DOI:
10.1039/D4VA00179F
(Critical Review)
Environ. Sci.: Adv., 2024,
3, 1351-1363
A brief review on flue gas desulfurization gypsum recovery toward calcium carbonate preparation
Received
30th May 2024
, Accepted 4th September 2024
First published on 4th September 2024
Abstract
The past several years have witnessed great progress in utilization of industrial waste gypsum. Newly developed carbonation technology toward CaCO3 preparation also reveals a significant utilization way to recover high-value products from waste gypsum, whereas there is a shortage of systematic reviews reporting the most recent progress in carbonation of flue gas desulfurization gypsum (FGDG). This review provides a timely and comprehensive summary of major achievements regarding FGDG carbonation and calcium carbonate production to address future investigation directions. We start with a brief introduction of FGDG production and utilization approaches in practical use with their advantages and disadvantages. Then we systematically summarize two types of carbonation, including a direct way and an indirect way. The direct way typically involves three steps: CO2 capture and CO32− formation; CaSO4·2H2O dissolution; CaCO3 crystallization. High purity CaCO3 is prepared and the polymorph of precipitated CaCO3 is affected by many factors, such as the Ca2+/CO32− ratio, reaction conditions, impurities, and additives. The indirect way involves gypsum thermal reduction, carbonation, and sulfur recovery. Finally, challenges of current work and perspectives are presented to expedite future industrialization progress and provide a promising research direction for FGDG carbonation.
Environmental significance
The accumulation of a large amount of flue gas desulfurized gypsum (FGDG) not only occupies valuable land resources, but also the harmful substances such as fluorine and heavy metals in FGDG may increase environmental risks. In addition, plants need large quantities of limestone as a desulfurizer. Limestone mainly comes from mining, and the mining process causes significant damage to the regional ecological environment and easily causes personal injury and death accidents. Newly developed technology can transform FGDG into calcium carbonate while capturing CO2 in the flue gas. The development of FGDG carbonation can recover Ca and S resources and contribute to the circular economy.
|
1. Introduction
Flue gas desulfurization gypsum is produced from the capture of SO2 in the flue gas of fossil fuel combustion using Ca-based absorbents.1–3 Limestone or lime slurry is a widely used Ca-based absorbent.4 This desulfurization process is as follows:5,6| | | SO2 + H2O = 2H+ + SO32− | (1) |
| | | CaSO4 + 2H2O = CaSO4·2H2O | (4) |
Finally, FGDG is produced from the suspension liquid after dewatering and washing. With strict limits on SO2 emissions, every year a huge amount of FGDG is produced.7,8 According to statistics, FGDG production is about 255 million tons.9 In China, the yield of FGDG has exceeded 100 million tons since 2017.5 In India, approximately 20 million tons of FGDG are expected to be generated by the year 2040.10 Due to the huge amount of production, the treatment of FGDG has become a hot issue in solid waste management in recent years.
The most common application of FGDG is to directly use it in civil infrastructure materials.11–14 The main component of FGDG is CaSO4·2H2O with purity more than 90%.15,16 Thus, it is an ideal material to replace natural gypsum.17,18 Currently, about 70% of the total FGDG is used as raw material to produce wallboard, gypsum board, whitewashing, cement and concrete in the USA and China.19–24 However, the supply of FGDG significantly exceeds the demand for building materials, leading to FGDG price dropping. Another application of FGDG in practice is as a resource material in agriculture and soil improvement.25–28 For example, FGDG can provide S, P, and K fertilizers for plant growth and amend saline–alkali soil by exchange of Na.29,30 Compared to natural gypsum, FGDG has a higher level of heavy metals.31,32 The content ranges of Hg, Pb, Sb and Zn in FGDG are 0.198–1.33, 1.33–1.84, 4.57–10.9, and 4.26–29 mg kg−1, respectively. Hence there is an environmental risk that heavy metals will migrate from FGDG to soil and plants.33 Nowadays, the consumption of FGDG in agriculture is very limited.
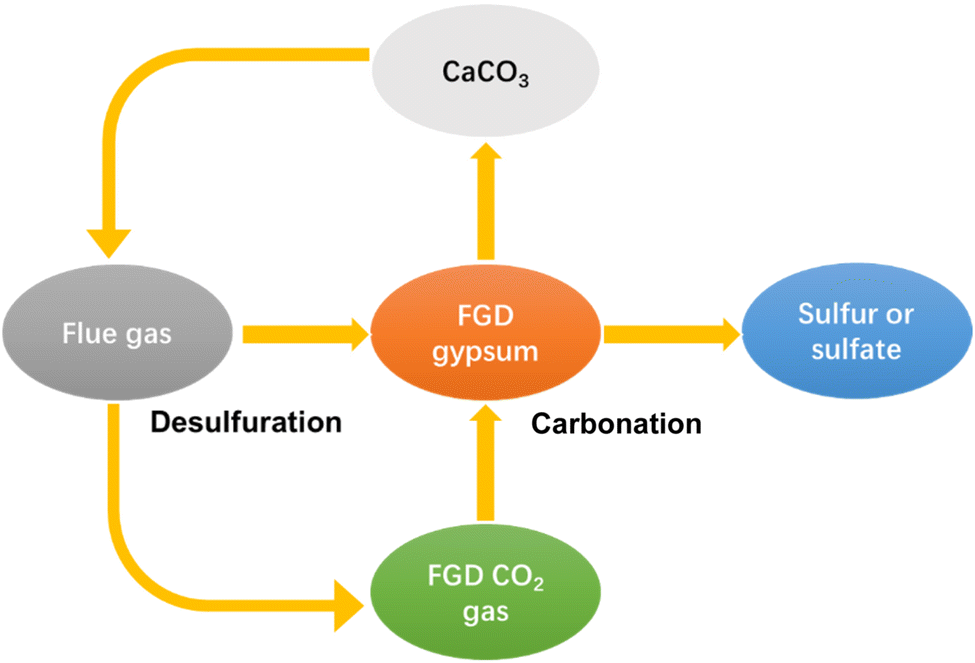
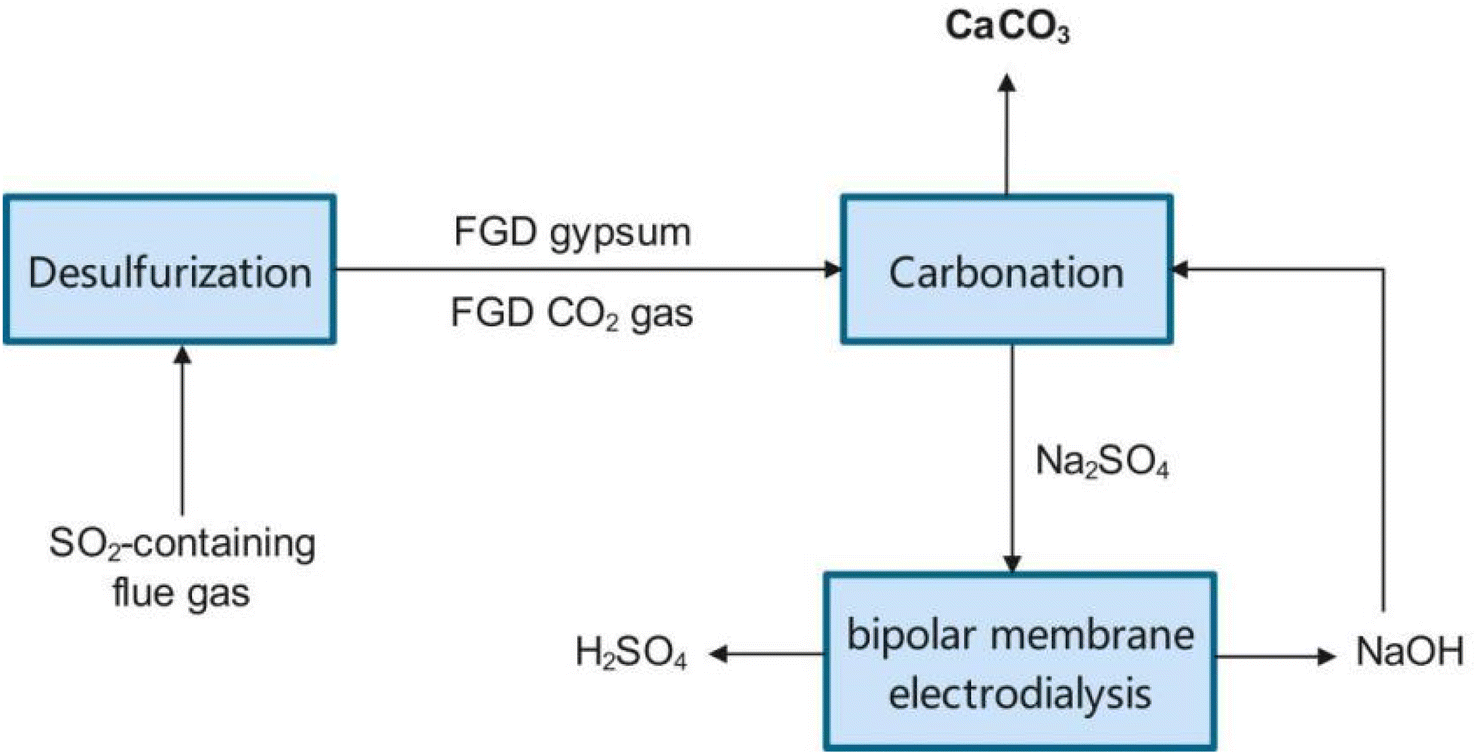
The lack of a utilization way gives rise to serious economy and environmental problems. FGDG treatment is going to be an extra expense for plants. Besides, the accumulation of excessive FGDG will occupy a lot of land, as well as increase the risk of heavy metal leaching. To solve these problems, new approaches have been developed recently.34 New approaches are aimed at preparing high-value chemicals such as hemihydrate, calcium carbonate, adsorptive materials and composites.35–42 Among them, preparation of calcium carbonate from FGDG has received great attention. Calcium carbonate is widely used as a filler in polymer composites, paper, plastic, rubber, and pharmaceutical applications.43–49 Besides, it can also function as a SO2 absorbent in the FGD process. In this way, FGDG is regenerated by the carbonation reaction and then reused in desulfurization, as shown in Fig. 1. Preparation of calcium carbonate from FGDG not only solves the problem of FGDG accumulation and saves the natural resources of CaCO3, but also provides by-products such as sulfur and ammonium sulfate. Furthermore, carbonation with FGDG is conducive to reducing carbon emission, by using CO2 in flue gas as a carbon source. There are mainly two kinds of methods developed for FGDG carbonation, i.e. a direct way and an indirect way. The direct way refers to the direct carbonation reaction of FGDG with CO2 and an alkaline reagent at low temperature. The indirect way is first to reduce CaSO4 to CaS or CaO at high temperature and then react it with CO2 to prepare CaCO3.50,51 However, a summary of recent progress in flue gas desulfurization gypsum recovery toward calcium carbonate preparation is still lacking.
 |
| | Fig. 1 Schematic diagram of the FGD process with calcium recycling. | |
Here we mainly focus on the most advanced development of FGDG application toward CaCO3 preparation. Specifically, we exclusively focus on the carbonation reaction under different conditions and the prepared CaCO3. To deepen comprehension of influence factors, the corresponding mechanism is discussed. We first introduce the direct way to prepare calcium carbonate from FGDG. Then the indirect way with the recovery of sulfur is presented. Finally, a succinct summary of this review and perspectives for future development are provided to lead to an investigation of cost-effective and high-efficiency technology for industrial application.
