
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal synthesis strategy for preparation of transition metal oxide electrocatalysts doped with noble metal single atoms for oxygen evolution reaction†
Jingyao
Wang
a,
Yiming
Zhu
a,
Xuepeng
Zhong
a,
Zhiwei
Hu
b,
Wei-Hsiang
Huang
c,
Chih-Wen
Pao
c,
Hongfei
Cheng
 *a,
Nicolas
Alonso-Vante
*d and
Jiwei
Ma
*a
*a,
Nicolas
Alonso-Vante
*d and
Jiwei
Ma
*a
aShanghai Key Laboratory for R&D and Application of Metallic Functional Materials, Institute of New Energy for Vehicles, School of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: cheng_hongfei@tongji.edu.cn; jiwei.ma@tongji.edu.cn
bMax Planck Institute for Chemical Physics of Solids, Nöthnitzer Strasse 40, 01187 Dresden, Germany
cNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 30076, Taiwan
dIC2MP, UMR-CNRS 7285, University of Poitiers, 4 rue Michel Brunet, 86073 Poitiers, France. E-mail: nicolas.alonso.vante@univ-poitiers.fr
First published on 4th July 2024
Abstract
Electrochemical water splitting is expected to be a promising solution to the growing problem of fossil fuel depletion, but is limited by the slow anodic oxygen evolution reaction (OER). Currently, nanomaterials such as Ru/Ir-based noble metal compounds have been used as highly active electrocatalysts for OER, but the high cost and scarcity hinder their wide application. Therefore, it is crucial to develop OER electrocatalysts that combine economic efficiency with high catalytic performance. In this work, we propose a universal synthesis strategy for the preparation of various noble metals-doped 3d-transition metal oxides (NM-TMO) electrocatalysts by the salt-template method. Our characterization analyses demonstrate that the noble metals are homogeneously dispersed as single atoms in transition metal oxides. Notably, Ir-doped Co3O4 catalysts, with Ir content as low as 1.35 at% (Ir–Co3O4), exhibit excellent OER performance in acidic, alkaline, and neutral media, compared to commercial IrO2 as well as undoped Co3O4. This work demonstrates that the synthesis method is applicable to a wide range of noble metals and 3d-transition metal oxide matrix. This method results in reduced costs by significantly decreasing the noble metal, but improving catalytic performance.
Introduction
Hydrogen is a promising source of clean energy because of its zero-carbon emissions and high specific energy density.1,2 Currently, hydrogen is produced industrially in three ways: coal gasification, steam-methane reforming, and water electrolysis. The first two methods consume a large amount of fossil fuels, causing significant carbon dioxide emissions and contributing significantly to the greenhouse effect.3 Electrochemical water decomposition has been widely recognized as a clean, sustainable, and efficient strategy for hydrogen production, which is expected to replace hydrogen production from fossil fuels in the future.4–7 In water electrolysis, the cathodic and anodic reaction is the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), respectively.8,9 In particular, OER is a remarkably intricate multi-step reaction, involving the transfer of four-electrons, and its slow kinetics and high overpotential become important factors limiting the overall efficiency of water decomposition.10–14 In pursuit of effectively reducing the energy barrier and improving energy conversion efficiency, researchers are committed to developing OER electrocatalysts that are both highly active and stable.15–21 Currently, noble metals like Ru, Ir, and their oxides exhibit excellent electrocatalytic OER properties over a wide pH range and have been used as commercial catalysts.22–26 However, it is impractical to widely use precious metal catalysts due to their scarcity and high price. Researchers have explored alternative candidates with abundant reserves and low cost for electrocatalytic OER, such as 3d-transition metal compounds, which have high abundance and unique electronic structures.27–32 Among them, 3d-transition metal oxides (TMOs), such as Co, Fe, and Ni, have been widely investigated for alkaline OER, but they have the disadvantages of high overpotential, poor activity, strong metal dissolution, and poor durability in acidic electrolytes.28,33–36 The working pH of the electrocatalysts severely limits their practical applications.37To balance the performance and cost of OER electrocatalysts, researchers have found that combining noble and nonprecious metals by doping or surface anchoring can achieve high catalytic efficiency while reducing the amount of noble metals.38–42 An effective strategy is to incorporate noble metal-based nanoparticles into 3d-transition metal oxides serving as support, which can promote electronic metal-support interactions.17,43–46 Hou et al.17 designed a multi-heterostructure, which features an Ir core wrapped in an IrO2 shell anchored on a Co3O4 structure. This design serves as a competent electrocatalyst for acidic OER, demonstrating superior OER activity and stability compared to unsupported IrO2 and Co3O4, due to the unique electron transfer from the IrO2 shell and Co3O4 support to Ir.
In addition to the composition of the catalyst, the size of the electrocatalyst also has a great influence on its catalytic performance.47 For example, common granular catalysts suffer from problems such as inefficient utilization of atoms.17,48–52 Reduction of noble metals to ultrafine clusters or even single atoms often results in catalysts with significant activity improvement.53–55 These small-sized catalysts are collectively referred to as atomically dispersed catalysts (ADCs).56 Within the field of catalysts, a burgeoning area of research is the exploration of ADCs. These catalysts comprise single atoms, double atoms, and clusters, obtained by various synthetic approaches. They stand out as a new frontier, as they show increased atom utilization efficiency and catalytic selectivity, which is achieved by precisely manipulating the configuration and composition surrounding the central metal atoms.57 Due to the Gibbs–Thomson effect, the central metal atoms tend to aggregate into nanoclusters/particles.58 The difficulty in fabricating a uniform and stable dispersion of metal atoms prevents the development of single-atom catalysts (SACs).56,59 Traditional approaches to precious metal-based SACs fabrication methods often involve intricate procedures.57 Hu et al.60 proposed a method to prepare one-dimensional coral-like Ru-doped cobalt oxide nanofibers possessing high specific surface area by electrostatic spinning and subsequent heat treatment. The overpotential of Ru-doped cobalt oxide nanofibers is 300 mV in 1.0 M KOH electrolyte at room temperature, lower than that of ruthenium oxide, with the same catalyst loading. However, the electrostatic spinning process requires a very high voltage to be carried out and strict control of the feed rate of the precursor solution as well as the feed distance, and the obtained fiber membrane must subsequently be oxidized at 300 °C and 450 °C in air. In addition, different metal centers have different chemical and electronic properties, and the way one SAC is synthesized cannot be simply extended to another SAC. The comprehensive exploration of SACs has been significantly restricted by the absence of efficient and widely applicable synthetic methodologies.
To overcome these challenges, we propose here a simple and energy-efficient salt-template method to prepare a series of electrocatalysts consisting of 3d-transition metal oxides doped with atomically dispersed noble metals. The atomic ratios of noble metals are around 0.93–3.44%. In this way, it can be said that the performance of the catalysts increases and therefore contributes to significantly reduced cost to meet the requirements of practical applications. The catalyst represented by Co3O4 doped with Ir (Ir–Co3O4) shows excellent OER performance over a wide pH range (acidic, alkaline, as well as neutral electrolytes). Particularly, in 0.5 M H2SO4 solution, Ir–Co3O4 exhibits an ultra-low overpotential of 268 mV, at a current density of 10 mA cm−2, along with a remarkably low Tafel slope of 38.0 mV dec−1, and can operate stably for 16 h at 10 mA cm−2, which are far superior to those of commercial IrO2, undoped Co3O4, and most recently reported Ir-based OER electrocatalysts. This work not only establishes a facile synthesis route for SACs, but also highlights their potential as high-performance and cost-effective electrocatalysts for OER applications.
