographs of the sample on different scales. Inset: nanorod Fe3S4 crystals surrounded by an ensemble of ultrafine nanoparticles on a scale of 20 nm with the interplanar spacings corresponding to Fe3S4. Bottom: histogram for ferrihydrite nanoparticles (Fe2O3·nH2O). The point reflections in the inset correspond to the Fe3S4 phase. | ||
In our previous studies on the biomineralized composite produced by another SRB, Desulfovibrio sp. A2,43 we observed a similar situation. Here, we obtained nearly identical size distribution of ultrafine ferrihydrite nanoparticles (average size of 2.5 nm), but we also observed the formation of additional siderite and vivianite crystal phases synthesized during the bacterial life cycle. The EDS analysis suggested the presence of these iron compounds.
The EDS study indicated the possibility of the occurrence of the aforementioned iron compounds. The obtained EDS data are presented in Fig. 4. The EDS spectrum is shown in the lower panel (Fig. 4c). Mapping shows that the distribution of the atoms is almost uniform throughout the studied area. However, it is clearly seen that the type of the chemical element atoms inside the local areas is significantly different. According to the TEM image, vivianite crystallites are coarse, while nanorod-shaped greigite is characterized by lower crystallinity. The Al (around 1.5 keV) and Cu (around 8 keV) lines are caused by the sample holder and should therefore be ignored.
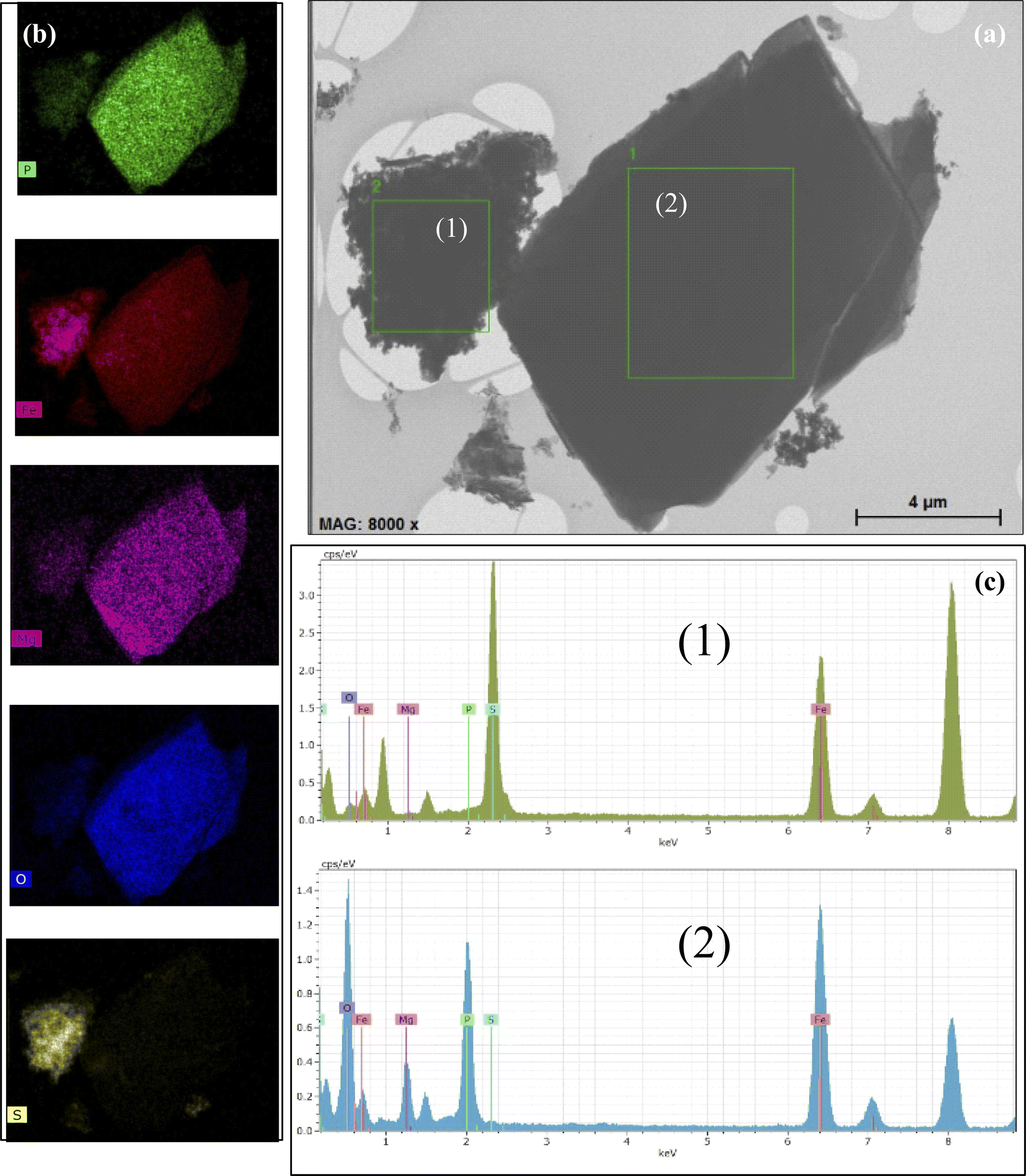 | ||
| Fig. 4 EDS mapping of the local field for the sample on a scale of 4.0 μm. (a) Two analyzed local areas and (b) EDS elemental mapping images of Fe, O, P, Mg, and S atoms. (c) Corresponding EDS spectra of two local fields. | ||
The mapping of the selected areas, as illustrated in Fig. 4b, shows a significantly higher concentration of oxygen atoms compared to iron, although the sulfur concentration appears relatively low. It is also important to note that vivianite contains a certain amount of Mg atoms. The EDS data are detailed in Table S2.† For selected area 1, the presence of Fe, P, O, Mg, and S atoms has been confirmed, while selected area 2 contains only Fe, O, and S atoms.
3.3 X-ray absorption spectroscopy
X-ray absorption spectroscopy (XAS) technique has been employed to directly probe the electronic structure of ligands and their metal bonding. Sulfur is crucial because it acts as an electron acceptor for sulfate-reducing bacteria. Therefore, identifying the electronic states of sulfur present in the samples is essential. The sulfur K-edge X-ray absorption spectrum, displayed in Fig. 5, reveals that the near-edge region of the XAS spectrum is dominated by dipole-allowed bound-state transitions of the 1s electron (at a K-edge- to vacant electronic orbitals with significant p-character), providing a sensitive measure of the electronic structure.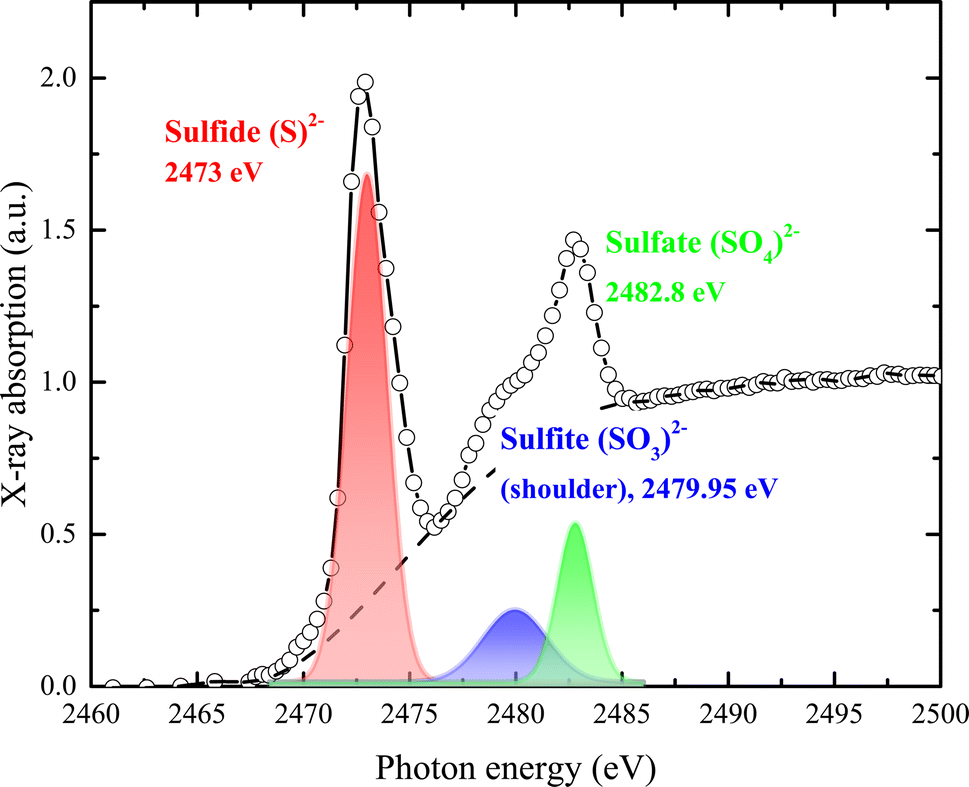 | ||
| Fig. 5 Normalized S K-edge X-ray absorption spectrum with assigned pre-edge transitions characteristic of the sulfide (S2−), sulfate (SO4)2−, and sulfite (SO3)2− oxidation states. | ||
As shown in Fig. 5, sulfur exhibits a rich K-edge spectrum consisting of a complex array of peaks with two main characteristic X-ray absorption features at approximately 2473 eV and 2482 eV, arising from different sulfur species. The primary feature at around 2473 eV corresponds to the S2− sulfide oxidation state, with the first peak arising from the electron transition of S 1s electrons to unoccupied hybridized orbitals of S 3p. Additional peak features represent sulfur in several oxidation states, including sulfate (SO4)2− (i.e., S6+) and sulfite (SO3)2− (i.e., S4+). Given that the edge energy is influenced by factors beyond simple electronic or valence states, no quantitative models were attempted. Instead, we modeled the experimental spectrum as the sum of one or two arctangent functions and several Gaussian functions. The relative concentration of each sulfur species was calculated based on the area under each Gaussian peak relative to the total area under all peaks. Our assumptions were: (1) the arctangent step functions represent the transition of ejected photoelectrons to the continuum; (2) the Gaussian functions represent 1s to 3p transitions; (3) the full width at half-maximum (FWHM) of each Gaussian function is constrained by their intensity and resolution; and (4) sulfur speciation was determined by the energy position of the Gaussian peak, with the relative abundance quantified from the peak area. The relative strength of absorption peaks is proportional to the quantity of the various sulfur species, providing a quantitative measure for K-edge XANES spectra. Sulfur contents were estimated from the peak area after subtracting the background, using a trigonometric function closely approximating the conventional arctangent function. The relative content of each species was quantified as 4 for S2−, 0.9 for (SO3)2−, and 1.11 for (SO4)2−.
