
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous covalent organic frameworks in photocatalytic ROS-mediated processes
Nikolaos
Karousis
 * and
Dimitrios
Tasis
*
* and
Dimitrios
Tasis
*
Department of Chemistry, University of Ioannina, Ioannina 45110, Greece. E-mail: nkarousis@uoi.gr; dtassis@uoi.gr
First published on 28th February 2024
Abstract
Porous carbon-based frameworks represent an emerging class of nanostructured materials with variable crystallinity. In this family, a large number of bonding configurations lead to the growth of either graphitic carbon nitride, amorphous covalent organic polymers, metal–organic frameworks or covalent organic frameworks. The latter nanostructures have attracted a great deal of attention owing to their porous crystalline character, originating from the linking of monomeric units by strong covalent bonds for the development of extended conjugated networks. The simultaneous existence of the conjugation and spatial separation of electron donor–acceptor moieties are critical factors for enhanced photocatalytic efficiency. In this review, we aim to summarize the diversity of covalent organic frameworks, highlighting their different synthetic protocols, most importantly, giving rise to various building blocks that interact with each other through diverse linkages, thereby forming novel architectures. Due to the aforementioned structural and semiconductive properties, porous covalent organic frameworks exhibit promising photocatalytic performance in the generation of reactive oxygen species. Our goal is to shed some light on critical factors leading to the enhanced generation of either transient intermediates or neutral oxygen-containing substances. We have directed our discussion to the spatial separation of electron donor–acceptor moieties within a carbon network and extracted some helpful data on the structure–activity relationship for hydrogen peroxide production. Furthermore, we have described recent progress in the development of efficient photocatalytic systems for organic substance degradation using ROS-mediated redox processes.
1. Introduction
In recent centuries, the exploitation of energy resources on our planet by humankind has been focused on the hidden chemical energy of fossil fuels. After centuries of exhaustive mining, severe environmental issues have been raised due to the emission of greenhouse gases in the atmosphere, with global warming being the main side-effect.1 Furthermore, wastewater derived from industrial activities contains harmful pollutants, such as organic dyes and ionic species. Modern society has recently made efforts to diminish the utilization of fossil fuels. Great necessity has been raised for harnessing the power of sustainable energy resources. One such resource is solar energy, which can be used for powering the planet.2 In parallel, photon energy can be converted and stored in the form of chemical energy, a process which is highly associated with environmental issues. The mechanistic scheme of photosynthesis is the best example to describe such kind of photoinduced processes, where water is oxidized to oxygen gas. Through this complex process, various transient intermediates participate and certain photo-adducts may be formed, even under dark conditions. In recent decades, there have been numerous attempts to develop multifunctional catalytic systems, which could play the role of a photosensitizer in certain redox processes.3,4 Specifically, such nanostructured materials should present a semiconducting character. Although inorganic semiconducting nanostructures have been primarily assessed as solar energy harvesters and simultaneous photocatalytic systems,5 carbon-based nanostructures have recently paved the way for energy conversion and related environmental remediation applications.6 The semiconducting character of carbon-based nanostructures stems from their optimal conjugated network, which gives rise to enhanced charge carrier delocalization. An additional structural advantage of a functional photocatalyst is the engineered building of a homogeneous porosity profile within the lattice. Such sophisticated development of porous carbon frameworks, along with their tailored decoration with appropriate functionalities, can contribute to the optimization of interfacial interactions between a photosensitizer surface and any substrate. These interactions are the basis to a wide range of energy conversion applications including a variety of topics, such as hydrogen gas evolution, carbon dioxide reduction, nitrogen fixation and oxidative/reductive transformations of substances.7 The latter processes are directly associated with the participation of reactive oxygen species (ROS) formed through various redox reactions. Such transients are vital components in mammalian and plant processes, strongly associated with life itself.8 In photocatalysis, these oxygen-based intermediates are generated by the interaction of photoexcited semiconductors (catalytic system) and the chemical environment.In this context, a novel family of porous carbon-based semiconducting architectures have been recently developed and are considered as a hot field in photocatalysis. This family contains nanostructured materials, such as graphitic carbon nitride,9 covalent organic polymers (COPs),10,11 metal–organic frameworks (MOFs)12 and covalent organic frameworks (COFs).13,14 The latter nanostructures have drawn intense interest due to their exceptional optical and carrier separation properties. Built on covalent linkages between appropriate monomers with variable symmetries, the obtained carbon networks possess tuned porosity patterns, which ensures a fairly high crystalline character. Moreover, the co-existence of spatially separated chromophores, acquiring electron donor–acceptor interactions, may play a vital role in the inhibition of carrier recombination in the excitonic form and the subsequent charge separation. In recent years, various synthetic approaches have been proposed in order to engineer the band gap of a carbon-based porous semiconductor to develop functional heterojunctions as well as to integrate appropriate functional groups into the carbon-based network as adsorption sites for any substrate.15
Regarding photocatalytic processes in the liquid phase, typical substrates may be either the dissolved oxygen gas, a sacrificial agent or the medium itself (H2O). The abovementioned components, which participate in the redox reactions with semiconductor's electrons and holes, need to be strongly adsorbed onto the photocatalyst lattice. The strong adsorption of molecular oxygen onto a semiconducting photocatalyst is critical due to the low solubility of O2 gas in aqueous media.
Due to the exotic optical and structural properties of nanostructured systems based on COFs, these carbon nanomaterials have participated in numerous studies, spanning a wide range of topics. Some excellent reviews and seminal works have recently focused on the chemistry of COFs as well as the potential applications of such systems. These involve the utilization of covalent organic frameworks as functional components in diverse catalytic applications,16,17 in energy storage,18–21 in organic electronic devices,22–24 in gas storage,25 as porous adsorbents,26,27 as chemical28 and fluorescent sensors,29 in thermocatalytic CO2 conversion,30 in photocatalytic evolution of oxygen,31,32 in photocatalytic evolution of hydrogen,33–35 in photocatalytic reduction of CO2,36,37 in electrochemical energy conversion,38–40 in nitrogen fixation,41 in photocatalytic organic synthesis42–45 as well in the photodegradation of pollutants.46,47
In this review, first, we describe the fundamental principles of photocatalytic processes towards the generation of reactive oxygen species, involving either transients or neutral substances. Taking into account the vast literature in the field of porous carbon-based semiconducting architectures, this work will not include carbon nitrides or metal-based frameworks but will be limited only to carbon-based COFs, with variable crystallinity. A systematic introduction of synthetic methods and structural motifs of COFs are presented. Then, the particular advantages of COFs in photocatalysis are discussed. Furthermore, the photocatalytic applications of COF-based materials are introduced in topics related to ROS-mediated hydrogen peroxide evolution and degradation of organic substances in aqueous environment. In particular, various approaches are discussed in detail towards the revealing of structure–activity relationship in photoactive COFs. Finally, a perspective on unsolved challenges and research opportunities of COF-based materials for photocatalytic applications is also presented.
2. Fundamentals of photocatalysis
2.1. General principles
When a semiconducting nanostructure interacts with light of sufficient energy, electronic transitions take place.48 Specifically, electrons migrate from the valence band (VB) to the empty conduction band (CB) of the semiconductor. This photoinduced process creates positive holes in the valence band and excited electrons to the conduction band, respectively. It is critical for the initially bound pair of charge carriers (exciton) to separate. This charge separation will give rise to the evolution of various oxygen-based transient intermediates through various redox processes (Fig. 1). Thus, photon energy catalyses the generation of reactive transients, which may subsequently decompose a pollutant in oxidative conditions. Regarding the charge carriers, the positive holes may participate in oxidative processes, whereas it is the excited electrons in the reductive ones. In typical nanostructured semiconductors, such as the inorganic ones, the fast charge recombination process is the main drawback, which leads to an appreciable decrease of the apparent quantum yield of redox processes. This is mainly solved through the utilization of an appropriate cocatalyst, a component which forms multiple heterojunctions with the main semiconductor.49 Actually, the cocatalyst plays the role of an electron acceptor, and this is highly correlated to its relatively large value of work function (e.g., metallic nanoparticles). As an additional scenario, the charge carriers may be trapped in the defect sites of the semiconductor lattice. Thus, all these major issues need to be taken into serious consideration when an efficient photocatalytic system is to be developed. For successful and efficient charge separation within the lattice of a photocatalyst, the semiconducting nanostructure should contain a conjugated network with spatially separated electron donor and electron acceptor domains. This would drive the process of recombination of the formed charge carriers to be appreciably inhibited. Thus, a successful separation of electrons and holes may be achieved. Beside the construction of a conjugated network with spatially decorated donor–acceptor moieties, the structural defects should be in minimum content.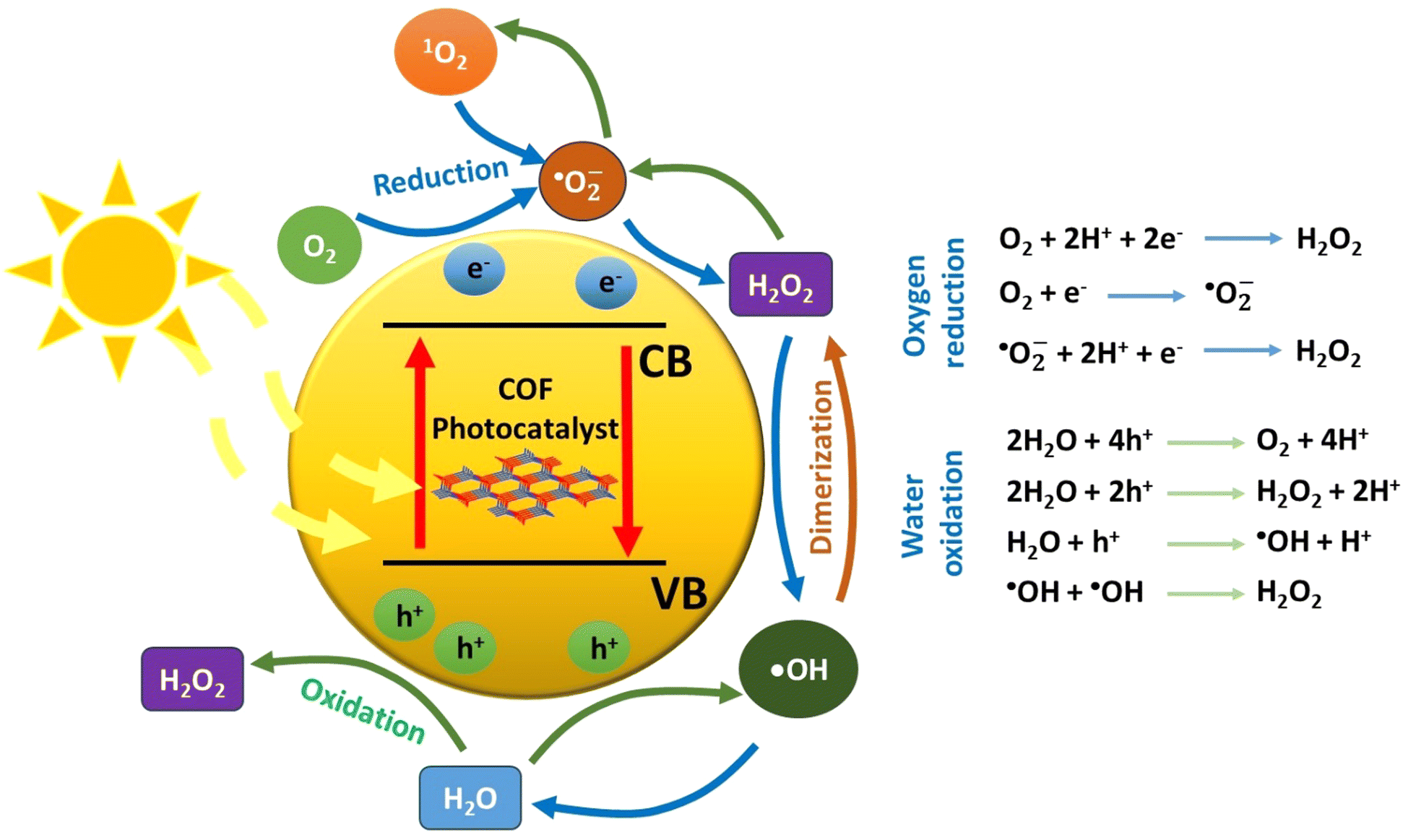 | ||
| Fig. 1 Underlying mechanistic pathways of reactive oxygen species-mediated processes. | ||
This implies that highly crystalline nanostructures may facilitate charge transfer and minimize any charge trapping phenomena. Furthermore, the development of highly porous frameworks may give rise to the rapid diffusion and adsorption of substrates in the catalytic sites of the semiconductor surface.
The aforementioned characteristics are met in nanostructures, such as COFs.50 The latter porous components possess a high photochemical stability due to the strong covalent bonding of the monomer units, which gives rise to durable nanostructures. Furthermore, the conjugated backbone in a COF lattice plays a key role in the light harvesting of visible spectrum wavelengths.
As stated above, two general redox schemes take place after the electron–hole separation step. These lead to the generation of reactive oxygen species (ROS), which eventually participate in the photocatalytic degradation of a pollutant. The first scheme involves the participation of excited electrons in reductive processes. The driving force for the reductive activity of a semiconductor stems from the energy difference between its conduction band level and the corresponding reduction potential of a specific half reaction. Specifically, the conduction band level should be higher (more negative potential) than the one of a half reaction. Typical electron acceptors, such as dissolved oxygen gas, may be reduced by either a one-, two-, three- or four-electron mechanism. The experimental evidence that O2 molecules behave as an efficient electron trap towards the formation of superoxide anion (one-electron reduction) comes from ESR measurements at low temperature (77 K). It was found that the intensity of the electron spin resonance (ESR) signal of trapped electrons was decreased in the presence of dissolved oxygen gas.51
In total, the stepwise reduction of oxygen gas may deliver superoxide anion (O2˙−), hydrogen peroxide (H2O2), hydroxy radical (˙OH) and water, respectively. As clearly seen, the hydrogen peroxide photo-adduct is generated via a two-electron reduction of oxygen gas. An additional pathway for H2O2 evolution involves the so-called disproportionation. This is actually a reaction between O2˙−, HOO˙ and water, yielding H2O2, O2 and OH−.52 All the aforementioned ROS, except water, demonstrate a highly oxidative character, leading to the appreciable degradation of a pollutant through the so-called advanced oxidation processes (AOPs). In the ideal case of mineralization, the organic pollutant may be quantitatively converted to carbon dioxide and water.
Beside oxygen gas, other electron acceptors may take part in the reductive processes. The latter may lead to the generation of fuel substances, such as the reduction of protons to molecular hydrogen as well as the reduction of CO2 to methane.53 Alternatively, chemical transformations may take place through the reductive path, such as the photoreduction of nitroarenes to aniline derivatives54 and the conversion of harmful metal cations to a different oxidation state. A typical example of the latter case is the reduction of Cr(VI) to Cr(III).55
On the other hand, the second scheme involves oxidative processes taking place in the valence band of the semiconducting photocatalyst. Positive holes may oxidize water molecules of the reaction medium. The stepwise hole-oxidation of water results in the generation of hydroxy radical (˙OH), hydrogen peroxide (H2O2), superoxide anion radical (O2˙−), and singlet oxygen (1O2), respectively.4 Regarding a one-hole oxidative process, the interaction of a water molecule with a positive hole results in the generation of a hydroxy radical and a proton. From a thermodynamic aspect, it is noted here that the energy level of singlet oxygen is higher than the one of superoxide anion radical; thus, the most probable pathway for the formation of O2 excited state involves the energy transfer between the excited semiconducting photocatalyst and the ground state molecular oxygen.
Alternative hole scavengers are the so-called sacrificial agents, which demonstrate an electron donor character. Such substances may be low molecular weight organic/inorganic species, polymeric substances such as biomass as well as the pollutant itself.56 Regarding the feasibility of the half reactions, leading to the photogeneration of the abovementioned ROS, these may be favoured in specific pH ranges.17 In alkaline environments, the relatively high concentration of hydroxy anions may lead to the formation of hydroxy radicals by one-hole oxidation mechanism. This specific pathway for hydroxy radical formation would not be favoured in acidic media. Beside the effect of acid–base equilibrium, thermodynamic aspects govern the driving force for oxidative processes. In an analogous rule as in the case of reductive processes, the valence band level of the semiconductor should be lower than the oxidation potential of a specific half reaction. It is noted here that both reductive and oxidative processes need to take place simultaneously in order for the photocatalytic activity to be optimal. The advantage of the electron/hole-mediated photocatalysis is that the approach generates a wide range of ROS for advanced oxidation processes. It is quite clear that such intermediates may be classified as either transients with short lifetime or neutral molecules.
Beside the direct interaction of molecular substances with either electrons or holes, there is an alternative pathway that involves energy transfer between the excited semiconductor and a substrate. This process belongs to the family of photosensitization, which leads to the generation of an excited state of a substrate. The more reactive excited species is able to react with another substrate and subsequently result in efficient chemical transformations. The excitation of molecular oxygen from its ground state (triplet multiplicity) to the corresponding singlet excited state (1O2) is a typical example of ROS generation by energy transfer mechanism. The advantage of the latter mechanistic scheme is that it permits the occurrence of redox reactions, which do not take place due to unfavourable reduction/oxidation potentials of specific half reactions from the thermodynamic point of view.
2.2. ROS detection techniques
Due to the short lifetime of the reactive oxygen species, it is quite difficult to detect them by steady-state techniques. Thus, the monitoring of such transients may take place either at low temperatures (77 K) or by indirect approach (radical trapping etc.). Certain techniques have demonstrated drawbacks for the detection of ROS. For example, the utilization of UV-Vis absorption spectroscopy does not provide safe quantitative data due to the low values of absorption coefficient of the corresponding ROS.4 The main physicochemical characterization techniques of ROS widely utilized in the photocatalytic studies of porous COFs are described in the following section.Besides ESR trapping experiments, absorption spectroscopy has been used in order to monitor the rates of hydroxy radical formation. Various chemical reactions of transient and organic substances have been studied, such as the decolouration of p-nitrosodimethylaniline (λmax at 440 nm)64 as well as the oxidation of 1,5-diphenyl carbazide to 1,5-diphenyl carbazone (λmax at 560 nm).65 However, the possibility of the involvement of trapped holes via the oxidation of the aforementioned substances should not be ruled out.
The time-dependent generation of singlet oxygen has also been monitored by UV-Vis spectroscopy. Upon the photoirradiation of a photocatalytic system in the presence of an oligonuclear hydrocarbon label (9,10-dimethylanthracene), the monitoring of its 375 nm peak indirectly shows the rate of endoperoxide formation.68,69 Analogous endoperoxides may be formed through the reaction of singlet oxygen with either furan or α-terpinene derivatives. In the former case, a typical probe for the detection of 1O2 species was 1,3-diphenylisobenzofuran (DPBF).70 The reaction between the two components forms an unstable peroxide that decomposes into colourless 1,2-dibenzoylbenzene. By monitoring the spectral changes during the photoinduced generation of singlet oxygen, quantitative data may be extracted. Specifically, the absorption of furan derivative at 420 nm was diminished, whereas clear isosbestic points suggest the generation of an organic adduct absorbed in the UV region (benzene derivative).
When using terpinene as the quencher, ascaridole photoadducts are expected to form through the trapping reaction.71 The conversion of α-terpinene was followed by the drop in the absorption intensity of its UV band at 267 nm.
