
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical hydrogen generation by a four-coordinate square-planar Ni(II) complex with an N2P2-type ligand†
Hidenori
Miyake
a,
Satomi
Hirasawa
a,
Yurika
Uno
a,
Kenichi
Nakao
a,
Takuma
Kato
a,
Yuko
Wasada-Tsutsui
a,
Yoshikuni
Hara
a,
Tomohiro
Ozawa
a,
Tomohiko
Inomata
a and
Hideki
Masuda
 *ab
*ab
aDepartment of Life Science and Applied Chemistry, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya 466-8555, Japan
bDepartment of Applied Chemistry, Faculty of Engineering, Aichi Institute of Technology, 1247 Yachigusa, Yakusa-cho, Toyota 470-0392, Japan
First published on 9th August 2024
Abstract
A Ni(II) complex with an N2P2-type ligand, [Ni(LH)2](BF4)2 (LH = 2-((diphenylphosphino)methyl)-pyridine), was prepared and characterized structurally, spectroscopically, and electrochemically. Its electrochemical hydrogen production capability was investigated and compared with that of a previously reported Ni(II) complex bearing an amino group in the ligand, [Ni(LNH2)2](BF4)2 (LNH2 = 6-((diphenylphosphino)methyl)-pyridin-2-amine). The X-ray crystal structure was revealed to be a four-coordinate square planar structure (τ4 = 0.25) in the cis form, with the counter anion BF4− weakly coordinated to the Ni(II) ion. The structure in the solution was assessed on the basis of UV-vis and NMR spectral features, which showed a four coordinate square planar structure in dichloromethane and a five- or six-coordinate structure bound with solvent molecules in acetonitrile. The electrochemical hydrogen production reaction using AcOH as a proton source showed a similar behaviour to that of [Ni(LNH2)2](BF4)2, with the catalytic current (icat) proportional to the square root of the concentration of AcOH added. This indicates that the reaction mechanism is EECC and that the rate-determining step is the reaction of the two-electron reduced Ni(0) species with the approaching proton to form the Ni(II)–H− species. The TOF and overpotential values, when evaluated under the same conditions as in a previous study (complex: 1 mM, electrolyte [n-Bu4N](ClO4): 0.1 M in MeCN (3 mL), AcOH = 145 equiv. (pKa = 22.3 in MeCN)), were found to be 1060 s−1 and 710 mV, respectively. These values were higher for the overpotential and smaller for TOF, as compared to those of [Ni(LNH2)2](BF4)2 (TOF 8800 s−1, overpotential 430 mV). The structure of the starting material [NiII(LH)2]2+ and the formation of the hydride Ni(II) complex [NiII(LH)2H]+, a reaction intermediate in the hydrogen evolution reaction, were evaluated by DFT calculations. The results of the hydrogen evolution behaviour of these two complexes show that the electron-donating amino group plays an important role in the hydrogen evolution reaction, not only capturing protons but also increasing the basicity of the pyridyl N atom.
Introduction
Hydrogen is a substance that has recently attracted attention as a clean energy source that can replace fossil fuels, because hydrogen produces only water during combustion. However, the current industrial method of hydrogen production, steam reforming,1 is not a clean energy source because it is based on fossil fuels and produces greenhouse gases. Therefore, in recent years, the use of electrolysis of water to achieve a completely clean energy cycle has been attracting attention.2 This is also attracting attention as a method that can preserve electrical energy, which is not suitable for long-term storage, in the form of chemical substances. Electrochemical hydrogen generation requires the use of a catalyst. Since precious metals such as platinum are currently required,3 there is a need for development of methods to use more abundant metals as catalysts.On the other hand, in nature, the enzyme hydrogenase catalytically produces hydrogen using water-derived protons under mild conditions.4,5 [FeFe]-hydrogenase, one of the hydrogenase enzymes, has two iron atoms in its active site and produces molecular hydrogen with a TOF of 6000–9000 s−1.4c Recent structural and functional studies of the active site of [FeFe]-hydrogenase have revealed that its catalytic activity is achieved by the following mechanism, that is, a base is present near one of the two iron centres, which transfers and supplies protons one after another, and then another iron centre reduces the proton to hydride, which combines with the next proton to produce H2. In addition, [NiFe]-hydrogenase has the same hydrogen production function as [FeFe]-hydrogenase.4 In the case of [NiFe]-hydrogenase, Ni acts primarily as a proton trapping site, and protons are reduced to hydride species due to the flexible valence character of the Ni atom.5
Based on these studies, electrochemical hydrogen production catalysts have been developed using transition metal complexes of iron,6 nickel,7 and other abundant metals such as manganese,8 cobalt,9 copper,10 and molybdenum.11 For example, DuBois et al. reported electrochemical hydrogen production using a phosphine-type nickel complex with a secondary amine as the proton-transferring site, mimicking the structure of [FeFe]-hydrogenase.7c The Ni complex showed high catalytic activity with TOF exceeding 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 in MeCN containing a small amount of water. However, the catalyst developed by DuBois et al. required the use of a strong acid, [(DMF)H]+ (pKa 6.1 in MeCN). Jones et al. also developed a Ni complex of an S2P2-type ligand with a ferrocene substituent that mimics [NiFe]-hydrogenase and achieved electrochemical hydrogen production under weakly acidic conditions using acetic acid (AcOH) (pKa = 22.48 in THF).7e Their excellent work also revealed a reaction intermediate in which the nickel centre/site is protonated. However, its TOF was only 1240 s−1, which was not comparable to the case of hydrogen production by the strong acid used by Dubois et al., as described above.
000 s−1 in MeCN containing a small amount of water. However, the catalyst developed by DuBois et al. required the use of a strong acid, [(DMF)H]+ (pKa 6.1 in MeCN). Jones et al. also developed a Ni complex of an S2P2-type ligand with a ferrocene substituent that mimics [NiFe]-hydrogenase and achieved electrochemical hydrogen production under weakly acidic conditions using acetic acid (AcOH) (pKa = 22.48 in THF).7e Their excellent work also revealed a reaction intermediate in which the nickel centre/site is protonated. However, its TOF was only 1240 s−1, which was not comparable to the case of hydrogen production by the strong acid used by Dubois et al., as described above.
There are some other reports on hydrogen generation catalysis using nickel complexes. Some of the studies, mainly based on their experimental approach, are presented as follows: Helm et al. reported that a square-planar Ni(II) electrocatalyst, [Ni(7PPh2NC6H4X)2](BF4)2, featuring seven-membered ring cyclic diphosphine ligands with an amine base, in MeCN in the presence of [(DMF)H]+, at the potential of Ni(II/I) couple, showed efficient electrocatalytic hydrogen evolution.12 TOFs of the reactions were accelerated by addition of water. The computational studies have indicated that the catalytic reaction rate is maximised when the pKa of the Ni(I) protonated pendant amine matches the pKa of the acid used to supply the protons, due to the electron-donating nature of X. Hammes-Schiffer et al. also investigated the electrocatalytic hydrogen evolution of Ni-based complexes with P2PhN2Bn type ligands bearing a pendant amine, [Ni(P2PhN2Bn)2]2+, in the presence of strong acids in MeCN and found a linear dependence of the NiII/I reduction potential on pH.13 Based on the experimental and theoretical studies, it was suggested to proceed by a proton-coupled electron transfer (PCET) process. It was previously demonstrated that the Ni catalyst with pyridine 2-thiolate, Ni(PyS)3−, showed good stability and activity as a H2 generation catalyst for use in solar energy storage.14 Based on the DFT analysis, it was found that the initial reduction occurs at the nickel site rather than via a non-innocent ligand and that protonation of the pyridyl nitrogen causes de-chelation, but water coordination is not favored.15 The Ni(DQPD) complex with an N4-type polypyridine ligand with a quinoline moiety as a proton relay has also been reported to act as a visible light-driven active catalyst for proton reduction from water in combination with the photosensitising agent fluorescein and triethylamine.16 Sakai et al. also recently reported that a square-planar Ni(II)(bpy)(dcbdt) hydrogen evolution catalyst undergoes consecutive two electron reduction reactions to form [NiI(bpy−˙)(dcbdt)]2−, followed by an increase in the basicity of the filled Ni dz2 orbital, which greatly accelerates the proton abstraction rate.17
We have also previously focused on the amino group near the metal of the [FeFe] hydrogenase enzyme and synthesized the Ni(II) complex with a phosphinopyridine-type ligand bearing an amino group, [Ni(LNH2)2](BF4)2 (LNH2 = 6-((diphenylphosphino)methyl)-pyridin-2-amine), as a functional model for hydrogen evolution. The electrochemical hydrogen evolution using this Ni(II) complex proceeded through an EECC mechanism (two electrons and two chemical steps) and resulted in a large TOF despite the low overpotential in the presence of a weak acid such as AcOH.18 The elucidation of this effect has important implications for the role of this complex and other catalysts with proton transfer sites in hydrogen evolution by [FeFe] hydrogenase. It is very important to determine at this stage whether the amino group plays a role as a proton transfer site in the catalytic activity of hydrogen production. In Scheme 1, a schematic diagram of the proton transferring behaviours of the nickel complexes with and without amino groups is shown; the Ni(II) complex is an electrochemically reduced to a Ni(0) species, where the Ni(0) atom is presumed to be considerably electron rich, and the added proton is considered to approach the electron-rich Ni(0). If the amino group has any influence on the behaviour of the protons, it is likely to affect the formation of the Ni–hydride complex. Therefore, the next step is to synthesize Ni(II) complexes without amino groups in the second coordination sphere and to investigate their effect on hydrogen production behaviour in order to elucidate the role of the amino groups.
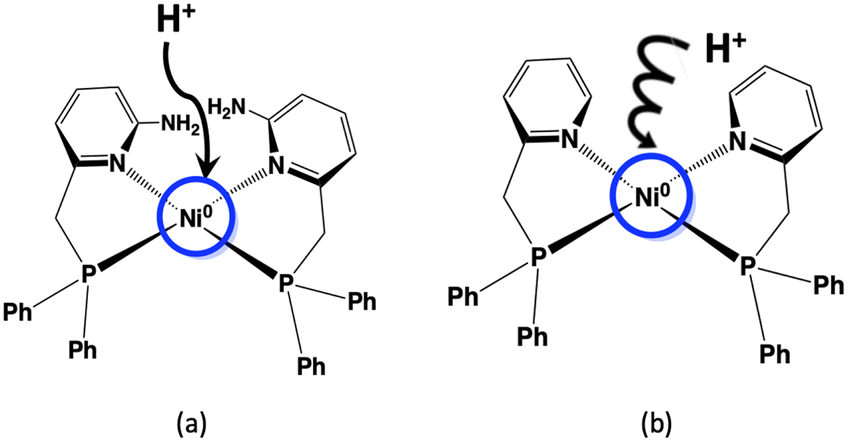 | ||
| Scheme 1 Effects of the (a) presence and (b) absence of amino groups in the second coordination sphere on the first step in hydrogen production using Ni complexes. | ||
In this paper, the Ni(II) complex [Ni(LH)2](BF4)2 (1) with a phosphinopyridine type ligand LH (LH = 2-((diphenylphosphino)methyl)pyridine) without any amino groups as a proton transfer site was synthesized and its electrocatalytic hydrogen evolution behaviour was studied. The results were compared with those reported previously for the Ni(II) complex with an amino group, [Ni(LNH2)2](BF4)2, and the role of the amino group was discussed. The hydrogen production mechanism of this system was also proposed.
