
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical and X-ray absorption interrogation of selenium-based redox in Li–SxSey batteries†
Ryan H.
DeBlock
 a,
Matthew J.
Lefler
b,
Zachary G.
Neale
b,
Corey T.
Love
a,
Jeffrey W.
Long
a and
Rachel
Carter
*a
a,
Matthew J.
Lefler
b,
Zachary G.
Neale
b,
Corey T.
Love
a,
Jeffrey W.
Long
a and
Rachel
Carter
*a
aSurface Chemistry Branch (Code 6170), U.S. Naval Research Laboratory, Washington, DC, USA. E-mail: rachel.carter@nrl.navy.mil
bNational Research Council Postdoctoral Associate, U.S. Naval Research Laboratory, Washington, DC, USA
First published on 12th January 2024
Abstract
In situ X-ray absorption spectroscopy and optical imaging confirm the role of selenium additives for enhancing power performance, increasing utilization, and suppressing undesirable side reactions in Li–sulphur batteries.
The ongoing renewable-energy revolution requires efficient, sustainable, and reliable electrochemical energy storage to bridge generation and demand cycles. Advanced batteries that use sulphur (S) as a cathode material (e.g., in room-temperature Li–S or Na–S configurations) are promising candidates for such applications due to high redox capacity (1675 mA h g−1) via the S/S2− reaction and the global abundance of S.1–3 Sulphur batteries have been the subject of intense investigation over the past decade, with research focused on improving cycle life and power by optimizing cathode construction, electrolyte formulation, and separator permeability. The poor conductivity of sulphur is a major limitation of this battery chemistry, a drawback that can be partially mitigated by applying thin sulphur coatings onto conductive, porous carbons.4–6
Another route to improving conductivity and electrode kinetics involves blending sulphur with a more electrically conductive chalcogen—selenium (Se)—(10−4 S m−1 compared to 10−14 S m−1) which displays similar electrochemical redox characteristics, albeit with lower theoretical capacity (678 mA h g−1).7–9 Selenium exhibits better capacity retention than sulphur upon extended cycling, ascribed to the higher conductivity of the former and lower prevalence of polyselenide formation (versus analogous polysulfide). To harness the best characteristics of abundant sulphur and conductive, cyclable selenium, we use a glassy sulphur–selenium composite10 as a vapor-deposition precursor in the fabrication of cathodes based on porous carbon nanofoam paper (CNFP) substrates, as recently demonstrated with sulphur-only deposition.11
The resulting “SSe@CNFP” cathodes, along with S@CNFP and Se@CNFP controls, are electrochemically evaluated in coin cells with Li metal anodes, testing for such performance parameters as specific capacity and rate retention. In situ X-ray absorption spectroscopy of the Se K-edge is used to track Se/Se2− reactions during charge–discharge in related pouch cells. Because the formation of soluble polysulfides is a common challenge for Li–S batteries, we also image the adjacent electrolyte in special optical cells as the chalcogen-containing electrodes are cycled through their operating voltage range.
Chalcogen powders (Fig. 1a) ranging from S (yellow) and Se (grey) to a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 atomic ratio SSe blend (red) serve as precursors for vapor-phase deposition within a custom-built, PTFE chamber (Fig. 1b). In this chamber, we also include pieces of carbon nanofoam paper (CNFP) to serve as a porous conductive substrate, as recently reported for sulphur-only deposition to form effective Li–S cathodes.11 The SSe blend deposits to ∼50 wt% chalcogen in the CNFP after 4 h at 225 °C (forming SSe@CNFP), thermal conditions determined by thermal gravimetric analysis and differential scanning calorimetry (Fig. S1, S2 and Table S1, ESI†). Chalcogen loading was determined by taking the mass of the CNFP before and after infiltration. Elemental mapping by EDS confirms that both S and Se deposit at the interior surfaces of the CNFP throughout the ∼90 μm thickness of this substrate (Fig. 1c and d).