Results and discussion
3d-transition metal oxides doped with noble metals (NM-TMOs) were synthesized by the salt-template method. First, noble metal precursors and 3d-transition metal precursors were mixed with KCl. Then, NM-TMOs grown on the salt template were obtained by calcination in air. After dissolving the template salt in water, we obtained the NM-TMO materials with high purity and high quality. In this process, the salt crystals act as templates to guide the growth of the oxides at high temperatures, so that the oxides are deposited on this template, which can endow these crystals with a great abundance of edges, corners, and surfaces, and yield oxides with large lateral dimensions.61 Salt templates can provide electrocatalysts with a large specific surface area and abundant active sites. These active sites are fully exposed on the surface of these oxides, which is conducive to improving the OER performance of the electrocatalysts.62 This synthesis method is very simple and energy-saving. To further extend the applicability of the synthetic method, a series of NM-TMO electrocatalysts were efficiently prepared by introducing the desired noble metal precursors and 3d-transition metal precursors.First, using Co3O4 as substrate and varying the noble metal precursors, Ir–Co3O4, Ru–Co3O4, and Pd–Co3O4 were synthesized, Fig. 1(a, e and i). The crystal structures of NM-TMOs and control samples are confirmed by X-ray diffraction (XRD). Diffraction peaks appear at 18.8°, 31.1°, 36.7°, 38.4°, 44.7°, 55.6°, 59.3°, and 65.1°, Fig. S1a (ESI†), belonging to the (111), (220), (311), (222), (400), (422), (511) and (440) crystalline planes of Co3O4 (face-centered cubic structure, JCPDS #42-1467), respectively. The XRD patterns of Ir–Co3O4, Ru–Co3O4, and Pd–Co3O4 show a standard cubic phase of Co3O4 with good crystallinity, and none of the diffraction peaks are associated with Ir or IrO2, Ru or RuO2, Pd or PdO, respectively.63 This result indicates that the crystal structure of Co3O4 remains unaltered by the presence of a small amount of Ir dopant, and further confirms that Ir is in the form of dispersed atoms. The high-resolution transmission electron microscopy (HR-TEM) image of Ir–Co3O4, Fig. 1b, shows the lattice fringes with a spacing of 0.488 nm, which is larger compared to the (111) facet of Co3O4 (0.467 nm). Moreover, no lattice fringes corresponding to metallic Ir or oxidized Ir nanoparticles are observed. The result indicates that Ir atoms are homogeneously dispersed in the Co3O4 lattice. Similarly, compared to the d-spacing of the (220) planes of Co3O4 (0.286 nm), the d-spacing of Ru–Co3O4 (0.298 nm) is larger, Fig. 1f. The d-spacing of (311) planes, of Pd–Co3O4 (0.267 nm), is also larger than that of Co3O4 (0.244 nm), Fig. 1j. Furthermore, by changing the transition metal precursors and using the Ir source, Ir–NiO and Ir–Fe2O3 can be synthesized by the same approach, Fig. 1(m and q). After doping with Ir atoms, the d-spacings of the (111) planes of Ir–NiO and the d-spacings of the (110) planes of Ir–Fe2O3 increase compared to NiO and Fe2O3, respectively, Fig. 1(n and r). The XRD patterns of Ir–NiO and Ir–Fe2O3 can be assigned to NiO and Fe2O3, respectively, in which diffraction peaks for neither Ir nor IrO2 crystals can be detected, Fig. S1b (ESI†). The uniform spatial distribution of noble metal elements, 3d-tradition metal elements, and oxygen in Ir–Co3O4, Ru–Co3O4, Pd–Co3O4, Ir–NiO, and Ir–Fe2O3 can also be confirmed by elemental mappings by energy-dispersive spectroscopy (EDS), Fig. 1(c, g, k, o and s).
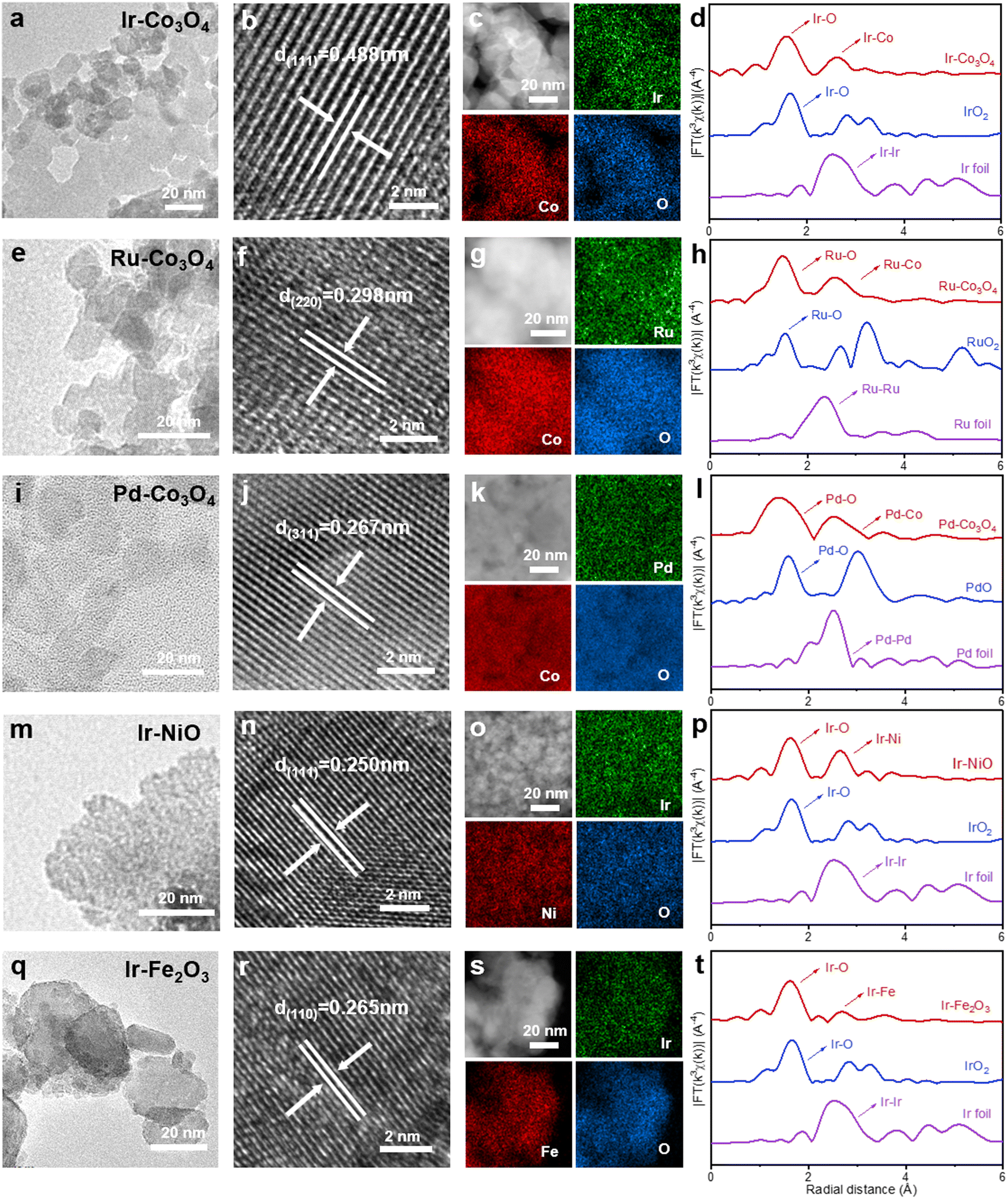 | ||
| Fig. 1 (a), (e), (i), (m) and (q) TEM images, (b), (f), (j), (n) and (r) HR-TEM images, (c), (g), (k), (o) and (s) HAADF-STEM images and the corresponding EDS elemental mappings, and EXAFS spectra of (d) Ir-L3 edge, (h) Ru-K edge, (l) Pd-K edge, (p) Ir-L3 edge, (t) Ir-L3 edge for Ir–Co3O4, Ru–Co3O4, Pd–Co3O4, Ir–NiO, and Ir–Fe2O3, respectively. | ||
In addition, we performed extended X-ray absorption fine structure (EXAFS) analyses to gain insight into the localized structures of NM-TMOs. The Fourier transformed-EXAFS (FT-EXAFS) spectrum of the Ir-L3 edge for Ir–Co3O4 displays two prominent peaks in R-space, denoting the first and second coordination shells of the central Ir atom, Fig. 1d. The main peak at about 1.56 Å corresponds to the Ir–O bond, compared to the Ir–O bond of IrO2 (1.66 Å). The second main peak of the Ir–Co (2.61 Å) bond is much smaller than the Ir–Ir bond in IrO2 (3.25 Å). The large reduced distance of the second shell originates from an edge-sharing network in Co3O4 as compared with a corner-sharing network in IrO2.64 The k3-weighted EXAFS spectrum of Ir-L3 was analyzed by wavelet transform, in Fig. S2(a–c) (ESI†). The wavelet transforms intensity maximum around 6.70 Å−1 arising from the Ir–O coordination is well resolved at 1.59 Å for Ir–Co3O4, which is similar to the Ir–O coordination in IrO2, while an intensity maximum at about 10.95 Å−1 associated with the Ir–Ir coordination in Ir foil is not observed in Ir–Co3O4.65 These results show that Ir–Ir scattering is absent in Ir–Co3O4 and, therefore, the formation of Ir cluster can be excluded. Similarly, FT-EXAFS spectra demonstrate that the noble metal species in Ru–Co3O4, Pd–Co3O4, Ir–NiO, and Ir–Fe2O3 are present as single atoms, Fig. 1(h, l, p and t) and Fig. S2(d–o) (ESI†). In summary, all catalysts showed a nanoparticular structure. Subsequent analysis of HR-TEM, EDS elemental mapping, and EXAFS spectra confirmed that the noble metal was uniformly doped into the lattice of the 3d-transition metal oxides in the form of single atoms. Undoubtedly, these characterization results fully confirm the universality of this synthesis method, which can achieve a diverse combination of atomically dispersed noble metals and 3d-transition metal oxides. This simple KCl method of preparing template, as well as the simple and nondestructive green transfer process, are of great importance for research in the preparation of single-atom noble metal doped TMO batches and the exploration of industrial applications.