Results and discussion
Synthesis and characterization
The synthesis of a Ni(II) complex with LH, [Ni(LH)2](BF4)2 (1), was carried out based on a previous study.18 The purity of complex 1 was confirmed by elemental analysis. Fortunately, the recrystallization of complex 1 in MeCN/Et2O yielded reddish-orange needle-like crystals with the Fdd2 (#43) space group, which were orthorhombic crystals, as confirmed by X-ray crystallography (Fig. 1 and Table S1, ESI†). In the crystal structure (Tables S2 and S3, ESI†), complex 1 had a four-coordinate square planar structure (τ4 = 0.25)19 with Ni–P1 = 2.1760(6) and Ni–N1 = 1.952(2) Å and existed only in the cis form in the unit cell. This shows almost the same coordination structure as that previously reported for [Ni(LNH2)2](BF4)2,18b and it is presumed that this coordination is the result of preventing repulsion due to the trans effect. The Ni–P1 and Ni–N1 bond lengths are in the range of previously reported bond lengths (Ni–P = 2.154–2.214 Å and Ni–N = 1.901–1.969 Å) of a low spin Ni(II) complex with chelating diphenylphosphinopyridine ligands, respectively.18,20 The closest distance between Ni and the counter anions was Ni⋯F = 3.394(2) Å, indicating that the BF4− anions were weakly coordinated to the Ni atom and were unlikely to be involved in the formation of the Ni–H bond in the catalytic cycle in solution.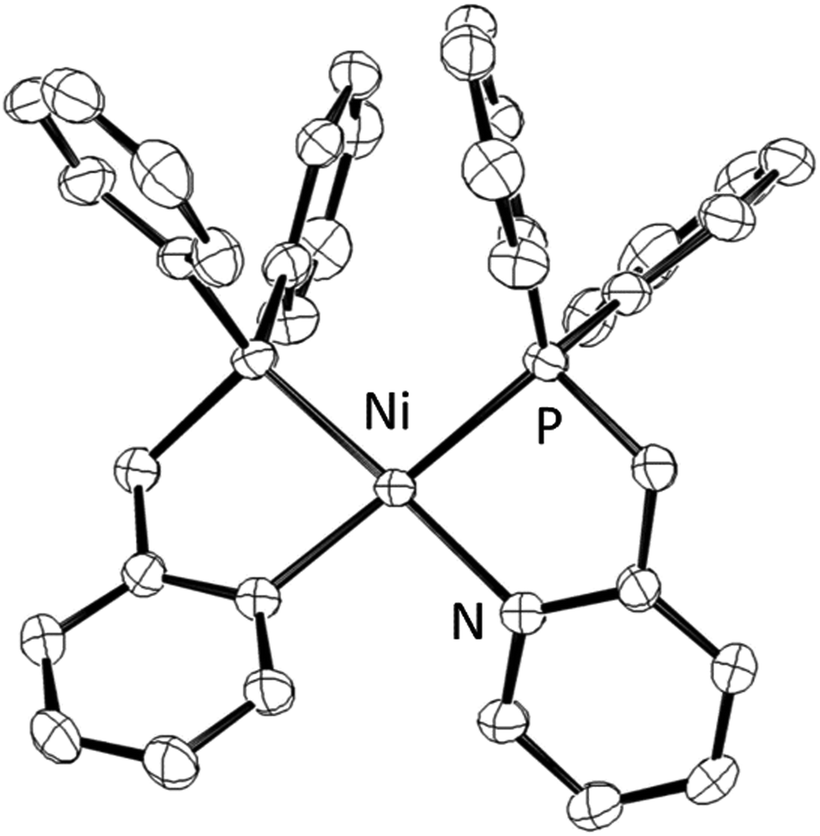 | ||
| Fig. 1 ORTEP drawing of the cationic part [Ni(LH)2]2+ of complex 1 with 50% ellipsoid probability. The counter anions and H atoms are omitted for clarity. | ||
Next, UV-vis absorption spectra were examined to study the structure of complex 1 in solution. In homogeneous catalysts, the structure in solutions is more relevant to the catalytic cycle than that in the crystalline state. UV-vis absorption spectra of complex 1 (1 mM) dissolved in MeCN and CH2Cl2 showed intense absorption bands at 428 nm (ε = 504 M−1 cm−1) (Fig. 2) and at 407 nm (ε = 335 M−1 cm−1) (Fig. S1, ESI†), respectively. Absorptions observed in the 400–550 nm region have been attributed by previous studies to d–d transitions originating from the square planar Ni(II) complex and LMCT band from the ligand to the Ni(II) ion.21 In the case of Ni(II) complexes, absorptions originating from octahedral and tetragonal pyramidal structures are usually observed around 900–1100 nm in addition to absorption around ∼400 nm.18b However, no specific absorption was observed in either solution for complex 1, while a slightly smaller one was visible in MeCN (inset in Fig. 2). This suggests that complex 1 maintains mainly a four-coordinate square planar structure in solutions.
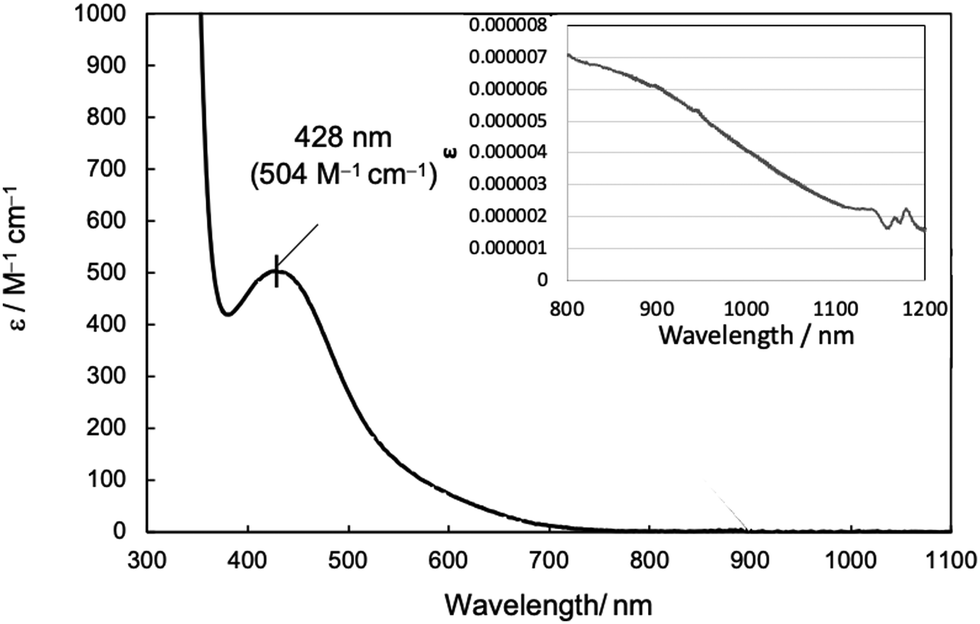 | ||
| Fig. 2 UV-vis spectrum of complex 1 (1 mM) in MeCN. The inset shows the spectrum measured at high concentration (8.58 mM) in the long wavelength region (800–1200 nm). | ||
In order to investigate the structure of the Ni(II) complex containing cis/trans isomers in solution, 1H and 31P{1H} NMR spectroscopy measurements were also carried out. Interestingly, 1H (Fig. S2, ESI†) and 31P{1H} NMR (Fig. S3, ESI†) of complex 1 in MeCN-d3 both showed broadened paramagnetic spectra, while those (Fig. S4 and S5, ESI†) in CD2Cl2 exhibited sharp diamagnetic ones. Based on the crystal field theory, the square planar structure is expected to be a low-spin Ni(II) complex, which is diamagnetic, while the octahedral or tetragonal pyramidal structure is predicted to be a high-spin Ni(II) complex, which is paramagnetic.18b Therefore, in MeCN, complex 1, which shows paramagnetic properties in MeCN-d3, seems to form a five- or six-coordinate structure bound with MeCN, while the diamagnetic properties exhibited in CD2Cl2 suggest that it is a square planar structure. Different results for the coordination structures of complex 1 in these solutions appear to have been obtained in the NMR and UV-vis spectra. Presumably, the MeCN molecule interacted weakly with the Ni(II) complex, but only to the extent that it was undetectable in the UV-vis spectrum, as discussed in the calculations below. By the way, 1H and 31P{1H} NMR spectra in both solutions showed that complex 1 is present as only one isomer each. These findings suggest that complex 1 exists only in one isomer in solution as well as in the crystal structure, and only in the cis-isomer in solution, although there may be differences in solvent coordination. Subsequent electrochemical and other measurements were performed in MeCN for comparison with the previously reported complex [Ni(LNH2)2](BF4)2.18b
Electrochemical measurements
Cyclic voltammetry (CV) measurements of complex 1 (1 mM) were carried out in MeCN solution containing 100 equiv. of electrolyte (0.1 M TBAP ([n-Bu4N](ClO4)) under inert gas conditions with sweep rates varying from 100 to 500 mV s−1 in steps of 100 mV s−1. The results are shown in Fig. 3, and the redox waves attributed to Ni(II/I) and Ni(I/0) are summarized in Table 1. Redox waves originating from Ni(II/I) and Ni(I/0) were observed as quasi-reversible and irreversible systems at E1/2 = −0.86 V (ΔE = 140 mV) and Epc = −1.73 V, respectively. An oxidation wave of Ni(I/0) was observed around −1.5 V, but it was much smaller than the reduction wave, which was interpreted as the Ni(I/0) reduction wave occurring almost irreversibly. The finding that the redox potentials of the Ni(II/I) and Ni(I/0) couples in the CV of complex 1 were attributed to one-electron reduction, respectively, was confirmed by the observation of waves corresponding to the Ni(II/I) and Ni(I/0) potentials at the same current values in the differential pulse voltammetry (DPV) method, carried out under the same conditions as the CV measurements. The DPV results are presented in Fig. S7 (ESI†) and the current integrals in Fig. 3 together with the CV of complex 1.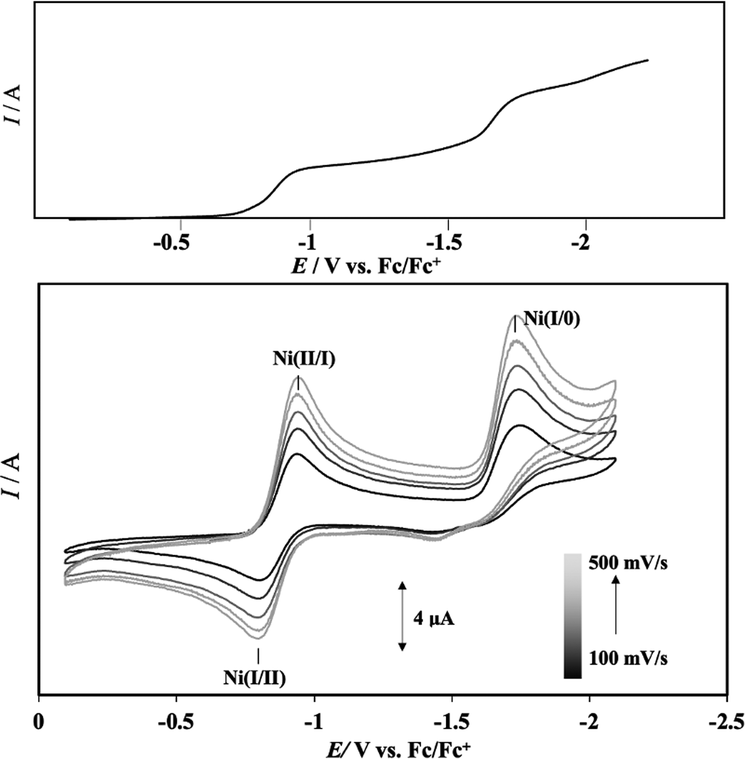 | ||
| Fig. 3 Cyclic voltammograms (bottom) of complex 1 in MeCN, as measured at several sweep rates and current integral (upper) estimated by DPV. Note that the redox potential of ligand LH may be observed in this region, but as shown in Fig. S6 of ESI,† no such wave was detected when the CV measurement of ligand LH was carried out under the same conditions as complex 1, and the wave is due to the Ni(I/0) couple of complex 1. | ||
| Ni(II/I) | Ni(I/0) | |
|---|---|---|
| E pc/V | −0.93 | −1.73 |
| E pa/V | −0.79 | |
| E 1/2/V | −0.86 | |
| ΔE/mV | 140 |
Each peak current (ip) is proportional to the square root of the sweep rate (Fig. 4), indicating that this system functions as a homogeneous system and a diffusion system rather than an adsorption system at the electrode surface.22 The redox waves of Ni(II/I) are observed at almost the same positions as the previously reported complex with amino groups, [Ni(LNH2)2](BF4)2 (E1/2 = −0.82 V, ΔE = 90 mV).18b On the other hand, the reduction wave of Ni(I/0) was observed to be more negative than that of the complexes reported in the previous study (Epa = −1.55 V).18b The finding that the Ni(II/I) redox potentials did not change significantly between Ni complexes with or without an amino group and the Ni(I/0) redox potentials of the Ni complex with the amino group shifted towards the positive side compared to that without the amino group suggests that the Lewis acidity of the Ni atom decreases when reduced to Ni(I) and the electron density of the amino group nitrogen increases in the Ni complex with the amino group. The interaction between the amino groups with the large electron density and the solvent molecules is considered to have strengthened and then the reduction potential of Ni(I/0) shifted to the positive side.