:40 atomic ratio SSe blend (red) serve as precursors for vapor-phase deposition within a custom-built, PTFE chamber (Fig. 1b). In this chamber, we also include pieces of carbon nanofoam paper (CNFP) to serve as a porous conductive substrate, as recently reported for sulphur-only deposition to form effective Li–S cathodes.11 The SSe blend deposits to ∼50 wt% chalcogen in the CNFP after 4 h at 225 °C (forming SSe@CNFP), thermal conditions determined by thermal gravimetric analysis and differential scanning calorimetry (Fig. S1, S2 and Table S1, ESI†). Chalcogen loading was determined by taking the mass of the CNFP before and after infiltration. Elemental mapping by EDS confirms that both S and Se deposit at the interior surfaces of the CNFP throughout the ∼90 μm thickness of this substrate (Fig. 1c and d).
 | ||
| Fig. 1 (a) Selenium (grey), 60:40 S:Se blend (red), and sulphur (yellow) precursor powders for vapor-phase infiltration. (b) Schematic of vapor-phase infiltration chamber (top) and a 5 cm × 5 cm carbon nanofoam paper (CNFP) electrode (bottom). (c) Top-down scanning electron micrograph and corresponding energy dispersive spectroscopy map of SSe@CNFP. (d) Cross-sectional scanning electron micrograph and corresponding energy dispersive spectroscopy map of SSe@CNFP. | ||
Control samples of S@CNFP and Se@CNFP were similarly fabricated for comparison (details found in the Materials and methods section of the ESI†). Because Se has a greater density than S (4.79 g cm−3vs. 2.07 g cm−3), the Se mass loading was limited to ∼30 wt% to achieve a similar volumetric loading of chalcogen compared to S@CNFP (Table S1, ESI†).
These device-ready chalcogen@CNFP cathodes were assembled into coin cells to assess their electrochemical characteristics. When tested with low current-density galvanostatic cycling, Li–S@CNFP and Li–Se@CNFP cells exhibit the multistage charge–discharge profiles typical for these chalcogen cathodes6 (Fig. 2a and c). Li–S and Li–Se cells exhibit a 2 discharge plateaus. For sulphur these occur at ∼2.3 and 2.1 V (Fig. 2a) and selenium at ∼2.2 and 2.0 V (Fig. 2c) due to lower standard reduction potential. The higher voltage peak is indicative of high order products (e.g. Li2Sx 8 ≥ x ≥ 4) and the lower voltage one, low order products (e.g. Li2Sx 2 ≥ x ≥ 1), respectively. The high order products are typically soluble in the electrolyte, whereas the low order products are large (compared to the pristine chalcogen), solid products. Further complexity is evident for the Li–SSe@CNFP cell where both S/S2− and Se/Se2− redox reactions may be active (Fig. 2b). The nuances of the discharge process may be viewed by replotting these data in a differential form (dV/dq), where many more peaks are observed (1.93, 2.05, 2.13, and 2.28 V) along the pathway to full discharge for the Li–SSe@CNFP cell (Fig. 2(d)–(f)).