We further investigated the structural and electronic properties of NM-TMO materials prepared by this method using Ir–Co3O4 as an example. XRD patterns of Ir–Co3O4 and undoped Co3O4 samples were refined, Fig. 2a and Table S1 (ESI†). The results showed that the lattice constant of Ir–Co3O4 (a = b = c = 8.10224 Å) increased after Ir doping compared to that of Co3O4 (a = b = c = 8.08579 Å), indicating that Ir was successfully dispersed into the Co3O4 lattice, causing the Co3O4 lattice to expand, Table S2 (ESI†). This result agrees with the HR-TEM results of Ir–Co3O4 mentioned above, Fig. 1c. The surface chemical states of the prepared materials were analyzed by X-ray photoelectron spectroscopy (XPS). The full XPS survey spectrum of Ir–Co3O4 confirms that Ir, Co, and O are present on the surface, Fig. 2b, which is in agreement with previous EDS mappings. Analyzing the high-resolution XPS spectra, Fig. 2c, the Ir 4f spectrum of commercial IrO2 shows a set of double peaks at 61.3 eV and 64.3 eV, which can be attributed to Ir4+. For Ir–Co3O4, the peak at about 60.1 eV corresponds to Co 3p, and the Ir 4f spectrum reveals two sets of double peaks, centered at 61.1/64.2 eV and 61.7/65.1 eV, and attributed to Ir4+ and Ir3+, respectively.65,66 The Co 2p spectra of Co3O4 and Ir–Co3O4 contain Co2+, Co3+, and satellite peaks, Fig. 2d. The Co2+ peaks are located at 796.1 eV (2p1/2) and 780.9 eV (2p3/2), and the Co3+ peaks are located at 794.6 eV (2p1/2) and 779.5 eV (2p3/2).22,60 The increase in the Co2+ to Co3+ ratio after Ir doping in Co3O4 indicates that Ir atoms replace Co3+ atoms at the octahedral sites, leading to the formation of more Co2+ to balance the surface oxygen vacancies.67 The chemical state of O 1s in Co3O4 and Ir–Co3O4 shows three peaks including lattice oxygen (M–O) at 529.5 eV, hydroxyl and oxygen vacancies (OH−/O2) at 531.2 eV, and adsorbed water molecules (H2O) at 532.8 eV, Fig. 2e.22 With Ir doping, the proportion of absorbed oxygen increases compared to lattice oxygen, suggesting that doping produces a large number of oxygen vacancies, providing more catalytically active sites.67
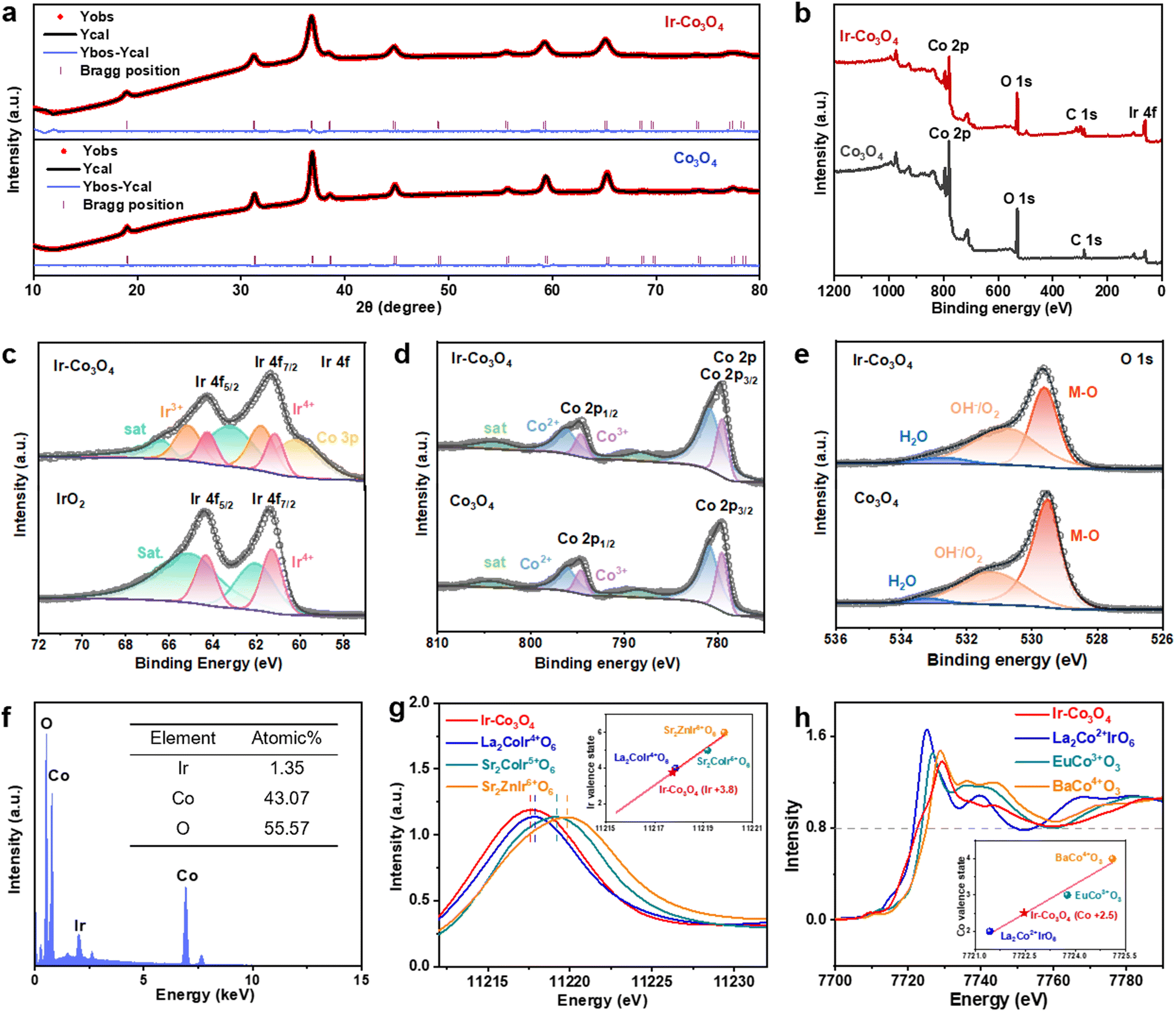 | ||
| Fig. 2 (a) Rietveld refinements of XRD patterns for Ir–Co3O4 and Co3O4. (b) XPS survey spectra of Ir–Co3O4 and Co3O4. XPS spectra of (c) Ir 4f for Ir–Co3O4 and IrO2, (d) Co 2p, and (e) O 1s for Ir–Co3O4 and Co3O4. (f) SEM-EDS spectrum of Ir–Co3O4. (g) The normalized XANES spectra of Ir-L3 edge for Ir–Co3O4, La2CoIr4+O6, Sr2CoIr5+O6, and Sr2ZnIr6+O6. The inset shows the Ir valence state. (h) The normalized XANES spectra of Co-K edge for Ir–Co3O4, La2Co2+IrO6, EuCo3+O3, and BaCo4+O3. The inset shows the Co valence state. | ||
The remaining NM-TMOs and their corresponding undoped TMOs were also analyzed by XPS to obtain the surface chemistry and valence states of the metal elements and oxygen, Fig. S3 (ESI†). The binding energies of Ru 3p1/2 and 3p3/2 at 487.3 eV and 465.3 eV suggest that Ru in Ru–Co3O4 occurs as Ru4+. Upon doping in Co3O4, the binding energy of Ru shifts about 1.0 eV for Ru 3p compared to RuO2, revealing a significant change in the electronic structure due to the doping effect, Fig. S3(a and d) (ESI†). In Pd–Co3O4, the deconvoluted spectrum of Pd 3d shows two sets of double peaks located at 336.7/342.0 eV and 338.4/343.9 eV, which are assigned to Pd2+ and Pd4+, respectively, Fig. S3(g and j) (ESI†).54 The binding energy level of Pd 3d after doping in Co3O4 is shifted by about 0.3 eV, compared to that of PdO. When Co3O4 were doped with Ru and Pd, the ratio of Co2+ also increased compared to that of Co3+, Fig. S3(b, e, h and k) (ESI†), suggesting that noble metal atoms replaced the octahedral sites of Co3+, resulting in the formation of more Co2+ to balance the surface oxygen vacancies. The O 1s spectra of all four materials show a higher proportion of absorbed oxygen compared to lattice oxygen, Fig. S3(c, f, i, l, o, r, u and x) (ESI†). Quantitative analysis of the SEM-EDS element spectra shows that Ir–Co3O4 contains 1.35 at% of Ir, Fig. 2f. For other NM-TMO materials, Ru–Co3O4 contains 1.4 at% of Ru, Pd–Co3O4 contains 0.9 at% of Pd, Ir–NiO contains 3.4 at% of Ir, and Ir–Fe2O3 contains 1.6 at% of Ir, Fig. S4(a–d) (ESI†). The above results are averages of multiple measurements and mostly consistent with the elemental content of the surface layer determined by XPS, Table S3 (ESI†).