All the aforementioned cases belong to the family of chemical quenching. This means that oxygen molecules are consumed towards the formation of a photo-adduct. There are also physical quenchers of singlet oxygen species. In physical quenching, no oxygen molecules are consumed. The only physical process is the deactivation of the excited species. Singlet oxygen may be regarded as an electrophile and thus may interact with species carrying charged heteroatoms. To identify the role of singlet oxygen in photocatalytic processes, sodium azide has been used as a physical quencher of the O2 excited state.72
3. COFs synthetic principles
COFs represent a class of polymer-like systems that combine both covalent bonding and non-covalent interactions in a well-defined structure, presenting periodic multi-angular frames and highly-ordered pores.73 Due to their higher binding energy through covalent bonding, COFs present superior stability than the corresponding similar frameworks, i.e., MOFs. The more stable the covalent linkage, the more crystalline and stable the COF. A proper selection of building blocks as precursors allows the design of COFs with specific chemical and physical properties and predictable structures following definite topologies and architectures (Fig. 2).74–76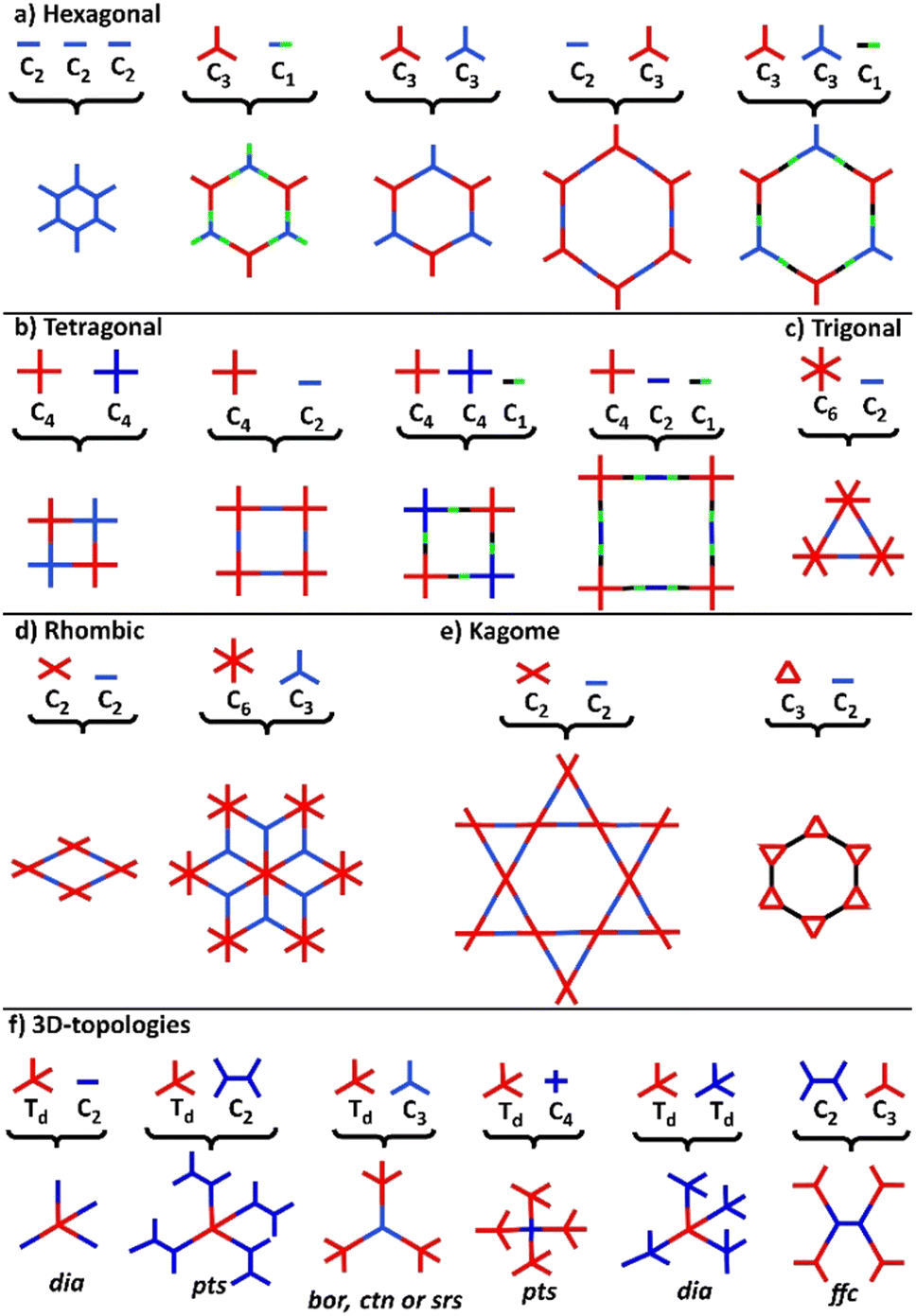 | ||
| Fig. 2 Illustrative topology diagrams of common 2D and 3D-COFs. | ||
Following the basic principles of reticular chemistry, two-dimensional hexagonal COFs with variable pore sizes are designed by the combination of monomers with variable symmetry patterns. Specifically, functional COFs have been shown to grow by the condensation of either C3- and C3-type monomers, C3- and C2-, C3 and C1-, as well as by the self-condensation of three C2-symmetric monomers (Fig. 2a). Likewise, the combinations of either C4- and C4- type monomers or C4- and C2-ones lead to symmetric tetragonal COFs, and the same architecture was achieved by the triple-component condensation of the above-mentioned combinations with a C1-symmetry monomer (Fig. 2b). Triangular architectures were designed by the combination of C6- and C2-monomers (Fig. 2c), while rhombic architectures come from the condensation of C3- and C2-monomers (Fig. 2d). More sophisticated two-dimensional, dual-pore, kagome-shaped COFs were proposed by combining asymmetric C2- and C2-type monomers and C3- and C2-monomers (Fig. 2e). In order to obtain three dimensional COFs, at least one monomer has to follow a non-planar architecture (Fig. 2f). Overall, the topology diagrams offer scientists the ability to predesign and predict a great plethora of COF structures.
The most important factor towards high-quality COFs is to control both nucleation and growth procedures. It is believed that the reversible formation of covalent bonds, the so called “dynamic covalent chemistry”, is the key to the construction of high-quality crystalline COFs, allowing the generation of highly ordered frameworks through self-healing.77 In this context, numerous reversible reactions were utilized in COF synthesis, starting from the highly reversible self-condensation of phenyl boronic acids or the reversible reaction between phenylboronic acid and catechols (Fig. 3a). The corresponding boroxine and boronate linkages were found to present limited stability, especially in acidic or basic conditions. On the other hand, COFs with carbon-nitrogen linkages, in the form of imine (Fig. 3b), imide and amide bonding, presented much higher stability and crystallinity. Recently, the more robust C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linked COFs gained increased interest of scientists due to their higher chemical and thermal stability (Fig. 3e).
C linked COFs gained increased interest of scientists due to their higher chemical and thermal stability (Fig. 3e).
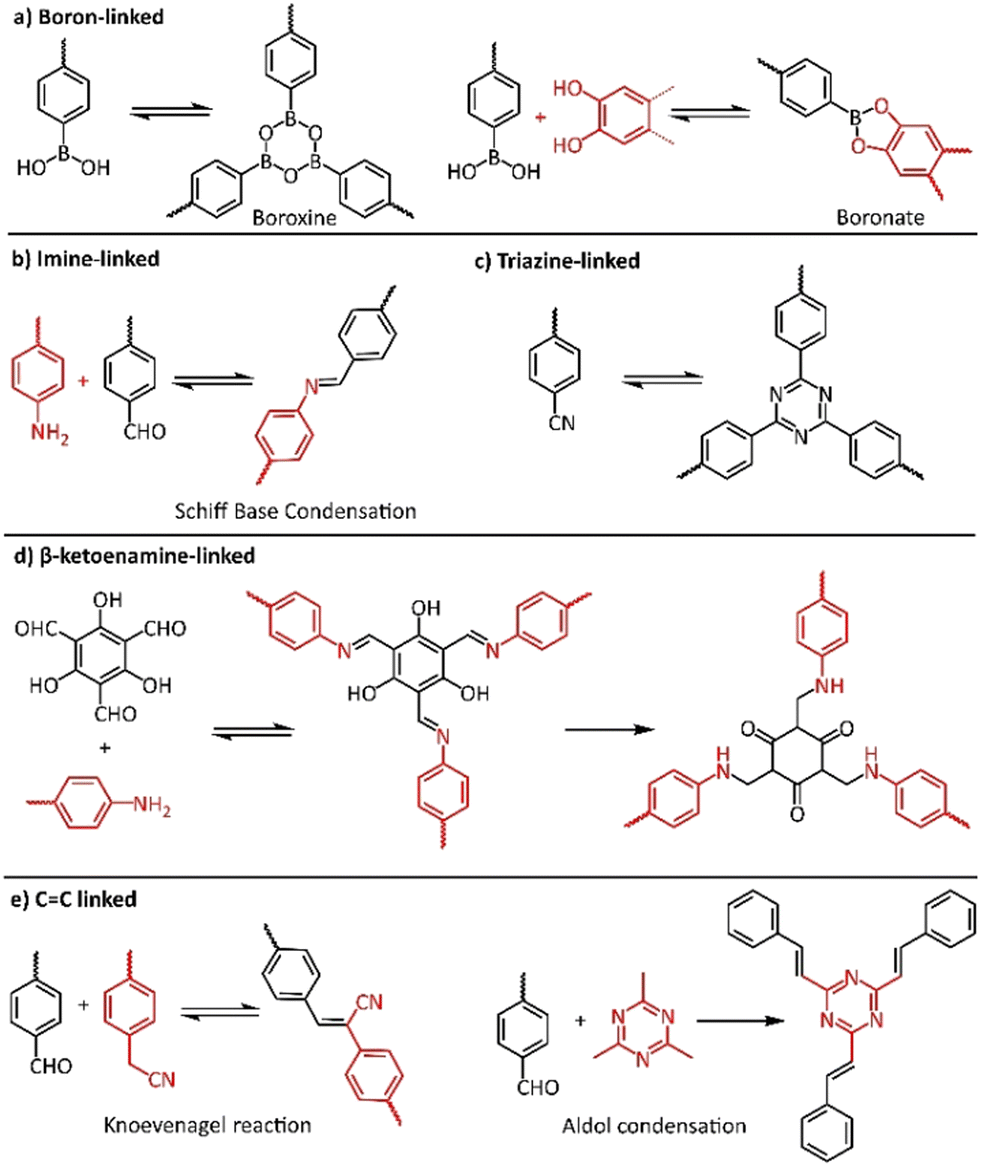 | ||
| Fig. 3 Typical covalent linkage reactions for COF synthesis. | ||
“Dynamic covalent chemistry” approach has been successfully utilized in the reversible condensation between 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP-2H) and the semiconductor 5,5′-(2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6 tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)dithiophene-2-carbaldehyde (DPP-1) under solvothermal conditions. It was surprisingly found that DPP–TAPP-COF crystallites agglomerate and self-organize to a unique microtubular structure, representing a nanotube, with an outer diameter of approximately 300 nm (Fig. 4).78
 | ||
| Fig. 4 Agglomeration of small DPP–TAPP-COF crystallites and tube formation by reversible imine condensations. Adapted with permission from ref. 78. Copyright 2018 Wiley. | ||
Later on, a comprehensive study by Xiong et al.79 revealed that amorphous non-porous polymers, which were prepared at room temperature from 1,3,5-tris(4-aminophenyl)benzene (TAPB) and benzene-1,3,5-tricarbaldehyde (BTCA), could undergo structural transformation after aging at higher temperatures. The resulting distinct hollow structures presented uniformly spherical morphology, which was highly dependent on the aging temperature, aging time and solvent. Several COFs with different linkages and tunable morphology have been produced following this strategy.
3.1. General synthetic protocols of COF construction
Although it has been almost 20 years since the first reference to porous COFs,73 there is still a strong debate as to whether appropriate synthetic protocols have been developed towards highly crystalline stable materials. As it is already mentioned, COFs are polymer-like materials; therefore, the nucleation and growth procedures play a critical role in their final structure and may determine their properties. In this context, the reaction conditions like time, temperature, pressure, the presence or absence of solvents and/or catalysts, and even the concentration of building blocks as starting materials are decisive in the quality of the final material.The most common procedure for the fabrication of COFs relates to conventional solvothermal conditions, including the reaction of the monomers in sealed vessels under vacuum, at high temperatures (100–180 °C) for several days (3–5 days). The structure and the properties of the as-prepared COFs are highly dependent on various parameters, such as temperature, time, and solvent medium.
Mechanochemistry is another important synthetic method that has been widely used in organic and materials chemistry, based on the direct absorption of mechanical energy by the reactants. Usually, it includes the grinding of solid reactants in the absence or with a minimal amount of liquid solvent. In comparison to the aforementioned solvothermal synthesis, this method avoids any additional heating and a large amount of solvent, thus being characterized as an environment-friendly and cost-effective synthetic procedure. In the field of COFs, mechanochemical synthesis was first introduced by Banerjee and co-workers,80 who proposed a rapid, solvent-free procedure by grinding 1,3,5-triformylphloroglucinol (TFP) and p-phenylenediamine (PA) or 2,5-dimethyl-1,4-phenylenediamine (MPA) or benzidine (BD) in a mortar with pestle to produce COF-TpPa-1 or COF-TpPa-2 or COF-TpBD, respectively. Although the as-prepared COFs presented increased chemical and thermal stability, however, their crystallinity and surface area were rather low.
Sonothermal procedures represent a milder alternative of solvothermal methods in COF synthesis. Yang et al.81 have introduced sonochemistry in the synthesis of boroxine-based COFs, reducing the reaction time from 3 days to 1 h, while the reaction took place at atmospheric pressure. More recently, Zhao et al.82 utilized sonochemistry to produce highly crystalline imine-based COFs in aqueous acidic conditions in the absence of any organic solvents and in less than 60 minutes. Their method was successfully tested in seven (7) already known COF derivatives, indicating improved crystallinity and porosity compared to COFs produced by solvothermal conditions.
Another approach relates to the combination of sonochemistry and ion induction, leading to amorphous materials. In this context, acridine-3,6-diamine (Acr) and 1,3,5-triformylbenzene (TFB) were ultrasonicated in acetic acid, and the resulting homogeneous solution was subsequently added to a solution of sodium chloride, magnesium chloride, or zinc nitrate to afford Acr–TFB–Na, Acr–TFB–Zn, and Acr–TFB–Mg amorphous precipitates, respectively.83 It was observed that by increasing he ion concentration, the particle size of the as-prepared materials was smaller. It was observed that the pore size of the zinc- and magnesium-coordinated frameworks Acr–TFB–Zn and Acr–TFB–Mg was 3.06 nm and 3.41 nm, respectively, while the salt-based Acr–TFB–Na was found to be much larger at 9.58 nm. Despite their amorphous character, these novel covalent organic polymer-like materials present efficient and reproducible photocatalytic activity in methylene blue degradation via superoxide radical anion generation.
Microwave-assisted procedures were employed in the synthesis of COFs to drastically reduce the reaction times without increasing the required energy consumption while, at the same time, affording high yield rates. Wei and co-workers84 applied microwave-assisted techniques to prepare TpPA-COF, which was already synthesized from TFP and PA by conventional solvothermal and mechanochemical procedures. It was found that this method resulted in materials with superior properties in terms of crystallinity, porosity and stability compared with both other methods, allowing the as-prepared material to be effectively used for CO2 storage.
In another approach, the use of light energy seemed to accelerate the reversible imine-condensation reaction, leading to COFs with high crystallinity. It was found that the reaction between 1,2,4,5-benzenetetramine (BTA) and hexaketocyclohexane (HCH) was advanced timely when the mixture was irradiated by simulated sunlight (200–2500 nm wavelength) in the presence of catalytic amounts of water and acetic acid.85 The hcc-COFs prepared by this method were organized in a highly uniform and atomically thin structure with high electrical conductivity. Overall, it seems that modern synthetic approaches such as sonochemical and light-assisted methods are gaining increased interest in COF synthesis, while the traditional solvothermal approach still predominates.
3.2. Linkages
It is already mentioned that the porous COFs were prepared via the covalent bonding of sp2-conjugated monomers carrying characteristic moieties. The choice of appropriate organic moieties and the corresponding linkages is quite important in order to achieve the desirable structure, porosity and symmetry of the resulting materials. In this context, the selection of photoactive components allows the construction of photoresponsive COFs, which can be effectively used in visible light-driven catalytic reactions. The key factor making a COF an attractive platform in photocatalysis is its conjugated structure and the formation of suitable channels that allow the undisrupted transfer of electrons, holes, and even molecules, while at the same time they prevent the fast recombination of charges. The presence of donor–acceptor and light-harvesting building blocks, including conjugated polyaromatic compounds, porphyrins, and triazines, further allows the fine-tuning of their optical and electronic properties.86,87 A great drawback that limits COFs’ excessive usage in practical applications is that most of the reported COFs are not isolated as single crystals but in the form of microcrystalline powder with several internal defect sites in their lattice, which are responsible for increased trapping phenomena. In this context, various linkage-types have been proposed in the synthesis of COFs towards this direction, and the most important will be discussed below.Dichtel and co-workers88 reported a significant development in the synthesis of 2D COF single crystals by introducing a two-step procedure, separating nucleation from the growth procedure. This was achieved by including acetonitrile in 80% vol in the mixture of the reaction solvents (1,4-dioxane and mesitylene) used in the solvothermal condensation reaction between 1,4-phenylenebis(boronic acid) (PBBA) and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP). This co-solvent system, along with a slow-rate initial mixing of the two monomers, resulted in a colloidal suspension of COF nanoparticles, which acted as the seeding core for the further growth of the COF system. Transient absorption spectroscopy verified the well-defined structure of this material by showing a three-fold signal-to-noise enhancement compared to polycrystallite materials obtained under traditional solvothermal conditions.
Following a similar acid-catalysed solvothermal procedure, Wang et al.90 proposed a benzodifuran-based donor–acceptor COF. In this context, the electron-rich 4,40-(benzo[1,2-b:4,5-b0]difuran-4,8-diyl)dibenzaldehyde (BDF-CHO) and the electron-deficient tris-(4-aminophenyl)triazine (TAPT) were reacted in a mixture of o-DCB/n-BuOH/3M acetic acid (5/15/2) in a sealed tube at 120 °C for 3 days to afford BDF–TAPT-COF. In addition to thermal and chemical stability, the as-obtained BDF–TAPT-COF presents high crystallinity and large porosity as well as an adsorption band with a maximum at ca. 460 nm, which is red-shifted compared with the starting materials due to electron push–pull interactions.
In another approach, Yaghi and co-workers91 suggested a new porous COF carrying free carboxylic groups, which was readily available for further post-synthetic modification. This COF prepared from p-terphenyl-2′,5′-dicarboxylic acid-4,4′′-dicarboxaldehyde (3P-COOH) and 1,1,2,2-tetraphenylethene (4PE) under typical solvothermal conditions proved an ideal platform for further amidation with secondary amines, esterification and thioesterification reactions, leading to a plethora of potential pollutant adsorbents.
With the same idea, a photosensitive highly stable and crystalline COF, as an effective photocatalyst for generating ROS, was reported. Taking advantage of the high photoactive properties of triphenylamine (TPA), Li et al.92 first prepared a multiple junction amine component, namely, hexakis(4-aminophenyl)-cyclotriphosphazene (HACP), in order to ensure an increased density of the photoactive moieties in the framework. Subsequently, HACP reacted under typical solvothermal conditions with 4,4′,4′′-nitrilotrisbenzaldehyde (NTBA), a multi-aldehyde precursor to form CPTPA-COF.
In the same context, two-dimensional porphyrin-based donor–acceptor type COFs were prepared for selective organic transformations. Following the well-established Schiff-base condensation under solvothermal conditions, the electron-rich TAPP-2H reacted effectively with three different electron-deficient aldehydes, specifically, a bi-carbazole-based (BTTZ), a poly-phenyl (FBQD) and a pyrazine-based (PTBC) tetra-aldehyde, forming highly crystalline BTTZ-Por COF, FBQD-Por COF and PTBC-Por COF, respectively, as proved by theoretical and X-ray diffraction measurements.93 All these COFs showed efficient charge separation under visible light irradiation, with PTBC-Por COF presenting the best optoelectronic properties as it showed stronger absorption intensity and a longer carrier lifetime (1.12 ns).
An imine-linked single-layer porphyrin-based framework (COF-420) with internal electronic heterojunctions was fabricated through a Schiff-based condensation between TAPP-2H and 5,10,15,20-tetrakis(4-formylphenyl)porphyrin (TFPP), which took place on an Au(111) substrate under ultra-high vacuum.94 This novel approach includes, first, the thermal evaporation (340 °C) and deposition of TFPP onto Au surface, followed by the deposition of TAPP-2H at 330 °C and subsequent annealing of the mixed layers at 180 °C for 45 minutes to complete the condensation reaction. The design of this process leads to a material with square lattice topology, where the cores have identical chemical composition, but they are electrically asymmetric due to the orientation of imine linkages, as proved by scanning tunneling microscopy (STM) (Fig. 5). This asymmetry causes the spatial separation of the conduction band and the valence band onto different distinct sublattices, thus enhancing the optoelectronic properties of the COF-420 material.
