
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriethanolamine-assisted surface reconstruction of nickel oxide for efficient oxygen evolution reaction†
Jiayun
Zhang
a,
Ruth
Knibbe
 *b and
Ian
Gentle
*a
*b and
Ian
Gentle
*a
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: i.gentle@uq.edu.au
bSchool of Mechanical and Mining Engineering, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: ruth.knibbe@uq.edu.au
First published on 26th September 2024
Abstract
Developing low cost and highly efficient electrocatalysts for the oxygen evolution reaction (OER) is highly desired for renewable energy production. Ni-based electrocatalysts have been widely investigated as candidates for the OER, but developing a low-cost, easily synthesized electrocatalyst with high activity and good stability remains elusive. Herein, we report the facile electrodeposition of triethanolamine-decorated Ni oxide on carbon paper (Ni/CP-TEA) as an efficient electrocatalyst for water oxidation. Structural and experimental analyses reveal that the electrode surface is modified by triethanolamine (TEA) through Ni–N coordination bonding. The leaching of TEA drives rapid in situ surface reconstruction, facilitating the generation of high-valence Ni (Ni3+) species, thereby accelerating the OER performance. The Ni/CP-TEA exhibits enhanced electrocatalytic OER performance with a low overpotential of 320 mV at 10 mA cm−2 and good long-term stability. This work presents a simple route for the rational design of cost-effective and highly efficient OER catalysts.
Introduction
Renewable energy sources are crucial for meeting growing energy demands and addressing environmental concerns.1,2 Among the many emerging technologies, electrocatalytic water splitting stands out as an efficient process for green hydrogen production.3,4 However, the oxygen evolution reaction (OER), a key component of water splitting, suffers from intrinsically sluggish kinetics, limiting the efficiency and practical application of water electrolysis. To date, Ru/Ir-based oxides are the benchmark OER electrocatalysts;5,6 however their cost, scarcity and unsatisfactory durability have hindered their widespread application.7 Significant efforts have been devoted to developing highly efficient and cost-effective OER electrocatalysts.8–10Earth-abundant Ni-based oxides have received considerable interest because of their promising OER performance and stability.11–18 However, the catalytic mechanism of Ni-based OER catalysts remains a topic of scientific debate.19 It has been demonstrated that during the OER under alkaline conditions, the surfaces of numerous transition-metal based catalysts including chalcogenides, nitrides, and phosphides undergo phase transition and reconstruction processes, transforming into the corresponding oxides.20–23 Generally, these in situ-generated higher valence oxide species display enhanced activities21,22 and are regarded as one of the reasons for the excellent catalytic activity.24,25 Therefore, more efforts to explore surface reconstruction and understand the catalytic origins are essential for the rational design of OER electrocatalysts.
Although binary or ternary metal-based compounds, such as S–FeCoNi LDH,26 N–NiMoO4 (ref. 27) and Fe–NiSOH (ref. 25) and others have been investigated, the structural complexity of these materials makes it difficult to gain a deeper mechanistic understanding. Also, strategies for fabricating these materials – including hydrothermal, solvothermal, and chemical vapor deposition synthesis methods – are complex and/or require high energy. A simple and effective synthesis strategy for a low-cost efficient electrocatalyst with high stability is desired.
Metal coordination complexes have received extensive attention as a class of OER electrocatalysts. It has been demonstrated that organic ligands can be electrochemically extracted under anodic potential, leaving the inorganic oxides as the active species.28 For example, Ni complexes can rapidly generate active NiOx species under an oxidation voltage.29 The tannin–NiFe complex film on carbon fibre paper showed much larger mass activity compared to the NiFe double layered hydroxide (LDH). The organic ligand tannic acid in tannin–NiFe was extracted under anodic potential, leaving behind NixFe1−xOyHz as the OER-active species.28 If the surface of Ni oxides is modified with a complexing agent, which can be easily removed under anodic potential, it could potentially facilitate the surface reconstruction process and promote the formation of high valence Ni species for the OER. Triethanolamine (TEA) is a ligand that is frequently used as a precipitant in the synthesis of Ni-based LDH electrocatalysts, along with other compounds.30–32 During sample preparation, TEA can also help modify the catalysts’ morphology.31,33 When introduced to the electrolyte in the electrodeposition process, TEA may modulate the morphology through forming Ni-complexes, which have a different deposition rate to the Ni ions. Additionally, TEA may coordinate with Ni through its N group and remain on the surface after synthesis, potentially enhancing the oxidation and reconstruction process under oxidation potential.
In the present work, we prepared a TEA-modified Ni oxide (Ni/CP-TEA) electrocatalyst, introducing Ni–N coordination that influences both the morphology and chemical structure of the electrocatalyst, which exhibits efficient OER activity. Structural and activity analysis of both fresh and post-reaction samples reveals that the TEA modifies the morphology and induces increased active surface area. The leaching of the TEA from the surface accelerates the in situ surface reconstruction process and promotes the generation of higher valence Ni species, thereby enhancing the OER performance.