2. Direct way
2.1 Carbonation system and pathway
The direct way is to prepare a calcium carbonate precipitate with waste gypsum at near room temperature. Soluble carbonates such as Na2CO3 and NH4HCO3 were used as CO32− sources for CaSO4 to CaCO3 conversion in the early research studies.52,53 Nowadays, with the strong demand for reducing carbon emission, more researchers are devoted to directly convert FGDG to CaCO3 using CO2 gas. The main component of FGDG is CaSO4·2H2O. The carbonation reaction of CaSO4·2H2O and CO2 proceeds only if the solubility product of CaSO4·2H2O is larger than that of CaCO3 in the solid–liquid system. At atmospheric CO2 pressure, the solubility products of CaSO4·2H2O and CaCO3 are affected by pH.54 According to theoretical calculations, the solubility product of CaSO4·2H2O is larger than that of CaCO3 at pH > 7.5.55 This will lead the reaction (5) to proceed rightwards. In contrast, if pH < 7.5, CaCO3 is going to dissolve and the reaction (5) proceeds leftwards.| | | CaSO4·2H2O + HCO3− ↔ CaCO3 + H+ + SO42− + 2H2O | (5) |
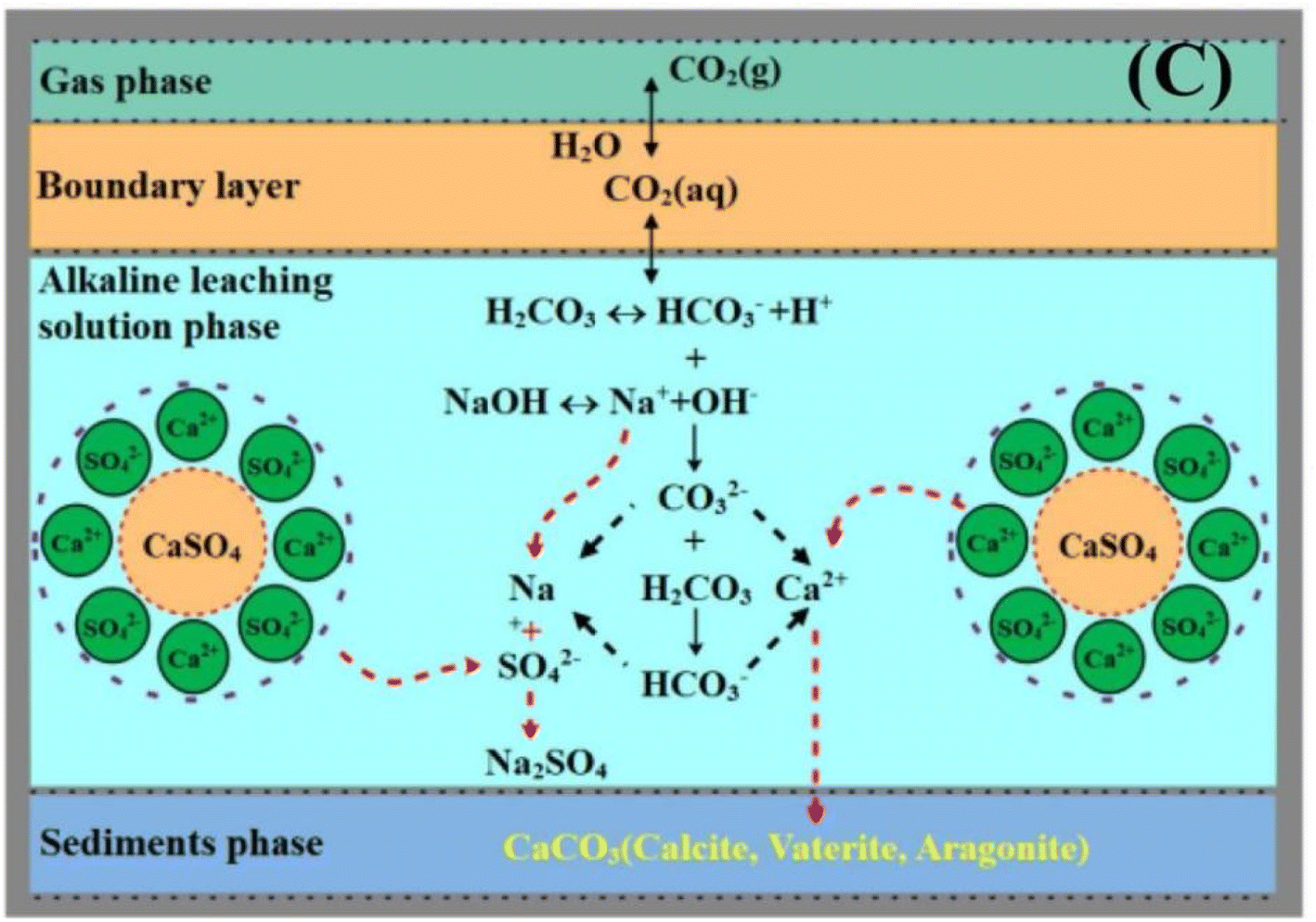
Hence, the control of pH conditions is very important for FGDG direct carbonation. In order to achieve high pH conditions, FGDG is mixed with alkaline solution. The alkaline solution is commonly made using sodium hydroxide or ammonia. Accordingly, carbonation systems are named NaOH-FGDG-CO2 and NH3-FGDG-CO2 respectively. As shown in Fig. 2, when using sodium hydroxide as an alkaline reagent, the carbonation processes involve three main steps:56
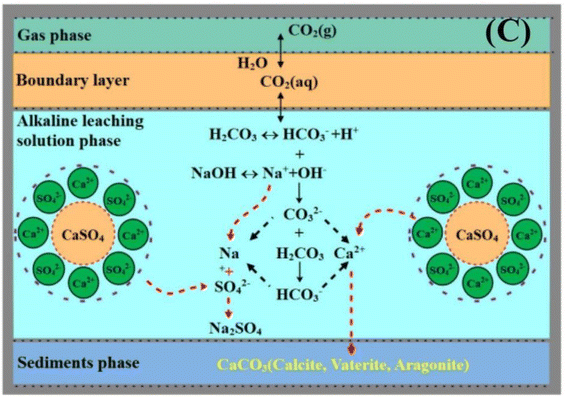 |
| | Fig. 2 Schematic diagram of FGDG carbonation processes with sodium hydroxide and CO2 (reprinted with permission from Luo et al.56 Copyright Elsevier 2023). | |
(1) The dissolution of FGDG in alkaline solution to produce Ca2+ and SO42−.
(2) The capture of CO2 gas using alkaline solution and generation of HCO3− and CO32−.
(3) The nucleation of CaCO3 and formation of supersaturation in the solution phase, followed by precipitation.
Similarly, Gong et al. put forward multistep carbonation of the NH3–gypsum–CO2 system.57 It involves CO32− formation, CaSO4·2H2O dissolution, and CaCO3 crystallization. CO2 is absorbed by ammonia to form NH2COO−, which then hydrolyzes to NH3 and CO32−. Ca2+ in the bulk solution is consumed rapidly with the formation of a CaCO30 ion pair. The concentration of the CaCO30 ion pair increases and reaches supersaturation. After that, the ion pair concentration decreases gradually, whereas CaCO3 precipitates obviously in the bulk solution. The rates of CaSO4·2H2O dissolution and CO2 absorption determine the carbonation rate. According to the recent work about FGDG carbonation as listed in Table 1, both sodium hydroxide and ammonia are efficient alkaline reagents for FGDG carbonation with more than a 98% conversion ratio achieved. The alkali concentration will affect the conversion ratio. As the NaOH concentration increased from 0.5 mol L−1 to 3 mol L−1, the conversion ratio increased from 96.4% to 99.94%.56 Compared to ammonia, sodium hydroxide is more environment-friendly, because it can avoid ammonium volatilization from ammonia.67 In addition, sodium hydroxide can shorten conversion time of FGDG. However, the by-product of the NaOH-FGDG-CO2 carbonation system is sodium sulfate, and its economic value is relatively low. In comparison, the ammonium sulfate by-product from the NH3-FGDG-CO2 system is a high-value chemical. Besides sodium hydroxide and ammonia, researchers developed FGDG carbonation methods by using amine as the alkaline reagent.66,68,69 Wang et al. tested several kinds of amine for FGDG carbonation, including monoethanolamine (MEA), diethanolamine (DEA), triethanolamine (TEA), 1,3-diaminopropane (DAP), piperazine (PZ), 3-amino-1-propanol (MPA), and N,N-dimethylethanolamine (DEEA).66 Amines can enhance the absorption of CO2 as well as promote gypsum dissolution by binding amino groups with Ca2+. The purity of CaCO3 reached 93.8% when using DAP as the alkaline reagent, which was comparable to 94.4% purity when using the NaOH alkaline reagent. Interestingly, after the carbonation process, the reagent solution was successfully treated by bipolar membrane electrodialysis for amine regeneration and recovery of H2SO4. This could largely reduce the cost of alkaline consumption.