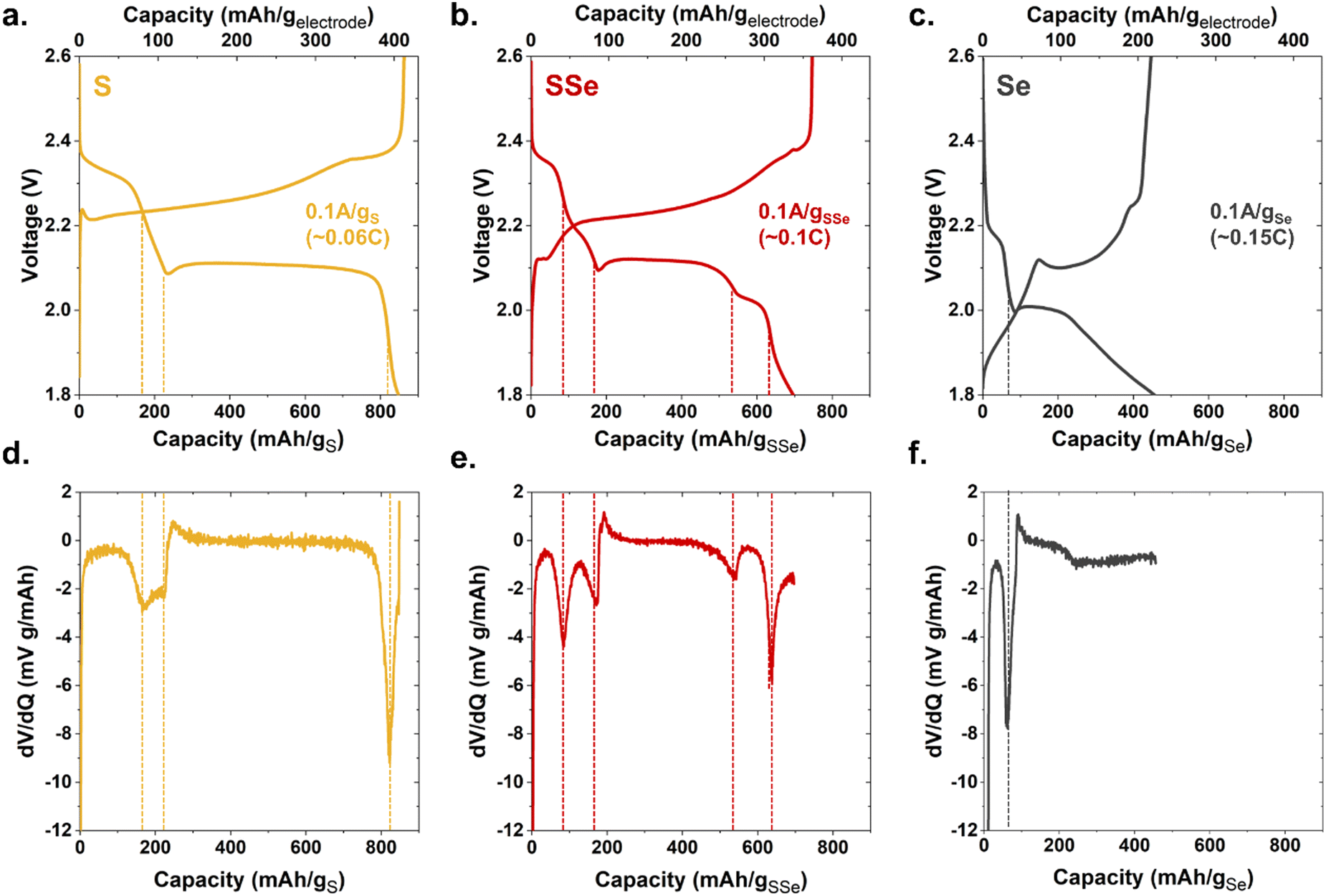 | ||
| Fig. 2 Galvanostatic charge/discharge curves at 0.1 A g−1 of chalcogen for (a) S@CNFP (b) SSe@CNFP (c) Se@CNFP and (d)–(f) the corresponding dV/dq plots. | ||
Total low-rate (0.1 A g−1) discharge capacity (normalized to chalcogen mass) qualitatively tracks the trend in theoretical values for S vs. Se (1675 and 678 mA h g−1, respectively). The Li–S@CNFP cell delivers the highest gravimetric capacity (∼820 mA h g−1) and Li–Se@CNFP the lowest (450 mA h g−1). When normalized to respective theoretical capacity, redox utilization for the Li–S@CNFP is 49% versus 66% for the Li–Se@CNFP cell, which can be ascribed to the higher conductivity of Se.7 Higher redox utilization for Se mitigates its impact on total capacity when blended with S. For example, low-rate capacity in the Li–S/Se@CNFP cell (700 mA h g−1) is only modestly lower than that for Li–S@CNFP control.