The electronic structure of Ir–Co3O4 is further analyzed using the Ir-L3 X-ray absorption near edge structure (XANES) spectrum. Fig. 2g shows Ir-L3 XANES of Ir–Co3O4 together with La2CoIr4+O6, Sr2CoIr5+O6, and Sr2ZnIr6+O6 for comparison. We obtained the valence state of Ir3.8+ in Ir–Co3O4 (inset, Fig. 2g). Based on the relationship between the absorption edge energy positions (at 0.8 normalized intensity), the XANES spectra of Co K in Ir–Co3O4 and in La2Co2+IrO6, EuCo3+O3, and BaCo4+O3 are presented in Fig. 2h, the estimated valence state of the Co ion in Ir–Co3O4, is about +2.5 (inset, Fig. 2h). The corresponding K-edge EXAFS of Co shows three main signals associated with the Co–O, Co–Cooct (octahedral site), and Co–Cotet (tetrahedral site) scattering paths, Fig. S5 (ESI†). These results are in good agreement with the XPS results.
The valences of Ir, Ru, and Pd are determined by Ir foil and IrO2 references, Ru foil and RuO2 references, Pd foil and PdO references, respectively. The white line peak position of the Ir-L3 edge in Ir–NiO and Ir–Fe2O3 is between those for Ir foil and IrO2, indicating that the Ir has a valence state between 0 to +4. Similarly, the oxidation state of Ru in the Ru–Co3O4 and Pd in Pd–Co3O4 is between 0 to +4 and 0 to +2, according to their energy position of absorption edge at the Ru- and Pd-K XANES spectra in Fig. S6(a–d), ESI.†
We evaluated the electrocatalytic OER performance of the representative Ir–Co3O4 sample using a standard three-electrode system in three different electrolyte solutions, i.e., 0.5 M H2SO4, 1.0 M KOH, and 1 × PBS solutions. All measurements were performed with 95% iR compensation, and the measured potentials were calibrated according to the reversible hydrogen electrode (RHE). Commercial IrO2 catalyst and undoped Co3O4 were used as references. First, we tested and discussed the performance of each catalyst in the acidic electrolyte (0.5 M H2SO4 solution). The linear scanning voltammetry (LSV) curves shown in Fig. 3a were recorded on a glassy carbon electrode (GCE) with a geometric area of 0.196 cm2. At the current density of 10 mA cm−2, Ir–Co3O4 showed an overpotential of 268 mV, which is lower than that of Co3O4 (466 mV) and commercial IrO2 (329 mV). The acidic OER properties of the catalysts prepared using this synthesis method are shown in Fig. S7(a and b), ESI.† These catalysts showed improved performance compared to both commercial IrO2 and undoped Co3O4. Among them, the overpotentials of these catalysts at a current density of 10 mA cm−2 follow the orders: Ir–Co3O4 < Ir–NiO < Ir–Fe2O3 < IrO2 (Fig. S7a, ESI†) and Ir–Co3O4 < Ru–Co3O4 < Pd–Co3O4 < Co3O4 (Fig. S7b, ESI†). The above results indicate that the synthesis method is a versatile method for the preparation of noble metal single-atom doped 3d-transition metal oxide electrocatalysts. Tafel plots are used to evaluate the kinetic behavior of the electrocatalysts, Fig. 3b, and show slopes of 38.0, 63.7, and 81.8 mV dec−1 for Ir–Co3O4, commercial IrO2, and Co3O4 catalysts, respectively, reflecting the fastest reaction rate for the Ir–Co3O4 catalysts, and indicating that the Ir doping could promote the OER kinetics. Electrochemical impedance spectroscopy (EIS) is a commonly used technique to understand OER activity. As shown in Fig. 3c, Ir–Co3O4 has a much smaller charge transfer resistance (Rct) (16.6 Ω) compared to commercial IrO2 (220.0 Ω) and Co3O4 (5271.0 Ω) values deduced from an equivalent circuit (Fig. S8, ESI†), revealing the fast electron transfer of Ir–Co3O4 during the OER reaction (Table S4, ESI†). Fig. S9 (ESI†) shows the TEM image of Co3O4, which demonstrates that Co3O4 has a similar morphology to Ir–Co3O4 (Fig. 1a), so the differences in catalytic performance are not due to morphology or size disparities. The charge density difference on Ir–Co3O4 indicates that incorporation of Ir into Co3O4 can effectively regulate charge redistribution, where Ir atoms tend to lose electrons and off-domain electrons accumulate around neighboring Ir–O bonds. This result also confirms the existence of strong electronic interactions between Ir single sites and the host Co3O4. The introduction of Ir single atoms endows Co3O4 with metallic properties, leading to better electrical conductivity and thus faster OER dynamics. Stronger interactions and more covalent properties between Co and Ir atoms suggest synergistic effects between Ir atoms and Co3O4.65
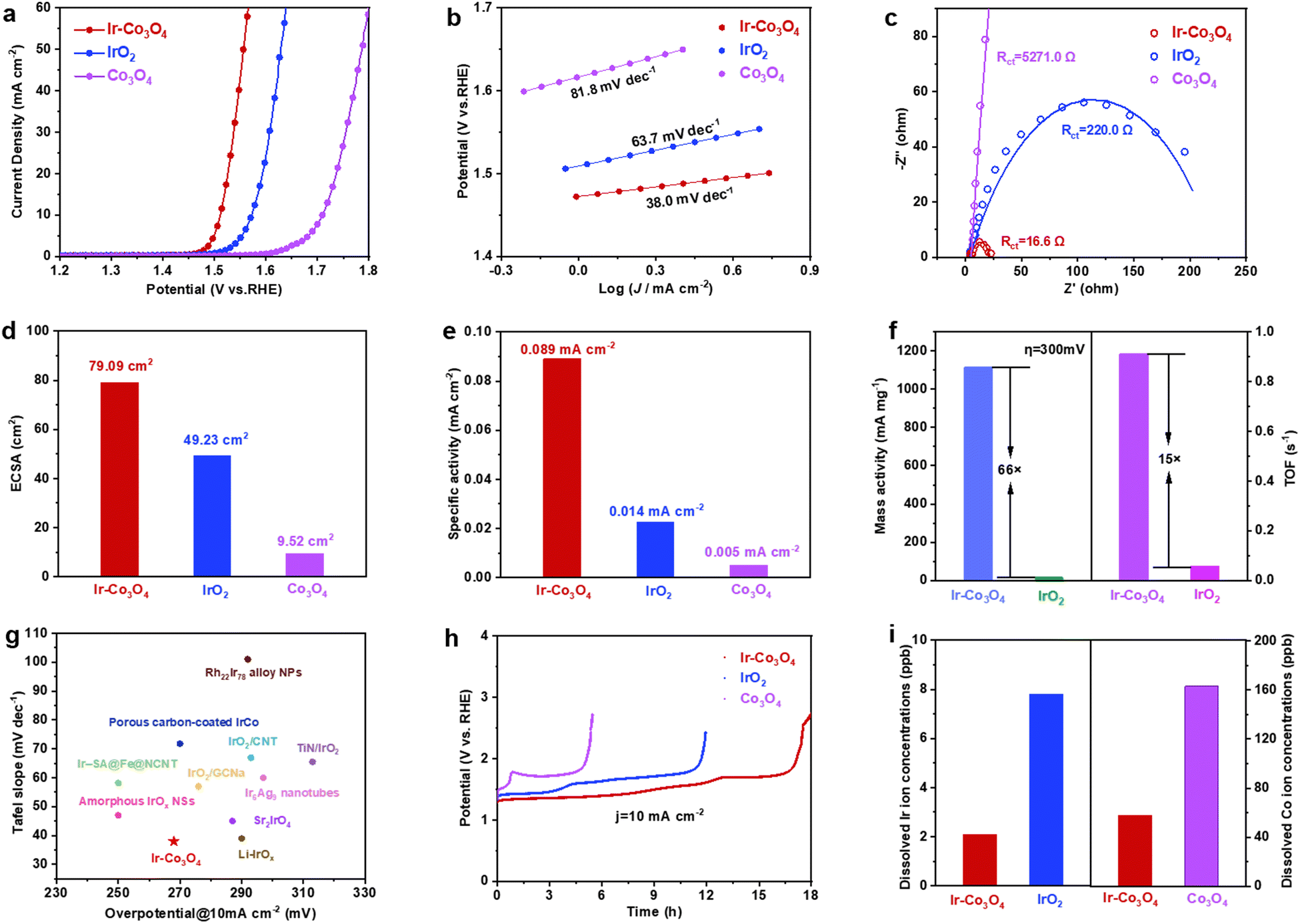 | ||
| Fig. 3 (a) LSV curves of Ir–Co3O4, IrO2, and Co3O4 collected at a scanning rate of 5 mV s−1 in 0.5 M H2SO4 solution. (b) The corresponding Tafel plots were obtained from the polarization curves in (a). (c) Nyquist plots and corresponding fitting curves of the samples obtained at 1.49 V vs. RHE for EIS analysis. (d) The calculated ECSA values for Ir–Co3O4, IrO2, and Co3O4 in 0.5 M H2SO4. (e) Specific activity of Ir–Co3O4, IrO2, and Co3O4. (f) Mass activities and TOF values of Ir–Co3O4, IrO2 at the overpotential of 300 mV. (g) Comparison of overpotentials at 10 mA cm−2 and Tafel slopes among reported Ir-based OER electrocatalysts. (h) Chronopotentiometric measurements of Ir–Co3O4, IrO2, and Co3O4 at 10 mA cm−2. (i) Dissolved Ir and Co (left and right y-axis, respectively) ion concentrations in the electrolyte measured by ICP-OES for Ir–Co3O4, IrO2, and Co3O4. | ||
