
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemically modulated separation of olefin–paraffin gas mixtures in membrane electrode assemblies†
Toshihiro
Akashige
a,
Adlai B.
Katzenberg
a,
Daniel M.
Frey
 a,
Debdyuti
Mukherjee
a,
César A.
Urbina Blanco
ab,
Brian
Chen
a,
Yoshiyuki
Okamoto‡
a and
Miguel A.
Modestino
*a
a,
Debdyuti
Mukherjee
a,
César A.
Urbina Blanco
ab,
Brian
Chen
a,
Yoshiyuki
Okamoto‡
a and
Miguel A.
Modestino
*a
aDepartment of Chemical and Biomolecular Engineering, New York University, Brooklyn, NY 11201, USA. E-mail: modestino@nyu.edu
bLaboratory for Chemical Technology (LCT), Department of Materials, Textiles and Chemical Engineering, University of Ghent, Ghent, 9052, Belgium
First published on 22nd March 2024
Abstract
Olefin–paraffin separations are large-volume energy-intensive processes for preparing purified monomers such as ethylene and propylene. Currently, these separations are performed using cryogenic distillations, accounting for approximately 250 trillion BTU per year of industrial energy consumption. This work demonstrates an alternative olefin–paraffin separation method based on an electrochemically modulated swing absorption system. Nickel maleonitriledithiolate, an electrochemically active organometallic complex, is dispersed in the ionogel binder of a membrane electrode assembly (MEA). When exposed to propylene–propane gas mixtures, propylene is selectively captured during the oxidation of the complex and is then released when the complex is reduced. Our results suggest that transport limitations of olefins to electrochemical active sites play an important role in determining separation efficacy. Experiments conducted under varying oxidative potentials (from 1 to 3 V) and a reductive potential of −2 V demonstrated the operational robustness of the MEA over multiple capture-and-release cycles. This proof-of-concept demonstration represents a new non-thermal route for purifying the large olefin commodities in the chemical industry.
1. Introduction
Separations are ubiquitous in most chemical processes. Up to 10% of the annual global energy consumption can be attributed solely to these energy-intensive processes.1–3 One of the most important chemical separation processes is cryogenic distillations of light olefin–paraffin gas mixtures, most notably ethylene–ethane and propylene–propane mixtures. Global ethylene and propylene production capacity exceeded 200 and 130 million metric tons per year in 2020, respectively, making these two gases the largest-volume petrochemical products.4,5 These light olefins are commonly produced via steam cracking, resulting in olefin–paraffin mixtures with 5 to 45 wt% of paraffin compositions.6 Olefins in these gas mixtures must be purified to at least 99.5–99.9 wt% purity – a level required for their use as feedstocks in producing polymers or chemical intermediates.6 Due to both the significant demand for purified olefin monomers and the high energy intensity of cryogenic distillations (≈3.6 MJ per kg of olefin product),7,8 the separation of olefin–paraffin mixtures is responsible for approximately 0.3% of the global energy consumption6 and more than 6% of thermal-based industrial separation processes in the U.S. alone.2 Therefore, more efficient and decarbonized olefin–paraffin separation technologies could help pave the way toward a more sustainable industry while meeting the increasing demands of purified olefins.Several alternative separation methods for olefin purifications have been demonstrated in the past century, but their scale-up and deployment have proven challenging. Membrane-based separations of gases are a promising non-thermal alternative with the potential to reduce the energy requirement by at least 85% relative to cryogenic distillation.9,10 Membrane separation often relies on a solution–diffusion mechanism.6,11 This separation method however often proves ineffective because of the similarity in molecular size and dipole moments of 2-carbon and 3-carbon olefins and paraffins.6,12 An improved version of this diffusion-based separation method involves molecular-sieving, in which carefully engineered metal–organic frameworks or zeolite structures, accurately designed to angstrom range, help improve selectivity between molecules of similar sizes.6,11,13 Alternatively, metallic species that selectively complex olefins can facilitate the separations. For example, silver(I) ions have been known to create weak pi-bond complexes with alkenes.14,15 Liquid solutions saturated with silver(I) ions are known to show increased sorption of olefin into the solution media when exposed to an olefin-rich environment.15 Similarly, polymer membranes doped or blended with silver(I) species exhibit an increase in selective sorption behavior towards olefin, enhancing the selectivity of olefin/paraffin separation.11,13,14,16 However, using silver ions has often been challenging because of possible poisoning by impurities found in gas mixtures (i.e., hydrogen sulfide or acetylene), reducing their lifespan.17,18 Recent review articles on membrane separations provide a comprehensive and in depth perspective on the strategies explored for olefin–paraffin mixtures.6,8
A promising yet underexplored nonthermal approach to olefin separations is electrochemical swing adsorption. In this process, olefins reversibly bind to redox-active organometallic complexes and their affinity can be electrochemically modulated. Similar electrochemical separation methods have been demonstrated for carbon dioxide (CO2) capture where quinone-based redox-active species selectively interact with CO2.19,20 The quinone species capture CO2 at its reduced state which can then be released when the species are oxidized. This selective capture-and-release mechanism can also be applied to olefin separation, as previously demonstrated using copper(I)-containing solutions which have high affinity to olefin complexation.21,22 However, because copper(I) ions are vulnerable to poisoning, a more robust olefin-complexing species (i.e., nonreactive to the impurities) is sought to facilitate this capture-and-release mechanism. To overcome this limitation, Wang and Steifel demonstrated that solutions containing Ni(mnt)2n complexes (mnt = [S2C2(CN)2]2−, maleonitriledithiolate; n = 0, −1, −2), where the nickel oxidation state (OS) can be IV, III, or II respectively, could be used to reversible bind olefins in its oxidized state, and release them in its reduced state.23 As shown in the reversible redox cycle depicted in Fig. 1, different oxidation states of Ni(mnt)2n species can be electrochemically accessed, in which one state has a high affinity to complex olefins (n = 0, OS = IV), while the other state has low affinity towards them (n = −2, OS = II). Each OS can be accessed via carefully tuning the electrode potential, enabling control over the olefin capture and release process. Since these early demonstrations, computational and experimental studies have sought to understand the mechanism of the reaction of Ni(mnt)2n species with olefins.24–29 This electrochemical swing separation approach is fundamentally different than that of membrane separations, as it relies on the differences in reactivity of olefins and paraffin towards redox-active species, rather than on their transport properties across a medium. Given that paraffins cannot react with Ni(mnt)2n complexes, this electrochemical separation has the potential to be highly selective towards the capture of olefins in mixtures. On the other hand, electrochemical separations present additional complexities for scale-up, as they require the integration of electrodes with large electrochemically-active surface areas and large interfaces with gas mixtures, such as those incorporated in membrane-electrode assemblies (MEAs).
