
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical characterization of γ-Fe2O3 and a reduced graphene oxide composite as a sustainable anode material for Na-ion batteries†
Antunes
Staffolani‡
*a,
Leonardo
Sbrascini
 a,
Luca
Bottoni
a,
Luca
Minnetti
a,
Hamideh
Darjazi§
a,
Angela
Trapananti
bc,
Francesco
Paparoni
b,
Seyed Javad
Rezvani
bd,
Marco
Minicucci
b,
Messaoud
Harfouche
e and
Francesco
Nobili
ac
a,
Luca
Bottoni
a,
Luca
Minnetti
a,
Hamideh
Darjazi§
a,
Angela
Trapananti
bc,
Francesco
Paparoni
b,
Seyed Javad
Rezvani
bd,
Marco
Minicucci
b,
Messaoud
Harfouche
e and
Francesco
Nobili
ac
aUniversity of Camerino, School of Science and Technology – Chemistry Division, Via Madonna delle Carceri, Camerino I-62032, Italy. E-mail: antunes.staffolani@unibo.it
bUniversity of Camerino, School of Science and Technology – Physics Division, Via Madonna delle Carceri, Camerino I-62032, Italy
cGISEL – Centro di Riferimento Nazionale per i Sistemi di Accumulo Elettrochimico di Energia, INSTM, Via G. Giusti 9, Firenze I-50121, Italy
dConsiglio Nazionale delle Ricerche (CNR), IOM-CNR, Laboratorio TASC, Basovizza SS-14, km 163.5, Trieste I-34149, Italy
eSynchrotron-light for Experimental Science and Applications in the Middle East (SESAME), 19252, Allan, Jordan
First published on 12th June 2024
Abstract
In this paper we report the synthesis and characterization of a γ-Fe2O3/reduced graphene oxide composite anode for Na-ion batteries. The nanocomposite anode is synthesized by a facile and green method. Structural and morphological characterization highlights a small γ-Fe2O3 particle size and their successful embedding in the carbonaceous matrix. Electrochemical characterization reveals a high specific capacity of ≈300 mA h g−1 at 1000 mA g−1, while at 5 A g−1 a capacity of 113 mA h g−1 is retained. Cyclic voltammetry at different scan rates, impedance spectroscopy, and ex situ Raman measurements evidence a redox pseudocapacitive behavior and full reversibility of the conversion reaction. The green synthesis coupled to the high specific capacity and rate capability make the proposed γ-Fe2O3/rGO nanocomposite a very promising candidate anode material for sustainable Na-ion batteries.
1. Introduction
Energy storage systems (ESSs) are playing a major role in the green transition from fossil fuels to renewable energy sources.1,2 Indeed, due to the fitful nature of these energy sources, efficient and sustainable ESSs are needed as a key enabling technology to ensure a constant supply of electrical energy. Li-ion batteries are playing a major role thanks to their high energy density, high energy efficiency, and cycle life compared to other available electrochemical energy storage technologies.1–3 However, further improvements are needed to reach complete electrification of transport and replacement of fossil fuels for energy production. In this regard, researchers focused their efforts on studying novel electrode materials able to satisfy the requirements in terms of performance and sustainability. Coupled to this, further concerns are related to availability of raw materials such as Li4,5 and Co.6 Furthermore, the recycling rate of lithium from spent batteries is still rather low (≈1%).7 For these reasons, alternative alkali-ion rechargeable batteries based on Na and K are being developed. Sodium is the 4th most abundant element in the earth crust, with reservoirs spread everywhere across the world. Furthermore, sodium and lithium share similar chemical properties, thus the know-how acquired on LIBs can be easily transferred to the sodium counterpart.8 However, some differences, such as the different standard reduction potential (−3.04 V vs. SHE for Li+/Li couple and −2.71 V vs. SHE for Na+/Na) and ionic radium (0.7 Å vs. 1 Å for Li+ and Na+, respectively), could lead to lower energy density and rate performances of NIBs compared to LIBs. These differences in the charge carriers also reflect on other electrochemical properties and processes, such as diffusion, electrode/electrolyte interphase formation and stability, and (in)activity towards certain materials. Indeed, while in LIBs graphite and silicon are considered the state-of-the-art anode materials, in NIBs their ability to accept Na+ ions is rather low, and thus poor performances are commonly observed.9,10 Commonly studied anode materials are hard carbons and transition metal oxides (TMOs). TMOs can react with Na+ by three different reaction paths, i.e.: (i) insertion reaction, (ii) mixed conversion-alloying reaction, and (iii) conversion reaction. In the first case, materials such as Li4Ti5O12 and Na2Ti3O7 can reversibly insert the alkali-ion ensuring a good capacity retention with small volume change.11,12 However, the specific capacity of these materials is low due to the low amount of cation which can be inserted in the crystal host structure. The second case includes materials such as SnO2, GeO2, and Sb2O3 which, after the conversion step, can form binary alloys with Na.13,14 As a result, very high specific capacities are observed; however, the structural stresses due to the volume expansion during Na-metal alloying can lead to extremely poor capacity retention. The latter case includes oxides of metals which are not able to form a binary alloy with sodium and can only exploit the multiple electron transfer given by the conversion reaction. However, poor capacity retention due to the structural rearrangement and high voltage hysteresis are commonly observed.Iron oxides have attracted much interest in both LIBs and NIBs systems thanks to their high theoretical specific capacity (ranging from 744 mA h g−1 for FeO15 up to 1007 mA h g−1 for Fe2O316) as well as their abundancy, cost, and nontoxicity.17 However, the issues connected with the continuous structural rearrangements, and the subsequent poor capacity retention, must be overcome to enable their practical application in commercial NIBs. Common strategies involve the preparation of nanostructured materials and/or nanocomposite materials. Nanostructured materials can offer the advantages of better rate performances and capacity retention given by higher specific surface area, shorter diffusion path, smaller volume changes. Several morphologies, such as 0-D nanoparticles,18 nanotubes,19 and chain-like particles,20 have shown remarkable improvements.
The preparation of nanocomposite materials with an active or inactive matrix can effectively mitigate the stresses due to the structural rearrangements upon cycling. Carbon-based materials are among the mostly used buffering matrixes, thanks to their remarkable electronic conductivity and low cost.21–23 Graphene has already been studied as an active material thanks to its high electronic conductivity, reliable mechanical properties, and high aspect ratio.24,25 However, its use in ESSs is still questioned by drawbacks such as high cost and tendency to restack to form amorphous carbon, and eventually graphite.26 Nevertheless, its use in nanocomposite formulations can buffer the volume changes and improve the mechanical stability of the electrode. Furthermore, its utilization in composites easily results in enhancement of overall electrode conductivity. For these reasons, graphene has already been used as containment matrix for conversion and conversion-alloying materials for both LIBs and NIBs systems.27 Nevertheless, some drawbacks regarding the synthesis of iron oxide/graphene-based composite materials need to be overcome. In most cases, hydrothermal and high temperature calcinations are used, which can hinder the potential scale-up of the synthesis process.28
Herein, we report a facile two-step synthesis of 5-nm γ-Fe2O3 particles by coprecipitation and their embedding into a matrix of reduced graphene oxide (rGO), without the use of high temperature treatments. The resulting composite material is a γ-Fe2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rGO ratio of 80:20 (m/m) which can ensure good electrochemical performances at a reasonable cost. Electrodes are prepared with PAA as a green and high-elastic-modulus binder.29 Thanks to the synergistic effect of the combination of γ-Fe2O3 with rGO, the composite material exhibits high specific capacity and excellent rate capability. An extensive electrochemical and structural characterization is reported. Finally, ex situ Raman measurements shed light on the reaction mechanism of γ-Fe2O3 in NIBs configuration.
:rGO ratio of 80:20 (m/m) which can ensure good electrochemical performances at a reasonable cost. Electrodes are prepared with PAA as a green and high-elastic-modulus binder.29 Thanks to the synergistic effect of the combination of γ-Fe2O3 with rGO, the composite material exhibits high specific capacity and excellent rate capability. An extensive electrochemical and structural characterization is reported. Finally, ex situ Raman measurements shed light on the reaction mechanism of γ-Fe2O3 in NIBs configuration.