Experimental
Chemicals and materials
NiSO4·6H2O (>99%) and triethanolamine (TEA) were purchased from Sigma-Aldrich and used as received. Potassium hydroxide (99% KOH, analytical grade, Fe content < 0.0003%) was purchased from Chem-Supply Pty Ltd. Carbon paper (CP, AvCarb carbon paper (P50)) was obtained from Fuel Cell Store.Preparation of Ni/CP-TEA and Ni/CP
Carbon paper (CP) was first cut into 1 × 1 cm pieces and subsequently treated in an oven at 250 °C for 2 h under Ar. Then the CP was ultrasonically cleaned in ethanol and deionized (DI) water for 10 min, respectively, and dried in an oven at 50 °C overnight. As shown in Scheme 1, the samples were synthesized using electrodeposition. The deposition electrolyte was prepared by dissolving NiSO4·6H2O (0.1 M) and TEA (5 mM) into 30 mL DI water. The pre-treated CP was used as the working electrode. Pt-mesh and an Hg/HgO (0.1 M KOH) electrode were used as the counter and reference electrode, respectively. Electrodeposition of Ni-TEA film on CP was carried out at −1.7 V vs. Hg/HgO for 5 min at room temperature. The final electrode was washed several times with ethanol and DI water and dried at room temperature for at least 12 hours. Meanwhile, Ni/CP-TEAx (x = 1, 3, 7, 9) was obtained by changing the TEA concentration in the deposition electrolyte to 1, 3, 7 and 9 mM. The Ni/CP sample was prepared using the same synthesis method but without the TEA electrolyte additive. To assess the impact of other additives, NH3·H2O, ethylenediaminetetraacetic acid (EDTA, [CH2N(CH2CO2H)2]2) (5 mM) and ethanol were used instead of TEA as the electrolyte additive yielding samples named Ni/CP–NH3, Ni/CP-EDTA and Ni/CP–ethanol, respectively.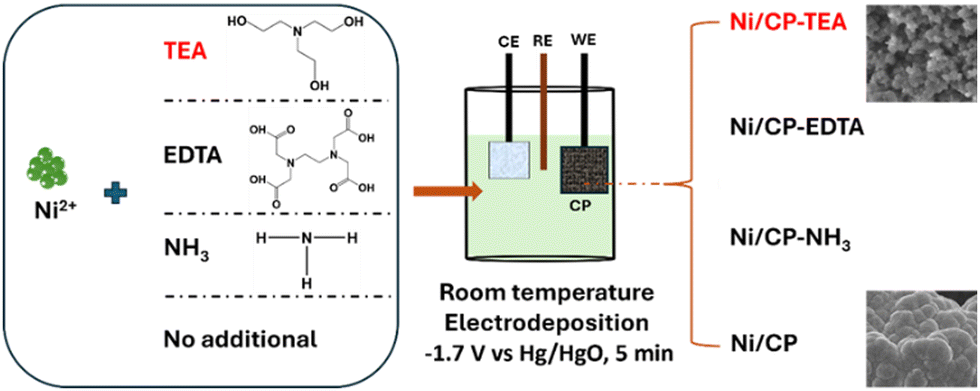 | ||
| Scheme 1 A schematic illustration of the preparation of Ni/CP and Ni/CP-TEA, Ni/CP-EDTA and Ni/CP–NH3 by adding TEA, EDTA and NH3·H2O in the Ni2+ solution, respectively. | ||
Catalyst characterization
The field emission scanning electron microscopy (FESEM) images were obtained on a JEOL JSM-7001F microscope operating at 10 kV. Transmission electron microscopy (TEM) and energy dispersive X-ray (EDX) spectroscopy mapping studies were performed on a Hitachi HF5000 at an accelerating voltage of 60 kV with Oxford dual 100 mm2 SDD EDX* detectors and a Hitachi HT7700 B operating at 80 kV. For TEM sample preparation, the electrocatalyst was scraped off the electrode surface and dispersed on the copper TEM grid (300 mesh). X-ray powder diffraction (XRPD) analysis was carried out on a Bruker D8 Advance MKII XRD using Cu Kα radiation (λ = 1.54 Å). Fourier-transform infrared (FTIR) spectra were collected using a PerkinElmer Spectrum 100 FTIR spectrometer. X-ray photoelectron spectrometry (XPS) was measured using a Kratos Axis Supra+ XPS instrument with a monochromatic Al Kα X-ray source (1486.6 eV). The C 1s peak at 284.8 eV was used for binding energy charge-correction and CasaXPS software was used for XPS spectra analysis. For XPS depth profiling analysis, the sample was etched using an Ar ion cluster beam at a 5 keV beam energy and a 2 × 2 mm raster size. With these etching parameters, the etch rate was about 4 nm min−1.Electrochemical measurements
Electrochemical tests were performed in a three-electrode setup connected to a BioLogic (VNP-300) potentiostat at room temperature. A Hg/HgO (0.1 M KOH) reference electrode and a Pt-mesh counter electrode were used. The as-prepared sample was used as the working electrode.Linear sweep voltammetry (LSV) measurements were performed at 5 mV s−1 in a 0.1 M KOH electrolyte. Four CV cycles were conducted before conducting the LSV. The potentials reported in this work are converted to the reversible hydrogen electrode (RHE) using the conversion equation: E(RHE) = E(Hg/HgO) + 0.0591 × pH + 0.165 V, unless stated otherwise. All electrochemical experiments were done without iR-correction. The electrochemical surface area (ECSA) was measured by the double-layer capacitance (Cdl, mF) of the catalytic surface according to the equation ECSA = Cdl/Cs.29 The Cdl was determined from scan-rate dependent cyclic voltammograms (CVs) at the non-Faradaic region, measured at scan rates of 20, 40, 60, 80, and 100 mV s−1 between 0.1–0.2 V Hg/HgO. Cs is the specific capacitance value and the value of 0.04 mF cm−2 was used in this work.30 To evaluate the durability of the electrocatalyst, chronopotentiometric measurements were conducted at a constant current density of 10 mA cm−2.