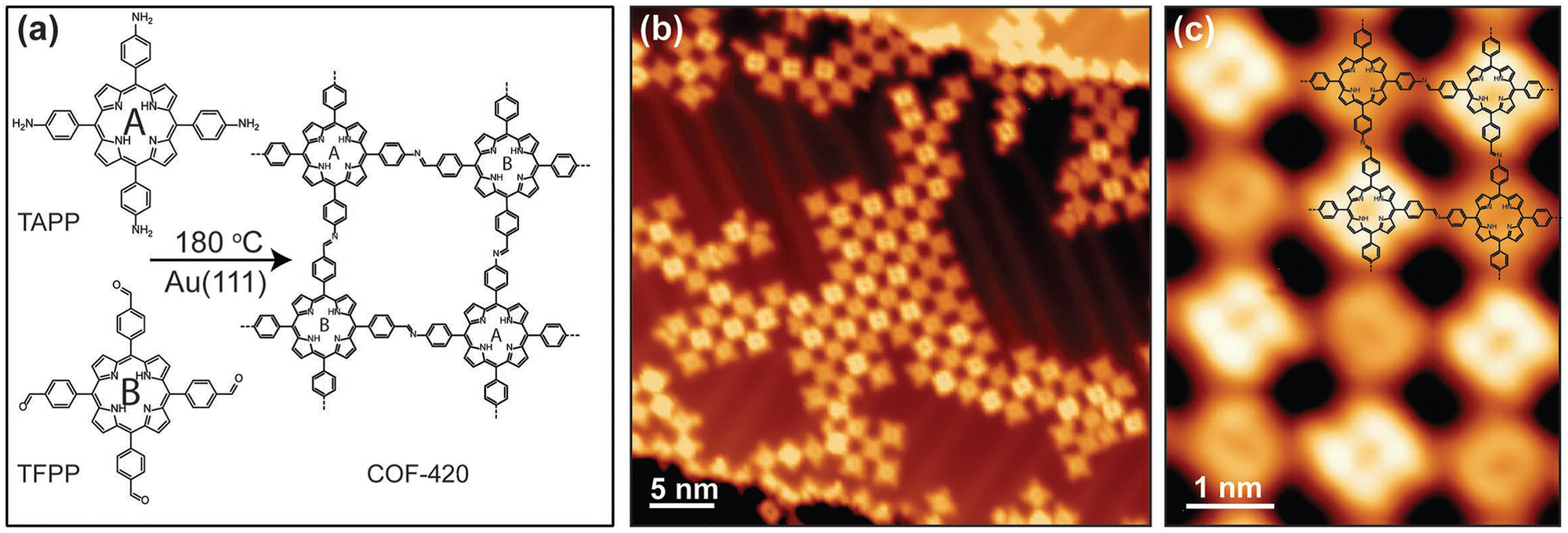 | ||
| Fig. 5 (a) Schematic representation of the synthesis of COF-420 from the molecular precursors TAPP and TFPP. (b) Representative large-scale STM topographic image of COF-420 on Au(111) (sample bias Vs = 0.8 V, tunnel current It = 10 pA). (c) Close-up STM image of COF-420 with the chemical structure overlaid in the top-right corner (Vs = 0.8 V, It = 10 pA). Adapted with permission from ref. 94. Copyright 2019 Wiley. | ||
Two imine-based 2-dimensional chiral COFs (CCOF-1 and CCOF-2) were synthesized from the condensation reaction of 4,4′-diaminodiphenylmethane (4,4′-DADPM) with tetraaryl-1,3-dioxolane-4,5-dimethanols TTA and TTPA, respectively, under solvothermal conditions and used as efficient heterogeneous catalysts after treatment with Ti(Oi-Pr)4.95 The unique propeller-like configuration of the dioxolane moieties combined with the angular conformation of DADPM provided to the as-prepared CCOFs heterojunctions enhanced the flexibility, which also affects their overall porosity.
Two novel triazine-based imine-linked COFs, namely, NL-COF-1 and NL-COF-2, were synthesized from 4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(oxy))trianiline (TTT-NH2) and 1,3,5-tris(p-formylphenyl)benzene (TFPB) and 1,3,5-tris(p-formyl-m-fluorophenyl)benzene (TFFBP), respectively, through a typical Schiff-base condensation reaction.96 These COFs show acceptable crystallinity in spite of the presence of their flexible triangular building blocks and uniform channels with elasticity and adaptability, making them suitable for high iodine capture.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) and diacetylene (–CC–CC–)-based β-ketoenamine COFs via an acid-catalysed solvothermal reaction between TFP and 4,4′-(ethyne-1,2-diyl)dianiline (EDDA) and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA), respectively. It seems that the keto–enol tautomerization due to ketoamine formation is responsible for the chemical stability in these materials. However, the crystallinity of TFB-BDDA is highly dependent on the reaction time, especially for the diacetylene-based analogues. The best crystallinity for TFB-BDDA was achieved by a 4-day solvothermal reaction, while these materials showed long-term (10 h) hydrogen evolution under visible light irradiation.
C–) and diacetylene (–CC–CC–)-based β-ketoenamine COFs via an acid-catalysed solvothermal reaction between TFP and 4,4′-(ethyne-1,2-diyl)dianiline (EDDA) and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA), respectively. It seems that the keto–enol tautomerization due to ketoamine formation is responsible for the chemical stability in these materials. However, the crystallinity of TFB-BDDA is highly dependent on the reaction time, especially for the diacetylene-based analogues. The best crystallinity for TFB-BDDA was achieved by a 4-day solvothermal reaction, while these materials showed long-term (10 h) hydrogen evolution under visible light irradiation.
Another TFP-based photoactive COF, wherein 1,3,5-triazine-2,4,6-triamine (Tt) plays the role of the electron acceptor building block, was proposed.98 The planar and conjugated structure of Tt enhanced the visible light absorption and charge separation and in connection with TFP as an efficient electron donor, the resulting two-dimensional conjugated TFP–Tt-COF exhibited exceptional photocatalytic activity towards oxidative coupling in air. However, TFP–Tt-COF suffered from low crystallinity due to the low electron density on the amine group of Tt, which reduced the reversible character of the condensation reaction and the corresponding COF's self-correction.
In another approach, a series of ketoenamine-linked COFs were prepared from TFP and several aromatic amines at ambient conditions with the aid of imidazolium-based ionic liquid as the solvent.99 This ionothermal method avoided the harsh conditions of the typical solvothermal procedure as it took place in an open vessel at lower temperatures, while at the same time, the ionic liquid acted both as a solvent and as a recyclable catalyst. The as-prepared COFs presented similar, though slightly inferior, properties compared to the corresponding ones prepared by the solvothermal method.
Xiong et al.100 suggested three β-ketoenamine-linked COFs with different linker lengths, which were synthesized with TFP as one component and 1,4-benzenediamine, 4,4′-biphenyldiamine, and 4,4′′-p-terphenyldiamine, respectively, as the second component. It was found that the increase of the linker length broadens the optical bandgap of the COFs, allowing them to activate O2 to superoxide anion radical.
The same researchers proposed an anthraquinone-based COF constructed from TFP and 2,6-diaminoanthraquinone (AQ) through a typical solvothermal synthesis.101 TFPAQ-COF presented one of the highest BET surface areas reported for β-ketoenamine-linked COFs, while at the same time possessed a broad absorption range, making it an ideal candidate for the selective photocatalytic oxidation of amines.
Highly crystalline and robust ketoenamine-based COFs were prepared following a synthetic protocol that separates the crystallization from the growth process in a similar approach as per Dichtel's suggestion for boron-based COFs.88 In this case, Zhang et al.102 suggested that pre-organized urea-based COFs, as already prepared by a vacuum-free solvothermal procedure from 1,1′-(1,4-phenylene)diurea with TFP,103 can reconstruct to the corresponding ketoenamine-based COF following a temperature and solvent-controlled protocol. These reconstructed COFs (RC–COFs) present enhanced crystallinity, increased BET surface area and narrow pore size distribution compared either to their urea-based COFs (Urea–COF-1) precursors or to the same ketoenamine COFs prepared by a direct solvothermal protocol (DP–COF-1) (Fig. 6).
 | ||
| Fig. 6 (a) and (b) Simulated and experimental PXRD patterns for urea–COF-1 (a) and RC–COF-1 (b). (c) Comparison of PXRD patterns for RC–COF-1 synthesized by the reconstruction protocol and DP–COF-1 synthesized by direct polymerization. (d). Nitrogen adsorption isotherm (filled symbols) and desorption isotherm (open symbols) for RC–COF-1, DP–COF-1 and urea–COF-1 recorded at 77.3 K; (e) SEM image of RC–COF-1. Scale bar, 1 μm. (f) HRTEM image of RC–COF-1. Insets show the FFT pattern taken from the regions highlighted by the dashed-line squares and the corresponding filtered inverse FFT image. Scale bars, 50 nm (main image); 10 nm (inset). Adapted with permission from ref. 102. Copyright 2022 Springer Nature. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C linked COFs.
Another important class in this family includes the CC linked COFs, which are prepared mainly following an acid- or base-catalysed aldol condensation reaction. These COFs are characterized by the presence of strong, irreversible and fully-conjugated sp2 hybridized CC bonding, ensuring high chemical stability, tunable porosity, high crystallinity and efficient π-electron delocalization, which is desirable in photocatalytic applications.
C linked COFs.
Another important class in this family includes the CC linked COFs, which are prepared mainly following an acid- or base-catalysed aldol condensation reaction. These COFs are characterized by the presence of strong, irreversible and fully-conjugated sp2 hybridized CC bonding, ensuring high chemical stability, tunable porosity, high crystallinity and efficient π-electron delocalization, which is desirable in photocatalytic applications.
Vinylene-bridged COFs were prepared via a typical aldol condensation reaction from tricyanomesitylene and a series of aromatic multi-aldehydes, namely, 4,4′′-diformyl-p-terphenyl (DFPTP), 4,4′-diformyl-1,1′-biphenyl (DFBP), or 1,3,5-tris-(4-formylphenyl)benzene (TFPB).104 The electron-withdrawing cyano-group facilitated the reaction between the acidic methyl groups of tricyanomesitylene and the aldehyde groups, thus resulting in crystalline fully-conjugated COF-p-3Ph, COF-p-2Ph, and COF-m-3Ph, respectively. The reactions took place under solvothermal conditions in the presence of secondary amines (piperidine, dimethylamine) as the base catalysts. According to their nitrogen sorption isotherms, only COF-m-3Ph presented a microporous structure while the other two vinylene-bridged COFs showed a mesoporous structure. TEM images indicated the honeycomb-like structure of the as-prepared COFs (Fig. 7).
 | ||
| Fig. 7 (a) TEM image of COF-p-3Ph. (b) Enlarged view of a selected area in the panel (a). The structural model is overlapped on the top. (c) TEM image of COF-p-3Ph from a different area, showing 1D channels with a width of 3 nm. (d) Illustrative structure of COF-p-3Ph. Adapted with permission from ref. 104. Copyright 2019 American Chemical Society. | ||
In a similar approach, another vinylene-bridged COF was prepared under solvothermal conditions using 6,14-bis(4-formyl-2,6-dimethylphenyl)dibenzo[hi,st]ovalene (DBOV-CHO) and 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) as building blocks and piperidine as the base catalyst.105 This high crystalline nanographene-based DBOV-COF showed strong light-harvesting ability and high charge-carrier mobility, which allowed its usage as an efficient photocatalyst in the hydroxylation of several arylboronic acids.
Following a different synthetic approach, Pastoetter et al.106 proposed the synthesis of two fully conjugated vinylene-based COFs (PPQV1 and PPQV2) through a Horner–Wadsworth–Emmons reaction between 2,3,8,9,14,15-hexa(4-formylphenyl)diquinoxalino[2,3-a:2′,3′-c]phenazine and 1,4-bis(diethylphosphonomethyl)benzene or 4,4′-bis(diethylphosphono methyl)biphenyl, respectively. The observed low optical energy band gap for the two COFs due to their planar conjugation resulted in increased π-delocalization, making these materials ideal catalysts in photoelectrochemical hydrogen evolution reactions.
Yaghi and co-workers107 proposed an unsubstituted olefin-based COF from the reaction of 2,4,6-trimethyl-1,3,5-triazine (TMT) with 4,4′-biphenyldicarbaldehyde (BPDA) under typical solvothermal conditions. The as-prepared COF-701 showed remarkable chemical stability over Brønsted and Lewis acids, bases and organolithium reagents and thermal stability up to 400 °C. COF-701 was successfully utilized as a scaffold to hold a strong Lewis catalyst like BF3OEt2 and participated effectively in acid-catalysed organic transformations.
More recently, Lei et al.108 used TMT and 1,2-di(4′-formylphenyl)acetylene (L-PH-A-CHO) to prepare an alkyne containing COF, COF-TMT-A. Despite the relatively low specific surface area (221 m2 g−1), the presence of the alkyne group and the conjugated π-electron character of COF-TMT-A optimized its electronic band structure, and the fluorescence lifetime was measured as 4.499 ns. COF-TMT-A showed improved selectivity and photocatalytic activity in CO2 reduction.
At the same period, another fully sp2-carbon conjugated COF was prepared under typical solvothermal conditions via the condensation of 1,3,6,8-tetrakis(p-formylphenyl)-pyrene (TFPPy) with 2,2′-(2′,5′-difluoro-[1,1′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4′,1′′-terphenyl]-4,4′′-diyl)diacetonitrile (FTPDAN).109 The as-prepared Py–FTP-COF presented high crystallinity, thermal stability and a carrier lifetime of 3.72 ns, which is considered efficient for charge separation phenomena.
:4′,1′′-terphenyl]-4,4′′-diyl)diacetonitrile (FTPDAN).109 The as-prepared Py–FTP-COF presented high crystallinity, thermal stability and a carrier lifetime of 3.72 ns, which is considered efficient for charge separation phenomena.
A new series of aza-based COFs (Hex-Aza-COFs) was reported via solvothermal condensation between hexaketocyclohexane and 1,2,4,5-tetraminobenzoquinone or 2,3,6,7-tetraamine-phenazine, respectively.111 Although this kind of 2D-COFs are fully aromatized, however, they showed moderate crystallinity due to the interlayer charge repulsion of the polarized nitrogen atoms, which prohibits long-range π–π stacking. Additionally, Hex-Aza-COF showed low BET surface area (124 m2 g−1), while at the same time, they showed high capacitance values when used as negative electrodes in asymmetric supercapacitors. Besides its electrochemical properties, the phenazine-based COF represented an electron donor–acceptor system with visible-light absorption ability and a strong emission band at 522 nm with an average lifetime of 4.6 ns, making it an efficient heterogeneous photocatalyst for the oxidative [3+2] cycloaddition of phenols with olefins.112
The widely used COF building blocks TAPB and DMTA were incorporated with diethyl phosphite in a simultaneous 3-component reaction at room temperature to construct an α-aminophosphonate-linked COF.113 Under the same conditions, the more sophisticated TAPP-2H was utilized to afford APCOF-1, which proved to be useful in heavy metal ion removal, while at the same time it showed solar-powered bactericidal activity. One of the most important advantages of this synthetic approach was that it could take place at the gram scale.
More recently, the well-established photocatalytic multi-component-based polymerization, known as Petasis reaction, has been successfully utilized for COF synthesis.114 The first COF, synthesized under these conditions, namely, Cy–N3-COF, included 1,3,5-tris(4-formyl-phenyl) triazine, hydrazine, and potassium cyclohexyltrifluoroborate as the components, and the reaction took place in the presence of [Ir(dtbbpy)-(ppy)2]PF6, acetic acid, and anhydrous 1,4-dioxane under blue LED irradiation for 24 h at ambient conditions. A series of porous crystalline COFs have been also synthesized following the same strategy by the condensation of potassium cyclohexyltrifluoroborate with different aldehydes and hydrazines or amines or hydrazides. The as-prepared COFs are expected to be efficient photocatalysts for the visible light-driven oxidative hydroxylation of arylboronic acids.
A solvent-free KCl-catalysed cyclotrimerization of carbonitrile monomers, namely, 1,4-dicyanobenzene (DCB), 1,3,5-tricyanobenzene (TCB) and 4,4′-dicyano-1,1′-biphenyl (DCBP), was reported, resulting in triazine-based 2D-polymers.115 The KCl planar lattice played the role of a scaffold that adsorbs the carbonitrile monomers and was subsequently distorted, thus gradually activating the cyano-groups allowing the cyano-cyclotrimerization reaction and finally the formation of a triazine ring. The as-prepared covalent triazine framework nanosheets (CTF–NSs) exhibit a remarkable photocatalytic hydrogen evolution rate and water splitting ability, and they might also be prepared at the gram scale.
A new and unpresented type of covalent organic frameworks bearing a nitrone linkage was prepared under solvothermal conditions from the condensation of multi-nitroso arenes, such as 1,3,5-tris(4-nitroso) benzene (TPB-3NO) and 1,3,5-tris-[4-nitroso-(1,1-biphenyl-4-yl)]benzene (TBPB-3NO), with 1,1′-[(2,5-dimethoxy-1,4-phenylene)bis(methylene)]bis[pyridinium] (BPS).116 These nitrone-based COFs present moderate crystallinity and good chemical stability after immersion in water and various organic solvents (ethanol, DMF, DMSO) but they proved to be unstable in strong acidic or basic conditions.
Overall, the recent literature suggests that COFs bearing chromophores in the form of fully extended conjugated systems such as porphyrins,78,93,94 anthracenes,101 and pyrenes,109 connected through robust linkages as imine bonds or carbon–carbon double bonds, are the most potential candidates in photocatalytic ROS-mediated applications. Table 1 summarizes the most important porous covalent organic frameworks divided by their linkage type as well as their structure and porosity parameters.
| a/a | COF | Linkage type | BET surface area (m2 g−1) | Pore size (nm) | Ref. |
|---|---|---|---|---|---|
| 1 | TPB–DMTP-COF | Imine-linked | 2105 | 3.26 | 89 |
| 2 | BDF–TAPT-COF | Imine-linked | 998 | 2.3 | 90 |
| 3 | CPTPA-COF | Imine-linked | — | 1.45 | 92 |
| 4 | BTTZ-Por COF | Imine-linked | 2678.2 | 1.93 | 93 |
| 5 | FBQD-Por COF | Imine-linked | 2067.9 | 1.68–2.22 | 93 |
| 6 | PTBC-Por COF | Imine-linked | 2793.6 | 1.61–2.20 | 93 |
| 7 | CCOF-1 | Imine-linked | 266 | 1.02 | 95 |
| 8 | CCOF-2 | Imine-linked | 335 | 1.06 | 95 |
| 9 | NL-COF-1 | Imine-linked | 797 | 2.24 | 96 |
| 10 | NL-COF-2 | Imine-linked | 1061 | 2.18 | 96 |
| 11 | TFP–EDDA-COF | Ketoenamine-linked | 523 | 3.0. | 97 |
| 12 | TFP–BDDA-COF | Ketoenamine-linked | 758 | 3.4 | 97 |
| 13 | TFP–Tt-COF | Ketoenamine-linked | — | 1.41 | 98 |
| 14 | TFP–PA-COF | Ketoenamine-linked | 446 | 1.2 | 99 |
| 15 | TFP–MPA-COF | Ketoenamine-linked | 604 | 1.2 | 99 |
| 16 | TFP–Azo-COF | Ketoenamine-linked | 809 | 2.6 | 99 |
| 17 | TFP–AHAn-COF | Ketoenamine-linked | 363 | 1.8 | 99 |
| 18 | TFPPA-COF | Ketoenamine-linked | 837 | 1.2 | 100 |
| 19 | TFPBD-COF | Ketoenamine-linked | 483 | 1.3 | 100 |
| 20 | TFPDT-COF | Ketoenamine-linked | 871 | 2.3 | 100 |
| 21 | TFPAQ-COF | Ketoenamine-linked | 1023 | 1.2 | 101 |
| 22 | RC–COF-1 | Ketoenamine-linked | 1712 | 1.6 | 102 |
| 23 | COF-p-3Ph | CC-linked |
963 | 3.0 | 104 |
| 24 | COF-p-2Ph | CC-linked |
1036 | 2.2 | 104 |
| 25 | COF-m-3Ph | CC-linked |
1231 | 1.7 | 104 |
| 26 | DBOV-COF | CC-linked |
581 | 1.9 | 105 |
| 27 | PPQV1 | CC-linked |
440 | 0.77/1.38 | 106 |
| 28 | PPQV2 | CC-linked |
100 | 0.88/1.45 | 106 |
| 29 | COF-701 | CC-linked |
1366 | 1.14 | 107 |
| 30 | COF-TMT-A | CC-linked |
221 | 1.4 | 108 |
| 31 | Py–FTP-COF | CC-linked |
1780 | 2.54 | 109 |
| 32 | APCOF-1 | a-Aminophosphonate-linked | 689 | 1.3 | 113 |
| 33 | Cy–N3-COF | Hydrazine-linked | 788 | 1.98 | 114 |
| 34 | CTF–NSs | Triazine-based | 304 | 1.1 | 115 |
4. ROS generation and detection in COF-based photocatalytic systems
4.1. Transient intermediates
It is well-established that the oxidative activity of semiconducting materials is mainly assessed by the extent of formation of reactive oxygen species. The latter intermediates may be generated by either energy- or electron transfer-related processes. A typical ROS, generated by energy transfer process, is singlet oxygen (1O2). Regarding the electron transfer- mediated reactions, ROS were shown to form through stepwise redox processes. These may involve the reduction of dissolved oxygen gas as well as the oxidation of water.The oxidative path may also take place using a sacrificial agent. Thus, superoxide radical anion and hydroxy radicals may be formed, simultaneously. Furthermore, the neutral molecule of hydrogen peroxide may be generated either by two-electron reduction or two-hole oxidation mechanism.
