
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbons derived from resole-type phenolic resins for use in lithium–sulfur batteries: templating the resins with sulfur leads to enhanced cell performance†
Luke D. J.
Barter
 a,
Irshad
Mohammad
a,
Steven J.
Hinder
b,
John F.
Watts
b,
Robert C. T.
Slade
a and
Carol
Crean
*a
a,
Irshad
Mohammad
a,
Steven J.
Hinder
b,
John F.
Watts
b,
Robert C. T.
Slade
a and
Carol
Crean
*a
aSchool of Chemistry and Chemical Engineering, University of Surrey, Guildford GU2 7XH, UK. E-mail: c.crean@surrey.ac.uk
bSchool of Mechanical Engineering Sciences, University of Surrey, Guildford GU2 7XH, UK
First published on 8th January 2024
Abstract
Pyrolysed resole-type phenol-formaldehyde resins were used as carbonaceous sulfur-hosts in the cathodes of lithium–sulfur batteries. Porosity was added through sulfur-depositing, via acidification of ammonium thiosulfate, within the polymerisation reaction. Micellar-templated and untemplated carbons were also synthesised as comparisons. The carbons that had been subjected to the lowest extremes of pyrolysis (600 °C) retained higher amounts of functional groups and greater pore volumes when characterised by IR and nitrogen sorption studies, respectively. The three carbon types behaved similarly giving performances of circa 500 mA h gsulfur−1 (by the 40th cycle at 0.05C) when melt-loaded with sulfur into cathode films. In contrast, when sulfur was deposited onto the same carbons, via thiosulfate acidification, the different porous designs led to changes in battery performance of between 500–800 mA h gsulfur−1 (after the 40th cycle at 0.05C). The structures afforded by the different sulfur-loading methods were analysed using SEM-EDS and Raman spectroscopy.
1. Introduction
Currently, energy-dense applications are being serviced by lithium-ion battery (LIB) chemistry. LIBs, as a class, work by intercalation and deintercalation (insertion and removal, respectively) of lithium-ions into and out of layered or porous structures leading to indirect-redox at the electrodes.1 The key metrics that make LIBs so heavily used in everyday life include their rechargeable nature with high cycle lifetimes (>2000, with some papers stating 85% of the initial capacity being retained after 3500 cycles) as well as being operable for high-power applications (e.g. 180 mA h gLi(Ni0.5Mn0.5)O2−1 at 6C [theoretically charging/discharging in 10 min]).2,3 However, LIBs are limited in terms of their energy storage performance (∼240–250 W h kg−1 for higher power capable cells), their safety involving thermal runaway, their high costs and environmental sustainability due to the large amounts of cobalt currently required for their cathodes.4–6| xLianode+ + xeexternal to the cell− + Li1−xCoO2cathode ↔ LiCoO2cathode | (1) |
Lithium–sulfur is an alternative to LIB-based technologies by providing a high specific energy storage performance of approximately 2500 W h kg−1 (driven by a specific capacity ≈1675 mA h gsulfur−1) and offering improvements in safety by avoiding thermal runaway. Lithium–sulfur also promises lower costs and fewer environmental impacts than LIBs by avoiding the use of cobalt in their manufacture.7 However, the lithium–sulfur battery (LSB) can suffer from low performances at high-powers as well as low cycle lifetimes especially under commercially relevant lean-electrolyte conditions.8 This is because LSBs do not operate by intercalation and deintercalation, but by lithium-ions from the anode reacting with sulfur at the cathode to yield lithium sulfide.9
| 16Lianode+ + 16eexternal to the cell− + S8cathode ↔ 8Li2Scathode | (2) |
During discharge the insoluble and electrically non-conductive elemental sulfur (S8) is converted via multiple redox reactions to insoluble and electrically non-conductive lithium sulfide (Li2S).10 These redox reactions produce several electrically conductive and soluble intermediate species – polysulfides.9
These lithium polysulfides (Li2Sx, where 3 ≤ x ≤ 8) can undergo a process termed ‘polysulfide shuttling’ where, caused by the polysulfides’ enhanced solubilities, the active-sulfur can migrate from the cathode to the separator or even as far as the anode.9 This leads to a reduction in battery charge-storage ability and chemical stability.11
Controlling the polysulfide-shuttle is still the major stumbling block to commercialisation. Thus, porous materials are being studied to be able to hold, retain, and even mediate charge-transfer of the sulfur to lithium polysulfides/lithium disulfide/lithium sulfide species.12 Porous carbons are a particularly broad class of materials that, are both relatively-cheap and cost-effective, and are most importantly incredibly scalable.13 Porosity can be tuned to the desired application by a variety of methods of designing precursor polymer materials and subsequent pyrolysis and calcining.14 The precursor polymer used will influence any heteroatom moieties to add functionality, which leads to a surface polarity change and potentially added benefits such as the hindering of polysulfide shuttling.15 However, the porous materials must be structurally resistant to volumetric changes that the sulfur-containing species go through during discharge (volumetric expansion) and charging (volumetric contraction) – during discharge the volume of sulfur (S8) expands by an extra 80% when converting to lithium sulfide (8 Li2S).16
Resoles [or resols], are a subset of phenol-formaldehyde resins and are thermoset polymers where cross-linking of polymer chains produces a robust polymeric material which cannot melt, after an initial curing step.17 Such materials, due to their dense cross-linking, can form structurally integral or structurally supportive porous carbons following pyrolysis (and porous templating by a pore former).
Currently employed pore-forming methods do not include using elemental sulfur, even though it is the humidity and air-stable species of interest during the active-material's operation within an LSB.18–20 All other final products/intermediate products other than elemental sulfur hydrolyse to form hydrogen sulfide (H2S) as an unwanted by-product. Sulfur can melt and vaporise, at approximately 119 °C and 445 °C respectively; below the temperatures required to convert the polymeric material into carbonaceous material by pyrolysis. Calcination could, therefore, be included in the same pyrolysis step producing an interlinked network.20 The rationale for the use of sulfur as a pore-former was that in addition to increased porosity and specific surface area, the sulfur may be able to lead to sulfur-specific shapes, dimensions, and morphologies. It was envisaged that they may impart memory-like sulfur impressions into the structure of porous carbons that would otherwise be inaccessible – allowing the coining of the term ‘memory-carbons’ when in the direct application of LSBs.
This report develops the idea of using sulfur to form pores in a carbon, to impart ‘memory’ effects. This material was fabricated into a cathode using both sulfur-deposition via acidification of thiosulfate and the established method of melt-loading. Stabilised sulfur nanoparticle production was based on citable literature where acidification of thiosulfate salts, in aqueous conditions, produced nanoscale colloidal sulfur.21 Surfactants are also already commonly used in the production of porous carbons, zeolites and other porous materials but this templating is generated solely by the formation of the surfactants’ own micellar structures.22 Therefore, combining the two methods would introduce a sulfur-specific porosity to a carbon.
2. Experimental
2.1. Chemicals and electrolyte composition
The starting materials utilised in this study were: phenol (Merck, 99%, ACS reagent grade); formaldehyde (Merck, 37% solution in water with 10% methanol content as stabiliser, ACS reagent grade); lithium carbonate (Merck, 99%, ACS reagent grade); cetyltrimethyl ammonium bromide, CTAB (Alfa Aesar, 98%); hydrochloric acid (Fisher, 37% hydrochloric acid in water); sulfur (Merck, 99.98% trace metals basis); oxalic acid dihydrate (Merck, 99%, ACS reagent grade); ammonium thiosulfate (Merck, 98%); Super P® carbon additional conductive additive (Timcal); N-methyl-2-pyrolidone, NMP (Merck, 99% anhydrous); polyvinylidene difluoride, PVDF as cathode binder (Merck, ca. 534![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MW by GPC); lithium ribbon for anode use (0.75 mm thickness, Merck, 99.9% metals basis).