 | ||
| Fig. 4 Plots of ip/μA vs. (scan rate)1/2/(mV s−1)1/2 of Ni(II/I) (left) and Ni(I/0) (right) of complex 1 in MeCN. | ||
Electrochemical hydrogen production
Electrochemical measurements were performed using AcOH as a proton source to realize electrochemical hydrogen production by the Ni complexes. However, it is necessary to consider the possibility that the coordination of AcOH may interfere with the hydrogen generation mechanism because of its coordination ability. Therefore, the possibility of AcOH coordination was investigated by tracing UV-vis spectra (Fig. S7, ESI†). As a result, in the case of complex 1, the absorption derived from the square planar structure of Ni(II) complex was not attenuated and that originating from the octahedral or square pyramidal structure did not rise by addition of AcOH in the range of 0–290 equiv. This means that, in this range, deactivation of the catalyst due to AcOH coordination does not need to be taken into account. Thus, electrochemical hydrogen production experiments were performed under the same conditions as the CV measurements described above, using AcOH (pKa = 22.3 in MeCN) added dropwise as the proton source. The results are shown in Fig. 5(a). The Ni(II/I) and Ni(I/0) reduction potentials did not change under the acidic conditions, and the onset potential of the catalytic current was found to be around −1.7 V, indicating that this catalyst facilitated a two-electron reduction through the EECC mechanism. As shown in Fig. 5(b), the catalytic current (icat) is proportional to the square root of the concentration of AcOH added. This is in good agreement with the case of [Ni(LNH2)2](BF4)2 reported in a previous study.18b As can be seen from eqn (2) and (3),7c,22icat is proportional to x/2 power of the acid concentration (x is the reaction order with respect to the acid). Thus, the hydrogen production reaction is estimated to be x = 1, indicating that the reaction is the first order. This also means that the rate-determining step of the reaction is the stage in which the first proton reacts with Ni(0) to form the hydride Ni(II) species.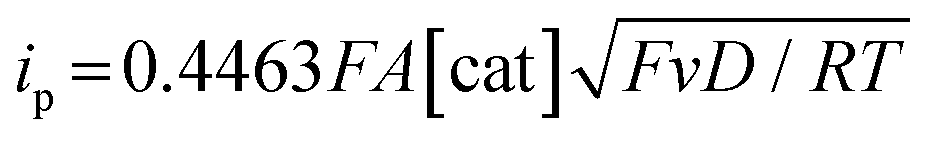 | (1) |
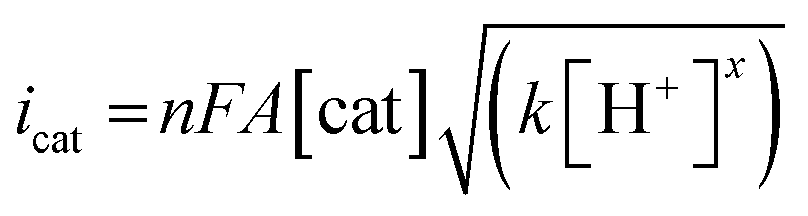 | (2) |
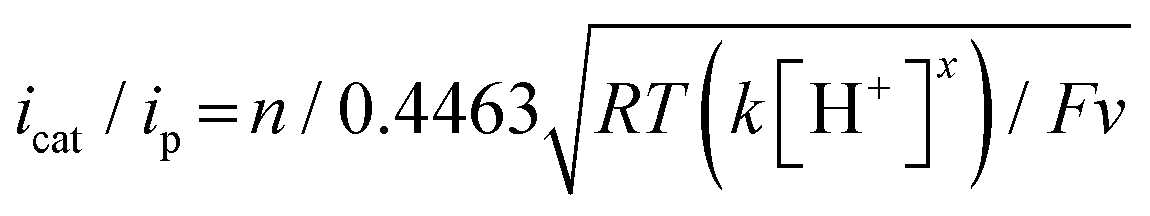 | (3) |
 | (4) |
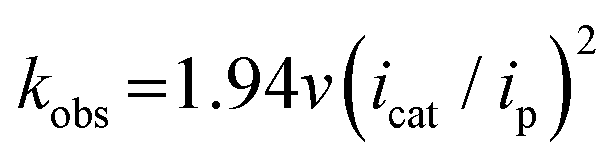 | (5) |
 | ||
| Fig. 5 (a) CVs and (b) plots of icat/ip A vs. (AcOH)1/2/(mmol L−1)1/2 of complex 1 (1.0 mM) in the presence of 0–203 equiv. of AcOH (pKa = 22.3 in MeCN) in MeCN containing 0.1 M TBAP at room temperature under an Ar atmosphere, recorded at a scan rate of 500 mV s−1. | ||
Turnover frequency (TOF) is calculated using a ratio of icat/ip (eqn (3) and (4)), where icat is the peak current of the reduction wave in the presence of acid (eqn (2)) and ip is the peak current applied to a reversible and one electron wave in the absence of acid (eqn (1)).7c,23 As defined in eqn (2), the value of icat is independent of the scan rate. Studies on the scan rate dependence of icat show that the scan rate independence is achieved only at a scan rate of 0.7 V s−1 and greater. Eqn (5) is obtained by simplifying eqn (3) and (4).
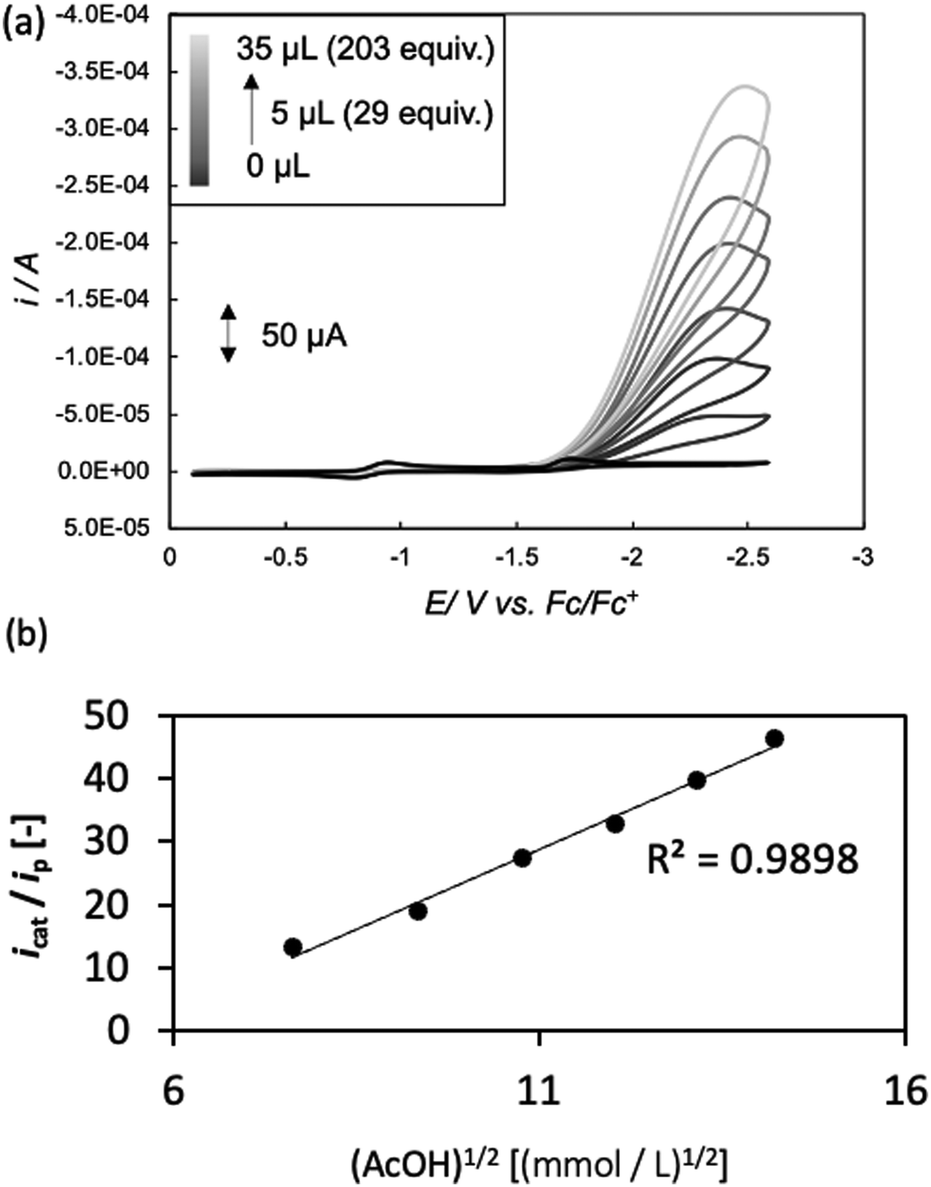
Next, in determining the overpotential, there is one more thing to note here. It is the problem of the homoconjugation of AcOH in MeCN, which was used as a proton source in this study. This depends on the concentration, which should be paid attention for evaluation of overpotential. It is thought in this system to mainly be a homoconjugate species under the evaluation conditions of the catalytic activity (25 mL in total 3 mL of MeCN; ca. 200 mmol L−1 of AcOH). Foumond et al. reported that it is not possible to exactly determine the overpotential of a system wherein a homoconjugate species is predominantly present.24 However, for convenience, we decided to use the equations shown above for the evaluation of the overpotential, just as the system with a non-homoconjugate acid, because the complexes compared in this study are structurally very similar to each other under the same reaction conditions. The overpotential can be calculated as the difference between EH+ and catalytic potential Ecat/2 (eqn (6) and (7)), where EH+ is the thermodynamic potential (for H+/H2 reduction) and Ecat/2 is the observed catalytic half-wave potential for AcOH reduction in the presence of complex 1.25a In MeCN, 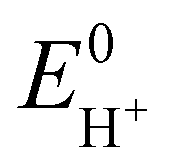 was recently determined to be −0.028 ± 0.008 V vs. Fc/Fc+ through open circuit measurements.25b
was recently determined to be −0.028 ± 0.008 V vs. Fc/Fc+ through open circuit measurements.25b
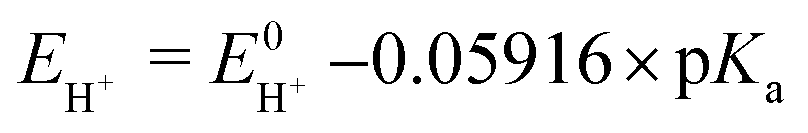 | (6) |
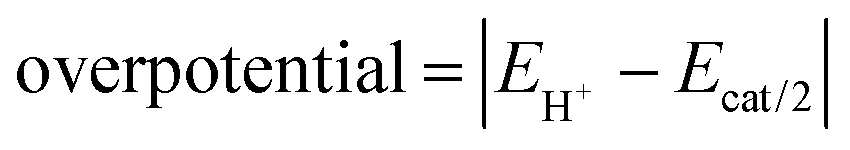 | (7) |
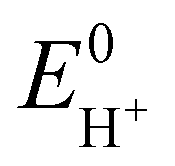 is the standard potential of the reduction of protons in V.
is the standard potential of the reduction of protons in V.
Using the above equations, the TOF and overpotential of complex 1 were evaluated to be 1060 s−1 and 710 mV, respectively, where they were evaluated under the same conditions as in the previous study (AcOH = 25 μL (145 equiv.), scan rate = 500 mV s−1) and are summarized together with them in Table 2. First, the TOF was found to be reduced to 1/8th of 8800 s−1 for [Ni(LNH2)2](BF4)2 in the previous study.18b This reduction in catalytic activity is thought to be due to the absence of amino groups, which are proton-transferring sites, in complex 1, resulting in a slower supply of protons from outside the system. More unfortunately, the overpotential became very large. This suggests that the amino group of [Ni(LNH2)2](BF4)218b has some significant effects, such as proton-transfer, on the decrease in the overpotential. Overpotential is made up of the sum of the following three components: activation overpotential, concentration overpotential, and resistance overpotential. In complex 1, there are no functional groups such as amino groups to transfer protons in the vicinity of the metal of the reaction centre. Of course, the other two effects could be considered, but they are unlikely to be significant. As a result, the concentration of raw material protons near the reaction centre is thought to be reduced due to the absence of amino groups. In other words, the concentration overpotential due to the absence of protons in the vicinity of the active centre is considered to have been higher in complex 1 than in the case with amino groups.26 This can also be supported by the fact that in the hydrogen production reaction using AcOD, the plots of the catalytic currents proportional to the square root of the acid concentrations showed a large heavy-atom effect (Fig. S9(a) and (b), ESI†).