Results and discussion
A simple electrodeposition procedure was used to prepare the Ni oxide electrocatalyst (Ni/CP-TEA), using a solution containing NiSO4·6H2O and triethanolamine (TEA) as the deposition electrolyte. Morphological analysis of the prepared samples was further conducted using field emission scanning electron microscopy (FESEM). As shown in Fig. 1a and Fig. S1a (ESI†), the Ni/CP-TEA has a coral-like structure. The Ni/CP (Fig. 1b) exhibits a rough surface made up of aggregated nanoparticles, averaging 1.07 ± 0.44 μm in size. X-ray diffraction (XRD) characterization was performed to explore the phase information of the as-prepared samples. As shown in Fig. 1c, the XRD pattern of Ni/CP displays characteristic diffraction peaks corresponding to the {111} and {200} crystal planes of metallic Ni (PDF#01-071-4655) as well as those of the CP substrate. In contrast, no obvious Ni or NiO diffraction peaks can be observed in the pattern for the Ni/CP-TEA sample. This could be due to a lower degree of Ni or NiO crystallinity or the CP substrate signals being too strong.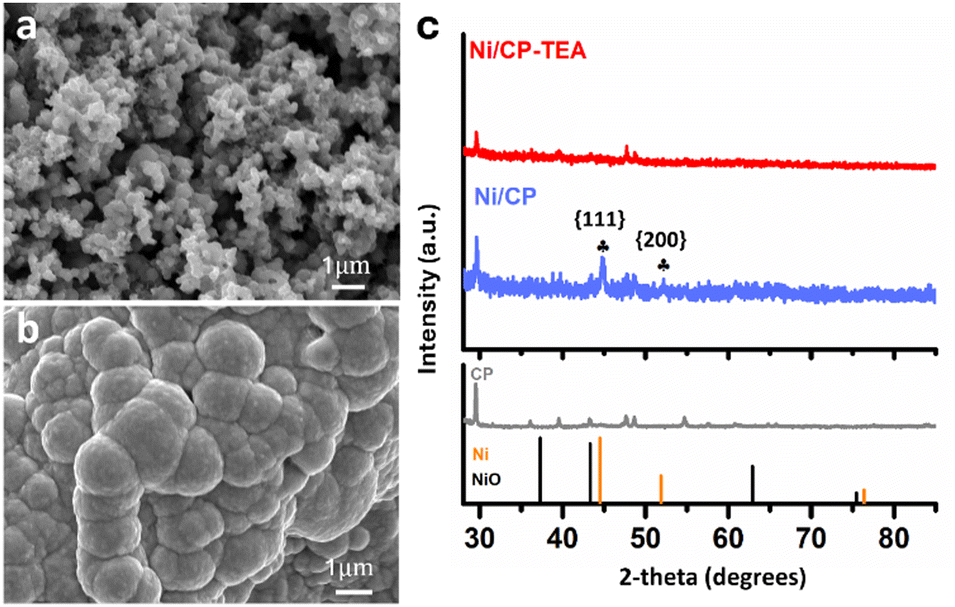 | ||
| Fig. 1 FESEM images of (a) Ni/CP-TEA and (b) Ni/CP. (c) XRD patterns of Ni/CP-TEA, Ni/CP and CP. NiO: PDF# 00-004-0835; Ni: PDF# 01-071-4655. | ||
Transmission electron microscopy (TEM) was also performed to study the structure. The high-resolution TEM (HRTEM) image of Ni/CP-TEA (Fig. S1b, ESI†) reveals some lattice fringes (marked out by yellow circles), with very small domain sizes. The selected area electron diffraction (SAED) pattern (Fig. S2a and b, ESI†) displays diffuse diffraction rings, which confirms the small domain sizes. The diffraction rings are ascribed to NiO, indicating Ni oxidation when the electrode is exposed to air. Fig. S2c (ESI†) exhibits the dark field STEM image of the Ni/CP-TEA and the related EDX elemental mapping of Ni and O showing uniform distribution of both elements.
The surface chemical composition of the as-prepared Ni/CP-TEA was analysed using XPS as shown in Fig. 2a, in which the main peaks of Ni 2p3/2 and Ni 2p1/2 are displayed. Both Ni/CP-TEA and Ni/CP exhibit Ni2+ doublets, indicating the presence of a Ni oxide surface layer due to exposure to air post-synthesis. Additionally, small metallic Ni doublets (labelled Ni0) are observed in the Ni/CP sample. The Ni2+ peak in the Ni/CP-TEA spectrum has a slightly higher binding energy than that of the Ni/CP (855.9 eV vs. 855.7, Ni 2p3/2). The positive shift indicates that the Ni2+ species in the Ni/CP-TEA have a lower electron density,34 suggesting an electronic structure modification by TEA, which could influence the catalytic properties of the Ni/CP-TEA. This interaction can be verified by the N 1s spectra. As can be seen in Fig. 2b, the N 1s signal is seen as a broad peak in the Ni/CP-TEA, showing features at 399.8 and 401.2 eV, attributed to metal–N (Ni–N) and O–N species,35 respectively. In addition, the main peak at 399.8 eV is negatively shifted compared with that of the TEA (400.3 eV), further suggesting the coordination bond between TEA and the Ni, specifically with the N. As expected, no N is detected in the Ni/CP (Fig. S3, ESI†).