000 MW by GPC); lithium ribbon for anode use (0.75 mm thickness, Merck, 99.9% metals basis).
The electrolyte used comprised of a solvent that was 50:50 v/v of 1,2-dimethoxyethane, DME, (Merck, anhydrous, 99.5% and free of inhibitors) and 1,3-dioxolane, DOL, (Merck, anhydrous, 99.8% with butylated hydroxytoluene (BHT) inhibitor [at a concentration within the solvent of 75 ppm]) and each was further dried by placing 4 Å molecular sieves (Merck, 8–12 mesh) into the solvent bottles at around a third of the solvent volume in order to dry for at least 3 days. Lithium bis(trifluoromethanesulfonyl)imide, LiTFSI, (Merck, 99.95% metals basis) was dried for 3 days at 120 °C on a Schlenk line before dissolution in the dried solvents to make an overall concentration of 1.0 mol dm−3 LiTFSI. Lithium nitrate (Merck, 99.99% trace metals basis) was also used as an additive to hinder dendrite formation in the electrolyte at a final concentration of 0.8 mol dm−3 LiNO3 in the solvent mixture, and was dried overnight in a vacuum oven at 80 °C.
2.2. Porous carbon synthesis
Following the scheme in Fig. 1, 10.6 g CTAB surfactant was dissolved in 900 cm3 of deionised water, heated and stirred at 70 °C. After 30 min, 10.0 g ammonium thiosulfate [(NH4)2S2O3] was dissolved in this solution and after 1 h approx. 8.5 g of oxalic acid dihydrate [H2C2O4·2H2O] was added, forming a white solution that became yellow and turbid. It should be noted that oxalic acid is toxic to human health.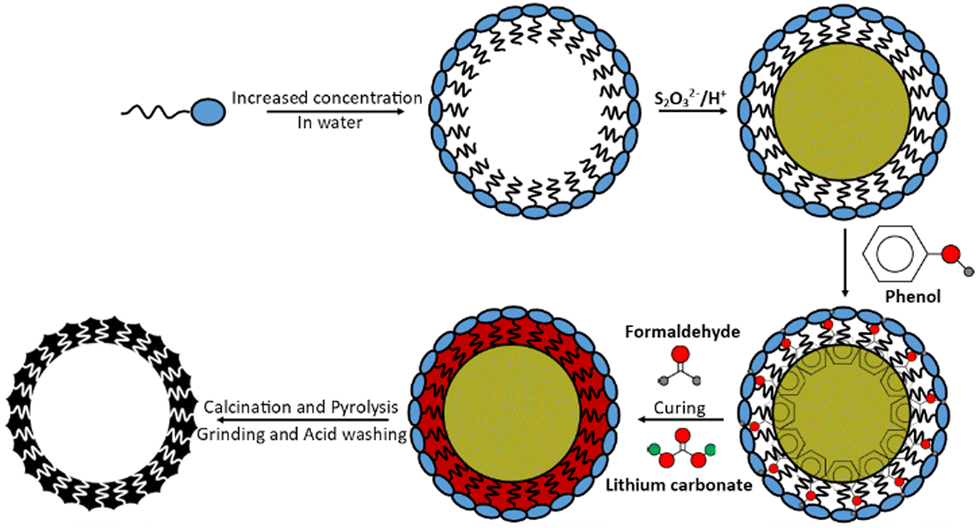 | ||
| Fig. 1 Synthesis procedure of a sulfur-particle-templated (“S”) resole resin. | ||
After 30 min of stirring, 20.0 g phenol was added followed by 32 cm3 formaldehyde. Finally, after 10 min of stirring 2.0 g lithium carbonate catalyst was added to form a white gum-like resin with yellow particles within. The resin mixture was heated to 180 °C for 4 h (to evaporate the excess water) followed by curing overnight at 120 °C. The yellow semi-solid polymer was finally cured for a further 90 min at 180 °C. The final resole was ground to a smooth yellow powder.
Two resole analogues were also synthesised without the prior deposition of sulfur (one analogue made with CTAB and one made without surfactant) for comparison; these analogues served as controls as simpler forms of porosity. All proportions of the remaining chemicals, timings, and heating temperatures remained constant irrespective of the resole formed (20.0 g phenol, 32 cm3 formaldehyde, 2.0 g lithium carbonate [and where micellar-templated 10.6 g CTAB]) but required fewer steps the less templated the end resole was. Both resoles were red colours and sample nomenclature is explained in Table 1.
| Sample | Abbreviation | Sample | Abbreviation |
|---|---|---|---|
| Sample prefix | Sample suffix | ||
| Sulfur-particle templated | “S” | Resole | Res |
| Regular/untemplated | Reg | Pyrolysed to 600 °C | 600 |
| Micellar-templated | Mic | Pyrolysed to 800 °C | 800 |
The resoles were pyrolysed to convert the resins to carbons with porous structures. Table 2 outlines the pyrolysis regimes used under 0.05 cm3 min−1 of a flowing nitrogen atmosphere. Two sets of samples were used – one pyrolysed at the lower temperature of 600 °C and the other pyrolysed at the higher temperature of 800 °C.
Following pyrolysis, the materials were milled (excluding the “S”-templated carbons) in an agate-walled Fritsch Pulverisette 0 mill with a 70 mm diameter agate grinding ball at 2 mm vertical oscillation for 15 min. All powders were then acid washed by refluxing in 4 mol dm−3 hydrochloric acid for 1 h. The powders were then washed several times with fresh deionised water and were evaporated to dryness. The “S”-templated carbons produced fine powders following acid washing without needing to be milled.
2.3. Sulfur-loading of pyrolysed materials
Composite materials were made with sulfur in a 7:1 mass ratio of elemental sulfur to pyrolysed resole. The sulfur was loaded via two methods: melt-loading and sulfur deposition.
:1 mass mixture of sulfur to pyrolysed porous host material was heated using an oil bath at 160 °C for 1 h to melt the sulfur.23 After 1 h, while still hot, the mixture was poured above an agate-walled mortar and milled in a Fritsch Pulverisette 0 mill with a 70 mm diameter agate grinding ball at 2 mm vertical oscillation for 15 min.
2.4. Cell production
:1:1:1 (defined by %mass), respectively. The sulfur and pyrolysed resole were added as a composite of 7:1 (either by melting or depositing the sulfur) of sulfur to pyrolysed resole, which became 80%mass of the total coating. Super P® was added as a 4.2%mass ink in NMP which became 10%mass of the coating. The coatings were cast on carbon-coated aluminium foil (MTI corp.) to a wet thickness of 400 μm and were vacuum dried at 50 °C overnight. Resulting electrodes were then cut to 14 mm diameter discs and used as the cathodes in CR2032 coin cells.
:1 v/v mixture of DME to DOL with 0.8 mol dm−3 LiNO3. The cathode was placed above the electrolyte followed by another 0.5 mm spacer. Coin cells were crimped before removal from the dry box.