2. Experimental
2.1 Materials
PAA (Mw 450000 g mol−1), FeCl2·4H2O (>99%), FeCl3·6H2O (<99%), propylene carbonate (PC, anhydrous 99.7%), dimethyl carbonate (DMC, anhydrous <99%), NaClO4 (>98%), hydrazine hydrate, and concentrated NH4OH were purchased from Sigma-Aldrich. Ethylene carbonate (EC, battery grade) was purchased from Solvionic (Tolouse, France). Graphene oxide (C:O ratio of 5:4.3) was purchased from Nanoinnova technologies SL. All reactants were used as received.
2.2 γ-Fe2O3/rGO synthesis
The nanocomposite anode material was prepared according to a similar method previously described in a previous report.30 Briefly, the synthesis involves firstly the coprecipitation of iron oxide nanoparticles, and then their embedding into reduced graphene oxide. Stoichiometric amounts of FeCl2·4H2O and FeCl3·6H2O (molar ratio 1:2) were dissolved in 40 mL of distilled water. After the complete dissolution of the salts, 100 mL of 10% NH4OH were added under vigorous stirring giving a black precipitate. The solution was then heated at 70 °C, and further 30 mL of concentrated NH4OH were added. The reaction was kept at 70 °C for 8 h. Eventually, the obtained iron oxide, in the form of a black powder, was washed thoroughly with distilled water, acetone, and ethanol. The powder was dried in vacuum oven at 50 °C. Afterwards, 200 mg of graphene oxide were dispersed in 150 mL of distilled water for 1 h to obtain a homogeneous dispersion. 500 mg of the synthesized iron oxide powder was subsequently added to the dispersion and further sonicated for 1 h. 10 mL of hydrazine hydrate were added to the above dispersion to reduce the graphene oxide matrix (an ice bath was used to control the heat of reaction). The dispersion was kept under sonication for another 2 h, followed by vacuum filtration with Millipore (0.2 μm GTTP) and washed with distilled water, acetone, and ethanol. Eventually, the obtained composite powder was dried under vacuum at 50 °C. Reference rGO was synthesized with the same procedure without the addition of iron oxide nanoparticles.
2.3 Structural, chemical, morphological characterization
The structure of the obtained materials was characterized by using a Horiba iH320 Raman spectrometer equipped with a 532 nm laser source, and a Philipps diffractometer equipped with a Cu-Kα source (λ = 1.540 Å). The carbon content in the composite material was assessed by using a Perkin-Elmer STA6000 TGA-DTA. Morphological characterization was performed by a ZEISS Sigma Series 300 field-emission scanning electron microscope (FE-SEM) and by a ZEISS EM 910 TEM microscope equipped with a tungsten thermionic electron gun operating at 100 kV.The chemical and structural features of the as prepared composite powder was assessed by X-ray absorption spectroscopy (XAS) measurements, performed at beamline XAFS/XRF beamline of the SESAME synchrotron source.31 The beamline was equipped with a fixed-exit double-crystal Si(111) monochromator installed between two cylindrically bent mirrors, both set to work at a grazing incidence angle of 2.8 mrad on the Si coated stripes for harmonic rejection. Fe K-edge spectra were collected in transmission mode by using ion chambers located downstream and upstream the sample. Sample powders, in amount giving an edge jump at the Fe K edge close to 1.0, were mixed with cellulose and measured in the form of 13 mm diameter pellets.
Measurements were compared with spectra of α-Fe2O3, γ-Fe2O3 and Fe3O4 commercial powders collected at SAMBA beamline (SOLEIL synchrotron), also in transmission mode. Spectra of metallic Fe foil were measured for initial energy calibration (first inflection point set at 7112 eV) and simultaneously with any sample (by using a third ion chamber) for the proper monitoring of the energy scale.
2.4 Electrode processing
The electrode layer was prepared by doctor blade technique. The slurry was prepared by mixing γ-Fe2O3/rGO active material:conductive carbon SUPERC 65 (Timcal C-Energy™): polyacrylic acid (PAA, Mw = 450000 g mol−1) in 70:20:10 m/m ratio. Firstly, the polymer binder was dissolved in ethanol. In the meantime, the active material and the carbon additive were finely grounded in an agata mortar. The mixed powders were added to the binder solution and stirred by a magnetic stirrer overnight. The obtained slurry was casted onto 10 μm copper foil by doctor-blade, with a wet thickness of 150 μm. The electrode layer was then dried at 70 °C for 2 h. Circular electrodes of ∅ = 9 mm were cut using an electrode puncher (EL-CELL). Eventually, the electrodes were pressed at 4.7 tons cm−2, weighted, and vacuum dried at 120 °C overnight. The electrodes resulted in an average loading of active material ≈1.5 mg cm−2.
2.5 Electrochemical characterization
For all the electrochemical measurements, the γ-Fe2O3/rGO electrodes have been used as working electrodes in three-electrode cells. Na metal disks have been used as counter and reference electrodes, 1 M NaClO4 EC:PC 1:1 (v:v) as electrolyte and Whatman GF/A glass fiber disc as separator. Swagelok-type cells have been used for cyclic voltammetry, galvanostatic cycling, and rate capability. Prior any electrochemical characterization, the cells underwent an open circuit voltage period of 12 h. Cyclic voltammetries (CV) have been acquired in the potential range 0.02 < E < 3 V at scan rates ranging from 100 μV s−1 up to 500 μV s−1 with 50 μV s−1 increment steps. Galvanostatic cycles have been performed with specific current ranging from 100 up to 5000 mA g−1 within the voltage window 0.020 < E < 3.000 V. ECC-ref cells (EL-CELL GmbH, Hamburg) have been used for electrochemical impedance spectroscopy (EIS) in potentiostatic mode. EIS measurements have been acquired every 10th cycle, by applying a sinusoidal perturbation of ΔE = ±5 mV over bias potential E = 0.5 V, in a frequency range 7 mHz < f < 200 kHz with 10 points per decade and logarithmic spacing. All the potential values are referred to the Na+/Na redox couple (E° = −2.71 vs. SHE).
Ex situ Raman measurements have been performed on samples which had been brought to selected states of charge (SOC) during the first cycle, by galvanostatic (dis)charge at 500 mA g−1 followed by 6 h-constant voltage step to ensure electrode equilibration. Subsequently, the cells were disassembled in glovebox under Ar atmosphere, then the electrodes were washed three times with dimethyl carbonate (anhydrous DMC, Sigma-Aldrich) to remove electrolyte components which could interfere with the Raman analysis, and finally put between two microscope slides sealed with epoxy resin.
All the cell assembly/disassembly procedures were carried out by using an Ar-filled glovebox (Jacomex GP-Campus) with H2O and O2 < 1 ppm.