A sharp peak at about 2482 eV typically indicates the presence of sulfate species in complex sulfur-bearing materials, with the peak size proportional to the amount of sulfate. When multiple sulfur species are present, the S K-edge XAS spectrum becomes a composite of the separate absorption edges, effectively approximating the weighted sum of these edges as the absorption cross-section of the absorber atom is relatively consistent regardless of the chemical state. Changes in the S K-edge XAS spectrum with variations in the proportions of S2− and S6+ are illustrated by the combined spectra in Fig. 5. Even when sulfate comprises only 10% (or less) of total sulfur species, it significantly impacts the spectrum, producing a recognizable signature feature at 2482 eV.
Phosphorus (P) K-edge XAS has proven to be an exceptionally effective spectroscopic method for studying materials containing phosphorus. This technique has been used to explore a wide range of chemical phenomena, including covalent metal–phosphorus bonding, redox non-innocence in ligands, spin state transitions related to molecular magnetism, and luminescence. Next, we will describe the XAS measurements performed at the P K-edge.
The experimental phosphorus K-edge XAS spectrum of our sample is displayed in Fig. 6. The spectrum features a pre-edge transition at 2150 eV, with the white line appearing at 2154.2 eV. A main broad peak, centered at 2170 eV, is observed at higher energies. The P K pre-edge signal, produced by transitions from P-1s to Fe(d,p) orbitals, indicates the presence of P–Fe molecular bonds.57 The spectrum reveals a K-edge absorption corresponding to a dipole-allowed 1s–3p transition, and a second feature at higher energy suggests a dipole-allowed transition of the 1s electron into conduction states that involve mixing of P 3p and Fe 4p character. Additionally, a pre-edge feature visible below the edge onset arises from mixing of P 2p and unoccupied metal 3d states.
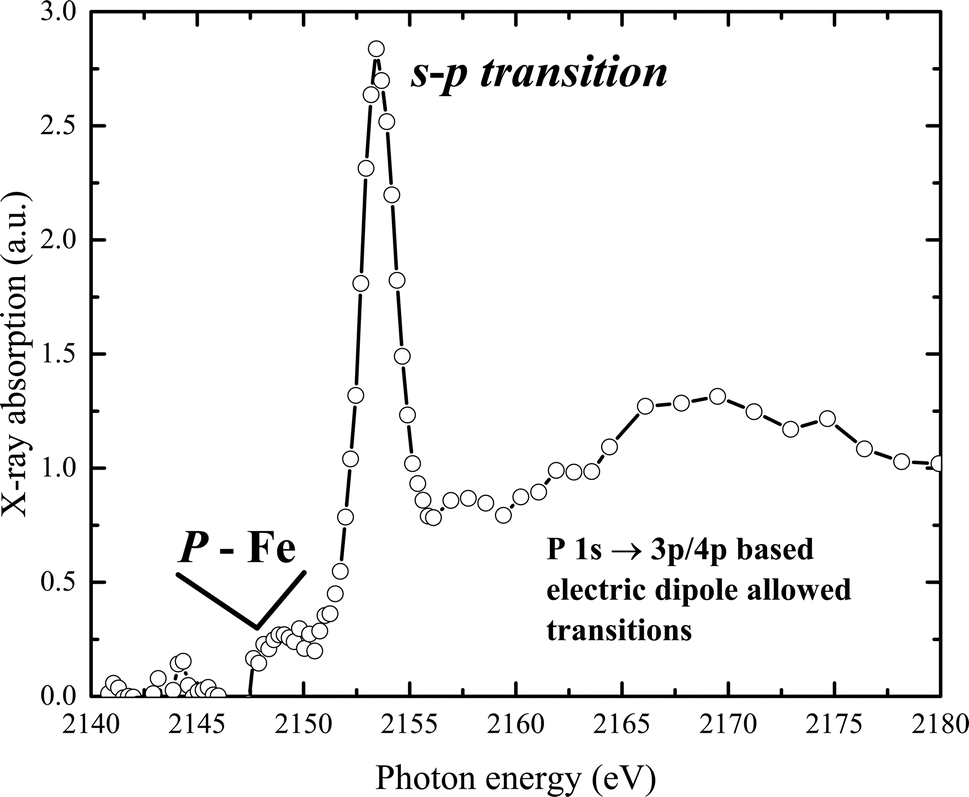 | ||
| Fig. 6 Normalized P K-edge X-ray absorption spectrum of the sample, measured in transmission mode. | ||
Interestingly, this distinct pre-peak feature is practically absent in the XAS spectra of elemental phosphorus (with P–P bonds), materials with metal–phosphorus bonds (such as CoP, FeP), phosphate-containing solids like sodium pyrophosphate, KH2PO4, or Co2(PO4)2, and in phosphate in an aqueous solution or related compounds.58–63 However, similar peak features have been observed in samples where phosphate ions are associated with metal (hydr)oxide species, for example, in crude soil or mud samples, and in phosphate in hydrated metal–phosphorus materials, including vivianite.64–66
3.4 Magnetic characterization
At room temperature, the sample exhibits magnetic hysteresis (Fig. 7a); its coercivity HC is approximately 300 Oe (see Subsection 3.4.2), corresponding to ferrimagnetic greigite particles of micron size. The M(H) dependence becomes irreversible in fields stronger than 3 kOe. The field-linear behaviour beyond 4 kOe reflects the paramagnetic responses of vivianite and siderite phases and the superparamagnetic (SPM) state of ferrihydrite nanoparticles, in contrast to the magnetization curve of the reference sample shown in Fig. S3†. Notably, the slope of the M(H) dependence for greigite in strong fields is typically much weaker.73
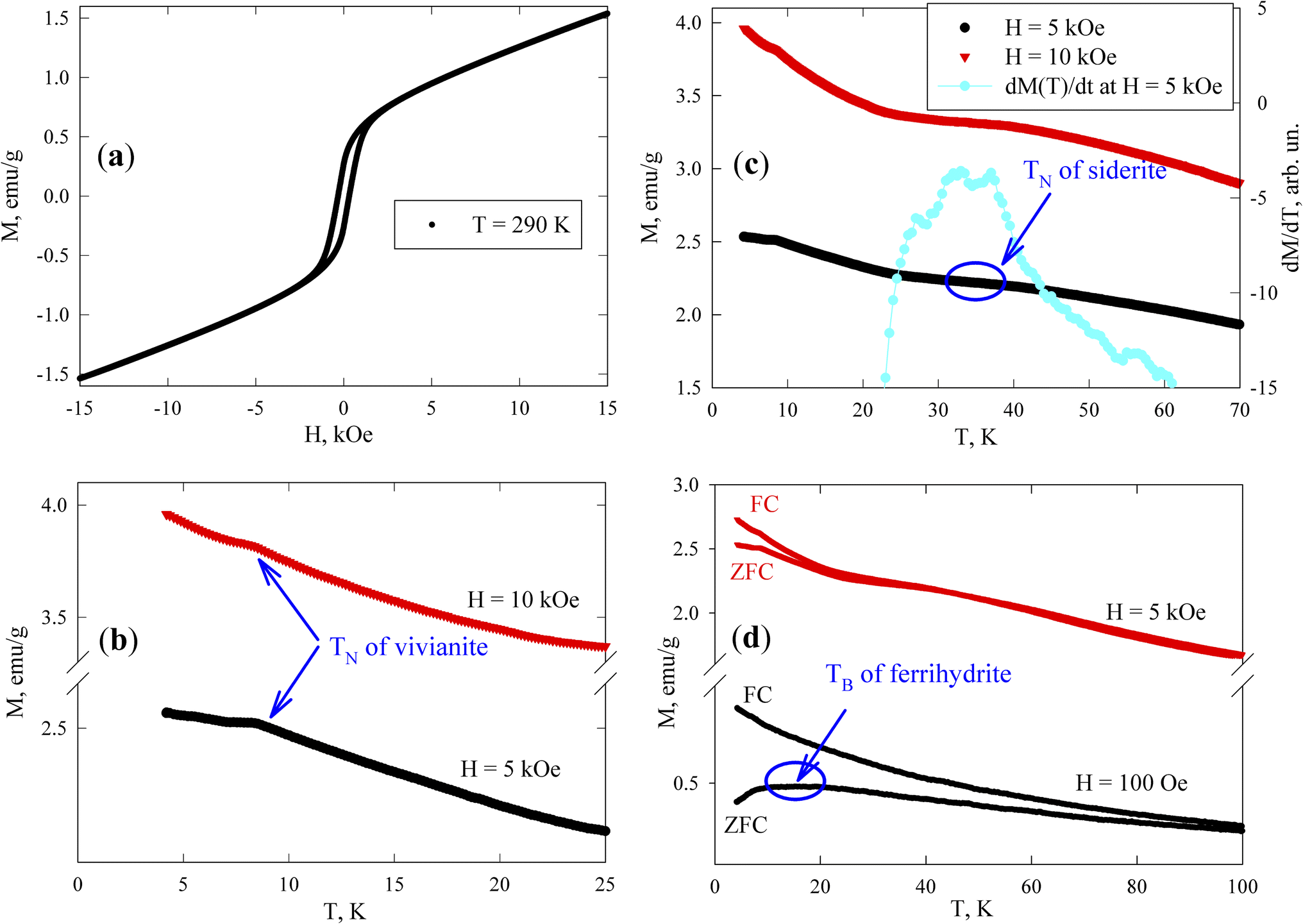 | ||
| Fig. 7 (a) Field and (b–d) temperature dependences of the magnetization. (c) Derivative dM(T)/dH at H = 5 kOe; the ordinate axis is on the right. The experimental parameters (temperature, external field, and thermomagnetic prehistory (FC and ZFC)) are indicated inside the figure. Characteristic temperatures corresponding to the Néel temperatures (TN) of (b) vivianite and (c) siderite and (d) the blocking temperature (TB) of ferrihydrite are shown. | ||
Fig. 7b presents the low-temperature M(T) dependences measured in fields of 5 and 10 kOe. Vivianite behaves as an antiferromagnet with a Néel temperature (TN) of about 8.8 K.72 Anomalies in M(T) (kinks) near this temperature (TN ≈ 8.4 K) suggest the typical behavior of a polycrystalline antiferromagnet where an M(T) peak is usually observed at the Néel point. Below 8 K, the M(T) dependences continue to rise as the temperature decreases, influenced by the behavior of ferrihydrite particles, while the magnetic response from greigite should remain almost constant in fields of 5 and 10 kOe, and siderite is already in the antiferromagnetic state at these temperatures.