COFs have been used as functional photocatalysts towards the generation of singlet oxygen by energy transfer processes. In the seminal work of Jiang and co-workers,117 a squaraine-linked porphyrin-based COF with a unique zigzag structure towards a distorted tetragonal architecture with a pore size of 2.1 nm was proposed. This material proved to be an excellent photocatalyst for the generation of singlet oxygen due to the presence of squaraine-linkage, which allows an extended resonance conjugation between the porphyrin and squaraine moieties.
Two years later, the same research group118 proposed the combination of 2,5-dihydroxyterephthalaldehyde (DHTA) and 5,10,15,20-tetrakis(4′-tetraphenylamino)porphyrin derivative (MP; M = Cu, H2, and Ni) for the construction of 2D COFs, locked by H-bonding interactions between adjacent hydroxy moieties, at variable population.
The excitation of the copper-based COF under 500 nm visible light resulted in the efficient generation of singlet oxygen. The latter species was detected using DPBF as a probe. The reaction of the aforementioned ROS with the furan derivative forms an unstable peroxide that decomposes into (colourless) 1,2-dibenzoylbenzene. By monitoring the spectral changes during the photoinduced generation of singlet oxygen, it was observed that the absorption of furan derivative at 420 nm was diminished, whereas clear isosbestic points suggested the generation of an organic adduct absorbing in the UV region (benzene derivative) (Fig. 8a). Yet, the metal-free framework was found to be the most efficient photocatalyst towards the evolution of the 1O2 species. It was observed that the furan derivative (50 μM) was quantitatively diminished within a time period of about 200 min using the protonated porphyrin-based COF (Fig. 8b). The photocatalytic activity of the COFs was directly correlated with the content of H-bonding functionalities, leading to intralayer docking interactions.
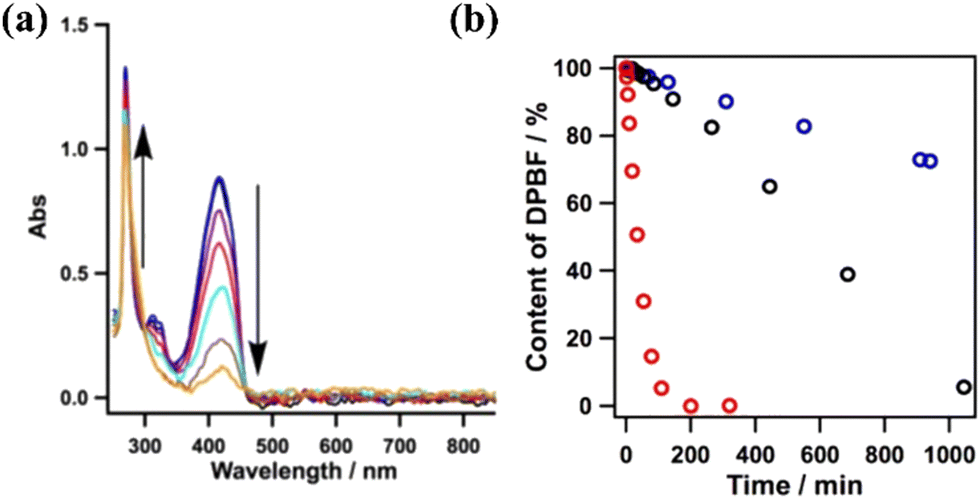 | ||
| Fig. 8 (a) Absorption spectral changes of DPBF in the presence of the Cu-based COF. (b) Effect of different COFs (0.1 mg) on the reaction (black: Cu-based COF; blue: Ni-based COF; red: metal-free COF). Adapted with permission from ref. 118. Copyright 2015 American Chemical Society. | ||
Analogously, 3D porphyrin-based COFs have been prepared by the condensation of tetra(p-aminophenyl)methane (TAPM) and 5,10,15,20-tetrakis(4-benzaldehyde)porphyrin (M: H2, Cu).119 Upon the photoirradiation (λ = 500 nm) of the H2-porphyrin-based COF in the presence of an appropriate label (9,10-dimethylanthracene), the authors have indirectly monitored the time-dependent generation of singlet oxygen by UV-Vis spectroscopy. By following the absorption of the 375 nm peak of the label, it was found that almost 99% of the starting concentration of anthracene derivative (10−2 M) was quenched within 90 min. On the contrary, the copper-porphyrin-based COF was comparatively inactive towards the generation of singlet oxygen since the anthracene probe concentration dropped to half its value after 12 hours photoirradiation. The poor photosensitizing effect was ascribed to the presence of paramagnetic metal ion within the porphyrin structure.
Using an analogous porphyrin–quinoline-based framework, the singlet oxygen-mediated dissociation of organic substances has been studied. Specifically, the aerobic C–C cleavage of cycloalkanone derivatives was demonstrated by Van Der Voort's group.120 The combination of ESR measurements as well as scavenging experiments with 1,3-diphenylisobenzofuran probe confirmed the involvement of the aforementioned ROS to the mechanistic scheme. The utilization of 2,2,6,6-tetramethyl-1-piperidine N-oxide (TEMPO) as an ESR trapping agent gave rise to a triplet peak signal with 1:1:1 intensity ratio. This implied the generation of singlet oxygen species. This was strongly supported by UV-Vis absorption spectroscopy data using the abovementioned furan derivative, which possesses an absorption maximum at about 412 nm. It was observed that a 70 μM scavenger concentration was fully converted to its colourless photoadduct within a time period of 6 min. These results suggested that singlet oxygen was the main ROS, which catalysed the oxidative cleavage of cycloalkanone derivatives.
Beside using furan derivatives as optical probes for singlet oxygen detection, α-terpinene was used as an alternative trapping substance. The cyclic diene may be oxidized by singlet oxygen, giving rise to variable yields of ascaridole photoadduct (Fig. 9a). Xiao and co-workers121 have developed a functional carbon framework, consisting of pyrene and electron-deficient thiadiazole moieties (Py–Td COF).
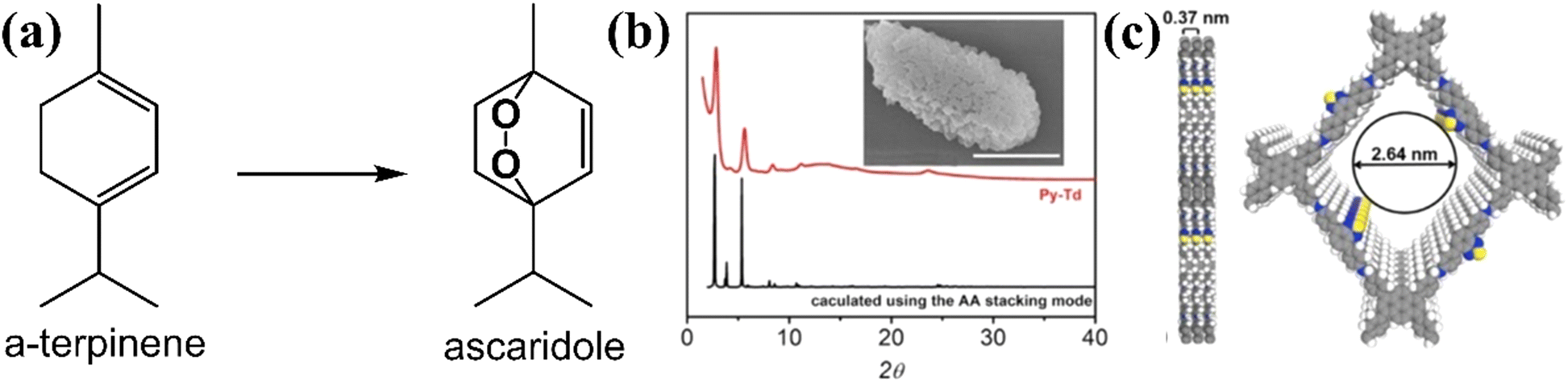 | ||
| Fig. 9 (a) Conversion of terpinene to ascaridole. (b) Experimental and calculated pXRD patterns. Inset: SEM image of Py–Td (scale bar, 1 μm). (c) Graphical view of the AA-stacking mode of Py–Td (gray, C; blue, N; yellow, S; white, H). Adapted with permission from ref. 121. Copyright 2020 Elsevier. | ||
The high crystallinity was assessed by powder X-ray diffraction technique (pXRD) (Fig. 9b), with the most intense peaks observed between 3 and 6 degrees, approximately. There was a striking similarity between the experimental and the calculated patterns. A graphic view of the framework demonstrated an A–A stacking mode (Fig. 9c). The homogeneity of the material was examined by SEM imaging (inset in Fig. 8b), demonstrating aggregate structures of irregular polyhedral nanostructures. It was expected that maximum donor–acceptor contrast between the structural units would give rise to enhanced photocatalytic activity. After the calculation of ascaridole yields by 1H-NMR spectroscopy, it was observed that the highest value was estimated for the crystalline Py–Td COF. Its amorphous counterpart demonstrated an appreciably lower ascaridole yield, about 60% of that estimated in the case of crystalline framework.
It is noted here that the Xiang group developed an analogous pyrene–quinoline-bridged COF, which demonstrated different photocatalytic activity.122 The authors observed that the superoxide radical anion was the sole mediator towards a wide range of photoreactions, such as the syntheses of either 2,4,6-triphenylpyridines, benzimidazole, or sulfoxide derivatives. The detection of the transient species took place through the utilization of an ESR trapping agent (DMPO probe) as well as p-benzoquinone. Regarding the latter case, the main product of the photoinduced reaction between p-benzoquinone and O2˙− is hydroquinone (H2Q). Both substances were quantitatively monitored through high-pressure liquid chromatography technique.
Han et al.123 have developed a functional COF by the condensation of diamine- and triformyl-substituted aromatic monomers. The photocatalytic activity of the COF material was assessed for its potential to oxidatively convert benzylamine to imine. To investigate the reaction mechanism, the imine yields of the main photocatalytic reaction were followed in the presence of certain scavengers for each of the reactive oxygen species. Specifically, p-benzoquinone has been widely used as an efficient scavenger for superoxide radical anion, a transient species that is originated by the interaction of photogenerated electron (at the photosensitizer conduction band) with the oxygen molecules. Analogously, an oxalate salt was used as an efficient hole scavenger. In both cases, the benzylamine conversion was appreciably diminished, implying the participation of the abovementioned transients in the mechanistic scheme of oxidative reaction. ESR measurements using DMPO trapping agent further supported the involvement of superoxide anion radical transient.
The involvement of superoxide radical anion in oxidative mechanistic schemes may be alternatively verified using a scavenger of photogenerated electron, such as AgNO3.124
Regarding the molecular oxygen activation process, related studies have shown that this may be regulated with the appropriate chemical decoration of the carbon framework. The Jiang group developed porphyrin-based frameworks through the hydrothermal treatment of DHTA and 5,10,15,20-tetrakis(4-aminophenyl)-21H, 23H-porphine derivatives (Tph–M; M = Ni and Zn).125 It was found that the Zn2+-based porphyrinic ring selectively led to energy transfer processes, thus giving rise to singlet oxygen generation. On the contrary, the Ni2+-based framework accelerated exciton dissociation towards the formation of isolated carriers and the subsequent generation of superoxide radical anion species (Fig. 10).
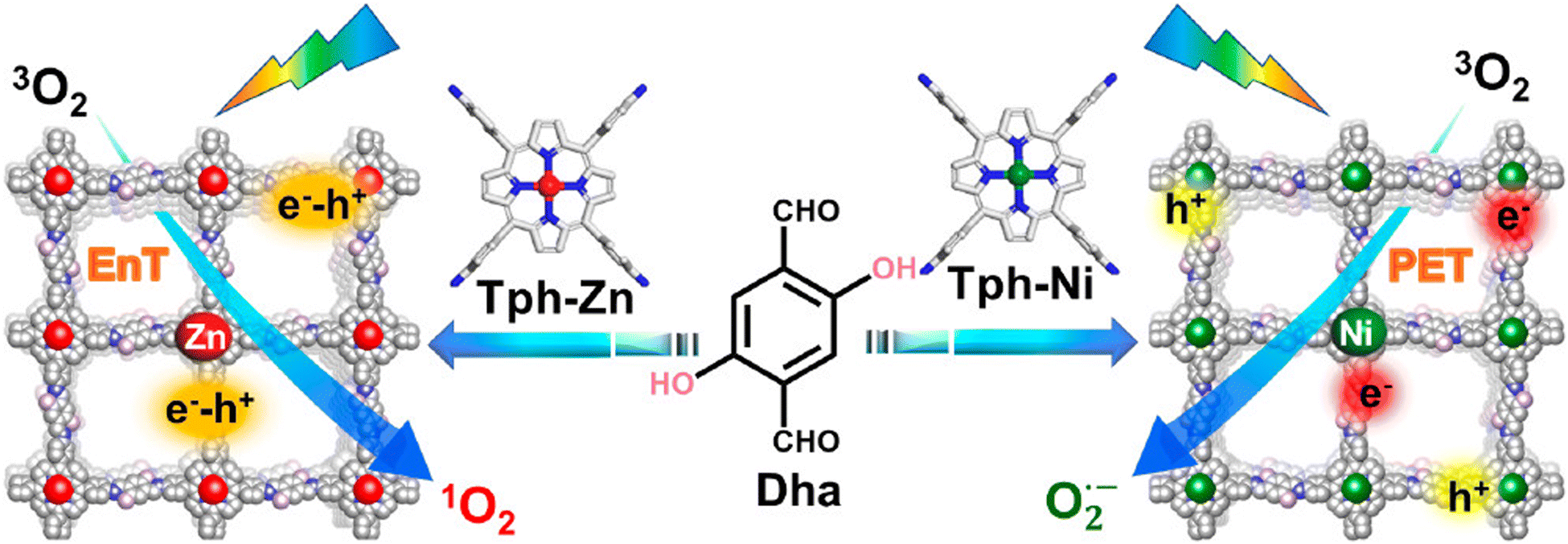 | ||
| Fig. 10 Synthesis and crystal structure of DhaTph–M (M = Zn and Ni) with selective oxygen activation to either singlet oxygen or superoxide radical under visible light irradiation. EnT: energy transfer. PET: photoinduced electron transfer. Adapted with permission from ref. 125. Copyright 2020 American Chemical Society. | ||
Using the model reaction of photosensitized terpinene oxidation, the authors observed adjustable selectivity regarding the composition of the photoadducts. Specifically, under identical visible light irradiation conditions, the utilization of DhaTph–Zn resulted in the conversion of terpinene to ascaridole, whereas DhaTph–Ni selectively produced p-cymene. To further support the selective oxygen activation mechanism, the authors performed oxidation experiments in benzidine derivatives. The involvement of 1O2 in the DhaTph–Zn-mediated photocatalytic reaction was proved through the utilization of carotene as an appropriate scavenger of the aforementioned ROS. The organic quencher completely suppressed the conversion of benzidine derivatives. The activation of molecular oxygen and the subsequent generation of energy transfer-mediated species was also observed in the metal-free DhaTph.126
As stated previously, the DMPO probe has been used as an ESR trapping agent for the detection of superoxide radical anion. The very same probe has been also used for the monitoring of hydroxy radicals through the recording of DMPO–˙OH adduct by the ESR technique.127,128 In an analogous study of Cheng and co-workers,129 an imine-linked framework was covalently decorated with both iron oxide and iron phosphide nanostructures. The synergetic effect of the dual cocatalyst was critical towards the efficient charge carrier separation. This was directly demonstrated by surface photovoltage spectroscopy (Fig. 11a). DMPO spin-trapping experiments under visible light irradiation demonstrated that both superoxide and hydroxy radical transient intermediates were formed in the case of the three-component hybrid (Fig. 11b and c). An indirect proof for efficient charge transfer in the formed heterojunctions was the enhanced intensity for each of the ESR spectra of the hybrid when compared with the corresponding ESR spectra of the parent components. According to the ESR data, a Z-scheme type mechanistic pathway was suggested to lead to efficient water splitting, with the iron phosphide component being the reduction catalytic site (Fig. 11d).
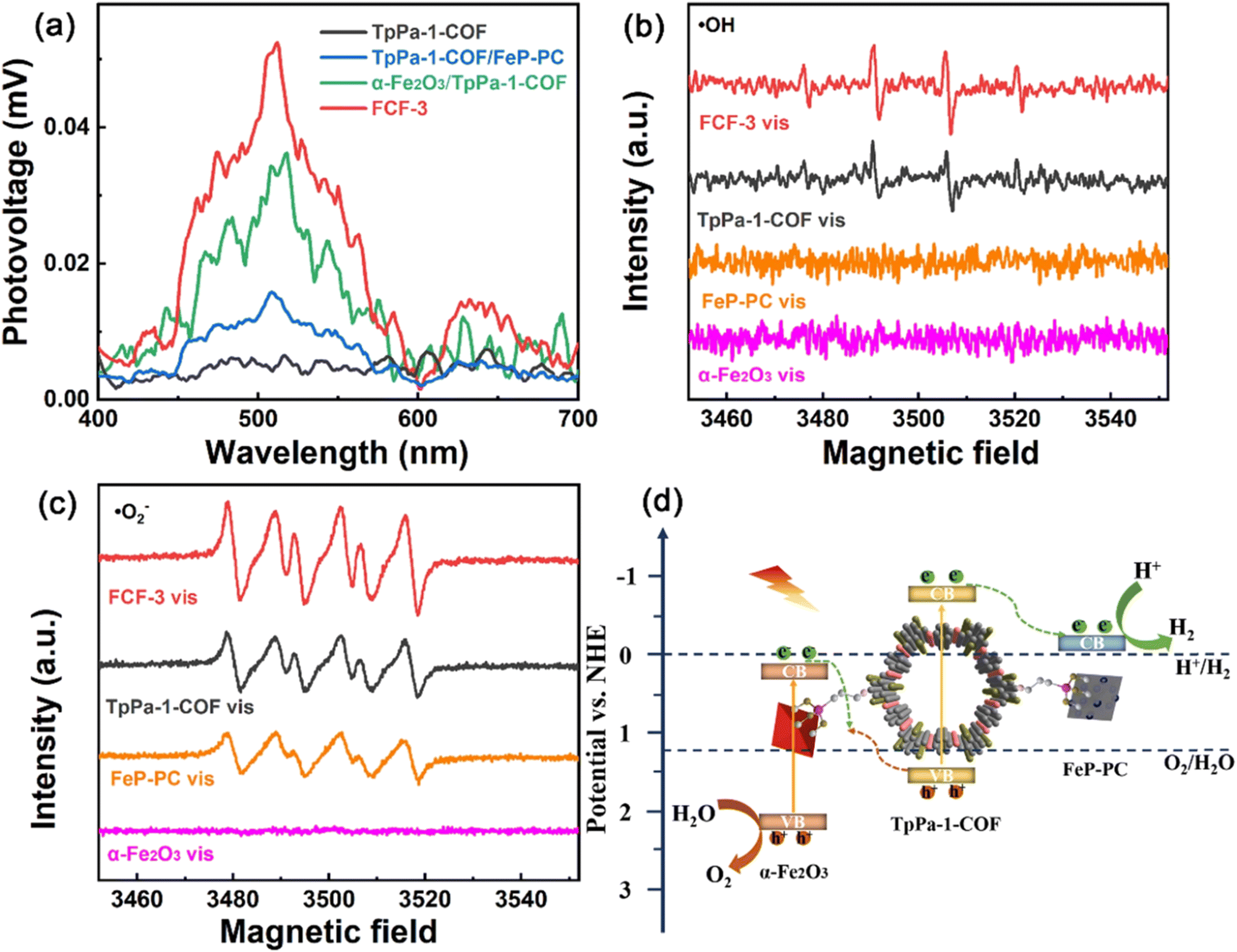 | ||
| Fig. 11 (a) Surface photovoltage spectra of TpPa-1-COF, α-Fe2O3/TpPa-1-COF, TpPa-1-COF/FeP-PC and FCF-3 hybrid. (b) and (c) DMPO spin-trapping EPR spectra of α-Fe2O3, FeP-PC, TpPa-1-COF and FCF-3. (d) Proposed mechanistic scheme of redox reactions in the hybrid. Adapted with permission from ref. 129. Copyright 2022 Elsevier. | ||
The simultaneous participation of both 1O2 and superoxide radical anion in COF-related photocatalytic processes was demonstrated in recent studies. The monitoring of these transients was assessed by ESR through trapping experiments with TEMPO and DMPO, respectively. The transients were involved in reactions such as the photocatalytic reduction of uranium(VI) in natural seawater130 as well as the synthesis of multi-substituted olefins.131 In the latter case, the authors performed additional trapping/quenching experiments using benzoquinone (superoxide radical anion scavenger) and sodium azide (singlet oxygen scavenger). All the supportive data strongly demonstrated the synergetic effect of the abovementioned ROS in the synthesis of multi-substituted olefins.