| TOF/s−1 | Overpotential/mV | |
|---|---|---|
| Complex 1 | 1060 | 710 |
| [Ni(LNH2)2](BF4)2 | 8800 | 440 |
Preparation of a hydride Ni(II) complex intermediate
In this study, electrochemical hydrogen production in MeCN has been investigated using AcOH as the proton source and using complex 1 without amino groups. Before discussing the reaction mechanism, it is necessary to confirm the formation and presence of a hydride Ni(II) complex intermediate, which is produced by the reaction between the Ni(0) species and the approaching protons. So, to demonstrate this possibility, the intermediate species produced by the reaction of a separately generated [Ni0(LH)2] complex with NH4PF6 as the H+ source were studied in THF-d8 in the NMR sample tube under anaerobic conditions. The sample for ESI-MS measurements was also prepared in the same way. First, in the ESI-MS spectrum (Fig. S10, ESI†), fortunately a main ion cluster was obtained at m/z = 613.49 in positive mode, which is in good agreement with the mass of the complex [NiII(LH)2H]+ in the simulation. The 1H NMR spectrum of the resulting solution (Fig. S11, ESI†) contained several chemical species, which cannot be accurately attributed, but a peak characteristic of the hydride species was observed at −23.9 ppm, which is in good agreement with values previously reported for similar hydride nickel complexes.27 This is also consistent with the values calculated from the DFT calculations described below, together with the confirmed formation of [NiII(LH)2H]+ from ESI-MS, which can be attributed to hydride species and is definitely the NMR peak for hydride species.Possible coordination of acetate ions to complex 1 and its electrochemical behaviour
Although no coordination to the Ni(II) ion of complex 1, as described above, was detected by addition of AcOH, AcO− remains when it reacts as an acid and the protons are removed during the hydrogen evolution process; AcO− may be coordinated to the Ni(II) complex. Therefore, the coordination of AcO− to complex 1 and its electrochemical behaviour were investigated using TBA+AcO−. As a result, tracing the absorption spectral changes due to the addition of TBA+AcO−, two broad peaks were observed in the range 500–900 nm with an isosbestic point (Fig. S12, ESI†). This suggests the formation of a five- or six-coordinated Ni(II) species with AcO− coordinated to complex 1. The CV measurements (Fig. S13, ESI†) were also carried out in this acid concentration range, and the Ni(II/I) reduction wave shifted slightly to a negative potential with the addition of TBA+AcO−, while the Ni(I/0) wave did not change much. This means that even if AcO− was coordinated to Ni(II) species in the electrochemical hydrogen production in this study, it was removed during the electrochemical reduction to Ni(0) species. Here, the source of protons for hydrogen production is a trace amount of water in the added TBA+AcO−.Electronic structure of [NiII(LH)2]2+ in MeCN as evaluated by DFT calculations
In order to study the electronic structures of the starting Ni(II) complex [NiII(LH)2]2+ in MeCN, the energies of the Ni(II) complexes with and without MeCN solvent as the ligand were calculated for the low- and high-spin states, respectively, using DFT calculations. The optimized structures of the MeCN-solvated Ni(II) complex, [NiII(LH)2(MeCN)2]2+, and the desolvated Ni(II) complex, [NiII(LH)2]2+, in the low- and high-spin states are shown in Fig. S14 (ESI†). The structural parameters around the Ni centre of these complexes and their Gibbs free energies with reference to the low-spin state at room temperature are listed in Table 3. The optimized structure of the desolvated Ni(II) complex [NiII(LH)2]2+ calculated in the low spin state, despite taking into account MeCN solution conditions, was a square planar structure as shown in Fig. S14(a) (ESI†), in which the Ni–P and Ni–N lengths were in good agreement with those of the crystal structure in the range of 0.03 Å and 0.06 Å, respectively. On the other hand, the optimized structure calculated in the high-spin state was a distorted four-coordinate geometry (Fig. S14(b), ESI†), where a solvent molecule has been desorbed from the equatorial site of the trigonal bipyramidal structure. The low spin state was 9.7 kcal mol−1 more stable than the high-spin state. These results indicate that [NiII(LH)2]2+ in the crystal is in the low-spin state.a
| [NiII(LH)2]2+ | [NiII(LH)2(MeCN)2]2+ | ||||
|---|---|---|---|---|---|
| Low-spin | High-spin | Crystallographic data | Low-spin | High-spin | |
| a At 298.15 K, 1 M. | |||||
| S (calc.) | 0 | 1.002 | 0 | 1.002 | |
| Energya (kcal mol−1) | |||||
| ΔE | 0.0 | 12.3 | 0.0 | 2.2 | |
| ΔH | 0.0 | 11.6 | 0.0 | 0.8 | |
| ΔG | 0.0 | 9.7 | 0.0 | −0.4 | |
| Solvation, ΔG0′ | 1.6 | 1.3 | |||
| Bond length (Å) | |||||
| Ni–P | 2.189 | 2.344 | 2.1771(6) | 2.199 | 2.432 |
| 2.192 | 2.341 | 2.1628(6) | 2.199 | 2.428 | |
| Ni–N(py) | 1.999 | 2.064 | 1.948(2) | 2.003 | 2.145 |
| 1.998 | 2.061 | 1.964(2) | 2.002 | 2.151 | |
| Ni–N(MeCN) | 3.555 | 2.106 | |||
| 3.560 | 2.121 | ||||
Next, the optimized structure of the Ni(II) complex with two MeCN molecules, [NiII(LH)2(MeCN)2]2+, was calculated. The optimized structure of the MeCN-solvated Ni(II) complex calculated in the low-spin state was a square-planar structure (Fig. S14(c), ESI†) with MeCN molecules loosely bound to Ni(II) with Ni–N(MeCN) = 3.555 and 3.560 Å, attracted by π–π interactions (∼3.3 Å) with aromatic rings of phenyl groups. The optimized structure of the MeCN-solvated Ni(II) complex in the high-spin state was octahedral (Fig. S14(d), ESI†), with the MeCN molecules strongly coordinated with Ni–N(MeCN) = 2.106 and 2.121 Å from the apex directions. Interestingly, the low-spin complex was slightly higher in energy than the high-spin one by 0.4 kcal mol−1 unlike the case of the desolvated complex, or rather the high-spin and low-spin complexes were almost equal. However, the stability of the low-spin di-MeCN complex (Fig. S14(c), ESI†) appears to be overestimated due to the π–π interactions as mentioned above. Although the solvation energy in the high-spin di-MeCN complex is slightly uphill by 1.3 kcal mol−1, considering the overestimation of entropy in the bimolecular reaction, the high-spin [NiII(LH)2(MeCN)2]2+ may be dominant in the MeCN solution.28 These results correspond well with the behavior observed in the aforementioned NMR spectra of the [NiII(LH)2](BF4)2 complex in the solution as diamagnetic in CD2Cl2 solution and paramagnetic in MeCN-d6 solution.
Structural characterization of a hydride Ni(II) complex intermediate, [NiII(LH)2H]+, by DFT calculations
It is expected that the two-electron reduction of [NiII(LH)2]2+ produces [Ni0(LH)2]0 prior to the reaction with protons. [Ni0(LH)2]0 has no ligand field stabilization energy due to the Ni centre with the d10-configuration. It is therefore not clear what kind of four-coordinate structure can be obtained. The optimized structures and energies of the four-coordinate and three-coordinate structures with a free pyridyl group, respectively, are shown in Fig. S15 (ESI†). The four-coordinate complex (Fig. S15(a), ESI†) was slightly lower in energy than the three-coordinate complex (Fig. S15(b), ESI†) by 1.6 kcal mol−1. This slight difference in energy would be sufficient to release the pyridyl group. This flexible coordination structure allows the formation of the hydride Ni(II) complex intermediate [NiII(LH)2H], which is formed by the initial proton attack on the Ni centre of [Ni0(LH)2]0.Gibbs free energies of hydride Ni(II) isomers, which could exist as several isomers, are given in Table 4. The optimized structures of the low-spin hydride Ni(II) complexes are shown in Fig. 6. The high-spin counterparts and mono-MeCN solvated low- and high-spin complexes are also shown in Fig. S16–S18 (ESI†), respectively. As mentioned in the Computational details section, no isomers with hydride ligands at the apex position were found.
a
| Trans atom of H− | N | P | ||
|---|---|---|---|---|
| Apex atom | N | P | N | P |
| a At 298.15 K, 1 M. b One pyridyl group leaves away from the Ni centre, and the other moves to the apex position, where the MeCN molecule is coordinated. | ||||
| Energy (kcal mol−1) desolvated | ||||
| Low-spin | 0.0 | 1.9 | 2.3 | 15.3 |
| High-spin | 13.5 | 9.4 | 12.3 | 10.4 |
| Mono-MeCN | ||||
| Low-spin | 0.0 | 1.5 | 4.7 | 5.5b |
| High-spin | 13.7 | 9.5 | 12.8 | 13.7 |
| Bond length (Å) desolvated | ||||
| Ni–H | 1.460 | 1.468 | 1.489 | 1.481 |
| Mono-MeCN | ||||
| Ni–H | 1.476 | 1.468 | 1.489 | 1.495 |
| Ni–N(CH3CN) | 3.687 | 3.898 | 4.015 | 1.880b |
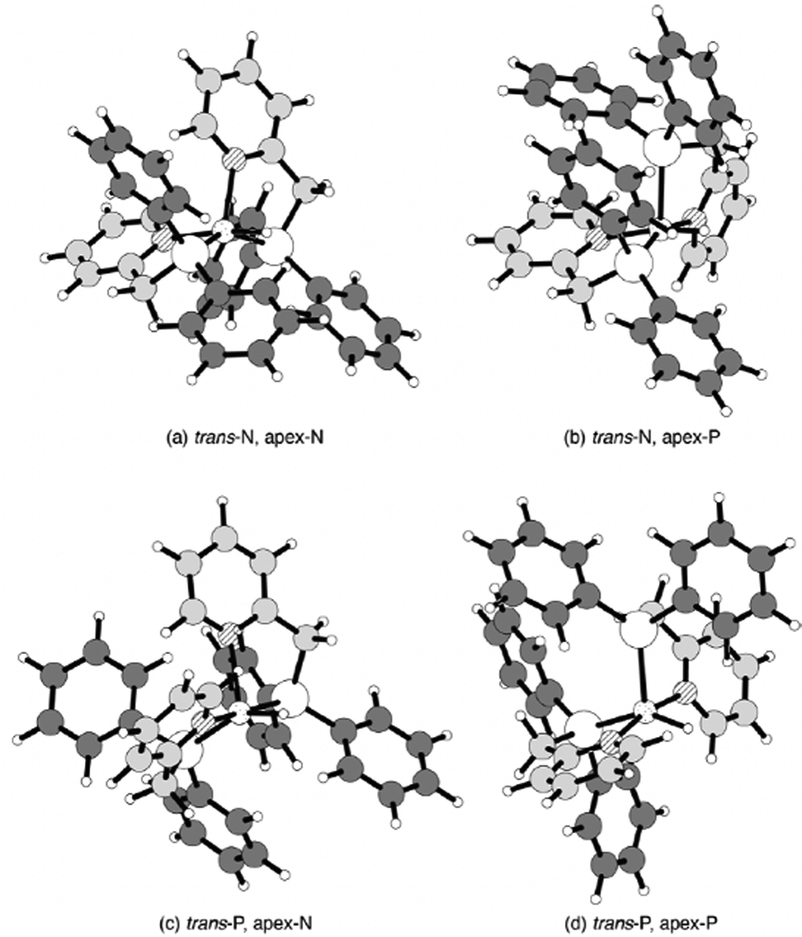 | ||
| Fig. 6 The optimized structures of the desolvated low-spin hydride Ni(II) complexes. Medium, large, shaded, grey, and small circles denote Ni, P, N, C, and H atoms, respectively. These square pyramidal structures are distinguished by atoms at the trans position of hydride and at the apex position: (a) N atoms occupy both positions, (b) N and P atoms occupy the trans position of the hydride and the apex position, respectively, (c) P and N atoms occupy the trans position of hydride and the apex position, respectively, and (d) P atoms occupy both positions. | ||
The low-spin isomers with low energies of around 2 kcal mol−1 seem to coexist with each other, except for the unstable isomer with P atoms at the apex- and the trans-positions of the hydride ligand. The MeCN molecules loosely bound to low-spin complexes have no significant effect on their coordination structure, except for the case of MeCN bound to the unstable isomer. In this mono-MeCN solvated complex, the MeCN molecule bound to the basal plane pushes one pyridyl group toward the apex position and moves the other pyridyl group away from the Ni centre. The isomer with N atoms at the apex and in the trans position of hydride (trans-N, apex-N in Fig. 6) was the most stable of all. A hydride ligand prefers a pyridyl group in the trans position for the stable short Ni–H bond.
Regardless of the binding of MeCN to the Ni centre, the low-spin complex has about 10 kcal mol−1 lower energy than the high-spin complex. In contrast to the case of [NiII(LH)2]2+, the MeCN coordination did not stabilize the high-spin state. The solvation energy of MeCN defined by eqn (16) in the most stable isomer (Fig. 6(a)) was positive, 1.6 kcal mol−1, and the bound MeCN molecule was out of the first-coordination sphere (Fig. S17(a), ESI†). Therefore, it can be said that solvation of MeCN has no obvious effect on the hydride Ni(II) complex.