According to other studies,72 the Néel temperature of siderite (FeCO3) is 38 K. However, given that our sample is a partially substituted mineral (as noted in Subsection 3.1 with the formula Ca(1−x−y)Mg(x)Fe(y)CO3), variations in Néel temperatures are expected among different particles (microcrystals) and within a single microcrystal. The data in Fig. 7c show that between 30 and 40 K, the M(T) behavior changes significantly, exhibiting a change in the curvature sign around 35 K, which appears as a broad maximum in the derivative dM(T)/dT (with the dM/dT axis on the right-hand scale in Fig. 7c, at H = 5 kOe). This temperature range also includes contributions to M(T) from vivianite and ferrihydrite (both varying approximately as 1/T), and a nearly constant contribution from greigite, leading to a manifestation of an almost pure siderite phase with low substitution levels in the magnetic behavior of the sample, as supported by our EDX analysis.
The magnetic moments of iron atoms in ferrihydrite are ordered antiferromagnetically.74 This mineral exists only at the nanoscale and contains defects that induce an uncompensated magnetic moment (where the number of spins up within one nanoparticle is unequal to the number of spins down), as predicted by Néel75 and confirmed in numerous experiments with various antiferromagnetic nanoparticles.76–78 Ferrihydrite particles, 2–4 nm in size, have a magnetic moment of 150–300 Bohr magnetons (30–60 uncompensated iron atom moments per particle).78–82 At sufficiently high temperatures, these moments exhibit SPM behavior, with SPM blocking occurring at low temperatures (10–50 K).79–84 The SPM blocking temperature (TB) typically manifests as a maximum in the ZFC M(T) dependence. Below TB, the magnetic moments of particles are in a blocked state, characterized by irreversible magnetization curves and influenced by thermomagnetic prehistory, as seen in the difference between M(T)FC and M(T)ZFC dependences. The effect of SPM blocking in ferrihydrite nanoparticles in our sample is evident as a broad M(T)ZFC maximum in a field of H = 1 kOe at temperatures of 10–20 K (Fig. 7d), a range inconsistent with the transition temperatures of vivianite and siderite phases to the antiferromagnetic state; additionally, micron-sized ferrimagnetic greigite particles should exhibit no features in a field of H = 1 kOe at temperatures up to 100 K, which is much stronger than the coercivity. The broad maximum in the M(T)ZFC curve in a field of H = 1 kOe might also indicate strong interparticle magnetic interactions.79–84 The presence of ferrihydrite nanoparticles in the magnetizing field of micron-sized greigite particles suggests that the SPM blocking processes might represent collective freezing of particle magnetic moments.84–89 This will be experimentally confirmed below, in Subsection 3.4.2.
The irreversible behavior of the magnetization of blocked ferrihydrite nanoparticles is evident in the differences between the M(T)ZFC and M(T)FC dependences in a strong field of H = 5 kOe, as shown in Fig. 7d. It is well known that an increasing external field lowers the temperature at which SPM blocking of magnetic nanoparticles occurs. Additionally, due to the distribution of particle sizes and magnetic moments, in a strong field, the peak can disappear from the M(T)ZFC curve, while a significant influence of thermomagnetic prehistory remains.79,84,87,90,91 As mentioned earlier, in a field of H = 5 kOe, ferrimagnetic greigite is saturated (also confirmed by low-temperature data73) and the discrepancy between the M(T)ZFC and M(T)FC curves in Fig. 7d is attributed to the blocking processes of ferrihydrite nanoparticles. Therefore, these magnetic measurements confirm the presence of all magnetically active phases previously identified: vivianite, siderite, greigite, and ferrihydrite.
Measuring the loops of field dependences of magnetization provides critical insights into both the magnetic order of a substance and its magnetic hardness. However, specialized techniques, such as the first-order reversal curve method, can significantly enhance the information gleaned from M(H) data. The method involves measuring and analyzing a series of partial magnetization reversal curves, which are generated by reducing the magnetic field to a specific value and then saturating the system. In our measurements, these partial magnetization curves were recorded in increments of 50 Oe.
The mathematical analysis was grounded in the classical Preisach model of a hysteresis loop, which models a single-domain particle (hysteron) with uniaxial anisotropy, unit coercivity, and unit magnetization.92 Using the experimental dataset, a 2D magnetization distribution was derived as a mixed second derivative of the magnetization with respect to the initial and current fields, allowing for the isolation of contributions from different subsystems to the magnetic properties of the multiphase samples. The resulting FORC diagrams (Fig. 8) are maps of the magnetic response of all crystalline phases within the sample, displaying their irreversible magnetizations in terms of coercivity and interaction field distribution (the Hc and Hu axes, respectively).