The indirect detection of superoxide radical anion may be achieved in triphenylamine-derived carbon frameworks.132 The irradiation of such materials in the solid state under oxygen atmosphere and subsequent ESR analysis afforded the spectrum of the triphenylamine radical cation. The authors suggested that the excited triphenylamine chromophore interacts with ground state oxygen and yields the charge-transfer adducts, namely, the triphenylamine radical cation and the superoxide radical anion.
4.2. Photocatalytic H2O2 evolution
As stated previously, hydrogen peroxide formation may take place either by two-electron oxygen reduction or by two-hole water oxidation mechanistic paths (Fig. 1). During the last three years or so, COF-assisted H2O2 photosynthesis has been thoroughly studied and discussed.133,134 In the seminal work of Xu and co-workers,135 it was shown that beyond the two-electron oxygen reduction mechanistic scheme, the direct formation of H2O2 was also accomplished via the elusive two-electron water oxidation process. Specifically, the authors developed covalent triazine frameworks (CTFs) modified with either acetylene or diacetylene moieties. The frameworks were prepared through the trimerization of 1,4-dibenzonitrile monomers, bearing the aforementioned unsaturated units. The extended conjugated network in the diacetylene-modified CTF resulted in red-shifted absorption up to about 850 nm.The exfoliated acetylene-bearing CTF nanosheets (thickness < 10 nm) exhibited high H2O2 production rates via both the aforementioned redox pathways under visible light irradiation (λ > 420 nm). Exfoliated nanosheets of the diacetylene-containing framework in an O2-saturated aqueous environment exhibited a H2O2 production rate of about 97 μmol g−1 h−1. Using NaIO3 as a sacrificial electron acceptor in inert atmosphere, the authors have demonstrated the appreciable formation of H2O2 for either acetylene- or diacetylene-modified CTFs (89 μmol g−1 h−1). This implied the involvement of the water oxidation path in the photoinduced formation of peroxide adduct. It is noted that in the absence of acetylene moieties in the backbone of CTF, H2O2 may be exclusively generated by two-electron oxygen reduction.
Almost at the same time period, Van Der Voort and co-workers136 developed crystalline frameworks via the condensation of C4 amino-modified and C2 carboxaldehyde-modified aromatic monomers. Using ethanol as the hole scavenger, the proposed COFs demonstrated H2O2 production rates of about 97 ± 10 μmol g−1 h−1 through the oxygen reduction pathway.
Subsequent works have achieved pronounced progress towards the development of efficient photocatalytic systems. Specifically, Li and co-workers137 have successfully integrated active heptazine moieties with aromatic linkers, both being trifunctional, through the condensation reaction between amine and aldehyde functions. The reaction took place in DMSO within a sealed glass tube at 120 °C (Fig. 12a). The formed keto-amine crosslinks were found to strongly contribute to the extension of the conjugated network and the subsequent absorption shift to the whole window of the visible spectrum. A fibrous morphology with abundant voids was demonstrated by electron microscopy (Fig. 12b and c). The mean diameter of the fiber-like structures was about 50 nm. Two main diffraction peaks at 7.8° and 27.7° were assigned to the (100) and (002) crystallographic planes, respectively (Fig. 12d). Photocatalytic H2O2 evolution was assessed in the presence of benzyl alcohol as the sacrificial agent under visible light irradiation. Under the same conditions, the COF-TpHt sample was about 50 times more efficient than the reference C3N4. The H2O2 production rate for the porous carbon framework was estimated to be 11986 μmol g−1 h−1, much higher than the ones of analogous frameworks. Using an optimized photocatalyst content (0.25 g L−1), the peroxide rate was further increased to 23148 μmol g−1 h−1. The apparent quantum efficiency at 420 nm (λmax of photocatalyst absorption spectrum) was calculated to be as high as 38%, clearly reflecting the diminished electron–hole recombination phenomena and enhanced charge separation. Scavenging experiments have clearly demonstrated the involvement of superoxide radical anion as the predominant intermediate regarding the oxygen reduction to H2O2.
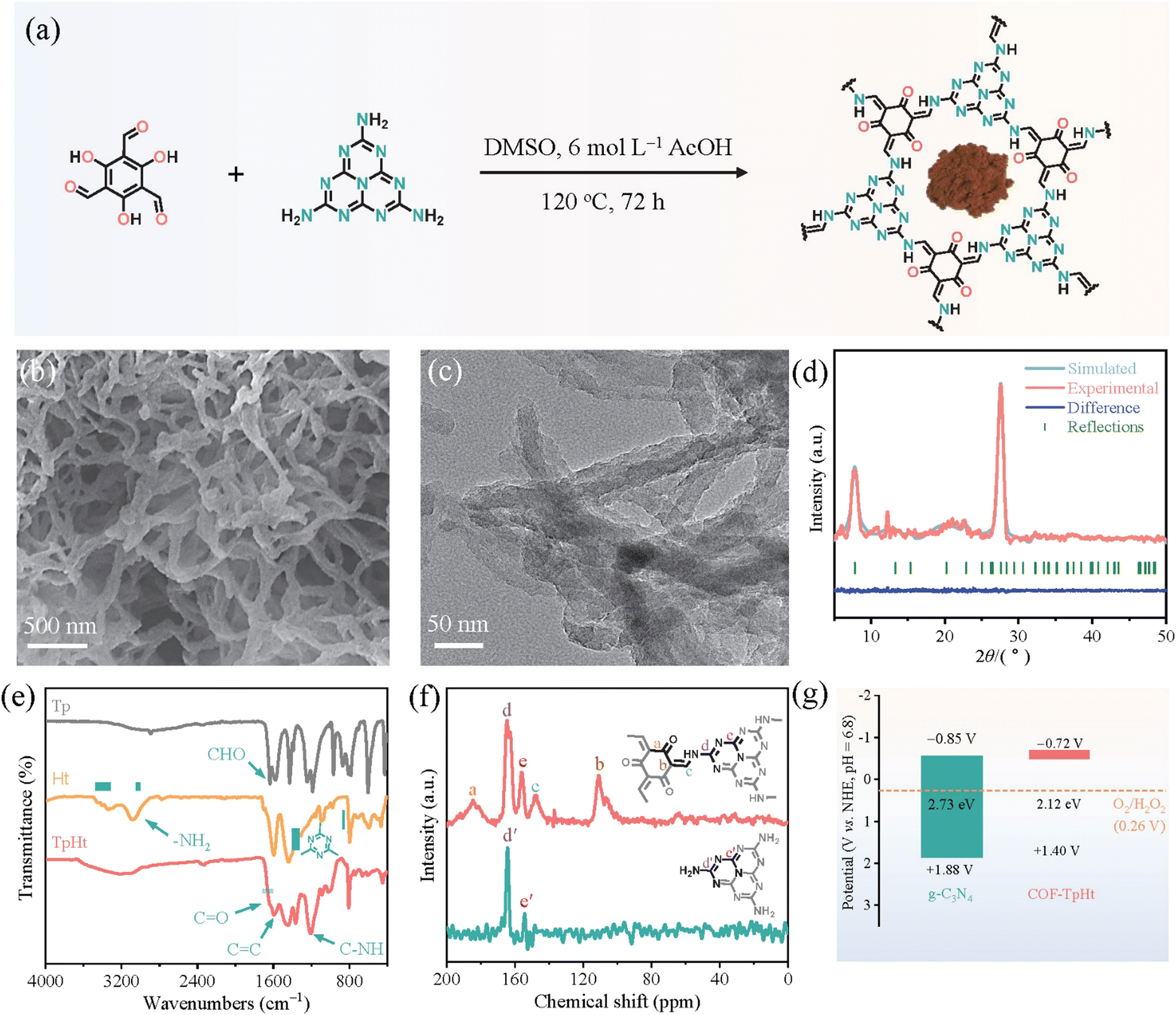 | ||
| Fig. 12 (a) Schematic synthetic protocol towards COF-TpHt together with its molecular structure and photograph of the powder product. (b) SEM imaging. (c) TEM imaging. (d) Experimental and simulated XRD patterns of COF-TpHt together with its structural refinement. Adapted with permission from ref. 137. Copyright 2023 Elsevier. | ||
Unprecedented hydrogen peroxide rate was achieved in the work of Gu and co-workers.138 The authors have developed protonation-induced dispersable porous frameworks by the trimerization of a photoactive benzonitrile derivative. The resulting protonated triazine moieties were found to induce the enhanced dispersability of the polymer framework in triflic acid. Under optimized conditions, the triazine-based photocatalyst yielded a peroxide rate up to 23700 μmol g−1 h−1 using 10 v/v% benzyl alcohol as a hole sacrificial agent. The apparent quantum efficiency at 450 nm was calculated to be as high as 11.3%. It is noted that the oxidation adduct, benzaldehyde, has been suggested to promote the auto-photocatalytic oxidation of aromatic alcohol, producing large quantities of H2O2.139
A sophisticated strategy was developed by the Wang group in order to engineer the polarization of COF frameworks.140 The authors integrated highly polar ionic liquid-type functionalities on the backbone of the COF lattice. The ionic network was prepared by the condensation of a triformyl-modified aromatic monomer with diimidium bromide under microwave conditions. The presence of ionic moieties gave rise to enhanced dielectric constant value, which is related to polarization contributions within the lattice. The successful charge carrier separation resulted in a H2O2 production rate of 10000 μmol g−1 h−1via both reductive and oxidative mechanistic paths. The ionic COF demonstrated a photocatalytic activity of about 7 times higher than the corresponding one of neutral COF counterparts.
A different synthetic strategy was utilized by Zheng and co-workers at room temperature conditions.141 The authors adopted a liquid crystal-directed production of crystalline frameworks. Specifically, the liquid crystal medium consisted of Triton X-100 surfactant, n-decyl alcohol and water in a certain composition. In the aforementioned medium, a trifunctional amine-terminated triazine monomer was crosslinked with a bifunctional formyl-modified arene. Beside using aldehyde-modified arenes of the C2 symmetry, trifunctional linkers were also used such as tris-formyl modified triazine (C3 symmetry). Highly crystalline frameworks were grown within the lamellar liquid crystal phase region. The highest crystallinity was achieved when the ratio of Triton X-100, n-decyl alcohol and water was 25%, 10%, and 65%, respectively. Regarding the morphology of the porous structure, electron imaging showed entangled ribbons with a width of about 50 nm. In the presence of ethanol as the hole scavenger, the optimized porous COF demonstrated a H2O2 production rate of 4347 μmol g−1 h−1. The hole oxidation of ethanol was suggested to provide protons for the two-electron reduction of oxygen. Both precursor monomers were of C3 symmetry and contained triazine as the core component.
Gu and co-workers142 have developed a functional porous framework (Bpy-TAPT) using a tetraformyl-modified bipyridine and a triamino-modified triazine as active monomers. Using nonstoichiometric crosslinking conditions, a certain number of carboxaldehyde moieties remained as pendant groups. The latter were found to accelerate carrier separation due to their electron-rich properties and helped to improve the adsorption process of molecular oxygen on the porous framework. In the absence of any hole scavenger or cocatalyst, Bpy-TAPT showed a H2O2 production rate of 4038 μmol h−1 g−1, exceeding that of all previously reported COF-based photocatalysts.
In the absence of a sacrificial agent, analogous H2O2 evolution rates were achieved by simultaneous oxygen reduction and water oxidation in the work of Yue et al.143 The authors have constructed a functional imine-linked COF bearing both photo-oxidation sites (benzene moieties) as well as photo-reduction sites (thiophene rings). A thiophene-containing triformyl-modified monomer was associated with either a triamino-modified pyridine derivative or a triazine one. In the former case, the yield of the peroxide reached a production rate of about 4060 μmol h−1 g−1 in deionized water. Slightly lower performance was observed in natural sea-water (3364 μmol h−1 g−1).
Han and co-workers144 have developed a functional photocatalytic system, by which the mechanism of one-step two-electron oxygen reduction reaction was regulated properly. The authors decorated sulfone functionalities onto the backbone of a porous carbon framework. In the absence of any hole scavenger and in air atmosphere, an efficient photocatalyst for H2O2 generation was demonstrated. Under visible light irradiation, the sulfone-modified COFs have shown pronounced photocatalytic activity with an H2O2 yield of about 3900 μmol h−1 g−1, outperforming analogous metal-free systems under similar conditions. The sulfone functionalities were shown to inhibit the electron/hole recombination and act as functional adsorption sites for the oxygen substrate. The dominance of one-step two-electron oxygen reduction mechanistic scheme resulted in efficient H2O2 generation with high selectivity.
In order to further promote the separation of charge carriers and its subsequent transport in the corresponding catalytic sites, Wu et al.145 have accomplished to chemically engineer the polarization of the conjugated networks. Specifically, the authors attached thiourea functionalities onto covalent triazine frameworks (CTFs). The thiourea-modified CTF demonstrated an appreciable enhancement in the photocatalytic H2O2 production rate, reaching a value of about 3270 μmol h−1 g−1. In the absence of sacrificial agents, the H2O2 generation quantum efficiency was 8.6% at 400 nm. It was suggested that oxygen molecules were adsorbed onto the triazine units and formed endoperoxide species, by which the reduction process evolved rapidly. On the contrary, the holes were gathered at the thiourea site. Also, molecular oxygen was generated in situ via four-electron water oxidation. This afforded an additional boost to the reductive mechanistic paths for the two-electron formation of H2O2 (Fig. 13).
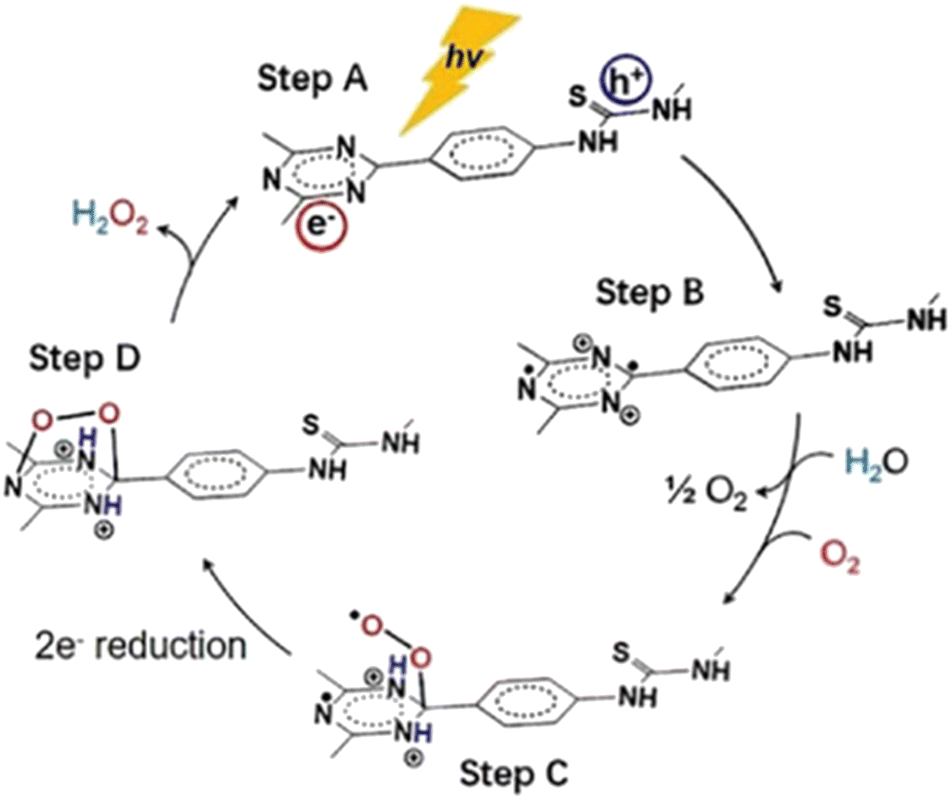 | ||
| Fig. 13 Mechanism for photocatalytic H2O2 production on the surface of Bpt-CTF. Adapted with permission from ref. 145. Copyright 2022 Wiley. | ||
Due to both low solubility and slow diffusion of molecular oxygen in aqueous systems, Li et al.146 developed a sophisticated concept, in which H2O2 formation took place in a solid–liquid–gas interface (Fig. 14). As it is known, gas-phase oxygen concentration (≈ 8.7 μmol cm−3) at the triphase interface is much higher than that in oxygen-saturated aqueous system (0.25 μmol cm−3). Furthermore, the low-rate adsorption of dissolved oxygen to the catalyst surface may be overcome due to the enhanced diffusion rate of gas-phase oxygen reactant. The concept involved the loading of the COF component onto a porous carbon fiber paper to form a catalyst layer. Due to the hydrophobicity of the polymeric semiconductor, water may partially wet the catalyst layer but cannot penetrate the carbon paper layer, leaving the porous structure acting as the oxygen gas diffusion paths. At the solid–liquid–gas interface, the critical components, such as the charge carriers of photocatalyst, gas-phase O2 as well as aqueous protons, were found to be in close contact. At optimized conditions, in the absence of any sacrificial agent, the H2O2 production rate reached a value of about 2882 μmol g−1 h−1 under visible light irradiation (>420 nm). This production rate was found to be 15 times higher than that observed in a diphasic system.
 | ||
| Fig. 14 Schematic illustration of diphase and triphase photocatalytic conditions. Adapted with permission from ref. 146. Copyright 2021 Wiley. | ||
The ideal case of H2O2 generation through both oxygen reduction and water oxidation was achieved in the work of Lan and co-workers.147 The authors have constructed a functional COF bearing both photo-oxidation site (tetrathiafulvalene moieties) as well as photo-reduction site (benzothiazole moieties). The conjugated network appeared to absorb photons in the whole region of the visible spectrum, whereas the catalytic sites resulted in efficient H2O2 formation via either oxidative or reductive pathways. Specifically, the yield of the peroxide reached a production rate of about 2760 μmol h−1 g−1 in the absence of any sacrificial agent.
An alternative strategy to increase the H2O2 yield is to indirectly produce singlet oxygen (1O2) species, the reactivity of which is spin-allowed for the O2 reduction reaction. This may be achieved by designing functional frameworks, carrying either heavy nuclei or moieties favouring donor–acceptor interactions. Thus, nonradiative processes, such as intersystem crossing (ISC) are enhanced, resulting in a successful pre-sensitization of the carbon framework in its triplet excited state. The excited semiconducting photocatalyst may be involved in the generation of singlet oxygen via an energy transfer mechanistic scheme. Di et al.148 have designed COF structures by the condensation of a triformyl-modified triazine monomer and a cyano-arene with C2 symmetry. The cyano moieties resulted in enhanced charge separation and subsequent photosensitization, giving rise to the activation of O2 to 1O2 under visible light irradiation. The CN-COF exhibited a high H2O2 yield rate of 2623 μmol h−1 g−1 and an appreciable apparent quantum efficiency of 9.8% at 420 nm.
Using a multi-component thermal reaction between trifunctional triazine derivatives, bearing amino- and formyl moieties, Thomas and co-workers149 have developed a functional carbon framework based on quinoline rings. In parallel, partially aromatized rings of dihydroquinoline were shown to form within the framework structure by revising the reaction conditions. As expected, the absorption spectrum of the conjugated quinoline-based COF was red-shifted relative to the one of dihydroquinoline. The estimated H2O2 production rate in an O2 atmosphere was 2588 and 2264 μmol g−1 h−1 for the dihydroquinoline- and quinoline-based COF, respectively. The relatively higher performance in the former case was attributed to the optical properties of the carbon framework and specifically in its more negative conduction band energy level (vs. NHE). Both quenching experiments and ESR spin traps strongly supported the involvement of superoxide radical anion in the mechanistic scheme towards the formation of H2O2.