The practical benefits of the SSe-blend cathode become more apparent when challenging this series of cells with increasing current demand, in this case ranging from 0.1 to 5 A g−1 (corresponding to C-rates of 0.06 to 7.4) as normalized to chalcogen mass (Fig. 3 and Fig. S3, S4, ESI†). Capacity from the Li–S@CNFP cell rapidly diminishes at C-rates ≥0.3; at 1C no appreciable capacity is observed. Although starting with lower initial capacity at 0.1C, the Li–SSe@CNFP cell delivers 402 mA h g−1 at 1C, and 221 mA h g−1 at 2C. As anticipated, the inclusion of Se with S in the cathode vastly improves rate retention, mitigating one of the key drawbacks of Li–S cells. Impedance spectra for Li–S@CNFP and Li–SSe@CNFP cells also show ∼2.5-fold lower charge-transfer resistance in the latter case (Fig. S5, ESI†), consistent with the enhanced power performance noted above.
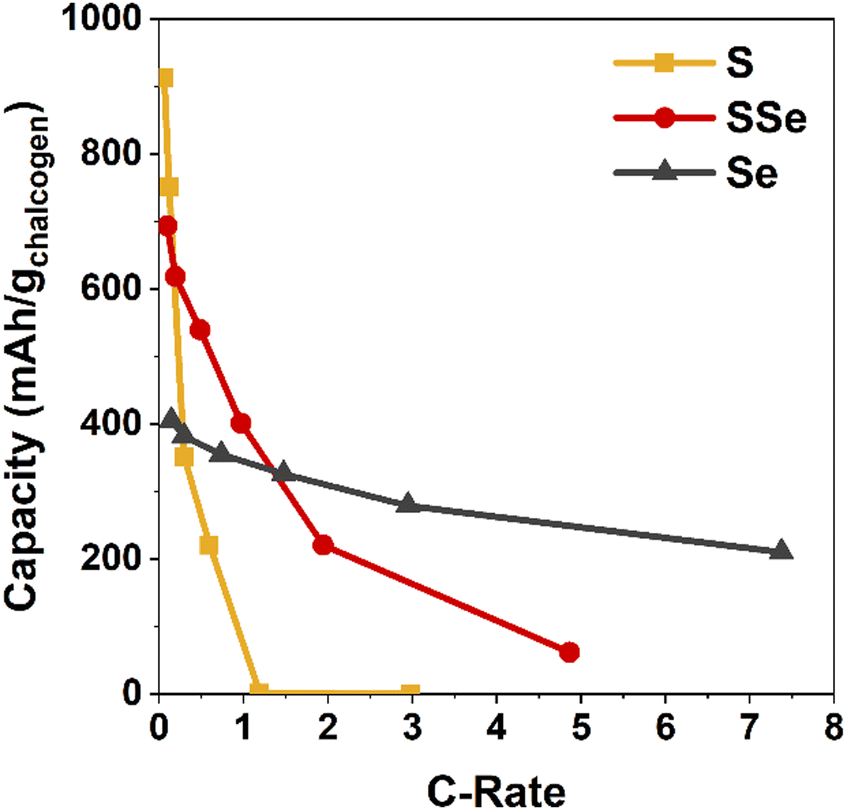 | ||
| Fig. 3 Discharge capacity of chalcogen@CNFP electrodes normalized to the active chalcogenide mass as a function of C-rate. | ||
Selenium serves not only as a conductivity enhancer, but also contributes its own redox reactions for charge storage, occurring within a similar voltage window to S-based redox. To differentiate the redox contributions of Se, we turned to X-ray absorption spectroscopy (XAS), a powerful tool to analyse the chemical state and local structure of battery-relevant materials that is also amenable to in situ/operando measurements. While historically requiring synchrotron sources for high-quality measurements, recent advancements with X-ray optics and instrument design now make XAS accessible in a laboratory setting.12 For this investigation, we use a commercial, bench-scale XAS with an energy window (∼5–20 keV) that encompasses the Se K-edge at ∼12.65 keV (note that the S K-edge at ∼2.47 keV is not accessible with the same instrument). The innate X-ray transmissivity of the CNFP-supported chalcogen electrodes is ideal for measurements of Se K-edge absorption with this instrument, even when testing in a functional pouch-cell battery with ancillary components (Li anode, electrolyte, packaging).