3. Results and discussion
3.1 Structural, morphological, and thermal characterization
The Raman spectrum of γ-Fe2O3/rGO composite powder is shown in Fig. 1a. The observed Raman pattern is consistent with the literature findings of maghemite Fe2O332 and of carbonaceous materials.33 Specifically, the peak located at 667 cm−1 can be ascribed to the A1g mode of γ-Fe2O3,32 while the peak located at 271 cm−1 can be ascribed the T1g mode of γ-Fe2O3.32 Three characteristic peaks of carbonaceous materials are observed in the Raman pattern. Specifically, the D-band and G-band located at 1302 and 1581 cm−1, respectively, clearly show the recovery, upon GO reduction, of the sp2 carbon pattern with defects.33 The high ID/IG ratio (≈1.31) suggests a high number of structural defects on rGO surface.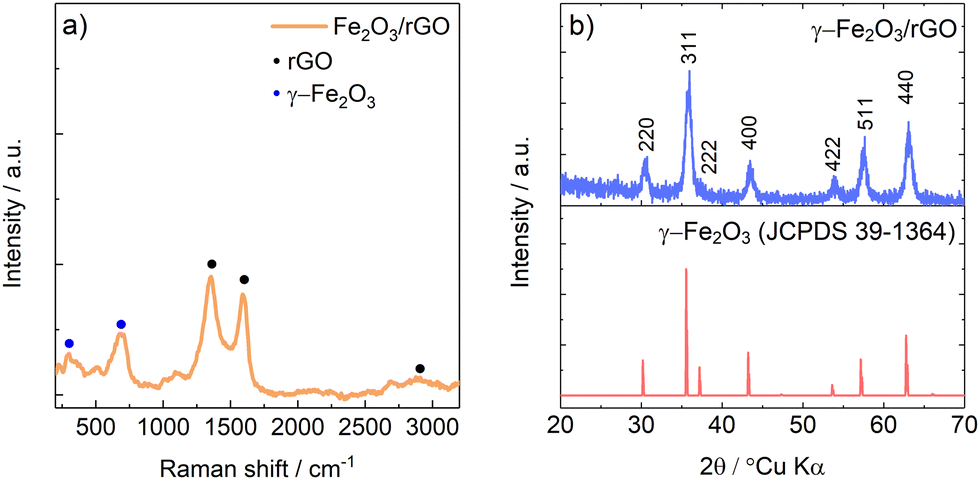 | ||
| Fig. 1 (a) Raman spectra of γ-Fe2O3/rGO. (b) Experimental diffractograms of γ-Fe2O3/rGO with reference JCPDS card of γ-Fe2O3. | ||
In Fig. 1b, the experimental diffractogram of γ-Fe2O3/rGO shows a diffraction pattern which can be ascribed to spinel γ-Fe2O3 maghemite with space group P4132 (JCPDS 39-1364). No signals of rGO were detected by XRD probably because of the larger scattering factor of Fe respect to C and, as shown in the TGA in Fig. S2 (ESI†), by the low amount of rGO compared to iron oxide.
It should be noted that, as reported in literature, Fe3O4 and γ-Fe2O3 share an almost identical structure, with the latter being considered an Fe(II)-deficient magnetite.32 This means that neither Raman spectroscopy nor X-Ray diffraction can differentiate the two different oxidation state. Thus, X-ray absorption spectroscopy in the near edge region (XANES) has been performed to assess the oxidation state of iron in the oxide powder. The results are shown in Fig. 2. The comparison of XANES spectra measured on the as prepared composite, with those of reference compounds, confirms that γ-Fe2O3 is the main phase formed within the sample, ruling out the presence of α-Fe2O3 or Fe3O4.
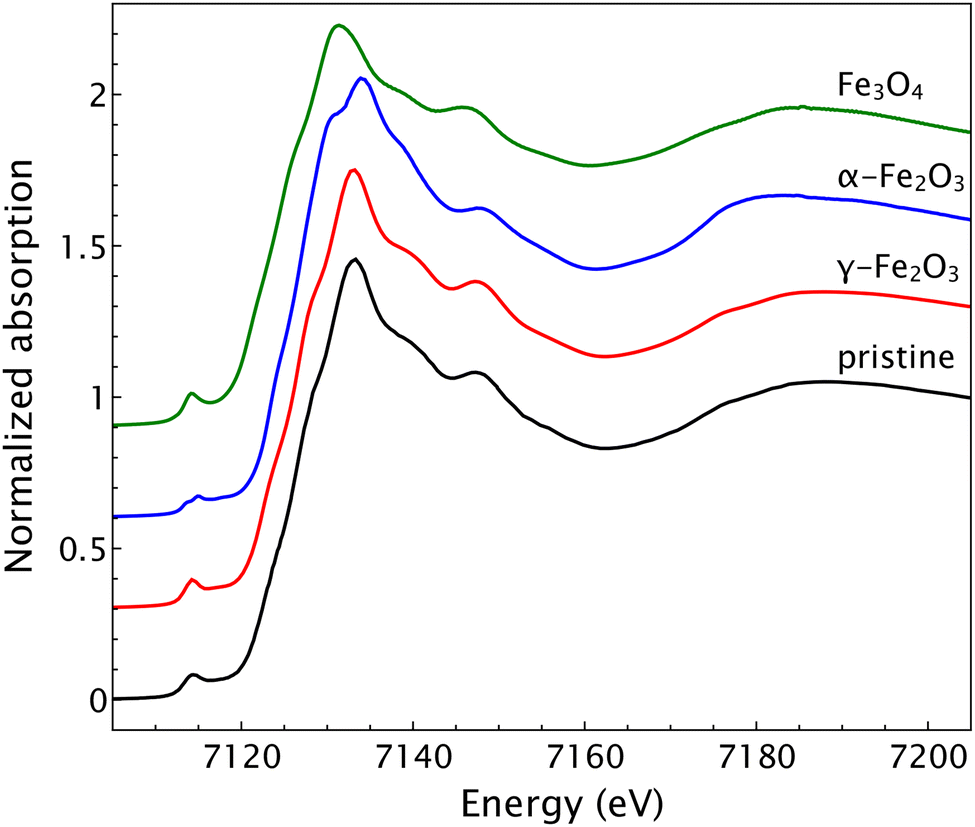 | ||
| Fig. 2 XANES spectra of the pristine composite powder with spectra of reference compounds γ-Fe2O3, α-Fe2O3 and Fe3O4. | ||
In Fig. 3a the SEM micrographs of γ-Fe2O3/rGO are shown. Formation of large aggregates of γ-Fe2O3 nanoparticles is evidenced, probably given by the magnetic stirring. However, at higher magnification the particle size can be visually estimated in the range 5–10 nm. The morphology of the bare γ-Fe2O3 nanoparticles has been confirmed also by TEM, shown in Fig. 3e, in which aggregates of spherical particles with a 10 nm diameter were observed.
 | ||
| Fig. 3 SEM micrographs of γ-Fe2O3/rGO at (a) 15000x and (b) 60000x. EDS mapping at 15000x with highlighted (c) carbon and (d) iron. TEM micrographs of (e) bare γ-Fe2O3 and (f) γ-Fe2O3/rGO taken at 63000x. | ||
On the other hand, at high magnification (Fig. 3b) it is possible to confirm the effective embedding of the nanoparticles into the rGO matrix. Furthermore, as depicted in Fig. S1 (ESI†), the carbon flakes observed in Fig. 3a and b are mainly composed by stacked carbon layers and embedded γ-Fe2O3 particles. The distribution of iron oxide through the carbonaceous matrix has been confirmed also by both EDS elemental mapping and TEM (Fig. 3c–f). Especially on Fig. 3f, it is possible to observe both small and large clusters of γ-Fe2O3 embedded in the carbon nanosheet.
The thermal gravimetric analysis, shown in Fig. S2 (ESI†), evidenced a weight loss of 19% above approximately 375 °C due to the oxidation of rGO to CO2, resulting in an estimated of 79:21 γ-Fe2O3:rGO mass ratio.
3.2 Electrochemical characterization
Fig. S3 (ESI†) shows the cyclic voltammetry of γ-Fe2O3/rGO. In the first cathodic scan two features of electrochemical processes involving the composite active material are evidenced: a broad peak at 0.75 V (A), which can be assigned to the conversion reaction of γ-Fe2O3 (eqn (1)),34,35 and a sharp peak (B) at low potential assigned to Na+ ions storage by rGO (eqn (2)).36| γFe2O3 + 6e− + 6Na+ ⇄ 2Fe0 + 3Na2O | (1) |
| rGO + xe− + xNa+ ⇄ NaxrGO | (2) |
The charge/discharge capability of the γ-Fe2O3/rGO nanocomposite and capacity retention for more than 120 cycles were assessed by galvanostatic cycles in the 0.02 < E < 3 V potential range. Fig. 4a and b reports the specific capacity values obtained at 500 and 1000 mA g−1 specific currents, as well as the Coulombic efficiency. At both specific currents applied, the capacity decay evidenced during the initial cycles is related to the irreversible SEI formation and the partly irreversible γ-Fe2O3 conversion which take place during electrode discharge. Specifically, the largest irreversibility is evidenced in both conditions during the first cycle, where the Coulombic efficiency values are 53.75% and 42.85% for 500 and 1000 mA g−1, respectively. A progressively decrease of irreversibility is observed through the following cycles, up to around the 20th cycle where the reversible capacity stabilizes for both currents applied at ≈300 mA h g−1, with an average efficiency of 98.8% and 97.7% at 500 and 1000 mA g−1, respectively. The E vs. Q galvanostatic profiles (Fig. 4c and d) confirm the first-cycle irreversibility of the electrodes. No clear plateaus are evidenced by the charge/discharge profiles of the first two cycles, suggesting that completion of the several phase transitions, related to the (de)conversion processes, is kinetically hindered. To better characterize the several processes taking place, reversibly and irreversibly, on rGO and γ-Fe2O3 components, differential analysis has been performed by plotting the dQ dE−1vs. E graphs calculated on the data of the galvanostatic E vs. Q profiles. The dQ dE−1vs. E profiles in Fig. 4e and f confirm that in the first cycle, at both current applied, a large contribution to the specific capacity at E < 1 V (peak A) is given by SEI formation and conversion reaction of γ-Fe2O3; on the other hand, during the oxidation the electrode does not yield the same amount of charge, confirming irreversibility of SEI formation and, partially, of the conversion reaction.