Table 1 List of published work about FGDG carbonation
| Works |
Alkaline reagent |
Condition |
Solid-to-solution ratio |
CO2 gas |
Conversion rate |
CaCO3 crystal |
| Luo et al.56 |
3 mol per L NaOH |
Stirring |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 :8 |
0.0 MPa CO2 |
99.94% |
Calcite |
| Luo et al.56 |
3 mol per L NaOH |
Stirring |
1:8 |
0.2 MPa CO2 |
99.95% |
Aragonite |
| Wang et al.58 |
25 wt% ammonia solution |
Stirring + ultrasound |
— |
99.99% CO2 |
98% |
Vaterite |
| Wang et al.59 |
25 wt% ammonia solution |
Stirring for 1 h |
10% |
99.99% CO2 |
— |
60% vaterite and 40% calcite |
| Tan et al.60 |
Aqueous ammonia |
— |
molar ratio of NH3 to CaSO4 = 2.0 |
99.99% CO2 |
90% at 40 °C, close to 100% at 80 °C |
— |
| Lee et al.61 |
25 wt% ammonia solution |
Stirring |
15% |
15 wt% CO2, 85 wt% N2 |
96% |
Calcite (40–90%) |
| Altiner et al.62 |
NaOH or NH4OH |
Stirring |
1:13, 1:9, and 1:7 |
99.99% CO2 |
— |
Calcite in NaOH and calcite and vaterite in NH4OH |
| Ding et al.63 |
3 mol per L ammonia solution |
Stirring |
20% |
75% N2, 15% O2, 10% CO2 |
90% |
Mainly calcite with some vaterite |
| Lee et al.64 |
NH4OH, 25 wt% |
Stirring |
15–50% |
15% CO2 and 85% N2 |
95% |
vaterite and calcite |
| Song et al.51 |
Ammonia + ethanol |
Stirring |
20% |
99.9% CO2 |
— |
Calcite and vaterite |
| Liu et al.65 |
Glycine + ammonia |
Stirring for 1 h |
— |
Pure CO2 |
97% purity |
Vaterite |
| Wang et al.66 |
1.0 mol per L amine |
Stirring |
6.8% |
40 vol% CO2 in N2 |
59.8–96.4% |
Vaterite (DAP), calcite (PZ) |
2.2 Mineralization principle and modeling
In an alkaline medium, the driving force of CO2 mineralization of desulfurized gypsum is due to the difference in solubility products between CaCO3 and CaSO4·2H2O in water. As the solubility product of CaCO3 (Ksp = 4.8 × 10−9, 298 K) is much smaller than that of CaSO4·2H2O (Ksp = 2.6 × 10−5, 298 K), the driving force of the reaction is large, and the theoretical conversion rate can reach more than 99%. Several studies have been conducted on modeling the CO2 adoption, ammonium bicarbonate hydrolysis and CaCO3 formation process. Tan et al. studied the process parameters of direct wet mineralization of CO2 with desulfurized gypsum and established a reaction model of direct wet mineralization of CO2 with desulfurized gypsum in an ammonia medium system.60 There is a competition between the CO2 absorption reaction and NH2COO− hydrolysis reaction, in which the CO2 absorption reaction dominates over the latter. The HCO3− (or CO32−) from NH2COO− hydrolysis is consumed fast to form calcium carbonate, which would enable the HCO3− (or CO32−) concentration to vary little. Liu et al. investigated CaCO3 crystal nucleation and growth processes in the gas (CO2)–liquid (NH3·H2O)–solid (CaSO4) three-phase system.70 The research revealed that temperature affected CaCO3 crystal growth more than the nucleation process. They established a model to predict the CaCO3 particle size. Gong et al. studied modeling of multistep Ca2+ transfer in CaSO4 mineralization using a gypsum disk.57 The CaCO30 ion pair was the intermediate product in CaCO3 induction and nucleation periods. The ion pair determined nucleation and formation of supersaturation. The modeling revealed that NH2COO− hydrolysis was the rate-limiting step in induction and nucleation periods, and the dissolution of CaSO4·2H2O became dominant in the growth period. An increase in the CO2 flow rate can improve NH2COO− hydrolysis, leading to more nuclei and high supersaturation. The impeller speed can affect the dissolution rate in the growth period, but it had no remarkable effect on the concentrations of components in induction and nucleation periods.
2.3 CaCO3 crystal and utilization
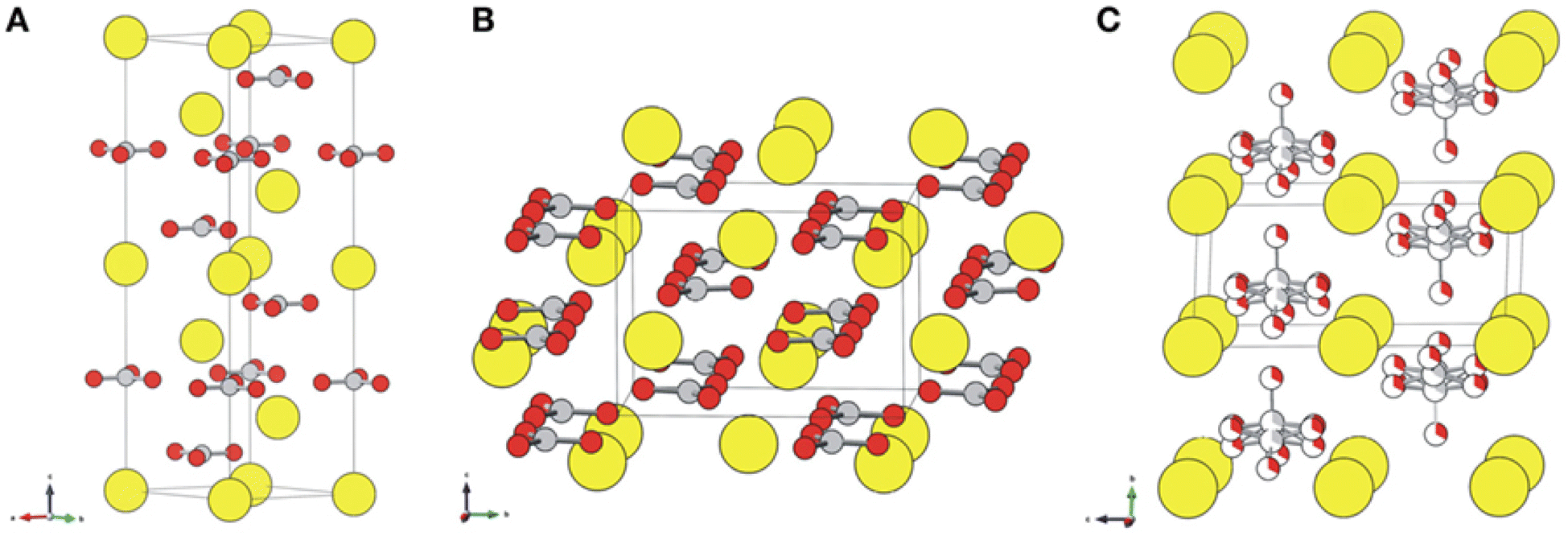
Calcium carbonate is an important raw material in many industrial processes, such as plastics, rubber, tires, paper, building materials, coatings, food, medicine and feed. Certainly, calcium carbonate from FGDG mineralization can be directly functioned as a desulphurization sorbent without impurity separation, because a desulphurization sorbent does not require highly purified CaCO3.71 Considering the large amount of FGDG, it is a promising way to consume the CaCO3 products. Besides, it can avoid long distance transportation. It's worth noting that calcium carbonate has three polymorphs, namely, calcite, aragonite, and vaterite,72,73 as shown in Fig. 3. The crystal structure of CaCO3 for FGDG carbonation has received a lot of attention because it determines specific purposes. Because vaterite CaCO3 has good smoothness, fluidity, dispersion and wear resistance, it is widely used in the fields of rubber, paint, ink, medicine, toothpaste and cosmetics.74
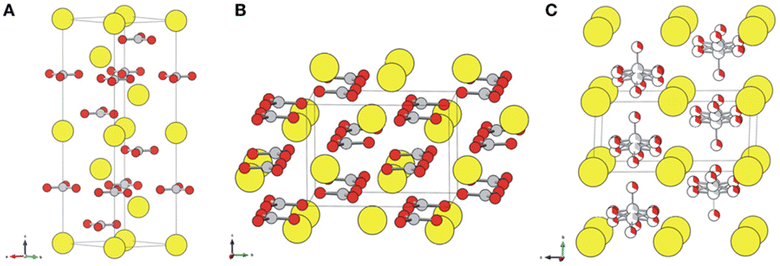 |
| | Fig. 3 Crystal structures of (A) calcite, (B) aragonite, and (C) vaterite. Ca atoms are displayed as large yellow balls, and carbonate groups are illustrated with gray (carbon) and red (oxygen) balls. Vaterite is depicted with a hexagonal P63/mmc structure that accounts for a partial occupancy of one-third of the carbonate groups. (Reprinted with permission from Chang et al.72 Copyright American Chemical Society 2017.). | |
It can be concluded from Table 1 that vaterite will be produced from FGDG mineralization when using ammonia. The possible principle might be as follows:58 In the first stage, the initial Ca2+ concentration in the system is low (the solubility of CaSO4·2H2O is 2 g L−1@20 °C, so the initial Ca2+ concentration in the solution is about 0.01 M). In the ammonia medium, when CO2 is introduced into the reaction system, CO2 and ammonia will react quickly to produce a large amount of CO32−; at this time, a very low Ca2+/CO32− ratio is conducive to the formation of vaterite.75 In the second stage, the mixture of (NH4)2CO3 and NH4HCO3 is the main substance.76 In addition, the concentration of SO42− gradually increases with the extension of reaction time, and a higher concentration of SO42− can stabilize vaterite in aqueous solution.77 Therefore, the synergistic effect of low Ca2+/CO32− with a higher concentration of SO42− may be a key factor in the formation of vaterite in the second stage. In the third stage, the reaction of HCO3− with Ca2+ to produce vaterite may be due to the high concentration of SO42−, because a high concentration of SO42− can stabilize vaterite in aqueous solution and inhibit its conversion to calcite. Therefore, the combined effect of the above factors promotes the formation of vaterite in the process of FGDG mineralization in the ammonia medium system.