2.5. Characterisation
X-ray diffraction (XRD) profiles were recorded using a Malvern Panalytical X’pert powder diffractometer at 45 kV and 40 mA with non-monochromatic Cu X-ray radiation for a 2 h measurement between 2θ angles of 10° and 70°. X-ray photoelectron spectroscopy (XPS) data were obtained using a Thermo Scientific K-Alpha+ spectrometer with a monochromatised Al Kα (hν = 1486.6 eV, 1 eV = 1.6 × 10−19 J) X-ray source. An X-ray spot of ∼400 μm radius was employed in the acquisition of all spectra. Survey spectra were acquired employing a pass energy of 200 eV. High resolution, core level spectra were acquired with a pass energy of 50 eV. All core level (high-resolution) spectra were charge referenced against the C1s peak at 285.0 eV to correct for charging effects.
Infrared (IR) spectroscopy was carried out on a PerkinElmer Spectrum Two FT-IR spectrometer with a UATR ZnSe/diamond accessory. Each analysis consisted of 32 Fourier transform (FT) measurements between 4000 and 400 cm−1, with a 2 cm−1 resolution. Scanning electron microscopy (SEM) analysis used a Thermo Scientific Apreo S electron microscope, that used a secondary electron detector, at 5 kV beam energy and 1.6 nA beam current with an energy-dispersive X-ray spectroscopy (EDS) detector for elemental mapping. All samples were coated with a 3 nm thickness gold coating to improve the samples’ conductivities. Nitrogen sorptiometry for surface-area and porosity studies for each sample was performed with a Belsorp mini II sorptometer at 77 K. CHNS elemental microanalyses were carried out using a Thermo Scientific Thermo FlashEA 1112 under dynamic flash combustion (DFC) at 1800 °C in a helium carrier gas and using a gas chromatography (GC) column (to separate species that were produced) were eluted into a thermal conductivity detector (TCD).
3. Results and discussion
XRD was used to compare the resole resins and their pyrolysed counterparts (Fig. S1, ESI†). Reg–Res and Mic–Res share peaks consistent with the starting catalyst lithium carbonate. “S”-Res shows sharp peaks, due to ammonium bromide and urea (reaction side products). All profiles feature a broad amorphous peak (more of a base line for “S”-Res) between 10° and 30° of 2θ caused by laterally stretched phenolic chains.24The peaks that are present in the pyrolysed carbon profiles are from the amorphous carbon (200) and (001) centred between 22°–24° and 44° of 2θ respectively. The greater low-angle scattering shown for the pyrolysed resoles approaching 10° (excluding Reg-800 and Mic-800) could suggest an enhanced microporous region relative to the other carbon samples.25
Fig. 2 shows N2 sorption–desorption isotherms for the carbons used in this study. The modelled pore data is shown in Fig. S2 (ESI†). All samples, apart from Mic-800, had sizable quantities of adsorbate in the microporous region (P/P0 < 0.1). The pyrolysed Reg-600 and Reg-800 carbons have porosity due to water and volatile organic components (VOCs) evaporation during the curing process. This implies unordered and less controlled porosity compared to materials using soft templates. There has clearly been a collapse of the micropores when increasing the pyrolysis temperature of the Reg-600 and Reg-800 carbons from 600 °C to 800 °C leading to a reduced specific surface area for the latter. A pyrolysis temperature of 600 °C is advantageous to preserving microporosity and functional groups [vide infra] of the carbon. A similar trend is observed for the micellar templated samples. Mic-800 carbon had a low specific surface area of 89 m2 g−1 (Table 3) and in direct comparison, Mic-600 had a specific surface area (driven by a greater micropore extent) of 468 m2 g−1.
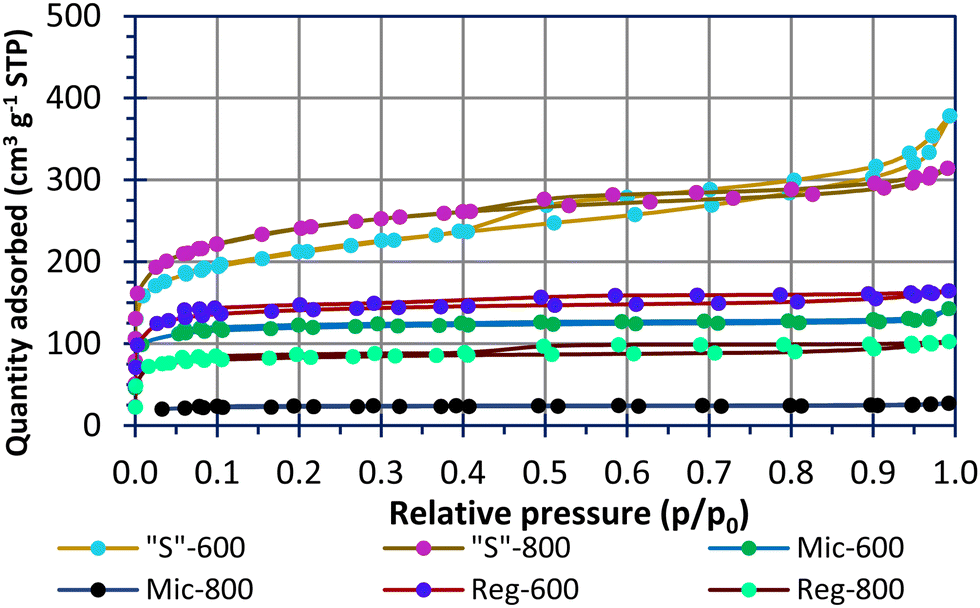 | ||
| Fig. 2 The nitrogen sorption isotherms of the carbonaceous materials. | ||
| Sample | BET (m2 g−1, 0.02 < P/P0 < 0.1) |
|---|---|
| Reg-600 | 548.3 ± 0.9 |
| Reg-800 | 322 ± 1.2 |
| Mic-600 | 468.1 ± 0.3 |
| Mic-800 | 88.5 ± 0.9 |
| “S”-600 | 774.3 ± 0.6 |
| “S”-800 | 890.8 ± 1.1 |
The sulfur templating in “S”-600 and “S”-800 was designed to direct the micellar structures in an effort to connect the pores into a porous network following calcination. This has resulted in materials with specific surface areas of 774 m2 g−1 (“S”-600) and 891 m2 g−1 (“S”-800) as well as significant microporous and mesoporous contributions which the other samples lack. In this instance the higher pyrolysis temperature has not led to a lower specific surface area. Microporosity is thought to be favoured for polysulfide absorption and high mesoporosity is favoured for high sulfur-loadings within a composite.15,26
Reviewing the literature, carbons pyrolysed above 600 °C can lead to extensive microporous networks, but this typically requires the etching of a hard-template of silica as the pore former.27 The methodology put forward in this work used sulfur that had been deposited from ammonium thiosulfate and ultimately could be removed during a single step of calcining which takes place within the pyrolysis process.