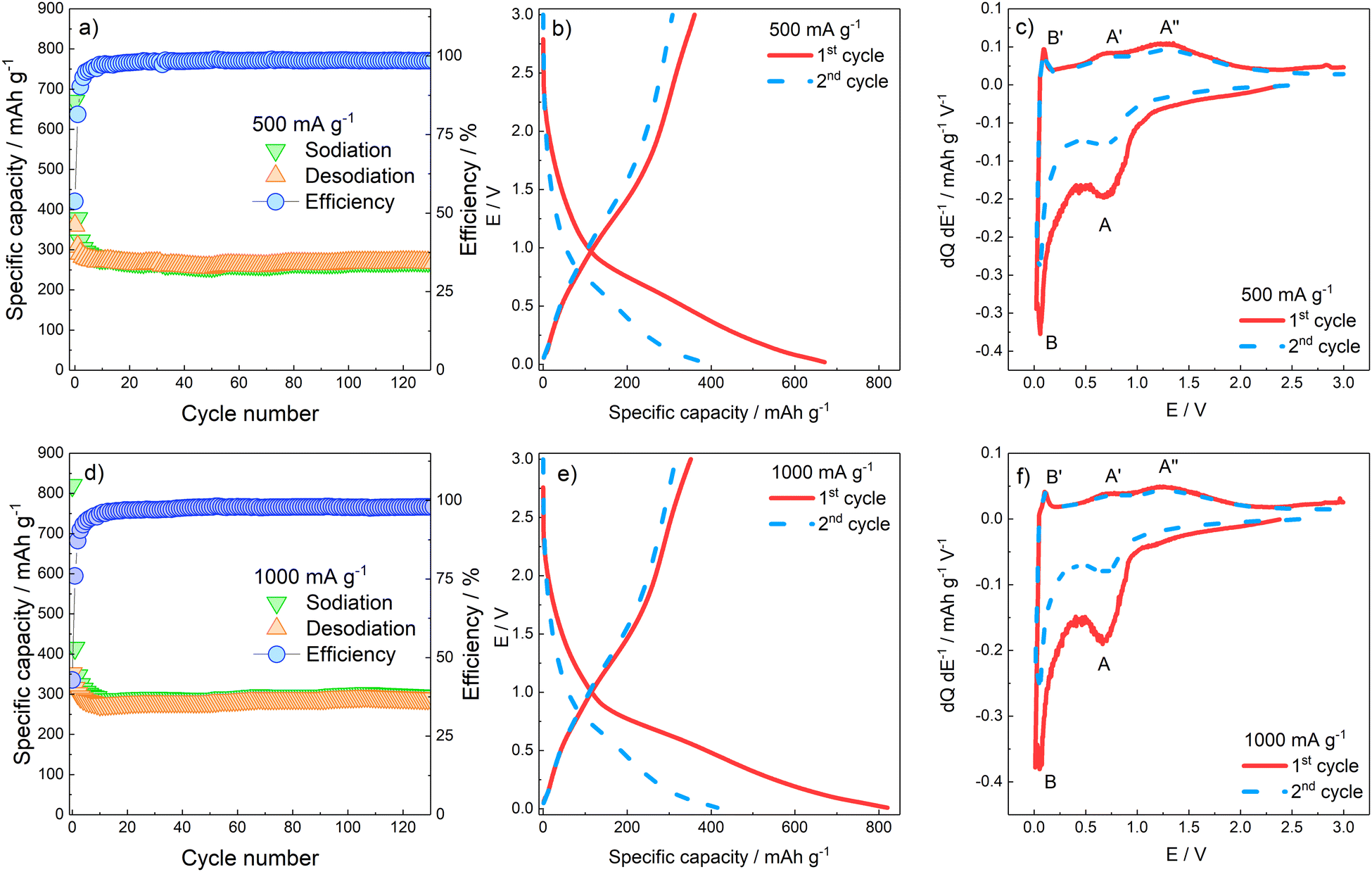 | ||
| Fig. 4 (a) Galvanostatic cycles obtained at (a) 500 and (b) 1000 mA g−1. Galvanostatic E vs. Q profiles obtained at (c) 500 mA g−1 and (d) 1000 mA g−1. Galvanostatic differential E vs. dQ dE−1 profiles obtained at (e) 500 mA g−1 and (f) 1000 mA g−1. | ||
The rate performance of γ-Fe2O3/rGO was assessed by applying a rate capability protocol. The electrode underwent charge/discharge at specific current values of 100, 200, 500, 1000, 2000, 5000 mA g−1 (5 cycles at each current). Subsequently, a specific current of 100 mA g−1 was applied to verify the capacity retention of the material. The results of the rate capability tests are shown in Fig. 5 and summarized in Table 1.
