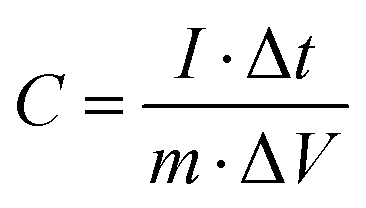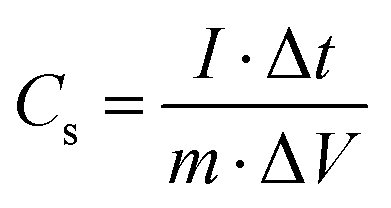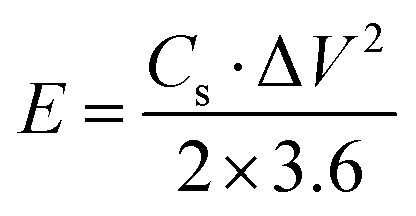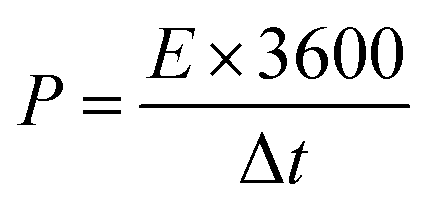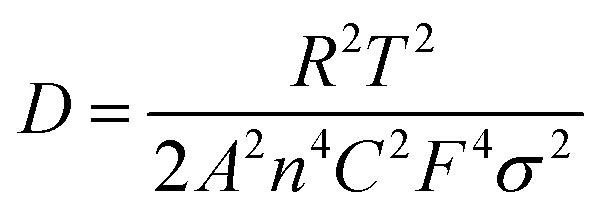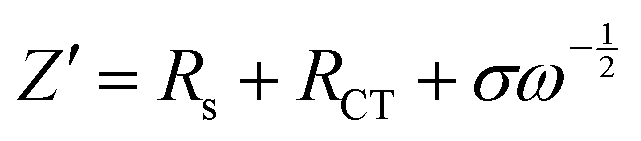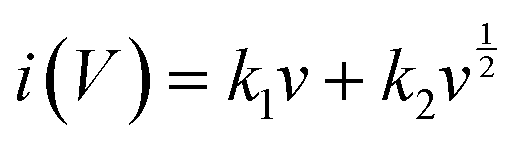DOI:
10.1039/D4YA00076E
(Paper)
Energy Adv., 2024,
3, 904-915
Oxygen-rich hierarchical porous carbon nanosheets derived from the KOH/KNO3 co-activation treatment of soybean straw for high-performance supercapacitors†
Received
3rd February 2024
, Accepted 17th March 2024
First published on 20th March 2024
Abstract
It is attractive to transform waste biomass into active carbon materials for energy storage via green chemical methods. In this study, we successfully prepared porous carbon nanosheets (PCNs) for carbon-based supercapacitors with a hierarchical porous structure and abundant oxygen-containing functional groups via a novel KOH/KNO3 co-activation method as an alternative to the common single KOH activation method, adopting soybean straw (SS) as a precursor. PCNs were obtained via a mild two-step conversion method with a high specific surface area of 1658.8 m2 g−1, a large mesopore and macropore ratio of 34.61%, and a considerable oxygen content of 16.77 at%, mainly consisting of hydrophilic carbonyl, carboxyl, and N–O functional groups compared to that in the single KOH activated sample, and are also outstanding for SS-based carbon nanomaterials. In a three-electrode system, the PCN electrode exhibited a high specific capacitance of 438 F g−1 at a current density of 1 A g−1, as well as an excellent capacitance retention of 91.32% when the current density increased to 20 A g−1. Moreover, the symmetrical supercapacitor based on the PCN electrode provided a large energy density of 16.09 W h kg−1 at a power density of 249.55 W kg−1, and a significant energy density of 9.39 W h kg−1 was achieved at a relatively high power density of 9141 W kg−1. In addition, the PCN-based supercapacitor exhibited excellent stability with 96.6% initial capacitance retention after 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a large current density of 10 A g−1. Therefore, the novel KOH/KNO3 co-activation method can not only improve the specific surface area and the porosity of SS-derived active carbon materials, but also implant abundant oxygen-containing functional groups for superior charge storage. Our work provides a compact co-activation method for the high-valued utilization of waste biomass resources.
000 cycles at a large current density of 10 A g−1. Therefore, the novel KOH/KNO3 co-activation method can not only improve the specific surface area and the porosity of SS-derived active carbon materials, but also implant abundant oxygen-containing functional groups for superior charge storage. Our work provides a compact co-activation method for the high-valued utilization of waste biomass resources.
Introduction
Nowadays, the shortage of fossil fuels and environmental pollution are the two major challenges facing the world. Accordingly, the search for new renewable energy sources and exploiting advanced energy storage techniques are important routes to alleviate these two crises. In the case of electrical energy storage, supercapacitors have the unique advantages of large power density, short charging time, long cycle life and an environment-friendly nature, thereby receiving widespread research interest in recent years.1 Currently, commercial supercapacitors are mainly constructed based on carbon-based electric double-layer capacitance (EDLC) to provide a large power density and excellent cycling stability.2 However, such a device generally exhibits a low energy density, mainly, owing to its lack of sufficient charge storage sites, making it difficult to meet the increasingly high electric energy demand of electronic devices. In this case, by introducing heteroatoms such as N, O, P, and S, or their compounds into the carbon lattice of existing high-power-density carbon electrode materials, active functional groups can be constructed to provide supplemental pseudocapacitance sites for reversible redox reactions.3–7 Among them, in the case of the oxygen heteroatom, it is easy to introduce abundant C–O (hydroxyl and ether) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (quinone, carbonyl and ester)-type functional groups considering the high affinity between the C atom and O atom.8 Active redox functional groups and hydrophilic moieties can be injected by suitably manipulating doping conditions, simultaneously boosting charge storage capacitance and electrode wettability.9,10 Nevertheless, the achievement of high-performed supercapacitors with a high energy density, while effectively balancing ion transport and electron migration is still a huge challenge.11 Alternatively, besides a large specific surface area, the rational pore size distribution on the surface of the electrode material is equally significant for achieving a large capacitance performance as possible.12,13 The pores of carbon materials can be categorized into micropore (<2 nm), mesopore (2–50 nm), and macropore (>50 nm) according to their diameter. For their functions, micropores are the main charge storage sites, mesopores can offer fast ion transport channels, and macropores create additional buffering space for ion transport.14 In the ideal situation, hierarchical porous carbon materials with a uniform pore size distribution are desirable, which can promote the charge storage capacitance without sacrificing the ion transport and interface contact.
O (quinone, carbonyl and ester)-type functional groups considering the high affinity between the C atom and O atom.8 Active redox functional groups and hydrophilic moieties can be injected by suitably manipulating doping conditions, simultaneously boosting charge storage capacitance and electrode wettability.9,10 Nevertheless, the achievement of high-performed supercapacitors with a high energy density, while effectively balancing ion transport and electron migration is still a huge challenge.11 Alternatively, besides a large specific surface area, the rational pore size distribution on the surface of the electrode material is equally significant for achieving a large capacitance performance as possible.12,13 The pores of carbon materials can be categorized into micropore (<2 nm), mesopore (2–50 nm), and macropore (>50 nm) according to their diameter. For their functions, micropores are the main charge storage sites, mesopores can offer fast ion transport channels, and macropores create additional buffering space for ion transport.14 In the ideal situation, hierarchical porous carbon materials with a uniform pore size distribution are desirable, which can promote the charge storage capacitance without sacrificing the ion transport and interface contact.
Waste lignocellulose resources are naturally abundant, and thus their rational utilization can not only save living space but also provide added value. In recent years, the conversion of biomass into carbon nanomaterials with a large specific surface area for electrical energy storage via simple chemical treatment has received significant attention because of is high compatibility with the concept of green chemistry, typically involving three steps of “pyrolysis-carbonization-activation”.15–17 Moreover, the natural texture of biomass can be utilized as a template to replicate the target product, resulting in the formation of a well-organized structure.18 With the advancement of synthetic techniques, a two-step or even one-step conversion method has been successfully developed recently to construct the target products with a shorter treatment period, reduced preparation cost, and improved utilization rate of raw materials.19–21 Soybean is a major source of vegetable protein and planted widely around the globe. It is noteworthy that a large amount of waste lignocelluloses is produced during the harvesting of this crop, such as bean shell, straw and root.22,23 Also, among them, soybean straw (SS) has a distinct framework including honeycomb-like epidermis, leaf vein, and fibrous tissue, which can promote the formation of well-structured carbonized products, and therefore the rational utilization of SS during soybean harvesting is not only beneficial for environmental protection, but also brings significant economic benefits. In this respect, researchers have reported several high-efficient three-step conversion methods for the utilization of waste SS resources. For example, Xu et al. prepared a porous-interlinked N/O co-doped nanographite sheet employing a “nickel salt co-hydrothermal treatment-carbonization-activation” process.24 The use of Ni(NO3)2 as a transition metal salt for the co-hydrothermal pyrolysis of SS can significantly improve the graphitization of the product and introduce abundant N/O functional groups and pores, meanwhile maintaining a high specific surface area. As a result, the supercapacitor fabricated with the obtained NNiSSC nanographite sheet exhibited an excellent specific capacitance of 407 F g−1 at a current density of 0.5 A g−1, good capacitance retention of 78.62% when loaded at 20 A g−1, high energy density of 36.11 W h kg−1 at a power density of 517.8 W kg−1, and good stability with 87.5% capacitance retention after 10000 cycles. Recently, Jiao et al. proposed an advanced “liquid phase transformation-hydrothermal carbonization-activation” method to process the SS precursor.25 The liquid phase transformation of SS in the initial stage can efficiently break the tight-binding covalent networks attached to the solid-state precursor, resulting in an enhancement in the subsequent activation step and promotion of the N/O co-doping level for the obtained layered porous carbon nanoflowers. Regarding its benefits, the symmetric supercapacitor provided a high specific capacitance of 220 F g−1 at a current density of 0.5 A g−1, a large energy density of 11.24 W h kg−1 at a power density of 400 W kg−1, and excellent cycle stability with a capacitance retention rate of 99.2% after 10000 cycles. The above-mentioned works indicate that it is highly feasible to construct superior porous carbon materials as supercapacitor electrodes adopting rich SS derived from soybean harvesting. Nevertheless, the three reaction steps are still tedious from the viewpoint of green chemistry, which is disadvantageous to shorten the handling period and reduce the cost for industrial mass production. Therefore, it is still a significant challenge to finely manipulate the pore size distribution and heteroatom doping level of SS-based porous carbon electrode materials through cost-effective, green and time-saving processing techniques, and further for application as superior supercapacitors with efficiently balanced energy density, specific capacitance, rate performance, and cycle stability.