We track the oxidation state of Se in a Li–SSe@CNF pouch cell using K-edge X-ray absorption near-edge spectroscopy (XANES). The dV/dq plots in Fig. 2 provide guidance on voltages at which to collect XANES data, targeting nominal points of transition between different redox processes. Cell voltage is swept between these points of interest and then held during XANES measurements (Fig. 4a). During discharge, the Se absorption edge begins shifting significantly only when the cell voltage is held below 2.1 V, indicating that Se/Se2− redox occurs primarily toward the latter stages of discharge. These Se K-edge shifts are fully reversible upon recharge (Fig. S6, ESI†).
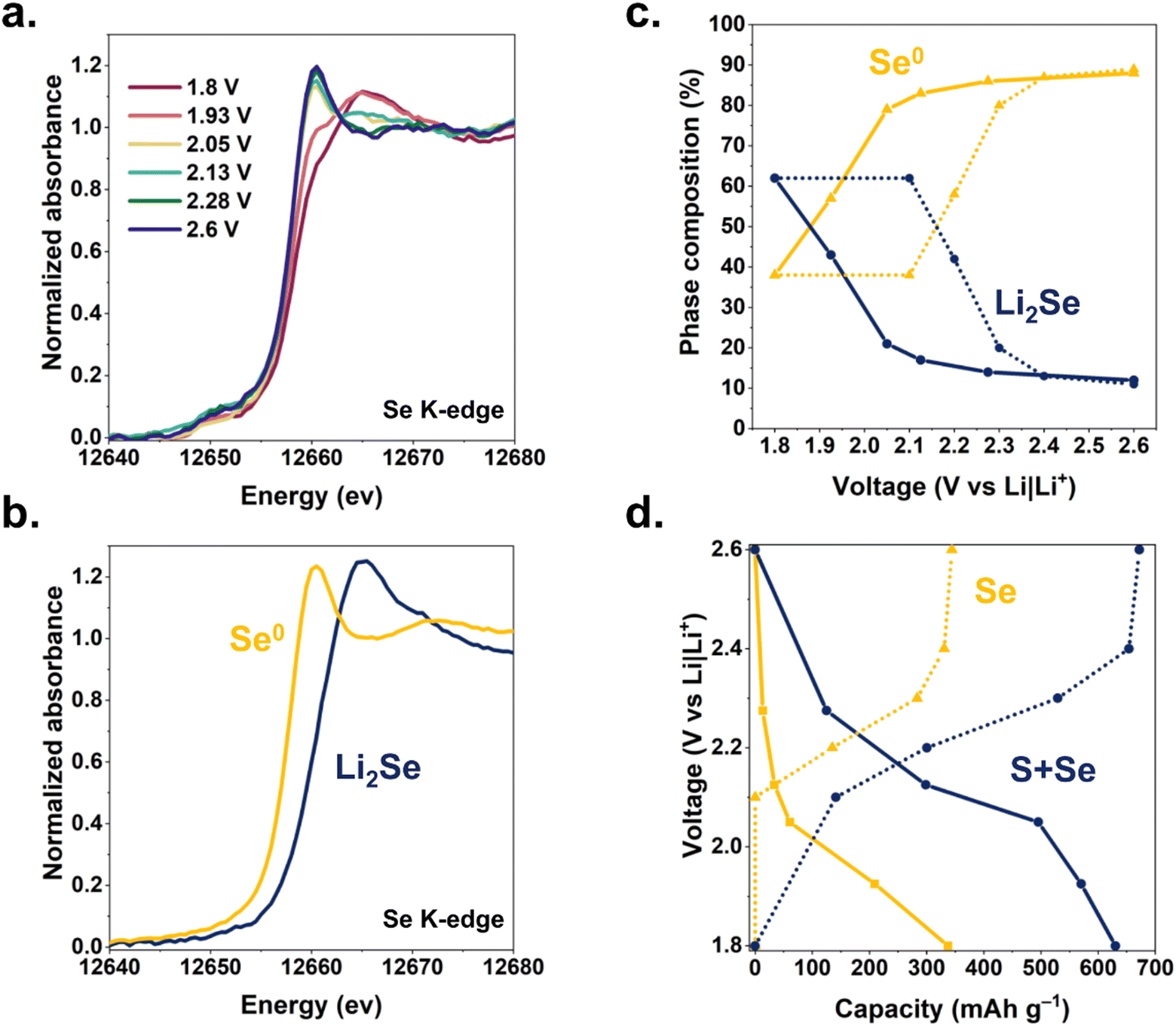 | ||
| Fig. 4 (a) in situ Se K-edge XANES spectra at selected cell voltages during discharge of a Li–SSe@CNFP cell. (b) Phase composition of the SSe@CNFP electrode as a function of voltage (solid lines: lithiation, dotted lines: delithiation). Ratios determined by linear combination fitting of the Se K-edge XANES spectra with (c) selenium metal and Li2Se XANES standards. (d) Capacity derived from XANES (Se redox only) compared to capacity derived from integration of current during voltammetric sweeps and holds (S + Se redox). Solid lines: lithiation, dotted lines: delithiation. | ||
Using principal component analysis, we determine that these XANES spectra are best fit by the superpositions of two components (Fig. S7, ESI†), which we ascribe as Se and Li2Se. Experimental XANES spectra of Se foil and Li2Se powder (Fig. 4b) were used for linear combination fitting (LCF) of spectra from Li–SSe@CNFP cells to deconvolve the Se/Li2Se phase composition (Fig. 4c) as a function of cell voltage. The resulting analysis reveals (i) that ∼50% of the Se in the cathode participates in the Se/Se2− redox during (de)lithiation in the SSe@CNFP cathode, and (ii) that Se/Se2− redox