The SEM image in Fig. 3 shows that when a carbon is “S”-templated (specifically shown by “S”-800) the structure forms a surface with visible macropores. The cross-section of “S”-800 appears mottled with uneven surfaces consistent with the presence of small pores (such as micropores and mesopores) which is supported by the sorptiometry data (shown in Fig. 2 and Fig. S2, ESI†).
 | ||
| Fig. 3 An SEM micrograph of the “S”-templated carbon “S”-800. | ||
SEM-EDS mapping results from the different sulfur-loading methods (both Reg-600 and “S”-600) are shown in Fig. 4. The carbon hosts both contain significant portions of oxygen (red) which leads to the false magenta colour when overlaid with carbon (blue). When melted, the composite surface is continuously covered in sulfur (yellow). The deposition method produced coatings of sulfur on the carbons’ surfaces evidenced by greater extents of the carbon surfaces being observed in those composites, relative to melt-loading. When melting the sulfur there seems to be less control in accessing porous surfaces; in contrast, the sizes of the deposited sulfur (relative to the melted sulfur) appear to be smaller which should be more accessible during cycling in a LSB (shown in Fig. 4c and f, and higher magnification SEM micrographs in Fig. S3, ESI†).
 | ||
| Fig. 4 False-coloured SEM micrographs of (a) Reg-600, (b) melt-loaded Reg-600/S composite and (c) deposit-loaded Reg-600/S composite, as well as the false-coloured SEM micrographs of (d) “S”-600, (e) melt-loaded “S”-600/S composite and (f) deposit-loaded “S”-600/S composite. Of the false-colours, carbon is blue, oxygen is red (producing magenta were carbon and oxygen co-exist), and sulfur is yellow. The dashed teal outline separates (where present) the carbon sulfur-host from the carbon tape used to adhere the samples to the SEM sample stubs. | ||
High-resolution XPS spectra (Fig. 5), show the carbon, nitrogen, sulfur, and oxygen environments of the sample surfaces. Other elemental impurities are shown (if present) in the survey spectra present in Fig. S4 (ESI†). From Fig. 5, Reg-600, Reg-800 and Mic-800 carbons gave a different carbon peak at 284.5 eV compared to the other templated carbons at 285.0 eV. The carbon environment at 284.5 eV is consistent with sp2 carbons and 285.0 eV is consistent with sp3 carbons. The sp2 carbon presence found for the Reg carbons suggests either incomplete conversion of the phenolic units (as the rings are aromatic) prior to pyrolysis or complete graphitisation of the carbon. However, templating and pyrolysis appear to complete the conversion of the phenolic rings to amorphous carbon. Mic-800 seems to contain sp2 carbons likely due to graphitisation at greater pyrolysis extremes. “S” composites loaded with sulfur (via either melting or depositing S onto “S”-600) show a further difference between the high-resolution C 1s spectra. When melted, the C 1s spectrum is indifferent to that of the bare carbon; depositing the sulfur however produced an additional peak at 286.4 eV consistent with oxalate C–O bonds.28
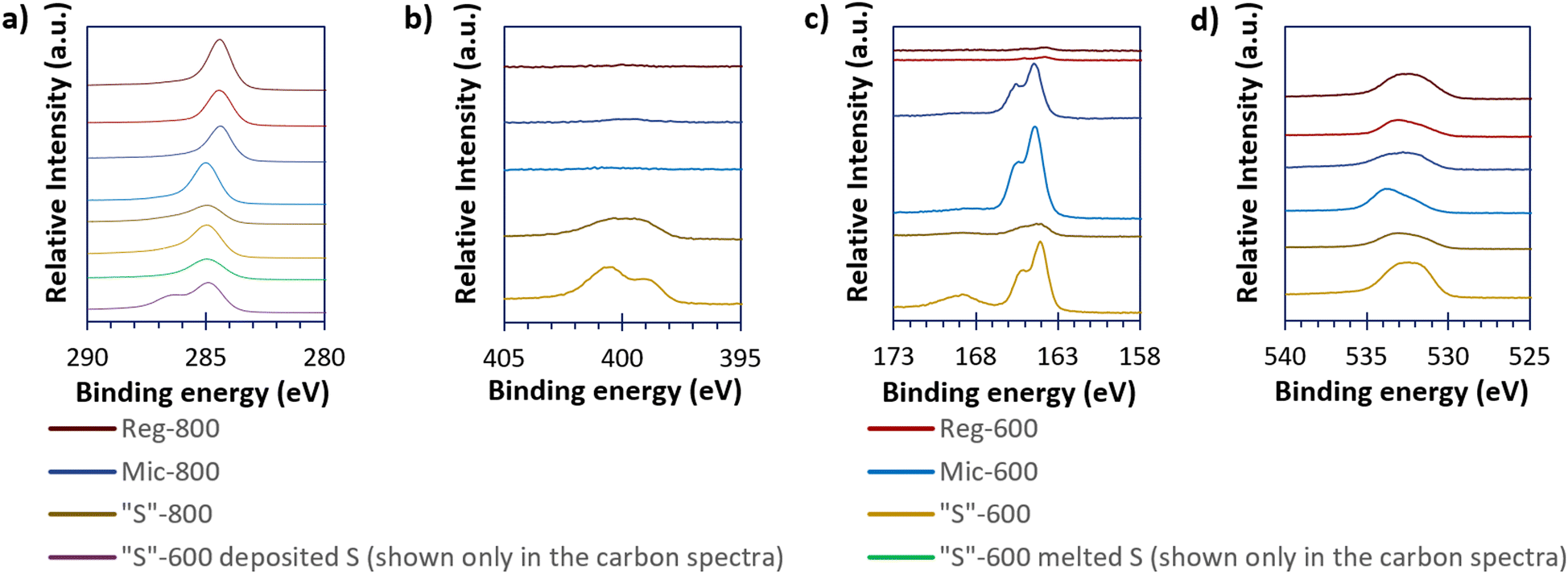 | ||
| Fig. 5 The high resolution XPS spectra of the carbonaceous (and the “S”-600/S composites) materials of C 1s (a), N 1s (b), S 2p (c), and O 1s (d). | ||
The Reg-800, Mic-600 and Mic-800 provided minimal nitrogen XPS responses but “S”-600 and “S”-800 have significant peaks at 398.8 and 401.0 eV suggesting that the nitrogen is pyridinic and graphite-like respectively.29 Upon further pyrolysis to 800 °C there was a reduction in the proportion of pyridinic nitrogen at the benefit of the graphite-like nitrogen. Doping porous carbons with nitrogen has been found in the literature to better allow surface chemisorption of the active material species in LSBs.30 Nitrogen was not detected for Reg-600.
Similarly, Reg-600 and Reg-800 give minimal sulfur XPS peaks, but all the other samples (“S”-600, “S”-800, Mic-600 and Mic-800) give the 2p3/2 and 2p1/2 peaks of elemental/bridging sulfur (S0B) at 164.0 and 165.6 eV as well as the broad and unresolved 2p3/2/2p1/2 sulfur peak of oxygenated sulfur groups at 168.5 eV. The oxygen peaks at 533.0 eV imply ether/hydroxyl groups and the peak at 532.0 eV implies oxygenated carboxyl groups (both occurring at the carbon surface).31 The ether/hydroxyl and carboxyl surface groups are thought to be beneficial in the hindering of the polysulfide-shuttle by immobilisation of the polysulfides; oxygen surface concentrations for all carbon samples are shown in Table 4.32,33
| Sample | Oxygen surface concentration/%atom |
|---|---|
| “S”-600 | 19.0 |
| “S”-800 | 9.3 |
| Mic-600 | 7.7 |
| Mic-800 | 7.2 |
| Reg-600 | 8.0 |
| Reg-800 | 8.9 |
The IR of the samples (see Fig. S5, ESI†) becomes far less complex with increasing pyrolysis temperature, due to the thermal etching of the resins’ functional groups. All resins have broad peaks 3600–3000 cm−1 corresponding to O–H stretches from phenolic groups. Both the Mic-Res and “S”-Res have peaks at 2915 cm−1 and 2850 cm−1 which correspond to C–H stretches from CTAB's methyl and methylene groups, respectively. Reg–Res (as the resin made without CTAB) did not share these peaks. “S”-Res also has broad peaks at 3200 cm−1 and 3400 cm−1 due to a carboxyl O–H stretch, from the oxalic acid, and the ammonium ion +N–H of ammonium thiosulfate, respectively. “S”-Res also has a strong carboxylate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1666 cm−1 and a smaller peak at 1715 cm−1 which is the CO stretch of a carboxylic acid dimer. The peaks in all resins at 1600 cm−1 are consistent with aromatic CC stretches and bends and C–O phenol stretches 1000–1400 cm−1. “S”-600 displays a very broad peak between 3600–3000 cm−1 as well as very strong peaks at approx. 1600 cm−1 and 1200 cm−1, suggesting that alcohol (O–H and C–O stretches) and aromatic groups (CC stretches) remain post pyrolysis. Reg-600 and Mic-600, meanwhile, retain their C–H bonds (2915 cm−1 and 2850 cm−1) and carbon–oxygen functionalities (1000–1400 cm−1). The spectra of materials pyrolysed at 800 °C are relatively featureless due to extensive thermal etching of the functional groups.