In certain cases, parent carbon frameworks present poor photocatalytic activity due to electron/hole recombination phenomena. Zhang et al.150 have developed a carbon framework (TpPa–Cl) by the condensation of a triformyl-modified arene and a chlorinated aromatic diamine. In order to further enhance its photocatalytic activity, the authors constructed a heterojunction by assembling a nanostructured inorganic semiconductor, such as ZnO. The components were assembled through electrostatic interactions. It was shown that the band structures of ZnO match TpPa–Cl properly, indicating the possibility of forming an S-scheme heterojunction. The hybrids demonstrated enhanced H2O2 production rates, with the maximum yield reaching 2443 μmol g−1 h−1. This value is about 9 times higher than that corresponding to the parent carbon framework.
A series of imine-linked COFs, composed of TAPB and modified terephthaldehydes (PDA–X, X = H2, –CH3, –OH, and –OCH3), were synthesized through open-air thermal reaction.151 The superior activity of the hydroxy-modified framework was attributed to the effective trapping of the holes, which subsequently leads to the recombination inhibition of photo-generated carriers. In addition, the estimated band gap of the hydroxy-modified network was the lowest (2.10 eV) compared with the other ones reported above (Eg spanning between 2.29 and 2.51 eV). It is noted that the surface area values of the H2- and OH-modified frameworks (900–1050 m2 g−1) were estimated to be approximately half that of the corresponding ones of CH3- and CH3O-modified networks (2200–2600 m2 g−1). In oxygen-saturated aqueous solution, the H2O2 production rate reached a value of 2118 μmol g−1 h−1 when using ethanol as the sacrificial agent.
Fluorine-derived cobalt phthalocyanine was effectively assembled with two different tetrafunctional amine-terminated arenes (benzene- and biphenyl-type) to form piperazine-linked crystalline frameworks through nucleophilic substitution reaction.152 The formation of C–NH bonds in the piperazine structure was demonstrated by FTIR spectroscopy, by which a band at 1301 cm−1 was detected. All the tested frameworks were found to be structurally stable in both organic as well as aqueous media (acidic and alkaline). The estimated band gaps were between 1.45 and 1.55 eV, respectively. The lower band gap in the framework derived from tetra-amino-benzene monomer was attributed to the more efficiently fused conjugated structure when compared with the biphenyl-derived COF. At a wavelength of 630 nm, the crystalline frameworks demonstrated an excellent quantum yield of 7.2%. In oxygen-saturated aqueous solution, the H2O2 production rate reached a value of 2096 μmol g−1 h−1 for the tetra-amino-benzene-based COF when using ethanol as the sacrificial agent. The corresponding rate for the biphenyl-based network was 1851 μmol g−1 h−1.
Amorphous covalent organic polymers (COPs) have been also assessed as potential photocatalytic systems for H2O2 generation. Kong and colleagues153 have developed covalent aromatic polyimides by the condensation of TAPT and 1,4,5,8-naphthalene-tetracarboxylic acid dianhydride (NTCDA) monomers. Thioether functionalities were grafted onto the formed COP backbone through the in situ decomposition of the organic medium (dimethylsulfoxide) in solvothermal conditions. The grafting of thioether groups was found to efficiently enhance visible light absorption and facilitate the separation of photogenerated electron/hole pairs during H2O2 photogeneration. In oxygen-saturated aqueous solution, the H2O2 production rate reached a value of 2240 μmol g−1 h−1, which was about 14 times higher than the one of nitrogen-purged solution. The corresponding yield of the control sample (sulfur-free COP) was about 234 μmol g−1 h−1, implying the critical role of the thioether moieties in the charge separation and transport processes.
Similarly, amorphous polymeric microfibers were developed by the joint collaboration of Kong, Wei and Yu groups.154 A combination of the hard template strategy, followed by its etching away, was developed to produce furan-benzimidazole-linked polymer hollow microfibers, bearing both O- and N-based functionalities (Fig. 15). The template microfibers, consisting of Al-diethylenetriamine pentaacetic acid coordination polymer, had an average diameter of about 500 nm. XRD analysis demonstrated the amorphous character of the polymeric network. SEM and TEM imaging assessed the dimension range of the formed hollow microfibers. The length was estimated between 30 and 100 μm, whereas the mean diameter and shell thickness were about 500 and 35 nm, respectively. The formed microfibers were subsequently assembled into macroscopic membranes. In a sacrificial agent-free oxygen-saturated aqueous environment, the membrane catalyst afforded an enhanced H2O2 production rate of 2200 μmol g−1 h−1.
 | ||
| Fig. 15 Illustration of the synthesis of the hollow Furan-BILP microfiber photocatalytic membrane. Adapted with permission from ref. 154. Copyright 2023 Wiley. | ||
In Table 2, general details about the chemical, optical and photocatalytic properties towards the H2O2 production of COF-based systems are illustrated. These carbon-based nanostructures demonstrate H2O2 evolution rates below 2000 μmol per gram of the catalyst per hour of irradiation.
| a/a | Sample | Details | E g (eV) | H2O2 yield (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|---|
| 1 | EBA-COF | CC-linked COF based on triazine-styrene-acetylene sequence |
2.39 | 1830 | 155 |
| 2 | TpMa/g-C3N4 | β-Ketoenamine-based TFP/melamine and carbon nitride type II heterojunction | 2.32 | 1760 | 156 |
| 3 | HEP–TAPT-COF | Imine-linked heptazine and triazine-based photocatalysts | 2.30 | 1750 | 157 |
| 4 | TF50-COF | Imine-linked fluorine-substituted triazine-based COFs | 2.09 | 1739 | 158 |
| 5 | CHF–DPDA COF | Trichloroheptazine-diacetylene-linked frameworks | 2.35 | 1725 | 159 |
| 6 | CTF–NSs & CTF–NS-xBT 0 ≤ x ≤ 10 | Triazine-phenylene and benzothiazole-doped triazine-phenylene frameworks | 2.45 | 1630 | 160 |
| 7 | 4PE–N & 4PE–N–S | Imine-linked COFs based on tetraaniline (4PE) and naphthalene (N) post-functionalized to thiazole-linked COFs | 1.79 | 1574 | 161 |
| 8 | Nx-COFs, x = 0 or 3 | Imine-linked COFs based on TAPB or TAPT and benzo[c][1,2,5]thiadiazole-4,7-dicarbaldehyde | 1.90 | 1570 | 162 |
| 9 | TAPT–HTA-COF | Imine-linked COFs with free hydroxy-groups allowing keto–enol tautomerism | 1.94 | 1482 | 163 |
| 10 | TiO2@BTTA | S-scheme heterojunction by encapsulating TiO2 nanofibers with an imine-linked triazine-based COF | 2.53 | 1480 | 164 |
| 11 | TaptBtt-COF | Benzotrithiophene-triazine-linked COF | 2.29 | 1407 | 165 |
| 12 | Py–Py-COFs | Imine-linked pyrene-based COFs with bi- or tetra-functional amine derivatives | 2.53 | 1242 | 166 |
| 13 | COF-TAPB–BPDA | Imine-linked benzene–acetylene-based COFs | 2.58 | 1240 | 167 |
| 14 | COF–NUST-16 | 3D-COF based on an octa-aldehyde aromatic monomer and a tetra-aminated pyrene derivative | 2.25 | 1081 | 168 |
| 15 | DETH-COF, BBT-COF, BHT-COF and BOT-COF | Hydrazone-linked frameworks based on hydrazine and aldehyde monomers | 2.88 | 1000 | 169 |
| 16 | TDB-COF | Hydrazone-linked thioether-triazine-COFs | 2.67 | 724 | 170 |
| 17 | COF–TfpBpy | Imine-linked bipyridine-based COFs | 2.37 | 695 | 171 |
| 18 | COF-N32 | Triamino-modified triazine coupled with triformyl-arene | 2.42 | 605 | 172 |
| 19 | MRF-250 | Mesoporous resorcinol–formaldehyde bowl-like frameworks | n.d. | 588 | 173 |
| 20 | TpAQ-COF | Ketoenamine-linked anthraquinone–TFP based COFs. | 1.74 | 420 | 174 |
| 21 | Sono-COF-F2 | Imine-linked triazine-based COFs | 2.86 | 415 | 175 |
4.3. Photocatalytic degradation of organic substances
During the last century or so, typical pollutants, such as dyes, drugs and fertilizers, have been discharged into aqueous environments to a large extent. Such accumulation of persistent organic substances, derived from the textile/pharmaceutical industry, is considered as a huge threat for the preservation of living organisms. In this regard, porous carbon-based frameworks have been recently considered as a functional platform for the photocatalytic degradation of organic substances. In this general family, the structural regularity of COFs is expected to give rise to enhanced photodegradation rates. Various pollutants have been studied as analytes for degradation essays. These include either dyes (rhodamine B, methyl blue, methyl orange, etc.) or drugs (tetracycline, paracetamol, etc.). Below, various examples of covalent organic polymer networks are discussed as potential photocatalytic systems for pollutant removal in aqueous environments. In order to compare the photodegradation efficiency of each photocatalytic system, the t50 parameter was calculated for each case. This involves the time needed for the starting analyte concentration to drop to its half value. Thus, the lower the t50 parameter value, the steeper the decay curve of the analyte.Crystalline carbon frameworks have been used as functional supports for the deposition of optically active components. Lu and co-workers177 developed a carbon framework, acquiring 2.8 nm pores. The condensation of 2,5-divinylterephthalaldehyde and 1,3,5-tris(4-aminophenyl)benzene afforded the crystalline nanostructure. The decoration of the lattice by vinyl groups was utilized to graft thiol functionalities by post-modification. Such thiol groups were actually a chemical anchor between the porous framework and the in situ-formed Au nanoclusters. Adsorption experiments in dark conditions confirmed the enhanced potential of both parent COF and COF-Au hybrid towards the removal of RhB in aqueous medium. It is noted that about 99.5% of the dye was adsorbed within 30 min within the pores of the parent COF. Concerning the photodegradation activities, the hybrid demonstrated higher rates. For the parent COF, the dye photodegradation rate was estimated to be 0.0579 min−1, whereas the t50 parameter was about 23 min. On the contrary, the COF-Au hybrid demonstrated a decay rate of about 0.0904 min−1, whereas the t50 parameter was about 9 min. Scavenging experiments implied that both superoxide radical and holes participated in the degradation of RhB dye.
In a similar study, a functional MOF/COF heterostructure was developed by Zhang and co-workers.178 Specifically, a cobalt-based MOF was grown within the 3.8 nm pores of a carbon framework, originated from the condensation of triamino-triazine and diformyl-bipyridine monomers (Fig. 16). The incorporation of MOF promoted photon harvesting. The crystalline hexagonal lattice of the carbon framework was assessed by the intense diffraction peak at 2.8° assigned to the (100) facet. RhB was a model dye analyte that participated in the photodegradation experiments. The porous character of the hybrid photocatalyst was strongly supported by adsorption–desorption measurements in dark conditions. It was found that about 70% of the dye was adsorbed within 60 min of incubation in an aqueous environment. The photodegradation rate was estimated to be 0.22 min−1, whereas the t50 parameter was about 4 min. Scavenging experiments demonstrated that the holes and the superoxide radical anions were the main species that contributed to the degradation of the dye.
 | ||
| Fig. 16 Scheme depicting the confinement synthesis of MOFs in the pores of the COF. Adapted with permission from ref. 178. Copyright 2022 Wiley. | ||
Zhan and co-workers179 have designed an efficient heterojunction between a carbon framework and an inorganic semiconductor BiOBr. The 2D/2D Z-scheme heterostructure was synthesized by the solvothermal reaction at autoclave conditions. The COF component, denoted as TzDa, was prepared by the condensation of TAPT and DHTA monomers. The theoretical weight fraction of the carbon component within the hybrid structure was approximately 9 wt%. The strong diffraction peak at 2.960 (100) implies the high crystallinity of the carbon component. Adsorption–desorption equilibrium data showed that about 25% of the RhB dye was adsorbed within 40 min of incubation in the aqueous environment under dark conditions. Concerning the photocatalytic activity, the dye photodegradation rate was estimated to be 0.090 min−1, whereas the t50 parameter was about 10 min. Scavenging experiments implied that both superoxide radical and holes participated in the degradation of the RhB dye.
Huang and co-workers180 developed a functional heterojunction consisting of CuS and a carbon framework prepared by the condensation of 2,6-diaminoanthraquinone (DAAQ) and TFP monomers. The hybrid assembly was supported onto cellulose fibers, which were decorated by a hyperbranched polyamide-amine polymer, acting as a heterojunction stabilizer. A Z-scheme model was demonstrated among the heterojunction components. The hybrid was assembled in the form of a macroscopic membrane and was used as a functional photocatalyst for dye removal. In the presence of 1% H2O2, the catalyst/RhB solution was continuously irradiated with near infrared light (808 nm). The monitoring of the dye concentration by UV-Vis absorption spectroscopy showed the rapid degradation of the dye. Half of the initial concentration was degraded within one minute, approximately (t50 ∼1 min). The first-order kinetics for dye concentration decay was estimated to be 0.5549 min−1. The absence of either CuS or COF component resulted in a deteriorated reaction rate by about 6–7 times. It is noted that no photocatalytic test in the absence of H2O2 was demonstrated in this study.
Instead of using cellulose fibers as a substrate component, Wang et al.181 adopted a combination of sacrificial template method and a pyrolysis step in order to prepare macroporous chitosan-based microspheres. The latter were used as a functional substrate for the liquid-phase deposition of a hybrid, consisting of Pd nanoparticles and a COF. The framework was originated by the condensation of TFP and triamino-triazine monomers. The XRD pattern of the carbon framework demonstrated two peaks at 3.3° and 27.1°. These corresponded to diffractions from the (100) and (002) facets, respectively. The adsorption–desorption equilibrium data showed that about 10% of the dye was adsorbed within 20 min of incubation in an aqueous environment. The photodegradation rate was estimated to be 0.106 min−1, whereas the t50 parameter was about 8 min. Scavenging experiments implied that both superoxide and hydroxy radicals mainly contributed to the degradation of the RhB dye.
The effect of the density of active centres as well as the conjugation degree of triazine-based frameworks was researched by the group of Cai.183 The authors synthesized various frameworks by the condensation of TFPT with three triamino-aromatic systems in separate experiments. Among the three COFs, the one containing the TAPT monomer unit demonstrated the highest photocatalytic activity towards water purification. Under irradiation, the methyl orange decay kinetics showed that the degradation rate of the dye was 0.090 min−1, whereas the t50 parameter was approximately 10 min. It is noted here that the red-shifted absorption response might play some role in the photocatalytic performance but does not seem to be the critical factor. The band gap of the optimized framework was the highest (2.56 eV) when compared with the ones of the other two COFs (2.49 and 2.21 eV). Scavenging experiments demonstrated that superoxide radical was the main contributor towards dye degradation, whereas some partial assistance was given by hydroxy radical species.
A further enhancement of the photocatalytic activity of the abovementioned framework was accomplished through its integration into functional heterojunctions. Cai and co-workers184 constructed a Z-scheme heterojunction between a titanium-based MOF and a crystalline carbon framework. As described above,183 the latter was synthesized using TFPT and TAPT monomers. The integration of the MOF promoted the charge separation and transfer processes. Either the parent COF or the hybrid demonstrated enhanced the adsorption capacity of organic dyes. Specifically, methyl orange could be efficiently removed from aqueous environments in short periods of time. Within 60 min, the hybrid was able to remove about 65% of the initial concentration of the organic dye, whereas the corresponding percentage for the neat COF was about 25%. This difference in the adsorption capability was reflected in the photodegradation rates. The hybrid photocatalyst demonstrated a dye decay rate of about 0.200 min−1, which was two times higher than that of the parent COF. The dye concentration was diminished to its half value after 5 min of irradiation, while the neat COF needed about 10 min. The crucial roles of both superoxide and hydroxy radicals in the degradation of methyl orange dye were affirmed by scavenging experiments.
The same group developed a crystalline carbon framework based on triazine units.185 This was prepared by the condensation of TFPT and 2,4,6-trimethyl-1,3,5-triazine monomers. The fully conjugated network, consisting of triazine-styrene sequences, gave rise to a pronounced absorption at the visible wavelengths, with an estimated band gap of about 2.46 eV. The planarity of both the monomers resulted in a fibrous morphology, which was assessed by electron microscopy imaging. Although the parent COF demonstrated a rather poor adsorption capability for methyl orange dye (5% within 60 min), the crystalline nanostructure was an efficient photocatalyst towards the degradation of the organic substance. Within the first 5 min of irradiation, the concentration of methyl orange decreased to half its value. Trapping experiments indicated that superoxide radical anion was the main species, which contributed to the photodegradation process.
 | ||
| Fig. 17 (a) XRD spectra of the ball-milled material (TpMA) and the solvothermally prepared one (TpMA-1). (b) Band structures of commercial g-C3N4, self-prepared g-C3N4 and TpMA, and the mechanism of photo-degradation of sulfathiazole catalysed by TpMA. Adapted with permission from ref. 186. Copyright 2021 Elsevier. | ||
These structural differences strongly reflect the adsorption capability of the porous materials under dark conditions. Specifically, a 40 min incubation of the sulfathiazole drug in an aqueous environment resulted in a 20% decrease in the starting substance concentration in the presence of the framework prepared by solvothermal conditions. In comparison, 47% of sulfathiazole was adsorbed within the same time period in the presence of the ball-milled material. The sulfathiazole drug was used as an analyte for comparative photodegradation tests. Under irradiation, the decay kinetics showed that the drug degradation rate in the presence of the ball-milled framework was 0.1498 min−1, whereas the t50 parameter was approximately 5 min. On the contrary, the utilization of the catalyst prepared by the solvothermal protocol gave rise to a lower degradation rate of 0.0385 min−1, with the t50 parameter being approximately 20 min. The proposed mechanism of photo-degradation of sulfathiazole catalysed by TpMA is shown in Fig. 17b.
Crystalline carbon frameworks have been used as functional supports for the deposition of single metal cation catalytic sites.
Zhao and co-workers187 have developed a carbon framework, acquiring 2.0 nm pores. The condensation of 1,4-diaminobenzene and a tetraformyl-modified 1,4-phenylene-bis(2,2′:6′,2′′-terpyridine) derivative afforded the crystalline nanostructure. The decoration of terpyridine moieties through the complexation of copper(II) cations was utilized to enhance the visible light absorption and achieve the desired charge carrier separation. Adsorption experiments in dark conditions confirmed the enhanced potential of Cu-coordinated COF towards the removal of sulfamethoxazole antibiotic in aqueous medium. It is noted that about 57% of the organic substance was adsorbed within 30 min. Concerning the photodegradation activity, the copper-modified COF demonstrated a decay rate of about 0.135 min−1, whereas the t50 parameter was about 10 min.
The similar coordination of cobalt ions in triazine functionalities of CTFs afforded enhanced photocatalytic activity towards the degradation of methylene blue dye.188 The cobalt-modified framework demonstrated a decay rate of about 0.1008 min−1, whereas the t50 parameter was about 10 min. However, it is noted that the photocatalytic degradation of the organic dye took place in the presence of H2O2 oxidant. The optimized dosage of peroxide was about 73 mmol L−1. The concept of metal-doped frameworks as potential photocatalysts for pollutant removal was also studied by Zhu et al.189 The authors have mixed CTF and FeCl3 in the liquid phase. After solvent evaporation, gentle grinding and pyrolysis to 700 °C, a porous Fe–N–C network was grown. Such iron-based carbon was shown to activate the peroxymonosulfate-mediated oxidative degradation of organic substances. The authors have used paracetamol drug as the analyte. The adsorption capability of the Fe–N–C network was moderate as only 5% of paracetamol was adsorbed within 60 min under dark conditions. The iron-modified nanostructure demonstrated a decay rate of about 0.3 min−1, whereas the t50 parameter was about 5 min.
In a different approach, Ma and co-workers190 developed a series of carbon networks derived by the condensation of TFP and five different p-phenylenediamine derivatives. The latter possessed a variable number or position of nitrogen atoms within the aromatic ring. The framework prepared using 3,6-diamine-pyridazine as the monomer showed the highest photocatalytic activity towards the degradation of paracetamol drug. The so-called PDZ-COF demonstrated a decay rate of about 0.128 min−1, whereas the t50 parameter was about 8 min. Thus, it was concluded that the photocatalytic activity of carbon frameworks may be tuned by adjusting the position of heteroatoms as well as their total number within an aromatic ring of the network.