primarily occurs at the lowest voltages during both charge and discharge. The lower-voltage reduction of the Se/Se2− reaction13 in the SSe blend is fortuitous in that elemental Se is still present through at least the first half of the discharge, providing enhanced conductivity within the cathode before ultimately converting to Li2Se. Using the change in Se/Li2Se ratio derived from XANES multiplied by the theoretical capacity for this reaction (2Li+ + Se + 2e− ⇆ Li2Se; 678 mA h g−1), we extract an expected capacity from Se lithiation at each voltage of interest. We compare this XANES-calculated Se capacity to the measured cell capacity at each voltage step (Fig. 4d). For example, when discharging the Li–SSe@CNF pouch cell to 2.13 V, significant lithiation (>300 mA h g−1) has already occurred, yet only ∼34 mA h g−1 can be attributed to Se redox according to LCF component determination. Below 2.13 V, >86% of the remaining cell capacity (∼300 mA h g−1) is attributed to Se redox. Considering the total discharge of a Li–SSe@CNF cell, over 50% of capacity can be ascribed to Se/Se2− redox even though Se is only present at 40 at% in the chalcogen blend and Se theoretical capacity is >2-fold lower than for S. Chalcogen-based redox may involve not only solid-state reactants/products (e.g., Se and Li2Se), but also soluble intermediates that complicate cell operation.14 To assess the implications of polysulfide/polyselenide generation, we turn to optical cell microscopy with which electrolyte bathing the CNFP-based cathodes under electrochemical control can be observed. Cells containing S@CNFP, SSe@CNFP, or Se@CNFP cathodes and a Li metal anode were scanned by cyclic voltammetry while changes in electrolyte colour between the electrodes were simultaneously recorded. Images corresponding to the same voltages identified in Fig. 2via dV/dq and used for XANES are collated in Fig. 5.