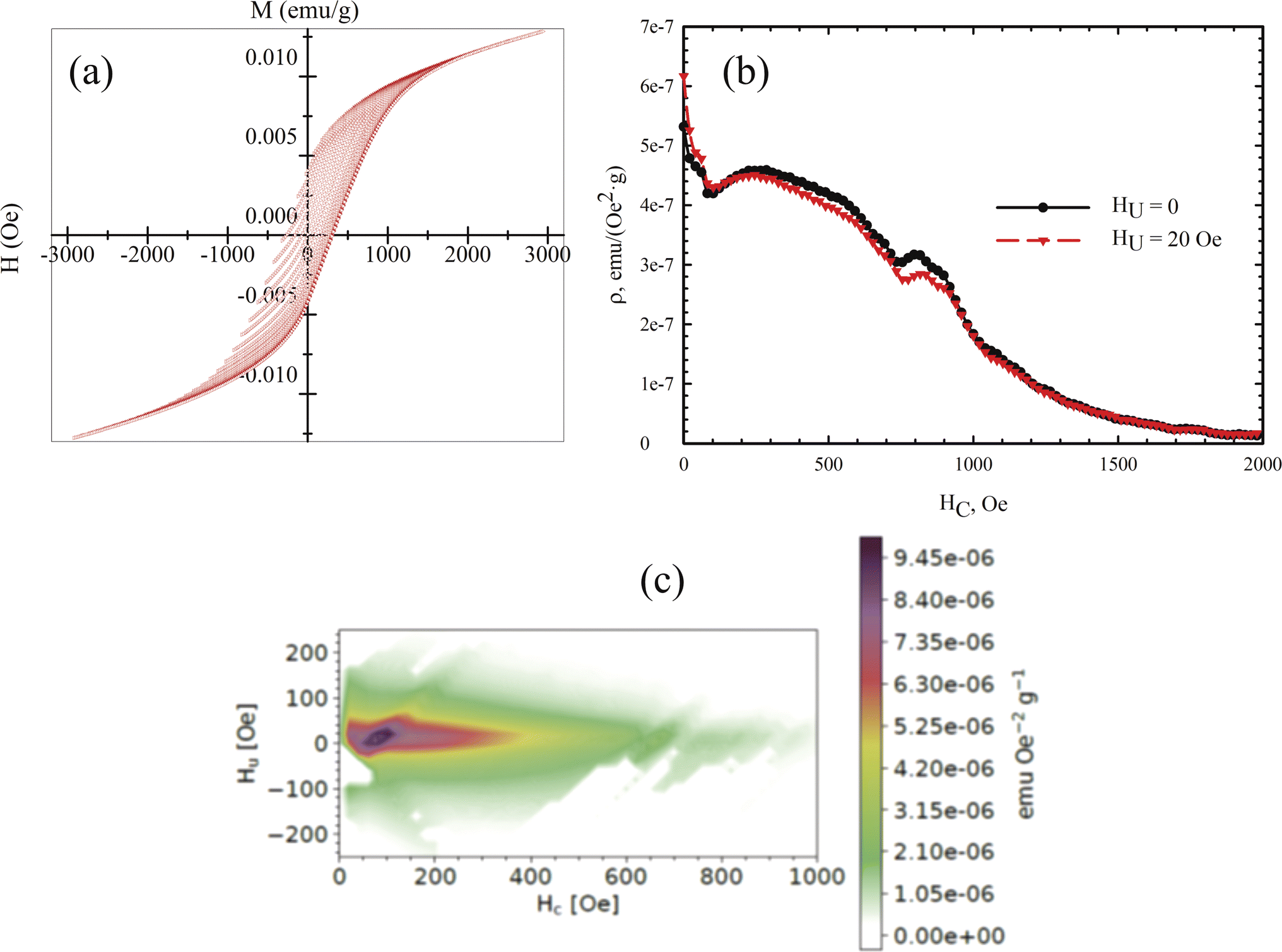 | ||
| Fig. 8 (a) Partial M(H) loops obtained with a step of 50 Oe at a temperature of 300 K. (b) Profile of the coercivity distribution in the sample. (c) Calculated FORC diagram of the sample. | ||
Given that the magnetic ordering temperatures of the main crystalline phases in vivianite and siderite are around 8 and ∼38 K respectively,72 these phases do not contribute to the coercivity of the samples. Mössbauer spectroscopy has shown that ferrihydrite nanoparticles are in the SPM state, indicating that the primary contribution to coercivity is made by nanorod-shaped greigite (Fe3S4) crystallites.
According to the FORC diagram obtained, the sample exhibits a wide coercivity distribution along the Hc axis, suggesting a varied crystallite size distribution. The FORC diagram profile along the sample interaction axis (Fig. 8b) shows a narrow distribution, with the maximum located around 300 Oe, consistent with our previous studies of biomineralized greigite.43 FORC diagrams of noninteracting single-domain particles typically feature a central spike at a certain coercivity value.93 In our case, the FORC diagrams are elongated with a predominant vertical distribution, and the signal intensity decreases as the coercivity exceeds 400–500 Oe. Similar FORC diagrams have been observed for multi- and pseudo-single-domain natural particles,94–96 aligning with the size of the greigite crystallites. A similar extension along the HC axis was reported for noninteracting bacterial magnetosomes in ref. 97 though these did not include the region near zero HC value. The interaction of superparamagnetic ferrihydrite nanoparticles likely contributes to the near-zero coercivity region, qualitatively aligning with results from previous studies.97,98 The presence of SPM nanoparticles can explain the second local maximum near zero HC value in the FORC diagram.
3.5 Mössbauer measurements
Fig. 9 displays the experimental spectra recorded at temperatures between 4.2 and 300 K. The processing of the spectra is represented by a solid line, and the Mössbauer parameters of the components are listed in Table S3.† The room-temperature spectrum consists of a combination of quadrupole doublets with varying chemical shifts and a broadened sextet. The parameters of this sextet are attributed to the greigite (Fe3S4) crystalline phase,43,99 align with electron microscopy findings, and indicate a relative atomic content of this mineral of no more than 5%. This low concentration, combined with a significant X-ray amorphous contribution, could obscure its detection by XRD.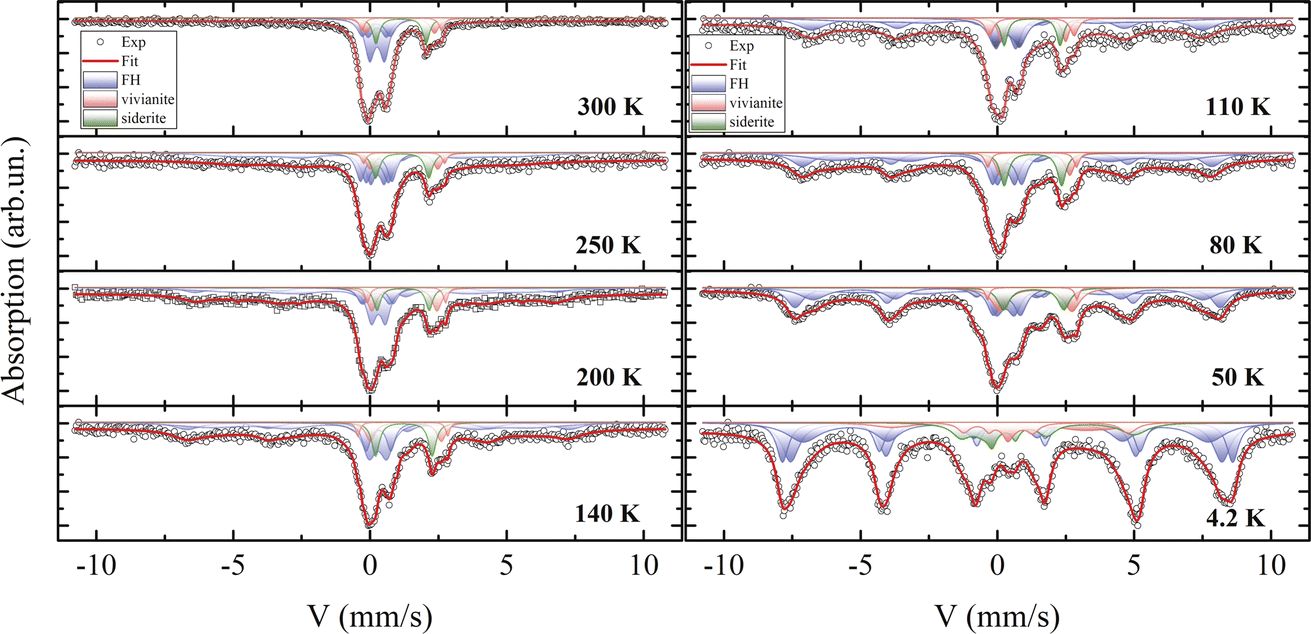 | ||
| Fig. 9 Mössbauer spectra of the sample across a temperature range from 4.2 to 300 K. The components associated with different crystalline phases are indicated in various colors. The solid line represents the mathematical modeling of the spectra. | ||
As reported in the literature,100 vivianite and siderite contain iron in the Fe2+ charge state and are paramagnetic at 300 K, aligning with both Mössbauer and magnetization measurements. The mathematical processing of the spectra reveals the presence of iron in two charge states, Fe3+ and Fe2+. Based on X-ray diffraction and electron microscopy data, the Mössbauer parameters for Fe2+ correlate well with previous reports for vivianite7,101 and siderite.102 Electron microscopy suggests that the second quadrupole doublet in the vivianite spectrum may be due to partial substitution of iron by Mg2+ atoms, adding disorder to the structure. The phase area ratios in the spectra correspond closely with Rietveld refinement results from XRD data.
Notably, most of the spectrum area is associated with an iron charge state of Fe3+ with a chemical shift δ of about 0.35 mm s−1,103 observable in three distinct states at 300 K. These states differ in the quadrupole splitting Δ and relative site occupancy. The parameters of these quadrupole doublets match those of iron oxyhydroxide (ferrihydrite), which exists in the SPM state due to the extremely small size of its nanoparticles, a condition well-studied104 and often occurring under aerobic conditions via microbial–mineral interactions.102
Mössbauer spectroscopy studies on vivianite, including biogenic types, have occasionally detected a minor presence of iron in the +3 charge state.101,105 Similar Fe3+ impurities have been noted in siderite phases in several studies,7,106 though often unidentified. However, the averaged Mössbauer parameters for ferrihydrite107,108 closely resemble those reported in both cited and current studies.