As a common inorganic salt, KNO3 has been commonly employed as an oxidant or a pore-forming reagent during the pretreatment period of various biomass precursors.26–28 The critical roles of KNO3 in these works are summarized as follows: (1) after heating, KNO3 is strongly oxidative to suppress the dense stacking of the precursor due to cyclization, facilitating the sufficient penetration of the following activation reagent, which is beneficial for the formation of an ordered porous texture. (2) At high temperature, KNO3 easily undergoes violent redox reactions with amorphous carbon, leading to significant volume expansion of the precursor in a short time, which can greatly elevate the specific surface area of the obtained product. Due to these characteristics, biomass-derived carbon materials with KNO3 pretreatment commonly exhibit a superior capacitance performance compared to that without KNO3 pretreatment. Nevertheless, there are few works reporting the use of KNO3 as a post-treatment reagent in the activation step to date to the best of our knowledge.29–31 It can be reasonably predicted that the texture, heteroatom content, and the resulting relevant electrochemical performance of the obtained carbon materials will be greatly adjusted when employing KOH/KNO3 as a co-activator instead of single KOH because of the potential oxidative doping, violent volume expansion and enhanced etching effect. Moreover, the addition of suitable KNO3 to KOH as a co-activator does not require additional reaction steps or treatment process, and therefore it is simple, and meanwhile highly efficient, fully matching with the concept of green chemistry. Due to these considerations, we attempted to exploit a compact KOH/KNO3 co-activation method adopting widespread SS as the biomass precursor in this work. Employing a two-step process consisting of a carbonization reaction and gentle co-activated treatment at 800 °C, desirable porous carbon nanosheets (PCNs) were obtained with a large specific surface area (1658.8 m2 g−1), abundant hierarchical pore structures (mesopore and macropore ratio of 34.61%), and high oxygen doping level (16.77%). Notably, the doped oxygen element mainly consisted of hydrophilic carbonyl, carboxyl, and N–O functional groups, which promoted the electrode kinetics and ion migration rate in the aqueous electrolyte, meanwhile offering beneficial pseudocapacitance activity for greater charge storage.9,11 Therefore, our work provides a useful KOH/KNO3 co-activation method for the simple preparation of superior biomass-derived porous carbon materials with suitable hierarchical porous textures and high oxygen doping contents.
Experimental
Materials
Soybean straw (SS) was collected from a suburban area in Wuhan, China. KOH (99.9%) and KNO3 (99.9%) were purchased from Sinopharm Chemical Reagent Co., China. All chemical reagents and raw materials were used as received without any treatment unless otherwise specified.
Preparation of SS-derived PCNs
The SS (10 g) was pulverized, and then carbonized under an argon atmosphere at a temperature of 600 °C for 3 h to obtain the carbonized product (labeled as SSC) with the weight of 2.7 g. Following this step, SSC (2 g) was further grounded and mixed evenly with KOH and KNO3 in ionized water at the weight ratios of 1:2:0, 1:2:0.5, 1:2:0.6, and 1:2:0.7, respectively. The mixture was dried at 80 °C after reacting for 6 h, and then heated at a temperature of 800 °C for 3 h in a tubular furnace under an argon atmosphere. The resulting power was washed with 20 wt% HCl and deionized water until to neutrality, and then dried at 80 °C overnight to obtain the final products, which were labeled as SSC2-X (where X = 0, 0.5, 0.6, and 0.7 represent the weight ratio of SSC to KNO3, and 2 represents the mass ratio of SSC to KOH).
Material characterization
The morphology of the sample was characterized using a JSM-7100F field emission scanning electron microscope (FE-SEM) at the accelerating voltage of 5 kV and JEM-2010 transmission electron microscope (TEM) at the accelerating voltage of 200 kV. The element content of the sample was collected with an XPS-VG Multilab2000 X-ray photoelectron spectrometer. The Raman spectra of the sample were recorded with a LABRAM-HR Laser Raman spectrometer at a wavelength of 532 nm. The specific surface area and pore size distribution of the sample were analyzed using an Autosorb-iQ analyzer via N2 adsorption–desorption isotherms measured at room temperature.
Electrochemical measurement
In the three-electrode system, the working electrode was made as follows: the ink was firstly prepared by mixing 5 mg of SSC2-X (X = 0, 0.5, 0.6, and 0.7), 40 μL of Nafion solution (DuPont, China) with a resin solid content of 5 wt%, 400 μL of deionized water, and 400 μL of ethanol. Then, the working electrode was prepared by dropping the ink onto a glassy carbon electrode with a diameter of 3 mm. In a 6 M KOH solution, the electrochemical performance of the sample was tested using the three-electrode system with Pt foil as the counter electrode and Hg/HgO electrode as the reference electrode. The voltage windows for cyclic voltammetry and galvanostatic charge–discharge (GCD) testing were −1 to 0 V, while the frequency range for electrochemical impedance spectroscopy was 10−2 Hz to 103 kHz with an amplitude of 5 mV. A CHI760E electrochemical workstation (Chenhua, China) was used for the electrochemical performance measurements.
The specific capacitance (C, F g−1) of the working electrode was calculated using the GCD curve and eqn (1), as follows:
| | 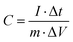 | (1) |
where
I (A) is the discharge current, Δ
t (s) is the discharge time, Δ
V (V) is the potential window, and
m (g) is the total mass of active substance on the working electrode.
In the two-electrode system, the symmetrical supercapacitor was made as follows: the biomass product, acetylene black, and PVDF binder were evenly mixed and ground in a mortar with the weight ratio of 8:1:1. Then, an appropriate amount of NMP solution was added into the mortar and stirred thoroughly to achieve a homogeneous slurry. After that, the slurry was uniformly coated on a foam nickel sheet (1 × 1 cm2) as the supporting substrate and dried in an oven at 80 °C for 8 h for use as the working electrode of the symmetrical supercapacitor. The volume density of the active materials in each electrode was about 703.5 mg cm−3. At last, the symmetrical supercapacitor was fabricated by placing the two foam nickel sheets with the same coatings on the two sides of a fibrous filter paper as the electrode separator. The button-shaped cell was assembled by adding 6 M KOH as the electrolyte, packing with a pair of 2032 stainless steel shells and pressing for 3 s at a pressure of 6 MPa.
The specific capacitance (Cs, F g−1) of the symmetrical supercapacitor was using the GCD curve and eqn (2), as follows:30
| | 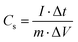 | (2) |
where
m (g) is the total mass of active substance in both electrodes,
I (A) is the discharge current, Δ
t (s) is the discharge time, and Δ
V (V) is the voltage window. The energy density (
E, W h kg
−1) and power density (
P, W kg
−1) of the symmetric supercapacitor were determined using
eqn (3) and (4), as follows:
| | 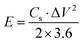 | (3) |
| | 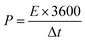 | (4) |
where
Cs (F g
−1) is the specific capacitance obtained from
eqn (2), Δ
V (V) is the voltage window during the discharge period, and Δ
t (s) is the discharge time.
Results and discussion
Morphology and pore structure
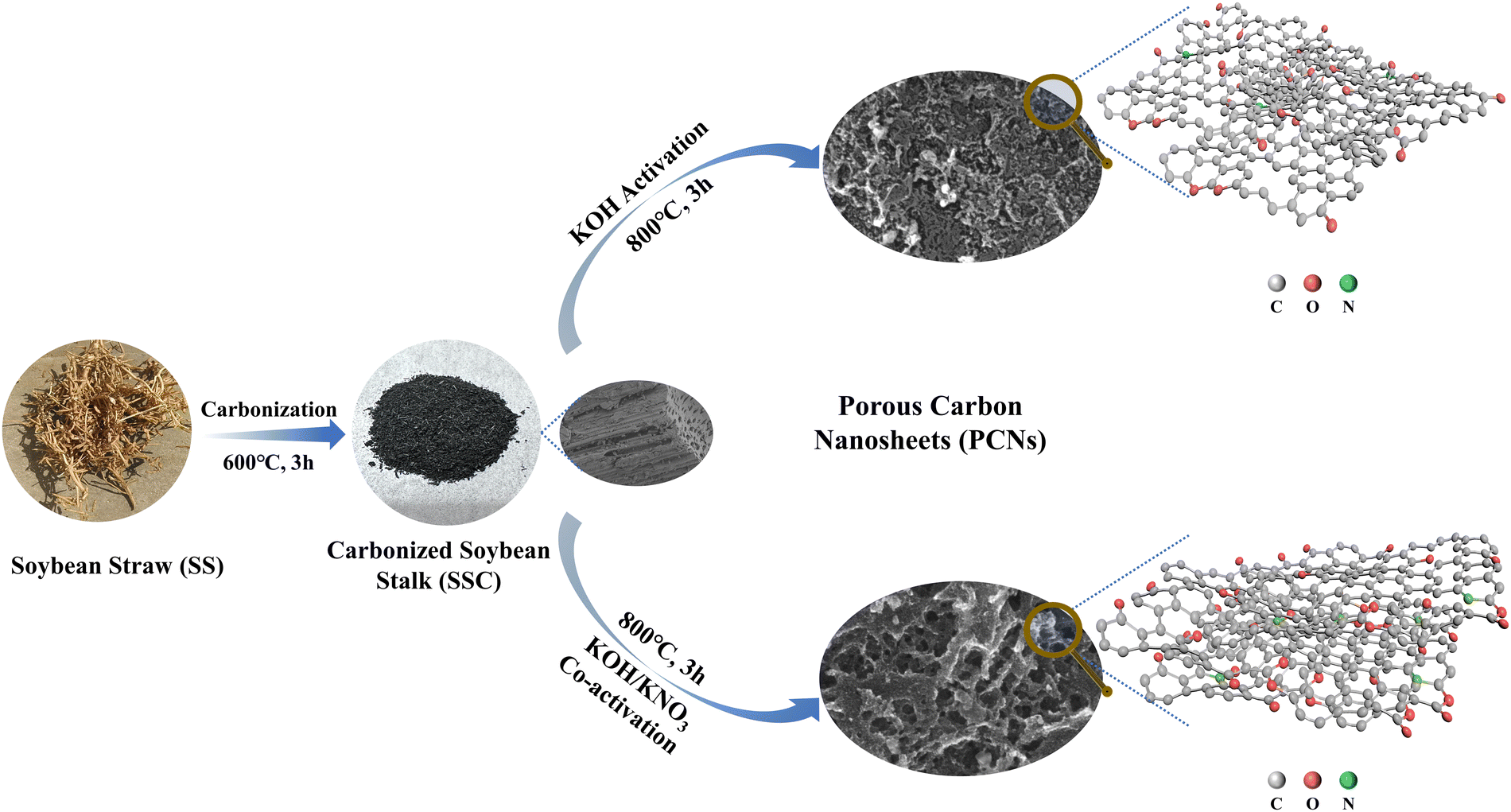
As shown in Scheme 1, a gentle two-step “carbonization-activation” method was employed to prepare porous carbon materials. The carbonized sample SSC (Fig. S1(a) and (b), ESI†) exhibited a close-stacked structure with intact macropore channels, which is probably caused by the natural templates within the SS.2 To improve the electrochemical activity of the carbonized product, chemical activation was further conducted. In general, the typical three-step treatment method was adopted for the biomass SS precursor with independent pyrolysis, carbonization and activation steps. Because of the shortened reaction steps in this work without the pyrolysis procedure, the strong oxidative KNO3 was introduced in the activation step for the purpose of strengthening the etching and activating effects. As calculated, the weight yield of the samples involving the two reaction steps was 16.74%, 14.58%, 13.50% and 12.96%, respectively. The reduced yield of the co-activated samples was caused by additional etching due to the introduction of KNO3. As can be observed in Fig. 1(a) and (b), the SSC2-0 sample activated with single KOH partially retained the compact texture inherent in the carbonized product; moreover, there were many protruding graphite sheets and a few irregular pore structures attached on its surface, which can be attributed to the concurrent catalysis and etching processes by KOH.15,16 In comparison, the SSC2-0.6 sample prepared with KOH/KNO3 co-activation showed a fluffy lamellar structure with interconnected pores. Therefore, the addition of KNO3 could significantly alter the morphology of the resultant activated carbon product. The reason for this is similar with previous works adopting KNO3 as a pretreating reagent. KNO3 reacts violently with unstable amorphous carbon after heat absorption, thus releasing a large amount of gas quickly for the huge volume expansion of the carbon lattice.28 Subsequently, the released K+ ions and oxidizing gases strongly etched the carbon framework at high temperature, and moreover the etching effect was promoted with an increase in the content of KNO3. To investigate the influence of the addition of KNO3 on the appearance of the carbonized product, co-activators with varying mass ratios of mKOH:mKNO3 = 1:0.5, 1:0.6, and 1:0.7 were used, and the corresponding samples were labeled as SSC2-0.5, SSC2-0.6, and SSC2-0.7, respectively. Both the density and the size of pores on the surface of the fluffy porous carbon materials were significantly enhanced with an increase in the content of KNO3 from 1:0.5 to 1:0.6 (Fig. S1(c) and (d), ESI†), respectively. However, with a further increase in the KNO3 content to 1:0.7, the amount of micropores decreased significantly, while the macropore size further expanded (Fig. S1(e) and (f) in the ESI†), respectively, which is likely due to the apparent pore collapse caused by excessive etching, as verified by the following BET analysis (Table 1). Therefore, the optimal KOH/KNO3 ratio in this study was 1:0.6. Moreover, it is obvious from the TEM imaging (Fig. 1(e) and (f)) that the SSC2-0.6 sample was composed of thin and textured nanosheets with size-dispersed nanoscale pores. Comparatively, the SSC2-0 sample was tight and lacked pores. The above-mentioned analysis indicates that the KOH/KNO3 co-activation is an effective method to prepare PCNs with abundant hierarchical porous structures.