 | ||
| Fig. 5 (a) Optical cell configuration and (b) the corresponding images of electrolyte colour as a function of voltage for S@CNFP, SSe@CNFP, and Se@CNFP electrodes during discharge and charge. Inset in the SSe@CNFP electrode at 1.8 V shows the precipitation of Li2Se crystals. | ||
The Li–S@CNFP optical cell reveals a similar discharge mechanism to what we previously reported with Na-S@CNTs.15 In that report, we demonstrated the ability to identify polysulfide species with optical images based on RGB pixels alone, eliminating the need for UV-visible spectroscopy analysis. In summary, high-order polysulfides (Sx2−; 8 ≥ x ≥ 6) are dark red/brown in colour, lower-order polysulfides (Sx2−; 4 ≥ x ≥ 2) are lighter red/yellow, and undesirable radicalized polysulfides (S3−) are blue. When chemical recombination to the radicalized ion occurs, the desired electrochemical pathway is hindered, limiting capacity and cyclability.
In the present case, the Li–S@CNFP electrode shows an initially transparent electrolyte that quickly saturates with soluble polysulfides. Early stages of discharge are known to proceed through high-order polysulfides,13 consistent with the optical image at 2.28 V. As the discharge continues, convective mixing of the electrolyte commences and red, brown, yellow, and green are all observed, indicating that chemical recombination is occurring. These colour changes correspond to temporal variation of RGB values (Fig. S8, ESI†). Upon charge, high-order polysulfides dissipate up to 2.4 V. At this voltage, the highest “G” pixelation and concentration of S3− is observed (combination of its blue colour and the remaining yellow, low-order polysulfides). Finally, at 2.6 V high-order polysulfides are reformed and a burst of red/brown is emitted from the cathode.
The onset of electrolyte coloration for the Li–Se@CNFP cell (Fig. 5b) is delayed until 2.13 V and below on discharge, in agreement with the lower discharge voltage for Se reduction (Fig. 2c). As discharge progresses, polyselenides (also red/brown in colour as previously reported by Xu et al.16) dissipate from the top of the Se@CNFP cathode and fill the electrolyte gap. Convective mixing of the polyselenides species is observed, much like the polysulfides in Fig. 4a, but the colour of the former remains constant as red/brown with minimal change in RGB (Fig. S8, ESI†). This colour persists as the Li–Se@CNFP cell charges, but with diminution of intensity.
The Li–SSe@CNFP optical cell shows further distinction versus either of the pure chalcogen cathodes. Dissolution is first observed at 2.28 V, but with uniform colour throughout the electrolyte. At this voltage, Se should not yet reduce, such that we can assume only polysulfides are present. The colour steadily intensifies to a dark red at 2.05 V. Under such conditions, fractal deposits are observed on the quartz window and within the CNFP, with further growth as voltage drops to 1.93 V. Raman scattering spectroscopy was used to identify these products as Li2Se (Fig. S9, ESI†), consistent with the XANES analysis noted above when operating in this voltage region (1.8–2.05 V).
Upon charging, the crystal growths dissipate, and the electrolyte colour lightens to a steady tone above 2.2 V, where Se2− is fully oxidized according to XANES analysis. Therefore, the remaining colour in the electrolyte likely arises from residual polysulfides, with no evidence of chemical recombination and radical formation or “G” pixelation (Fig. S8, ESI†). This distinction from the Li–S@CNFP is consistent with the difference from Li–SSe@CNFP in long-term cycle stability under galvanostatic conditions (Fig. S10, ESI†). Overall, observations from the Li–SSe@CNFP optical cell show that the SSe@CNFP cathode shows less propensity for chemical recombination of polysulfides or polyselenides and promotes a more direct discharge pathway that is conducive to higher rate capability and capacity retention.