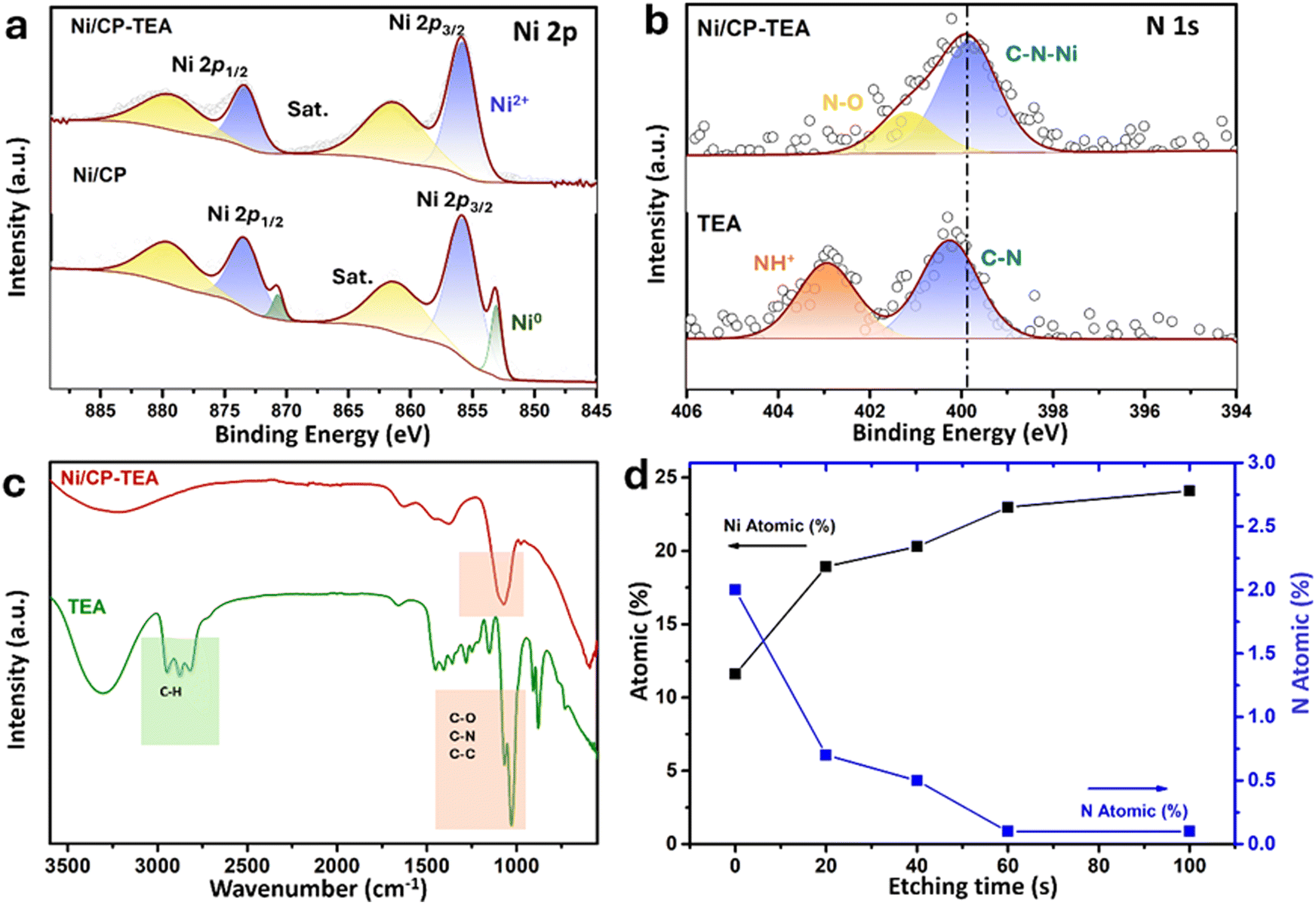 | ||
| Fig. 2 XPS spectra of (a) the Ni 2p region of Ni/CP-TEA and Ni/CP and (b) the N 1s region of Ni/CP-TEA and Ni/CP. (c) FTIR spectra of Ni/CP-TEA and TEA. (d) Surface N and Ni atomic concentration (at%; atom contains: Ni, C, N and O) as a function of etching time of Ni/CP-TEA from XPS depth profile analysis. | ||
This interaction is also verified through Fourier transform infrared (FTIR) spectroscopy of the Ni/CP-TEA, Ni/CP and TEA samples. As shown in Fig. 2c, both the Ni/CP-TEA and the reference TEA samples have a typical broad band around 3300 cm−1, which corresponds to the stretching vibration of the –OH groups.31–33 Notably, the band peaks associated with C–H stretching and C–H bending in the region of 2814–2953 cm−1 and 880 cm−1 respectively,36,37 observed in TEA, cannot be detected in Ni/CP-TEA. The vibrational peaks attributed to C–N/C–C/C–O at around 1028 cm−1 in TEA, are detectable in the Ni/CP-TEA, but show a blue shift of around 50 cm−1 (1070 cm−1). The lack of C–H peaks, coupled with the shift of the C–N/C–C/C–O peak, indicates an interaction between TEA and the Ni.38,39 For Ni/CP (Fig. S4, ESI†), no obvious band can be detected, indicating that the chemical environment of the Ni/CP-TEA is different to that of the Ni/CP.