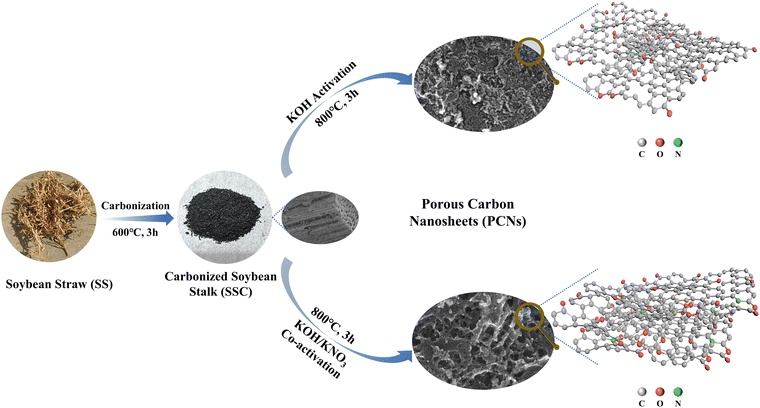 |
| | Scheme 1 Synthesis routes of the samples in this work. | |
 |
| | Fig. 1 (a) and (c) FE-SEM images of the SSC2-0 sample; (b) and (d) FE-SEM images of the SSC2-0.6 sample; (e) TEM image of the SSC2-0 sample; (f) TEM image of the SSC2-0.6 sample. | |
Table 1 Pore distribution characteristics of the samples
| Sample |
S
BET
(m2 g−1) |
Pore volume (cm3 g−1) |
V
n<2nm/VTot(%) |
V
n≥2nm/VTot(%) |
D
ave
(nm) |
|
V
Tot
|
V
n<2nm
|
V
n≥2nm
|
|
The specific surface area is calculated from the BET method.
The total pore volume is obtained at P/P0 = 0.99.
The micropore volume is calculated from the DFT method.
The mesopore and macropore volume of Vn≥2nm is calculated from VTot − Vn<2nm.
The average pore size is calculated by the BET method.
|
| SSC2-0 |
1158.43 |
0.55 |
0.45 |
0.10 |
81.79 |
18.21 |
1.89 |
| SSC2-0.5 |
1456.17 |
0.70 |
0.53 |
0.17 |
75.68 |
24.32 |
1.93 |
| SSC2-0.6 |
1658.80 |
0.84 |
0.55 |
0.29 |
65.39 |
34.61 |
2.02 |
| SSC2-0.7 |
1643.26 |
0.77 |
0.62 |
0.15 |
80.88 |
19.12 |
1.87 |
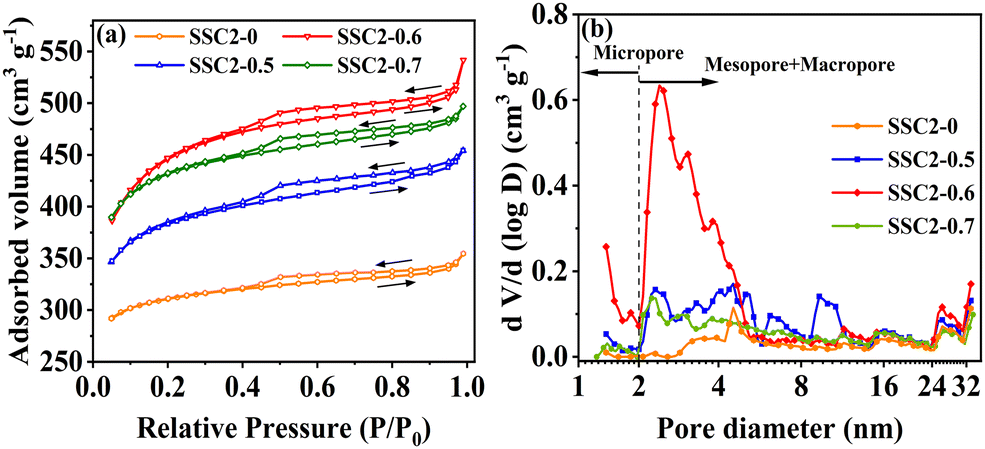
The specific surface area and pore characteristics of the samples were investigated by N2 adsorption–desorption isotherms. The carbonized sample SSC exhibited low N2 adsorption and a type II adsorption–desorption isotherm (Fig. S2, ESI†), implying the retained macropores in the initial SS precursor. In contrast, the KOH-activated sample (SSC2-0) showed completely different N2 adsorption–desorption properties (Fig. 2(a)). The total N2 adsorption amount not only increased significantly, but also the adsorption–desorption isotherm shifted to type IV, indicating the formation of abundant superficial micropores and mesopores. Moreover, the isotherm exhibited a type H4 hysteresis loop at relatively high pressures (0.4 < P/P0 < 0.99), indicating the presence of hierarchically porous structures.12,32 Similar to SSC2-0 activated with single KOH, the samples co-activated with KOH/KNO3 (SSC2-X (X = 0.5, 0.6, and 0.7)) also exhibited a hierarchical porous structure with type IV adsorption–desorption isotherm and type H4 hysteresis loop. Differently, the N2 adsorption amount at all the tested pressures increased remarkably after co-activation, and among them SSC2-0.6 showed the largest increment of more than 1.5 times because of its suitable KNO3 content. However, with a further increase in the KNO3 content (mKOH:mKNO3 = 1:0.7), the total N2 adsorption amount significantly decreased, indicating probable pore collapse because of the excessive KNO3, which is consistent with the morphology feature of the SEM image (Fig. S1(d), ESI†). Adopting the DFT model, the pore size distribution of the activated/co-activated samples was calculated. The varying trends of the samples are plotted in Fig. 2(b), and the calculated specific surface areas are listed in Table 1. Compared to single KOH activation (SSC2-0), the KOH/KNO3 co-activated samples (SSC2-X (X = 0.5, 0.6, and 0.7)) showed a significant increase in specific surface area, pore volume, and ratio of mesopores and macropores, implying the effective morphology regulating role of KNO3.27,28 It is noteworthy that SSC2-0.6 prepared with the optimal ratio of mKOH:mKNO3 = 1:0.6 in this work provided the largest specific surface area of 1658.80 m2 g−1, total pore volume of 0.84 cm3 g−1, considerable mesopore and macropore volume of 0.29 cm3 g−1, and average pore size of 2.02 nm, which are all outstanding for soybean biomass-derived carbon electrodes (as summarized in Table 2). Therefore, the KOH/KNO3 co-activation is superior to single KOH activation in terms of improving specific surface area and porosity for target carbon electrode materials. It is expected that the optimized structure can offer rich charge storage sites, and at the same time, favorable ion transport channels and buffering regions for supercapacitors.33
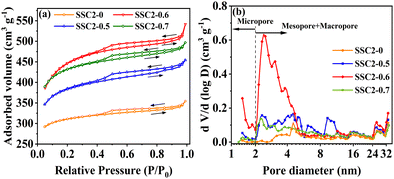 |
| | Fig. 2 (a) N2 adsorption–desorption isotherms. (b) Pore size distributions of the samples. | |
Table 2 Comparison of the performance of various soybean biomass-derived carbon electrode materials
| Biomass precursor |
Activation reagent |
Reaction steps |
Specific surface area (m2 g−1) & mesopore and macropore ratio (vol%) |
Heteroatom content (at%) |
Specific capacitance in three electrodes (F g−1@A g−1) |
Energy density (W h kg−1)@power density (W kg−1) |
Cycling stability |
Ref. |
| Soybean dregs |
KOH |
Two |
2313.3/— |
O (14.46)/N (2.07) |
321@1 (6 M KOH) |
25.9@859 (1 M Na2SO4) 9@490 (6 M KOH) |
90.8% (5000@5 A g−1) |
23
|
| Soybean stalk/urea/Ni(NO3)2 |
KOH |
Three |
2226.29/8.1 |
O (9.4)/N (5.08) |
407@0.5 (6 M KOH) |
36.11@517.8 (6 M KOH) |
87.5% (10000@10 A g−1) |
24
|
| Soybean straw/thiourea |
C6H5K3O7 |
Three |
1457.69/— |
O (14.7)/N (1.64)/S (0.53) |
220@0.5 (1 M KOH) |
11.24@400 (1 M KOH) |
99.2% (10000@5 A g−1) |
25
|
| Soybean straw |
KOH |
Two |
2012.62/— |
O (9.89)/N (1.7) |
380.5@0.5 (1 M KOH) |
8.95@25 (1 M KOH) |
73.97% (10000@10 A g−1) |
32
|
| Soybean straw |
KOH/KMnO4 |
Two |
348.59/— |
O (24.45)/Mn (22.52) |
482@0.5 (6 M KOH) |
14.5@250 (6 M KOH) |
99.5% (10000@10 A g−1) |
55
|
| Soybean protein |
ZnCl2 |
One |
1034.33/45 |
O (13.58)/N (6.37) |
191.15@0.1 (6 M KOH) |
8.74@8440 (6 M KOH) |
93.02% (10000@20 A g−1) |
56
|
| Soybean protein |
Zn(NO3)2·6H2O |
One |
1350/79 |
O (10.53)/N (5.63) |
248@0.5 (6 M KOH) |
15.75@200 (6 M KOH) |
90.61% (20000@30 A g−1) |
57
|
| Soybean shell |
KOH |
Two |
2802/15.4 |
O (10.64)/N (0.11) |
465@1 (6 M KOH) |
9.9@24 (PVA/6 M KOH) |
86.2% (10000@5 A g−1) |
58
|
| Soybean pods |
KOH |
Two |
1807.56/— |
O (9.86)/N (0.91) |
366.1@1 (6 M KOH) |
8.34@247.95 (PVA/KOH) |
96.03% (50000@5 A g−1) |
59
|
| Soybean protein |
Fe(NO3)3·9H2O/Zn(NO3)2·6H2O |
Two |
616.46/61 |
O (10.35)/N (3.99) |
271@0.2 (6 M KOH) |
16.83@199.36 (6 M KOH) |
88.14% (10000@2 A g−1) |
60
|
| Soybean straw |
KOH/KNO3 |
Two |
1658.8/34.6 |
O (16.77)/N (2.35) |
438@1 (6 M KOH) |
14.89@249.44 (6 M KOH) |
96.94% (16000@10 A g−1) |
This Work |
Microstructure and surface chemical property
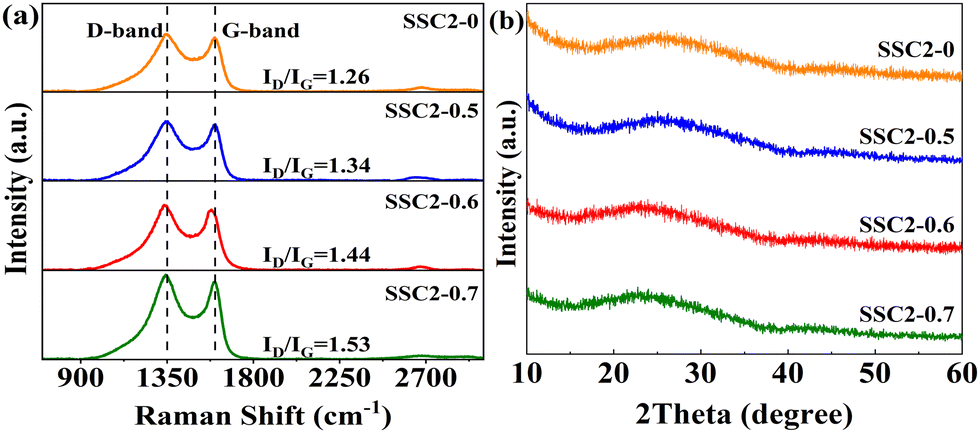