In addition, cyclic voltammetry (CV) scans, at different scan rates, were performed to measure the electrochemical double-layer capacitance (Cdl), which was used to calculate the effective electrochemical surface area (ECSA), Fig. S10(a–d), ESI.† It can be seen that the ECSA of Ir–Co3O4 is 79.09 cm2, much higher than that of commercial IrO2 (49.23 cm2), as well as that of Co3O4 (9.52 cm2), Fig. 3d. To further investigate the intrinsic activity of the catalyst, the specific activity of Ir–Co3O4, IrO2, and Co3O4 is determined by normalizing the catalytic current to the ECSA of the catalysts. At an overpotential (η) of 300 mV, Ir–Co3O4 exhibits the highest specific activity (0.089 mA cmECSA−2), which exceeds the specific activities of IrO2 (0.014 mA cmECSA−2) and Co3O4 (0.005 mA cmECSA−2) by approximately 6.4 and 17.8 times, respectively, Fig. 3e. As shown in Fig. 3f, Ir–Co3O4 has 66 times higher mass activity (MA) as well as 15 times higher turnover frequency (TOF) than that of commercial IrO2 at η = 300 mV. The above results suggest that the doping with Ir contributes to a significant increase in the utilization of Ir atoms as well as the active sites of the Co3O4 catalyst, thus improving its intrinsic activity. It is noteworthy that the overpotential of Ir–Co3O4 catalyst at 10 mA cm−2 and its Tafel slope in acidic electrolyte (0.5 M H2SO4) is lower than those of most Ir-based OER catalysts, Fig. 3g and Table S5 (ESI†).
Apart from activity, the durability of OER electrocatalysts is also a crucial parameter to evaluate their performance. Ir–Co3O4 demonstrates stability over 16 h at a current density of 10 mA cm−2, exceeding that of commercial IrO2 and Co3O4 and showing the lowest degradation of its OER activity, Fig. 3h. This suggests that doping a small amount of Ir can obviously improve the stability of Co3O4.68 Nevertheless, Ir–Co3O4 still has some limitations due to the intrinsic instability of Co3O4 under harsh OER conditions (e.g. oxidation potentials; acidic electrolytes).65 Following the OER stability test, inductively coupled plasma-atomic emission spectrometry (ICP-OES) measurement showed severe leaching of Ir ions in IrO2 and Co ions in Co3O4 compared to Ir–Co3O4, Fig. 3i. The inhibitory effect of Ir-doped Co3O4 on the dissolution of Ir ions and Co ions is illustrated, which further explains the higher stability of the Ir–Co3O4 catalyst. Morphological characterization by low-magnification TEM shows that Ir–Co3O4 retains nanoparticles after chronopotentiometric measurement at 10 mA cm−2, Fig. S11a (ESI†), and the HR-TEM image shows no obvious structural changes, Fig. S11b (ESI†). EDS elemental mappings show that the distribution of Co, Ir, and O elements, after the stability test, remained uniform throughout Ir–Co3O4, Fig. S11c (ESI†). However, the XPS spectra of Ir 4f and Co 2p show an increase in the valence of Ir and Co after chronopotentiometry of Ir–Co3O4 at 10 mA cm−2, which is common in OER catalysts at oxidizing applied electrode potentials, Fig. S11(d and e) (ESI†). The XPS spectrum of O 1s shows an increase in the proportion of absorbed oxygen, providing further confirmation of the increase in the overall catalyst valence after the stability test, Fig. S11f (ESI†).
Next, the electrochemical performance of each catalyst in the alkaline electrolyte (1.0 M KOH solution) is analyzed. In the LSV curves, Fig. 4a, Ir–Co3O4 has a lower overpotential (289 mV) at 10 mA cm−2, compared to IrO2 (377 mV) and Co3O4 (406 mV), demonstrating a good OER activity of Ir–Co3O4 in alkaline electrolyte. The Tafel slopes of Ir–Co3O4, commercial IrO2, and Co3O4 catalysts are 72.4, 81.4, and 126.9 mV dec−1, respectively, Fig. 4b. The lower Tafel slope of Ir–Co3O4 suggests that it has a higher reaction rate than commercial IrO2 and Co3O4 catalysts. In the EIS Nyquist plot displayed in Fig. 4c, the interfacial charge transfer resistance of Ir–Co3O4 (14.6 Ω) was reduced compared with that of IrO2 (102.2 Ω) and Co3O4 (445.3 Ω) (Table S6, ESI†), indicating that the doping of the noble metal Ir led to a decrease of the electron conduction activation energy, which improved the conductivity of the catalysts, thus promoting the catalytic activity. The ECSA of Ir–Co3O4, 58.52 cm2, was higher than that of IrO2 (20.44 cm2) and Co3O4 (24.04 cm2), Fig. S12(a–e) (ESI†). By normalizing the OER current to ECSA at η = 300 mV, Ir–Co3O4 exhibits higher specific activity (0.039 mA cmECSA−2) than the commercial IrO2 (0.021 mA cmECSA−2) and Co3O4 (0.013 mA cmECSA−2), Fig. 4d. In addition, the MA and TOF were calculated at η = 300 mV, Fig. 4e. Ir–Co3O4 showed a MA (358.0 mA mg−1) 36 times higher than that of IrO2 (9.9 mA mg−1), and a TOF (0.1821 s−1) 37 times higher than that of IrO2 (0.0049 s−1). The above results showed that Ir doping also greatly enhances the catalytic activity of Co3O4 in alkaline electrolytes, and its activity is superior to that of IrO2 with the same mass loading. Meanwhile, Ir−Co3O4 also shows better stability than IrO2, Fig. 4f. Ir−Co3O4 can operate stably for 200 h at 10 mA cm−2 and the potential only increased slightly by 0.08 V, while the IrO2 catalyst was deactivated after only 44 h. This reflects that Ir doping in Co3O4 can substantially improve the durability of Ir-based catalysts.