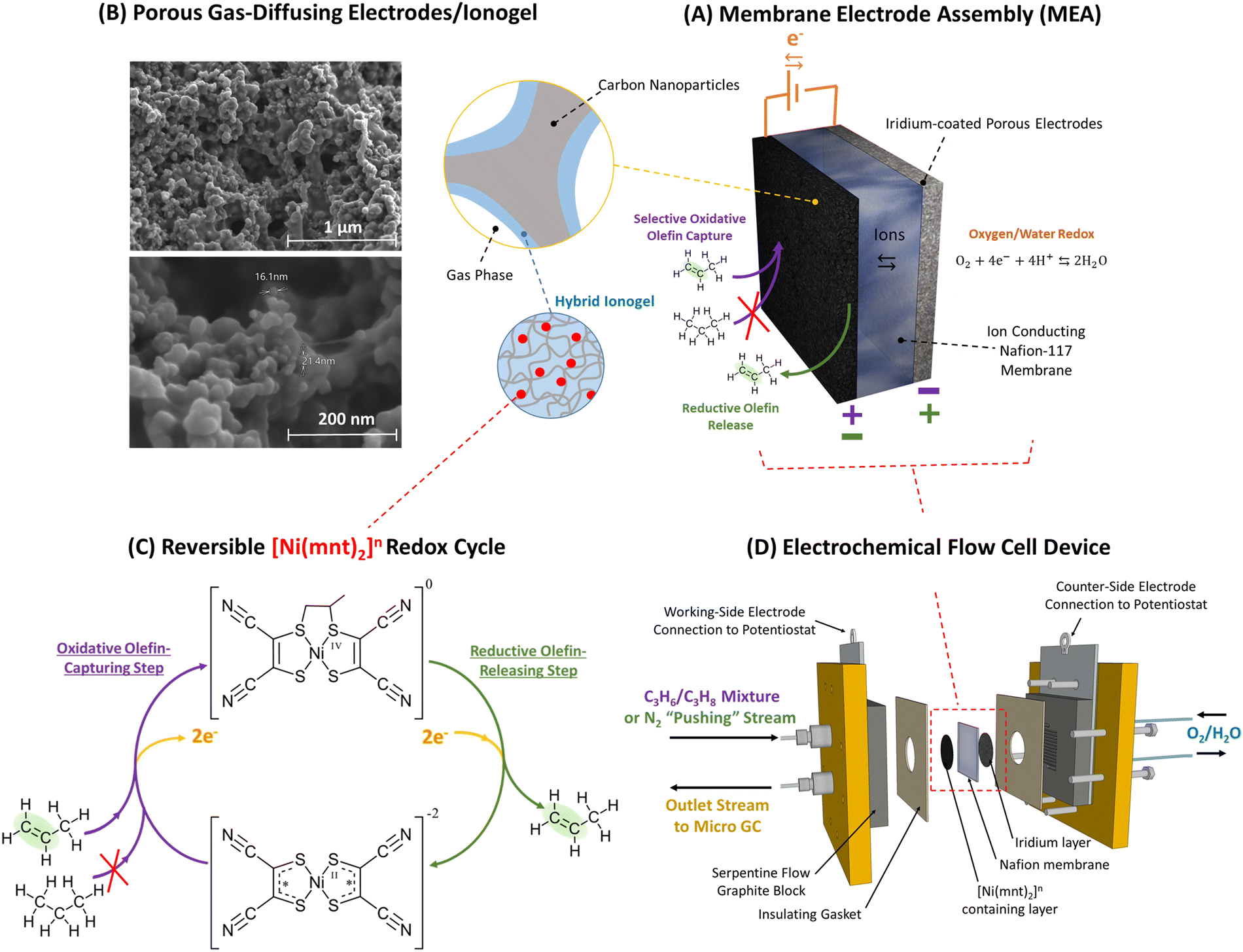 | ||
| Fig. 1 (A) Propylene can be separated from propane-containing gas mixture via selective capture and release of alkene molecules facilitated by a membrane electrode assembly (MEA) as depicted above in the top-right. (B) The MEA's reaction surface consists of a layer of porous gas-diffusing electrodes that is made primarily of carbon nanoparticles scaffolded by a hybrid ionogel mixture of [BMIM][PF6] ionic liquid and Nafion™ polymers. SEM images of the porous electrode/ionogel structures are shown in the top-left. (C) Ni(mnt)2n species are dispersed within the ionogel whose close vicinity to both the carbon electrode surface and the gas-phase interface containing propane/propylene mixture allows electrochemically modulated complexation and de-complexation with propylene via a reversible redox olefin separation process. The selective pi-bond complexation affinity between propylene and Ni(mnt)2n is dependent on the oxidation state of Ni(mnt)2n as depicted in the lower-left. At an oxidation state of 0, propylene can complex to the Ni(mnt)2 species, while at an oxidation state of −2, complexation is not favored. (D) The sample MEA is installed into an electrochemical flow cell device, as shown in the lower-right, which is connected to a potentiostat (for modulating potential in the MEA), a mass flow control meter feeding propane/propylene gas mixture or nitrogen into the MEA during appropriate phases of the experiment (refer to Fig. 2), and a GC to measure the concentration of the outlet gas stream. Because the flow cell device requires a counter electrode and a balanced ion transfer in the central Nafion-117 membrane, water oxidation and reduction were utilized on the side opposite to the gas-separating side of the flow cell. | ||
In this work, we demonstrate that Ni(mnt)2n species can be effectively utilized for propylene–propane gas separation when appropriately embedded into a membrane electrode assembly (MEA) as depicted in Fig. 1. The Ni(mnt)2n species can be distributed within a hybrid ionogel medium composed of an ionic-liquid-swollen ionomer. This ionogel acts as a binder for electrically conductive carbon particles deposited on porous gas diffusion electrodes (GDEs). The porous nature of the GDEs provides a large interface between the gas phase and the redox sites (Fig. 2). The incorporation of Ni(mnt)2n species into the ionogel composite electrode layer was partly inspired by works involving porous coordination polymers (PCP) with integrated metal bis(dithiolene) units that have been used in electrochemically modulated separation of olefins.30 PCP can be utilized as a scaffold to hold olefin-complexing metal dithiolene species in thin, porous layers that can be electrochemically modulated with oxidative and reductive charge to capture and release olefins. MEA-based devices are advantageous over solution-based electrochemical separation systems because the latter usually operates in a liquid organic media prone to evaporation requiring frequent replenishment, and imposes significant mass transport resistances to olefin gases with low solubility. Unlike solution-based systems, ionogel-containing MEAs are not prone to evaporative losses of media, and the presence of ionic liquids in the gels enhances the solubility of olefins.31–33
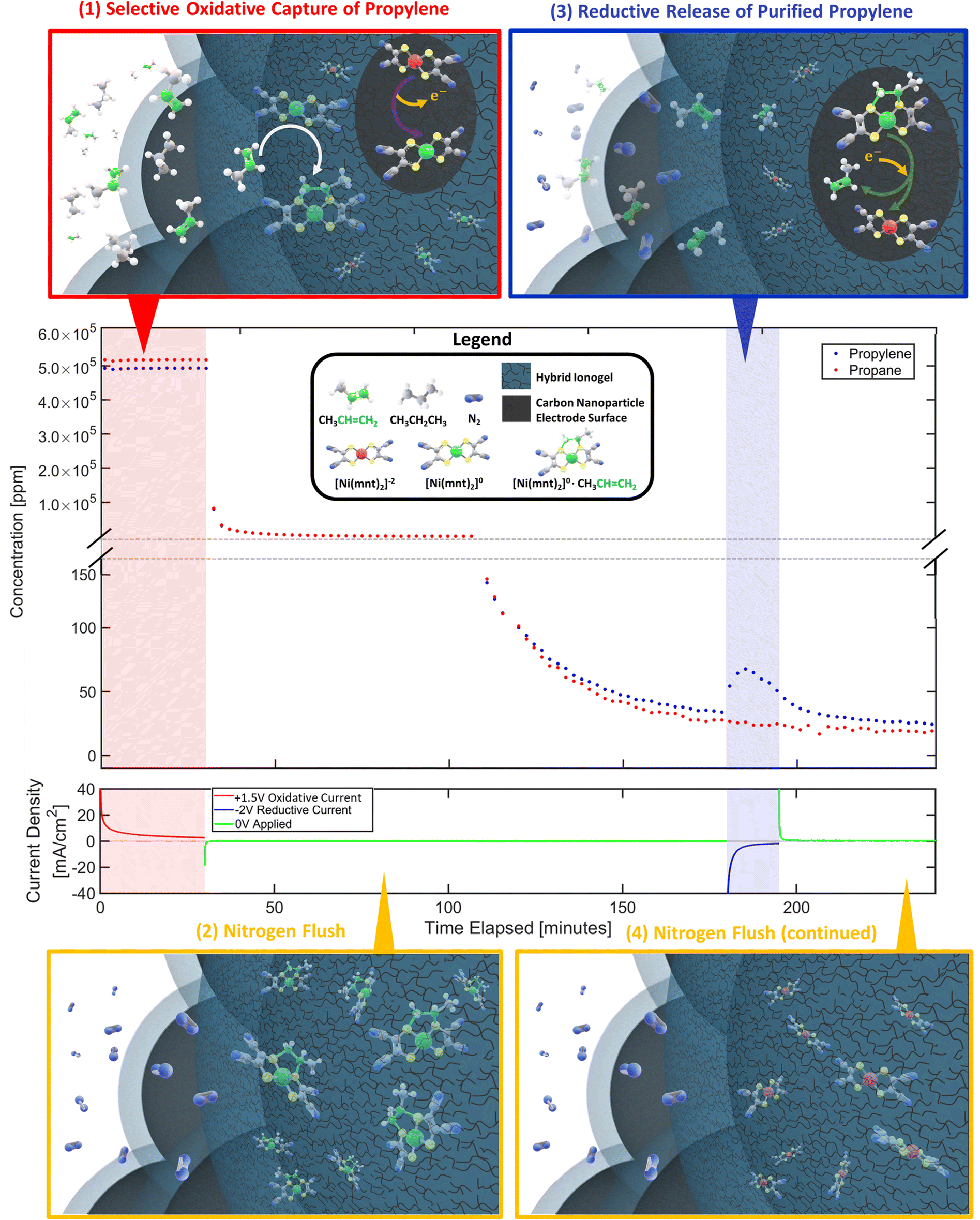 | ||