Fig. 3(a) shows the Raman spectra of the samples prepared with different activator ratios (SSC2-X (X = 0, 0.5, 0.6, 0.7)). All the samples exhibited two distinct peaks at approximately 1350 cm−1 and 1600 cm−1, corresponding to the D-band and G-band of obtained carbon materials, respectively. In general, the D-band is caused by the breathing modes of sp2 carbon atoms within the rings, which directly verifies the presence of defective carbon. Alternatively, the G-band originates from the stretching vibrations of the sp2 carbon atom pairs, which represents the degree of graphitization of the carbon lattice.34,35 As a result, the higher intensity ratio of the D-band to G-band (ID/IG), the larger the degree of defects. The calculated ID/IG for the four samples is 0.96, 1.34, 1.44, and 1.53, respectively. The degree of surface defects increased significantly with the introduction of KNO3 and continuously improved with the accumulation of KNO3. This is consistent with the results of the BET analysis, which suggests that KNO3 not only caused significant volume expansion in the co-activated products but also introduced abundant pores as defects. Moreover, no obvious 2D-band peak was observed at about 2700 cm−1 in the Raman spectra of the samples, implying that the prepared carbon materials were mainly defect-enriched few-layered PCNs. This structural feature is beneficial to achieve a high specific surface area and provide abundant active sites for charge storage. In addition, the X-ray diffraction (XRD) patterns of the samples (Fig. 3(b)) exhibit a broad (002) peak together with faint (100) and (101) peaks, indicating the amorphous feature of the obtained carbon materials. In comparison to the SSC2-0 sample, the (002) peaks of the three co-activated samples shifted slightly towards lower 2θ angles (26.1° vs. 25.0°, 23.9°, and 22.7°), reflecting an enlarged interlayer distance in the graphite structure. Applying the Bragg equation (2d002sinθ = nλ) to the XRD spectra, the average (002) interlayer distances for SSC2-0, SSC2-0.5, SSC2-0.6, and SSC2-0.7 were calculated to be 0.34 nm, 0.36 nm, 0.37 nm, and 0.39 nm, respectively.12,22 The larger interlayer distances of the three co-activated samples are attributed to the abundant nucleation sites because of the explosive effect of the KNO3 reagent,26,28 and the loose structure caused by the large interlayer distance probably contributed to the higher capacitance performance.
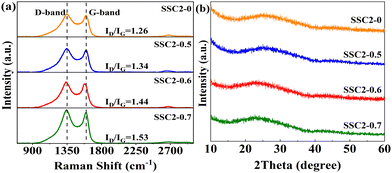 |
| | Fig. 3 (a) Raman spectra and (b) XRD patterns. | |
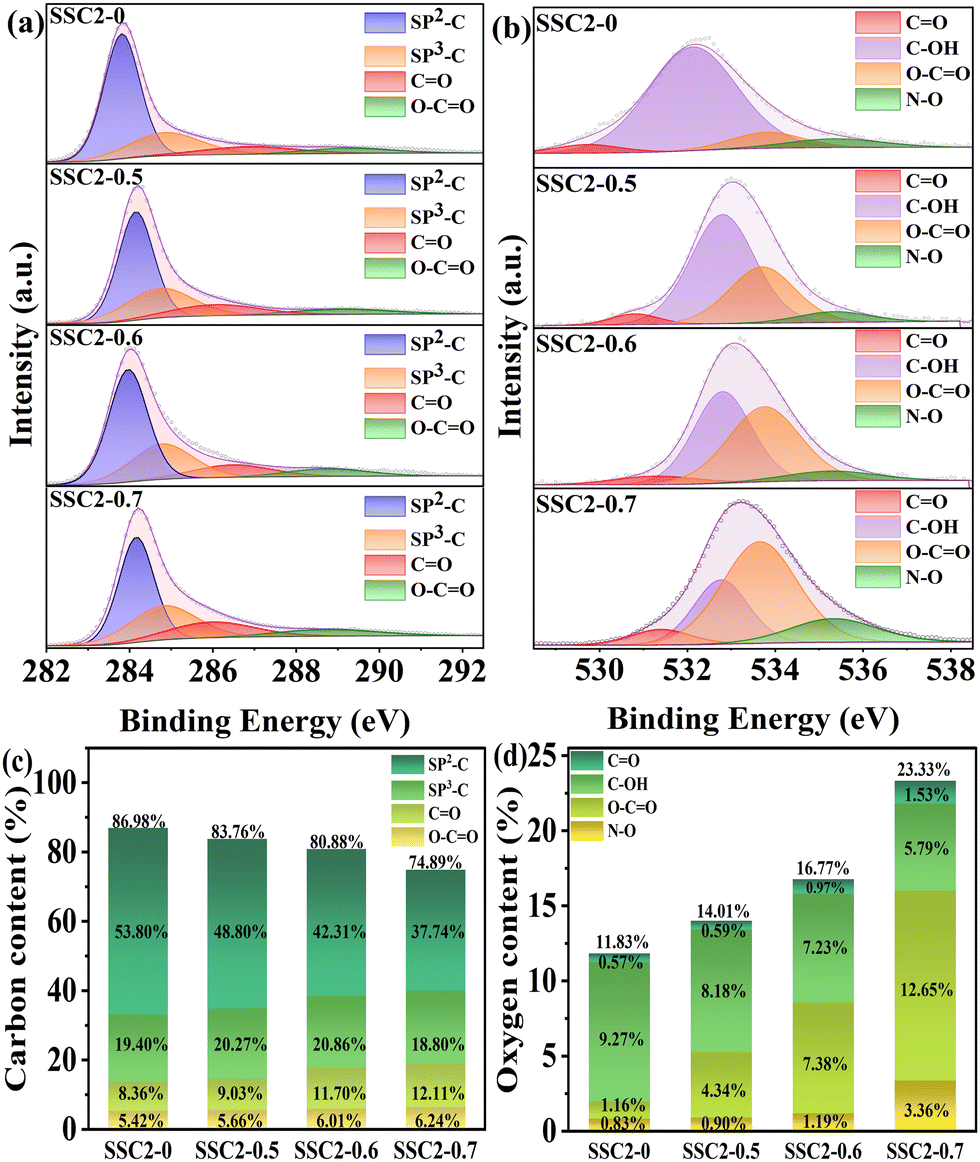
XPS measurement was employed to further investigate the effect of different KNO3 contents on the chemical composition and functional groups attached to the surface of the samples. As shown in Fig. S3 (ESI†), all the samples exhibited intense emitting peaks at approximately 284 eV and 532 eV, corresponding to the C and O elements, respectively. Additionally, a weak N characteristic peak was detected at about 400 eV. The elemental contents of the samples are also summarized in Table S1 (ESI†), which indicate that the O content was continuously enhanced with an increase in the content of KNO3, implying the significant oxidative doping effect of KNO3. In the case of SSC2-0.6 with the highest specific surface area and total pore volume, its O and N contents remained as high as 16.77 at% and 2.35 at%, respectively. To deeply understand the doping role of KNO3, the full spectra of C 1s and O 1s were further analyzed. Fig. 4(a) and (c) depict the deconvoluted C 1s spectra and relevant statistical diagrams of the C-containing functional groups. The spectra could be split into four types of functional groups including sp2-C, sp3-C, CO, and O–CO, corresponding to the peaks located at 283.98 eV, 284.80 eV, 286.11 eV, and 288.76 eV, respectively.8,36 Among them, sp2-C and sp3-C individually indicate basal plane carbon and defective carbon, respectively, while CO refers to carbonyl, ketone, or aldehyde; and O–CO represents carboxyl, carboxylic anhydride, or ester.9,10 Compared to SSC2-0, the three co-activated samples (SSC2-X (X = 0.5, 0.6, and 0.7)) exhibited significantly increased sp3-C, CO and O–CO contents together with a remarkable reduction in sp2-C moieties, which can be attributed to the breaking of the chemical bond in the carbon lattice caused by the rapid volume explosion and strong oxidative etching effect originating from KNO3. This explanation can also be supported by continuously decreased sp2-C moieties accompanied with the addition of KNO3 and the above-mentioned Raman spectra analysis. Furthermore, the high-resolution O 1s spectra (Fig. 4(b) and (d)) indicate that a conversion process occurred from the carbon–oxygen single bonds into carbon–oxygen double bonds due to their strong oxidation by KNO3.37 In addition, the introduction of N–O functional groups originated from both the self-doping of SS and KNO3 doping. The previous investigations suggested that quinone-type CO bonds and N–O functional groups can provide effective pseudocapacitance in alkaline electrolyte due to their relatively high redox activity in comparison to other O-containing functional groups.8,10
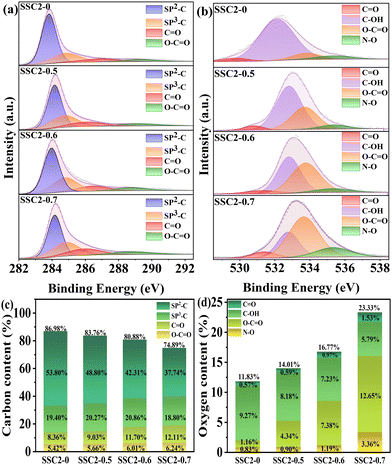 |
| | Fig. 4 (a) C 1s XPS peaks and (b) O 1s XPS peaks. Contents of the functional groups of (c) carbon and (d) oxygen. | |
Based on the above-mentioned analysis, the possible benefits of the KOH/KNO3 co-activation as an alternatively to single KOH activation can be summarized as follows: (1) the rapid volume expansion of the precursor in the initial activation stage caused by the explosive KNO3, allowing the uniform distribution of the activators to create a well-ordered hierarchical porous structure with more pores, and simultaneously enhanced mesopore and macropore ratio, resulting in abundant charge storage sites, meanwhile accelerating the ion transfer kinetics. (2) The strong oxidative doping effects of KNO3 promote the production of quinone-type CO and N–O functional groups for reversible charge storage, as well as hydrophilic carboxyl and ester functional groups to reduce the electrode/electrolyte interface resistance. The role of the KNO3 was further verified via the following electrochemical performance measurements.