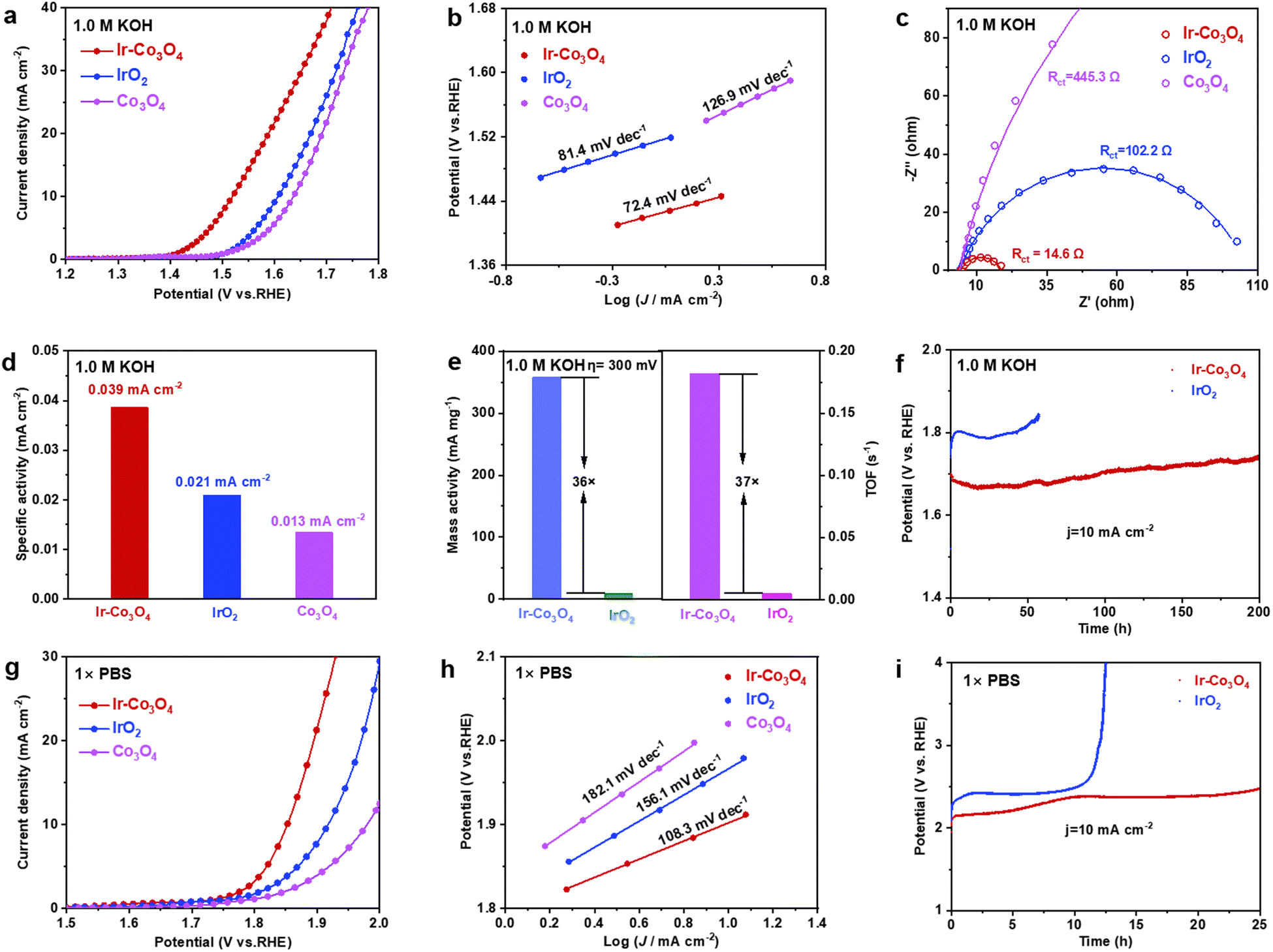 | ||
| Fig. 4 (a) LSV curves of Ir–Co3O4, IrO2, and Co3O4 in 1.0 M KOH at a scanning rate of 5 mV s−1. (b) The corresponding Tafel plots were obtained from the polarization curves in (a). (c) Nyquist plots and corresponding fitting curves of samples obtained at 1.52 V vs. RHE. (d) Specific activity of Ir–Co3O4, IrO2, and Co3O4. (e) Mass activities and TOF values of Ir–Co3O4, and IrO2 at 300 mV overpotential. (f) Chronopotentiometric measurements, in 1.0 M KOH, of Ir–Co3O4 and IrO2 at 10 mA cm−2 using carbon paper as catalyst support. (g) LSV curves of Ir–Co3O4, IrO2, and Co3O4 in 1 × PBS at a scanning rate of 5 mV s−1. (h) The corresponding Tafel plots were obtained from the LSV curves in (g). (i) Chronopotentiometric measurements, in 1 × PBS, of Ir–Co3O4 and IrO2 at 10 mA cm−2. | ||
To better understand the electrochemical performance of the Ir−Co3O4 catalyst over the entire pH range, we tested the OER performance in 1 × PBS neutral electrolyte. As shown in Fig. 4(g and h), the overpotential of Ir–Co3O4 at current density = 10 mA cm−2 (622 mV) was lower than that of IrO2 (687 mV) and Co3O4 (749 mV), and the Tafel slope of Ir–Co3O4 (108.3 mV dec−1) was the lowest of the three. This suggests that Ir–Co3O4 has a better catalytic activity compared to IrO2 and Co3O4 in neutral electrolytes. To investigate the stability in neutral electrolyte, the catalysts were tested for stability under the same conditions at 10 mA cm−2. Fig. 4i shows that the Ir–Co3O4 catalyst remained stable for 25 h, while the IrO2 catalyst started to deactivate after 10 h of testing. These results indicate that Ir–Co3O4 has a long-term durability in neutral electrolyte. The above results suggest that Ir doping significantly improves the OER performance of Co3O4 in acidic, alkaline, and neutral electrolytes.
Conclusions
In summary, we present a universal salt-template method to prepare 3d-transition metal oxides doped with single noble metal atoms. In addition to Ir–Co3O4, a wide variety of electrocatalysts, including Ru–Co3O4, Pd–Co3O4, Ir–NiO, and Ir–Fe2O3, can be obtained by simply substituting the noble metal or the 3d-transition metal precursor. EXAFS spectroscopy reveals that the noble metals in the NM-TMO electrocatalysts synthesized by this method are all in the monoatomic form and uniformly dispersed in the nanoparticles, which greatly enhances the atomic utilization of the noble metals and provides more active sites for the catalysts. Surprisingly, the Ir–Co3O4 synthesized by this method showed excellent electrocatalytic performance and stability compared with commercial IrO2 and Co3O4 in acidic, alkaline, and neutral media. The present study demonstrates the versatility of the synthesis method over a wide pH range, which not only reduces the catalyst cost by decreasing the precious metals content but also improves the catalytic performance. The simplicity and energy-saving characteristics of the synthesis method will be of great reference value for the practical application of various noble metal-based catalysts.Experimental
Materials
Iridium(III) trichloride (IrCl3, solid, 99.9%), cobalt chloride hexahydrate (CoCl2·6H2O, solid, AR), nickel chloride hexahydrate (NiCl2·6H2O, solid, 98%), potassium chloride (KCl, solid, ≥99.0%), and sodium hydroxide (NaOH, solid, ≥98%) were purchased from Aladdin. Palladium(II) chloride (PdCl2, solid, Pd 59–60%), ruthenium(III) chloride (RuCl3, solid, 99.5%), iridium(IV) oxide (IrO2, solid, 99.9%), ruthenium dioxide (RuO2, solid, 99%), palladium(II) oxide (PdO, solid, 98%), and 1 × phosphate buffered saline (1 × PBS, solution, pH 7.2–7.4, 0.01 M) were purchased from Innochem. Potassium chloride (KCl, solid, 99.8%), iron trichloride hexahydrate (FeCl3·6H2O, solid, ≥99.0%), sulfuric acid (H2SO4, liquid, ≥96%), and potassium hydroxide (KOH, solid, 97%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Nafion® solution (5 wt%) was purchased from Alfa Aesar.Synthesis of Ir–Co3O4 electrocatalyst
Ir–Co3O4 was synthesized by the salt template method. 200 mg KCl, 0.5 mmol CoCl2·H2O, and 0.0375 mmol IrCl3 (the molar ratio of CoCl2·6H2O to IrCl3 is 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were dissolved in 30 mL of deionized water and stirred at 90 °C for 4 h to obtain a 10 mL solution. To the above solution, 40 mg NaOH dissolved in 10 mL of deionized water was added and the mixture was stirred at 90 °C for 3 h. The dried powder was scraped to grind, spread evenly over a crucible, and heated to 350 °C in the air at a heating rate of 5 °C min−1 and kept at 350 °C for 6 h. After cooling naturally, the powder was washed and filtered by pumping and dried at 60 °C for 3 h to finally obtain the Ir–Co3O4 catalyst.