2.4 Effect of factors on the CaCO3 polymorph
Much effort has been made to control the polymorph of calcium carbonate from carbonation. The polymorph of CaCO3 is affected by many factors, such as the Ca2+/CO32−ratio, reaction conditions, impurities, and additives. The Ca2+/CO32− ratio is a key factor related to CaCO3 crystallization growth.78,79 A high ratio of Ca2+/CO32− favors the formation of the calcite.75 Experiments demonstrated that NaOH solution has a better Ca2+ extraction effect than NH3·H2O solution, resulting in a higher Ca2+/CO32− ratio in NaOH solution.80 Thus, it has been found that CaCO3 tends to be pure calcite in the NaOH system, whereas vaterite was achieved in the NH3·H2O system.81 Another way to reduce the Ca2+/CO32−ratio is using high CO2 pressure. Experiments demonstrated that calcite is generated by carbonation with NaOH and atmospheric CO2, while aragonite is generated at CO2 pressure 0.2 MPa.56 As CO2 pressure increases, CaCO3 particle size becomes larger and purity is 0.7% lower.
Using additives is an effective way to control the CaCO3 polymorph. Liu et al. added glycine in the NH3-FGDG-CO2 system.65 As a result, calcite content in CaCO3 reduced and vaterite content increased. When no glycine is added, around 40% is calcite and 60% is vaterite. When glycine concentration is 20 wt%, vaterite purity reached 97%. They attributed it to the formation of a Ca(NH2CH2COO)2 intermediate via reactions (6) and (7), which reduced the concentration of Ca2+ in bulk solution and provided a lower local Ca2+/CO32− ratio. Song et al. introduced ethanol into the NH3-FGDG-CO2 system during the induction period.51 Ethanol might block the surface and inhibit the perfect growth of calcite, by binding more strongly at the calcite surface than with water. Under stoichiometric ammonia conditions, the addition of ethanol gave rise to polymorph change from calcite to vaterite. Under excess-ammonia conditions, peanut-like aragonite crystals with dandelion-like heads were formed when 30 and 50 vol% ethanol were used. However, with ethanol more than 70 vol%, the reaction products were not CaCO3, but were rather compounds composed of (NH4)2SO4 and (NH4)2Ca(SO4)2·H2O. Polyacrylic acid is proved to enhance gypsum dissolution.82 With the addition of 2.7 g per L polyacrylic acid in the NH3-FGDG-CO2 system, the amount of dissolved Ca2+ increased to 60% of the gypsum. The prepared CaCO3 was amorphous, which could completely crystallize to calcite after exposure to air for 2.5 hours.
| | | NH2CH2COOH + NH4OH → NH2CH2COONH4 + H2O | (6) |
| | | 2NH2CH2COONH4 + CaSO4·2H2O → Ca(NH2CH2COO)2 + (NH4)2SO4 + 2H2O | (7) |
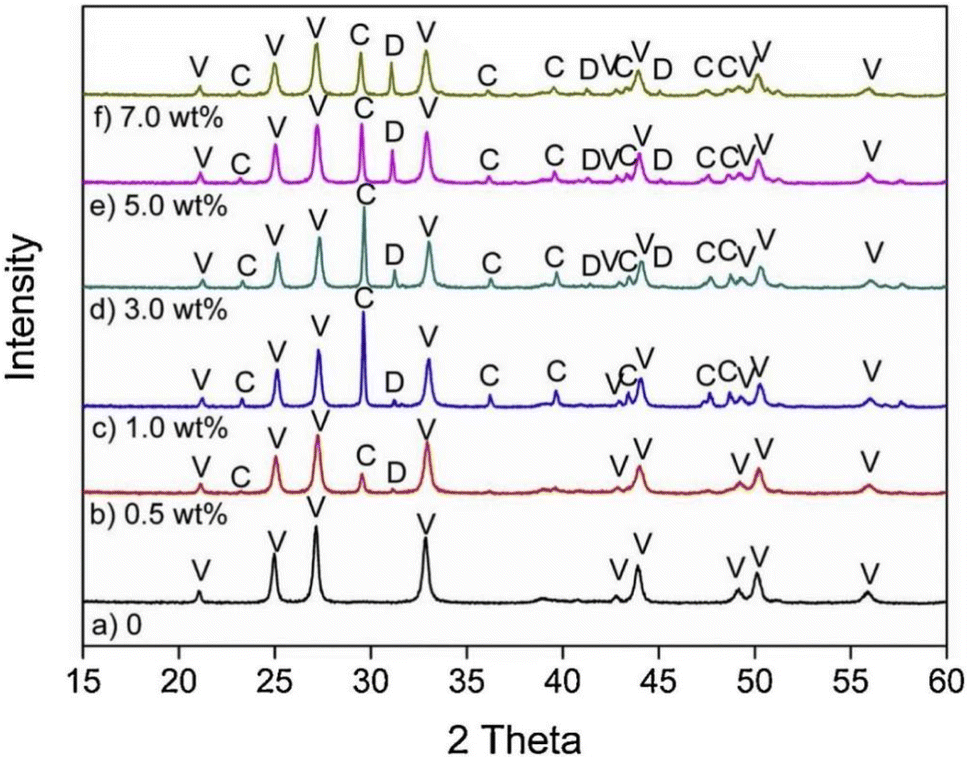
In general, impurity elements such as Si, Mg, Al, Fe, F and K exist in FGDG,83 and it's worth noting the influence of impurities. CaCO3 generated from pure CaSO4·2H2O with NH3–CO2 is 100% vaterite, while from FGDG-NH3-CO2 it is a mixture of vaterite and calcite.59 According to the XRD results in Fig. 4,59 dolomite (CaMg(CO3)2) in CaSO4·2H2O will lead to the formation of calcite using NH3–CO2. The dolomite particles have a negative charge in the process of carbonation of CaSO4·2H2O, which preferentially adsorb Ca2+ through the electrostatic attraction force leading to a higher local ratio of Ca2+/CO32−. It is reported that CaSO4·2H2O carbonation at a high ratio of Ca2+/CO32− tends to form calcite.84,85 This may be the reason why different polymorphs are observed. High content of Mg2+ or MgO can stabilizing the amorphous CaCO3 produced from precipitate crystallization.86–88 The presence of F, Fe, and Mg accelerates the conversion rate of calcium carbonate from thermodynamically unstable vaterite to thermodynamically stable calcite.89,90 The effect of F is more obvious, in all samples with F, and only calcite and no vaterite are present. In the reaction of FGDG carbonation with ammonium carbonate, CO32− in ammonium carbonate replaces SO42− in gypsum to form CaCO3. Therefore, the greater concentration of CO32− ions in the solution will be conducive to the reaction. F, Fe and Mg impurities will affect the progress of ammonium carbonate hydrolysis. The hydrolysis of F− produces OH−, leading to the increase of CO32− ions, and the conversion rate is increased. The hydrolysis of Mg2+ and Fe3+ produces H, which reduces the concentration of CO32−. Fe(OH)3 and Mg(OH)2 generated from hydrolysis will cover the surface of FGDG with precipitation and reduce the reaction rate and conversion rate. The higher the concentration of Mg2+ and Fe3+, the more serious the hydrolysis and deposition, and the more the conversion rate is reduced.91
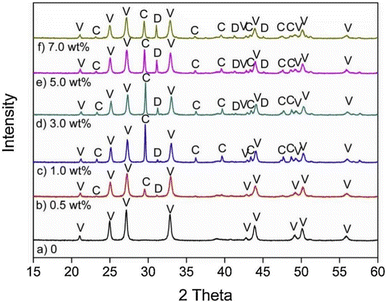 |
| | Fig. 4 XRD pattern of CaCO3 obtained in the presence of varying amounts of dolomite particles: (a) CaCO3 sample prepared in the absence of dolomite; (b–f) CaCO3 prepared with 0.5 wt%, 1.0 wt%, 3.0 wt%, 5.0 wt% and 7.0 wt% dolomite particles, respectively. Abbreviations: V, vaterite; C, calcite; D, dolomite. (Reprinted with permission from Wang et al.59 Copyright Elsevier 2019.). | |
Reaction conditions such as temperature, CO2 flow rate, solvent ratio, alkaline concentration and stirring rate are important factors. Temperature has a great influence on CaCO3 polymorphs according to the work by Lee et al.61 At 20 °C and 40 °C, the carbonation product is a mixture of spherical vaterite and calcite with poorly developed edges. At 60 °C, most vaterite will transform into calcite. At 80 °C, needle-flower-like aragonite is newly formed. The use of ultrasound can help control the crystallization process and the formation of CaCO3 polymorphs due to sonocrystallization.92,93 Application of ultrasound technology in FGDG carbanion can achieve high conversion efficiency and pure vaterite in the ammonia system.58,94 The CaSO4·2H2O conversion efficiency is about 60% at 25% ultrasonic amplitude (20 kHz, 650 W), while both conversion efficiencies at 50% and 75% ultrasonic amplitudes were around 98%. The stirring speed has a relatively minor effect on the formation of CaCO3 polymorphs. It mainly affects the particle size and morphologies. Increasing stirring speed can reduce CaCO3 particle size. The particle size at 450 rpm and 675 rpm is 23.45 μm and 18.38 μm, respectively.56
2.5 FGDG leaching and mineralization technology
As the low solubility of FGDG inhibited the carbonation reaction, researchers developed new mineralization technology with leaching. First, Ca2+ is leached from FGDG using a leaching agent. Secondly, the leachate and solid residue are separated. Finally, the Ca2+ in the leachate is carbonated to produce CaCO3. By leaching technology, the impurities in FGDG are removed, and thus high purity CaCO3 can be obtained. Several leaching agents have been developed, including inorganic salts and organic leaching agents.