A remarkable enhancement in the photocatalytic degradation of bisphenol A (BPA) was observed in the joint work of Li and Lu groups.191 The authors constructed a Z-scheme heterojunction between a titanium-based MOF and a crystalline carbon framework. The latter was synthesized using TFP and a trifunctional amine-terminated triazine monomer. The integration of MOF promoted the charge separation and transfer processes. The hybrid MOF@COF demonstrated enhanced adsorption capacity of organic substances. Specifically, 45% of BPA could be efficiently removed from aqueous environments within 30 min. The hybrid photocatalyst demonstrated a BPA decay rate of about 0.32 min−1, whereas the analyte concentration was diminished to its half value after 3 min of irradiation.
The already mentioned TzDA framework, integrated within a Z-scheme heterojunction with BiOBr,179 alternatively participated in a similar heterojunction with AgBr/Ag.192 The theoretical weight fraction of the carbon component within the hybrid structure spanned between 5 and 15 wt%. BET analysis demonstrated that the surface area of the carbon component was appreciable diminished due to the growth of AgBr nanoparticles. This reflected in the rather low adsorption capability of the hybrid structure. Specifically, a 30 min incubation of tetracycline antibiotic in aqueous environment resulted in just a 2% decrease of the starting substance concentration.
Tetracycline was used as an analyte for photodegradation tests. Under irradiation, the decay kinetics showed that the estimated t50 parameter was approximately 6 min in the presence of the optimized hybrid containing 9 wt% of carbon component.
In Table 3, numerous examples of porous carbon-based frameworks for dye removal are illustrated. These systems demonstrate the t50 parameter values above 10 min. Similarly, hybrid photocatalytic systems containing a low content of carbon-based nanostructures are illustrated in Table 4.
| a/a | Sample | Adsorption capability at dark conditions | Degradation rate in min−1 (degradation time t50) | Ref. |
|---|---|---|---|---|
| a Under visible light. | ||||
| 1 | TpMA | 12% of phenol within 60 min | 0.044 (16 min) | 193 |
| 2 | TpMA@MOF/persulfate | 10% of BPA within 60 min | 0.011 (50 min) | 194 |
| 3 | HBC–TFPN | 15% of methyl orange within 60 min | 0.016 (40 min) | 195 |
| 4 | COF–PD/AgI | 25% of RhB within 30 min | 0.0466 (17 min) | 196 |
| 5 | Py–POP | 32% of RhB within 30 min | Not reported (25 min) | 197 |
| 6 | TFP-based POP-1a | 100% RhB degradation within 30 min at 40 °C | 0.015 (28 min) | 198 |
| 7 | g-C18N3-COF | 35% of RhB within 120 min | 0.0134 (30 min) | 199 |
| 8 | TPA–PTH COFs | No adsorption-related loss of Sudan Red III | n.d. (∼20 hours) | 200 |
| 9 | T-COF | 8% of methylene blue within 90 min | 0.008 (75 min) | 201 |
| 10 | T-COF nanosheets | 30% of methylene blue within 90 min | 0.066 (20 min) | 201 |
| 11 | A–Fe2O3/g-C3N4/COF | 30% of tetracycline within 60 min | 0.0056 (40 min) | 202 |
| 12 | NiFe-LDH/CTF-1 | 50% of tetracycline within 60 min | 0.0100 (50 min) | 203 |
| 13 | Keto-CTF | 15% of tetracycline within 40 min | 0.0351 (18 min) | 204 |
| 14 | 30% TiO2/MDCTF | 15% of 2,2′,4,4′-tetrahydroxybenzophenone within 60 min | 0.0091 (90 min) | 205 |
| 15 | Anthraquinone-based COP/g-C3N4 | 15% of fast Green dye within 60 min | 0.032 (80 min) | 206 |
| 16 | 50% BiVO4/CTF/KHSO4 | 55% of paracetamol within 30 min | 0.04469 (22 min) | 207 |
| 17 | Chitosan-COF films | 2% of paracetamol within 30 min | 0.0177 (30 min) | 208 |
| 18 | SQ-COF-1 | 100% of sulfadimidine within 60 min | 0.034 (33 min) | 209 |
| a/a | Sample | Adsorption capability at dark conditions | Degradation rate in min−1 (degradation time t50) | Ref. |
|---|---|---|---|---|
| 1 | BiOBr | 13% of tetracycline hydrochloride within 30 min | 0.02214 (30 min) | 210 |
| 2 | BiOBr/CTF-3D 2% | 25% of tetracycline hydrochloride within 30 min | 0.04122 (12 min) | 210 |
| 3 | MoS2@COF 20% | 98% RhB degradation within 30 min | 0.118 (12.5 min) | 211 |
| 4 | Fe–TiO2@COF | 65% of methylene blue within 30 min | n.d. (60 min) | 212 |
5. Conclusions
There is an increasingly growing interest in the scientific community for the development of novel porous carbon-based frameworks with extended π-conjugated structure that show great potentials in diverse environment-related photocatalytic applications, including oxygen production, CO2 reduction and pollutant degradation. In this review, we have outlined the synthetic protocols that were frequently followed for the construction of porous carbon-based frameworks, focusing on their structural characteristics. Subsequently, a comprehensive study of COF-mediated photocatalytic reactive oxygen species generation was presented.Porous carbon-based frameworks are polymer-like materials with high crystallinity, large specific surface area and good chemical and thermal stability. Their major advantage over similar materials and polymers is the ability to tailor their skeleton to a great number of pre-designed architectures and topologies, leading to highly ordered structures.
In terms of synthetic methods, the well-established solvothermal approach is predominant in the synthesis of porous covalent organic framework, leading to thermodynamically stable materials; however, it is a time-consuming and an energy-demanding method. Therefore, it is of high importance to develop new, environment-friendly protocols that will allow the growth of highly crystalline COFs with tunable structure and porosity in high yields and in a reproducible manner. In this context, sonochemical, microwave and mechanochemical methods should be further explored in the future for the preparation of porous COFs as they can provide short reaction times, high yields and can be easily upscaled.
As it is well known, COFs consist of building blocks that are linked covalently, forming highly organized polygons. In most cases, the chemical reactions forming this covalent bonding are reversible, thus allowing the self-healing of the as-prepared porous framework from any possible defects. This allows to afford high crystalline materials; however, these materials are relatively sensitive to strong acidic or basic environments, thus limiting their usability in potential applications. Therefore, there is a need of a gold standard to face the inconsistency between crystallinity and stability observed for COFs so far.
In terms of their structural conformation, the development of single crystal COFs would facilitate the understanding of growth and nucleation mechanisms, further allowing their direct characterization. Another challenge would be the development of functional 2D-thin films of porous COFs, which would make them ideal candidates in photocatalytic applications, as an alternative of graphene, which is considered the best example of 2D-conjugated polymers. The introduction of highly crystalline 3D frameworks with high surface areas and tunable channels is still challenging the scientific community.
Regarding the photocatalytic activity of COFs, it seems that a number of critical parameters need to be simultaneously adjusted. These parameters are strongly correlated with both electronic and structural aspects. In the former case, the adjustment of both the VB and CB energy level redox potentials will produce the right driving forces towards the molecular activation process (O2, H2O, etc.) through either reductive or oxidative paths. The energy level engineering along with the polarization of the conjugated carbon framework by chemical means constitute the holy grail of photocatalytic processes in such systems. It has been widely accepted that this may be regulated with the proper selection of building units for constructing the carbon framework. It is clearly understood that efficient charge carrier separation needs to be accomplished. Electron–hole dissociation may take place either by spatially designing a donor–acceptor network or by building an engineered heterojunction with a cocatalyst system. In the former case, the donor–acceptor interactions within the framework will give rise to polarization contributions. A sophisticated strategy leading to such processes involves the integration of highly polar ionic liquid-type functionalities on the backbone of the COF lattice.
Regarding the utilization of an appropriate cocatalyst and the subsequent fabrication of multiple heterojunctions, it has been demonstrated that band structure matching between the semiconducting components should be achieved in order to form functional configurations (Z-scheme, S-scheme, etc.).
Concerning the structural issues, it was clearly demonstrated that intra-layer interactions may be substantially enhanced using H-bonding functionalities (hydroxy, etc.). The latter act as supramolecular docking sites, whereas their precisely designed positions and contents may lead to optimized stacking motifs and subsequent crystallinity enhancement. An alternative approach towards the development of homogeneously organized stackings is the utilization of soft templates. A sophisticated strategy involves the liquid crystal-directed production of crystalline frameworks. In certain liquid mixtures, forming lamellar liquid crystal phase region at specific composition, crystalline carbon-based frameworks were grown using this templated approach. Concerning the surface area of the formed carbon-based nanostructures, it is noted that this structural parameter is not considered as a priority factor for obtaining photocatalytically-active porous frameworks. Further work needs to be devoted towards the optimization of this multi-parameter task.
Author contributions
Nikolaos Karousis and Dimitrios Tasis equally contributed in conceptualization, writing-review & editing of this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support of this work by the project “Advanced Nanostructured Materials for Sustainable Growth: Green Energy Production/Storage, Energy Saving and Environmental Remediation” (TAEDR-0535821) which is implemented under the action “Flagship actions in interdisciplinary scientific fields with a special focus on the productive fabric” (ID 16618), Greece 2.0 – National Recovery and Resilience Fund and funded by European Union NextGenerationEU.References
- F. Martins, C. Felgueiras, M. Smitkova and N. Caetano, Energies, 2019, 12, 964 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- Y. Nosaka and A. Nosaka, Chem. Rev., 2017, 117, 11302 CrossRef CAS PubMed.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, G. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 28, 9454 CrossRef PubMed.
- I. Ahmad, S. Shukrullah, M. Y. Naz, M. Ahmad, E. Ahmed, Y. Liu, A. Hussain, S. Iqbal and S. Ullah, Adv. Colloid Interface Sci., 2022, 304, 102661 CrossRef CAS PubMed.
- W. Droge, Physiol. Rev., 2002, 82, 47 CrossRef CAS PubMed.
- D. Masih, Y. Ma and S. Rohani, Appl. Catal., B, 2017, 206, 556 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kroger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168 CrossRef CAS.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodriguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- X. Lin, C. Chen, T. Zhou and J. Zhang, Solar RRL, 2021, 5, 2000458 CrossRef.
- L. Huang, J. Yang, Y. Asakura, Q. Shuai and Y. Yamauchi, ACS Nano, 2023, 17, 8918 CrossRef CAS PubMed.
- L. Wang and H. Xu, Prog. Polym. Sci., 2023, 145, 101734 CrossRef CAS.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814 CrossRef CAS PubMed.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001 RSC.
- R. Zhuang, X. Zhang, C. Qu, X. Xu, J. Yang, Q. Ye, Z. Liu, S. Kaskel, F. Xu and H. Wang, Sci. Adv., 2023, 9, eadh8060 CrossRef CAS PubMed.
- R. Shah, S. Ali, F. Raziq, S. Ali, P. M. Ismail, S. Shah, R. Iqbal, X. Wu, W. He, X. Zu, A. Zada, F. Mabood, A. Vinu, S. H. Jhung, J. Yi and L. Qiao, Coord. Chem. Rev., 2023, 477, 214968 CrossRef CAS.
- Q. Hao, Z.-J. Li, B. Bai, X. Zhang, Y.-W. Zhong, L.-J. Wan and D. Wang, Angew. Chem., 2021, 133, 12606 CrossRef.
- H. Yu, P.-K. Zhou and X. Chen, Adv. Funct. Mater., 2023, 33, 2308336 CrossRef CAS.
- P.-K. Zhou, H. Yu, W. Huang, M. Y. Chee, S. Wu, T. Zeng, G. J. Lim, H. Xu, Z. Yu, H. Li, W. S. Lew and X. Chen, Adv. Funct. Mater., 2024, 34, 2306593 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- P. Samanta, A. V. Desai, S. Let and S. K. Ghosh, ACS Sustainable Chem. Eng., 2019, 7, 7456 CrossRef CAS.
- J. Gan, X. Li, K. Rizwan, M. Adeel, M. Bilal, T. Rasheed and H. M. N. Iqbal, Chemosphere, 2022, 286, 131710 CrossRef CAS PubMed.
- G. Das, B. Garai, T. Prakasam, F. Benyettou, S. Varghese, S. K. Sharma, F. Gándara, R. Pasricha, M. Baias, R. Jagannathan, N. Saleh, M. Elhabiri, M. A. Olson and A. Trabolsi, Nat. Commun., 2022, 13, 3904 CrossRef CAS PubMed.
- J. Hu, J. Zhang, Z. Lin, L. Xie, S. Liao and X. Chen, Chem. Mater., 2022, 34, 5249 CrossRef CAS.
- J. Liang, Q. Wu, Y.-B. Huang and R. Cao, EnergyChem, 2021, 3, 100064 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135 RSC.
- H. Li, L. Wang and G. Yu, Nano Today, 2021, 40, 101247 CrossRef CAS.
- G.-B. Wang, S. Li, C.-X. Yan, F.-C. Zhu, Q.-Q. Lin, K.-H. Xie, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2020, 8, 6957 RSC.
- S. Wu, Y. Pan, H. Lin, L. Li, X. Fu and J. Long, ChemSusChem, 2021, 14, 4958 CrossRef CAS PubMed.
- W.-T. Chung, I. M. A. Mekhemer, M. Gamal Mohamed, A. M. Elewa, A. F. M. EL-Mahdy, H.-H. Chou, S.-W. Kuo and K. C.-W. Wu, Coord. Chem. Rev., 2023, 483, 215066 CrossRef CAS.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- K. Chen, A. Cai and T.-T. Li, ChemSusChem, 2023, 16, e202300021 CrossRef CAS PubMed.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382 RSC.
- Y. Cao, W. Peng, Y. Li, F. Zhang, Y. Zhu and X. Fan, Green Energy Environ., 2023, 8, 360 CrossRef CAS.
- Z. Alsudairy, N. Brown, A. Campbell, A. Ambus, B. Brown, K. Smith-Petty and X. Li, Mater. Chem. Front., 2023, 7, 3298 RSC.
- C. Xia, K. O. Kirlikovali, T. H. Chuong Nguyen, X. Cuong Nguyen, Q. Ba Tran, M. Khoa, D. M. T. Nguyen Dinh, D. L. T. Nguyen, P. Singh, P. Raizada, V.-H. Nguyen, S. Young Kim, L. Singh, C. C. Nguyen, M. Shokouhimehr and Q. Van Le, Coord. Chem. Rev., 2021, 446, 214117 CrossRef CAS.
- Q. Yang, M. Luo, K. Liu, H. Cao and H. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS.
- Y. Zhi, Z. Wang, H.-L. Zhang and Q. Zhang, Small, 2020, 16, 2001070 CrossRef CAS PubMed.
- P. Costa, A. Vega-Peñaloza, L. Cognigni and M. Bonchio, ACS Sustainable Chem. Eng., 2021, 9, 15694 CrossRef CAS.
- H. Chen, H. Sekhar Jena, X. Feng, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2022, 61, e202204938 CrossRef CAS PubMed.
- S.-Y. Hu, Y.-N. Sun, Z.-W. Feng, F.-O. Wang and Y.-K. Lv, Chemosphere, 2022, 286, 131646 CrossRef CAS PubMed.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- A. Li, X. Chang, Z. Huang, C. Li, Y. Wei, L. Zhang, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 13734 CrossRef CAS PubMed.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802 CrossRef CAS PubMed.
- Y. Nakaoka and Y. Nosaka, J. Photochem. Photobiol., A, 1997, 110, 299 CrossRef CAS.
- N. Karamoschos and D. Tasis, Energies, 2022, 15, 6202 CrossRef CAS.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- Y. Fu, T. Huang, B. Jia, J. Zhu and X. Wang, Appl. Catal., B, 2017, 202, 430 CrossRef CAS.
- Y. Ku and I. L. Jung, Water Res., 2001, 35, 135 CrossRef CAS PubMed.
- M. Yasuda, T. Matsumoto and T. Yamashita, Renewable Sustainable Energy Rev., 2018, 81, 1627 CrossRef CAS.
- J. R. Harbour and M. L. Hair, J. Phys. Chem., 1977, 81, 1791 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS PubMed.
- J. Xiao, M. Wang, Z. Pang, L. Dai, J. Lu and J. Zou, Anal. Methods, 2019, 11, 1930 RSC.
- K. Sahel, L. Elsellami, I. Mirali, F. Dappozze, M. Bouhent and C. Guillard, Appl. Catal., B, 2016, 188, 106 CrossRef CAS.
- Y. Wei, J. Zhang, Q. Zheng, J. Miao, P. J. Alvarez and M. Long, Chemosphere, 2021, 279, 130556 CrossRef CAS PubMed.
- S. Putt and R. B. Pugh, PLoS One, 2013, 8, e79218 CrossRef PubMed.
- Y.-S. Wang, J.-H. Shen and J.-J. Horng, J. Hazard. Mater., 2014, 274, 420 CrossRef CAS PubMed.
- I. Kraljic and C. N. Trumbore, J. Am. Chem. Soc., 1965, 87, 2547 CrossRef CAS.
- L. Zhu and W.-C. Oh, J. Korean Ceram. Soc., 2016, 53, 56 CrossRef CAS.
- V. Brezova, S. Gabcova, D. Dvoranova and A. Stasko, J. Photochem. Photobiol., B, 2005, 79, 121 CrossRef CAS PubMed.
- Y. Nosaka, H. Natsui, M. Sasagawa and A. Nosaka, J. Phys. Chem. B, 2006, 110, 12993 CrossRef CAS PubMed.
- E. Albiter, S. Alfaro and M. A. Valenzuela, Photochem. Photobiol. Sci., 2015, 14, 597 CrossRef CAS PubMed.
- J. Shin, D. W. Kang, J. H. Lim, J. M. An, Y. Kim, J. H. Kim, M. Sun Ji, S. Park, D. Kim, J. Yong Lee, J. Seung Kim and C. S. Hong, Nat. Commun., 2023, 14, 1498 CrossRef CAS PubMed.
- X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536 CrossRef CAS PubMed.
- F. Ronzani, N. Costarramone, S. Blanc, A. K. Benabbou, M. Le Bechec, T. Pigot, M. Oelgemöller and S. Lacombe, J. Catal., 2013, 303, 164 CrossRef CAS.
- M. Bancirova, Luminescence, 2011, 26, 685 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed.
- S. J. Lyle, P. J. Waller and O. M. Yaghi, Trends Chem., 2019, 1, 172 CrossRef CAS.
- X. Chen, K. Geng, R. Liu, K. Tian Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050 CrossRef CAS PubMed.
- N. Li, J. Du, D. Wu, J. Liu, N. Li, Z. Sun, G. Li and Y. Wu, Trends Anal. Chem., 2018, 108, 154 CrossRef CAS.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469 RSC.
- B. Gole, V. Stepanenko, S. Rager, M. Grüne, D. D. Medina, T. Bein, F. Würthner and F. Beuerle, Angew. Chem., Int. Ed., 2018, 57, 846 CrossRef CAS PubMed.
- Z. Xiong, B. Sun, H. Zou, R. Wang, Q. Fang, Z. Zhang and S. Qiu, J. Am. Chem. Soc., 2022, 144, 6583 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328 CrossRef CAS PubMed.
- S.-T. Yang, J. Kim, H.-Y. Cho, S. Kim and W.-S. Ahn, RSC Adv., 2012, 2, 10179 RSC.
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87 CrossRef.
- Q. Chen, Y. Wang and G. Luo, Langmuir, 2023, 39, 11731 CrossRef CAS PubMed.
- H. Wei, S. Chai, N. Hu, Z. Yang, L. Wei and L. Wang, Chem. Commun., 2015, 51, 12178 RSC.
- S. Kim and H. C. Choi, Commun. Chem., 2019, 2, 60 CrossRef.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806 CrossRef CAS PubMed.
- H. Vignesh Babu, M. G. Monika Bai and M. Rajeswara Rao, ACS Appl. Mater. Interfaces, 2019, 11, 11029 CrossRef PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS PubMed.
- G.-B. Wang, F.-C. Zhu, Q.-Q. Lin, J.-L. Kan, K.-H. Xie, S. Li, Y. Geng and Y.-B. Dong, Chem. Commun., 2021, 57, 4464 RSC.
- L. Guo, S. Jia, C. S. Diercks, X. Yang, S. A. Alshmimri and O. M. Yaghi, Angew. Chem., Int. Ed., 2020, 59, 2023 CrossRef CAS PubMed.