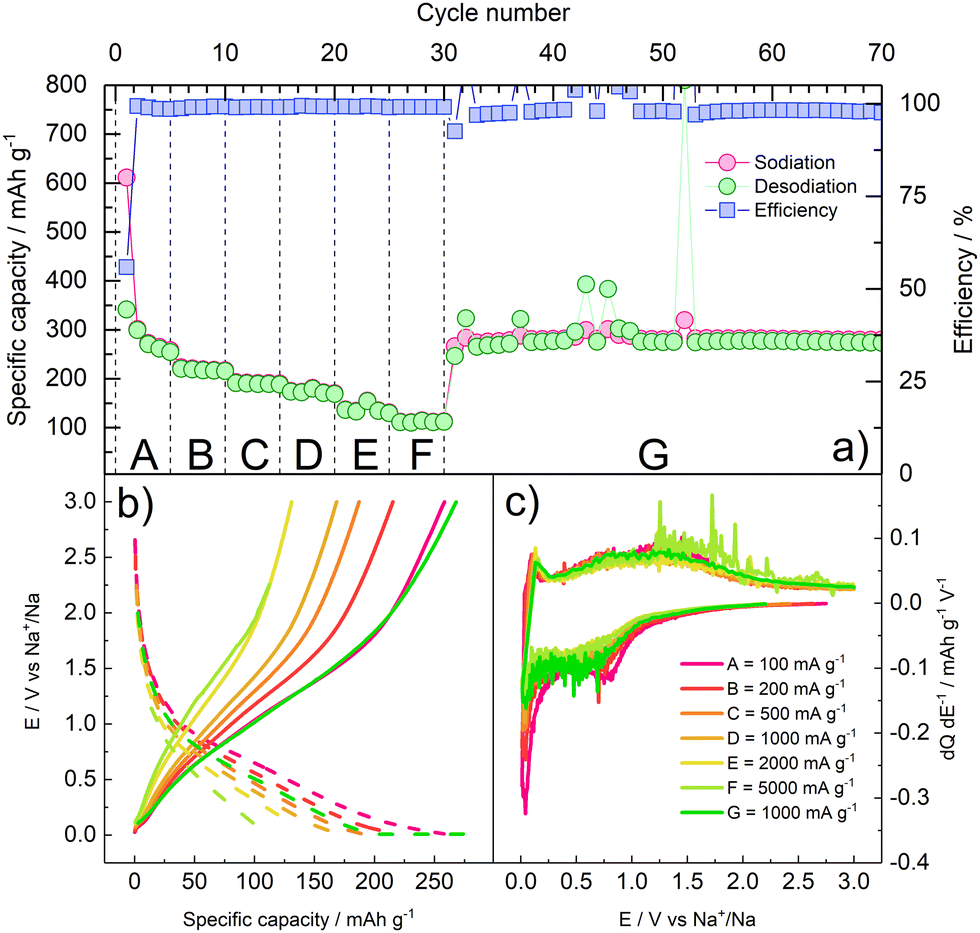 | ||
| Fig. 5 Rate capability of γ-Fe2O3/rGO. (a) cycle number vs. specific capacity/efficiency; (b) galvanostatic E vs. Q profiles at each current value; (c) differential E vs. dQ dE−1 profiles at each current values. The current values applied at each step are detailed in Table 1. | ||
| C-rate (mA g−1) | Capacity (mA h g−1) | Efficiency (%) | |
|---|---|---|---|
| A | 100 | 274.3 | 98.88 |
| B | 200 | 220.0 | 99.08 |
| C | 500 | 191.4 | 99.07 |
| D | 1000 | 174.1 | 99.27 |
| E | 2000 | 138.6 | 99.21 |
| F | 5000 | 112.6 | 99.11 |
| G | 100 | 280.6 | 99.66 |
The rate capability results confirm that the composite material can withstand high current rates. As expected, the specific capacity decreases as the specific applied current increases. Nevertheless, around 50% and 40% of the capacity initially delivered at 100 mA g−1 (A) is still delivered at the very high specific currents of 2000 mA g−1 (E) and 5000 mA g−1 (F), respectively. When the current is restored to the initial value (G) of 100 mA g−1, an average capacity of 280 mA h g−1 over 40 cycles is retained, corresponding to the initial reversible capacity. Some cycles exhibit high spikes in the delivered capacity, as well as in the galvanostatic differential profiles, during oxidation, suggesting minor breakage/reformation of the SEI, and thus release of Na+ ions which are then plated on the counter electrode. This behaviour is consistent with the well-known instability of passivation layer on NIB anodes.38 Furthermore, the capacity obtained at 0.5 and 1 A g−1 is lower than the one observed in Fig. 4, suggesting that cycling at lower current rates in the initial cycles may have fostered the formation of a thicker SEI, affecting the electrode performance.
Finally, long-term cyclability of the electrode has been assessed by running 1000 charge/discharge cycles at 1 A g−1 specific current in the potential range 0.02 V < E < 3 V. As shown in Fig. S4 (ESI†), the electrode delivers an initial discharge capacity around 300 mA h g−1, with slow fading upon long-term cycling. In fact, 250 mA h g−1 and 200 mA h g−1 are retained after 500 and 1000 cycles, respectively, resulting in a capacity retention of 66% at the end of the experiment. The Coulombic efficiency is stabilized at values approaching 100% after 25 cycles.
For sake of comparison, we have performed a long-term cycling test in the same condition for electrodes containing bare γ-Fe2O3 and bare rGO as active materials, obtained by the same synthetic procedure reported in Section 2.2 without the rGO embedding step. The experiments resulted in poorer cycling behaviour for the bare components than the composite (Fig. S5, ESI†), with measured specific capacities of ≈120 mA h g−1 for both γ-Fe2O3 and rGO, and failure of the rGO cell at the 350th cycle.
It should be noted that the charge/discharge capacity values obtained by the γ-Fe2O3:rGO 80:20 composite under investigation outperform the results reported by several authors36,39–41 with rGO-enriched compositions (up to pure rGO) at comparable cycling rates. These results suggest that the γ-Fe2O3:rGO 80:20 composition represents an optimal trade-off between performances and sustainability. The results are summarized in Table S1 (ESI†).
The electrode morphology was assessed before and after 100 cycles by SEM and EDS elemental mapping. The results are shown in Fig. S6 (ESI†). The pristine γ-Fe2O3/rGO electrode (Fig. S6 and S6c, ESI†) is characterized by the active material powder attached to the current collector. Several large aggregates are clearly visible in the micrograph, probably due to the magnetic stirring during the preparation of the slurry. The EDS elemental mapping revealed no unexpected elements apart from Cr, given by the metallization before the SEM measurement. However, the cycled γ-Fe2O3/rGO electrode (Fig. S6b, ESI†) presented a very different morphology, with the active material being covered by an almost uniform layer. This layer could be both the SEI layer and the Na2O coming from the conversion reaction. However, by only SEM is not possible to discern among the two options. In Fig. S6d (ESI†), it is possible to confirm the presence of the elements Na and Cl coming from the electrolyte and, probably, from the Na2O from the conversion reaction. Thus, the mentioned layer could be both the SEI layer along with the irreversibly formed Na2O, which can explain the large irreversible capacity observed in the initial cycles in Fig. 4.
3.3 Interfacial and transport properties characterization
The interfacial and transport properties were characterized by means of cyclic voltammetry at different scan rates, potentiostatic electrochemical impedance spectroscopy, and ex situ Raman spectroscopy.As the scan rate increases, the exchanged current proportionally increases (Fig. 6a). Indeed, the I vs. ν1/2 plot (peak A has been considered for the calculation) underlines a linear relationship (R2 = 0.99838) which suggests a diffusion-controlled behaviour (Fig. 6b). However, if both faradaic charge transfer (iFar) and capacitive (iCap) contributions to the overall current are taken into account, the delivered current can be approximated by the empirical relationships reported in eqn (3), where the terms a and b are dimensionless adjustable parameters:42
| i(v) = iFar + iCap = avb | (3) |
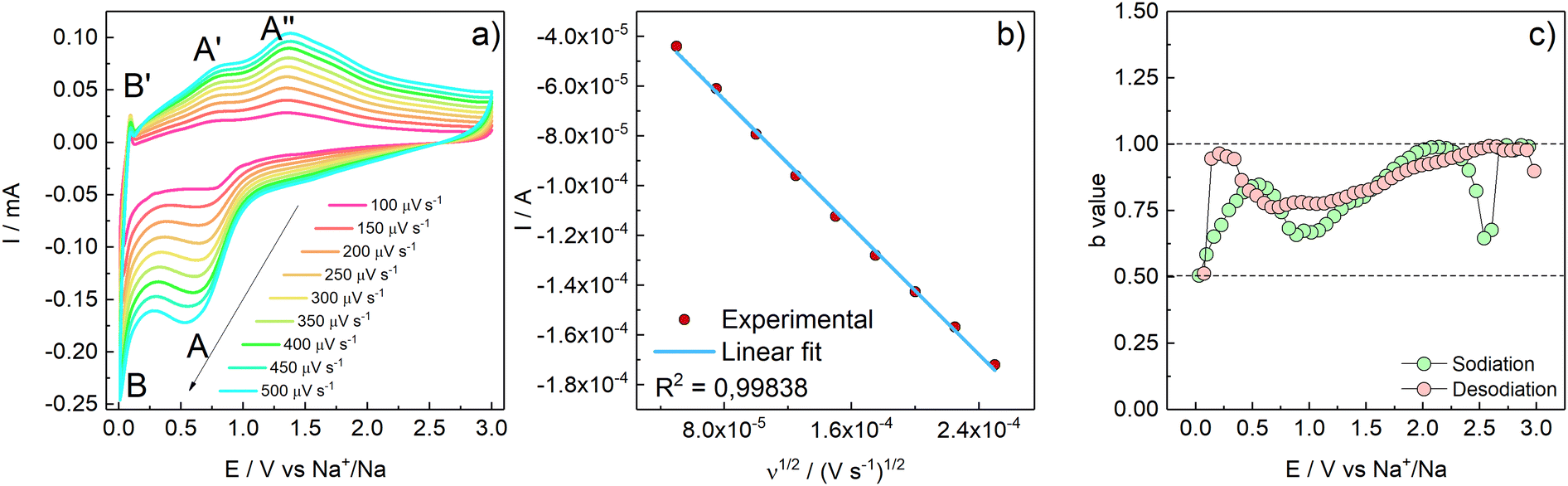 | ||
| Fig. 6 (a) Cyclic voltammetries acquired at different scan rates from 100 up to 500 μV s−1 (50 μV s−1 increment steps). (b) I vs. ν1/2 plot with linear fit of peak A. (c) calculated b-values vs. E. | ||
During the reduction (sodiation) b is between 0.5 and 1. Particularly, at E values corresponding to the CV peaks A and B, b approaches 0.5, consistently with a predominant faradaic process. At E values far from the faradaic reactions, b approaches 1, evidencing the occurrence of surface charge storage processes. During the oxidation scan, at potentials close to 0, where desodiation does not take place, b is close to 1 evidencing a purely capacitive process; b approaches 0.75 in the E region of peaks A′ and A′′, meaning that the process is, at least partially, faradaic as expected.42 Furthermore, the contribution of diffusive processes vs. pseudocapacitive processes at each applied scan rate was calculated.43 The results are shown in Fig. S7 (ESI†). As demonstrated in Fig. 6, the peak current has a linear relationship with the increase of the ν1/2; however, the overall contribution to the capacity is given by the pseudocapacitive feature of the active material. Indeed, as the scan rate increases, also the pseudocapacitive contribution increases from 55% at 0.1 mV s−1 up to 81% at 0.5 mV s−1.