NaCl, NH4Cl and CH3COONH4 were inorganic salt based leaching agents. Chen et al. explored the mineral mineralization process of phosphogypsum under the action of NaCl and NH4OH.95 The dissolution efficiency of phosphogypsum under the optimal conditions was 49.42%. One ton of phosphogypsum can chelate 115 kg of CO2 during mineralization and produce 262 kg of CaCO3. In the whole preparation process, NaCl was recycled 4 times, and the corresponding reaction efficiency was above 60%. Ding et al. experimentally and theoretically studied the mineral mineralization of phosphogypsum with ammonium acetate.96 Under the optimal conditions, the dissolution rate of calcium is 98.1% and the mineralization efficiency is 98.32%. The structure of carbonated products was controlled by adjusting the reaction temperature and the amount of ammonia. The phosphogypsum leached with NH4Cl showed that the optimum dissolved amount of CaSO4·2H2O was 18.7 g L−1 and the carbonation rate was 98.22%.97
Organic leaching agents contain carboxyl (–COOH) and amino (–NH2) groups in their molecular structures that can form a soluble chelate with Ca2+. Using sodium gluconate as a phase transfer agent, Yang et al. synthesized calcium carbonate using phosphogypsum as a raw material at room temperature and atmospheric pressure through a simple and effective “phase transfer – precipitation” route.98 The results showed that the presence of sodium gluconate inhibited the nucleation and growth of calcite and promoted the formation of vaterite. Gong et al. used aspartic acid as the leaching agent, and the results showed the amount of dissolved CaSO4·2H2O and the total carbonation efficiency during cycling were determined to be 16.3 ± 0.4 g L−1 and 46.5 ± 1.9%, respectively.99
3. Indirect way
3.1 Thermal reduction of CaSO4
The indirect way for calcium carbonate preparation from FGDG is quite different from the direct way. Firstly, CaSO4 in gypsum is converted to CaS via thermal reduction. Secondly, calcium carbonate is produced via CaS carbonation. In the thermal reduction reaction, carbon, carbon monoxide gas or hydrogen gas is used as a reductant and mixed with gypsum at high temperature.100–102 Typically, carbon is more accessible and thus it has been widely used. The thermal reduction process using carbon as a reductant is present in reaction (8).| | | CaSO4·2H2O + 2C → CaS + 2CO2 + 2H2O | (8) |
The reduction reaction is an endothermic reaction. Enthalpy (ΔH°), entropy (ΔS°), and Gibbs-free energy (ΔG°) decrease over the temperature and the proportionality constant (k) increases, as shown in Table 2 (data from Tewo et al.103).
Table 2 The effect of temperature on ΔH°, ΔS°, ΔG°, and k (data are reproduced with permission from Tewo et al.103 Copyright Elsevier 2019)
|
T (°C) |
ΔH° (kJ mol−1) |
ΔS° (kJ mol−1) |
ΔG° (kJ mol−1) |
k
|
| 25 |
171.0 |
367.0 |
61.5 |
1.7 × 10−11 |
| 156 |
172.0 |
367.0 |
13.5 |
2.3 × 10−2 |
| 286 |
170.1 |
365.6 |
−34.3 |
1.6 × 103 |
| 417 |
167.3 |
361.2 |
−81.9 |
1.6 × 106 |
| 547 |
162.9 |
355.4 |
−128.7 |
1.6 × 108 |
| 678 |
157.0 |
348.8 |
−174.7 |
3.9 × 109 |
| 939 |
141.1 |
334.1 |
−263.8 |
2.4 × 1011 |
| 1069 |
131.0 |
326.2 |
−307.0 |
8.8 × 1011 |
| 1200 |
119.7 |
318.1 |
−349.0 |
2.4 × 1012 |
Tewo et al. used the Pyrosim Mintek model to predict that CaSO4 to CaS conversion could be increased from 26.8 to 85.0% when the temperature was raised from 500 to 1100 °C.103 The appropriate temperature range is 700−900 °C for a high conversion ratio.104 If the temperature exceeds 1000 °C, self-decomposition reactions and re-reactions probably occur according to reactions (9)–(13). Tan et al. conducted thermal reduction experiments with a 2:1 weight ratio of FGDG to carbon powder, and gypsum was completely decomposed into calcium sulfide by calcining at 900 °C for 30 min.105 However, only the oldhamite phase has been observed after in the temperature range of 900 to 1100 °C. Liu et al. achieved a FGDG conversion ratio of 97.89% and CaS purity of 90.42% with 30% carbon content after thermal reduction at 900 °C for 2 hours.104 They indicated that thermal reduction was completed via step-by-step reactions, and SO42− was transformed into SO32−–S22−–S2−. Confirmed by a scanning electron micrograph, a hollow structure was formed gradually from the outside to the inside. Phosphogypsum is also a major source of industrial waste gypsum. Laasri et al. tested thermal reduction of phosphogypsum by CO gas.106 At 600 °C, calcium sulfide (CaS) was mainly formed with high CO partial pressure (>50%), and calcium oxide (CaO) was mainly formed with lower CO partial pressure (<20%). At 1000 °C and above, CaSO4 was completely converted to CaS, CaO, and minor co-products due to the presence of impurities in phosphogypsum. One should note that other products were also found after thermal reduction apart from CaS and CaO, such as Ca2Fe2O5 and Ca2Al(AlSiO7).107–109 The reason is that CaSO4 will react with the impurities (Al, Fe, Si etc.) in gypsum at high temperature.
| | | CaSO4 → CaO + SO2 + 0.5O2 | (9) |
| | | CaS + 3CaSO4 → 4CaO + 4SO2 | (10) |
| | | 3CaS + CaSO4 → 4CaO + 2S2 | (11) |
| | | CaS + 1.5O2 → CaO + SO2 | (13) |
The carbon to gypsum ratio is a vital factor. Insufficient carbon content will lead to a low conversion efficiency. However, too much carbon will result in carbon waste and low CaS purity. It is reported that there is an increasing trend in conversion efficiency from 57.4 to 83.8% when the carbon ratio is increased from 1:1 to 4:1.103 Liu et al. investigated the relationship between carbon content and conversion.104 At 10% carbon content, conversion efficiency was about 41% with 25% CaS purity. The conversion efficiency increased to above 97% and the CaS purity was about 90% at 30% carbon content. Further increasing the carbon content had a slight influence on conversion efficiency, but the CaS purity reduced less than 80% due to carbon residues. Motaung et al. made pelletized gypsum from acid mine drainage (AMD) neutralization by adding starch or cellulose as a binder into a mixture of gypsum and coal.110 The CaS yield of pelletized gypsum thermo-reduction was improved from 60% without starch or cellulose to 71% or 67% with 8% starch or cellulose, respectively. The CaS yield could further reach above 90% with gypsum and starch at a ratio of 1:2.9 at 1050 °C for 20 min.
3.2 CaS carbonation and sulfur recovery
The produced CaS can react with H2O and CO2 for carbonation via reaction (14). However, the produced CaCO3 is low-grade (i.e. <90 mass%) which comprised a mixture of calcite and vaterite.102 Carbonation of CaS is difficult and sluggish in aqueous solution, and more than 3 hours are needed for CaCO3 to occur.105 This is due to CaS being a sparingly soluble salt in water.111 Accordingly, de Beer developed a new carbonation method by sparging H2S gas to aqueous CaS suspensions.50 H2S reacted with CaS to form water-soluble Ca(HS)2via reaction (15). Thus 91.7% CaS was extracted into the aqueous phase and separated from the impurities in the solid phase. Then CO2 gas is introduced for carbonation by reaction (16). High purity (99.5%) CaCO3 is produced. Rhombohedral structured calcites with different particle sizes were demonstrated after Ca(HS)2 carbonation with different initial Ca2+ concentrations, as shown in Fig. 5.| | | CaS + H2O + CO2(g) → H2S(g) + CaCO3 | (14) |
| | | CaS + H2O + H2S → Ca(HS)2 + H2O | (15) |
| | | Ca(HS)2 + H2O + CO2 → CaCO3 + H2S | (16) |
 |
| | Fig. 5 SEM images (top row 1000× and bottom row 5000× magnification) of high-grade CaCO3 produced at initial Ca2+ concentrations of (a) 450 mmol L−1, (b) 900 mmol L−1 and (c) 1800 mmol L−1. (Reprinted with permission from de Beer et al.50 Copyright Elsevier 2015.). | |
The H2S produced in reactions (14) and (16) can be further oxidized to high-value elemental sulfur.112,113 High pH (9.0) is required for the dissolution of hydrogen sulfide, and Fe-based chelates are widely used to promote the reaction. The main chemical reaction can be simplified in reaction (17):114
| | | H2S + 2Fe3+Ln− → S0 + 2Fe2+Ln− + 2H+ | (17) |
where L
n− denotes an organic ligand with
n− charges and S
0 represents a zero-valence sulfur product. Tan
et al. successfully recovered elemental sulfur with 0.5–3 μm in diameter from FGDG.
105
4. Perspectives
4.1 Technical and economic analyses
Although many studies have reported that a high FGDG conversion rate can be achieved, the results are obtained from bench and pilot scale units. There are still many challenges to further development of practical engineering projects. Cost and profit mainly depended on the raw materials (ammonia or sodium hydroxide) and byproducts (ammonium sulfate, sodium sulfate, and elemental sulfur). However, the prices of these chemicals fluctuate wildly, leading to an economic uncertainty. Raw material sources and byproduct utilization deserve further investigation for practical engineering projects. Advanced technologies to produce high quality CaCO3via impurity separation and polymorph control are also beneficial for practical economic viability. Table 3 summarizes the comparison of FGDG carbonation approaches. The challenges that each face in approaching practical application are discussed in detail in this section.