O stretch at 1666 cm−1 and a smaller peak at 1715 cm−1 which is the CO stretch of a carboxylic acid dimer. The peaks in all resins at 1600 cm−1 are consistent with aromatic CC stretches and bends and C–O phenol stretches 1000–1400 cm−1. “S”-600 displays a very broad peak between 3600–3000 cm−1 as well as very strong peaks at approx. 1600 cm−1 and 1200 cm−1, suggesting that alcohol (O–H and C–O stretches) and aromatic groups (CC stretches) remain post pyrolysis. Reg-600 and Mic-600, meanwhile, retain their C–H bonds (2915 cm−1 and 2850 cm−1) and carbon–oxygen functionalities (1000–1400 cm−1). The spectra of materials pyrolysed at 800 °C are relatively featureless due to extensive thermal etching of the functional groups.
Raman spectra of the different pyrolysed carbons are shown in Fig. S6 (ESI†). Both the regular and micellar pyrolysed materials showed an increase in ID/IG ratio (the D-band was centred at 1338 cm−1 and the G-band centred at 1592 cm−1 for all carbons) at the higher pyrolysis temperature (800 °C). This shows an increase in defects (sp3 vibrations), relative to graphitised carbon (sp2 vibrations), at the higher pyrolysis temperature. As previously reported for Raman spectra of carbons produced from phenolic resins it is not uncommon for the degree of disorder (ID/IG ratio in Table 5) to increase until 1000 °C where a glassy carbon forms from a carbon macrostructure.31
Fig. 6 shows the Raman scattering results from either melt-loading or deposit-loading sulfur with “S”-600 carbon. The most intense vibrations associated with elemental sulfur occur at frequencies 100–500 cm−1 and these vibrations are due to S–S bond vibration within the cyclo-octasulfur ring. The vibrations between S8 rings occur at frequencies below 100 cm−1 and produce subtle differences between the different phases (α-S8, β-S8 or γ-S8).34 The data is consistent with the α-phase S8 given by both sulfur-loading methods with no significant differences in any of the sulfur peaks relative to peak intensity of either (D or G-band) carbon peak.34,35
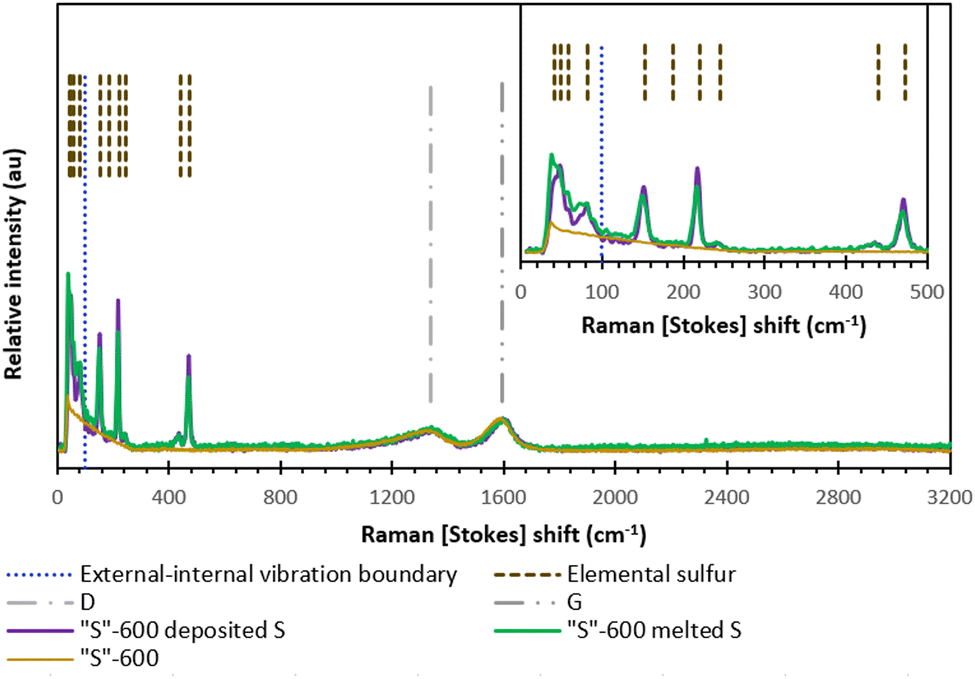 | ||
| Fig. 6 The Raman spectra of “S”-600 as well as the melt-loaded “S”-600/S composite and deposit-loaded “S”-600/S composite. | ||
From the CHNS data (Table 6) all the carbonaceous materials, not containing any elemental sulfur following pyrolysis, were primarily comprised of carbon. The “S”-600 and “S”-800 have additional components in the reaction mixture containing different heteroatoms leading to a reduction in carbon. However, Mic-600 gave a higher carbon content, and this was ascribed to the decomposition of the CTAB and potential occlusion/trapping of any organic components. The hydrogen contents align with what is expected for polymeric materials and generally decrease as a function of pyrolysis temperature again yielding a lower nitrogen content.
| Sample | C/%mass | H/%mass | N/%mass | S/%mass |
|---|---|---|---|---|
| Limit of detection (LOD) 0.10%mass. Error ± 0.30%mass. | ||||
| Reg-600 | 78.90 | 2.59 | <LOD | 0.32 |
| Reg-800 | 79.22 | 1.58 | <LOD | 0.11 |
| Mic-600 | 83.54 | 2.51 | <LOD | 0.13 |
| Mic-800 | 72.47 | 1.22 | 0.25 | <LOD |
| “S”-600 | 61.30 | 2.27 | 7.26 | 2.03 |
| “S”-800 | 62.07 | 1.26 | 4.89 | 1.07 |
| “S”-600 melted S | 5.83 | 0.71 | 0.69 | 89.93 |
| “S”-600 deposited S | 9.69 | 0.93 | 1.30 | 83.36 |
All carbons had detectable levels of sulfur, excluding Mic-800, which is not unexpected. The “S”-600 and “S”-800 carbons gave sulfur contents up to 18-fold higher than the other carbons driven by the deposition of sulfur from ammonium thiosulfate. Again when the pyrolysis temperature increased, there was a reduction in sulfur content.
Oxygen cannot be ascribed to the remaining %mass of the samples due to the addition of CTAB and lithium carbonate (used as pore-former/stabiliser) and the polymerisation catalyst. Therefore, bromine and lithium contents could exist at gravimetrically significant contents within the carbons. The surface concentrations of oxygen from the XPS results (in %atom) are given in Table 4.
The “S”-600 composites (melted and deposited) are predominantly sulfur (due to elemental sulfur). While the melt-loaded sample has increased sulfur relative to the deposit-loaded sample, the latter method incorporates enough sulfur to function as a cathode material. The nitrogen content of the deposit-loaded material is higher than the melt-loaded cathode due to the formation of the ammonium oxalate by-product.