For ferrous compounds vivianite and siderite, Zeeman splitting of their doublets is observed below 50 K, consistent with our magnetization measurements and published data.72 These results support earlier Mössbauer findings on the magnetically ordered state of these phases, where an extra electron compared to Fe3+ causes strong electron–nuclear interactions, leading to a significant asymmetry of the Zeeman sextet, a large quadrupole shift Δ, and a comparatively weaker hyperfine field Hhf on nuclei.
4. Discussion
Biomineralization can serve as an environmentally friendly technology for producing a wide range of functional materials.109,110 Previous studies have highlighted the significant role of microorganisms in the sorption of heavy metals and their impact on greenhouse gas emissions in natural aquatic systems.5,8,24,31,34 This is particularly relevant to our current research. From the data presented in the current paper and previous investigations,43 we have observed that greigite nanorods are a biomineralized crystal phase produced by the Desulfosporosinus metallidurans OLT species, while we cannot definitively state that the phases of vivianite (Fe3(PO4)2·8H2O) and siderite (FeCO3) are directly produced by these bacteria. Nevertheless, they are the major solid microbially mediated phases produced by the pure culture of sulfate-reducing D. metallidurans during dissimilatory iron reduction.111,112 Additionally, vivianite has been identified in steel corrosion products by SRB Desulfovibrio vulgaris113 and as a primary solid phase in cultures of sulfidogenic Tissierella.40Vivianite is commonly found in association with siderite in sedimentary deposits.106 Research by Lemos29 showed that siderite forms through the bioreduction of ferric iron compounds by microorganisms, and vivianite is sometimes found as a substitute for goethite and hematite in the siderite matrix. It has also been noted that poorly crystallized iron oxyhydroxide is the primary form of Fe(III) oxide reduced by bacteria in anoxic sediments.26,31,34,114 The 32 bioreduction of ferrihydrite to vivianite in a phosphate-rich medium, observed during the cultivation of S. putrefaciens CN32 species, was analyzed using XRD. This suggests that nanosized, X-ray amorphous ferrihydrite could be a nutrition source for bacteria during the bioreduction process.
To corroborate this point, consider a previous Mössbauer study of bacterial corrosion.102 The study revealed that the byproducts of bacterial corrosion contain a primary siderite component and an unidentified Fe2+ phase with a quadrupole splitting of about 2.4 mm s−1 at room temperature. This quadrupole splitting closely matches our data and aligns with the parameters for vivianite (Fe3(PO4)2·8H2O). Additionally, Sawicki and Brown102 observed that the spectrum collapsed around 20 K, consistent with our magnetization measurements, suggesting the presence of vivianite in the sediments. However, the composition of the microbial consortium used in these experiments remains unknown.
Considering that vivianite is a significant form of phosphorus storage in iron-enriched lacustrine sediments,13 we can highlight the crucial role of microbially mediated processes in forming sedimentary deposits. Furthermore, Einsele and Mortimer demonstrated that the strong interdependence of Fe and P cycles is largely due to the sorption capacity of iron oxides (oxyhydroxides), which efficiently bind orthophosphate ions (PO4)3−.115,116 Thus, it is plausible to conclude that bioreduction processes driven by microbial activity play a vital role in the Fe and P biogeochemical cycles.
Turning to the findings of this paper, vivianite is associated with other inorganic phases identified through XRD, Mössbauer spectroscopy, and magnetization measurements. These results align with observations from similar systems.117–119 Given that the metabolism of anaerobic bacteria facilitates the formation of magnetite and siderite,26,27 and considering previous observations of interaction between vivianite and siderite during microbial activity,28,29,102 it is likely that these interactions may enhance phosphorus extraction techniques. Significantly, our experiments noted the formation of a substantial fraction of SPM nanoparticles of poorly crystallized ferrihydrite.102,120 This form of iron oxyhydroxide, with its highly active and developed surface, exhibits a strong sorption capacity,43,107 which is pivotal in our studies.
As demonstrated by Prot,7 magnetic properties are crucial for utilizing biomineralized materials to prevent eutrophication in wetlands. Comparing our measurements with those from a previous study,7 we can assert that SPM ferrihydrite nanoparticles may play a key role in water sludge separation. This assertion is supported by Mössbauer measurements from the same study,7 which show that up to 70% of the compound matches the characteristics of ferrihydrite. Our findings indicate that the SPM blocking of magnetic moments in ferrihydrite nanoparticles occurs at an unusually high temperature of 250 K, as evidenced by the appearance of an additional sextet (Table S3† and Fig. 9). This observation aligns with our FORC diagram, which reveals a significant contribution of these nanoparticles to the coercivity and interaction distribution within our sample. The interacting regions are predominantly composed of ferrihydrite nanoparticles absorbed onto rod-type greigite crystallites, as observed in TEM studies. Regarding our concept for purifying water sludge, SPM ferrihydrite plays a crucial role because it is a highly sorptive material. Given the substantial magnetic moment of greigite, it enables the magnetic separation of wastewater sludge.
Given these insights, biomineralization processes can be leveraged to capture bound phosphorus for industrial applications. Processing settled industrial waste efficiently purifies water and prevents eutrophication7,9 in artificial reservoirs while also extracting phosphorus for industrial reuse. This challenge can be addressed by developing a technology to separate vivianite – the primary phosphorus-containing mineral – from the mass of biomineral deposits. Vivianite, which absorbs 70 to 90% of the phosphorus in sewage sludge if sufficient iron is present,7,13 is paramount due to its paramagnetic nature under normal conditions, making separation difficult.
Moreover, considering the Mössbauer spectroscopy data of the reference sample formed solely from ferrihydrite under abiotic conditions without SRB (Fig. S2 and Table S1†), we confidently infer that the Fe2+ compounds result from SRB activity. Our findings corroborate previous studies102 indicating that vivianite is commonly produced through bacterial sulfate reduction. The biochemical processes in the SRB lifecycle lead to the formation of H2S,24,42 a strong reducing agent, hence vivianite formation is predominantly observed in the presence of SRB.
5. Conclusions
In summary, under anaerobic conditions and a sulfate-rich medium of a laboratory pure culture Desulfosporosinus metallidurans forms crystalline greigite (Fe3S4) rods with adsorbed ferrihydrite nanoparticles. Additionally, the formation of vivianite (Fe3(PO4)2·8H2O) and siderite (FeCO3) appears to be a microbially mediated process facilitated by the vital activity of bacteria. Despite the presence of sulfates, the Desulfosporosinus strain can effectively reduce iron, as indicated by Mössbauer spectroscopy and XAS data which show iron in a reduced charge state of Fe2+ (in contrast to the Fe3+ in the reference sample). This strain not only acts as a sulfate reducer – using sulfate as an electron acceptor to produce energy – but also serves as an iron reducer.The obtained microphotographs reveal that ultrafine ferrihydrite nanoparticles adhere to vivianite, siderite, and greigite nanorod crystallites. FORC analysis has determined that SPM ferrihydrite nanoparticles and greigite nanorods dictate the magnetic properties of the synthetic sludge produced. The presence of highly magnetic greigite and SPM ferrihydrite nanoparticles in the biomineralized phase mixture can enhance the separation of already adsorbed phosphate compounds, facilitating the isolation of vivianite and the subsequent utilization of phosphorus. Thus, Desulfosporosinus metallidurans may be leveraged in advanced water purification techniques and phosphorus extraction from industrial waste sludge, helping both to prevent eutrophication of wastewater and to reclaim phosphorus for industrial application.
Studying the mechanisms of biochemical and biophysical processes in complex water systems, such as wetlands, is crucial for understanding the development and management of artificial water systems. Although this study is based on pure culture conditions, it illustrates the potential of Desulfosporosinus strain to influence the Fe and P cycles in tailing wetlands through biomineralization processes driven by the vital activity of microorganisms. This underscores the importance of biomineralization in the formation of wetland deposits and highlights the need for further in-depth investigations. This research not only provides insights into microbial interactions with environmental contaminants but also offers a blueprint for harnessing these processes to mitigate environmental challenges, such as the development of magnetic technologies for wastewater treatment.
Author contributions
Yu. V. Knyazev: conceptualization, investigation, data curation, visualization, writing – original draft, methodology, software, writing – review & editing. O. P. Ikkert: formal analysis, investigation, funding acquisition. S. V. Semenov: formal analysis, investigation, visualization. O. A. Bayukov: methodology, investigation, software. M. S. Platunov: investigation, data curation, visualization, writing – original draft, software, writing – review & editing; A. D. Nikolenko: investigation. V. P. Nazmov: investigation. A. A. Dubrovskiy: investigation. M. N. Volochaev: investigation. M. S. Molokeev: investigation. E. D. Smorodina: investigation, visualization. D. A. Balaev: visualization, writing – original draft, methodology, software, supervision. O. V. Karnachuk: methodology, data curation, formal analysis, supervision.Conflicts of interest
The authors declare herewith that they do not have any conflict of interest with any organization or individual.Acknowledgements
This study was supported by the Russian Science Foundation, project no. 22-24-00601 (https://rscf.ru/project/22-24-00601/). The electron microscopy and Mössbauer studies were carried out on the equipment of the Krasnoyarsk Territorial Center for Collective Use, Krasnoyarsk Scientific Center, Siberian Branch of the Russian Academy of Sciences. The research contribution of M. S. P. was partially supported by the Ministry of Science and Higher Education of the Russian Federation within the governmental assignment for Synchrotron radiation facility “SKIF”, Boreskov Institute of Catalysis (project FWUR-2024-0040).References
- V. Kumar, A. Sharma, Minakshi, R. Bhardwaj and A. K. Thukral, Hum. Ecol. Risk Assess.: Int. J., 2018, 24, 2162–2181, DOI:10.1080/10807039.2018.1440529.