| Fig. 2 Propylene gas separation performance was quantified for each MEA sample by keeping periodic GC measurements of the output gas stream's composition for a duration of 4 hours per experimental run cycle, a sample experimental cycle of which is demonstrated above. Each cycle consists of 4 distinct phases: (1) selective oxidative capture of olefin, (2) nitrogen flush, (3) reductive release of purified olefin, (4) final nitrogen flush. | ||
Ionogels have been extensively studied for electrochemical energy conversion, catalysis, and gas separations.34–38 Most importantly, ionogels are ionically conductive, allowing 3-dimensional transport of organometallic species and supporting ions. To demonstrate the concept, we prepared electrode layers with ionogels of Nafion and 1-n-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]) ionic liquid (IL) along with the Ni(mnt)2n species. These ionic liquids are known to provide high ionic conductivity exceeding 10 mS cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39,40 and have an inherently higher solubility for olefins than paraffins, further compounding the effective selectivity towards olefins in gas separations.32,33
39,40 and have an inherently higher solubility for olefins than paraffins, further compounding the effective selectivity towards olefins in gas separations.32,33
2. Results and discussions
2.1 Selectivity of olefin capture-and-release
To demonstrate the proposed electrochemical separation method, we developed an experimental protocol consisting of 4 phases as illustrated in Fig. 2: (1) selective oxidative capture of olefin. [Ni(mnt)2]−2 are oxidized into [Ni(mnt)2]0 on the carbon electrode surface by applying a constant positive voltage (typically +1.5 V vs. iridium counter-side). Simultaneously a mixture stream of propylene and propane is introduced to the gas phase interface. As [Ni(mnt)2]0 migrates toward the gas-phase interface, it can selectively complex to propylene turning into [Ni(mnt)2·C3H6] while propane behaves as a spectator molecule. (2) Nitrogen flush. After the MEA absorbs enough propylene, the flow cell is flushed with a stream of pure nitrogen. (3) Reductive release of purified olefin. [Ni(mnt)2·C3H6] is reduced to [Ni(mnt)2]−2 at the carbon electrode surface (typically by a reductive potential of −2 V), making the complex unstable and therefore decoupling the propylene from the [Ni(mnt)2·C3H6] species. The decoupled propylene would migrate through the hybrid ionogel and reach the gas-phase interface where they are carried away by a stream of nitrogen gas that is destined to reach the GC sample inlet. (4) Final nitrogen flush. The flow cell is flushed with additional nitrogen to remove any remaining hydrocarbon gases and prepare for the next experimental cycle.The separation performance of the device was characterized by GC analysis of the outlet gas stream. Our experimental results (Fig. 2 and 3) show strong evidence of olefin separation facilitated by our Ni(mnt)2n-containing MEA. Moreover, the device separated a 50:50 propylene–propane gas mixture to a higher propylene ratio in its final output, reaching as high as 80:20 propylene–propane ratio, excluding the nitrogen flush gas (see Fig. S4D in ESI†). The separation performance was quantified as the number of moles of propylene captured and released per MEA projected area from a propylene–propane gas mixture of known composition (i.e., usually of 50:50 by volume mixture unless otherwise stated). Carbon electrode nanoparticles and Nafion ionomer loadings were kept constant throughout this study to help minimize the number of parameters affecting the MEA's separation performance. Control experiments without Ni(mnt)2n species did not exhibit any sign of propylene gas separation from the propane mixture (see Fig. S1A in ESI†).