To evaluate the N location in the Ni/CP-TEA, XPS depth profiling was measured with a total etch duration of 100 s (Fig. 2d and Table S1, ESI†). Before Ar+ etching, the concentrations of Ni and N are 11.6 and 2.0 at%, respectively. During etching, the N content sharply decreased to a trace amount of 0.5 at% after 40 s, corresponding to an estimated etch depth of approximately 2.7 nm. This analysis indicates that the modification exists only at the surface of the Ni/CP-TEA.
The electrocatalytic performance of the samples for water oxidation was explored. A preliminary LSV test (Fig. S5, ESI†) was measured for the Ni/CP-TEAx prepared with different TEA concentrations (1, 3, 5, 7 and 9 mM). It shows that the optimal TEA concentration is 5 mM, exhibiting the highest OER activity, and so all Ni/CP-TEA samples subsequently discussed in this study were synthesized using 5 mM TEA. As shown in the LSV curve (Fig. 3a), the Ni/CP-TEA requires a notably lower overpotential to reach the same current density as that of Ni/CP, demonstrating that the surface modification enhances the OER activity. The bare CP substrate demonstrates negligible OER activity. The Ni/CP-TEA requires an overpotential (η) of about 320 mV to reach a current density of 10 mA cm−2, which is considerably lower than that of Ni/CP (η = 520 mV). This overpotential was determined from subsequent chronopotentiometry, where the overpotential stabilizes for the Ni/CP-TEA. This process is used as the Ni oxidation peak overlaps with the OER process, making it difficult to clearly separate the two in the preliminary LSV test.
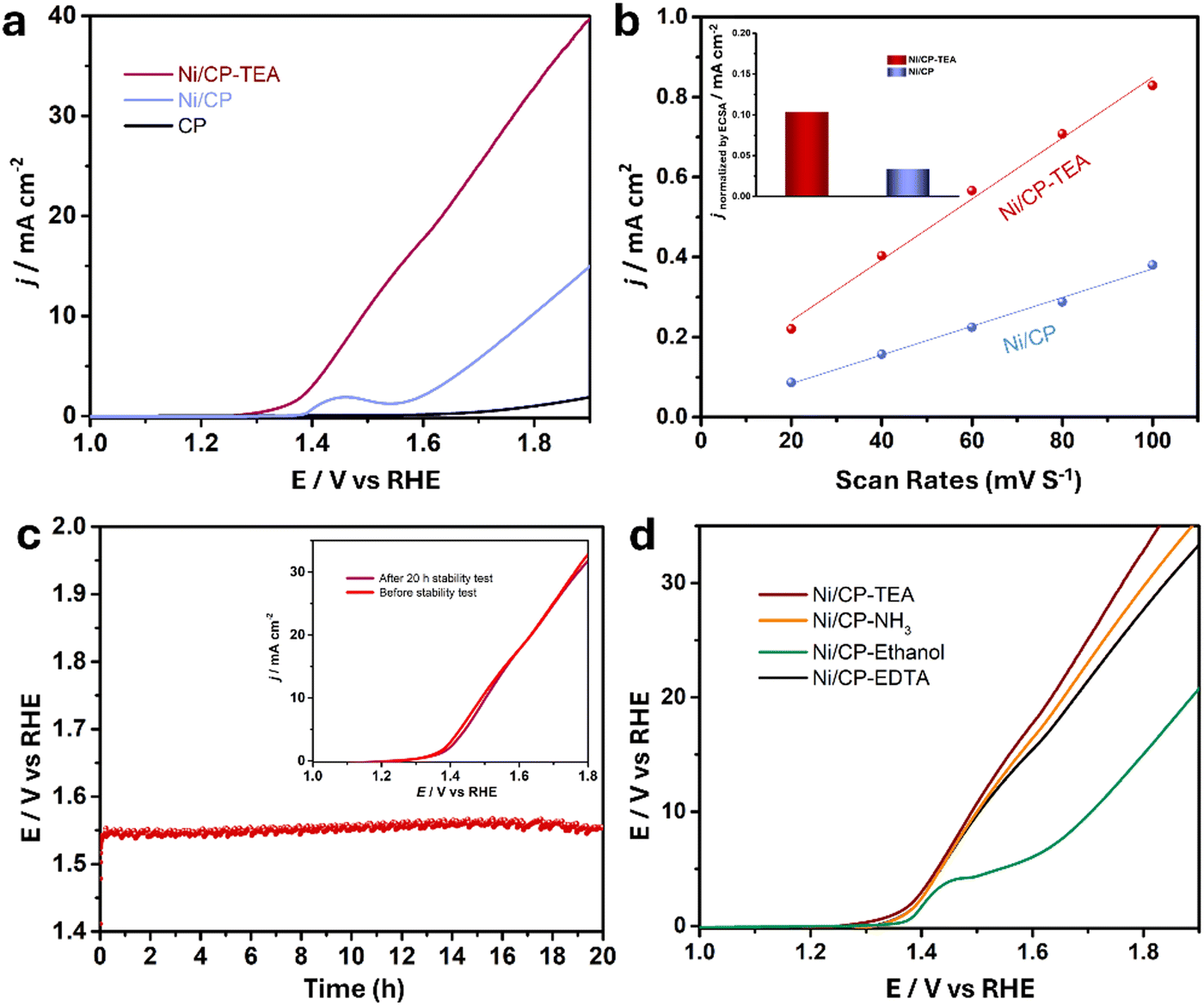 | ||
| Fig. 3 (a) Linear sweep voltammetry (LSV) polarization curve of Ni/CP-TEA, Ni/CP and CP at a scan rate of 5 mV s−1 in 0.1 M KOH. (b) The ratio of current density against different scan rates based on the CV curves. Inset: Comparison of the current density normalized by the ECSA at an overpotential of 400 mV. (c) Chronopotentiometry curve of Ni/CP-TEA at 10 mA cm−2. The inset shows the LSV polarization curves of Ni/CP-TEA before and after the stability test. (d) LSV polarization curve of Ni/CP-TEA, Ni/CP–NH3, Ni/CP-EDTA and Ni/CP–ethanol at 5 mV s−1 in 0.1 M KOH. | ||
The OER performance of the Ni/CP-TEA was also compared with various Ni-based electrocatalysts (Table S2, ESI†). The Ni/CP-TEA exhibits a much lower overpotential (320 mV) to achieve a current density of 10 mA cm−2 compared to the benchmark NiOx (430 mV) and is comparable to the active Ni-based electrocatalysts.