To further characterize the kinetics of the redox processes, impedance spectra were acquired over the bias potential E = 0.5 V vs. Na+/Na in the 2nd cycle and then every 10th cycles. The Nyquist plots, as well as the fit results, are shown in Fig. 7.
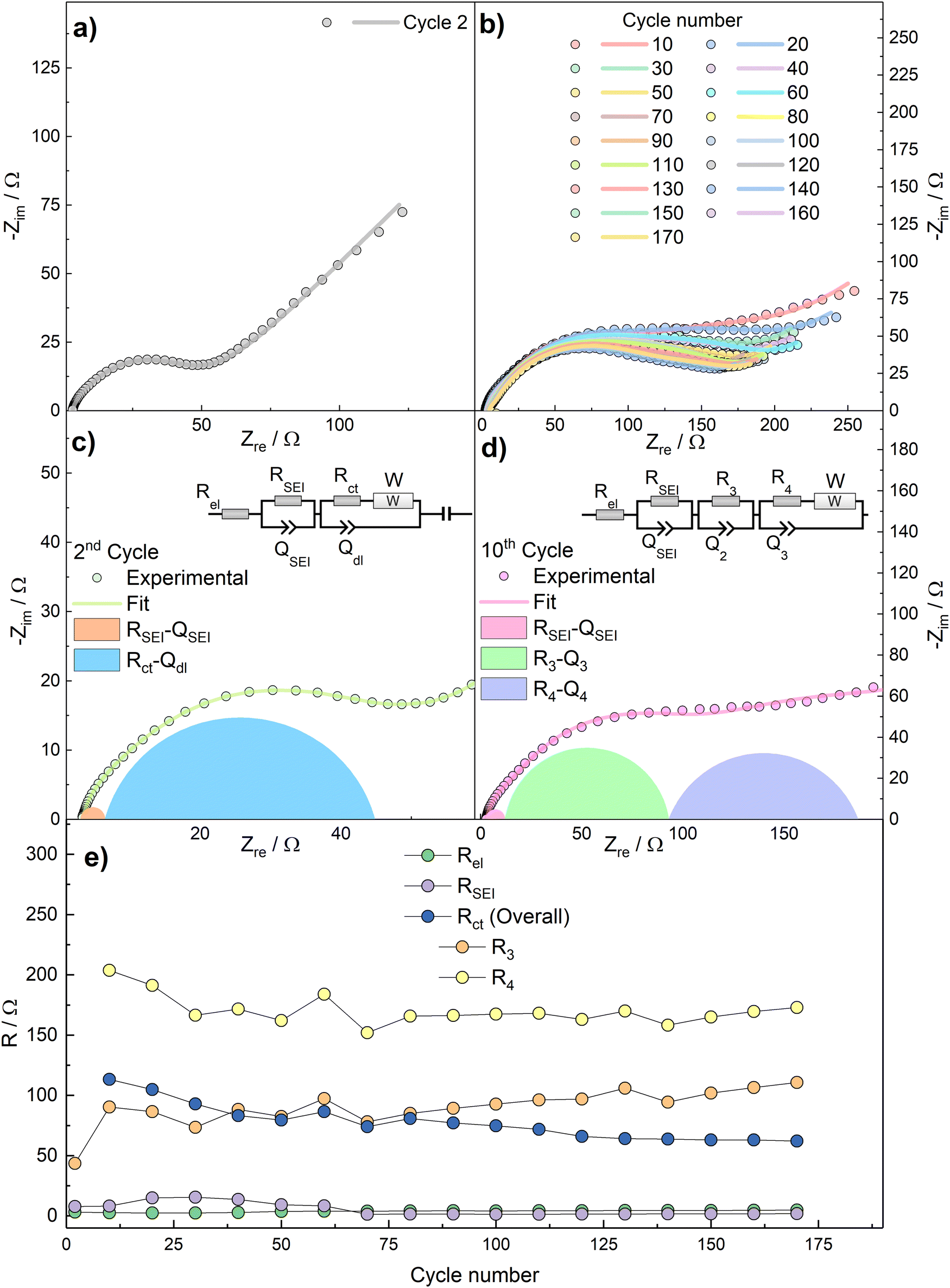 | ||
| Fig. 7 Nyquist plot as well as fit of (a) 2nd cycle and (b) from 10th cycle onwards. Panel (c) and (d) graphic demonstration of the fit of the 2nd and 10th cycles, respectively. (e) Fit results obtained by using the equivalent circuit models in panels (c) and (d). | ||
The Nyquist plot related to the 2nd cycle (Fig. 7a) is characterized, at a first approximation, by three main features, i.e.: (i) an intercept with the real axis at high frequencies, (ii) a large, suppressed semicircle at medium-to-high frequencies, and (iii) a straight line at low frequencies. The impedance dispersions have been modelled by the Equivalent Circuit method44 and the relevant parameters have been calculated by NLLS fitting performed with Relaxis3 software (rhd instruments). The high-frequency intercept, describing ion migration through the electrolyte, can be modelled as a purely resistive element Rel. The medium-to-high frequency arc describes the migration of Na+ ions through the SEI layer and the faradaic charge transfer process. These two processes can be modelled by two distinct resistive elements (RSEI and Rct, respectively), and are accompanied by an accumulation of charges around the surface of the SEI layer and of the active material particles, which can be modelled by parallel capacitors elements (CSEI and Cdl, respectively). Finally, the low-frequency straight line represents a semi-infinite diffusion to a blocking electrode (W impedance in series with Ci intercalation capacitance). This results into an equivalent circuit Rel(RSEICSEI)([RctW]Cdl)Ci, written in Boukamp's notation,45 which models the dispersion acquired in the 2nd cycle. During the data fitting procedure, the pure capacitive elements C have been substituted by constant phase elements Q, to consider electrode inhomogeneities and roughness.44 A drastic change in the shape of the impedance spectra occurs during subsequent cycles (Fig. 7b). This phenomenon, as well as the measured specific capacity lower than the theoretical one, can be explained by the irreversible formation of a dense Na2O layer over the active material during the conversion reaction, which acts as a hindrance to Na+ transport. This behaviour has already been evidenced for SnO2 electrodes in NIBs configuration,46 in which a specific capacity well below the theoretical one was obtained and ascribed to sodium oxide formation. Therefore, the data from 10th cycle onwards have been more accurately fitted by deconvoluting the (RctCdl) arc into two (R3C3) and (R4C4) features, which take into account the formation of a further interface between the active material and the Na2O layer, thus resulting into the equivalent circuit Rel(RSEICSEI)(R3C3)([R4W]C4)Ci. The deconvolution of the Nyquist plots for 2nd and 10th cycles are shown in Fig. 7c and d.