Natural atomic charges and 1H-NMR chemical shifts of the hydride in the low-spin hydride Ni(II) complexes are listed in Table 5. The calculated 1H-NMR chemical shift of the hydride of the isomer with the pyridyl group attached in the trans position was about −18 ppm, close to the measured value (−23.9 ppm). In contrast, the calculated chemical shift of the hydride of the isomer in which the hydride and a phosphino group are bound in the trans position was −8 ppm. The calculated chemical shifts are similar to those previously reported for Pt(II) hydride complexes and are in good agreement with the fact that ligands with a high trans influence on hydrides reduce the chemical shifts.29 This stable isomer is inferred to have a pyridyl group attached to the trans position of the hydride, based on the agreement between the observed and calculated chemical shifts and the calculated energies.
| Trans atom of H− | N | P | ||
|---|---|---|---|---|
| Apex atom | N | P | N | P |
| a At the trans position of the hydride ligand. b At the apex position. c The chemical shift of the hydride ligand referenced to 2- and 5-protons of THF. | ||||
| Desolvated | ||||
| Ni | 0.79 | 0.82 | 0.78 | 0.88 |
| P | 0.93 | 0.97 | 0.89a | 0.88a |
| P | 0.90 | 0.85b | 1.01 | 0.80b |
| N | −0.57a | −0.57a | −0.55 | −0.54 |
| N | −0.58b | −0.57 | −0.57b | −0.53 |
| H | −0.23 | −0.23 | −0.30 | −0.32 |
| δ (1H NMR, ppm)c | −18.2 | −16.9 | −8.2 | −8.2 |
| Mono-MeCN | ||||
| Ni | 0.76 | 0.84 | 0.73 | 0.75 |
| P | 0.94 | 0.95 | 0.90a | 0.93a |
| P | 0.92 | 0.85b | 0.98 | 0.99b |
| N | −0.57a | −0.56a | −0.57 | −0.52 |
| N | −0.54b | −0.58 | −0.49b | −0.50 |
| H | −0.26 | −0.23 | −0.29 | −0.25 |
| N(acetonitrile) | −0.43 | −0.43 | −0.42 | −0.50 |
The H ligand subjected to a large magnetic shielding effect is attributed to be hydride formed by electron transfer from the Ni(0) centre to the bound proton. Natural population analysis shows that the negative atomic charge of the coordinated H is small (−0.2 to −0.3), suggesting that the Ni–H bond in the low-spin complex has some polarized Ni(II)–H− feature. The positive charges on Ni and P atoms are due to the electron donation from phosphino ligands to the electron-deficient Ni centre. Phosphino ligands may act as a buffer for the positive charge generated during the electron transfer process.
The electron configuration of the low-spin (trans-N, apex-N) hydride Ni(II) complex with respect to the 3d-shell of the Ni centre and the hydride ligand is shown in Fig. S19 (ESI†). The HOMO−2, HOMO−1, and HOMO are attributed to 3dzx, 3dyz, and 3dz2 orbitals. The 3dxy orbitals are mixed with some occupied orbitals in the lower energy levels than them, as predicted from the square pyramidal coordination. The bonding and antibonding orbitals of Ni–H, corresponding to HOMO−14 and LUMO+12, respectively, are shown in Fig. S20 (ESI†). The fairly close weights of the 3d orbital on the Ni centre and the 1s orbital on the hydride ligand suggest the covalent nature of the Ni–H bond. However, the larger weight of the d-orbitals than the 1s orbital in the LUMO+12 as well as the LUMO+1 and the remarkable bulge on the hydride ligand in the HOMO−14 indicate that the donor–acceptor interaction attributed to the electron donation from the H− donor to the Ni(II) centre makes some contributions to the Ni–H bond.
Thermodynamic schemes of electrocatalytic hydrogen generation by the [NiII(LH)2]2+ system
In the previous section, the intermediates observed for the catalytic HER were assigned to calculated geometric and electronic properties using DFT calculations. In this section, thermodynamically possible reaction pathways are proposed based on calculation of the protonation and reduction steps connecting these intermediates.The thermodynamic diagrams of the HERs for complex 1 and its amino derivative, [NiII(LNH2)2]2+, are given in Fig. 7(a) and (b), respectively. The calculated Ni(II/I) reduction potentials are substantially less negative than the observed one. This is due to the underestimation of the solvation energy for highly charged ions by the polarized continuum model (PCM), and that of the real solvated [NiII(LH)2]2+ is considered to be more stable than the calculated value. The change in the Ni(II/I) reduction couple from the amino derivative to complex 1 is calculated to be −0.06 V, comparable to the observed one, −0.11 V, as errors in solvation energies are canceled out. The change in Ni(I/0) is calculated to be −0.03 V, which is evaluated as a substantially smaller change compared to the observed one, −0.18 V, but these tendencies with the substituent change are in the same direction. The desolvation energy of the Ni(I) species in complex 1 is estimated to be 2.4 kcal mol−1, which is thermodynamically unfavorable, while that for the amino derivative, 5.1 kcal mol−1, is more positive (Fig. S4, ESI†). Considering the underestimation of calculated solvation energies, the Ni(I) species in complex 1 would have a solvent molecule at the apex and be more stabilized in real solution, while the amino derivative would not.
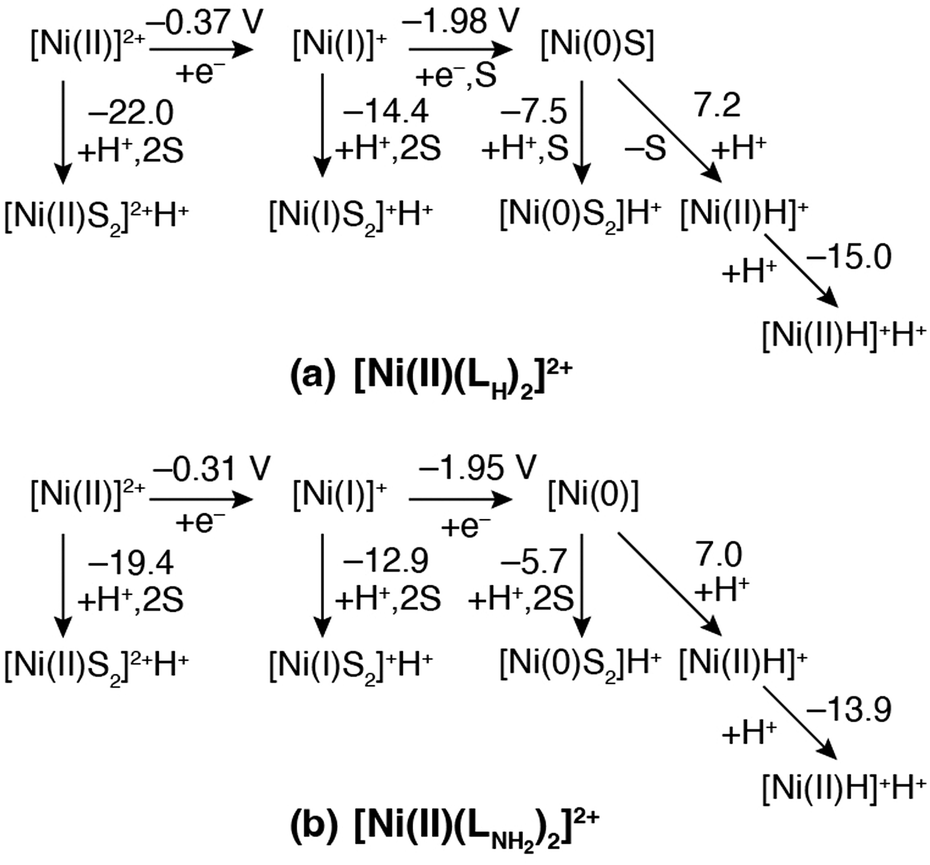 | ||
| Fig. 7 Thermodynamic diagrams for (a) complex 1, [NiII(LH)2]2+, and (b) [NiII(LNH2)2]2+ systems. The reduction potentials are for the ferrocene/ferrocenium couple as the reference electrode. The ΔpKa values for the protonation pathway are based on AcOH. The ‘S’ in the equations represent the solvated MeCN molecule. | ||
For the complex 1 system, the ΔpKa values with respect to AcOH for the Ni(II), Ni(I), and Ni(0) species corresponding to [NiII(LH)2]2+, [NiI(LH)2]+, and [Ni0(LH)2S], respectively, and the hydride complex of 1, [NiII(LH)2H]+, are −22.0, −14.4, −7.5, and −14.9, respectively, which are more acidic than AcOH. These values are roughly controlled by the electrostatic repulsion between a proton and the total charge of the conjugate bases. The reduction potential of the Ni(II/I) couple with reference to the Ni(I/0) couple was +0.80 V (−18.4 kcal mol−1), which was evaluated from the experimental Epc values of the Ni(II/I) and Ni(I/0) couples rather than the potentials calculated from the insufficient solvation model. Based on this value, the reactions starting from [NiII(LH)2]2+ to form the protonated Ni(0) species, [Ni0(LH)2]H+, and then the protonated hydride complex, [NiII(LH)2H]H+, are exergonic at the Ni(I/0) reduction potential, but those forming the protonated Ni(II) and Ni(I) species, [NiII(LH)2]2+H+ and [NiI(LH)2]+H+, respectively, are not. These calculation results suggest that the catalyst follows the EECC mechanism. The Gibbs free energy of the reaction starting from [NiI(LH)2]+ to form complex 1, which was recovered by the release of the H2 molecule, was −9.5 kcal mol−1 (Fig. S22, ESI†), and it was estimated to be −27.9 kcal mol−1 for the catalytic cycle by adding the Ni(II/I) reduction potential. This pathway of the catalytic cycle is entirely downhill via formation of a stable hydride complex intermediate.
On the other hand, the species in the [NiI(LNH2)2]+ system have more positive ΔpKa values than those in the complex 1 system. Like the complex 1 system, protonation of the N atom of a leaving pyridyl group is preferred to protonation of an amino group forming an intramolecular hydrogen bond with another pyridyl group, because the positive charges on the metal centre and the protons are far from each other (Fig. S23, ESI†). The role of the electron donating amino group is not only to capture protons but also to increase the basicity of the pyridyl N atom. The reduction potential of the Ni(II/I) couple with reference to the Ni(I/0) couple for the amino derivative was +0.82 V (−18.9 kcal mol−1) and was evaluated from the experimental Epa values in the same manner as in the complex 1 system. Considering this reduction potential, the protonation of [Ni(I)]+ with the reaction energy of 17.6 kcal mol−1 would also be available in the catalytic cycle if the reaction barrier raised by the repulsive interaction between positive charges on the [Ni(I)]+ cation and a proton is ignored. The Gibbs free energy of the reaction starting from [NiI(LNH2)2]+ to form [NiII(LNH2)2]+, restored by releasing the H2 molecule, is −7.7 kcal mol−1 (Fig. S22, ESI†) and that of the catalytic cycle was estimated to be −26.3 kcal mol−1. The difference in the total reaction energies between [NiII(LH)2]2+ and [NiII(LNH2)2]2+ systems is insignificant. It is considered that the high catalytic reactivity found in the [NiII(LNH2)2]2+ system comes from the low reaction barriers of proton transfer steps, owing to the electron donating amino group feasible for protonation at the pyridyl N atom.
The calculated thermochemical diagram suggests that the high-energy barriers of the catalytic cycle are located around the zero-valent [Ni0(LH)2]0 and the high energy protonated hydride complex, [NiII(LH)2 H]+H+ (Fig. S22, ESI†). In particular, the latter complex is the product of an unfavorable reaction between a positively charged hydride complex and an electrostatically repulsive proton. The H2 elimination reaction of [NiII(LH)2H]+H+ produces the HER product [NiII(LH)2]2+, which is also the reactant of the catalytic cycle. It is considered that the more negative the energy of H2 eliminated from [NiII(LH)2H]+H+ is, the higher the energy barrier for the catalytic HER is.
The previously reported thermodynamic analysis of the driving force of the HER catalyzed by bis(diphosphine)Ni(II) complexes shows that the driving force of the catalytic HER is evaluated from the reaction given in eqn (8),12
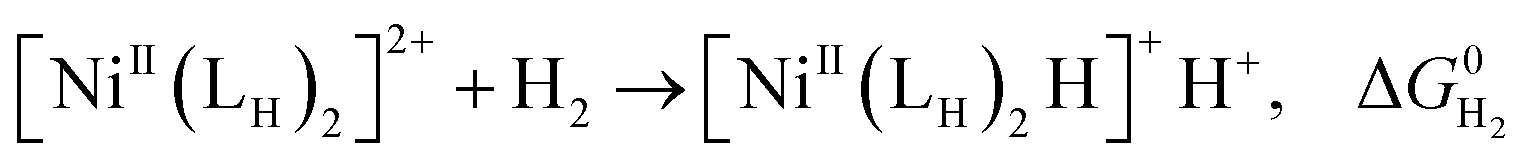 | (8) |
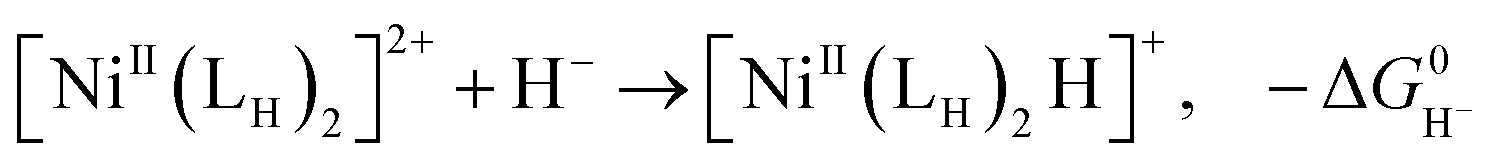 | (9) |
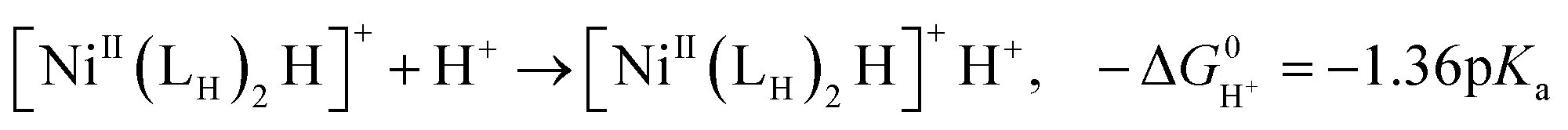 | (10) |
 | (11) |
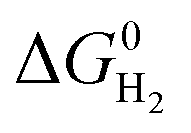 is represented as follows,
is represented as follows,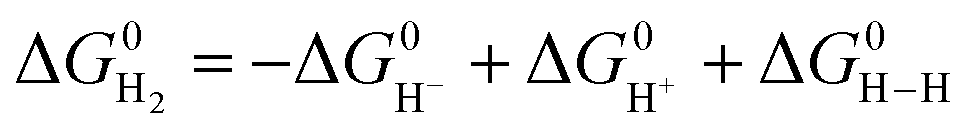 | (12) |
An unstable [NiII(LH)2H]+H+ with the low hydricity and basicity, namely, i.e. small pKa, causes largely exergonic H2 elimination, meaning that the energy barrier for the HER to form [NiII(LH)2H]+H+ from the [NiII(LH)2]2+ reactant is high.