- Y. Li, T.-X. Luan, Ke Cheng, D. Zhang, W. Fan, P.-Z. Li and Y. Zhao, ACS Mater. Lett., 2022, 4, 1160 CrossRef CAS.
- H. C. Shan, D. Cai, X. X. Zhang, Q. Zhu, P. Y. Qin and J. Baeyens, Chem. Eng. J., 2022, 432, 134288 CrossRef CAS.
- T. Joshi, C. Chen, H. Li, C. S. Diercks, G. Wang, P. J. Waller, H. Li, J.-L. Bredas, O. M. Yaghi and M. F. Crommie, Adv. Mater., 2019, 31, 1805941 CrossRef PubMed.
- X. Wang, X. Han, J. Zhang, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332 CrossRef CAS PubMed.
- Z. Wang and Y. Huang, New J. Chem., 2023, 47, 3668 RSC.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423 CrossRef CAS PubMed.
- J. Feng, J. Cheng, J. Pang, M. Tang, Z. Liu, C. Rong and R. Tan, Catal. Sci. Technol., 2022, 12, 6865 RSC.
- B. Dong, W. J. Wang, W. Pan and G. J. Kang, Mater. Chem. Phys., 2019, 226, 244 CrossRef CAS.
- K. H. Xiong, Y. X. Wang, F. L. Zhang, X. Li and X. J. Lang, Appl. Catal., B, 2023, 322, 122135 CrossRef CAS.
- K. H. Xiong, F. L. Zhang, Y. X. Wang, B. Zeng and X. J. Lang, J. Colloid Interface Sci., 2023, 643, 340 CrossRef CAS PubMed.
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X.-W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y.-B. Zhang, H. Tian, W.-H. Zhu and A. I. Cooper, Nature, 2022, 604, 72 CrossRef CAS PubMed.
- C. Zhao, C. S. Diercks, C. Zhu, N. Hanikel, X. Pei and O. M. Yaghi, J. Am. Soc., 2018, 140, 16438 CrossRef CAS PubMed.
- S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893 CrossRef CAS PubMed.
- E. Jin, S. Fu, H. Hanayama, M. A. Addicoat, W. Wei, Q. Chen, R. Graf, K. Landfester, M. Bonn, K. A. I. Zhang, H. I. Wang, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2022, 61, e202114059 CrossRef CAS PubMed.
- D. L. Pastoetter, S. Xu, M. Borrelli, M. Addicoat, B. P. Biswal, S. Paasch, A. Dianat, H. Thomas, R. Berger, S. Reineke, E. Brunner, G. Cuniberti, M. Richter and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 23620 CrossRef CAS PubMed.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848 CrossRef CAS PubMed.
- T. Lei, Y. Mi, Z. Wei, S. Li and S. Pang, Dalton Trans., 2023, 52, 1761 RSC.
- G.-B. Wang, K.-H. Xie, J.-L. Kan, H.-P. Xu, F. Zhao, Y.-J. Wang, Y. Geng and Y.-B. Dong, Chem. Commun., 2023, 59, 1493 RSC.
- Y. Yang, W. Zhao, H. Niu and Y. Cai, ACS Appl. Mater. Interfaces, 2021, 13, 42035 CrossRef CAS PubMed.
- S. Kandambeth, J. Jia, H. Wu, V. S. Kale, P. T. Parvatkar, J. Czaban-Jozwiak, S. Zhou, X. Xu, Z. O. Ameur, E. Abou-Hamad, A. H. Emwas, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Energy Mater., 2020, 10, 2001673 CrossRef CAS.
- P. T. Parvatkar, S. Kandambeth, A. C. Shaikh, I. Nadinov, J. Yin, V. S. Kale, G. Healing, A. H. Emwas, O. Shekhah, H. N. Alshareef, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2023, 145, 5074 CrossRef CAS PubMed.
- W. X. Wu, F. Li, B. J. Yao, L. G. Ding, J. L. Kan, F. Liu, G. Y. Zhao, S. Wang and Y. B. Dong, J. Hazard. Mater., 2022, 433, 128831 CrossRef CAS PubMed.
- G.-B. Wang, Y.-J. Wang, J.-L. Kan, K.-H. Xie, H.-P. Xu, F. Zhao, M.-C. Wang, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 4951 CrossRef CAS PubMed.
- C. Wang, P. Lyu, Z. Chen and Y. Xu, J. Am. Chem. Soc., 2023, 145, 12745 CrossRef CAS PubMed.
- F. Kang, X. Wang, C. Chen, C.-S. Lee, Y. Han and Q. Zhang, J. Am. Chem. Soc., 2023, 145, 15465 CrossRef CAS PubMed.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770 CrossRef CAS PubMed.
- X. Chen, M. A. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 3241 CrossRef CAS PubMed.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705 CrossRef CAS PubMed.
- H. Chen, W. Liu, C. Liu, J. Sun, L. Bourda, R. Morent, N. De Geyter, R. Van Deun, K. Van Hecke, K. Leus and P. Van Der Voort, Appl. Catal., B, 2022, 319, 121920 CrossRef CAS.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F.-S. Xiao, Matter, 2020, 2, 416 CrossRef CAS.
- X. Zhao, H. Pang, D. Huang, G. Liu, J. Hu and Y. Xiang, Angew. Chem., Int. Ed., 2022, 61, e202208833 CrossRef CAS PubMed.
- W. Han, L.-H. Shao, X.-J. Sun, Y.-H. Liu, F.-M. Zhang, Y. Wang, P.-Y. Dong and G.-L. Zhang, Appl. Catal., B, 2022, 317, 121710 CrossRef CAS.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y.-B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763 CrossRef CAS PubMed.
- G. Yu, W. Li, H. Gao, M. Zhang, Y. Guo and S. Chen, J. Phys. Chem. Lett., 2022, 13, 2814 CrossRef CAS PubMed.
- M.-L. Xu, M. Lu, G.-Y. Qin, X.-M. Wu, T. Yu, L.-N. Zhang, K. Li, X. Cheng and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210700 CrossRef CAS PubMed.
- Y. Yang, X. Chu, H.-Y. Zhang, R. Zhang, Y.-H. Liu, F.-M. Zhang, M. Lu, Z.-D. Yang and Y.-Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- M.-L. Xu, J.-R. Li, X.-M. Wu, T. Yu, G.-Y. Qin, F.-J. Wang, L.-N. Zhang, K. Li and X. Cheng, Appl. Surf. Sci., 2022, 602, 154371 CrossRef CAS.
- W.-R. Cui, F.-F. Li, R.-H. Xu, C.-R. Zhang, X.-R. Chen, R.-H. Yan, R.-P. Liang and J.-D. Qiu, Angew. Chem., Int. Ed., 2020, 59, 17684 CrossRef CAS PubMed.
- W.-K. An, S.-J. Zheng, X. Xu, L.-J. Liu, J.-S. Ren, L. Fan, Z.-K. Yang, Y. Ren and C. Xu, Appl. Catal., B, 2022, 316, 121630 CrossRef CAS.
- Y.-Z. Cheng, W. Ji, X. Wu, X. Ding, X.-F. Liu and B.-H. Han, Appl. Catal., B, 2022, 306, 121110 CrossRef CAS.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- D. Tan, R. Zhuang, R. Chen, M. Ban, W. Feng, F. Xu, X. Chen and Q. Wang, Adv. Funct. Mater., 2024, 34, 2311655 Search PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed.
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107 CrossRef CAS PubMed.
- C. Shao, Q. He, M. Zhang, L. Jia, Y. Ji, Y. Hu, Y. Li, W. Huang and Y. Li, Chin. J. Catal., 2023, 46, 28 CrossRef CAS.
- S. Wang, Z. Xie, D. Zhu, S. Fu, Y. Wu, H. Yu, C. Lu, P. Zhou, M. Bonn, H. I. Wang, Q. Liao, H. Xu, X. Chen and C. Gu, Nat. Commun., 2023, 14, 6891 CrossRef CAS PubMed.
- I. Krivtsov, A. Vazirani, D. Mitoraj and R. Beranek, ChemCatChem, 2023, 15, e202201215 CrossRef CAS.
- G. Li, P. Fu, Q. Yue, F. Ma, X. Zhao, S. Dong, X. Han, Y. Zhou and J. Wang, Chem Catal., 2022, 2, 1734 CrossRef CAS.
- F. Tan, Y. Zheng, Z. Zhou, H. Wang, X. Dong, J. Yang, Z. Ou, H. Qi, W. Liu, Z. Zheng and X. Chen, CCS Chem., 2022, 4, 3751 CrossRef CAS.
- Y. Liu, W.-K. Han, W. Chi, Y. Mao, Y. Jiang, X. Yan and Z.-G. Gu, Appl. Catal., B, 2023, 331, 122691 CrossRef CAS.
- J.-Y. Yue, L.-P. Song, Y.-F. Fan, Z.-X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202309624 CrossRef CAS PubMed.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- C. Wu, Z. Teng, C. Yang, F. Chen, H. B. Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 34, 2110266 CrossRef CAS PubMed.
- L. Li, L. Xu, Z. Hu and J. C. Yu, Adv. Funct. Mater., 2021, 31, 2106120 CrossRef CAS.
- J.-N. Chang, Q. Li, J.-W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS.
- X. Di, X. Lv, H. Wang, F. Chen, S. Wang, G. Zheng, B. Wang and Q. Han, Chem. Eng. J., 2023, 455, 140124 CrossRef CAS.
- P. Das, G. Chakraborty, J. Roeser, S. Vogl, J. Rabeah and A. Thomas, J. Am. Chem. Soc., 2023, 145, 2975 CrossRef CAS.
- Y. Zhang, J. Qiu, B. Zhu, M. V. Fedin, B. Cheng, J. Yu and L. Zhang, Chem. Eng. J., 2022, 444, 136584 CrossRef CAS.
- Y. Yang, J. Kang, Y. Li, J. Liang, J. Liang, L. Jiang, D. Chen, J. He, Y. Chen and J. Wang, New J. Chem., 2022, 46, 21605 RSC.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328 CrossRef CAS PubMed.
- J. Chu, Z. Liu, T. Yang and A. Kong, Appl. Surf. Sci., 2023, 611, 155717 CrossRef CAS.
- T. Yang, Y. Jin, Y. Wang, A. Kong, Y. Chen, Y. Zou, C. Liu, G. Wei and C. Yu, Adv. Funct. Mater., 2023, 33, 2300714 CrossRef CAS.
- L. Zhai, Z. Xie, C.-X. Cui, X. Yang, Q. Xu, X. Ke, M. Liu, L.-B. Qu, X. Chen and L. Mi, Chem. Mater., 2022, 34, 5232 CrossRef CAS.
- H. Wang, E. Almatrafi, Z. Wang, Y. Yang, T. Xiong, H. Yu, H. Qin, H. Yang, Y. He, C. Zhou, G. Zeng and P. Xu, J. Colloid Interface Sci., 2022, 608, 1051 CrossRef CAS PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS PubMed.
- H. Wang, C. Yang, F. Chen, G. Zheng and Q. Han, Angew. Chem., Int. Ed., 2022, 61, e202202328 CrossRef CAS PubMed.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Adv. Mater., 2022, 34, 2107480 CrossRef CAS.
- X. Yu, B. Viengkeo, Q. He, X. Zhao, Q. Huang, P. Li, W. Huang and Y. Li, Adv. Sustainable Syst., 2021, 5, 2100184 CrossRef CAS.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. Van Hecke, R. Morent, N. De Geyter, K. Leus, H. Chen and P. Van Der Voort, Green Chem., 2023, 25, 3069 RSC.
- S. Chai, X. Chen, X. Zhang, Y. Fang, R. S. Sprick and X. Chen, Environ. Sci.: Nano, 2022, 9, 2464 RSC.
- H. Hu, Y. Tao, D. Wang, C. Li, Q. Jiang, Y. Shi, J. Wang, J. Qin, S. Zhou and Y. Kong, J. Colloid Interface Sci., 2023, 629, 750 CrossRef CAS PubMed.
- Y. Yang, J. Liu, M. Gu, B. Cheng, L. Wang and J. Yu, Appl. Catal., B, 2023, 333, 122780 CrossRef CAS.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- J. Sun, H. Sekhar Jena, C. Krishnaraj, K. Singh Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y.-Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Angew. Chem., Int. Ed., 2023, 62, e202216719 CrossRef CAS PubMed.
- T. Yang, Y. Chen, Y. Wang, X. Peng and A. Kong, ACS Appl. Mater. Interfaces, 2023, 15, 8066 CrossRef CAS PubMed.
- M. Wu, Z. Shan, J. Wang, T. Liu and G. Zhang, Chem. Eng. J., 2023, 454, 140121 CrossRef CAS.
- G. Pan, X. Hou, Z. Liu, C. Yang, J. Long, G. Huang, J. Bi, Y. Yu and L. Li, ACS Catal., 2022, 12, 14911 CrossRef CAS.
- Z. Zhou, M. Sun, Y. Zhu, P. Li, Y. Zhang, M. Wang and Y. Shen, Appl. Catal., B, 2023, 334, 122862 CrossRef CAS.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng, J. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H. Huang and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14, 4344 CrossRef CAS PubMed.
- L. Yuan, C. Zhang, J. Wang, C. Liu and C. Y4u, Nano Res., 2021, 14, 3267 CrossRef CAS.
- X. Zhang, J. Zhang, J. Miao, X. Wen, C. Chen, B. Zhou and M. Long, Chem. Eng. J., 2023, 466, 143085 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902 CrossRef CAS PubMed.
- Y. Hou, C.-X. Cui, E. Zhang, J.-C. Wang, Y. Li, Y. Zhang, J.-C. Wang, Y. Li, Y. Zhang, Y. Zhang, Q. Wang and J. Jiang, Dalton Trans., 2019, 48, 14989 RSC.
- Y. Deng, Z. Zhang, P. Du, X. Ning, Y. Wang, D. Zhang, J. Liu, S. Zhang and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082 CrossRef CAS PubMed.
- Y. Deng, Y. Wang, Z. Di, M. Xie, F. Dai, S. Zhan and Z. Zhang, Small Methods, 2022, 6, 2200265 CrossRef CAS PubMed.
- Y. Zhang, Z. Chen, Z. Shi, T.-T. Lu, D. Chen, Q. Wang and Z. Zhan, Sep. Purif. Technol., 2021, 275, 119216 CrossRef CAS.
- J. Zhang, Y. Shi, X. Huang and X. Qian, Cellulose, 2023, 30, 1773 CrossRef CAS.
- G. Wang, Z. Hu, Z. Chen, J. Wang, J. Hu and X. Xu, Appl. Surf. Sci., 2023, 631, 157538 CrossRef CAS.
- S. He, Q. Rong, H. Niu and Y. Cai, Chem. Commun., 2017, 53, 9636 RSC.
- S. He, B. Yin, H. Niu and Y. Cai, Appl. Catal., B, 2018, 239, 147 CrossRef CAS.
- S. He, Q. Rong, H. Niu and Y. Cai, Appl. Catal., B, 2019, 247, 49 CrossRef CAS.
- Y. Yang, H. Niu, L. Xu, H. Zhang and Y. Cai, Appl. Catal., B, 2020, 269, 118799 CrossRef CAS.
- L. Niu, X. Zhao, F. Wu, H. Lv, Z. Tang, W. Liang, X. Wang and J. Giesy, Chem. Eng. J., 2021, 414, 128619 CrossRef CAS.
- Z. Dong, L. Zhang, J. Gong and Q. Zhao, Chem. Eng. J., 2021, 403, 126383 CrossRef CAS.
- J. Chen, G. Li, N. Lu, H. Lin, S. Zhou and F. Liu, Mater. Today Chem., 2022, 24, 100832 CrossRef CAS.
- J. Zhu, L. Wang, W. Hayat, Y. Zhang, S. Huang, X. Zhang and S. Zhou, Sep. Purif. Technol., 2023, 310, 123034 CrossRef CAS.
- F. Liu, Z. Ma, Y. Deng, M. Wang, P. Zhou, W. Liu, S. Guo, M. Tong and D. Ma, Environ. Sci. Technol., 2021, 55, 5371 CrossRef CAS PubMed.
- L. Yang, Y. Wang, J. Yuan, G. Wang, Q. Cao, H. Fei, M. Li, J. Shao, H. Li and J. Lu, Chem. Eng. J., 2022, 446, 137095 CrossRef.
- Z. Shi, Z. Chen, Y. Zhang, X. Wang, T. Lu, Q. Wang, Z. Zhan and P. Zhang, Sep. Purif. Technol., 2022, 288, 120717 CrossRef CAS.
- H. Lv, X. Zhao, H. Niu, S. He, Z. Tang, F. Wu and J. P. Giesy, J. Hazard. Mater., 2019, 369, 494 CrossRef CAS PubMed.
- S.-W. Lv, J.-M. Liu, C.-Y. Li, N. Zhao, Z.-H. Wang and S. Wang, Chemosphere, 2020, 243, 125378 CrossRef CAS PubMed.
- H.-Y. Yu, J.-S. Wang, F.-Y. Xie, Q. Yang, Y. Chen, L. Zhao, Y. Li and W.-J. Ruan, Chem. Eng. J., 2022, 445, 136713 CrossRef CAS.
- F. Liu, C. Nie, Q. Dong, Z. Ma, W. Liu and M. Tong, J. Hazard. Mater., 2020, 398, 122865 CrossRef CAS PubMed.
- M. Li, H. Zhao and Z. Lu, Microporous Mesoporous Mater., 2020, 292, 109774 CrossRef CAS.
- P. Liu, L. Xing, H. Lin, H. Wang, Z. Zhou and Z. Su, Sci. Bull., 2017, 62, 931 CrossRef CAS PubMed.
- T. Xia, Z. Wu, Y. Liang, W. Wang, Y. Li, Z. Sui, L. Shan, C. Li, R. Fan and Q. Chen, Mater. Today Chem., 2022, 25, 100962 CrossRef CAS.
- A. Jimenez-Almarza, A. Lopez-Magano, R. Cano, B. Ortín-Rubio, D. Díaz-García, S. Gomez-Ruiz, I. Imaz, D. Maspoch, R. Mas-Balleste and J. Aleman, Mater. Today Chem., 2021, 22, 100548 CrossRef CAS.
- S.-X. Gan, C. Jia, Q.-Y. Qi and X. Zhao, Chem. Sci., 2022, 13, 1009 RSC.
- W. Lu, T. Qin, B. Wang, J. Li, H. Zhu, G. Bai, Y. Li, L. Chen and X. Yan, ChemPhotoChem, 2023, 7, e202200230 CrossRef CAS.
- J. Zhang, X. Chen, Q. Chen, Y. He, M. Pan, G. Huang and J. Bi, Nanomaterials, 2022, 12, 4111 CrossRef CAS PubMed.
- X. Li, L. Zhang, S. Niu, Z. Dong and C. Lyu, J. Hazard. Mater., 2023, 444, 130366 CrossRef CAS PubMed.
- Y. Shen, J. Wu, C. Zhu, J. Zhao, Q. Fang, Y. Zheng, C. T. J. Ferguson and S. Song, Chem. Eng. J., 2023, 465, 143026 CrossRef CAS.
- N. Venkatesh, G. Murugadoss, A. Ashif Mohamed, M. Rajesh Kumar, S. Gouse Peera and P. Sakthivel, Molecules, 2022, 27, 7168 CrossRef CAS PubMed.
- Y. Yu, Y. Sun, B. Ge, J. Yan, K. Zhang, H. Chen, J. Hu, J. Tang, S. Song and T. Zeng, Environ. Sci. Poll. Res., 2023, 30, 27570 CrossRef CAS PubMed.
- B. Zhang, F. Liu, C. Nie, Y. Hou and M. Tong, J. Hazard. Mater., 2022, 435, 128966 CrossRef CAS PubMed.
- H. Ben, G. Yan, H. Liu, C. Ling, Y. Fan and X. Zhang, Adv. Funct. Mater., 2022, 32, 2104519 CrossRef CAS.
- S.-R. Zhu, Q. Qi, Y. Fang, W.-N. Zhao, M.-K. Wu and L. Han, Cryst. Growth Des., 2018, 18, 883 CrossRef CAS.
- K. K. Khaing, D. Yin, Y. Ouyang, S. Xiao, B. Liu, L. Deng, L. Li, X. Guo, J. Wang, J. Liu and Y. Zhang, Inorg. Chem., 2020, 59, 6942 CrossRef CAS PubMed.
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC.
| This journal is © The Royal Society of Chemistry 2024 |