- P. Mazinder Baruah and G. Singh, Environ. Sci. Pollut. Res., 2022, 29(18), 27366–27381, DOI:10.1007/s11356-021-17918-0.
- H. T. Odum and B. Odum, Ecol. Eng., 2003, 20(5), 339–361, DOI:10.1016/j.ecoleng.2003.08.008.
- N. Sharma, E. D. Flynn, J. G. Catalano and D. E. Giammar, Geochim. Cosmochim. Acta, 2022, 327, 96–115, DOI:10.1016/j.gca.2022.04.019.
- P. Lefebvre, P. Le Pape, A. Mangeret, A. Gourgiotis, P. Sabatier, P. Louvat, O. Diez, O. Mathon, M. O. J. Y. Hunault, C. Baya, L. Darricau, Ch. Cazala, J. R. Bargar, J. Gaillardet and G. Morin, Geochim. Cosmochim. Acta, 2022, 338, 322–346, DOI:10.1016/j.gca.2022.10.018.
- T. Xu, B. Weng, D. Yan, K. Wang, X. Li, W. Bi, M. Li, X. Cheng and Y. Liu, Int. J. Environ. Res. Public Health, 2019, 16, 1818, DOI:10.3390/ijerph16101818.
- T. Prot, V. H. Nguyen, P. Wilfert, A. I. Dugulan, K. Goubitz, D. J. De Ridder, L. Korving, P. Rem, A. Bouderbala, G. J. Witkamp and M. C. M. van Loosdrecht, Sep. Purif. Technol., 2019, 224, 564–579, DOI:10.1016/j.seppur.2019.05.057.
- B. Yu, X. Xiao, J. Wang, M. Hong, C. Deng, Y. Y. Li and J. Liu, Bioresour. Technol., 2021, 341, 125899, DOI:10.1016/j.biortech.2021.125899.
- A. Rapin, M. Grybos, M. Rabiet, B. Mourier and V. Deluchat, J. Environ. Sci., 2019, 77, 250–263, DOI:10.1016/j.jes.2018.07.016.
- T. Yamashita and R. Yamamoto-Ikemoto, Int. J. Environ. Res. Public Health, 2014, 11(9), 9835–9853, DOI:10.3390/ijerph110909835.
- K. C. Ruttenberg, Treatise Geochem., 2003, 8, 682, DOI:10.1016/B0-08-043751-6/08153-6.
- P. N. Froelich, Limnol. Oceanogr., 1988, 33(4part2), 649–668, DOI:10.4319/lo.1988.33.4part2.0649.
- M. Rothe, A. Kleeberg and M. Hupfer, Earth-Sci. Rev., 2016, 158, 51–64, DOI:10.1016/j.earscirev.2016.04.008.
- A. Bounaga, A. Alsanea, K. Lyamlouli, C. Zhou, Y. Zeroual, R. Boulif and B. E. Rittmann, Biotechnol. Adv., 2022, 57, 107949, DOI:10.1016/j.biotechadv.2022.107949.
- Z. Zhang, C. Zhang, Y. Yang, Z. Zhang, Y. Tang, P. Su and Z. Lin, J. Cleaner Prod., 2022, 376, 134109, DOI:10.1016/j.jclepro.2022.134109.
- Y. Zhang, J. Qin, Z. Chen, Y. Chen, X. Zheng, L. Guo and X. Wang, Environ. Res., 2023, 228, 115848, DOI:10.1016/j.envres.2023.115848.
- B. Li, C. Lai, G. Zeng, D. Huang, L. Qin, M. Zhang, M. Cheng, X. Liu, H. Yi, C. Zhou, F. Huang, S. Liu and Y. Fu, Small, 2019, 15(8), 1804565, DOI:10.1002/smll.201804565.
- R. Zhai, L. Zhang, M. Gu, X. Zhao, B. Zhang, Y. Cheng and J. Zhang, Small, 2023, 19, 2207840, DOI:10.1002/smll.202207840.
- M. Rothe, A. Kleeberg, B. Grüneberg, K. Friese, M. Pérez-Mayo and M. Hupfer, PLoS One, 2015, 10(11), e0143737, DOI:10.1371/journal.pone.0143737.
- P. Kraal, N. Dijkstra, T. Behrends and C. P. Slomp, Geochim. Cosmochim. Acta, 2017, 204, 140–158, DOI:10.1016/j.gca.2017.01.042.
- C. März, N. Riedinger, C. Sena and S. Kasten, Mar. Geol., 2018, 404, 84–96, DOI:10.1016/j.margeo.2018.07.010.
- N. Dijkstra, C. P. Slomp and T. Behrends, Mar. Geol., 2016, 438, 58–72, DOI:10.1016/j.chemgeo.2016.05.025.
- P. Kraal, C. P. Slomp, D. C. Reed, G. J. Reichart and S. W. Poulton, Biogeosciences, 2012, 9(7), 2603–2624, DOI:10.5194/bg-9-2603-2012.
- J. S. Berg, A. Duverger, L. Cordier, C. Laberty-Robert, F. Guyot and J. Miot, Sci. Rep., 2020, 10(1), 8264, DOI:10.1038/s41598-020-64990-6.
- G. A. Hood and D. G. Larson, Freshwater Biol., 2015, 60(1), 198–208, DOI:10.1111/fwb.12487.
- J. K. Fredrickson, J. M. Zachara, D. W. Kennedy, H. Dong, T. C. Onstott, N. W. Hinman and S. Li, Geochim. Cosmochim. Acta, 1998, 62, 3239–3257, DOI:10.1016/S0016-7037(98)00243-9.
- P. E. Bell, A. L. Mills and J. S. Herman, Appl. Environ. Microbiol., 1987, 53, 2610–2616, DOI:10.1128/aem.53.11.2610-2616.1987.
- H. G. Dill and A. Techmer, Sediment. Geol., 2009, 217, 95–111, DOI:10.1016/j.sedgeo.2009.03.014.
- V. P. Lemos, M. L. da Costa, R. L. Lemos and M. S. G. de Faria, Quim. Nova, 2007, 30, 36–40, DOI:10.1590/S0100-40422007000100008.
- P. Wilfert, P. S. Kumar, L. Korving, G. J. Witkamp and M. C. M. Van Loosdrecht, Environ. Sci. Technol., 2015, 49(16), 9400–9414, DOI:10.1021/acs.est.5b00150.
- K. Gao, Y. Hu, C. Guo, C. Ke, C. He, X. Hao, G. Lu and Z. Dang, Ecotoxicol. Environ. Saf., 2020, 202, 110921, DOI:10.1016/j.ecoenv.2020.110921.
- R. K. Kukkadapu, J. M. Zachara, J. K. Fredrickson and D. W. Kennedy, Geochim. Cosmochim. Acta, 2004, 68, 2799–2814, DOI:10.1016/j.gca.2003.12.024.
- S. Papirio, D. K. Villa-Gomez, G. Esposito, F. Pirozzi and P. N. L. Lens, Crit. Rev. Environ. Sci. Technol., 2013, 43, 2545–2580, DOI:10.1080/10643389.2012.694328.
- F. Di Capua, M. C. Mascolo, F. Pirozzi and G. Esposito, Chemosphere, 2020, 255, 126977, DOI:10.1016/j.chemosphere.2020.126977.
- A. Picard, A. Gartman, D. R. Clarke and P. R. Girguis, Geochim. Cosmochim. Acta, 2018, 2020, 367–384, DOI:10.1016/j.gca.2017.10.006.
- R. B. Herbert, S. G. Benner, A. R. Pratt and D. W. Blowes, Chem. Geol., 1998, 144, 87–97, DOI:10.1016/S0009-2541(97)00122-8.
- J. H. P. Watson, B. A. Cressey, A. P. Roberts, D. C. Ellwood, J. M. Charnock and A. K. Soper, J. Magn. Magn. Mater., 2000, 214, 13–30, DOI:10.1016/S0304-8853(00)00025-1.
- D. Rickard and J. W. Morse, Mar. Chem., 2005, 97, 141–197, DOI:10.1016/j.marchem.2005.08.004.
- J. P. Gramp, J. M. Bigham, F. S. Jones and O. H. Tuovinen, J. Hazard. Mater., 2010, 175, 1062–1067, DOI:10.1016/j.jhazmat.2009.10.119.