The hydricity of [NiII(LH)2H]+, 39.8 kcal mol−1, is smaller than that of [NiII(LNH2)2H]+, 41.4 kcal mol−1, and the pKa of [NiII(LH)2H]+H+, 7.4, is smaller than that of [NiII(LNH2)2H]2+H+, 8.4; these pKa values are calculated using the pKa value (22.3) of AcOH in MeCN. The H2 elimination of [NiII(LH)2H]+H+, −20.3 kcal mol−1, is more exergonic than that of [NiII(LNH2)2H]+H+, −17.2 kcal mol−1, which is consistent with the small hydricity degree and pKa value of the [NiII(LH)2]2+ system. The unstable Ni–H bond and the N–H bond of the proton raise the energy barrier of the HER from the reactant [NiII(LH)2]2+.
The bis(diphosphine)Ni(II) system with pendant amines has a large hydricity of 54.9 kcal mol−1, but the pKa value, 8.9, of the pendant amines30 close to our system leads to a  of 10.0 kcal mol−1, smaller than the systems reported in this study. As a result, the present P2N2 system requires an environment for the reduction process that promotes the stabilization of [Ni(II)H]+H+ compared to the P4 system.
of 10.0 kcal mol−1, smaller than the systems reported in this study. As a result, the present P2N2 system requires an environment for the reduction process that promotes the stabilization of [Ni(II)H]+H+ compared to the P4 system.
The slightly higher hydricity for protons for the hydride complex of the amino-substituted derivative compared to that of the unsubstituted form is due to the difference between their most stabilized structures, that is, the most stable structure of the amino-substituted derivative is the trans-P, apex-N structure, while that of the unsubstituted forms is the trans-N, apex-N structure. The population analysis of these structures showed that the Ni centre in the amino-substituted derivative is more electron-rich than that in the unsubstituted form due to the strong electron-donating properties of the amino group, and thus the trans-P structure, which can disperse the negative charge on the hydride H, leads to a stable structure. As a result, the Ni–H bond is elongated and polarized by the trans influence of P, which is considered to be the result of the electrostatically strengthened and stabilized Ni(II)–H− bond. In fact, the charges on H of these hydrides were −0.30 (Table S5, ESI†) for the amino-substituted derivative and −0.23 (Table 5) for the unsubstituted form. In fact, the charges on H of these hydrides were −0.30 (Table S5, ESI†) for the amino-substituted derivative and −0.23 (Table 5) for the unsubstituted form. The large negative charge on the hydride ligand in the amino-substituted derivative increases the hydride basicity (ΔpKa = −13.7 in Fig. 7), leading to the high catalytic activity in the hydrogen evolution reaction in comparison with the unsubstituted form (ΔpKa = −15.0 in Fig. 7).
Possible mechanism of electrocatalytic hydrogen generation by [NiII(LH)2](BF4)2 in MeCN
Based on the above experimental and computational results, a mechanism for the hydrogen production reaction in this system is proposed, as shown in Scheme 2, that is, (i) the Ni(II) complex is electrochemically reduced to the Ni(0) species by two-electrons. [Ni0(LH)2]0 has a pyridyl group that is easily cleaved from the metal centre (Table S4, ESI†), in contrast to [Ni0(LNH2)2]0 barely retaining tetra-coordination with electron-donating amino groups. Consequently, the Ni(0)–N(py) bond is weakened due to lowering of the Lewis acidity of the Ni centre in both complexes, which promotes the protonation of pyridyl nitrogen. (ii) The reduced, electron-rich Ni(0) species repulses the coordinating pyridine and the pyridine attracts H+ from AcOH, and (iii) the attracted H+ is immediately attracted to and bonds with the electron-rich Ni(0) species. (iv) The formed Ni–H bond is polarized to Ni(II)–H− and subsequently accepts H+ from AcOH to produce H2, which is released, and the Ni complex then reverts to the starting Ni(II) complex, followed by subsequent hydrogen evolution catalysis. (v) Some Ni complexes are coordinated with the free AcO− and deactivated, but subsequent electrochemical reduction is expected to revert to the Ni(0) species and return to the catalytic cycle of hydrogen production. Here, the greater catalytic capability for hydrogen generation exhibited by [Ni(LNH2)2]2+ compared to [Ni(LH)2]2+ is due to the following two effects. One is due to the increased basicity of the pyridine nitrogen in [Ni(LNH2)2]2+ with electron-donating amino groups, which contributes to a rise in proton concentration around the Ni(0) centre more than that in [Ni(LH)2]2+. Another is that the negative charge on the hydride H in the most stable structure of these hydride complexes is greater in the amino-substituted derivative than in the unsubstituted form.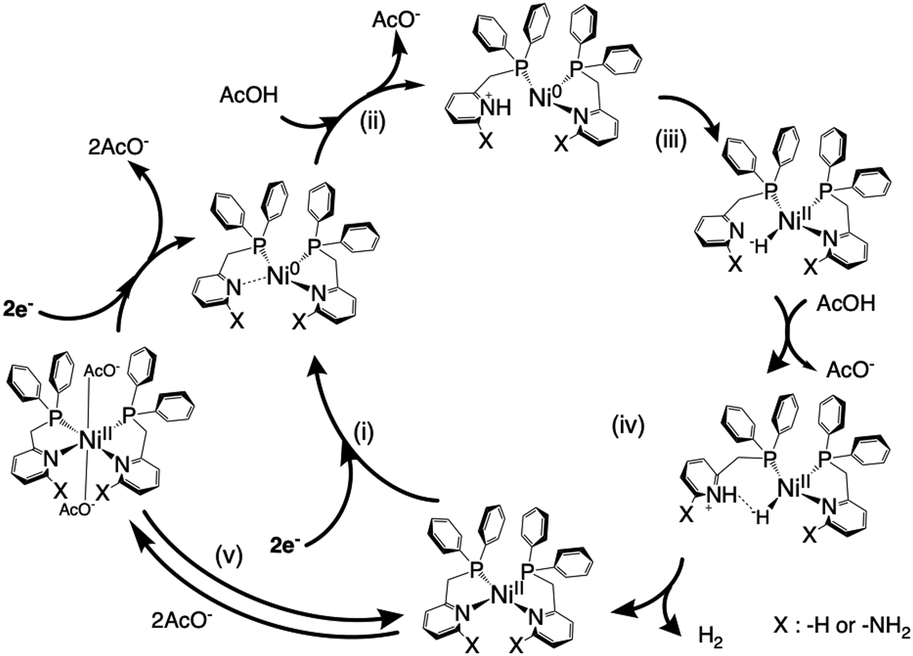 | ||
| Scheme 2 Possible reaction mechanism in the electrocatalytic hydrogen production by [NiII(LH)2]2+/[NiII(LNH2)2]2+ using AcOH in MeCN. | ||
Experimental
General procedure
All reagents were the purest available from Tokyo Kasei Co. and Fujifilm Wako Pure Chemicals Co. The solvents of THF, n-hexane, acetonitrile and dichloromethane were purified as the ultra-dehydrated ones using activated alumina columns. All processes were carried out under inert gas conditions. Ligand synthesis was carried out using the Schlenk technique and complex synthesis using MBrown and UniLab gloveboxes.Synthesis of the ligand LH (2-((diphenylphosphino)methyl)-pyridine)
Under a N2 atmosphere, α-picoline (1.02 g, 11.0 mmol) was added to 30 mL of dehydrated THF, cooled to 0 °C and 8.0 mL (11.2 mmol) of a hexane solution of 1.6 M n-BuLi was added dropwise, and the solution turned from yellow to red. When the solution was cooled to −78 °C, an orange-white solid precipitated. A solution of diphenylphosphine chloride (2.21 g, 10.0 mmol) in 10.0 mL of dehydrated THF was added dropwise to the suspension, which turned to an orange solution. After stirring for three days, it was subsequently deactivated using oxygen-free water and extracted with dichloromethane and sodium bicarbonate. Citric acid was then added to the organic phase until pH 2 was achieved, the resulting salts were degassed again and extracted using 10 mL of water, calcium hydroxide was added to the extracted aqueous phase until pH 14 was achieved, extraction was carried out with n-hexane and a white solid was obtained by decompression and concentration (yield: 673.87 mg, 2.43 mmol, 24.3%).700 MHz 1H-NMR (δ/ppm in C6D6) 3.63 (s, 2H); 6.49 (m, 1H); 6.80 (d, 1H); 6.90 (td, 1H); 7.01–7.48 (m, 6H); 7.48 (td, 1H); 8.40 (d, 1H), 282 MHz 31P{1H}-NMR (δ/ppm in MeCN-d3) = −10.10 (s).
Preparation of [Ni(LH)2](BF4)2
Under a N2 atmosphere, LH (166.30 mg, 0.60 mmol) was suspended in 10 mL of acetonitrile, and to the solution was added slowly dropwise a 5 mL MeOH solution containing Ni(BF4)2·6H2O (104.21 mg, 0.31 mmol). After stirring for 2 hours, the solution changed from light yellow to red, the solvent was removed by reduced pressure and an orange solid was obtained. The solid was dissolved in 10 mL of diethyl ether, filtered by suction and the precipitate formed was filtered off to give an orange powder. The powder was dissolved in acetonitrile and recrystallized using acetonitrile/diethyl ether by a vapor-liquid equilibrium or a liquid–liquid equilibrium method to obtain red-orange crystals (yield: 98.6 mg, 0.13 mmol, 41.7%). Elemental analysis [Ni(LH)2](BF4)2 (C36H32N2B2F8NiP2), calcd: C 54.95; H 4.10; N 3.56, found: C 54.73; H 4.20; N 3.56. 700 MHz 1H-NMR (δ/ppm in MeCN-d3) δ = 2.12 (s); 4.79 (br); 7.29–7.41 (m); 7.67 (br); 8.07 (s); 8.42 (br), 282 MHz 31P{1H}-NMR (δ/ppm in MeCN-d3) δ = 44.9 (br).Preparation of [NiII(LH)2H]+
The ligand LH (5.54 g, 20.0 mmol) dissolved in THF (10 mL) was added dropwise under anaerobic conditions to Ni(COD)2 (solid, 2.75 g, 10.0 mmol) and then cooled at −78 °C and stirred for 30 minutes. To the solution was added 10 mL of THF solution of NH4PF6 (1.67 g, 10.0 mmol) dropwise, and then a dark orange solution was obtained. Its ESI MS spectrum was measured in the positive mode. The same method was carried out to prepare the complex for NMR measurements at −90 °C, using THF-d8 as solvent. Its 1H NMR spectrum was obtained at room temperature. ESI MS: [NiII(LH)2H]+ 613.49; 1H NMR: (δ/ppm in THF-d8) δ = −23.9. Note that confirmation of the formation of Ni–H− by electrochemical or chemical two-electron reduction of the [NiII(LH)2](BF4)2 complex at low temperature and successive addition of AcOH was attempted, but was not obtained.Measurements
NMR spectra were measured using a JEOL JNM-ECZ-700 NMR spectrometer, for 1H NMR, in the range −4 to 14 ppm (diamagnetic range) and −50 to 50 ppm (paramagnetic range), and for 31P{1H}-NMR, in the range −120 to 120 ppm. UV-vis absorption spectra were recorded using a JASCO U-best V-720 spectrometer in the range 230–1100 nm, with the sample concentration adjusted to 1 mM. ESI-MS spectra were measured using a Micromass LCT ESI-TOF mass spectrometer.For X-ray crystallography, single crystals with a size of 0.1–0.2 mm were used to collect diffraction data, and measurements were performed at −100 °C with the crystals fixed with grease on a glass fiber. The measurements were carried out using a Rigaku VariMax RAPID II diffractometer, and the lattice parameters were analyzed by the direct method (SIR 2008) using diffraction points with appropriate intensities in the range of 2θ < 55° and refined by the least-squares method. The structural parameters of four-coordinate complexes, τ4 values,19 were calculated using the two largest bond angles.