2.2 Varying [Ni(mnt)2] loading
To derive material design guidelines that control the separation performance, we first explored the effects of varying loadings of bis(tetraethylammonium) bis(maleonitriledithiolato) nickel [N(Et)4]2[Ni(mnt)2] from 0 to 8 mg cm−2 at a constant IL loading of 4 mg cm−2 as depicted in Fig. 3A1. N(Et)4+ was assumed to be a spectator counterion species with respect to the overall capture-and-release mechanism. In our discussion, we consider the total [N(Et)4]2[Ni(mnt)2] loading instead of the [Ni(mnt)2]n loading because the preparation of the colloidal “ink” mixture was sensitive to the total mass of solute used during the spray deposition of MEA samples (see Methods section).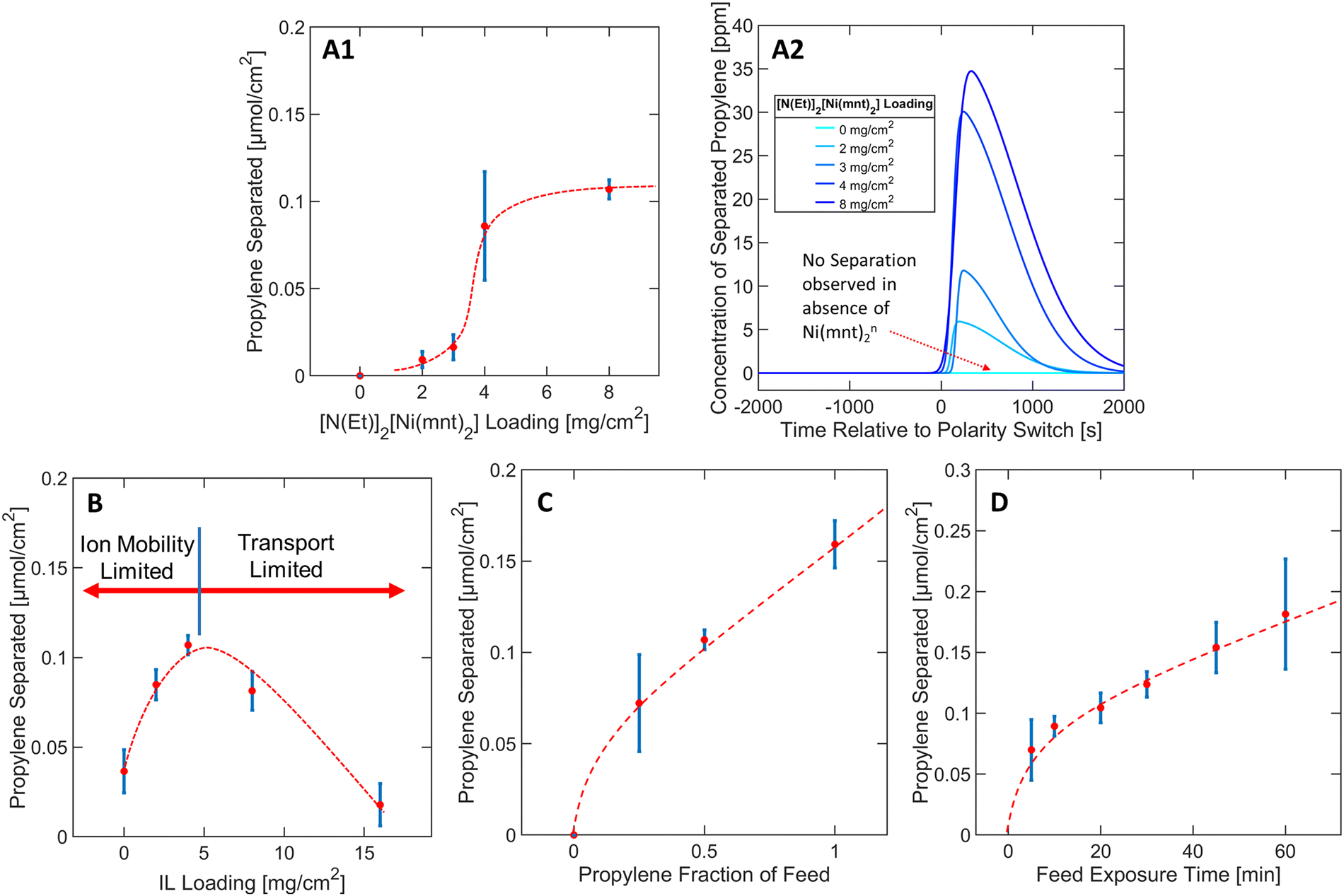 | ||
| Fig. 3 Propylene capture/released by the MEA (per unit planar area) as a function of (A1) [N(Et)4]2[Ni(mnt)2] loading, (B) ionic liquid loading, (C) propylene fraction in the feed, and (D) propylene/propane 50:50 feed exposure time. (A2) Deconvoluted propylene concentration profile per individual [N(Et)4]2[Ni(mnt)2] loading shows the spike in propylene concentration when a reductive potential (polarity switch) is applied to the MEA. The integrated area under the curve is proportional to the amount of propylene separated via the electrochemical capture-and-release mechanism of the MEA. Error bars on (A1) and (B)–(D) represent standard deviations (see Methods section). | ||
As depicted in Fig. 3A1, at [N(Et)4]2[Ni(mnt)2] loadings between 2–4 mg cm−2, the propylene separation performance is a strong function of the complex loading, but eventually approaches a plateau as the [N(Et)4]2[Ni(mnt)2] loading approaches >4 mg cm−2. This asymptotic behavior suggests that at high complex loadings, the separation is hindered due to transport limitations that limit the access of [Ni(mnt)2]n to the electrode surface, or the ability for propylene to reach all reaction sites. One of the transport limitation sources can be attributed to possible phase separation of [N(Et)4]2[Ni(mnt)2] crystals out of the ionogel medium due to oversaturation (see Fig. S7 in ESI†), which could be related to the plateauing separation performance since the ionogel (of a specified IL vs. polymer ratio) can only dissolve a finite maximum amount of [N(Et)4]2[Ni(mnt)2]. In subsequent experiments, the [N(Et)4]2[Ni(mnt)2] loading was kept constant at 8 mg cm−2 since it exhibited a sufficiently high throughput of purified propylene (for further details regarding Fig. 3A1 see Section S1 in ESI†).
2.3 Varying IL loading
The presence of the ionic liquid was found to be a crucial factor in improving the separation performance of the MEA. Nafion was used in the MEA as both a scaffold for the ionogel and a conductive ionomer. Nafion scaffolded the [Ni(mnt)2]n species within close vicinity of both the carbon electrode surface and the gas interface, allowing the [Ni(mnt)2]n to interact with both the electrode surface and the propylene gas stream for complexation. Only modest separation performance was observed in MEAs where no IL was used, likely due to lack of mobility of [Ni(mnt)2]n through the neat polymer matrix. Introducing an IL into the Nafion medium increased the separation performance by as much as 2.5 times (at an IL loading of 4 mg cm−2), as depicted in Fig. 3B.There is a local maximum in the relationship between IL loading and separation performance because of transport limitations induced by increased concentration of IL in the MEA. As shown in Fig. 3B, the separation performance decreased progressively after reaching an IL loading of 4 mg cm−2. It is possible that excessive IL in the ionogel can inadvertently lower the gas/ionogel interfacial area, consequentially restricting the amount of propylene that can be absorbed into the MEA. Furthermore, the ionogel can easily be swelled with an increased IL ratio, increasing the physical distance between the carbon electrode surface and the gas interface and mass transport resistance. Therefore, a carefully optimized IL loading should balance both the positive effect of increased mobility of [Ni(mnt)2]n species in the ionogel and the negative effect of both the ionogel swelling and the reduced interfacial area to achieve a high separation performance. Since an IL loading of 4 mg cm−2 demonstrated optimal functionality with 8 mg cm−2 of [N(Et)4]2[Ni(mnt)2] in terms of propylene separation performance, these parameters were kept constant in subsequent studies (for further details regarding Fig. 3B see Section S2 in ESI†).