Due to low specific surface area Mic-800 was not tested within a cell. The GDC data of the melt-loaded composites, in Fig. 7(a and b), shows an initial conditioning period for all samples that stabilised by the 10th cycle. The melt-loaded samples obtained stable specific capacities of beyond 500 mA h gsulfur−1 which is low compared to the theoretical specific capacity of LSBs (1675 mA h gsulfur−1). The Reg-800 gave a performance that is significantly lower than the other samples. This confirms the hypothesis that the greater porosity yields improved performance. Yet, where templated, the different templating and pyrolysis methods do not appear to lead to vastly different performances either through higher obtainable specific capacities or greater cycle stabilities. This may imply that melt-loading sulfur may not be able to completely access the benefits that different microporosities could provide. The GDC data of the deposit-loaded composites, in Fig. 7(c and d), show a broader spread of performances in terms of specific capacities. The higher performing cells, Mic-600 and “S”-600 gave the greatest specific capacities that gradually reduced over 40 cycles from a maximum of 900 mA h gsulfur−1 to approx. 800 mA h gsulfur−1 at 0.05C.
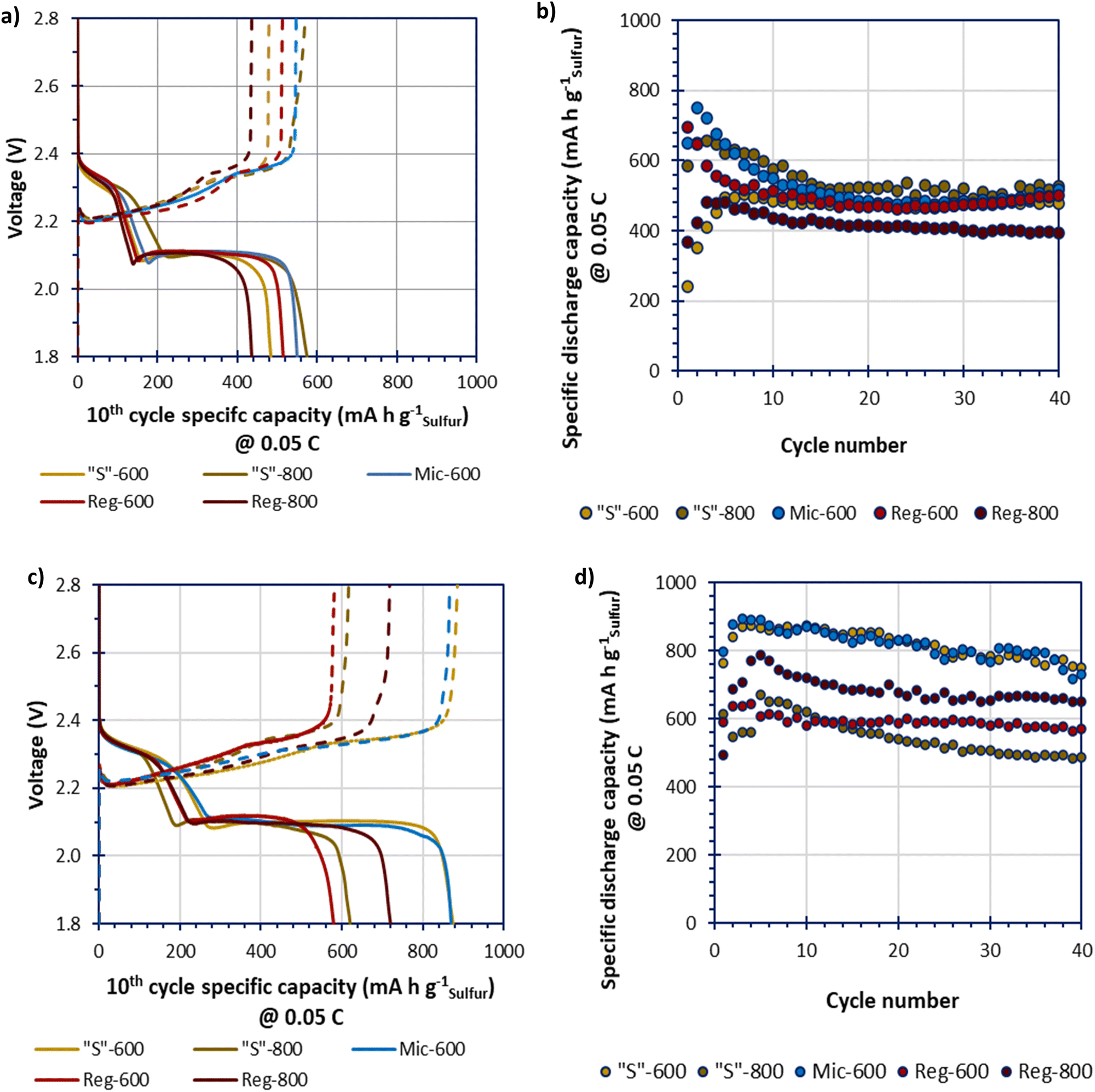 | ||
| Fig. 7 The 10th cycle discharge and charge profiles of cells at 0.05 (a) and (c) and the specific discharge capacity (relative to the sulfur active-material mass initially in the cathode) relationship over 40 cycles at 0.05C (b) and (d) for the melted-loaded cells (a) and (b) and the deposit-loaded cells (c) and (d). | ||
Further studying Fig. 7a and c, all deposit-loaded cells give improvements in their specific capacities at the 10th cycle, relative to their melt-loaded analogues. These improvements seem to be exclusively driven at the lower voltage plateau (ca. 2.10 V). This suggests that deposit-loading better facilitates polysulfide conversion instead of improving the sulfur utilisation (where improvements would instead/also be seen at the higher voltage plateau at ca. 2.30 V) relative to melt-loading.36
The discharge–charge profiles for “S”-600 and Mic-600 over 40 cycles, in 10 cycle intervals (at 0.05C), are shown in Fig. S7 (ESI†). The slow discharge–charge rate of 0.05C was selected to better monitor the sensitive electrochemistries of the LSBs. Of the two materials “S”-600 provides the most stable profile relative to Mic-600 as the polarisation (η) of “S”-600 was consistent at 0.19 V over 40 cycles. Mic-600 had a polarisation of η = 0.22 V at cycle 10 which increased to η = 0.27 V after 40 cycles representing greater internal losses to the cell as a function of cycling. “S”-600 has a greater microporous and mesoporous network relative to Mic-600, suggesting that better charge-transfer kinetics is responsible for the difference in polarisation during cycling (lower ηCT).37 “S”-600 also has a much higher surface oxygen content than Mic-600 which may reduce the loss of soluble polysulfides, therefore enhancing the electrochemical performance.
The low electrochemical performances of the LSBs in this study (given by the lower specific capacities achieved relative to the theoretical specific capacity of sulfur at 1675 mA h gsulfur−1) is thought to be caused by (i) the high proportion of sulfur in the cathode (70%mass of the cathode coating [i.e. not 70%mass of a composite that would be further diluted within the cathode]); (ii) the low proportion of porous carbon in the cathode (10%mass of the cathode) and (iii) the lean electrolyte-to-sulfur (E/S) ratio of 10 μLelectrolyte mgsulfur−1 for the coin cells tested.38–40 Thus, the lower than theoretical specific capacities obtained in part suggest the harshness of the composition during fabrication and assembly that the cells were subjected to.