Table 3 Comparison of the direct way and indirect way
| Approaches |
Reaction processes |
Technical and economic analyses |
| Direct way |
NaOH-FGDG-CO2 or NH3-FGDG-CO2 |
Advantages of simple processes, high conversion rates and mild reaction condition. Issues of ammonia escape, device corrosion and by-product application |
| Indirect way |
1. Thermal reduction; 2. CaS carbonation; 3. elemental sulfur preparation |
Insufficient conversion rate. High purity CaCO3. Suitable for factories with cheap energy sources |
The direct way has many advantages such as convenient operation, low energy cost and high conversion rates, and it is promising for scaling up. FGDG carbonation can be accomplished in a single step, which is very beneficial to reduce equipment investment and post-production operation. The direct way is carried out under mild reaction conditions, with temperature less than 100 °C and pressure less than 0.2 MPa. According to economic analysis, energy cost for 1 ton of FGDG is about 1.4 ton of steam and 150 kW h of electricity, including the crystallization process of the ammonium sulfate by-product.36 According to the data in Table 1, conversion rates of CaSO4 can reach more than 90%, and some even approach 100%. Developing large reactors on an industrial scale is a technical key point to achieve satisfactory conversion rates on a large scale. It is worth noting that Cl and F are enriched in FGDG from flue gas.115,116 The issues of equipment corrosion by Cl and F should be taken into consideration in practical use. Another challenge is the value of the sulfate by-product. For the NaOH-FGDG-CO2 approach, Na2SO4 is produced. For the NH3-FGDG-CO2 approach, (NH4)2SO4 is produced. We have conducted a market survey on the price of raw materials in China, and the price of (NH4)2SO4 is three to five times that of Na2SO4. Thus, the NH3-FGDG-CO2 approach is more scalable in economy. However, if a large amount of (NH4)2SO4 is produced from FGDG, there would be a risk of price drop. Besides, the ammonia escape is still a common environmental problem.117–119
The indirect way is a multistep approach, which is more complicated than the direct way. The FGDG thermal reduction step requires high temperature around 1000 °C, resulting in large energy consumption. The energy input is about 4060 kJ per kg-FGDG for thermal reduction.120 As for the indirect way, it is meaningful to combine it with other industrial processes which produce plenty of cheap methane, hydrogen, carbon dioxide and petroleum coke by-products.121–123 Beneficially, the indirect way can avoid the evaporative crystallization process and save energy. In comparison, the conversion rate of the indirect way is lower than that of the direct way. The FGDG to CaS efficiency is usually less than 90%. Efforts should be made to develop optimum reaction conditions as well as efficient reaction devices. One advantage of the indirect way is that it is possible to prepare high purity CaCO3 from CaS. By forming water-soluble Ca(HS)2, water-insoluble impurities are removed, and the CaCO3 purity reaches 99.5%.50
4.2 Direct way coupled with NaOH regeneration
The direct way has many technical advantages and is promising for practical use. But stakeholders are usually more concerned about economic feasibility. Raw materials and energy consumption are the main operation costs. Raw materials depend on the price of NaOH or NH3OH. Energy is mainly consumed in the crystallization of the Na2SO4 or (NH4)2SO4 by-product. Wang et al. proposed a way to apply amine for carbonation and recycle the protonated amine by bipolar membrane electrodialysis (BMED).66 Inspired by their work, it may be feasible to develop a NaOH–Na2SO4 circular system by bipolar membrane electrodialysis to reduce the cost of NaOH consumption and avoid energy consumption for crystallization.124 As shown in Fig. 6, by introducing bipolar membrane electrodialysis into the NaOH-FGDG-CO2 system, the by-product of Na2SO4 can be split into NaOH and H2SO4.125 In this way, the in situ regenerated NaOH is reused for FGDG carbonation, instead of raw material procurement. Operation cost comparisons are shown in Table 4. The data of energy consumption for direct FGDG mineralization without BMED were obtained from a pilot plant at a scale of 3 ton per day located in Zhejiang Province, China. For 1000 kg of FGDG, reaction heating consumes about 400 kg of steam and evaporative crystallization consumes about 1000 kg of steam. The quantities of raw materials and products are calculated according to the stoichiometric ratio. When 1000 kg of FGDG was used, 476 kg of CaCO3 and 676 kg of Na2SO4 were produced with the use of 381 kg of NaOH. Likewise, 629 kg of (NH4)2SO4 was obtained when using 334 kg of ammonia. The price of chemicals is the market price in China. The operation costs for NaOH-FGDG-CO2 and NH3-FGDG-CO2 approaches to process 1 ton of FGDG are $139 and $64, respectively. The difference is mainly due to the market price variance of Na2SO4 and (NH4)2SO4. Although the NH3-FGDG-CO2 approach demonstrates better cost-effectiveness, the use of ammonia increases environmental risks. Interestingly, by coupling BMED with the NaOH-FGDG-CO2 approach, operation cost is reduced to $67, close to that of NH3-FGDG-CO2. The energy consumption value of BMED is set to 3 kW h per kg-NaOH according to reported data.118,126 This is mainly due to the saving of raw materials and steam consumed by evaporation crystallization. Considering that raw material prices are wildly affected by the market, BMED is a promising way to obtain sustainable alkaline supply and withstand market risk.
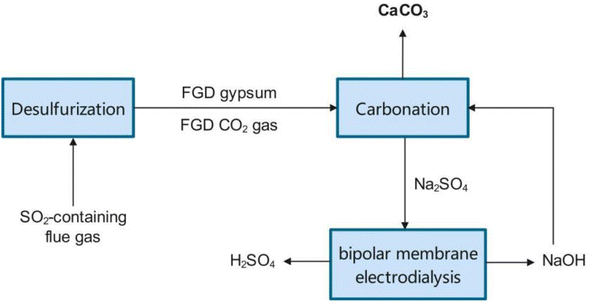 |
| | Fig. 6 Schematic diagram of FGDG carbonation coupled with bipolar membrane electrodialysis. | |
Table 4 Operation cost comparisons of the direct way with and without BMED
|
|
|
NH3-FGDG-CO2 |
NaOH-FGDG-CO2 |
NaOH-FGDG-CO2 with BMED |
|
Materials input
|
| Ammonia |
Input |
334 kg |
0 |
0 |
| Cost |
$175 USD |
0 |
0 |
| NaOH |
Input |
0 |
381 kg |
0 |
| Cost |
0 |
$164 USD |
0 |
|
|
Energy input
|
| Steam |
Input |
1400 kg |
1400 kg |
400 kg |
| Cost |
$59 USD |
$59 USD |
$17 USD |
| Electricity |
Input |
0 |
0 |
1143 kW h |
| Cost |
0 |
0 |
$114 USD |
|
|
Product output
|
| CaCO3 |
Output |
476 kg |
476 kg |
476 kg |
| Income |
$55 USD |
$55 USD |
$55 USD |
| (NH4)2SO4 |
Output |
629 kg |
0 |
0 |
| Income |
$115 USD |
0 |
0 |
| Na2SO4 |
Output |
0 |
676 kg |
0 |
| Income |
0 |
$29 USD |
0 |
| H2SO4 |
Output |
0 |
0 |
460 kg |
| Income |
0 |
0 |
$10 |
| Total cost |
|
$64 USD |
$139 USD |
$66 USD |
5. Conclusion
In this review, we mainly summarize the most recent developments of calcium carbonate recovery from FGDG. There are mainly two kinds of methods developed for FGDG carbonation. i.e. the direct way and indirect way. The direct way is gypsum carbonation with CO2 in alkaline solution at near room temperature. The indirect way is thermal reduction of gypsum first and then carbonation of the thermal reduction product.
The direct way typically involves three stages: CO2 capture and CO32− formation; CaSO4·2H2O dissolution; CaCO3 crystallization. NaOH-FGDG-CO2 and NH3-FGDG-CO2 are widely studied carbonation systems. The polymorph of CaCO3 is affected by many factors, such as the Ca2+/CO32− ratio, reaction conditions, impurities, and additives.
The indirect way involves gypsum thermal reduction, carbonation, and sulfur recovery. Thermal reduction is preferred to achieve a high conversion ratio at around 900 °C with the addition of carbon as the reductant. The carbonation of CaS is sluggish and produces low purity CaCO3 and H2S gas by-products. Transformation of CaS into Ca(HS)2 by H2S before carbonation can purify the CaCO3 product. Finally, high-value elemental sulfur can be recovered from H2S oxidation.
In comparison, the direct way has advantages of simple processes, a high conversion rate and mild reaction conditions. The indirect way can obtain high purity CaCO3 and is suitable for factories with cheap energy sources. The combination of the direct way and BMED technique has significant advantages. Although there are several approaches developed for FGDG carbonation, pilot and industrial applications are needed to study technical and economic feasibility in the future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Author contributions
Wei Xu contributed to conceptualization, writing of the original draft, review & editing. Chunhong Liu contributed to conceptualization, writing – review & editing. Kaimin Du contributed to conceptualization and writing of the original draft. Qiangsheng Gao contributed to writing – review & editing. Zheming Liu contributed to writing – review & editing. Weijian Wang contributed to writing of the original draft.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Zhejiang Energy Group Science and Technology Project.
References
- T. Cheng, X. C. Zhou, L. J. Yang, Z. Q. Sun and H. Wu, Energy Fuels, 2020, 34, 3836–3842 CrossRef CAS.
- Z. Xu, D. Hu, R. An, L. Lin, Y. Xiang, L. Han, Y. Yu, L. Ning and J. Wu, Case Stud. Constr. Mater., 2022, 17, e01549 Search PubMed.
- S. Jian, X. Yang, W. Gao, B. Li, X. Gao, W. Huang, H. Tan and Y. Lei, Constr. Build. Mater., 2021, 301, 124341 CrossRef CAS.
- T. Y. Qi, J. Z. Zhang, T. Li, S. L. An, Q. W. Li and L. D. Wang, Appl. Surf. Sci., 2024, 648, 158981 CrossRef CAS.
- S. Liu, W. Liu, F. Jiao, W. Qin and C. Yang, Environ. Pollut., 2021, 288, 117799 CrossRef CAS PubMed.
- S. J. Chen, T. Q. Jiang, H. W. Zhang, K. Kong and L. S. Bie, Energy Sci. Eng., 2020, 8, 2662–2679 CrossRef CAS.
- H. S. Guo, Q. P. Wang, W. F. Li, X. Feng, J. L. Yang, J. J. Cao, T. Z. Shen, X. M. Qin, Y. F. Liu, Y. H. Gui and L. M. Zhou, Constr. Build. Mater., 2023, 377, 130981 CrossRef CAS.
- M. J. Li, G. D. Huang, B. Wang, Y. Cui, B. B. Chang, Q. Q. Yin, M. Ge, S. W. Zhang, Q. Wang and J. C. Feng, Polymers, 2022, 14, 4761 CrossRef CAS PubMed.
- Aakriti, S. Maiti, N. Jain and J. Malik, Constr. Build. Mater., 2023, 393, 131918 CrossRef CAS.
- P. Bakshi, A. Pappu and M. K. Gupta, J. Mater. Cycles Waste Manage., 2022, 24, 49–62 CrossRef.
- N. H. Koralegedara, P. X. Pinto, D. D. Dionysiou and S. R. Al-Abed, J. Environ. Manage., 2019, 251, 109572 CrossRef CAS PubMed.
- J. Y. Hao, G. J. Cheng, T. Hu, B. Guo and X. J. Li, Constr. Build. Mater., 2021, 306, 124910 CrossRef CAS.