Electrochemical performance in three-electrode system
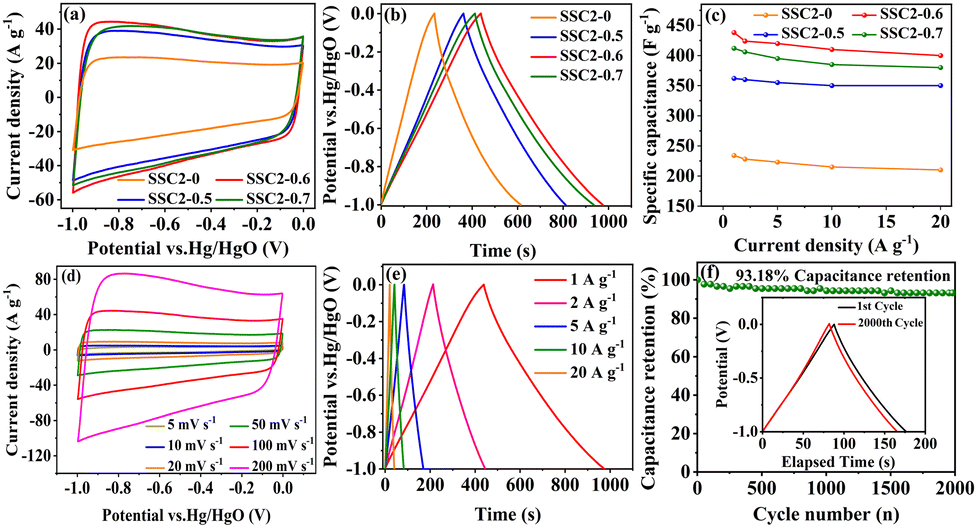
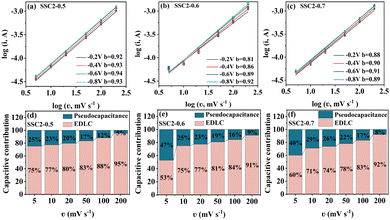
The CV curves of the activated/co-activated samples (SSC2-X (X = 0, 0.5, 0.6, and 0.7)) in 6 M KOH electrolyte are shown in Fig. 5(a). All the curves exhibited a quasi-rectangular shape at a large scanning rate of 100 mV s−1, reflecting their good EDLC behavior.38 Moreover, two broad potential peaks appeared in the range of −1 V to −0.5 V, suggesting concurrent pseudocapacitance behavior caused by the abundant oxygen-containing functional groups in the defective sites of the graphitic plane.39 The integral area of the CV curve can represent the amount of stored charge,40 and it is clear that the retained charges of the KOH/KNO3 co-activated samples (SSC2-X (X = 0.5, 0.6, and 0.7)) were much higher than that of the sample with single KOH activation (SSC2-0), and the highest charge capacitance was achieved for the SSC2-0.6 with the integral area of 360.0 C g−1, which can be attributed to its largest specific surface area, optimal pore distribution, and abundant oxygen-containing functional groups. Therefore, the addition of KNO3 can simultaneously contribute both the EDLC and the pseudocapacitance.41 The GCD curves of the samples at a current density of 1 A g−1 are shown in Fig. 5(b), and all curves exhibit highly symmetrical isosceles triangle shapes, indicating their excellent charge–discharge reversibility.42 According to the GCD curves, the specific capacitance, Cs, of the SSC2-X samples (X = 0, 0.5, 0.6, and 0.7) is 234, 362, 438, and 412 F g−1, respectively, apparently demonstrating that the Cs of the KOH/KNO3 co-activated samples is much higher than that of the single KOH activated sample (SSC2-0). With the addition of KNO3, the increased specific surface area created abundant active sites and the expanded porosity provided sufficient ion diffuse and buffered spaces, thereby promoting the specific capacitance.43 However, the excessive KNO3 in SSC2-0.7 caused over-etching toward the graphitic planes, leading to their undesirable pore collapse, thus suppressing the specific surface area and porosity (Table 1), reducing the number of active sites and accumulative ion traps.44 Moreover, suitable oxygen-containing functional groups on the surface can promote the desolvation process of ions through solvent adsorption effect as well, while excessive oxygen-containing functional groups (SSC2-0.7) may block the pore channels, leading to an increase in ion transport resistance.8,45 Consequently, It can be reasonably predicted that SSC2-0.6 has the optimal amount of oxygen-containing functional groups with the oxygen elemental ratio of 16.77 at%. We further investigated the charge–discharge rate performance of the samples (SSC2-X (X = 0, 0.5, 0.6, and 0.7)), and the specific capacitance at different current densities ranging from 1 A g−1 to 20 A g−1 is shown in Fig. 5(c). At a high current density of 20 A g−1, the capacitance retention rates of the samples are 89.74%, 96.69%, 91.32%, and 92.23%, respectively; and among them, the SSC2-0.6 also maintained the highest specific capacitance of 400 F g−1, verifying its superior hierarchical pore structure and rational distribution of oxygen-containing functional groups. Moreover, the CV curves of the SSC2-0.6 sample at different scanning rates (Fig. 5(d)) maintained a quasi-rectangular shape and the GCD curves at different current densities (Fig. 5(e)) kept a similar isosceles triangle at different current densities, demonstrating its excellent rate performance. Fig. 5(f) records the capacitance retaining rates of the SSC2-0.6 sample under continuous GCD cycles compared to the first cycle, where the rate was maintained at 93.18% (410 F g−1) after 2000 cycles and the GCD curve approximately kept the same shape as the first cycle (the inset of Fig. 5(f)), suggesting its good GCD stability.20,46 Therefore, the working electrode fabricated with the SSC2-0.6 sample exhibited an excellent electrochemical performance involving specific capacitance, rate performance, and cycle stability due to its suitable KNO3 content.
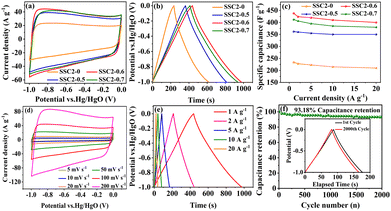 |
| | Fig. 5 (a) CV curves at 100 mV s−1; (b) GCD curves at 1 A g−1; (c) specific capacitance at different current densities; (d) CV curves of the SSC2-0.6 at different scanning rates; (e) GCD curves of the SSC2-0.6 under varying charging current densities; and (f) capacitance retention rate of SSC2-0.6 after 2000 GCD cycles at 5 A g−1 (the inset is the GCD curves for the 1st and 2000th cycles). | |
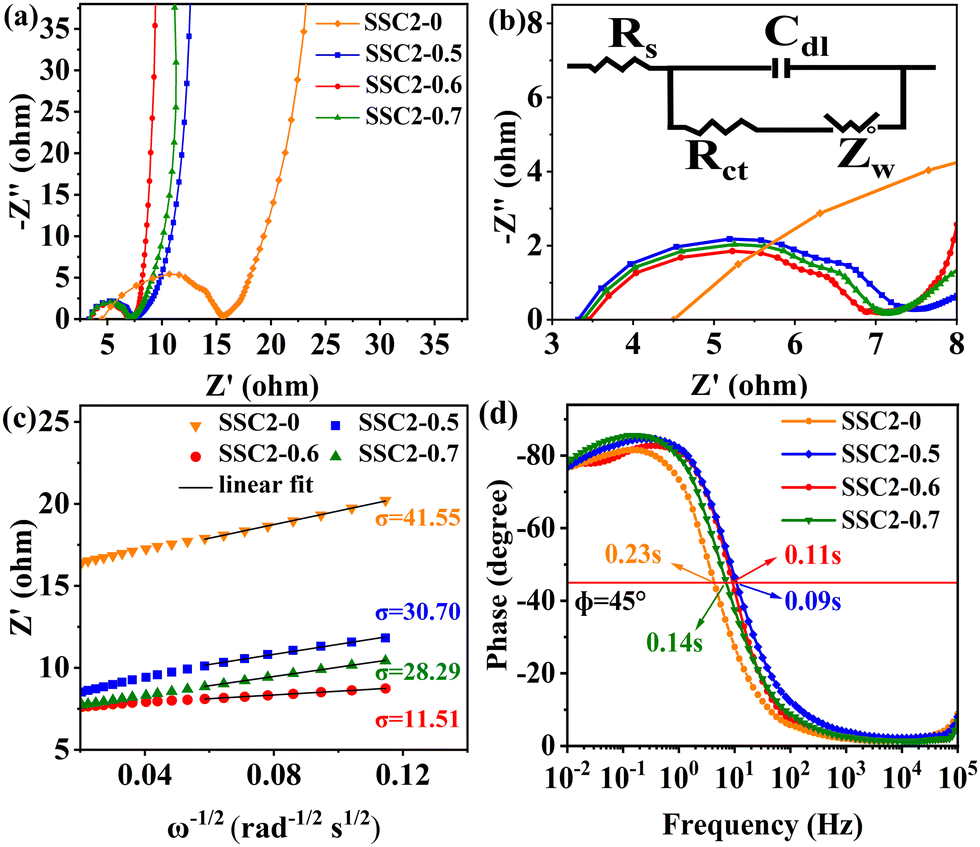
To investigate the electrochemical kinetic behavior of the obtained electrode materials, their AC impedance spectra were measured in the frequency range of 10−2 Hz to 103 kHz at the potential amplitude of 5 mV. As shown in the Nyquist plots (Fig. 6(a) and (b)) of the samples, the linear curves in the low-frequency region represents the Warburg impedance (Zw) determined by the mass transfer process, while the semi-circular curves in the high-frequency region is the charge transfer resistance (Rct), which is related to the superficial functional groups and as well as internal structural characteristics. It is obvious that the KNO3/KOH co-activated samples have linear slopes close to 90° in the low-frequency region, which is very different from the SSC2-0 sample with a slope approaching 45°, implying its better EDLC behavior due to the co-activation treatment. The measured resistance spectra were further fitted into the equivalent circuits using the Zview software, as shown in the inset of Fig. 6(b), and the values of the fitted Rct, Zw and Rs (the ionic resistance of electrolyte) are summarized in Table S3 (ESI†). The results indicate that the Rct for the samples is 10.73 Ω, 4.13 Ω, 3.40 Ω, and 3.84 Ω, respectively, demonstrating a significant reduction in Rct after the addition of KNO3 as the activator to create abundant pore channels for ion diffusion and buffering, resulting in an accelerated charge transfer rate. Additionally, the Rs of the co-activated samples (3.19–3.63 Ω) was also significantly reduced compared to that of SSC2-0 (4.57 Ω), reflecting the largely suppressed ion contact resistance between the electrode surface and electrolyte interface, which can be attributed to the favorable electrolyte permeation due to the accumulation of hydrophilic oxygen-containing functional groups.6 Comprehensively, the working electrode fabricated with SSC2-0.6 exhibited a superior electrode kinetic process including mass transfer, charge diffusion and electrolyte transport due to the optimization of its hierarchical pore structure and weakened ion transfer impedance. Furthermore, the diffusion kinetics of the electrode can be described by the diffusion coefficient, D, which was calculated using eqn (5) and (6), as follows:21
| | 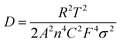 | (5) |
| | 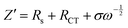 | (6) |
where
R is the thermodynamic constant,
T is the thermodynamic temperature,
A is the effective area of the electrode,
n is the number of electrons,
C is the ion concentration,
F is the Faraday constant,
σ is the Warburg factor, and
ω is the angular frequency in the low-frequency region. According to
eqn (5) and (6), the Warburg factor
σ is the reverse of the diffusion coefficient
D. The Warburg factors of the samples were calculated
via linear fitting (
Fig. 6(c)), which showed that SSC2-0.6 has the lowest
σ value (11.51), corresponding to the largest
D among the samples. This result is consistent with the impedance analysis, indicating the excellent hierarchical porous structure of the SSC2-0.6.