:3) were dissolved in 30 mL of deionized water and stirred at 90 °C for 4 h to obtain a 10 mL solution. To the above solution, 40 mg NaOH dissolved in 10 mL of deionized water was added and the mixture was stirred at 90 °C for 3 h. The dried powder was scraped to grind, spread evenly over a crucible, and heated to 350 °C in the air at a heating rate of 5 °C min−1 and kept at 350 °C for 6 h. After cooling naturally, the powder was washed and filtered by pumping and dried at 60 °C for 3 h to finally obtain the Ir–Co3O4 catalyst.
Synthesis of Ru–Co3O4 and Pd–Co3O4 electrocatalysts
Using the above synthesis method, Ru–Co3O4 and Pd–Co3O4 were synthesized using RuCl3 (0.0375 mmol) and PdCl2 (0.0375 mmol) as sources, respectively, maintaining all other conditions.Synthesis of Ir–NiO, Ir–Fe2O3 electrocatalysts
Using the above synthesis method, the 3d-transition metal Co source CoCl2·6H2O was replaced with Ni source NiCl2·6H2O (0.5 mmol) and Fe source FeCl3·6H2O (0.5 mmol) to synthesize Ir–NiO and Ir–Fe2O3 catalysts, respectively, and the rest of the conditions were kept unchanged.Synthesis of Co3O4, NiO, and Fe2O3 electrocatalysts
Co3O4, NiO, and Fe2O3 catalysts were synthesized using the above synthesis method without using the noble metal precursors, and the rest of the conditions were kept unchanged.Characterization
Scanning electron microscopy (SEM) images were acquired using a Zeiss Supra 55 at an acceleration voltage of 5 kV. Transmission electron microscopy (TEM) and High-resolution TEM (HR-TEM) images were obtained using a JEOL 2100F instrument, operating at a voltage of 200 kV. Aberration-corrected high-angle annular dark field-scanning TEM (AC HAADF-STEM) images, along with corresponding elemental mapping, were captured on an aberration-correction Hitachi 2700D microscope operated at 200 kV. The mass fraction of Ir in Ir–Co3O4 and the dissolved ion concentrations after stability tests were analyzed using ICP-OES on an Agilent ICP-OES 730.To further investigate the electronic structure, XPS measurements were conducted on a Thermo Scientific Escalab 250Xi with an Al-Kα source. The XPS spectra were deconvoluted by Avantage software, and the binding energy was calibrated by C 1s (284.8 eV). The baseline was added by selecting Smart type background, and Gauss–Lorentz fitting was used for peak fitting. The position of the peak to be added was selected by moving the three vertical lines on the spectrum screen where the peaks were to be split (single peak and double peaks were selected for different binding energies according to the references). XAS of Ir L3-edge, Ru K-edge, Pd K-edge, Co K-edge, Ni K-edge, and Fe K-edge were performed at the 44A beamline at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. All XAS experiments were conducted in ambient air at room temperature and analyzed using the standard program Demeter. For wavelet-transformed k3-weighted EXAFS, the χ(k) exported from Athena was processed using the Hama Fortran code designed by Harald Funke and Marina Chukalina. Parameters included a range of R (0–6 Å), k (0–15 Å−1), k weight (0), and the Morlet function with kappaMorlet = 10, sigmaMorlet = 1 as the mother wavelet for an encompassing distribution.
Structural analysis through XRD employed a Bruker D8 Advance powder diffractometer (operating at 40 kV, 40 mA) equipped with a Cu-Kα source (λ = 1.5405 Å) and fitted with a beryllium window at room temperature. Rietveld refinements for XRD data were executed with the Full-Prof program. The refined parameters comprise background parameters, line shift errors (zero shift), Caglioti coefficients (U, V, and W), scale factor, lattice parameters, atomic position, atomic rate occupancy, and isotropic atomic displacement parameters.
Electrochemical measurements
The electrochemical testing of the oxygen evolution reaction performance was conducted using an electrochemical workstation (Biologic) equipped with a rotating disk electrode (RDE) device. The workstation has a standard three-electrode cell, consisting of a carbon rod (CR) as the counter electrode, a glassy carbon electrode coated with a certain amount of catalyst as the working electrode, and a calomel electrode as the reference electrode. The tests were performed in electrolytes of 0.5 M H2SO4 solution, 1.0 M KOH solution, and 1 × PBS solution. The final potential was calibrated to the reversible hydrogen electrode (RHE) using the equation E(RHE) = E(Hg|HgCl2) + 0.0591 × pH + 0.241.The working electrode was prepared as follows: due to the poor conductivity of 3d-transition metal oxides, catalyst powder was mixed with Vulcan carbon (XC-72R, under N2 atmosphere, 400 °C, 5 °C min−1) in a 1:1 mass ratio. A homogeneous suspension was formed by dispersing 5 mg of catalyst powder and 5 mg of C powder in 1900 μL of isopropanol and sonicated for 30 min. 100 μL of 5.0 wt% Nafion® solution was added to the above suspension, and the catalyst ink was obtained by sonication of the mixed solution for 5 min. A glassy carbon electrode (with an area of 0.19625 cm2) was coated with a thin-film electrode by depositing 20 μL of the catalyst ink, followed by drying under a heat lamp.
To reach a steady state, CV measurements were performed for 10 cycles at a scan rate of 100 mV s−1. The LSV curves were later recorded at a scan rate of 5 mV s−1. The mass of all catalysts in the performance tests was 0.05 mg (254.8 μg cm−2). During the stability test, the catalyst was slowly dropped onto carbon paper, which was used as the substrate, and the catalyst ink was prepared in the same way as described above.
Data availability
All relevant data are available from the corresponding authors upon reasonable request.Author contributions
J. Wang and Y. Zhu contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work leading to these results has received funding from the National Natural Science Foundation of China (22179098). We acknowledge support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials. N. Alonso-Vante acknowledges financial support from the European Union (ERDF) and Région Nouvelle Aquitaine.References
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620 RSC.
- T. Zhang, Y.-P. Liu, Q.-T. Ye and H.-J. Fan, J. Electrochem., 2022, 28, 2214006 Search PubMed.
- W.-F. Xie and M.-F. Shao, J. Electrochem., 2022, 28, 22014008 Search PubMed.
- Y. Zhang, C. Wu, H. Jiang, Y. Lin, H. Liu, Q. He, S. Chen, T. Duan and L. Song, Adv. Mater., 2018, 30, 1707522 CrossRef PubMed.
- H. Yang, X. Han, A. I. Douka, L. Huang, L. Gong, C. Xia, H. S. Park and B. Y. Xia, Adv. Funct. Mater., 2021, 31, 2007602 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196 RSC.
- Y. Liu, D. Zhou, T. Deng, G. He, A. Chen, X. Sun, Y. Yang and P. Miao, ChemSusChem, 2021, 14, 5359 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- D. Tang, Y. Ma, Y. Liu, K. Wang, Z. Liu, W. Li and J. Li, J. Alloys Compd., 2022, 893, 162287 CrossRef CAS.
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- K. Zhang and R. Zou, Small, 2021, 17(37), 2100129 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- X. Zhang, C. Feng, B. Dong, C. Liu and Y. Chai, Adv. Mater., 2023, 35, 2207066 CrossRef CAS PubMed.