Trotochaud et al. reported that Fe impurities in KOH electrolyte could boost the OER activity of some Ni-based electrocatalysts by the incorporation of Fe-impurities during CV or aging. This was demonstrated by the observations that the Fe 2p XPS signal could be detected even after 5 CV cycles and that the OER performance improved with each CV cycle.40 To clarify the impact of Fe impurities on the Ni/CP-TEA, XPS spectra and CV analysis were conducted. As shown in the XPS spectra of the Fe 2p region for the Ni/CP-TEA after the stability test (Fig. S6a, ESI†), no Fe 2p signal can be detected. Additionally, in the CV cycles (Fig. S6b, ESI†), it can be observed that the OER activity shows a negligible change. These results indicate that the enhanced catalytic activity of the Ni/CP-TEA for the OER cannot be attributed to Fe contamination.
To gain a better understanding of the catalytic OER activity, the electrochemical surface area (ECSA) was measured. The ECSA was evaluated using the electrochemical double-layer capacitance (Cdl). For the Cdl measurement, CV curves were recorded with different sweep rates (Fig. S7, ESI†). As shown in Fig. 3b, the Cdl values for Ni/CP-TEA and Ni/CP are 7.6 and 3.6 mF, respectively, demonstrating a larger ECSA and more active sites in the Ni/CP-TEA. This result is consistent with the coral-like structure in the Ni/CP-TEA (Fig. 1a), in which more edges and surface area are exposed compared to the aggregated nanoparticles in the Ni/CP (Fig. 1b).
To reveal the intrinsic OER activity, the LSV curves of the Ni/CP-TEA and Ni/CP were normalised against their ECSA and mass values (calculated based on the I–t curve of the electrodeposition process, as shown in Fig. S8, ESI†). The ECSA-normalised LSV curves (Fig. S9, ESI†) show that the Ni/CP-TEA possesses a significantly higher current density than the Ni/CP at the same potential, suggesting an enhancement in the intrinsic OER activity through TEA modification. As shown in Fig. 3b inset and Fig. S10 (ESI†), at a given potential of 400 mV, the ECSA and mass normalised current density for the Ni/CP-TEA are 0.1 and 14.0 mA cm−2, respectively, which are 3.3 and 14 times higher than those of Ni/CP, respectively.
The long-term durability of the Ni/CP-TEA and Ni/CP samples was also measured in 0.1 M KOH. As shown in Fig. 3c, the Ni/CP-TEA showed negligible potential change over 20 h at a current density of 10 mA cm−2 (η = 320 mV). Furthermore, the LSV curves before and after stability testing (inset of Fig. 3c) also show negligible change. All the results demonstrate that the Ni/CP-TEA is an efficient OER electrocatalyst with high activity and good stability.
To verify whether the N or the –OH group in the TEA structure is primarily responsible for the observed effects and to assess whether other N-containing complex agents present similar effects, NH3·H2O, ethylenediaminetetraacetic acid (EDTA) and ethanol were used as TEA substitutes. As shown in Fig. 3d, adding NH3·H2O and EDTA results in a similar OER activity increase, whereas adding ethanol only slightly increases the OER performance. This indicates that the N group coordination is qualitatively responsible for the OER activity enhancement, rather than the –OH group. This also indicates that the use of surface modification with N group-containing ligands to enhance electrocatalytic OER activity can be expanded to other electrocatalysts.