- C. X. Wu, J. H. He, K. Wang, L. Yang and F. Z. Wang, Constr. Build. Mater., 2023, 387, 131565 CrossRef CAS.
- P. Maichin, P. Jitsangiam, T. Nongnuang, K. Boonserm, K. Nusit, S. Pra-ai, T. Binaree and C. Aryupong, Materials, 2021, 14, 1858 CrossRef CAS PubMed.
- P. Córdoba, Fuel, 2015, 144, 274–286 CrossRef.
- A. Myka, R. Łyszczek, A. Zdunek and P. Rusek, J. Therm. Anal. Calorim., 2022, 147, 9923–9934 CrossRef CAS.
- T. Sithole, T. Mashifana, D. Mahlangu and L. Tchadjie, Buildings, 2021, 11, 500 CrossRef.
- E. Baran, S. Czernik, M. Hynowski, B. Michałowski, M. Piasecki, J. Tomaszewska and J. Michalak, Sustainability, 2021, 13, 4298 CrossRef CAS.
- J. Li, X. Zhuang, C. Leiva, A. Cornejo, O. Font, X. Querol, N. Moeno, C. Arenas and C. Fernández-Pereira, Constr. Build.
Mater., 2015, 95, 910–921 CrossRef.
- L. Xu, K. Wu, N. Li, X. Zhou and P. Wang, J. Cleaner Prod., 2017, 161, 803–811 CrossRef CAS.
- S. Wansom, P. Chintasongkro and W. Srijampan, Cem. Concr. Compos., 2019, 103, 134–148 CrossRef CAS.
- Q. Wu, H. Ma, Q. Chen, Z. Huang, C. Zhang and T. Yang, Constr. Build. Mater., 2019, 214, 318–325 CrossRef CAS.
- L. Yang, M. Jing, L. Lu, X. Zhu, P. Zhao, M. Chen, L. Li and J. Liu, Constr. Build. Mater., 2020, 257, 119519 CrossRef CAS.
- H. A. Nguyen, C. T. Chen, T. P. Chang and J. Y. Shih, J. Therm. Anal. Calorim., 2023, 13761–13773, DOI:10.1007/s10973-023-12715-y.
- H. Kaur, K. W. J. Williard, J. E. Schoonover and G. Singh, Water, Air, Soil Pollut., 2022, 233, 72 CrossRef CAS.
- S. F. Acuña-Guzman and L. D. Norton, Sustainability, 2023, 15, 1977 CrossRef.
- D. B. Watts, G. B. Runion and H. A. Torbert, Horticulturae, 2021, 7, 199 CrossRef.
- J. Wang, A. Q. Zhao, F. Ma, J. L. Liu, G. J. Xiao and X. Xu, Sustainability, 2023, 15, 8658 CrossRef CAS.
- J. Wang and P. Yang, Renewable Sustainable Energy Rev., 2018, 82, 1969–1978 CrossRef CAS.
- W. C. Zhang, Y. G. Zhao, S. J. Wang, Y. Li, Y. Q. Zhuo and J. Liu, Land Degrad. Dev., 2021, 32, 4193–4202 CrossRef.
- P. Sundha, R. Mukhopadhyay, N. Basak, A. K. Rai, S. Bedwal, S. Patel, S. Kumar, H. Kaur, P. Chandra, P. C. Sharma, S. K. Saxena, S. S. Parihar and R. K. Yadav, Sci. Rep., 2023, 13, 19787 CrossRef CAS PubMed.
- Z. X. Liu, Y. Hao, J. Zhang, S. M. Wu, Y. Pan, J. Z. Zhou and G. R. Qian, Fuel, 2020, 271, 117515 CrossRef CAS.
- Y. M. Huang, J. L. Liu, G. Wang, X. Y. Bi, G. Y. Sun, X. Wu, Q. F. Wang and Z. G. Li, Int. J. Environ. Res. Public Health, 2022, 19, 12617 CrossRef CAS PubMed.
- J. Gao, Q. Li and F. L. Liu, Chin. J. Chem. Eng., 2020, 28, 2221–2226 CrossRef CAS.
- G. Chen, R. Liu, C. Song and J. Luo, J. Cryst. Growth, 2021, 576, 126360 CrossRef CAS.
- C.-U. Kang, S.-W. Ji and H. Jo, Sustainability, 2022, 14, 4436 CrossRef CAS.
- Y. Yan, X. Dong, X. Sun, X. Sun, J. Li, J. Shen, W. Han, X. Liu and L. Wang, J. Colloid Interface Sci., 2014, 429, 68–76 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, Y. Guo, P. K. Chu and S. Tu, Waste Biomass Valorization, 2017, 8, 203–207 CrossRef CAS.
- J. Kang, X. Gou, Y. Hu, W. Sun, R. Liu, Z. Gao and Q. Guan, Sci. Total Environ., 2019, 649, 344–352 CrossRef CAS PubMed.
- P. Bakshi, A. Pappu and D. K. Bharti, Mater. Lett., 2022, 308, 131177 CrossRef CAS.
- D. M. Wang, C. Chen, Y. B. Wang, S. Jiu and Y. X. Chen, Constr. Build. Mater., 2023, 394, 132280 CrossRef CAS.
- X. M. Chen, J. M. Gao, Y. Wu, Q. H. Wu and L. Luo, Materials, 2022, 15, 2691 CrossRef CAS PubMed.
- C.-q. Li, C. Liang, Z.-m. Chen, Y.-h. Di, S.-l. Zheng, S. Wei and Z.-m. Sun, J. Cent. South Univ., 2021, 28, 2589–2611 CrossRef CAS.
- M. Baláž, E. V. Boldyreva, D. Rybin, S. Pavlović, D. Rodríguez-Padrón, T. Mudrinić and R. Luque, Front. Bioeng. Biotechnol., 2021, 8, 612567 CrossRef PubMed.
- J. Luo, F. Kong and X. Ma, Cryst. Growth Des., 2016, 16, 728–736 CrossRef CAS.
- K. Longkaew, A. Gibaud, W. Tessanan, P. Daniel and P. Phinyocheep, Polymers, 2023, 15, 4287 CrossRef CAS PubMed.
- O. Ersoy and H. Köse, Polym. Compos., 2020, 41, 3483–3490 CrossRef CAS.
- Z. L. Liu, W. Y. Liu, L. Y. Ma, M. Mo, Y. F. Huang, W. Q. Lai, M. X. Xu, Y. M. Li, Q. F. Mo, X. J. Bai, C. Chen, Z. M. Huang and Y. H. Zheng, Polym. Compos., 2023, 1494–1507, DOI:10.1002/pc.27869.
- T. Ahmed, H. H. Ya, R. Khan, A. Lubis and S. Mahadzir, Materials, 2020, 13, 3333 CrossRef CAS PubMed.
- M. de Beer, F. J. Doucet, J. P. Maree and L. Liebenberg, Waste Manage., 2015, 46, 619–627 CrossRef CAS PubMed.
- K. Song, W. Kim, J.-H. Bang, S. Park and C. W. Jeon, Mater. Des., 2015, 83, 308–313 CrossRef CAS.
- A. Azdarpour, M. A. Karaei, H. Hamidi, E. Mohammadian, M. Barati and B. Honarvar, Int. J. Miner. Process., 2017, 169, 27–34 CrossRef CAS.
- L. Fernández-Díaz, C. M. Pina, J. M. Astilleros and N. Sánchez-Pastor, Am. Mineral., 2009, 94, 1223–1234 CrossRef.
- Y. Zarga, H. Ben Boubaker, N. Ghaffour and H. Elfil, Chem. Eng. Sci., 2013, 96, 33–41 CrossRef CAS.
- L. Yu, L. M. Daniels, J. J. P. A. Mulders, G. D. Saldi, A. L. Harrison, L. Liu and E. H. Oelkers, Chem. Geol., 2019, 525, 447–461 CrossRef CAS.
- X. Luo, C. Wei, X. Li, Z. Deng, M. Li and G. Fan, Fuel, 2023, 333, 126305 CrossRef CAS.
- Y. Gong, L. Wu, J. Li and J. Zhu, J. Cryst. Growth, 2019, 522, 128–138 CrossRef CAS.
- B. Wang, Z. Pan, H. Cheng, Y. Guan, Z. Zhang and F. Cheng, Environ. Chem. Lett., 2020, 18, 1369–1377 CrossRef CAS.
- B. Wang, Z. Pan, Z. Du, H. Cheng and F. Cheng, J. Hazard. Mater., 2019, 369, 236–243 CrossRef CAS PubMed.
- W. Tan, Z. Zhang, H. Li, Y. Li and Z. Shen, Environ. Sci. Pollut. Res., 2017, 24, 8602–8608 CrossRef CAS PubMed.
- M. G. Lee, K. W. Ryu, S. C. Chae and Y. N. Jang, Environ. Technol., 2015, 36, 106–114 CrossRef CAS PubMed.
- M. Altiner, Int. J. Coal Prep. Util., 2019, 39, 113–131 CrossRef CAS.
- W. Ding, H. Yang and J. Ouyang, RSC Adv., 2015, 5, 67184–67194 RSC.
- M. g. Lee, Y. N. Jang, K. w. Ryu, W. Kim and J.-H. Bang, Energy, 2012, 47, 370–377 CrossRef CAS.
- X. Liu, B. Wang, Z. Zhang, Z. Pan, H. Cheng and F. Cheng, Environ. Chem. Lett., 2022, 20, 2261–2269 CrossRef CAS.
- Y. Wang, X. Zheng, Y. Wang, Q. He, S. Yan and L. Ji, Chem. Eng. J., 2023, 478, 147335 CrossRef CAS.
- W. Liu, B. B. Wu, X. X. Bai, S. H. Liu, X. Y. Liu, Y. Hao, W. Z. Liang, S. M. Lin, H. J. Liu, L. N. Luo, S. Zhao, C. Y. Zhu, J. M. Hao and H. Z. Tian, Environ. Sci. Technol., 2020, 54, 390–399 CrossRef CAS PubMed.
- W. Liu, L. Teng, S. Rohani, Z. Qin, B. Zhao, C. C. Xu, S. Ren, Q. Liu and B. Liang, Chem. Eng. J., 2021, 416, 129093 CrossRef CAS.