- R.-Y. Fan, Y.-N. Zhou, M.-X. Li, J.-Y. Xie, W.-L. Yu, J.-Q. Chi, L. Wang, J.-F. Yu, Y.-M. Chai and B. Dong, Chem. Eng. J., 2021, 426, 131943 CrossRef CAS.
- J.-Y. Xie, Z.-Z. Liu, J. Li, L. Feng, M. Yang, Y. Ma, D.-P. Liu, L. Wang, Y.-M. Chai and B. Dong, J. Energy Chem., 2020, 48, 328 CrossRef.
- N. Yu, Y. Ma, J.-K. Ren, Z.-J. Zhang, H.-J. Liu, J. Nan, Y.-C. Li, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 478, 147415 CrossRef CAS.
- H.-J. Liu, S. Zhang, W.-Y. Yang, N. Yu, C.-Y. Liu, Y.-M. Chai and B. Dong, Adv. Funct. Mater., 2023, 33, 2303776 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, Nano Energy, 2020, 78, 105270 CrossRef CAS.
- L. Hou, H. Jang, H. Liu, Z. Li, M. G. Kim, Q. Qin and X. Liu, ACS Sustainable Chem. Eng., 2022, 10, 15950 CrossRef CAS.
- L. Zhang, Y. Wang, Y. Wang, H. Liu, Q. Qin and X. Liu, ACS Sustainable Chem. Eng., 2022, 10, 10658 CrossRef CAS.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- G. Wu, X. Zheng, P. Cui, H. Jiang, X. Wang, Y. Qu, W. Chen, Y. Lin, H. Li, X. Han, Y. Hu, P. Liu, Q. Zhang, J. Ge, Y. Yao, R. Sun, Y. Wu, L. Gu, X. Hong and Y. Li, Nat. Commun., 2019, 10, 4855 CrossRef PubMed.
- Y. Xie, Y. Su, H. Qin, Z. Cao, H. Wei, F. Wu and G. Ou, Int. J. Hydrogen Energy, 2023, 48, 14642 CrossRef CAS.
- S. Kalantarifard, S. I. Allakhverdiev and M. M. Najafpour, Int. J. Hydrogen Energy, 2020, 45, 33563 CrossRef CAS.
- S. Mehrabani, R. Bikas, Z. Zand, Y. Mousazade, S. I. Allakhverdiev and M. M. Najafpour, Int. J. Hydrogen Energy, 2020, 45, 17434 CrossRef CAS.
- Z. Lei, W. Cai, Y. Rao, K. Wang, Y. Jiang, Y. Liu, X. Jin, J. Li, Z. Lv, S. Jiao, W. Zhang, P. Yan, S. Zhang and R. Cao, Nat. Commun., 2022, 13, 24 CrossRef CAS PubMed.
- T. G. Yun, Y. Heo, H.-B. Bae and S.-Y. Chung, Nat. Commun., 2021, 12, 824 CrossRef CAS PubMed.
- A. L. Bhatti, A. Tahira, A. Gradone, R. Mazzaro, V. Morandi, U. Aftab, M.-I. Abro, A. Nafady, K. Qi, A. Infantes-Molina, A. Vomiero and Z. H. Ibupoto, Electrochim. Acta, 2021, 398, 139338 CrossRef CAS.
- Q. Wang, Z. Zhang, C. Cai, M. Wang, Z. L. Zhao, M. Li, X. Huang, S. Han, H. Zhou, Z. Feng, L. Li, J. Li, H. Xu, J. S. Francisco and M. Gu, J. Am. Chem. Soc., 2021, 143, 13605 CrossRef CAS PubMed.
- Y. Tong, P. Chen, L. Chen and X. Cui, ChemSusChem, 2021, 14, 2576 CrossRef CAS PubMed.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973 CrossRef CAS.
- Y. Dai, J. Yu, J. Wang, Z. Shao, D. Guan, Y.-C. Huang and M. Ni, Adv. Funct. Mater., 2022, 32, 2111989 CrossRef CAS.
- M. Xiao, J. Zhu, S. Li, G. Li, W. Liu, Y.-P. Deng, Z. Bai, L. Ma, M. Feng, T. Wu, D. Su, J. Lu, A. Yu and Z. Chen, ACS Catal., 2021, 11, 8837 CrossRef CAS.
- Z. Shi, Y. Wang, J. Li, X. Wang, Y. Wang, Y. Li, W. Xu, Z. Jiang, C. Liu, W. Xing and J. Ge, Joule, 2021, 5, 2164 CrossRef CAS.
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C.-H. Yan, J. Am. Chem. Soc., 2020, 142, 18378 CrossRef CAS PubMed.
- X. Liang, L. Shi, R. Cao, G. Wan, W. Yan, H. Chen, Y. Liu and X. Zou, Adv. Mater., 2020, 32, 2001430 CrossRef CAS PubMed.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346 CrossRef CAS.
- W. Hu, S. Chen and Q. Xia, Int. J. Hydrogen Energy, 2014, 39, 6967 CrossRef CAS.
- H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552 CrossRef CAS PubMed.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425 CrossRef CAS PubMed.
- Y. Zhu, J. Wang and J. Ma, Small Sci., 2023, 3, 2300010 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Small Struct., 2021, 2, 2000051 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34 CrossRef CAS.
- Q. Wang, Y. Lei, Y. Wang, Y. Liu, C. Song, J. Zeng, Y. Song, X. Duan, D. Wang and Y. Li, Energy Environ. Sci., 2020, 13, 1593 RSC.
- Y. Cui, Z. Cao, Y. Zhang, H. Chen, J. Gu, Z. Du, Y. Shi, B. Li and S. Yang, Small Sci., 2021, 1, 2100017 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740 CrossRef CAS PubMed.
- C.-W. Tung, Y.-Y. Hsu, Y.-P. Shen, Y. Zheng, T.-S. Chan, H.-S. Sheu, Y.-C. Cheng and H. M. Chen, Nat. Commun., 2015, 6, 8106 CrossRef CAS PubMed.
- K. Fominykh, G. C. Tok, P. Zeller, H. Hajiyani, T. Miller, M. Döblinger, R. Pentcheva, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2017, 27, 1605121 CrossRef.
- W. Wang, S. Xi, Y. Shao, X. Gao, J. Lin, C. Meng, W. Wang, X. Guo and G. Li, ChemElectroChem, 2019, 6, 1846 CrossRef CAS.
- Y. Wang, X. Cui, J. Zhang, J. Qiao, H. Huang, J. Shi and G. Wang, Prog. Mater. Sci., 2022, 128, 100964 CrossRef CAS.
- L. Wang, M.-X. Chen, Q.-Q. Yan, S.-L. Xu, S.-Q. Chu, P. Chen, Y. Lin and H.-W. Liang, Sci. Adv., 2019, 5, eaax6322 CrossRef CAS PubMed.
- S. Zhao, R. Jin and R. Jin, ACS Energy Lett., 2018, 3, 452 CrossRef CAS.
- D. Hu, R. Wang, P. Du, G. Li, Y. Wang, D. Fan and X. Pan, Ceramics Int., 2022, 48, 6549 CrossRef CAS.
- Y. Huan, J. Shi, X. Zou, Y. Gong, C. Xie, Z. Yang, Z. Zhang, Y. Gao, Y. Shi, M. Li, P. Yang, S. Jiang, M. Hong, L. Gu, Q. Zhang, X. Yan and Y. Zhang, J. Am. Chem. Soc., 2019, 141, 18694 CrossRef CAS PubMed.
- X. Xiao, H. Song, S. Lin, Y. Zhou, X. Zhan, Z. Hu, Q. Zhang, J. Sun, B. Yang, T. Li, L. Jiao, J. Zhou, J. Tang and Y. Gogotsi, Nat. Commun., 2016, 7, 11296 CrossRef CAS PubMed.
- T.-Y. Jeon, S.-H. Yu, S.-J. Yoo, H.-Y. Park and S.-K. Kim, Carbon Energy, 2021, 3, 8 CrossRef.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek, M. Zhu, X. Wu, C.-W. Pao, C.-K. Chang, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J.-M. Chen, S.-Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2022, 13, 7754 CrossRef CAS PubMed.
- V. Pfeifer, T.-E. Jones, J.-J. Velasco Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143 RSC.
- B. Guo, R. Ma, Z. Li, J. Luo, M. Yang and J. Wang, Mater. Chem. Front., 2020, 4, 1390 RSC.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4rp00144c |
| This journal is © The Royal Society of Chemistry 2024 |