2.4 Varying feed composition and feed exposure time
Propylene separation was observed over the whole propylene fraction range, as depicted in Fig. 3C, indicating that the MEA can reliably separate any ratio of propylene–propane mixture consistently. Separation performance was observed initially at 0.25 propylene fraction, followed by a gentler linear increase at higher propylene fraction, which may be due to finite saturation of propylene molecules complexing in the MEA after approximately 0.25 propylene fraction. The slight linear slope at higher fractions may be due to the increased concentration gradient that can increase the transport rate of propylene into the ionogel (for further details regarding Fig. 3C see Section S3 in ESI†). Fig. 3D depicts a similar trend except now in terms of the total amount of time the MEA was exposed to the propylene/propane feed mixture during the oxidative olefin-capture phase. Although 30 minutes of feed exposure was utilized in a typical experimental run, the separation performance did not improve significantly after the first 10 minutes of feed exposure, plateauing into a relatively linear slope of approximately 0.002 μmol minute−1. The 10-minute exposure separation performance was approximately 72% of that observed under the 30-minute exposure condition according to Fig. 3D. In most experimental runs, 70–80% of the total charge was applied in the first 10 minutes of the oxidative phase, indicating that most of the electrochemical processes occurred in this window of time, including the oxidation of [Ni(mnt)2]−2 to [Ni(mnt)2]0 (for further details regarding Fig. 3D see Section S6 in ESI†).2.5 Varying potentials
The effect of varying the oxidative potential at a constant reductive potential (of −2 V) was studied and depicted in Fig. 4A. A minimum thermodynamic potential of +0.82 V is required to trigger the generation of Ni(IV) ([Ni(mnt)2]0) species to initiate the selective capture mechanism (see Section S14 in ESI†). An applied potential of at least +1 V was required to detect separation, suggesting that [Ni(mnt)2]0 species are not accessible below this potential, most likely due to overpotential losses. A general decrease in separation performance can be observed between +1.5 V and +3 V, likely arising from parasitic oxidation of carbon or other organic components in the ionogel (see Section S4 in ESI†). These results underscore the need to carefully tune the oxidative potential to achieve optimal conditions for propylene complexation.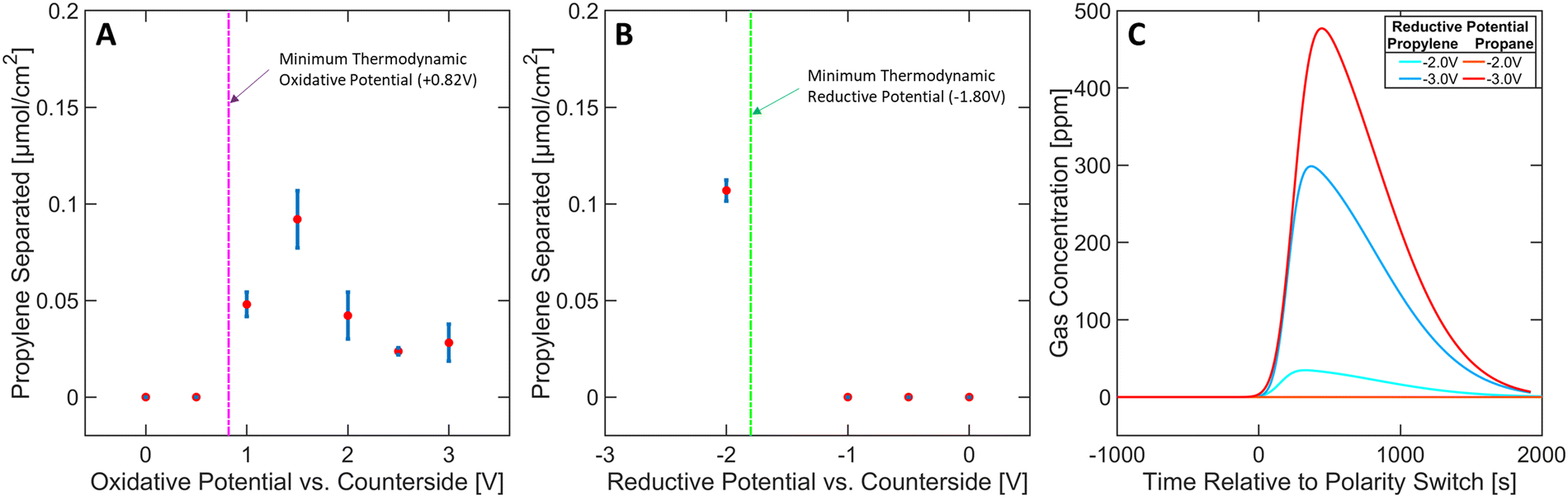 | ||
| Fig. 4 Propylene capture/released by the MEA as a function of (A) the oxidative potential vs. the counter electrode and (B) the reductive potential vs. the counter electrode. (C) Concentration profiles of both propylene and propane at varying reductive potentials. Error bars on (A) and (B) represent standard deviations (see Methods section). | ||
Furthermore, the effect on separation performance from varying reductive potentials at a constant oxidative potential (of +1.5 V) was studied (Fig. 4B). A minimum thermodynamic potential of −1.80 V is required to reduce the [Ni(mnt)2·C3H6] species back to [Ni(mnt)2]2− and propylene, which agrees with the experimental data shown in Fig. 4B (see Section S14 in ESI†). However, it must be noted that excessive reductive potential can defeat the targeted goal of olefin purification due to parasitic propylene hydrogenation. In Fig. 4C, propylene separation is achieved at −2 V where propane's concentration profile exhibited no activity upon applying the reductive potential (polarity switch) to the MEA. However, applying −3 V reductive potential released propylene and a significant amount of propane, likely arising from hydrogenation side reactions (for further details regarding Fig. 4B and C see Section S5 in ESI†). Furthermore, hydrogen gas was commonly observed as a side product during reductive phases in almost all experimental runs. Approximately 10–20% of reductive charge contributed to hydrogen gas production (see Sections S1–S3 in ESI†). Therefore, carefully controlling the reductive potential is necessary to ensure the released propylene is not hydrogenated.