To gain a deeper insight into the structure–function relationships and to understand the role of the N group in enhancing OER performance, the used electrodes were further characterized and compared to the as-prepared electrodes. As presented in Fig. S11 (ESI†), the XRD patterns of the used and fresh Ni/CP-TEA are identical. Similarly, the XRD patterns of the used and fresh Ni/CP sample display similar diffraction peaks – assigned to Ni metal. The overall surface morphology (Fig. S12, ESI†) is preserved in both the used Ni/CP-TEA and Ni/CP. However, the used Ni/CP shows a smoother surface and a larger particle size (Fig. S12b, ESI†). In the TEM image (Fig. 4a), the selected area electron diffraction (SAED) pattern only shows weaker diffraction rings ascribed to Ni oxide compared to the fresh sample. The structural reconstruction of Ni oxides under OER conditions can derive a large number of defects/vacancies, enhancing OER performance.41,42 The weaker diffraction rings and unchanged XRD profile observed in the used Ni/CP-TEA perhaps result from a lower degree of crystallinity with numerous defects/vacancies formed during the surface reconstruction under OER conditions, which could contribute to the enhanced activity. EDX mapping (Fig. S13, ESI†) of the used Ni/CP-TEA displays the distribution of Ni and O.
 | ||
| Fig. 4 (a) HRTEM image of the used Ni/CP-TEA (after LSV polarization curve) and the inset is the corresponding SAED pattern. (b) XPS spectra of the N 1s region of fresh Ni/CP-TEA and used Ni/CP-TEA. (c) XPS spectrum of the Ni 2p region of used Ni/CP-TEA. (d) XPS spectra of the O 1s region of the used Ni/CP-TEA and used Ni/CP. | ||
Further XPS analysis was conducted to analyze the surface chemical states of the used electrodes. As shown in Fig. 4b, the N 1s signal cannot be detected in the used Ni/CP-TEA, suggesting the complete leaching of the N group from the surface of the used Ni/CP-TEA. Additionally, in the FTIR spectra (Fig. S14, ESI†), the vibrational peak corresponding to C–N/C–C/C–O present in the fresh Ni/CP-TEA cannot be detected in the used sample, further confirming the complete dissociation of TEA in the used sample. The disappearance of N signal is accompanied by a shift in the Ni 2p XPS spectrum in the used Ni/CP-TEA. As shown in Fig. 4c, for the used Ni/CP-TEA the Ni3+ species can be observed at 858.0 eV (Ni 2p3/2) and 875.5 eV (Ni 2p1/2). The Ni3+ species play a vital role for OER activity and are reported as the actual active center in the Ni-based electrocatalysts.43 For Ni/CP (Fig. S15, ESI†), the intensity of the Ni0 peaks in the used Ni/CP is much lower than that of the fresh Ni/CP and the Ni2+ peaks remain well preserved. These results suggest that the in situ leaching of TEA ligands induces the formation of high-valence Ni species in the Ni/CP-TEA. As for the O 1s spectra (Fig. 4d), the used Ni/CP-TEA and used Ni/CP have the same components, whereas the O 1s peaks of the used Ni/CP-TEA shift negatively to a lower binding energy compared to those of the used Ni/CP. This negative shift demonstrates an increased electron density at the O atoms and indicates a higher Ni valence state in the Ni/CP-TEA, consistent with the Ni 2p XPS spectra.
Based on all experimental and characterization results presented in this study, the possible surface reconstruction and transformation process of the Ni/CP-TEA electrode during the OER is presented in Fig. 5. In the initial state, TEA interacts with surface Ni sites via coordination bonding between the N in TEA and the Ni. Under the anodic potential, the TEA ligands are completely leached. The Ni species on the surface are in situ transformed into stable high-valent species, which finally serve as the OER-active sites. These high-valent Ni species endow the single-metal Ni oxide electrocatalyst with higher activity than the benchmark NiOx and make it comparable to other multi-metal and heteroatom-doped OER electrocatalysts. This exemplifies the effect of N coordination bonding surface modification in accelerating the reconstruction process and facilitating the generation of stable active species for the OER.
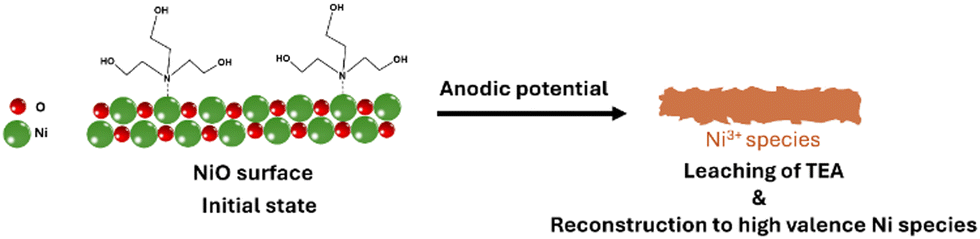 | ||
| Fig. 5 Proposed transformation of the electrode surface of Ni/CP-TEA during the OER. | ||
Conclusions
An N-group modified Ni electrocatalyst (Ni/CP-TEA) supported on a carbon paper substrate via a simple electrodeposition method has been fabricated and characterized, and its performance as an OER electrocatalyst has been evaluated. The Ni/CP-TEA electrocatalyst displays enhanced OER performance, with an overpotential of 320 mV at 10 mA cm−2 in 0.1 M KOH electrolyte, comparable to the best Ni–oxide based electrocatalysts reported to date for the OER, and shows good stability. The coral-like structure of the Ni/CP-TEA possesses larger surface area compared to the Ni/CP and more active sites for the OER. The in situ leaching of the N group facilitates the rapid formation of high valence Ni species, specifically Ni3+, thereby endowing the Ni/CP-TEA with enhanced OER activity. This work demonstrates the effect of modification of the Ni oxide surface with an N-group containing ligand to accelerate the rapid formation of high valence Ni species. This simple strategy may provide a new path for the low-cost and highly efficient design of OER electrocatalysts.Author contributions
Jiayun Zhang: conceptualization; experiment; formal analysis, writing – original draft. Ruth Knibbe and Ian Gentle: conceptualization, supervision, writing – review & editing, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by The University of Queensland (UQ) and Australian Research Council through grants of DP200102573 and FT220100666. The authors gratefully acknowledge the use of facilities and the valuable technical assistance at the Centre for Microscopy and Microanalysis (CMM) facilities at The University of Queensland, Australia.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed
.