Elemental analysis was carried out on samples purified and isolated by recrystallization using a Vario EL cube CHNS elemental analyzer. After a gas blank measurement prior to sample measurement, approximately 2 mg of the sample sealed in approximately 40 mg of silver foil was measured twice. The data were corrected with the acetanilide standard material for elemental analysis to obtain the mass content (%) of C, H, N and S elements.
CV and DPV measurements were performed using a HOKUTO DENKOH SV-110 electrochemical analyzer. Glassy carbon (d = 1 mm) was used as working electrode, Ag/Ag+ (MeCN) as the reference electrode, Pt coil as the counter electrode, and TBAP ([(n-Bu)4N](ClO4)), 0.1 M in MeCN) as the electrolyte. Deaeration was performed by bubbling with Ar for 20 min before the measurements to maintain anaerobic conditions in the measurements, and argon flow was maintained during the measurements. The solvent was MeCN, the concentration of the complex was 1 mM, and the solution was unified to 3 mL. All potentials given in this work were quoted relative to the ferrocene/ferricenium (Cp2Fe/Cp2Fe+) couple, whose potential was measured in the cell at the end of each experiment. All potentials are presented based on Cp2Fe/Cp2Fe+. The results were taken from the steady state, where the cycles were repeated and the waveform no longer changed.
Caution
Perchlorate used as an electrolyte is toxic and can be explosive upon impact, so it should be handled with care.Computational details
Density functional theory (DFT) calculations were performed using the B3LYP functional30a with D3 version of Grimme's dispersion.30b The solvent effect of MeCN on structures and thermodynamic properties was evaluated using the polarized continuum model (PCM).31 The geometry optimizations and the frequency calculations were carried out using the basis sets used for the respective atoms as follows: 6-311G for Ni,32a,b P,32c and N32d atoms and 6-31G(d)32e,f for C and H atoms. Two p functions scaled by 1.532a were added as 4p orbitals on Ni atoms. Single-point energy calculation using the def2-TZVP basis set33 at each optimized structure was performed to reduce the basis set superposition error (BSSE) on the Ni centre bound to the hydride and explicit MeCN molecules. The Gibbs free energy of each species at ambient temperature was determined by adding the thermal correction derived from frequency calculation to the single-point energy.Both high- (S = 1) and low-spin (S = 0) states were examined for all Ni complexes. The initial structure of geometry optimization for [NiII(LH)2]2+ was taken from the crystal structure. [NiII(LH)2]2+ in MeCN was considered as a di-MeCN complex, [NiII(LH)2(MeCN)2]2+, with six-coordinate octahedral geometry, which has MeCN molecules at both axial positions. The stability of [NiII(LH)2(MeCN)2]2+ was also examined using the solvation energy of MeCN. The solvation energy for two equiv. of MeCN was calculated for the reaction (eqn (13))
| [NiII(LH)2]2+(low-spin) + 2MeCN ⇄ [NiII(MeCN)2(LH)2]2+ | (13) |
The low-spin desolvated complex, which is more stable than the high-spin complex (Table 3), is considered as the reactant complex of the solvation.
The Gibbs free energy of the reaction (eqn (13)) in MeCN solution, ΔG0′, is described as eqn (14),
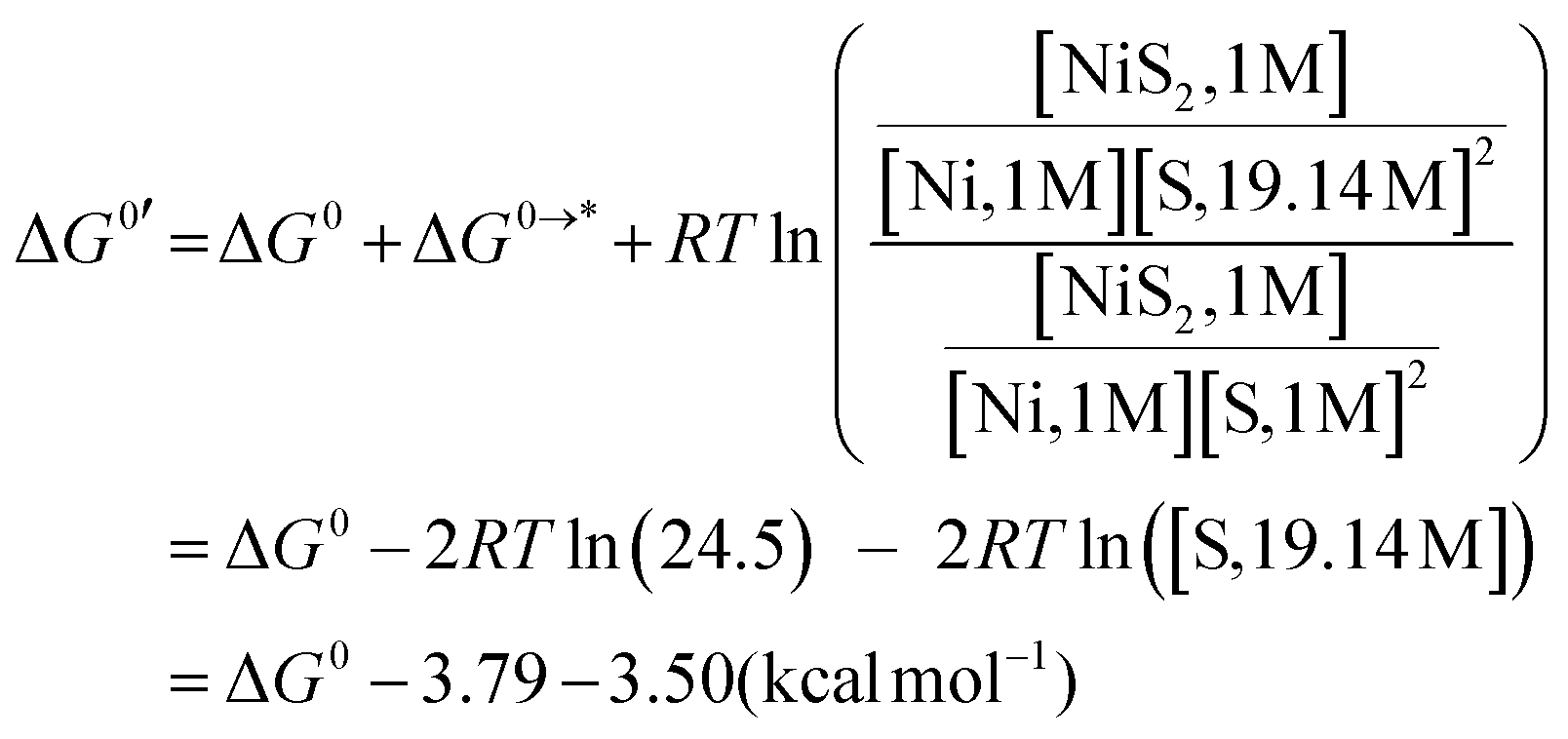 | (14) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(24.5) = 3.79 kcal mol−1. From a density of 0.7857 g cm−3 at 293.15 K, the mole of MeCN in 1 L is found to be 19.14.35 ΔG0s were also calculated for optimized structures obtained by calculations without empirical dispersion to estimate solute–solvent interactions.
ln(24.5) = 3.79 kcal mol−1. From a density of 0.7857 g cm−3 at 293.15 K, the mole of MeCN in 1 L is found to be 19.14.35 ΔG0s were also calculated for optimized structures obtained by calculations without empirical dispersion to estimate solute–solvent interactions.
Ni(0) and hydride Ni(II) complexes, [Ni0(LH)2]0 and [NiII(LH)2H]+, were calculated as the assumed products of the two-electron reduction of [NiII(LH)2]2+ and subsequent protonation process, respectively. The initial structure of the Ni(0) complex was the optimized structure of the low-spin Ni(II) complex without MeCN molecules because the neutral metal centre is not attractive enough to directly coordinate solvent molecules. In addition, the three-coordinate structure with a free pyridyl group was calculated to examine the stability of the four-coordinate structure. The structural optimization of hydride Ni(II) complexes was carried out using two different initial structures. One was the optimized tetrahedral Ni(0) complex with a proton on the Ni centre from a direction perpendicular to the chelate ring, and the other was the optimized low-spin square planar Ni(II) complex with the hydride at the apex position. First, the low-spin hydride Ni(II) complexes suggested by the aforementioned NMR spectra were examined. The N and P donor atoms and hydride ligands are available as apex atoms in the square pyramidal complex. Moreover, there are two different orientations of the basal LH ligand for each apex donor atom, giving six possible initial structures. However, four of the optimized hydride Ni(II) isomers could be distinguished from the other optimized isomers by the transformation of the initial structure with a hydride ligand at the apex to an N-apex enantiomer during geometry optimization (Fig. S21, ESI†). We also calculated geometries of the four isomers in the high-spin state and mono-MeCN solvated counterparts, where a MeCN ligand fills the opposite site to the apex donor to form a feasible octahedral geometry. In addition, geometry optimization of mono-MeCN solvated low-spin complexes was performed using the optimized structure of the six-coordinate high-spin complex as the initial structure.
The solvation energy of the hydride Ni(II) complex for reaction (eqn (15)) was calculated using eqn (16).
| [NiII(LH)2H]+ + MeCN ⇄ [NiII(LH)2H(MeCN)]+ | (15) |
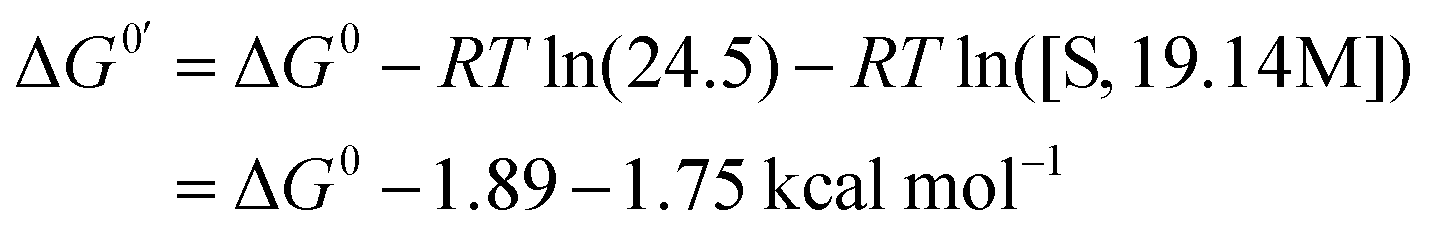 | (16) |
1H NMR chemical shift of the hydride ligand in the four low-spin hydride Ni(II) isomers in THF was evaluated with NMR shielding tensors calculated using the Gauge-Independent Atomic Orbital (GIAO) method36 with the def2TZVP basis set. Atomic charges in the hydride complexes were calculated using natural population analysis.37
Thermodynamic energy diagrams38 were constructed from reduction and protonation steps for plausible catalytic mechanisms of [NiII(LH)2]2+ and [NiII(LNH2)2]2+ systems. Solvation energies of all the intermediates with explicit solvent molecules were calculated in the same manner as [NiII(LH)2H]+ and [NiII(LH)2]2+ for mono- and disolvation, respectively. Possible binding sites of solvent molecules in the intermediates of [NiII(LNH2)2]2+ systems are the axial positions of a square-planar P2N2 coordination structure, the empty site left by a pyridyl group, and a proton of ammonium or pyridinium groups to form a hydrogen bond. The electrostatic potential generated using the default scaling factor for the van der Waals radius, α = 1.1, in the PCM calculation is too sensitive to geometrical change to optimize the structure of [NiII(LH)2]2+H+·S2 (S = MeCN). Thus, α = 1.2, which reduces the generated potential, was used for the geometry optimization and frequency calculation to obtain the thermochemical correction of the free energy and α = 1.1 was used for the energy calculation at the optimized structure in the case of [NiII(LH)2]2+H+·S2. Calculated solvation energies are listed in Table S4 (ESI†), and calculated solvation structures are shown in Fig. S24 and S25 (ESI†) for [NiII(LH)2]2+ systems. Only the disolvation model for protonated Ni(II) and Ni(I) complexes with a leaving pyridinium group in [NiII(LH)2]2+ systems, [Ni(II/I)]2+/+H+, was calculated because the protonated analogues of [NiII(LNH2)2]2+ systems are considerably lower in energy than that with a coordinated pyridyl group over 10 kcal mol−1 (Table S4, ESI†). The positive solvation energies of deprotonated species suggest that their P2N2 chelation structures are stable against ligand-dissociation and exchange processes except for [Ni0(LH)2]0, which has a coordinated MeCN molecule replaced with a leaving pyridyl group (Fig. S24, ESI†). In contrast, the negative disolvation energies of protonated species, except for the protonated hydride complex [Ni(II)H]+H+, have one MeCN molecule H-bonded to the leaving pyridinium group and the other bound to the left site (Fig. S25, ESI†). Thus, the reasonable solvation models are desolvation for deprotonated complexes and hydride complexes except for monosolvation of Ni(0), [Ni0(LH)2]0, and disolvation for protonated complexes except for desolvation of protonated hydride complexes. The reduction potential and pKa values derived from free energies of reduction and protonation steps were calculated between reactant and product complexes with the most exergonic solvation as discussed above. The reduction potentials relative to the reference electrode, the ferrocene/ferrocenium couple, were calculated from the Gibbs free energies of the cell reactions coupled with the ferrocene oxidation reaction. The pKa values were calculated from the Gibbs free energies of the reaction of the bases with the proton source, AcOH, and the absolute pKa values were obtained from adding the pKa value of AcOH in MeCN (22.3). The basis set used for geometry optimization of ferrocene and ferrocenium complexes was 6-311G augmented with two p functions for Fe atoms like Ni atoms and 6-311G(d) for the other atoms, and that of an AcOH molecule and AcO− anion was 6-311G(d) for the O atom and 6-31G(d) for the other atoms. H2 at 1 atm in the gas phase was considered for the free energy of the H2 elimination reactions.