- O. P. Ikkert, A. L. Gerasimchuk, P. A. Bukhtiyarova, O. H. Tuovinen and O. V Karnachuk, Antonie van Leeuwenhoek, 2013, 103, 1221–1234, DOI:10.1007/s10482-013-9900-x.
- C. Zhou, R. Vannela, K. F. Hayes and B. E. Rittmann, J. Hazard. Mater., 2014, 272, 28–35, DOI:10.1016/j.jhazmat.2014.02.046.
- O. V. Karnachuk, O. P. Ikkert, M. R. Avakyan, Y. V. Knyazev, M. N. Volochaev, V. S. Zyusman, V. L. Panov, V. V. Kadnikov, A. V. Mardanov and N. V. Ravin, Microorganisms, 2021, 9, 2558, DOI:10.3390/microorganisms9122558.
- Y. V Knyazev, O. P. Ikkert, S. V Semenov, M. N. Volochaev, M. S. Molokeev, M. S. Platunov, E. V Khramov, A. A. Dubrovskiy, N. P. Shestakov and E. D. Smorodina, J. Alloys Compd., 2022, 923, 166346, DOI:10.1016/j.jallcom.2022.166346.
- C. Lippens and J. De Vrieze, Water Res., 2019, 163, 114859, DOI:10.1016/j.watres.2019.114859.
- I. A. Panova, O. Ikkert, M. R. Avakyan, D. S. Kopitsyn, A. V. Mardanov, N. V. Pimenov, V. A. Shcherbakova, N. V. Ravin and O. V. Karnachuk, Int. J. Syst. Evol. Microbiol., 2021, 71, 004876, DOI:10.1099/ijsem.0.004876.
- F. Widdel and F. Bak, The Prokaryotes: a Handbook on the Biology of Bacteria: Ecophysiology, Isolation, Identification, Applications, Springer, New York, NY, 1992, pp. 3352–3378, DOI:10.1007/978-1-4757-2191-1_21.
- O. V. Karnachuk, N. V. Pimenov, S. K. Yusupov, Y. A. Frank, Y. A. Puhakka and M. V. Ivanov, Microbiology, 2006, 75, 82–89, DOI:10.1134/S0026261706010152.
- O. V. Karnachuk, Y. A. Frank, A. P. Lukina, V. V. Kadnikov, A. V. Beletsky, A. V. Mardanov and N. V. Ravin, ISME J., 2019, 13, 1947–1959, DOI:10.1038/s41396-019-0402-3.
- P. S. Zavertkin, D. V Ivlyushkin, M. R. Mashkovtsev, A. D. Nikolenko, S. A. Sutormina and N. I. Chkhalo, Optoelectron. Instrum. Data Process., 2019, 55, 107–114, DOI:10.3103/S8756699019020018.
- V. V Zabrodsky, P. N. Aruev, V. L. Sukhanov, N. V Zabrodskaya, B. J. Ber, D. Y. Kasantsev and A. G. Alekseyev, in Proceedings of the 9th International Symposium on Measurement Technology and Intelligent Instruments, Saint-Peterburg, Russia, 2009, vol. 3, pp. 243–247 Search PubMed.
- V. P. Nazmov, A. V. Varand, M. A. Mikhailenko, B. G. Goldenberg, I. Y. Prosanov and K. B. Gerasimov, Рolymethyl Metacrylate with a Molecular Weight of 107 g/mol for X-ray Lithography, J. Surf. Invest., 2023, 17(3), 652–655, DOI:10.1134/s102745102303028x.
- A. D. Balaev, Y. V Boyarshinov, M. M. Karpenko and B. P. Khrustalev, Instrum. Exp. Tech., 1985, 26, 167–168 Search PubMed.
- D. Heslop, A. P. Roberts, H. Oda, X. Zhao, R. J. Harrison, A. R. Muxworthy, P.-X. Hu and T. Sato, J. Geophys. Res.: Solid Earth, 2020, 125, e2020JB020418, DOI:10.1029/2020JB020418.
- A. P. Roberts, D. Heslop, X. Zhao and C. R. Pike, Rev. Geophys., 2014, 52, 557–602, DOI:10.1002/2014RG000462.
- H. Effenberger, K. Mereiter and J. Zemann, Z. für Kristallogr. - Cryst. Mater., 1981, 156, 233–244, DOI:10.1524/zkri.1981.156.14.233.
- P. Fejdi, J.-F. Poullen and M. Gasperin, Bull. Mineral., 1980, 103, 135–138 CAS.
- J. Prietzel and W. Klysubun, J. Synchrotron Radiat., 2018, 25, 1736–1744, DOI:10.1107/S1600577518013334.
- M. Yoshida, T. Mineo, Y. Mitsutomi, F. Yamamoto, H. Kurosu, S. Takakusagi, K. Asakura and H. Kondoh, Chem. Lett., 2016, 45, 277–279, DOI:10.1246/cl.151073.
- R. Franke, Spectrochim. Acta, Part A, 1997, 53, 933–941, DOI:10.1016/S1386-1425(97)00040-1.
- Z. Yin, M. Kasrai, G. M. Bancroft, K. H. Tan and X. Feng, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 742, DOI:10.1103/PhysRevB.51.742.
- J. Prietzel, A. Dümig, Y. Wu, J. Zhou and W. Klysubun, Geochim. Cosmochim. Acta, 2013, 108, 154–171, DOI:10.1016/j.gca.2013.01.029.
- F. Werner and J. Prietzel, Environ. Sci. Technol., 2015, 49, 10521–10528, DOI:10.1021/acs.est.5b03096.
- C. Vogel, C. Rivard, V. Wilken, A. Muskolus and C. Adam, Ambio, 2018, 47, 62–72, DOI:10.1007/s13280-017-0973-z.
- S. Beauchemin, D. Hesterberg, J. Chou, M. Beauchemin, R. R. Simard and D. E. Sayers, J. Environ. Qual., 2003, 32, 1809–1819, DOI:10.2134/jeq2003.1809.
- A. A. Rouff, S. Rabe, M. Nachtegaal and F. Vogel, J. Phys. Chem. A, 2009, 113, 6895–6903, DOI:10.1021/jp811276t.
- C. M. Donahue and S. R. Daly, Comments Inorg. Chem., 2018, 38, 54–78, DOI:10.1080/02603594.2018.1465938.
- H. Stanjek, J. W. E. Fassbinder, H. Vali, H. Wägele and W. Graf, Eur. J. Soil Sci., 1994, 45, 97–103, DOI:10.1111/j.1365-2389.1994.tb00490.x.
- A. P. Roberts, L. Chang, C. J. Rowan, C. S. Horng and F. Florindo, Rev. Geophys., 2011, 49, RG1002, DOI:10.1029/2010RG000336.
- D. Y. Rogozin, D. A. Balaev, S. V. Semenov, K. A. Shaikhutdinov and O. A. Bayukov, Dokl. Earth Sci., 2016, 469, 819–823, DOI:10.1134/S1028334X16080067.
- R. Day, M. Fuller and V. A. Schmidt, Phys. Earth Planet. Inter., 1977, 13, 260–267, DOI:10.1016/0031-9201(77)90108-X.
- S. V. Semenov, D. A. Balaev, K. A. Shaykhutdinov and D. Y. Rogozin, J. Sib. Fed. Univ. - Math. Phys., 2017, 10, 252–256, DOI:10.17516/1997-1397-2017-10-2-252-256.
- T. Frederichs, T. von Dobeneck, U. Bleil and M. J. Dekkers, Phys. Chem. Earth, Parts A/B/C, 2003, 28, 669–679, DOI:10.1016/S1474-7065(03)00121-9.
- L. Chang, A. P. Roberts, Y. Tang, B. D. Rainford, A. R. Muxworthy and Q. Chen, J. Geophys. Res.: Solid Earth, 2008, 113, B06104, DOI:10.1029/2007JB005502.
- M. S. Seehra, V. S. Babu, A. Manivannan and J. W. Lynn, Phys. Rev. B, 2000, 61, 3513, DOI:10.1103/PhysRevB.61.3513.
- L. Néel, C. R. Acad. Sci., 1961, 253, 203–208 Search PubMed.
- S. Mørup, D. E. Madsen, C. Frandsen, C. R. H. Bahl and M. F. Hansen, J. Phys.: Condens.Matter, 2007, 19, 213202, DOI:10.1088/0953-8984/19/21/213202.
- Y. L. Raikher and V. I. Stepanov, J. Phys.: Condens. Matter, 2008, 20, 204120, DOI:10.1088/0953-8984/20/20/204120.
- A. A. Krasikov and D. A. Balaev, J. Exp. Theor. Phys., 2023, 136, 97–105, DOI:10.1134/S1063776123010132.
- Y. V. Knyazev, D. A. Balaev, S. V. Stolyar, A. A. Krasikov, O. A. Bayukov, M. N. Volochaev, R. N. Yaroslavtsev, V. P. Ladygina, D. A. Velikanov and R. S. Iskhakov, J. Alloys Compd., 2021, 889, 161623, DOI:10.1016/j.jallcom.2021.161623.