2.6 Pathways for scalable deployment
The work presented here demonstrates an early-stage proof-of-concept MEA device that utilizes electrochemical cycles to separate olefin–paraffin mixtures. In scaling this concept for implementation into industrial separation processes, a multi-stage electrochemical separation cycle would be required to achieve polymer-grade purity of olefin. It is anticipated that the nitrogen gas stream used as a carrier during the reductive olefin-release phase of our experiments, would be replaced by an enriched olefin stream. This way, olefins would be constantly released and gradually enrich this stream, continuously increasing their concentration as a function of the number of electrochemical cycles. Industrial electrochemical reactors based on this concept would need to be based on large-scale MEAs arranged in a multi-stack device (similar to chloro-alkali systems or water electrolyzers). Leveraging the multi-stack arrangement, olefin-enriched streams can travel to multiple olefin-releasing cells in continuous succession, achieving a high concentration of olefin at the outlet of the reactor while allowing a total olefin throughput equal to the total amount of olefins captured in the electrodes in each cycle. A possible mode of scalable operation is presented below in Fig. 5.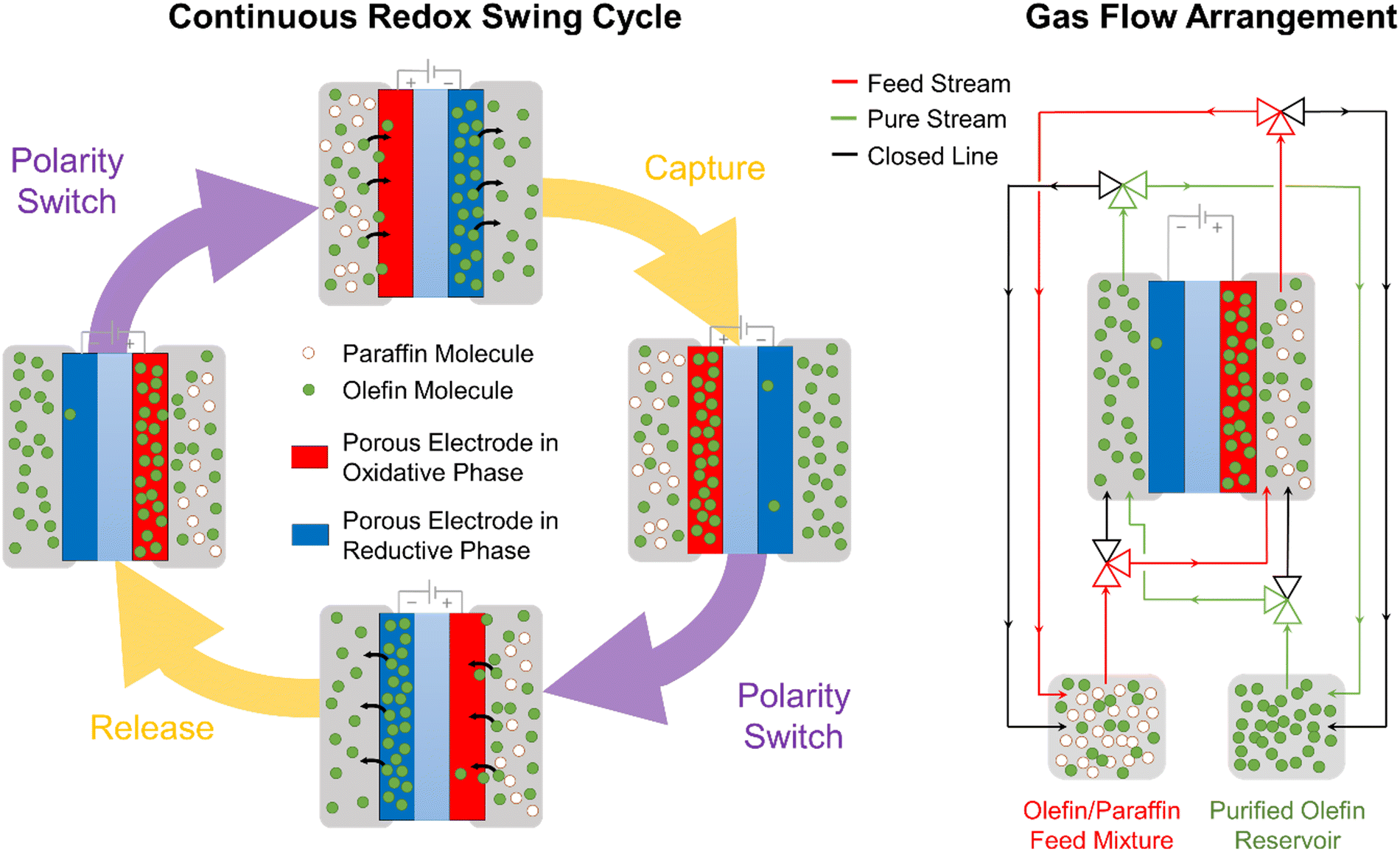 | ||
| Fig. 5 Configuration of olefin/paraffin separating MEA device for continuous scalable operation. Under this design, both electrodes interchangeably perform functions for olefin-capture or olefin-release, allowing olefin separation to continuously occur in each cell and operating similarly to swing absorbers. Stacking multiple parallel cells, an olefin-enriched stream can travel from one olefin-releasing electrode to another in a consecutive manner, continuously increasing olefin concentration of the product stream. A sample schematic in how to arrange the gas flow of the olefin-enriched stream is shown in gas flow arrangement. | ||
Realizing the proposed scalable concepts will rely in overcoming the scientific and technical barriers identified in our study, including faradaic efficiency, utilization fraction of [Ni(mnt)2]n species, mass transport limitations, and stability. Typically, a low faradaic efficiency (FE) was observed (≤1%), which is calculated as the fraction of the total transferred charge used for the electrochemical modulated capture and release of propylene from a 50:50 propylene–propane mixture. The utilization fraction (UF) of the embedded [Ni(mnt)2]n species in the MEA was typically <2% when separating propylene from a 50:50 mixture. The low FE and UF arise from parasitic reactions (e.g., HER or OER from water molecules that diffused from the counter electrode) and mass transport limitation in the ionogel (see Fig. 3B), impinging the accessibility of [Ni(mnt)2]n species to the interfacial area with the gas phase. These limitations can be addressed by controlling electrode side reactions and developing highly porous electrodes that can scaffold a high loading of [Ni(mnt)2]n species such as by using PCP30 or by using alternative ionogel materials and compositions that exhibit high solubility and ion mobility of embedded [Ni(mnt)2]n species with minimal negative effect on porosity and interfacial area. Lastly, long-term stability is an important factor in the scale-up of this separation process. Degradation of separation performance has been observed over consecutive cycles during our study (see Fig. S1C and D in ESI†). Even though [Ni(mnt)2]n can undergo reversible redox processes, excess voltage coupled with the presence of water, oxygen, and hydrogen (from the counter-side of the MEA) can lead to undesirable parasitic reactions that irreversibly degrade [Ni(mnt)2]n species. Previous computational studies have also suggested that a [Ni(mnt)2]− anion participates as a catalyst for the complexation of olefins, and that without the presence of this anion the complex can undergo degradation.26 Strategies such as the swing cycle shown in Fig. 5 as well as minimizing overpotentials in the cell can help mitigate the irreversible degradation of the Ni complex.