- Z. Yan, Y. Wang, H. Yue, C. Liu, S. Zhong, K. Ma, W. Liao, S. Tang and B. Liang, ACS Sustain. Chem. Eng., 2021, 9, 8238–8248 CrossRef CAS.
- H. Liu, P. Lan, S. Lu and S. Wu, J. Cryst. Growth, 2018, 492, 114–121 CrossRef CAS.
- M. Altiner, S. Top, B. Kaymakoğlu, İ. Y. Seçkin and H. Vapur, J. CO2 Util., 2019, 29, 117–125 CrossRef CAS.
- R. Chang, D. Choi, M. H. Kim and Y. Park, ACS Sustain. Chem. Eng., 2017, 5, 1659–1667 CrossRef CAS.
- A. Katsman, I. Polishchuk and B. Pokroy, Faraday Discuss., 2022, 235, 433–445 RSC.
- D. B. Trushina, T. V. Bukreeva, M. V. Kovalchuk and M. N. Antipina, Mater. Sci. Eng., C, 2014, 45, 644–658 CrossRef CAS PubMed.
- Y. S. Han, G. Hadiko, M. Fuji and M. Takahashi, J. Eur. Ceram. Soc., 2006, 26, 843–847 CrossRef CAS.
- H. Chen, B. Dou, Y. Song, Y. Xu, X. Wang, Y. Zhang, X. Du, C. Wang, X. Zhang and C. Tan, Int. J. Greenhouse Gas Control, 2012, 6, 171–178 CrossRef CAS.
- I. Cuesta Mayorga, J. M. Astilleros and L. Fernández-Díaz, Minerals, 2019, 9, 178 CrossRef.
- S. Seepma, S. E. Ruiz-Hernandez, G. Nehrke, K. Soetaert, A. P. Philipse, B. W. M. Kuipers and M. Wolthers, Cryst. Growth Des., 2021, 21, 1576–1590 CrossRef CAS PubMed.
- M. Wolthers, G. Nehrke, J. P. Gustafsson and P. Van Cappellen, Geochim. Cosmochim. Acta, 2012, 77, 121–134 CrossRef CAS.
- S. M. Pérez-Moreno, M. J. Gázquez and J. P. Bolívar, Chem. Eng. J., 2015, 262, 737–746 CrossRef.
- R. Chang, S. Kim, S. Lee, S. Choi, M. Kim and Y. Park, Front. Energy Res., 2017, 5, 17 CrossRef.
- K. Song, W. Kim, S. Park, J.-H. Bang, C. W. Jeon and J.-W. Ahn, Chem. Eng. J., 2016, 301, 51–57 CrossRef CAS.
- K. Song, Y.-N. Jang, W. Kim, M. G. Lee, D. Shin, J.-H. Bang, C. W. Jeon and S. C. Chae, Chem. Eng. J., 2012, 213, 251–258 CrossRef CAS.
- Y. I. Svenskaya, H. Fattah, O. A. Inozemtseva, A. G. Ivanova, S. N. Shtykov, D. A. Gorin and B. V. Parakhonskiy, Cryst. Growth Des., 2018, 18, 331–337 CrossRef CAS.
- M. Abebe, N. Hedin and Z. Bacsik, Cryst. Growth Des., 2015, 15, 3609–3616 CrossRef CAS.
- A. E. Morandeau and C. E. White, Chem. Mater., 2015, 27, 6625–6634 CrossRef CAS.
- Z. Zhang, Y. Xie, X. Xu, H. Pan and R. Tang, J. Cryst. Growth, 2012, 343, 62–67 CrossRef CAS.
- H. Tang, J. Yu and X. Zhao, Mater. Res. Bull., 2009, 44, 831–835 CrossRef CAS.
- H. Lu, B. Zhong, B. Liang and Y. Zhang, J. Chem. Eng. Chin. Univ., 2002, 16, 97–100 CAS.
- Q. Chen, J. Wang, Z. Zhang, X. Wang, L. Yang, B. Zhong and X. Yang, Chem. Ind. Eng. Prog., 2016, 35, 3714–3719 Search PubMed.
- J. Zhao, X. Song, Y. Sun, B. Chen and J. Yu, Cryst. Res. Technol., 2015, 50, 277–283 CrossRef CAS.
- B. Njegić Džakula, J. Kontrec, M. Ukrainczyk, S. Sviben and D. Kralj, Cryst. Res. Technol., 2014, 49, 244–256 CrossRef.
- M. D. Luque de Castro and F. Priego-Capote, Ultrason. Sonochem., 2007, 14, 717–724 CrossRef CAS PubMed.
- H. Cheng, X. Wang, B. Wang, J. Zhao, Y. Liu and F. Cheng, J. Cryst. Growth, 2017, 469, 97–105 CrossRef CAS.
- Q. Chen, W. Ding, H. Sun, T. Peng and G. Ma, Energy, 2020, 206, 118148 CrossRef CAS.
- W. Ding, Q. Chen, H. Sun and T. Peng, J. CO2 Util., 2019, 34, 507–515 CrossRef CAS.
- Q. Chen, W. Ding, H. Sun, T. Peng and G. Ma, ACS Sustain. Chem. Eng., 2020, 8, 11649–11657 CrossRef CAS.
- B. Yang, M. Yang, B. Wang, X. Fang and Q. Wan, Mater. Res. Express, 2019, 6, 045042 CrossRef.
- Y. Gong, X. Zhu, Z. Yang, X. Zhang and C. Li, RSC Adv., 2022, 12, 26556–26564 RSC.
- L. Ma, X. Niu, J. Hou, S. Zheng and W. Xu, Thermochim. Acta, 2011, 526, 163–168 CrossRef CAS.
- Z. Miao, H. Yang, Y. Wu, H. Zhang and X. Zhang, Ind. Eng. Chem. Res., 2012, 51, 5419–5423 CrossRef CAS.
- M. de Beer, J. P. Maree, L. Liebenberg and F. J. Doucet, Waste Manage., 2014, 34, 2373–2381 CrossRef CAS PubMed.
- R. K. Tewo, J. P. Maree, S. Ruto, H. L. Rutto and L. K. Koech, Chem. Eng. Commun., 2017, 204, 1412–1419 CrossRef CAS.
- S. Liu, C. Yang, T. Zhang, W. Liu, F. Jiao and W. Qin, Sep. Purif. Technol., 2023, 314, 123537 CrossRef CAS.
- H. Tan, M. Ye, X. Su, X. Ma, F. Dong and F. Yang, J. Therm. Anal. Calorim., 2022, 147, 14115–14121 CrossRef CAS.
- F. Laasri, A. Carrillo Garcia, M. Latifi and J. Chaouki, Waste Manage., 2023, 171, 482–490 CrossRef CAS PubMed.
- Y. Yu and L. Li, ISIJ Int., 2020, 60, 2291–2300 CrossRef CAS.
- J. Zhao, H. Su, H. Zuo, J. Wang and Q. Xue, J. Cleaner Prod., 2020, 267, 122002 CrossRef CAS.
- N. Mihara, D. Kuchar, Y. Kojima and H. Matsuda, J. Mater. Cycles Waste Manage., 2007, 9, 21–26 CrossRef CAS.
- S. R. Motaung, J. N. Zvimba, J. P. Maree and A. V. Kolesnikov, Water SA, 2015, 41, 369–374 CrossRef CAS.
- K. Uiga, E. Rikmann, I. Zekker and T. Tenno, Proc. Est. Acad. Sci., 2020, 69, 323–330 CrossRef.
- S. J. Eng, R. J. Motekaitis and A. E. Martell, Inorg. Chim. Acta, 2000, 299, 9–15 CrossRef CAS.
- K. Wang, S. Lin and Z. Z. Ran, J. Therm. Anal. Calorim., 2023, 148, 7357–7368 CrossRef CAS.
- S. Piché and F. Larachi, Chem. Eng. Sci., 2006, 61, 7673–7683 CrossRef.
- P. Córdoba, R. Ochoa-Gonzalez, O. Font, M. Izquierdo, X. Querol, C. Leiva, M. A. López-Antón, M. Díaz-Somoano, M. Rosa Martinez-Tarazona, C. Fernandez and A. Tomás, Fuel, 2012, 92, 145–157 CrossRef.
- E. Álvarez-Ayuso, X. Querol and A. Tomás, Chemosphere, 2006, 65, 2009–2017 CrossRef PubMed.
- M. Zhu, B. H. Tian, S. Luo, Y. Z. Chi, D. Aishajiang, Y. Zhang and M. Yang, Resour. Conserv. Recycl., 2022, 186, 106556 CrossRef CAS.
- O. S. L. Bruinsma, D. J. Branken, T. N. Lemmer, L. van der Westhuizen and S. Rossouw, Desalination, 2021, 511, 115096 CrossRef CAS.
- Kuldeep, W. D. Badenhorst, P. Kauranen, H. Pajari, R. Ruismäki, P. Mannela and L. Murtomäki, Membranes, 2021, 11, 718 CrossRef CAS PubMed.
- X. Xia, L. Q. Zhang, X. L. Yuan, C. Y. Ma, Z. L. Song and X. Q. Zhao, Environ. Eng. Sci., 2021, 38, 886–898 CrossRef CAS.
- L. L. Guo, Z. Wu, H. Wang, H. L. Yan, F. S. Yang, G. X. Cheng and Z. X. Zhang, Chem. Eng. J., 2023, 455, 140689 CrossRef CAS.
- J. X. Xu and W. S. Lin, Energy, 2021, 227, 120416 CrossRef.
- H. Hu and M. B. Wu, J. Mater. Chem. A, 2020, 8, 7066–7082 RSC.
- L. Monat, S. Chaudhury and O. Nir, ACS Sustain. Chem. Eng., 2020, 8, 2490–2497 CrossRef CAS.
- N. V. Kovalev, T. V. Karpenko, N. V. Sheldeshov and V. I. Zabolotskii, Membr. Membr. Technol., 2020, 2, 391–398 CrossRef CAS.
- W. Gao, Q. Fang, H. Yan, X. Wei and K. Wu, Membranes, 2021, 11, 152 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Chunhong
Liu
a,
Kaimin
Du
a,
Qiangsheng
Gao
a,
Zheming
Liu
b and
Weijian
Wang
b
*a,
Chunhong
Liu
a,
Kaimin
Du
a,
Qiangsheng
Gao
a,
Zheming
Liu
b and
Weijian
Wang
b