- M. S. D. I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- J. Turner, G. Sverdrup, M. K. Mann, P.-C. Maness, B. Kroposki, M. Ghirardi, R. J. Evans and D. Blake, Int. J. Energy Res., 2008, 32, 379–407 CrossRef CAS
- M. E. Lyons and S. Floquet, Phys. Chem. Chem. Phys., 2011, 13, 5314–5335 RSC
- T. Kwon, H. Yang, M. Jun, T. Kim, J. Joo, J. Kim, H. Baik, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 14352–14362 RSC
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC
- Y. P. Zhu, C. Guo, Y. Zheng and S. Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed
- K. Zhang and R. Zou, Small, 2021, 17, e2100129 CrossRef
- Y. Li, X. Bao, D. Chen, Z. Wang, N. Dewangan, M. Li, Z. Xu, J. Wang, S. Kawi and Q. Zhong, ChemCatChem, 2019, 11, 5913–5928 CrossRef CAS
- M. Yu, E. Budiyanto and H. Tuysuz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed
- W. Yuan, C. Li, M. Zhao, J. Zhang, C. M. Li and S. P. Jiang, Electrochim. Acta, 2020, 342, 136118 CrossRef CAS
- M. Zhao, H. Li, W. Yuan and C. M. Li, ACS Appl. Energy Mater., 2020, 3, 3966–3977 CrossRef CAS
- M. Gorlin, P. Chernev, J. Ferreira de Araujo, T. Reier, S. Dresp, B. Paul, R. Krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS
- H. Sun, X. Xu, Y. Song, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2009779 CrossRef CAS
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS
- D. Liu, H. Ai, J. Li, M. Fang, M. Chen, D. Liu, X. Du, P. Zhou, F. Li, K. H. Lo, Y. Tang, S. Chen, L. Wang, G. Xing and H. Pan, Adv. Energy Mater., 2020, 10, 2002464 CrossRef CAS
- C. Huang, Q. Zhou, D. Duan, L. Yu, W. Zhang, Z. Wang, J. Liu, B. Peng, P. An, J. Zhang, L. Li, J. Yu and Y. Yu, Energy Environ. Sci., 2022, 15, 4647–4658 RSC
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC
- L. An, J. Feng, Y. Zhang, R. Wang, H. Liu, G. C. Wang, F. Cheng and P. Xi, Adv. Funct. Mater., 2018, 29, 1805298 CrossRef
- Y. Shi, Y. Yu, Y. Liang, Y. Du and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 3769–3773 CrossRef CAS
- E. Cossar, M. S. E. Houache, Z. Zhang and E. A. Baranova, J. Electroanal. Chem., 2020, 870, 114246 CrossRef CAS
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed
- R. M. Zhaoping Liu, Minoru Osada, Kazunori Takada and Takayoshi Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef
- D. P. Dubal, V. J. Fulari and C. D. Lokhande, Microporous Mesoporous Mater., 2012, 151, 511–516 CrossRef CAS
- L. A. Saghatforoush, M. Hasanzadeh, S. Sanati and R. Mehdizadeh, Bull. Korean Chem. Soc., 2012, 33, 2613–2618 CrossRef CAS
- J. Hao, W. Luo, S. Wang, K. Zhao, J. Hou, L. Li, B. Ge, W. Yang and W. Shi, Angew. Chem., Int. Ed., 2021, 60, 20042–20048 CrossRef CAS PubMed
- J. Shen, X. Zheng, L. Peng, G. I. N. Waterhouse, L. Tan, J. Yang, L. Li and Z. Wei, ACS Appl. Nano Mater., 2020, 3, 11298–11306 CrossRef CAS
- T. A. Mohamed, I. A. Shaaban, R. S. Farag, W. M. Zoghaib and M. S. Afifi, Spectrochim. Acta, Part A, 2015, 135, 417–427 CrossRef CAS PubMed
- B. Song, C. Sizemore, L. Li, X. Huang, Z. Lin, K.-s Moon and C.-P. Wong, J. Mater. Chem. A, 2015, 3, 21789–21796 RSC
- A. Jawad, Y. Li, X. Lu, Z. Chen, W. Liu and G. Yin, J. Hazard. Mater., 2015, 289, 165–173 CrossRef CAS PubMed
- M. B. Jensen, S. Morandi, F. Prinetto, A. O. Sjåstad, U. Olsbye and G. Ghiotti, Catal. Today, 2012, 197, 38–49 CrossRef CAS
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS
- R. Gao, M. Deng, Q. Yan, Z. Fang, L. Li, H. Shen and Z. Chen, Small Methods, 2021, 5, e2100834 CrossRef
- T. Meng, Q. Li, M. Yan, D. Wang, L. Fan, X. Liu, Z. Xing and X. Yang, Chem. Eng. J., 2021, 410, 128331 CrossRef CAS
- Y. Liu, Z. Liu, L. Jia, D. Gao and Z. Tang, Appl. Surf. Sci., 2022, 606, 154897 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00420e |
| This journal is © The Royal Society of Chemistry 2024 |