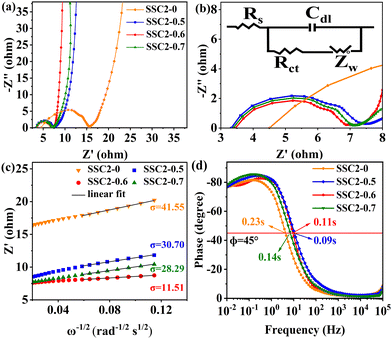 |
| | Fig. 6 (a) Nyquist plots; (b) enlarged view in the high-frequency region and the corresponding equivalent circuit model in the inset; (c) relationships between Z′ and ω−1/2; and (d) Bode plots. | |
To inspect the ion transfer kinetics of the electrode, the relationship between frequency and phase angle was established (Fig. 6(c)) to obtain the Bode plots (Fig. 6(d)). It can be observed that at the low limit of 10−2 Hz, the phase angles of all the samples were below 90° (76.6°–78.1°), suggesting dominant pseudocapacitance behavior. The characteristic frequency at the 45° phase angle reflects the transition speed of the electrode from resistive behavior to capacitive behavior.47 Thus, the response time, τ0, can be calculated using the formula τ0 = 1/f0, where a low response time represents a fast ion transfer rate.48 The τ0 of the four samples is 0.23 s, 0.09 s, 0.11 s, and 0.14 s, respectively, indicating a significantly reduced response time of the co-activated samples due to their abundant hierarchical pore structure and large specific surface area, which allows the electrolyte to quickly access the electrode surface. Therefore, the electrode fabricated with the SSC2-0.6 sample has a beneficial diffusion coefficient and low response time, providing a fast kinetic route for electrolyte ions.
To verify the charge storage mechanism of the samples, their electrochemical behaviors were investigated using Dunn's power law equation. In general, there are two charge storage mechanisms in the working electrode, i.e., one is capacitance-controlled static adsorption–desorption process of electrolyte ions on the surface of the electrode, which corresponds to EDLC behavior; the other is a diffusion-controlled redox reaction process occurring at the electrode/electrolyte interface, corresponding to pseudocapacitance behavior.49 By fitting the CV curves of the working electrode at different scanning rates, the charge storage behavior was analyzed using eqn (7) and (8), as follows:3
where
a and
b are variable parameters, and
v is the scanning rate. Thus, the slope of the curve of log
i vs. log
v can be used to calculate the
b value. Typically, when
b = 1, it indicates that the energy storage is mainly capacitive; when
b = 0.5, dominant diffusive behavior is implied; and when
b is between 0.5 and 1, it suggests that the charge storage is a mixed kinetic-controlled process.
7 According to the calculation, the
b values for all samples are between 0.5 and 1 (
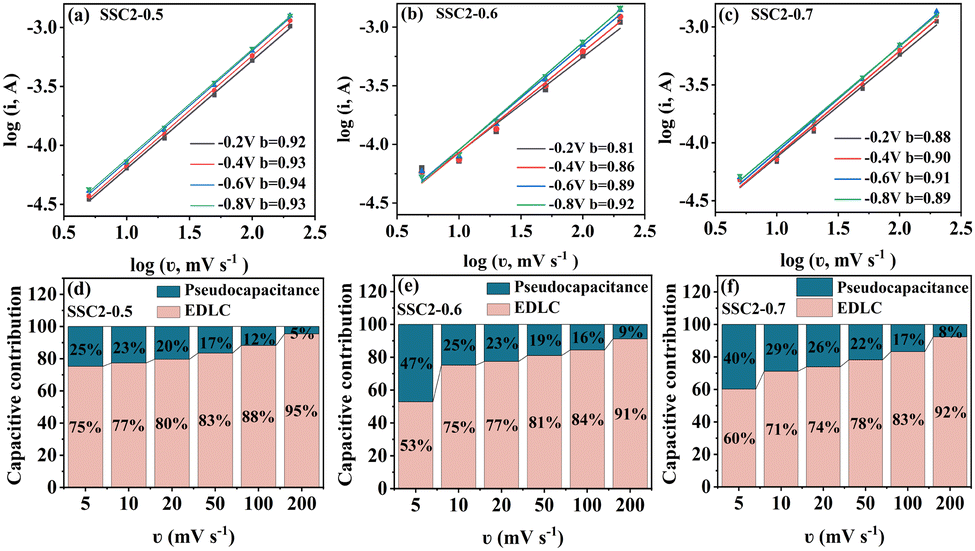
Fig. 7(a)–(c)), demonstrating that the charge storage is double controlled by both capacitance and diffusive processes. Moreover, the dominant capacitance process is deduced because the
b value is closer to 1. Specifically, the equation from Dunn and colleagues was employed to calculate the proportion of capacitance behavior (
k1v) and diffusion behavior (
k2v1/2) involving the total stored charges, as follows:
37| | 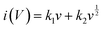 | (9) |
where
k1 and
k2 are variable parameters. The ratios of the EDLC (capacitive) to the pseudocapacitance (diffusive) contribution in the total stored charges at different scanning rates are plotted in
Fig. 7(d)–(f). The portion of diffusion-determining capacitance decreased with an increase in the scan rate, implying the faster charge transfer rate of the EDLC process. For the three samples at the same scanning rate, the portion of the pseudocapacitance improved with an increase in the heteroatom content, which is consistent with the trend of KNO
3 content and resultant oxygen atom doping level due to the close contribution of reactive oxygen-containing functional groups containing quinone-type C
O bonds and N–O moieties. Specifically, the absolute values of EDLC capacitance for the three co-activated samples at the same scanning speed of 50 mV s
−1 are 280.2 F g
−1, 302.2 F g
−1 and 282.5 F g
−1, while that of the pseudocapacitance are 55.7 F g
−1, 70.7 F g
−1 and 79.1 F g
−1, respectively. The results imply that about 20% of the total stored charges for the co-activated samples originated from the pseudocapacitance behaviour. Given that the EDLC is mainly dominated by the porosity, while the pseudocapacitance is mainly determined by the amount of active oxygen-containing functional groups, as analyzed above (
Fig. 2 and 4(c) and (d)), the SSC2-0.6 sample possessed a higher pseudocapacitance ratio at various scanning rates compared to the SSC2-0.5 sample because of its relatively large reactive oxygen content. Therefore, comprehensively considering the porosity and active oxygen-containing functional groups, the SSC2-0.6 sample obtained with moderate KOH/KNO
3 activation provided the largest specific capacitance.