All the electronic structure calculations were performed using Gaussian 16, revision C.01,39 and the molecular orbitals were drawn using the MOPLOT and MOVIEW programs40 on the HPE ProLiant DL560 system at the Nagoya University Information Technology Centre. The natural population analyses were performed using the NBO7 program41 in Gaussian 16, revision C.01,39 in Research Centre for Computational Science, Okazaki (Project: 23-IMS-C24).
Conclusions
We have previously focused on the roles of the amino group in [FeFe]-hydrogenase, which is considered to be a proton-transferring site, and have synthesized the phosphinopyridine ligand with an amino group in the second coordination sphere of the metal complex and the Ni(II) complex thereof, [Ni(LNH2)2](BF4)2, and examined its hydrogen-producing ability. We have demonstrated that it has a very high hydrogen production capability. In this study, as a comparison, we synthesized the Ni(II) complex of the phosphinopyridine ligand without the amino group, [NiII(LH)2](BF4)2 (1), and studied its hydrogen production ability. Complex 1 is a four-coordinate square planar geometry with only a cis-isomer in the crystal structure, and the structure in the solution state was also revealed by UV-vis and NMR spectra. The crystal structure is a four-coordinate square planar structure in the cis-form and is similar to that of [Ni(LNH2)2](BF4)2 reported previously.18b However, in solution it was slightly different: in dichloromethane it took a four-coordinate square planar structure, whereas in acetonitrile solution it formed a five- or six-coordinate structure with weakly bound MeCN molecules, according to the spectral features. The electrocatalytic current of complex 1, indicative of its ability to produce H2, increased with increasing amounts of AcOH as the proton source under the same conditions as those for the previously reported [Ni(LNH2)2](BF4)2 complex.18b The catalytic current increased in proportion to the square root of the drop of AcOH, in a similar manner to that of the previously reported [Ni(LNH2)2](BF4)2 complex. However, as expected, the TOF in electrochemical hydrogen reduction was reduced and the overpotential increased compared to the case of [Ni(LNH2)2](BF4)2. This clearly indicates that the rate-determining step in the catalytic cycle is affected by the amino groups in the second coordination sphere of [Ni(LNH2)2](BF4)2. It was also found that an amino group in the complex system, which is a proton-transferring moiety, improves the catalytic turnover frequency (TOF) and contributes to the reduction of the overpotential. The above findings were explained and discussed based on the results evaluated from the DFT calculations, and the hydrogen generation mechanism was also proposed based on these findings. The DFT calculations supported the EECC mechanism for both systems with and without amino groups. These results suggest that the amino group of the ligand stabilizes the protonated intermediate, which causes the pyridyl group to leave in order to capture the proton. Thus, the electron-donating properties of the amino group help in the capture of protons from the reduced species. The results obtained in this study show not only the significance of the amino group in the [FeFe]-hydrogenase enzyme, but also the role of the amino group introduced in the second coordination sphere in the previously reported [Ni(LNH2)2] complex as the functional model. In the future, these results will be applied to develop catalysts for more efficient hydrogen production.Data availability
Experimental details, UV-vis spectral data and 1H and 31P{1H} NMR spectra of Ni(II) complexes, CVs and scan rate dependence of the complex, DFT calculation results, and X-ray data (PDF).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Culture, Sports, Science, and Technology, Japan (MEXT/JSPS KAKENHI Grant Number (B) 20H02752 for H. M. and 17K05808 and 22K05191 for T. O., respectively) and the Yashima Environment Technology Foundation also provided in part financial support, which is hereby gratefully acknowledged.Notes and references
- L. García, Compendium of Hydrogen Energy, Hydrogen Production and Purification, Woodhead Publishing Series in Energy, 2015, p. 83 Search PubMed.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shahd and K. R. Ward, Energy Environ. Sci., 2019, 12, 463 RSC.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933 RSC.
- (a) J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS; (b) K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366 CrossRef CAS PubMed; (c) M. Frey, ChemBioChem, 2002, 3, 153 CrossRef CAS; (d) C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245 CrossRef CAS PubMed.
- (a) H. Ogata, K. Nishikawa and L. Wolfgang, Nature, 2015, 520, 571 CrossRef CAS; (b) H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577 RSC.
- (a) M. E. Ahmed, S. Dey, S. M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 1245 Search PubMed; (b) I. Bhugun, D. Lexa and J. M. Saveant, J. Am. Chem. Soc., 1996, 118, 3982 CrossRef CAS; (c) S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033 CrossRef CAS; (d) M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310 CrossRef CAS PubMed.
- (a) S. Wiese, U. J. Kilgore, D. I. DuBois and R. M. Bullock, ACS Catal., 2012, 2, 720 CrossRef CAS; (b) A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5, 1397 CrossRef CAS; (c) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863 CrossRef CAS PubMed; (d) W. A. Hoffert, J. A. S. Roberts, R. M. Bullock and M. L. Helm, Chem. Commun., 2013, 49, 7767 RSC; (e) L. Gan, T. L. Groy, P. Tarakeshwar, S. K. S. Mazinani, J. Shearer, V. Mujica and A. K. Jones, J. Am. Chem. Soc., 2015, 137, 1109 CrossRef CAS.
- (a) V. Kaim and S. Kaur-Ghumaan, Eur. J. Inorg. Chem., 2019, 5041 CrossRef CAS; (b) S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54 CrossRef CAS; (c) H. Hou, H. T. Poh and Y. Fan, Chem. Commun., 2014, 50, 6630 RSC; (d) M. D. Sampson and C. P. Kubiak, Inorg. Chem., 2015, 54, 6674 CrossRef CAS PubMed; (e) V. Kaim, M. Natarajan and S. Kaur-Ghumaan, Chemistry Select, 2019, 4, 1789 CAS.
- (a) T. Straistari, R. Hardré, J. Fize, S. Shova, M. Giorgi, M. Regliér, V. Artero and M. Orio, Chem. – Eur. J., 2018, 24, 8779 CrossRef CAS; (b) N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274 CrossRef CAS; (c) M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786 CrossRef CAS; (d) J. W. Jurss, R. S. Khnayzer, J. A. Pantier, E. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954 RSC; (e) G. M. Jacobsen, J. Y. Yang, B. Twamley, A. D. Wilson, R. M. Bullock, M. R. DuBois and D. L. DuBois, Energy Environ. Sci., 2008, 1, 167 RSC; (f) X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723 RSC.
- (a) H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5, 5145 CrossRef CAS; (b) K. Sudhakar, A. Mahammed, Q.-C. Chen, N. Fridman, B. Tumanskii and Z. Gross, ACS Appl. Energy Mater., 2020, 3, 2828 CrossRef CAS; (c) S. Nestke, M. Kügler, J. Scholz, M. Wilken, C. Jooss and I. A. Siewert, Eur. J. Inorg. Chem., 2017, 3376 CrossRef CAS; (d) S. Majumder, A. A. Haleem, P. Nagaraju and Y. H. Naruta, Dalton Trans., 2017, 46, 9131 RSC; (e) D. Khusnutdinova, B. L. Wadsworth, M. Flores, A. M. Beiler, E. A. R. Cruz, Y. Zenkov and G. F. Moore, ACS Catal., 2017, 8, 9888 CrossRef.
- (a) H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. A. Chang, Science, 2012, 335, 698 CrossRef CAS PubMed; (b) H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329 CrossRef CAS PubMed.
- M. P. Stewart, M.-H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033 CrossRef CAS PubMed.
- S. Horvath, L. E. Fernandez, A. M. Appel and S. Hammes-Schiffer, Inorg. Chem., 2013, 52, 3643 CrossRef CAS PubMed.
- (a) Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659 CrossRef CAS PubMed; (b) Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667 CrossRef CAS PubMed.
- C. N. Virca and T. M. McCormick, Dalton Trans., 2015, 44, 14333 RSC.
- K. Majee, J. Patel, S. Rai, B. Das, B. Panda and S. K. Padhi, Phys. Chem. Chem. Phys., 2016, 18, 21640 RSC.
- K. Koshiba, K. Yamauchi and K. Sakai, ChemElectroChem, 2019, 6, 2273 CrossRef CAS.
- (a) R. Tatematsu, T. Inomata, T. Ozawa and H. Masuda, Angew. Chem., Int. Ed., 2016, 55, 5247 CrossRef CAS PubMed; (b) T. Kato, R. Tatematsu, K. Nakao, T. Inomata, T. Ozawa and H. Masuda, Inorg. Chem., 2021, 60, 7670 CrossRef CAS PubMed.
- τ4 is defined as a simple geometry indicator for quantitatively evaluating the geometry in a four-coordinate complexes and compounds. For square-planar, τ4 = 0 and for tetrahedral, τ4 = 1, L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- H. W. Haase, Z. Anorg. Allg. Chem., 1974, 404, 273–283 CrossRef.
- (a) M. O. Workman, G. Dyer and D. W. Meek, Inorg. Chem., 1967, 6, 1543 CrossRef CAS; (b) G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181 CrossRef CAS PubMed; (c) C. Costentin and J.-M. Saveant, ChemElectroChem, 2014, 1, 1226 CrossRef CAS; (d) S. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Comprehensive Coordination Chemistry II, Pergamon Press, 1987, vol. 5, p. 65 Search PubMed.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, New York, 2nd edn, 2001 Search PubMed; (b) R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706 CrossRef CAS; (c) J. M. Saveant and E. Vianello, Electrochim. Acta, 1965, 10, 905 CrossRef CAS.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, New York, 2nd edn, 2001 Search PubMed; (b) R. S. Nicholson and I. Shain, Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems, Anal. Chem., 1964, 36, 706–723 CrossRef CAS; (c) J. M. Saveant and E. Vianello, Potential-sweep chronoamperometry: Kinetic currents for first-order chemical reaction parallel to electron-transfer process (catalytic currents), Electrochim. Acta, 1965, 10, 905–920 CrossRef CAS.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338 CrossRef CAS PubMed.
- (a) M. Aaron, M. Appel and L. M. Helm, ACS Catal., 2014, 4, 630 CrossRef; (b) J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 7, 3823 CrossRef PubMed.
- O. H. Ryan, C. Suk-Won, C. Whitney and B. P. Fritz, Fuel Cell Fundamentals, John Wiley & Sons’ Inc., 2016, vol. 35, p. 452 Search PubMed.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116(15), 8373–8426 CrossRef CAS PubMed.
- W. L. Jorgensen, D. Lim and J. F. Blake, J. Am. Chem. Soc., 1993, 115, 2936–2942 CrossRef CAS.
- D. S. Moore and S. D. Robinson, Chem. Soc. Rev., 1983, 415–452 RSC.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 52, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- (a) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS; (b) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef; (c) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (d) K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef; (e) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (f) P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Lewis, J. A. Bumpus, D. G. Truhlar and C. J. Cramer, J. Chem. Educ., 2004, 81, 596–603 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton FL, 90th edn, 2010 Search PubMed.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- (a) H. Wasada and Y. Tsutsui, Bull Coll. Gen. Edu. Gifu Univ., 1996, 33, 145–158 CAS; (b) I. Takahashi, H. Wasada and Y. Tsutsui, MOVIEW: Program of Nagoya University Information Technology Centre Representing Molecular Orbitals and Electron Density Maps by Isosurfaces Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0.7, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2019.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV-vis spectral data and 1H and 31P{1H} NMR spectra of Ni(II) complexes, CVs and scan rate dependence of the complex, DFT calculation results, and X-ray data (PDF). CCDC 2302422 for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ya00345d |
| This journal is © The Royal Society of Chemistry 2024 |