Fig. 8 shows the impedance behaviour, and the differences before and after 40 cycles of 0.05C, between “S”-600 and Mic-600 cells where the cathodes had been loaded with 70%mass sulfur via the deposit-loading method. In both cases, the impedance data shows a significant change in the impedance behaviour before and after cycling, manifesting as a greater number of better resolved but lower resistive parallel resistor-capacitor (RC) pairs given as arcs in the Nyquist plots. Each solid line (green or red) is generated by a model that has been fitted to the data points within the before cycling (green) or after cycling (red) data series. There are two contributing reasons that are thought to be the driving force in the change of impedance spectra: the first is the reduction in the resistance to charge transfer (RCT), given by the diameter of the arc before cycling reducing, due to the sulfur becoming soluble, mobile, and shuttling reducing the amount of active-sulfur.41 The second reason is that, initially (pre cycling) a solid–electrolyte interface (SEI) is not yet fully formed but following cycling (e.g. after 40 cycles at 0.05C) the SEI has more completely formed. The electrolyte solvent was composed of a 50% (v/v) solvent mixture containing DME because LiTFSI has a great affinity for dissolution within DME (even at high concentrations).42 The other 50% (v/v) of the solvent mixture contained DOL because the rings open on contact of the metallic anode to form a stable SEI of polydioxolanes on the electrode surface.43 The lithium nitrate was also added to grant stability to an SEI formation while also deterring the formation of dendrites by forming layers comprised of oxidised polysulfides and LiNxOy in addition to the polydioxolanes.43
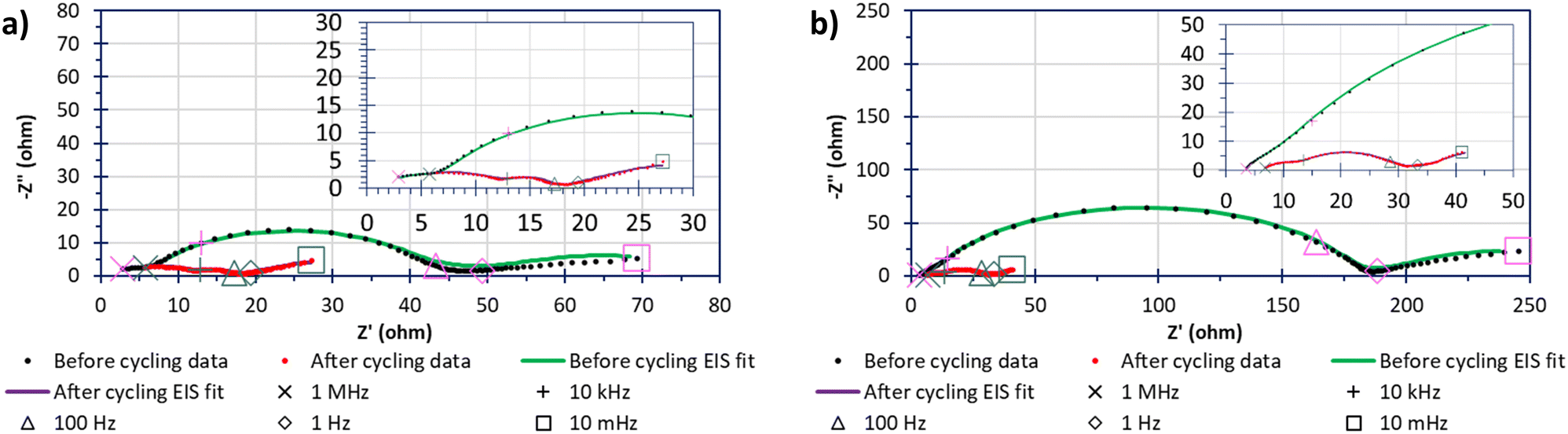 | ||
| Fig. 8 EIS spectra, pre and post 40 cycles at 0.05C data points and fitted models (green and red solid lines, respectively) of the same data points in their respective series, of the same deposit-loaded cells of “S”-600 (a) and Mic-600 (b). | ||
The values of the elements of the equivalent circuits are present within Table S1 (ESI†). However, the surface imperfections led to the capacitance response laying within the periphery of a resistor and a capacitor; this required the capacitors to be replaced with constant phase elements (CPEs) and the resulting RC pairs appeared as arc-like ‘depressed’ semicircles/arcs.44 The arcs closest to the negative imaginary impedance axis (−Z′′) after cycling were attributed to either the cathode's bulk contribution of the porous carbon's microstructure before cycling and the formation of the SEI within the LSB after cycling.45 The next arc along the real impedance axis (Z′) was associated to the charge transfer in the sulfur-based active material of the cell. The RCT of the polysulfide species (Li2S6 as the open circuit voltages were around 2.3 V for 2+ h) were shown to have far lower resistances after cycling than before cycling which is even more interesting considering that the Mic-600 cell gave an RCT that was 5–6-fold in magnitude relative to the RCT of “S”- 600 which may be reasoned as due to the higher surface areas and porosities that the “S”-templated materials can provide relative to the micellar-templated materials to be able to spread the sulfur throughout the porous network (as evidenced in the sorptiometry information).46 The next arc generated after cycling was attributed to the formation of Li2S/Li2S2 films which form an extra RC pair, contributing to extra arcs in the EIS spectra.41 The final arc of Mic-600 is caused at the interface of solid/liquid polysulfides The linear regimes at low frequencies is termed Warburg impedance where observed and describes the Li+ into a carbon's pores.
4. Conclusion
In summation, “S”-templating as a proof-of-concept was shown to be an effective technique for imparting porous architectures into sulfur-host materials within cathodes of LSBs. By depositing sulfur (by scalable thiosulfate acidification) prior to polymerisation, pores are formed that can be accessed following calcining/pyrolysis. Compared to the micellar-templated resole analogues, the “S”-templated materials gave higher surface areas that were more robust to higher pyrolysis temperatures. The Reg materials were more stable to higher degrees of pyrolysis but are of low specific surface area and specific pore volumes. Lower pyrolysis temperatures preserved greater extents of porosity as well as preserving the functionality (particularly alcohol, ether, and carboxyl groups) which in tandem resulted in favourable interactions between the sulfur-species and the surface. Deposit-loading (further to the templating differences) leads to typically higher specific capacities for the different carbons relative to melt-loading with sulfur at 0.05C.Author contributions
Conceptualisation, R. C. T. S. and C. C.; methodology, L. D. J. B.; investigation, L. D. J. B., I. M. and S. J. H.; writing, original draft preparation, L. D. J. B.; writing, review and editing, R. C. T. S., J. F. W. and C. C.; XPS assignment review, S. J. H. and J. F. W. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support for conducting Li–S battery research under the Lithium–sulfur Technology Accelerator (LiSTAR) programme funded by The Faraday Institution (grant FIRG014). We also thank Medac Ltd for CHNS analyses and access to the SEM microscope at the local at the local School of Mechanical Engineering Sciences’ MicroStructural Studies Unit (MSSU).References
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef
.