 |
| | Fig. 7 (a)–(c) Curves of logi vs. logv for the co-activated samples at different scanning rates. (d)–(f) Contribution ratios of the EDLC and the pseudocapacitance at different scanning rates. | |
Electrochemical performance in symmetric supercapacitor
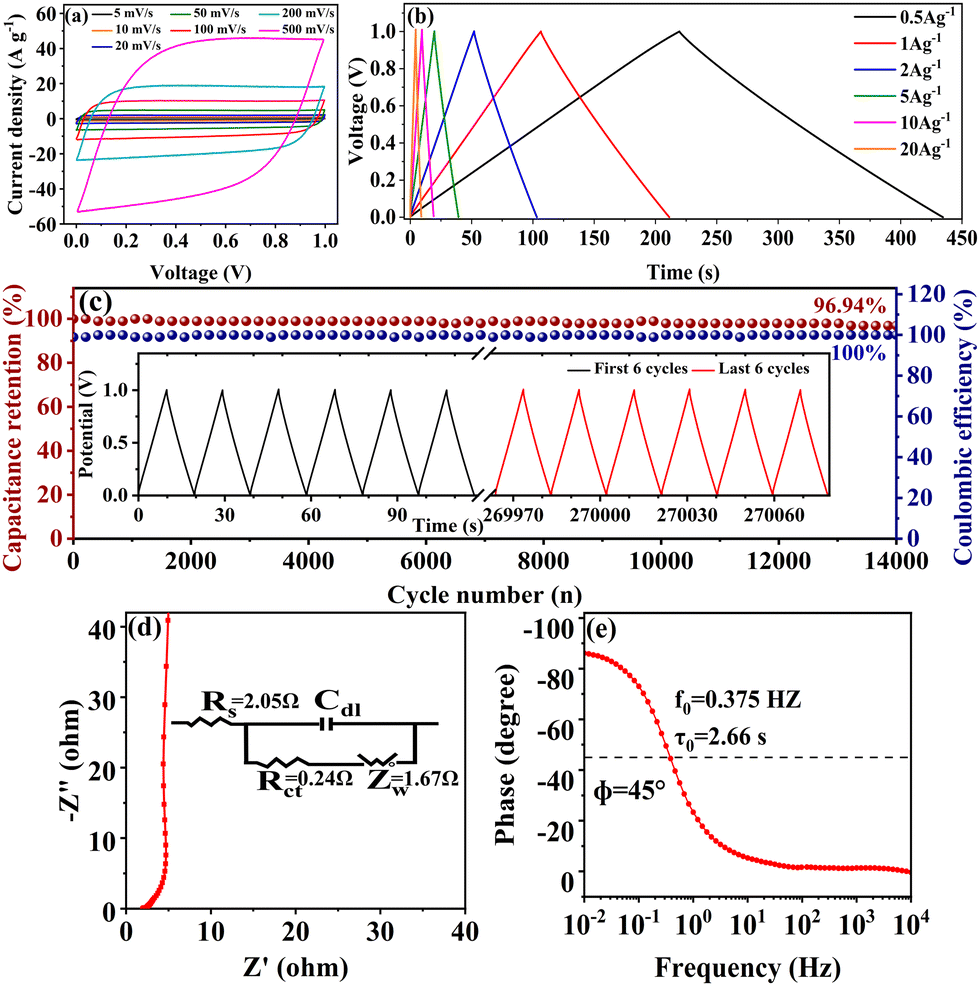
To verify the practical application of the co-activated samples, a symmetric supercapacitor with two-electrode system was fabricated using foam nickel as the electrode substrate and 6 M KOH as the electrolyte. Considering the collective impacts of the electrode material and the electrolyte stability, the potential window of −1 V to 0 V was applied on the working electrode as commonly adopted in previous works.50–52 Different from the three-electrode system with a negative potential window input on the working electrode in relation to the counter electrode, the absolute value of the potential difference between the positive electrode and the negative electrode was recorded as the potential window in the two-electrode system with a symmetric supercapacitor device.53 As shown in Fig. S4(a) (ESI†), the CV curves of the symmetric supercapacitors fabricated with the three co-activated samples SSC2-X (X = 0.5, 0.6 and 0.7) at the scan rate of 100 mV s−1 exhibited a quasi-rectangular shape, suggesting nearly ideal charge–discharge behavior as the three-electrode systems. The small hump at the potential of 0.2 V is probably caused by the pseudocapacitance, as analyzed above. The GCD curves at a current density of 1 A g−1 also showed a symmetrical triangular shape (Fig. S4(b), ESI†), demonstrating excellent charge–discharge reversibility. According to the discharge curves, the specific capacitances of the three supercapacitors are 82.3 F g−1, 104.7 F g−1 and 87.8 F g−1, respectively, which are consistent with the trend in the three-electrode testing and indicate the presence of beneficial hierarchical pore structures and oxygen-containing functional groups in the co-activated samples. Due to its superior performance among the three samples, the supercapacitor fabricated with SSC2-0.6 was further investigated. The CV curves of the sample at wide scanning rates (5–500 mV s−1) are shown in Fig. S4(c) (ESI†), where it is noteworthy that even at an ultra-high scanning rate of 200 mV s−1, the curve could still maintain an approximately rectangular shape and only had slight distortion with a further increase in the scanning rate to 500 mV s−1. This testing suggests the excellent capacitance retention capacity of SSC2-0.6 to offer fast ion migration even at ultra-high scan rates. Fig. 8(a) depicts the GCD curves of SSC2-0.6 at wide current densities from 0.5 A g−1 to 20 A g−1, and it can be observed that the curve maintained a high linearity and symmetrical triangular shape with a capacitance retention rate of 85.58% at the large current density of 20 A g−1, further demonstrating its excellent rate performance. In addition, it exhibited a relatively large specific capacitance at all the tested current densities (Fig. 8(b)), which is consistent with the three-electrode testing results. The capacitance retention of the supercapacitor under continuous GCD cycling is depicted in Fig. 8(c), which demonstrates that after 14000 cycles at a large current density of 10 A g−1, the supercapacitor still had a high capacitance retention rate of 96.94%; meanwhile, with a nearly ideal Coulombic efficiency of 100% and a highly symmetrical triangular shape (the inset of Fig. 8(c)), suggesting its excellent cycling stability. The Nyquist plot and the corresponding equivalent circuit model (the inset) of the supercapacitor fabricated with SSC2-0.6 are shown in Fig. 8(d). It also exhibits a small semi-circle with a low Rs of 2.05 Ω and Rct of 0.24 Ω in the high-frequency region, while a straight line with the slope close to 90° in the low-frequency region, with a Zw of 1.67 Ω, implying favorable EDLC behavior with fast ion migration similar that in the three-electrode system. Furthermore, the Bode plot of the supercapacitor (Fig. 8(e)) indicates that the calculated f0 is 1.84 Hz at a phase angle of 45°, which corresponds to the relaxation time of 0.54 s. The short relaxation time suggests that the supercapacitor has a rapid kinetic response characteristic.54
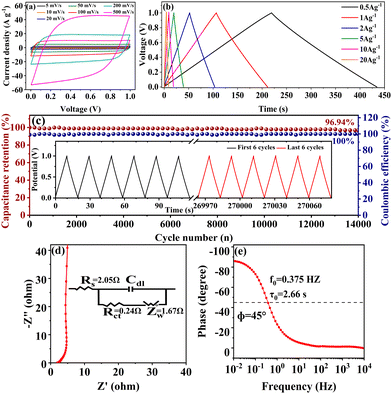 |
| | Fig. 8 Capacitive properties of the symmetric supercapacitor with the SSC2-0.6 electrode: (a) CV curves at different scan rates; (b) GCD curves of at different current densities; (c) the stability of capacitance retention with 14000 cycles at a current density of 10 A g−1 (the inset is the GCD curves of the first 6 cycles and the last 6 cycles before and after continuous cycling); (d) the Nyquist plot (the inset shows the corresponding equivalent circuit model); (e) the Bode plot. | |
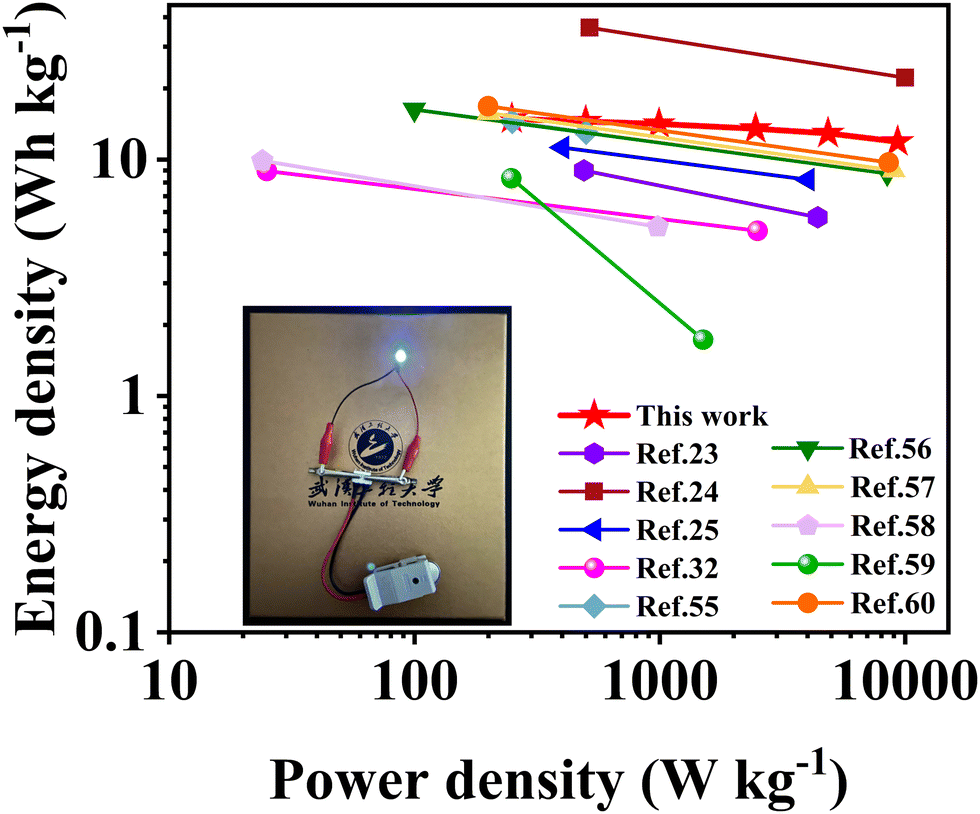
Energy density is a key performance parameter for electrochemical supercapacitors, which has become a bottleneck limiting their development at present. Fig. 9 shows the Ragone plots of the symmetric supercapacitors in alkaline KOH electrolyte fabricated with SSC2-0.6 and other soybean biomass-derived carbon electrode materials reported in the last three years. The supercapacitor fabricated in this work exhibited a high energy density of 14.89 W h kg−1 at a power density of 249.44 W kg−1; even at a large power density of 9313.04 W kg−1, it could provide an outstanding energy density of 11.90 W h kg−1. Compared to the other biomass-based carbon materials without transition metal atoms (Table 2), large specific capacitance, excellent energy density and good cycle stability of SSC2-0.6 can be attributed to its large specific surface area, high mesopore and macropore ratio and abundant oxygen-containing functional groups, indicating the advantage of the KOH/KNO3 co-activation method proposed in this work.
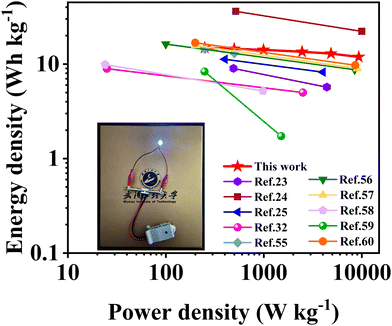 |
| | Fig. 9 Ragone plots of the symmetric supercapacitor fabricated with the SSC2-0.6 electrode in this work and the other soybean biomass-derived carbon electrode materials reported in the literature. | |
Conclusions
In this work, we reported a novel KOH/KNO3 co-activation method to treat waste biomass of soybean straw. The hierarchical porous nanosheets were obtained with a large specific surface area of 1658.8 m2 g−1, high mesopore and macropore ratio of 34.61% and considerable oxygen doping content of 16.77 at%. The critical role of KNO3 was also verified. Structurally, it could improve the specific surface area, increase the mesopore and macropore ratio and promote the uniform etching of the activator. Compositionally, it could introduce abundant quinone-like CO and C–N functional groups with high pseudocapacitive activity, as well as hydrophilic carbonyl and ester groups for efficient ion transport. Due to these advantages, the obtained electrode material exhibited a significant specific capacitance of 438 F g−1 at the scanning rate of 1 A s−1 and capacitance retention rate of 91.32% at the current density of 20 A g−1 in a three-electrode system. Moreover, the symmetric supercapacitor provided a high energy density of 14.89 W h kg−1 at a power density of 249.44 W kg−1, an energy density of 11.90 W h kg−1 even at an ultrahigh power density of 9313.04 W kg−1, and excellent stability with the capacitance retention rate of 96.94% after 14000 cycles. This work provides a compact KOH/KNO3 co-activation method as an alternative to the common single KOH activation method to effectively transform waste biomass resources into high-value electrode materials for supercapacitors.
Author contributions
Conceptualization: S. W. and Z. D.; data curation: Y. L.; formal analysis: S. W., Z. D., Y. L., and C. K.; funding acquisition: S. W., Z. D. and J. Z.; investigation: Y. L. and C. K.; methodology: S. W., C. K. and Y. L.; project administration: S. W., Z. D. and J. Z.; resources: Z. D., C. K., and Y. L.; software: C. K., J.-W. Z., C. L. and Y. J.; supervision: S. W. and Z. D.; validation: Y. L. and C. K.; visualization: Z. D. and Y. L.; writing – original draft: Y. L.; writing – review & editing: Z. D., X. Q., J. Z., J.-W. Z., C. L. and Y. J. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of China (no. 51272187). Z. D. acknowledges the financial support by PhD research startup foundation of Hubei University of Automotive Technology (BK202351). J. Z. acknowledges the financial support by the Open Fund of the Hubei Key Laboratory of Automotive Power Train and Electronic Control, Hubei University of Automotive Technology (ZDK1201902).