Conclusion
The inclusion of Se in S-based cathodes addresses one of the remaining challenges of Li–S batteries: poor rate capability. X-ray absorption spectroscopy confirms the contributions of Se redox to total capacity and the important persistence of conductive elemental Se in early stages of discharge. The formation of undesirable reaction intermediates is also thwarted by the presence of Se, as confirmed by in situ optical imaging. The findings of this investigation further illuminate the role of Se in achieving important performance enhancements in terms of rate retention and redox utilization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Office of Naval Research (ONR) Grant N0001424WX00013. The authors thank Dr Junghoon Yeom (Naval Research Laboratory) for SEM characterization of the CNFP materials. This project was supported in part (Z. G. N. and M. J. L) by an appointment to the NRC Research Associateship Program at the U.S. Naval Research Laboratory, administered by the Fellowships Office of the National Academies of Sciences, Engineering, and Medicine.Notes and references
- A. Fotouhi, D. Auger, L. O’Neill, T. Cleaver and S. Walus, Energies, 2017, 10, 1937 CrossRef.
- K. Zhu, C. Wang, Z. Chi, F. Ke, Y. Yang, A. Wang and L. Miao, Front. Energy Res., 2019, 7, 123 CrossRef.
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41 CrossRef CAS.
- A. Eftekhari and D. Kim, J. Mater. Chem. A, 2017, 5, 17734 RSC.
- Q. Xiao, J. Yang, X. Wang, Y. Deng, P. Han, N. Yuan, L. Zhang, M. Feng, C. Wang and R. Liu, Carbon Energy, 2021, 3, 271 CrossRef CAS.
- F. Sun, H. Cheng, J. Chen, N. Zheng, Y. Li and J. Shi, ACS Nano, 2016, 10, 8289 CrossRef CAS PubMed.
- Y. Cui, A. Abouimrane, J. Lu, T. Bolin, Y. Ren, W. Weng, C. Sun, S. Heald and K. Amine, JACS, 2013, 135, 8047 CrossRef CAS PubMed.
- Z. Zhou, X. Huo, Y. Liu, S. Wang, W. Guan, Z. Dun and W. Ai, ACS Appl. Eng. Mater., 2023 DOI:10.1021/acsaenm.3c00587.
- D. Boyd, C. Baker, J. Myers, V. Nguyen, G. Drake, C. McClain, F. Kung, S. Bowman, W. Kim and J. Sanghera, Chem. Commun., 2017, 53, 259 RSC.
- Z. Neale, M. Lefler, J. Long, D. Rolison, M. Sassin and R. Carter, Nanoscale, 2023, 15, 16924 RSC.
- P. Zimmerman, S. Peredkov, P. Macarena Abdala, S. DeBeer, M. Tromp, C. Müller and J. van Bokhoven, Coord. Chem. Rev., 2020, 423, 213466 CrossRef.
- J. Sun, Z. Du, Y. Liu, W. Ai, K. Wang, T. Wang, H. Du, L. Liu and W. Huang, Adv. Mater., 2021, 33, 2003845 CrossRef CAS PubMed.
- Q. Zou and Y. Lu, J. Phys. Chem. Lett., 2016, 7, 1518 CrossRef CAS PubMed.
- R. Carter, A. NewRingeisen, D. Reed, R. Atkinson III, P. Mukherjee and C. Love, ACS Sustainable Chem. Eng., 2020, 9, 92 CrossRef.
- G. Xu, J. Liu, R. Amine, Z. Chen and K. Amine, ACS Energy Lett., 2017, 2, 605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, thermogravimetric analysis, additional electrochemical experiments, supplementary XAS data, colorimetric deconvolution, Raman spectroscopy. See DOI: https://doi.org/10.1039/d3ya00548h |
| This journal is © The Royal Society of Chemistry 2024 |