3. Experimental procedure
3.1 Synthesis of [N(Et)4]2[Ni(mnt)2]
A modified synthesis protocol for tetraethyl ammonium salt of [Ni(mnt)2]−2 was adapted from Davidson et al.,41 details of which can be found in Section S8 in ESI.† X-Ray diffraction (XRD) and elemental analysis were employed to verify the chemical makeup and purity of synthesized [N(Et)4]2[Ni(mnt)2] salt as shown in Section S9 in ESI.† Cyclic voltammetry of [N(Et)4]2[Ni(mnt)2] in varying organic media solution environments helped confirm the electrochemical behavior of [Ni(mnt)2]n species as shown in Section S10 in ESI.†3.2 Spray deposition
An MEA catalyst ink dispersion spray system with a pair of automated programmable 2-dimensional displacement motorized stages was developed to fabricate reproducible MEA samples with uniform porous dispersion coating on a carbon gas diffusion layer (GDL) as shown in Fig. S12A in ESI.† The motorized stages (linear long travel 150 mm stages) were acquired from ThorLabs. The spray nozzle was acquired from Grainger (Exair atomizing spray nozzle with manufacturing model number SR1010SS). The spraying system was fed a colloidal “ink” solution containing the relevant components of the sample MEA, including carbon nanoparticles, [N(Et)4]2[Ni(mnt)2], IL, and Nafion. Vulkan XC-72R activated carbon powder and 10% Nafion solution in isopropanol were utilized as the primary source of carbon nanoparticles and Nafion polymer for the ink solution respectively. The carbon and the Nafion solution were acquired from Fuel Cell Store (Bryan, Texas). The ink solution mainly consisted of 70% isopropanol in water solvent while the aforementioned components comprised less than 0.2% by weight of the overall solution. Keeping the ink solution within this weight percent proved crucial in maintaining a homogenous spray texture onto the substrate GDL. The deposited porous layer was vacuum dried at room temperature for 24 hours after spray deposition to remove residual solvent used during the process.3.3 MEA preparation
Nafion-117 was used as the primary ion-exchange membrane in the MEA. The counter-side of the electrochemical flow cell device consisted of iridium coating to perform water oxidation or reduction as a counter reaction to the [Ni(mnt)2]n reactions. Iridium was used in the counter electrode because of its high activity towards water oxidation, and its demonstrated stability under water splitting conditions in the MEA. Both the porous electrode layer containing [N(Et)4]2[Ni(mnt)2] and the iridium counter-side electrode layer were each hole-punched of the center of its ink-sprayed area into a circular disc of 1.5875 cm (5/8-in.) diameter. The ion-exchange Nafion-117 membrane was then placed between these two disc-shaped electrode layers, as shown in Fig. 1. Because the same circular size area was used throughout this work, the active geometric area of the device was 1.98 cm2 for all the experiments.The MEA components of specified compositions were then carefully installed into an electrochemical flow cell device like the one depicted in Fig. 1 with torque wrenches (set to 6.00 N m) to ensure good contact between different components of the device. The composition of each deposited layer can be found in the figure captions in Sections S1–S6 in ESI.† Workable electric potentials to induce the selective olefin complexation mechanism were also identified for the MEA through cyclic voltammetry as shown in Section S11 in ESI.†
3.4 Characterization of electrochemical redox cycle
As shown earlier in Fig. 1, the outlet gas is directly connected to a gas chromatographer (Agilent Micro GC 990) for gas composition analysis. The Micro GC 990 has 2 channels to record the thermal conductivity detector (TCD) signal coming from 2 pressure columns, a Molecular Sieve 5A Column (for hydrogen detection) and a PoraPlotQ Column (for propylene and propane detection), both running with ultra-high purity helium carrier gases purchased from Air Gas. Calibration measurements with low concentration standards of propylene and propane gases (purchased from Advanced Specialty Gases in Reno, Nevada) were periodically conducted (along with maintenance purpose baking of GC column) to ensure reasonably accurate conversion from the GC TCD signals to actual concentrations in the analyzed gas sample. Three Brooks Instrument G40 electrically controlled Mass Flow Controllers were utilized to provide accurate gas feed mixture or nitrogen carrier gas into the electrochemical flow cell device for each experiment. The three main gases used in any given experiment are propylene, propane, and nitrogen; each has its own dedicated flow meter. Propylene and propane, at a purity of approximately 99.5%, were purchased from Sigma-Aldrich in small 2-L gas cylinders. High-purity nitrogen was purchased from Airgas in 200-L cylinders. Electrical potentials were modulated using the Agilent Potentiostat SP-40 in a 2-electrode configuration. Deionized water was fed to the iridium counter-electrode side with a peristaltic pump at 10 ml min−1. Labview software was utilized to automate the flow meters in sync with the Potentiostat and the Micro GC 990 collecting data in real-time (Micro GC took measurements approximately once every 2 minutes). Each MEA sample of unique loading compositions is tested for a minimum of 4 cycles, with the first cycle acting as a stabilizing run while the last 3 cycles are used in our data analysis and final summary graphs shown in Fig. 3 and 4. Error bars have been plotted, representing the standard deviations of the last 3 measurements (cycles) per MEA sample of specified loading composition and experimental conditions.Each separation test cycle typically lasts for 4 hours and consists of four distinct phases to enable the electrochemical flow cell device to separate propylene–propane gas mixture in an experimentally observable manner. Temperature and pressure inside the flow cell are kept constant at approximately 25 degrees celsius and 1 atm, respectively. Fig. 2 depicts a typical separation test cycle. In phase (1), a mixture of propylene and propane gas is fed into the flow cell for 30 minutes at a combined total flow rate of 20 sccm. Simultaneously, an oxidative potential is applied to the gas-diffusing electrode layer (+1.5 V at the standard procedure) vs. counter iridium-coated electrode. The oxidative current converts [Ni(mnt)2]−2 to [Ni(mnt)2]0 near the surface of the carbon nanoparticles within the ionogel.
After enough propylene complexation takes place, phase (2) involves the removal of propylene and propane gas molecules in the environment of the MEA through a steady nitrogen flush at a constant flow rate of 5 sccm. Propylene and propane feed flow ceases to 0 sccm at start of phase (2). The main purpose of this flush is to lower the concentration of the propylene and propane molecules in the gas stream and increase the GC sensitivity to detect small changes in the concentration of these gases. Furthermore, sufficient time was required to flush the system of propylene and propane molecules that were absorbed into the MEA system via concentration-gradient-driven sorption and avoid non-electrochemically complexed propylene and propane to substantially interfere with our measurements. Nitrogen carrier flow rate will maintain at 5 sccm for the remainder of the cycle including phases (3) and (4).
Phase (3) can be initiated once the propylene–propane concentration is low enough. A reductive potential is applied to the gas-diffusing electrode layer (−2 V vs. counter-side at the standard procedure) so that the complexed species can decouple the propylene at the carbon electrode surface. Concentration-gradient-driven diffusion would allow more of the propylene-containing complex to travel to the electrode surface to get reduced and decoupled. The propylene concentration should theoretically increase initially near the carbon electrode surface and eventually diffuse towards the gas-phase interface where a bulk nitrogen stream can carry the propylene into the GC sampling port. By measuring the concentration of the propylene at periodic time intervals after applying this polarity switch in electric potential, the total moles of propylene gas that were captured and released by this MEA system can be quantified through area integration of the characteristic “peak” as shown in deconvoluted propylene concentration profiles in Sections S1–S6 in ESI.† Phase (4) was a maintenance-purpose flush to ensure the flow cell device restarts in a similar condition for the next experimental cycle. Data that support the findings of this study have been deposited in GitHub publicly accessible (requiring no access codes) via web link: https://github.com/ta1535/Nickel_Dithiolene_Research_Experimental_Data.git.