Notes and references
- L. Zheng, X. Dai, Y. Ouyang, Y. Chen and X. Wang, J. Energy Storage, 2021, 33, 102152 CrossRef.
- P. Manasa, S. Sambasivam and F. Ran, J. Energy Storage, 2022, 54, 105290 CrossRef.
- M. Jalalah, H. Han, M. Mahadani, A. K. Nayak and F. A. Harraz, J. Electroanal. Chem., 2023, 935, 117355 CrossRef CAS.
- W. Yang, W. Yang, L. Kong, A. Song, X. Qin and G. Shao, Carbon, 2018, 127, 557–567 CrossRef CAS.
- F. Chen, J. Yang, T. Bai, B. Long and X. Zhou, Electrochim. Acta, 2016, 192, 99–109 CrossRef CAS.
- L. Miao, D. Zhu, M. Liu, H. Duan, Z. Wang, Y. Lv, W. Xiong, Q. Zhu, L. Li, X. Chai and L. Gan, Chem. Eng. J., 2018, 347, 233–242 CrossRef CAS.
- X. Yang, Y. Zheng, C. He, Y. Qiu, W. Hou, B. Lu, Y. Chen, B. Huang, J. Lv and G. Lin, J. Anal. Appl. Pyrolysis, 2023, 169, 105822 CrossRef CAS.
- C. Qiu, L. Jiang, Y. Gao and L. Sheng, Mater. Des., 2023, 230, 111952 CrossRef CAS.
- L. H. Zheng, M. H. Chen, S. X. Liang and Q. F. Lü, Diamond Relat. Mater., 2021, 113, 108267 CrossRef CAS.
- D. Qiu, N. Guo, A. Gao, L. Zheng, W. Xu, M. Li, F. Wang and R. Yang, Electrochim. Acta, 2019, 294, 398–405 CrossRef CAS.
- B. Jia, Q. Mian, D. Wu and T. Wang, Mater. Today Chem., 2022, 24, 100828 CrossRef CAS.
- D. He, Z. Huang and M. Wang, J. Mater. Sci.: Mater. Electron., 2018, 30, 1468–1479 CrossRef.
- B. Yang, D. Zhang, W. She, J. Wang, S. Gao, Y. Wang and K. Wang, J. Power Sources, 2021, 492, 229666 CrossRef CAS.
- M. Liu, J. Niu, Z. Zhang, M. Dou and F. Wang, Nano Energy, 2018, 51, 366–372 CrossRef CAS.
- G. Jiang, R. A. Senthil, Y. Sun, T. R. Kumar and J. Pan, J. Power Sources, 2022, 520, 230886 CrossRef CAS.
- R. Chakraborty, V. K, M. Pradhan and A. K. Nayak, J. Mater. Chem. A, 2022, 10, 6965–7005 RSC.
- M. Jalalah, H. Han, A. K. Nayak and F. A. Harraz, Adv. Compos. Hybrid Mater., 2024, 7, 20 CrossRef CAS.
- L. Qin, Z. Xiao, S. Zhai, S. Wang, H. Lv, W. Niu, Z. Zhao and Q. An, Mater. Chem. Phys., 2021, 260, 124121 CrossRef CAS.
- Z. Li, Q. Xu, L. Zhang, X. Wang, F. He, J. Cheng and H. Xie, Sustainable Energy Fuels, 2020, 4, 3418–3427 RSC.
- W. Zhang, J. Xu, D. Hou, J. Yin, D. Liu, Y. He and H. Lin, J. Colloid Interface Sci., 2018, 530, 338–344 CrossRef CAS PubMed.
- X. Xu, K. Sielicki, J. Min, J. Li, C. Hao, X. Wen, X. Chen and E. Mijowska, Renewable Energy, 2022, 185, 187–195 CrossRef CAS.
- C. Zhao, Y. Ding, Y. Huang, N. Li, Y. Hu and C. Zhao, Appl. Surf. Sci., 2021, 555, 149726 CrossRef CAS.
- X. Wu, G. Lei, Y. Xu and H. Liu, J. Mater. Sci.: Mater. Electron., 2019, 31, 728–739 CrossRef.
- R. X. Xu, Y. P. Zhao, G. H. Liu, J. S. Zhu, R. Y. Wang, J. P. Cao and X. Y. Wei, J. Colloid Interface Sci., 2020, 558, 211–219 CrossRef PubMed.
- S. Jiao, Y. Yao, J. Zhang, L. Zhang, C. Li, H. Zhang, X. Zhao, H. Chen and J. Jiang, Appl. Surf. Sci., 2023, 615, 156365 CrossRef CAS.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1–31 Search PubMed.
- Y. Li, Y. Liang, H. Hu, H. Dong, M. Zheng, Y. Xiao and Y. Liu, Carbon, 2019, 152, 120–127 CrossRef CAS.
- D. Wang, J. Nai, L. Xu and T. Sun, J. Energy Storage, 2019, 24, 100764 CrossRef.
- D. Dong, Y. Zhang, Y. Xiao, T. Wang, J. Wang, C. E. Romero and W. Pan, J. Colloid Interface Sci., 2020, 580, 77–87 CrossRef CAS PubMed.
- Y. Tao, W. Liu, Z. Li, Y. Zheng, X. Zhu, H. Wang, Y. Wang, Q. Lin, Q. Wu, Y. Pang, Z. Shen and H. Chen, J. Colloid Interface Sci., 2021, 602, 636–645 CrossRef CAS PubMed.
- K. Liang, Y. Chen, D. Wang, W. Wang, S. Jia, N. Mitsuzakic and Z. Chen, Sustainable Energy Fuels, 2023, 7, 3541–3559 RSC.
- S. Jiao, L. Zhang, C. Li, H. Zhang, J. Zhang, P. Li, Y. Tao, X. Zhao, H. Chen and J. Jiang, Ind. Crops Prod., 2022, 183, 114906 CrossRef CAS.
- X. Luo, Y. Chen and Y. Mo, New Carbon Mater., 2021, 36, 49–68 CrossRef CAS.
- A. R. Selvaraj, H. J. Kim, K. Senthil and K. Prabakar, J. Colloid Interface Sci., 2020, 566, 485–494 CrossRef CAS PubMed.
- A. Deshpande, S. Rawat, I. M. Patil, S. Rane, T. Bhaskar, S. B. Ogale and S. Hotha, Carbon, 2023, 214, 118368 CrossRef CAS.
- M. Wang, D. He, M. Zhu, L. Wu, Z. Wang, Z.-H. Huang and H. Yang, J. Power Sources, 2023, 560, 232703 CrossRef CAS.
- M. Jalalah, S. S. Sivasubramaniam, B. Aljafari, M. Irfan, S. S. Almasabi, T. Alsuwian, M. I. Khazi, A. K. Nayak and F. A. Harraz, J. Energy Storage, 2022, 54, 105210 CrossRef.
- C. Costentin, T. R. Porter and J. M. Savéant, ACS Appl. Mater. Interfaces, 2017, 9, 8649–8658 CrossRef CAS PubMed.
- L. Yang, Y. Feng, M. Cao and J. Yao, Mater. Chem. Phys., 2019, 238, 121956 CrossRef CAS.
- Z. Shang, X. An, H. Zhang, M. Shen, F. Baker, Y. Liu, L. Liu, J. Yang, H. Cao, Q. Xu, H. Liu and Y. Ni, Carbon, 2020, 161, 62–70 CrossRef CAS.
- N. Guo, M. Li, X. Sun, F. Wang and R. Yang, Green Chem., 2017, 19, 2595–2602 RSC.
- R. A. Senthil, V. Yang, J. Pan and Y. Sun, J. Energy Storage, 2021, 35, 102287 CrossRef.
- Julnaidi, E. Saputra, Nofrizal and E. Taer, J. Chem. Technol. Biotechnol., 2022, 98, 45–56 CrossRef.
- J. Yang, H. Wu, M. Zhu, W. Ren, Y. Lin, H. Chen and F. Pan, Nano Energy, 2017, 33, 453–461 CrossRef CAS.
- P. Waribam, S. D. Ngo, T. T. V. Tran, S. Kongparakul, P. Reubroycharoen, N. Chanlek, L. Wei, H. Zhang, G. Guan and C. Samart, Waste Manage., 2020, 105, 492–500 CrossRef CAS PubMed.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30, 09265 Search PubMed.
- B. Xue, J. Xu and R. Xiao, ACS Sustainable Chem. Eng., 2021, 9, 15925–15934 CrossRef CAS.
- S. Shanmugapriya, S. Surendran, Y. S. Lee and R. K. Selvan, Appl. Surf. Sci., 2019, 492, 896–908 CrossRef CAS.
- H. Shen, X. Kong, P. Zhang, X. Song, H. Wang and Y. Zhang, J. Alloys Compd., 2021, 853, 157357 CrossRef CAS.
- M. Yu, Y. Lu, H. Zheng and X. Lu, Chem. – Eur. J., 2018, 24, 3639–3649 CrossRef CAS PubMed.
- N. Wu, X. Bai, D. Pan, B. Dong, R. Wei, N. Naik, R. R. Patil and Z. Guo, Adv. Mater. Interfaces, 2020, 8, 2001710 CrossRef.
- J. Sun, C. Wu, X. Sun, H. Hu, C. Zhi, L. Hou and C. Yuan, J. Mater. Chem. A, 2017, 5, 9443–9464 RSC.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- Q. Zhang, K. Han, S. Li, M. Li, J. Li and K. Ren, Nanoscale, 2018, 10, 2427–2437 RSC.
- H. Li, H. Yang, H. Sun, Y. Huang, P. An, Y. Yunhua and H. Zhao, Electrochim. Acta, 2024, 473, 143514 CrossRef CAS.
- L. Feng, B. Yan, J. Zheng, J. Chen, R. Wei, S. Jiang, W. Yang, Q. Zhang and S. He, New J. Chem., 2022, 46, 10844–10853 RSC.
- L. Feng, B. Yan, J. Zheng, Q. Zhang, R. Wei, C. Zhang, J. Han, S. Jiang and S. He, Diamond Relat. Mater., 2023, 133, 109767 CrossRef CAS.
- Y. Wu, Y. Wang, L. Pan and X. R. Wu, RSC Adv., 2022, 12, 32600–32610 RSC.
- F. Wu, X. Ren, F. Tian, G. Han, J. Sheng, Y. Yu, Y. Liu and W. Yang, New J. Chem., 2022, 46, 19667–19674 RSC.
- Q. Zhang, L. Feng, Z. Liu, L. Jiang, T. Lan, C. Zhang, K. Liu and S. He, Molecules, 2023, 28, 6994 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bd,
Ju
Zhang
b,
Xuan
Qin
b,
Jiwei
Zhang
b,
Chulin
Li
b,
Yang
Jin
b and
Shenggao
Wang
*bd,
Ju
Zhang
b,
Xuan
Qin
b,
Jiwei
Zhang
b,
Chulin
Li
b,
Yang
Jin
b and
Shenggao
Wang