Fig. 7e reports the values of Rel, RSEI, Rct as calculated by using the models in Fig. 7c (2nd cycle) and Fig. 7d (10th and subsequent cycles). In the latter case, the distinct R3 and R4 contributions to overall Rct are reported as well.
R el electrolyte resistance is practically constant upon cycling, as expected. RSEI shows an increasing trend during the initial cycles which can be assigned to breakage and reconstruction of a partly unstable SEI layer. This phenomenon can explain the decreasing capacity and the poor coulombic efficiencies observed in the initial cycles in Fig. 4. However, as the Coulombic efficiency increases, the values of RSEI decrease and finally stabilize. This behaviour could be tentatively explained by: anisotropic growth and subsequent stabilization of the passivation layer, and the lower stability of the Na+-ion SEI than the Li+-ion counterpart.38 The sharp increase of overall Rct after the 2nd cycle can be explained with the aforementioned formation of the dense Na2O interlayer.
In order to verify the reversibility of the Na+ uptake/release processes, ex situ Raman has been performed at selected potential values during the first discharge and charge half-cycles. The points highlighted in Fig. 8b represent the following electrode states: (i) fresh electrode before conversion process and SEI formation (E = 1.5 V); (ii) initial stages of γ-Fe2O3 conversion and SEI formation (E = 0.72 V); (iii) final stages of SEI formation and initial stages of Na–C processes (E = 0.25 V); (iv) electrode fully discharged and sodiated by γ-Fe2O3 conversion and Na–C uptake (E = 0.02 V); (v) electrode after release of most of Na by C matrix (E = 0.12 V); (vi) intermediate stages of Fe oxides formation (E = 0.81 V); (vii) final stages of active material oxidation (E = 1.4 V); (viii) fully charged and desodiated electrode (E = 3 V).
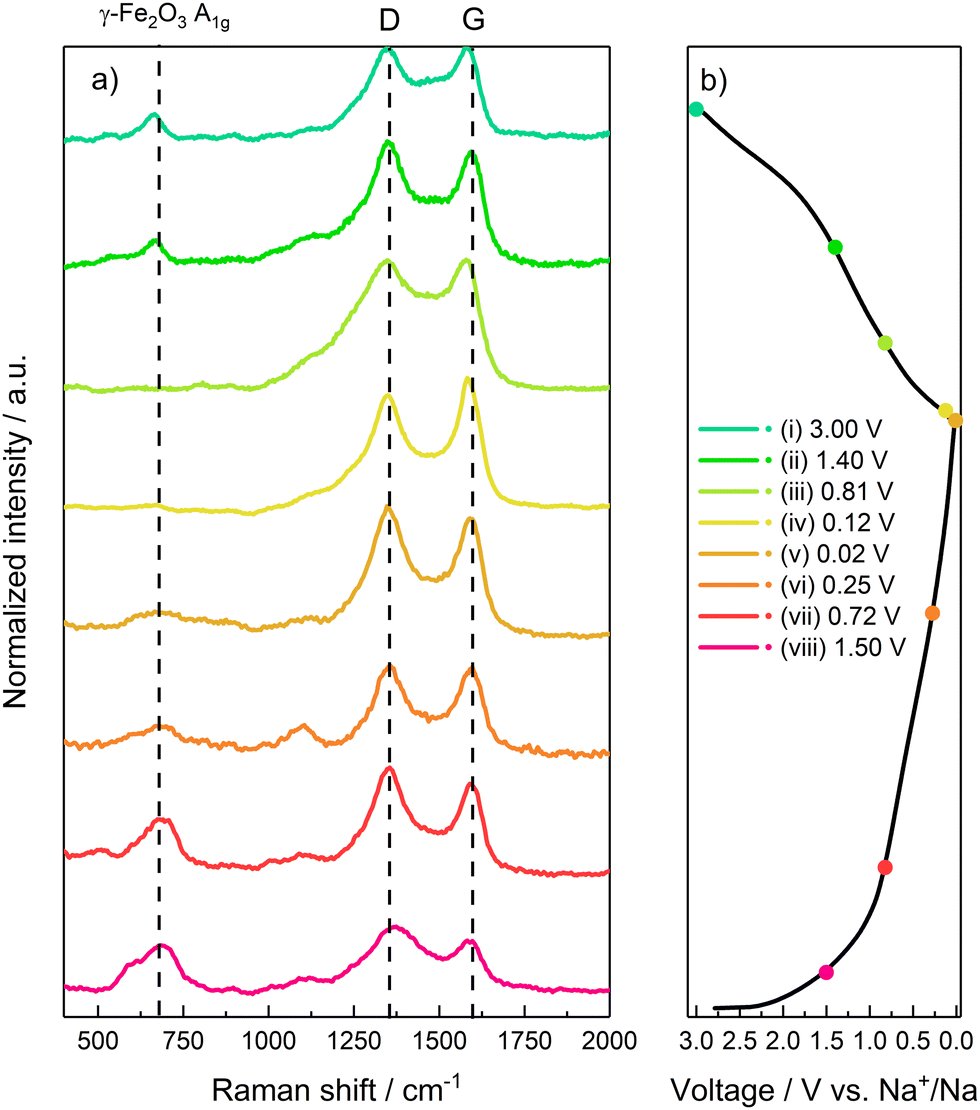 | ||
| Fig. 8 Ex situ Raman characterization. (a) Raman spectrum acquired during the sodiation/desodiation. (b) Galvanostatic profiles with highlighted acquisitions points. | ||
During the electrode discharge/sodiation (Fig. 8a), all the spectra show the carbon-related D and G bands, given by graphene and SuperC65 additive. In spectra (i) and (ii) a peak located at 667 cm−1 can be ascribed to the A1g mode of γ-Fe2O3, due to the stretching of the Fe–O bond in the FeO4 tetrahedra.
Upon further discharge, as evidenced at points (iii) and (iv), the intensity of the γ-Fe2O3-related peak A1g progressively decreases, becoming only a small hump at full discharge (E = 0.02 V). This result confirms the conversion of γ-Fe2O3 to Fe0 nanoparticles and Na2O. However, the A1g signal is still present even at the low cut-off voltage, thus evidencing that the conversion reaction does not go to completion, as confirmed by a practical capacity lower than the theoretical value. Once again, this behaviour could be tentatively explained by the formation of the Na2O interlayer, as suggested by EIS, which could increase electrode polarization, thus forcing the cell to reach the low-potential cut-off before the conversion reaction is completed. The two constant peaks located at 250 cm−1 and 1000 cm−1 (labelled as *) can be indexed to both inorganic SEI components such as Na2CO3 or organic alkyl carbonates.47 Specifically, the band located at 1100 cm−1 can be ascribed to the stretching ν C–O–C bonds in alkyl carbonates.
During charge (desodiation step) (Fig. 8a) a reverse behaviour can be observed through points (v) to (viii). Particularly, points (vii) and (viii) evidence the growth of the γ-Fe2O3-related A1g peak, suggesting an oxidation of Fe0 to mixed Fe2+/Fe3+ oxidation state, and thus the reversibility of the conversion reaction to γ-Fe2O3.
4. Conclusion
We have demonstrated that γ-Fe2O3 nanoparticles, synthesized by a green and facile coprecipitation method, are a suitable anode material for Na-ion batteries. When composite electrodes with high iron oxide loading (γ-Fe2O3:rGO ratio of 80:20) are cycled in Na-ion half-cells with NaClO4-based electrolyte, specific capacities values about 300 mA h g−1 are obtained at specific currents in the order of 500–1000 mA g−1. Furthermore, outstanding rate capability is demonstrated, with specific capacity values of 140 and 110 mA h g−1 obtained at 2 and 5 A g−1, respectively.
The investigation of transport properties pursued by cyclic voltammetry at different scan rates evidenced a diffusion-controlled behaviour, with predominant faradaic mechanism concurrent with a near-surface adsorption of ionic species. Impedance analysis revealed a relevant contribution to the electrode polarization by a Na2O interlayer, formed upon conversion, and its interface with active material. Ex situ Raman spectroscopy shed light onto the reversibility of the conversion process, evidencing that the conversion of iron oxide to metal iron is not complete upon discharge, while γ-Fe2O3 is formed back as the end member of the charge process.