4. Conclusions
The proof-of-concept MEA device presented in this study demonstrated the effective separation of propylene from propane mixtures. Because of the lack of affinity between [Ni(mnt)2]n complexes and propane, the system exhibited remarkable selectivity in its olefin capture-and-release mechanism, opening a path to electrochemically modulated olefin–paraffin separation processes. The results show that careful tuning of [Ni(mnt)2]n and IL loading in the ionogel can improve ion mobility and capacity to store additional redox sites. Intermediate IL loadings resulted in optimal performance, highlighting the trade-offs between the need for increased [Ni(mnt)2]n mobility while maintaining fast gas transport to the electrodes. Through this study, we have identified plausible routes for scale-up and key technical challenges that must be addressed to translate this separation concept to industrial processes that can compete with state-of-the-art cryogenic distillations or membrane separation methods. These include designing materials and operation strategies to enhance FE, gas mass transport, and stability. Future work on these strategies may result in high-performing electrochemically-modulated olefin–paraffin separation units that can integrate renewable electricity sources directly and help decarbonize the chemical manufacturing industry.Conflicts of interest
All authors report no conflict of interest in any part of this study.Acknowledgements
The authors would like to acknowledge the financial support provided by New York University, Tandon School of Engineering, through the MAM startup fund. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant no. DGE1839302 and DGE2234660. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund under Grant #59824-DNI5 for partial support of this research. The authors acknowledge NYU Nanofabrication Facility for SEM imaging done with Hitachi SU8600, and the NYU Tandon Makerspace for fabrication support.References
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- O. R. N. Laboratory, Materials for Separation Technologies. Energy and Emission Reduction Opportunities, United States, 2005 Search PubMed.
- B. Dudley, BP Statistical Review, London, UK, accessed Aug, 2018, vol. 6, p. 00116.
- A. Khanunthong, Industry Outlook: 2020-2022: Petrochemical, Krungsri Research, accessed April, 2022.
- GlobalData, Ethylene Industry Installed Capacity and Capital Expenditure (CapEx) Forecast by Region and Countries including details of All Active Plants, Planned and Announced Projects, 2021–2025, 2021.
- Y. Ren, X. Liang, H. Dou, C. Ye, Z. Guo, J. Wang, Y. Pan, H. Wu, M. D. Guiver and Z. Jiang, Adv. Sci., 2020, 7, 2001398 CrossRef CAS PubMed.
- J. B. Greenblatt, D. J. Miller, J. W. Ager, F. A. Houle and I. D. Sharp, Joule, 2018, 2, 381–420 CrossRef CAS.
- A. Roy, S. R. Venna, G. Rogers, L. Tang, T. C. Fitzgibbons, J. Liu, H. McCurry, D. J. Vickery, D. Flick and B. Fish, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022194118 CrossRef CAS PubMed.
- A. Van Miltenburg, W. Zhu, F. Kapteijn and J. A. Moulijn, Chem. Eng. Res. Des., 2006, 84, 350–354 CrossRef CAS.
- J. A. Moulin, M. Makkee and A. V. Diepen, Chemical process technology, John Wiley & Sons, New York, 2005 Search PubMed.
- J. Hou, P. Liu, M. Jiang, L. Yu, L. Li and Z. Tang, J. Mater. Chem. A, 2019, 7, 23489–23511 RSC.
- J. Prausnitz, E. G. d Azevedo and R. N. Lichtenthaler, Molecular thermodynamics of fluid-phase equilibria, Prentice-Hall PTR, Upper Saddle River, NJ, 1999 Search PubMed.
- C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- T. C. Merkel, R. Blanc, I. Ciobanu, B. Firat, A. Suwarlim and J. Zeid, J. Membr. Sci., 2013, 447, 177–189 CrossRef CAS.
- J. Solodar and J. P. Petrovich, Inorg. Chem., 1971, 10, 395–397 CrossRef CAS.
- D. Yang, R. S. Barbero, D. J. Devlin, E. L. Cussler, C. W. Colling and M. E. Carrera, J. Membr. Sci., 2006, 279, 61–69 CrossRef CAS.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- R. Mohtadi, W.-K. Lee, S. Cowan, J. Van Zee and M. Murthy, Electrochem. Solid-State Lett., 2003, 6, A272 CrossRef CAS.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- B. Gurkan, F. Simeon and T. A. Hatton, ACS Sustainable Chem. Eng., 2015, 3, 1394–1405 CrossRef CAS.
- T. Suzuki, R. D. Noble and C. A. Koval, Inorg. Chem., 1997, 36, 136–140 CrossRef CAS.
- P. A. Terry, R. D. Noble, D. Swanson and C. A. Koval, AIChE J., 1997, 43, 1709–1716 CrossRef CAS.
- K. Wang and E. I. Stiefel, Science, 2001, 291, 106–109 CrossRef CAS PubMed.
- J. Pitchaimani, S.-F. Ni and L. Dang, Coord. Chem. Rev., 2020, 420, 213398 CrossRef CAS.
- L. Dang, X. Yang, J. Zhou, E. N. Brothers and M. B. Hall, J. Phys. Chem. A, 2012, 116, 476–482 CrossRef CAS PubMed.
- L. Dang, M. F. Shibl, X. Yang, A. Alak, D. J. Harrison, U. Fekl, E. N. Brothers and M. B. Hall, J. Am. Chem. Soc., 2012, 134, 4481–4484 CrossRef CAS PubMed.
- R. K. Raju, D. N. Sredojevic, S. Moncho and E. N. Brothers, Inorg. Chem., 2016, 55, 10182–10191 CrossRef CAS PubMed.
- L. Dang, M. F. Shibl, X. Yang, D. J. Harrison, A. Alak, A. J. Lough, U. Fekl, E. N. Brothers and M. B. Hall, Inorg. Chem., 2013, 52, 3711–3723 CrossRef CAS PubMed.
- H. Kunkely and A. Vogler, Inorg. Chim. Acta, 2001, 319, 183–186 CrossRef CAS.
- L. Mendecki, M. Ko, X. Zhang, Z. Meng and K. A. Mirica, J. Am. Chem. Soc., 2017, 139, 17229–17232 CrossRef CAS PubMed.
- J. H. Lee, S. W. Kang, D. Song, J. Won and Y. S. Kang, J. Membr. Sci., 2012, 423, 159–164 CrossRef.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef CAS.
- B.-C. Lee and S. L. Outcalt, J. Chem. Eng. Data, 2006, 51, 892–897 CrossRef CAS.
- T. Ueki and M. Watanabe, Macromolecules, 2008, 41, 3739–3749 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- Y. Gu and T. P. Lodge, Macromolecules, 2011, 44, 1732–1736 CrossRef CAS.
- K. H. Lee, M. S. Kang, S. Zhang, Y. Gu, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2012, 24, 4457–4462 CrossRef CAS PubMed.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- J. Fuller, A. Breda and R. Carlin, J. Electrochem. Soc., 1997, 144, L67 CrossRef CAS.
- R. T. Carlin and J. Fuller, Chem. Commun., 1997, 1345–1346 RSC.
- A. Davison, N. Edelstein, R. Holm and A. Maki, Inorg. Chem., 1963, 2, 1227–1232 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00508a |
| ‡ This project is dedicated to the late Dr Yoshiyuki Okamoto who devoted much of his life to polymer science research. Achieving a practical system for olefin–paraffin separation was his last dream. |
| This journal is © The Royal Society of Chemistry 2024 |