- S. A. Makhlouf, F. Parker and A. Berkowitz, Phys. Rev. B, 1997, 55, R14717, DOI:10.1103/PhysRevB.55.R14717.
- N. J. O. Silva, V. S. Amaral and L. D. Carlos, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 184408, DOI:10.1103/PhysRevB.71.184408.
- D. A. Balaev, A. A. Krasikov, A. A. Dubrovskiy, S. I. Popkov, S. V. Stolyar, O. A. Bayukov, R. S. Iskhakov, V. P. Ladygina and R. N. Yaroslavtsev, J. Magn. Magn. Mater., 2016, 410, 171–180, DOI:10.1016/j.jmmm.2016.02.059.
- A. Punnoose, T. Phanthavady, M. S. Seehra, N. Shah and G. P. Huffman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 054425, DOI:10.1103/PhysRevB.69.054425.
- D. A. Balaev, S. V. Stolyar, Y. V. Knyazev, R. N. Yaroslavtsev, A. I. Pankrats, A. M. Vorotynov, A. A. Krasikov, D. A. Velikanov, O. A. Bayukov, V. P. Ladygina and R. S. Iskhakov, Results Phys., 2022, 35, 105340, DOI:10.1016/j.rinp.2022.105340.
- S. Mørup, M. F. Hansen and C. Frandsen, Beilstein J. Nanotechnol., 2010, 1, 182–190, DOI:10.3762/bjnano.1.22.
- K. Nadeem, M. Kamran, A. Javed, F. Zeb, S. S. Hussain, M. Mumtaz, H. Krenn, D. V. Szabo, U. Brossmann and X. Mu, Solid State Sci., 2018, 83, 43–48, DOI:10.1016/j.solidstatesciences.2018.07.006.
- W. C. Nunes, L. M. Socolovsky, J. C. Denardin, F. Cebollada, A. L. Brandl and M. Knobel, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 212413, DOI:10.1103/PhysRevB.72.212413.
- Y. V. Knyazev, D. A. Balaev, S. A. Skorobogatov, D. A. Velikanov, O. A. Bayukov, S. V. Stolyar, R. N. Yaroslavtsev and R. S. Iskhakov, Phys. Rev. B, 2023, 107, 115413, DOI:10.1103/PhysRevB.107.115413.
- M. Suzuki, S. I. Fullem, I. S. Suzuki, L. Wang and C. J. Zhong, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 024418, DOI:10.1103/PhysRevB.79.024418.
- M. Knobel, W. C. Nunes, L. M. Socolovsky, E. De Biasi, J. M. Vargas and J. C. Denardin, J. Nanosci. Nanotechnol., 2008, 8, 2836–2857, DOI:10.1166/jnn.2008.15348.
- A. A. Krasikov, Y. V Knyazev, D. A. Balaev, D. A. Velikanov, S. V Stolyar, Y. L. Mikhlin, R. N. Yaroslavtsev and R. S. Iskhakov, Phys. B, 2023, 660, 414901, DOI:10.1016/j.physb.2023.414901.
- F. Preisach, Z. Phys., 1935, 94, 277–302, DOI:10.1007/bf01349418.
- R. Egli, A. P. Chen, M. Winklhofer, K. P. Kodama and C.-S. Horng, Geochem., Geophys., Geosyst., 2010, 11, 1–22, DOI:10.1029/2009gc002916.
- C. R. Pike, A. P. Roberts, M. J. Dekkers and K. L. Verosub, Phys. Earth Planet. Inter., 2001, 126, 11–25, DOI:10.1016/S0031-9201(01)00241-2.
- A. R. Muxworthy and D. J. Dunlop, Earth Planet. Sci. Lett., 2002, 203, 369–382, DOI:10.1016/S0012-821X(02)00880-4.
- A. V Smirnov, Geochem., Geophys., Geosyst., 2006, 7, 1–19, DOI:10.1029/2006GC001397.
- A. P. Roberts, L. Chang, D. Heslop, F. Florindo and J. C. Larrasoaña, J. Geophys. Res.: Solid Earth, 2012, 117, B08104, DOI:10.1029/2012JB009412.
- A. P. Roberts, C. R. Pike and K. L. Verosub, J. Geophys. Res.: Solid Earth, 2000, 105, 28461–28475, DOI:10.1029/2012JB009412.
- R. D. Ivantsov, C.-R. Lin, O. S. Ivanova, R. R. Altunin, Y. V Knyazev, M. S. Molokeev, S. M. Zharkov, Y.-Z. Chen, E.-S. Lin and B.-Y. Chen, Curr. Appl. Phys., 2021, 25, 55–61, DOI:10.1016/j.cap.2021.02.013.
- K. Ôno and A. Ito, J. Phys. Soc. Jpn., 1964, 19, 899–907, DOI:10.1143/JPSJ.19.899.
- A. Lancok and L. Volfova, Pure Appl. Chem., 2017, 89, 461–470, DOI:10.1515/pac-2016-1102.
- J. A. Sawicki and D. A. Brown, Hyperfine Interact., 1998, 117, 371–382, DOI:10.1023/A:1012626923056.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer Science & Business Media, 2010, DOI:10.1007/978-3-540-88428-6.
- E. Murad, L. H. Bowen, G. J. Long and T. G. Quin, Clay Miner., 1988, 23, 161–173, DOI:10.1180/claymin.1988.023.2.04.
- K. M. Ø. Jensen, H. P. Gunnlaugsson, M. Christensen and B. B. Iversen, Hyperfine Interact., 2014, 226, 73–78, DOI:10.1007/s10751-013-0992-5.
- M. McNeil and J. McKay, MRS Online Proceedings Library (OPL), 1993, vol. 333, p. 699, DOI:10.1557/proc-333-699.
- T. S. Berquó, J. J. Erbs, A. Lindquist, R. L. Penn and S. K. Banerjee, J. Phys.: Condens. Matter, 2009, 21, 176005, DOI:10.1088/0953-8984/21/17/176005.
- X. Wang, M. Zhu, L. K. Koopal, W. Li, W. Xu, F. Liu, J. Zhang, Q. Liu, X. Feng and D. L. Sparks, Environ. Sci.: Nano, 2016, 3, 190–202, 10.1039/C5EN00191A.
- Y. Lai, Z. Xu, X. Hu, L. Lei, L. Li, L. Dong, H. Yu and W. Zhang, Small, 2019, 15, 1904397, DOI:10.1002/smll.201904397.
- L. Cheng, Z. Chen, Z. Cai, J. Zhao, M. Lu, J. Liang, F. Wang, J. Qi, W. Cui and L. Deng, Small, 2020, 16, 2005433, DOI:10.1002/smll.202005433.
- X. Luo, L. Wen, L. Zhou and Y. Yuan, Int. J. Environ. Res. Public Health, 2023, 20, 4247, DOI:10.3390/ijerph20054247.
- F. Jorand, B. M. R. Appenzeller, M. Abdelmoula, P. Refait, J. C. Block and J. M. R. Génin, Environ. Technol., 2000, 21, 1001–1005, DOI:10.1080/09593330.2000.9618985.
- I. P. Pankhania, A. N. Mossavi and W. A. Hamilton, Microbiology, 1986, 132, 3357–3365, DOI:10.1099/00221287-132-12-3357.
- Z. Chen, Y. Zhang, Q. Luo, L. Wang, S. Liu, Y. Peng, H. Wang, L. Shen, Q. Li and Y. Wang, J. Environ. Sci., 2019, 78, 193–203, DOI:10.1016/j.jes.2018.09.021.
- C. H. Mortimer, J. Ecol., 1941, 29, 280–329, DOI:10.2307/2256395.
- W. Einsele, Arch. Hydrobiol., 1936, 29, 664–686 CAS.
- M. Egger, T. Jilbert, T. Behrends, C. Rivard and C. P. Slomp, Geochim. Cosmochim. Acta, 2015, 169, 217–235, DOI:10.1016/j.gca.2015.09.012.
- L. J. Kubeneck, W. K. Lenstra, S. Y. Malkin, D. J. Conley and C. P. Slomp, Mar. Chem., 2021, 231, 103948, DOI:10.1016/j.marchem.2021.103948.
- L. Joëlle Kubeneck, L. K. ThomasArrigo, K. A. Rothwell, R. Kaegi and R. Kretzschmar, Geochim. Cosmochim. Acta, 2023, 346, 231–244, DOI:10.1016/j.gca.2023.01.029.
- S. V. Stolyar, D. A. Balaev, V. P. Ladygina, A. A. Dubrovskiy, A. A. Krasikov, S. I. Popkov, O. A. Bayukov, Y. V. Knyazev, R. N. Yaroslavtsev, M. N. Volochaev, R. S. Iskhakov, K. G. Dobretsov, E. V. Morozov, O. V. Falaleev, E. V. Inzhevatkin, O. A. Kolenchukova and I. A. Chizhova, J. Supercond. Novel Magn., 2018, 31, 2297–2304, DOI:10.1007/s10948-018-4700-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4va00040d |
| This journal is © The Royal Society of Chemistry 2024 |