- S. Liu, L. Xiong and C. He, J. Power Sources, 2014, 261, 285–291 CrossRef CAS
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS
- W. Cao, J. Zhang and H. Li, Energy Storage Mater., 2020, 26, 46–55 CrossRef
- Z. Gao, X. Zhang, Y. Xiao, H. Wang and N. Li, Int. J. Hydrogen Energy, 2018, 43, 5261–5271 CrossRef CAS
- S. H. Farjana, N. Huda and M. A. P. Mahmud, J. Sustainable Min., 2019, 18, 150–161 CrossRef
-
S. Perathoner and G. Centi, in Emerging Materials for Energy Conversion and Storage, ed. K. Y. Cheong, G. Impellizzeri and M. A. Fraga, Elsevier, Amsterdam, 1st edn, 2018, pp. 305–325 Search PubMed
- C. Luo, E. Hu, K. J. Gaskell, X. Fan, T. Gao, C. Cui, S. Ghose, X. Q. Yang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14712–14720 CrossRef CAS PubMed
- S. Lang, S. H. Yu, X. Feng, M. R. Krumov and H. D. Abruña, Nat. Commun., 2022, 13, 1–11 Search PubMed
- T. Danner and A. Latz, Electrochim. Acta, 2019, 322, 134719 CrossRef CAS
- L. Dong, J. Liu, D. Chen, Y. Han, Y. Liang, M. Yang, C. Yang and W. He, ACS Nano, 2019, 13, 14172–14181 CrossRef CAS PubMed
- B. Liu and V. S. Thoi, Chem. Commun., 2022, 58, 4005–4015 RSC
- N. Diéz, G. A. Ferrero, M. Sevilla and A. B. Fuertes, Sustainable Energy Fuels, 2019, 3, 3498–3509 RSC
- G. He, S. Evers, X. Liang, M. Cuisinier, A. Garsuch and L. F. Nazar, ACS Nano, 2013, 7, 10920–10930 CrossRef CAS PubMed
- F. Hippauf, W. Nickel, G. P. Hao, K. Schwedtmann, L. Giebeler, S. Ostwald, L. Borchardt, S. Doerfler, J. J. Weigand and S. Kaskel, Adv. Mater. Interfaces, 2016, 3, 1–9 Search PubMed
- L. K. J. Ting, Y. Gao, H. Wang, T. Wang, J. Sun and J. Wang, ACS Omega, 2022, 7, 40682–40700 CrossRef CAS PubMed
-
A. Pizzi and C. C. Ibeh, in Handbook of Thermoset Plastics, ed. H. Dodiuk and S. H. Goodman, Elsevier Inc., Boston, MA, 3rd edn, 2014, pp. 13–44 Search PubMed
-
P. Nagy, in Hydrogen Sulfide in Redox Biology, ed. E. Cadenas and L. Packer, Academic Press, 2015, vol. 554, pp. 3–29 Search PubMed
- T. Kimura, A. Kato, C. Hotehama, A. Sakuda, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2019, 333, 45–49 CrossRef CAS
-
J. Räisänen, in Encyclopedia of Analytical Science, ed. P. Worsfold, A. Townshend and C. Poole, Elsevier, Oxford, 2nd edn, 2005, pp. 415–423 Search PubMed
- R. G. Chaudhuri and S. Paria, J. Colloid Interface Sci., 2010, 343, 439–446 CrossRef CAS PubMed
- M. M. Bruno, N. G. Cotella, M. C. Miras, T. Koch, S. Seidler and C. Barbero, Colloids Surf., A, 2010, 358, 13–20 CrossRef CAS
- G. O. Sofekun, E. Evoy, K. L. Lesage, N. Chou and R. A. Marriott, J. Rheol., 2018, 62, 469–476 CrossRef CAS
- S. K. Sharma, J. Prakash, K. Sudarshan, P. Maheshwari, D. Sathiyamoorthy and P. K. Pujari, Phys. Chem. Chem. Phys., 2012, 14, 10972–10978 RSC
- L. Shao, Y. Sang, N. Liu, J. Liu, P. Zhan, J. Huang and J. Chen, ACS Omega, 2020, 5, 17450–17462 CrossRef CAS PubMed
- M. Wang, Z. Bai, T. Yang, C. Nie, X. Xu, Y. Wang, J. Yang, S. Dou and N. Wang, Adv. Energy Mater., 2022, 12, 2201585 CrossRef CAS
- Q. Zhao, X. Bao, L. Meng, S. Dong, Y. Zhang, C. Qing, T. Zhu and H. E. Wang, J. Colloid Interface Sci., 2023, 644, 546–555 CrossRef CAS PubMed
- S. Chenakin and N. Kruse, Appl. Surf. Sci., 2020, 515, 146041 CrossRef CAS
- R. Jia, J. Chen, J. Zhao, J. Zheng, C. Song, L. Li and Z. Zhu, J. Mater. Chem., 2010, 20, 10829–10834 RSC
- Y. W. Tian, Y. Yu, L. Wu, M. Yan, W. Da Dong, C. Y. Wang, H. S. H. Mohamed, Z. Deng, L. H. Chen, T. Hasan, Y. Li and B. L. Su, J. Energy Chem., 2023, 85, 1–10 CrossRef CAS
- T. H. Ko, W. S. Kuo and Y. H. Chang, J. Appl. Polym. Sci., 2001, 81, 1084–1089 CrossRef CAS
- J. Li, L. Dai, Z. Wang, H. Wang, L. Xie, J. Chen, C. Yan, H. Yuan, H. Wang and C. Chen, J. Energy Chem., 2022, 67, 736–744 CrossRef CAS
- R. Zhe, T. Zhu, X. Wei, Y. Ren, C. Qing, N. Li and H. E. Wang, J. Mater. Chem. A, 2022, 10, 24422–24433 RSC
- C. Nims, B. Cron, M. Wetherington, J. Macalady and J. Cosmidis, Sci. Rep., 2019, 9, 1–12 CrossRef CAS PubMed
- R. Fang, G. Li, S. Zhao, L. Yin, K. Du, P. Hou, S. Wang, H. M. Cheng, C. Liu and F. Li, Nano Energy, 2017, 42, 205–214 CrossRef CAS
- L. Zhou, H. Li, Y. Zhang, M. Jiang, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Mater. Today Commun., 2021, 26, 102133 CrossRef CAS
- Z. X. Chen, Q. Cheng, X. Y. Li, Z. Li, Y. W. Song, F. Sun, M. Zhao, X. Q. Zhang, B. Q. Li and J. Q. Huang, J. Am. Chem. Soc., 2023, 145, 16449–16457 CrossRef CAS PubMed
- Y.-S. Su, Y. Fu, T. Cochell and A. Manthiram, Nat. Commun., 2013, 4, 2985 CrossRef PubMed
- S. Choudhury, B. Krüner, P. Massuti-Ballester, A. Tolosa, C. Prehal, I. Grobelsek, O. Paris, L. Borchardt and V. Presser, J. Power Sources, 2017, 357, 198–208 CrossRef CAS
- E. Cha, M. Patel, S. Bhoyate, V. Prasad and W. Choi, Nanoscale Horiz., 2020, 5, 808–831 RSC
- J. Yan, X. Liu and B. Li, Adv. Sci., 2016, 3, 1600101 CrossRef PubMed
- V. Nilsson, A. Kotronia, M. Lacey, K. Edström and P. Johansson, ACS Appl. Energy Mater., 2020, 3, 200–207 CrossRef CAS
- T. Jaumann, J. Balach, M. Klose, S. Ostwald, J. Eckert and L. Giebeler, J. Electrochem. Soc., 2016, 163, A557–A564 CrossRef CAS
- Z. Lukács and T. Kristóf, Electrochim. Acta, 2020, 363, 137199 CrossRef
- S. Waluś, C. Barchasz, R. Bouchet and F. Alloin, Electrochim. Acta, 2020, 359, 136944 CrossRef
- X. Yu, S. Feng, M. J. Boyer, M. Lee, R. C. Ferrier, N. A. Lynd, G. S. Hwang, G. Wang, S. Swinnea and A. Manthiram, Mater. Today Energy, 2018, 7, 98–104 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00481c |
| This journal is © The Royal Society of Chemistry 2024 |