These findings evidence that the proposed γ-Fe2O3/rGO composite is a very promising, sustainable, and reliable candidate anode material for Na-ion batteries.
Author contributions
All authors contributed to this work. All authors approved the final version of the manuscript. A. S. and F. N. conceptualized the work and wrote the manuscript draft. A. S. and L. S. performed the synthesis of the material. L. M. performed the TEM measurements. L. S., L. M., L. B., H. D. contributed to the analysis and discussion. A. S. and M. M. performed the ex situ Raman measurements. A. T., M. H., F. P., and S. J. R. performed the synchrotron measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of ENEA and MiSE (Agenzia Nazionale per le Nuove Tecnologie, l’Energia e lo Sviluppo Sostenibile e Ministero per lo Sviluppo Economico) is gratefully acknowledged under the Project ‘Sistemi di Accumulo di Energia per il Sistema Elettrico’, funded by PAR2019-2021 Program. This study was carried out within the MOST e Sustainable Mobility Center and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) e MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 e D.D. 1033 17/06/2022, CN00000023). This manuscript reflects only the authors’ views and opinions, neither the European Union nor the European Commission can be considered responsible for them. Prof. Jusef Hassoun and Dr. Vittorio Marangon are kindly acknowledged for the TEM measurements.References
- A. Kalair, N. Abas, M. S. Saleem, A. R. Kalair and N. Khan, Energy Storage, 2021, 3, e135 CrossRef.
- A. B. Gallo, J. R. Simões-Moreira, H. K. M. Costa, M. M. Santos and E. Moutinho dos Santos, Renewable Sustainable Energy Rev., 2016, 65, 800–822 CrossRef CAS.
- J. Arteaga, H. Zareipour and V. Thangadurai, Curr. Sustainable/Renewable Energy Rep., 2017, 4, 197–208 CrossRef CAS.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- J. M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- X. Fu, D. N. Beatty, G. G. Gaustad, G. Ceder, R. Roth, R. E. Kirchain, M. Bustamante, C. Babbitt and E. A. Olivetti, Environ. Sci. Technol., 2020, 54, 2985–2993 CrossRef CAS PubMed.
- J. Speirs, M. Contestabile, Y. Houari and R. Gross, Renewable Sustainable Energy Rev., 2014, 35, 183–193 CrossRef.
- S. Fang, D. Bresser, S. Passerini, S. Fang, D. Bresser and S. Passerini, Adv. Energy Mater., 2020, 10, 1902485 CrossRef CAS.
- A. Mauger and C. M. Julien, Mater., 2020, 13, 3453 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y. S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1–10 Search PubMed.
- P. Senguttuvan, G. Rousse, V. Seznec, J. M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- Y. Liu, A. H. Siddique, H. Huang, Q. Fang, W. Deng, X. Zhou, H. Lu and Z. Liu, Nanotechnology, 2017, 28, 465401 CrossRef PubMed.
- S. Liang, Y.-J. Cheng, J. Zhu, Y. Xia, P. Müller-Buschbaum, S. Liang, P. Müller-Buschbaum, Y. Cheng, J. Zhu, Y. Xia and P. Müller-Buschbaum Heinz Maier-Leibnitz Zentrum, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- M. Gao, P. Zhou, P. Wang, J. Wang, C. Liang, J. Zhang and Y. Liu, J. Alloys Compd., 2013, 565, 97–103 CrossRef CAS.
- L. Xu, Y. Tian, T. Liu, H. Li, J. Qiu, S. Li, H. Li, S. Yuan and S. Zhang, Green Energy Environ., 2018, 3, 156–162 CrossRef.
- Chemistry of the Element, Elsevier, 1997, ch. 25, pp. 1070–1112.
- N. Zhang, X. Han, Y. Liu, X. Hu, Q. Zhao and J. Chen, Adv. Energy Mater., 2015, 5, 1401123 CrossRef.
- J. Ni, M. Sun and L. Li, Adv. Mater., 2019, 31, 1902603 CrossRef CAS PubMed.
- G. Qin, J. Duan, Y. Yang and F. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6441–6452 CrossRef CAS PubMed.
- S. Wei, D. Di Lecce, R. Messini D’Agostini and J. Hassoun, ACS Appl. Energy Mater., 2021, 4, 8340–8349 CrossRef CAS PubMed.
- C.-G. Han, N. Sheng, C. Zhu and T. Akiyama, Mater. Today Energy, 2017, 5, 187–195 CrossRef.
- Y. Liu, Y. Dai, X. Jiang, X. Li, Z. Yan and G. He, Mater. Today Energy, 2019, 12, 269–276 CrossRef.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- B. Marinho, M. Ghislandi, E. Tkalya, C. E. Koning and G. de With, Powder Technol., 2012, 221, 351–358 CrossRef CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2014, 14, 271–279 CrossRef PubMed.
- L. Wang, Z. Wei, M. Mao, H. Wang, Y. Li and J. Ma, Energy Storage Mater., 2019, 16, 434–454 CrossRef.
- Y. Fu, Q. Wei, X. Wang, G. Zhang, H. Shu, X. Yang, A. C. Tavares and S. Sun, RSC Adv., 2016, 6, 16624–16633 RSC.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CrossRef CAS PubMed.
- A. Staffolani, H. Darjazi, G. Carbonari, F. Maroni, S. Gabrielli and F. Nobili, Mol., 2021, 26, 4316 CrossRef CAS PubMed.
- M. Harfouche, M. Abdellatief, Y. Momani, A. Abbadi, M. Al Najdawi, M. Al Zoubi, B. Aljamal, S. Matalgah, L. U. Khan, A. Lausi and G. Paolucci, J. Synchrotron Radiat., 2022, 29, 1107–1113 CrossRef CAS PubMed.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CrossRef CAS.
- J. Bin Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- P. R. Kumar, Y. H. Jung, K. K. Bharathi, C. H. Lim and D. K. Kim, Electrochim. Acta, 2014, 146, 503–510 CrossRef CAS.
- J. Ming, H. Ming, W. Yang, W. J. Kwak, J. B. Park, J. Zheng and Y. K. Sun, RSC Adv., 2015, 5, 8793–8800 RSC.
- Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- M. A. Muñoz-Márquez, M. Zarrabeitia, E. Castillo-Martínez, A. Eguía-Barrio, T. Rojo and M. Casas-Cabanas, ACS Appl. Mater. Interfaces, 2015, 7, 7801–7808 CrossRef PubMed.
- H. Qi, L. Cao, J. Li, J. Huang, Z. Xu, Y. Jie and C. Wang, ChemistrySelect, 2019, 4, 2668–2675 CrossRef CAS.
- G. Wei, J. Zhou and M. Xie, J. Phys. Conf. Ser., 1637, 2020, 012079 Search PubMed.
- S. Zhang, W. Li, B. Tan, S. Chou, Z. Li and S. Dou, J. Mater. Chem. A, 2015, 3, 4793–4798 RSC.
- Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 CrossRef.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- E. Barsoukov and J. Ross Macdonald, Impedance Spectroscopy: Theory, Experiment, and Applications, Wiley, 2005, ch. 3, pp. 107–174 DOI:10.1002/9781119381860.
- B. A. Boukamp, Solid State Ionics, 1986, 20, 31–44 CrossRef CAS.
- D. Dixon, M. Ávila, H. Ehrenberg and A. Bhaskar, ACS Omega, 2019, 4, 9731–9738 CrossRef CAS PubMed.
- J. Nanda, G. Yang, T. Hou, D. N. Voylov, X. Li, R. E. Ruther, M. Naguib, K. Persson, G. M. Veith and A. P. Sokolov, Joule, 2019, 3, 2001–2019 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00335g |
| ‡ Current address: University of Bologna, Department of Chemistry “Giacomo Ciamician”, Via Francesco Selmi 2, Bologna 40126, Italy. |
| § Current address: Group for Applied Materials and Electrochemistry - GAME Lab, Department of Applied Science and Technology - DISAT, Politecnico di Torino, Corso Duca degli Abruzzi 24, Torino 10129, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |