
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication methods, pseudocapacitance characteristics, and integration of conjugated conducting polymers in electrochemical energy storage devices
Meysam
Heydari Gharahcheshmeh
 *a and
Kafil
Chowdhury
ab
*a and
Kafil
Chowdhury
ab
aDepartment of Mechanical Engineering, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182, USA. E-mail: mheydari@sdsu.edu
bDepartment of Mechanical and Aerospace Engineering, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
First published on 15th October 2024
Abstract
Among the diverse range of modern renewable energy storage technologies, electrochemical energy storage devices have been rapidly adopted across various applications owing to their superior characteristics, including high coulombic efficiency, elevated energy and power densities, scalability, modularity, and rapid response capabilities. Conjugated conducting polymers have recently attracted significant attention in electrochemical energy storage devices due to their unique pseudocapacitive behavior, hybrid ionic/electronic conduction, rapid doping/de-doping dynamics, bulk intercalation of ionic species, high specific capacity, and exceptional structural and thermal stability. Conducting polymers exhibit pseudocapacitance through reversible redox reactions coupled with doping/de-doping processes, facilitating the movement of counterion dopants and ionic species between the polymer matrix and the electrolyte. The size and nature of counterion dopants significantly influence the electrochemical performance of these polymers. Small counterion dopants like chloride enhance redox exchange with the electrolyte and broaden the electrochemical potential window, which is advantageous for electrochemical energy storage devices. The pseudocapacitive properties can be further enhanced by increasing the semi-crystalline characteristics and attaining longer polymer chains. This review article focuses on the fabrication methods, fundamental aspects of ionic and electrical conductivity, and pseudocapacitance characteristics of conjugated conducting polymers, as well as their applications in Li–ion batteries, supercapacitors, and redox flow batteries.
 Meysam Heydari Gharahcheshmeh | Dr Meysam Heydari Gharahcheshmeh is currently an Assistant Professor in the Department of Mechanical Engineering at San Diego State University (SDSU). He completed his postdoctoral training in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT) in 2021 and received his PhD in Materials Science and Engineering from the University of Houston in 2017. His research focuses on texture and nanostructure engineering of advanced functional materials, such as conducting polymers and superconductors, fabricated using chemical vapor deposition (CVD) for renewable energy and biocompatible device applications. |
 Kafil Chowdhury | Kafil Chowdhury is currently pursuing his PhD as a joint doctoral program student in the Department of Mechanical Engineering at San Diego State University (SDSU) and Department of Mechanical and Aerospace Engineering at the University of California, San Diego (UCSD). His research focuses on manufacturing conducting polymers using the chemical vapor deposition (CVD) method and their implementation in various renewable-energy-related applications including solar cells, supercapacitors, and redox flow batteries. |
1. Introduction
To mitigate the negative impacts of greenhouse gas emissions on the global climate, policymakers are increasingly prioritizing sustainable energy technologies with reduced carbon footprints.1–4 Nowadays, a diverse range of energy storage technologies, including chemical, electrochemical, mechanical, thermal, and electromagnetic systems, are being developed to meet specific application needs.5 Among these renewable energy technologies, electrochemical energy storage devices, such as lithium-based batteries (lithium–ion and lithium–metal), redox flow batteries, and supercapacitors, are particularly promising due to their high energy conversion efficiency, scalability, and portability.3,6The growing interest in utilizing conjugated conducting polymers in various electrochemical energy storage devices is driven by their pseudocapacitive properties, unique hybrid ionic/electronic conduction, rapid doping/de-doping behavior, relatively high specific capacity, high electrical conductivity, exceptional structural and thermal stability, low oxidation potential, and superior mechanical flexibility.7–10 The presence of mixed ionic and electronic conduction (MIEC) in conjugated conducting polymers, despite its inherent complexity, has attracted substantial interest in the field of electrochemical energy storage devices.3,10,11 In recent years, a diverse range of conjugated conducting polymers, including polypyrrole (PPy), polyaniline (PANI), polythiophene (PT), and poly(3,4-ethylenedioxythiophene) (PEDOT), as well as their composites and derivatives, have been extensively employed in electrochemical energy storage devices.9,10,12–17
The pseudocapacitive characteristics of conjugated conducting polymers arise from reversible redox reactions coupled with doping/de-doping processes, which facilities the movement of counterion dopants and ionic species between the polymer matrix and electrolyte.8,18,19 The doping process in conjugated conducting polymer thin films can be electrochemically induced by applying an external electric field while the conjugated conducting polymer thin film is in contact with an electrolyte. In the doped state, conjugated polymer chains typically adopt an extended conformation to minimize repulsion between similar charges. Decreasing the magnitude of the applied electric field leads to the reduction of polycationic polymer chains and the expulsion of counterions from the conjugated polymer into the electrolyte, resulting in dedoping. In this de-doped state, the conjugated polymer chains often adopt a compact conformation.3 The doping process in conducting polymers leads to the formation of charged species such as polarons and bipolarons along the conjugated backbone.8 During de-doping process in p-type conducting polymers, bipolarons reduce to polarons, and polarons to neutral segments, while positively charged ions from the electrolyte enter the polymer thin film to maintain overall charge balance.10,20
To be effective as pseudocapacitive materials and achieve high specific capacitance, conjugated polymers must meet several criteria, including low molecular weight, an appropriate electrochemical potential window, high thermal and chemical stability, good mechanical flexibility, excellent charge mobility, and high electrical conductivity.3,10 The theoretical capacitance of a material represents the maximum charge it can hold relative to its mass.21 Consequently, the maximum theoretical capacitance in conjugated conducting polymers corresponds to one charge per monomer unit, making low molecular weight polymers such as PANI, PPy, and P3HT generally preferred for achieving high specific capacitance.3 However, conducting polymers with high molecular weights, such as PEDOT, also demonstrate excellent pseudocapacitance due to their superior ionic charge mobility, outstanding electrical conductivity, exceptional thermal and chemical stability, and resilience to expansion and contraction during electrochemical reactions.7,9,10,22–24 Conjugated conducting polymers undergo expansion and contraction in response to electrochemical reactions, where redox processes induce elasticity. The inclusion or exclusion of anions from the polymer matrix causes strain, which varies with the amount of anionic redox species involved.9,25,26 Conducting polymers with high molecular weights, such as PEDOT, effectively withstand the expansion and contraction that occur during electrochemical reactions.
The storage of electrochemical energy is governed by two principal mechanisms: the formation of electrochemical double layers on electrode surfaces through adsorption and faradaic charge storage in pseudocapacitive materials.8,27 To enhance the storage capacity of electrochemical double layers, it is crucial to increase the electrode surface area available for adsorption. Various porous carbon materials have been investigated as high-surface–area electrodes for electrochemical double-layer capacitors (EDLCs).28 The incorporation of a pseudocapacitive layer onto the carbon electrodes provides an additional reservoir for electrochemical charge. This faradaic charge storage is facilitated by reversible redox reactions occurring within the volume of the pseudocapacitive layer, leading to a substantial increase in total storage capacity.3,10 The intercalation of ions within conjugated conducting polymer chains enables the use of the entire polymer volume for charge transfer, extending beyond the electrode/electrolyte interface and thereby enhancing pseudocapacitance.9,10,18 High surface area is essential for efficient pseudocapacitive charge storage, as thinner layers over larger interfacial surface areas accelerate the charging and discharging rates for a given volume of conjugated polymer.
The size and nature of counterion dopants are critical in determining the electrochemical performance of conjugated conducting polymers.8,10,29 Conjugated conducting polymers can be doped with a various range of counterions, from small monoatomic anions like chloride (Cl−) to large macromolecular polyanions such as poly(styrene sulfonate) (PSS), tosylate, and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ). Using small counterion dopants like chloride in conjugated conducting polymers facilitates redox exchange with the electrolyte and offers a broader electrochemical potential window, which is advantageous for electrochemical energy storage devices.8,10,30,31 Enhancing the pseudocapacitive properties of conducting polymers can be achieved by increasing their semi-crystalline characteristics and attaining longer polymer chains.7,8 Ion diffusivity differs between crystalline domains and amorphous regions,3,19 and conducting polymer thin films with an edge-on orientation exhibit higher charge storage capacities during charge/discharge cycles.10,31 Additionally, smaller counter-ion dopants enhance ionic conductivity and accelerate redox reactions.
The large-scale adoption of electric vehicles (EVs), electronics, and drones has intensified the demand for high-energy, durable, and cost-effective lithium–ion batteries (LIBs).32 Recent advancements in conjugated conducting polymers within Li-based batteries have significantly improved performance and stability.33,34 Optimizing the anode surface area in LIBs is crucial: reducing anode surface area enhances energy density by minimizing lithium consumption for solid electrolyte interphase (SEI) formation, while increasing anode surface area boosts power density but lowers energy density due to higher lithium consumption.35 The artificial SEI film, replicating the natural SEI layer, enhances Li–metal battery performance by regulating Li+ deposition and suppressing Li dendrite growth. Conducting polymers with excellent ionic conductivity, superior processability, and outstanding flexibility are ideal for use as artificial SEI.36 To address the volumetric expansion of Si anodes in LIBs and prevent interfacial side reactions, stable SEI development is essential. Conducting polymer coatings on porous Si anodes enhance electrochemical performance by stabilizing the SEI layer, improving electronic conduction, rate capability, cycling stability, volumetric tolerance, electrolyte uptake, and electron transport.12,37,38
As a promising cathode for next-generation LIBs, Ni-rich NCM, particularly NCM811 (LiNi0.8Co0.1Mn0.1O2), offers high energy density with a specific capacity of ∼210 mA h g−1, good power densities, and reduced cost but faces challenges such as low thermal stability, poor cycle life, and rapid capacity degradation.32,39 To address these issues, intensive research has focused on surface coating of NCM cathodes to enhance cycling performance using various materials, including metal oxides,40,41 metal fluorides,42,43 metal phosphates,44,45 Li–ion conductive coatings (e.g., LiTi2O4),46,47 and conducting polymers.48,49 Compared to inorganic coatings, conducting polymers offer high electronic conductivity, environmental stability, and low cost, thus stabilizing cycling and improving rate capability. The lithium–sulfur (Li–S) battery is a promising Li–metal based technology, offering a high energy density of ∼2600 W h kg−1 and low-cost. However, the practical application of Li–S is hindered by challenges such as poor rate capability and limited rechargeability due to the insulating nature of S8 and Li2S, rapid capacity degradation from the dissolution of lithium polysulfides (Li2Sn, 3 ≤ n ≤ 6), and significant volume expansion (∼80%) during charging.33,50–52 To improve sulfur cathode utilization and cycling stability, various sulfur composites are used, including sulfur dispersed in conductive mediums like carbon-based materials,53,54 and conducting polymers.55,56 Coating the sulfur cathode in Li–S batteries with conducting polymers enhances capacity retention, improves overall capacity by restricting the shuttling of polysulfide intermediates, and mitigates electrode degradation from sulfur's volume expansion due to the polymers’ self-healing properties.55,57
Supercapacitors have attracted significant attention for their exceptional capacitance (100–1000× higher than conventional capacitors), high power density, rapid charge/discharge times (1–10 seconds), and long cycling life (>30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours), making them ideal for flexible electronics, electric vehicles, and renewable energy devices.2,7,58 Enhanced capacitance is achieved by increasing electrode surface area, leading to the use of high-surface–area materials such as activated carbon (AC), carbon nanotubes (CNTs), graphene, graphene oxide (GO), carbon cloths (CCs), carbon fibers (CFs), and other nanoporous structures.2 Pseudocapacitors, which typically offer higher energy density than EDLCs, bridge the gap between EDLCs and rechargeable batteries.2,20 Conducting polymers and transition metal compounds ((TMCs)), such as metal oxides/sulfides/nitrides/carbides, are commonly used as electrode materials in pseudocapacitors.2 The mechanical stability of conducting polymers, combined with their straightforward fabrication, redox properties, and mixed ionic and electrical conductivity, makes them ideal for use in flexible supercapacitors.2,3,20 PANI stands out as a leading candidate among conducting polymers, noted for its lower molecular weight and impressive specific capacitance of 1284 F g−1.59 Conducting polymers generally show high specific capacitances, such as 480 F g−1 for PPy,60 and 210 F g−1 for PEDOT.10,61
000 hours), making them ideal for flexible electronics, electric vehicles, and renewable energy devices.2,7,58 Enhanced capacitance is achieved by increasing electrode surface area, leading to the use of high-surface–area materials such as activated carbon (AC), carbon nanotubes (CNTs), graphene, graphene oxide (GO), carbon cloths (CCs), carbon fibers (CFs), and other nanoporous structures.2 Pseudocapacitors, which typically offer higher energy density than EDLCs, bridge the gap between EDLCs and rechargeable batteries.2,20 Conducting polymers and transition metal compounds ((TMCs)), such as metal oxides/sulfides/nitrides/carbides, are commonly used as electrode materials in pseudocapacitors.2 The mechanical stability of conducting polymers, combined with their straightforward fabrication, redox properties, and mixed ionic and electrical conductivity, makes them ideal for use in flexible supercapacitors.2,3,20 PANI stands out as a leading candidate among conducting polymers, noted for its lower molecular weight and impressive specific capacitance of 1284 F g−1.59 Conducting polymers generally show high specific capacitances, such as 480 F g−1 for PPy,60 and 210 F g−1 for PEDOT.10,61
Redox flow batteries (RFBs) are ideal for large-scale, cost-effective electrochemical energy storage due to their simple and modular design, scalability, long-duration storage, rapid load response, high efficiency, safety, and long cycle life.62,63 Common electrode materials in RFBs include low-cost porous carbons like carbon fiber paper (CFP), carbon cloth (CC), and graphite felt (GF).9,64,65 Carbon cloth shows promise for RFB electrodes but suffers from low specific capacitance, limited surface area, and poor wettability due to its hydrophobic nature.9,10,66 Similarly, graphite felt faces challenges like low electrochemical activity, limited surface area, poor wettability, and suboptimal chemical kinetics which results in low mass transfer.67 To improve the electrochemical performance of carbon-based electrodes, various modification techniques have been explored, including wet chemical oxidation,68,69 electrochemical oxidation,70,71 thermal treatment,72,73 plasma enhancement,74,75 and utilization of conducting polymer coatings.9,10 Applying thin layers of conducting polymers and their composites onto carbon electrodes has proven to be an effective method for improving the electrochemical performance of carbon-based electrodes in RFBs.9,10,16,76 PEDOT, specifically, can be chemically tailored to regulate the diffusion of electroactive species within its structure, making it an excellent ion-selective material.16
This review focuses on the electrochemical characteristics of conjugated conducting polymers and their applications in electrochemical energy storage devices, such as Li–ion batteries, supercapacitors, and redox flow batteries. Various solution-based and vapor-based fabrication methods for conjugated conducting polymers are also discussed. Additionally, the impact of texture and nanostructure on the electrical and ionic conductivity of conjugated polymers is examined, emphasizing their importance in the development of next-generation materials with diverse counterion dopants to meet future demands.
2. Fabrication methods for conducting polymers
Fabrication methods and corresponding process parameters can significantly influence various physical and chemical properties of conjugated conducting polymers, such as crystallinity, glass transition temperature, molecular weight, and morphology. Fabrication techniques for conducting polymers can be divided into two broad categories: solution-based and vapor-based, as illustrated in Fig. 1. Solution-based methods include: spin coating,77,78in situ chemical polymerization (ICP),79–81 electropolymerization (electrochemical polymerization (ECP)).82–85 Vapor-based techniques includes: vapor phase polymerization (VPP),86–93 oxidative chemical vapor deposition (oCVD).2,9,94–99 In addition to these established methods, other polymerization techniques such as hydrothermal,100 electrospinning,101 self-assembly,102 interfacial,103 and plasma polymerization,104 have also been used for fabricating conducting polymer thin films and composites.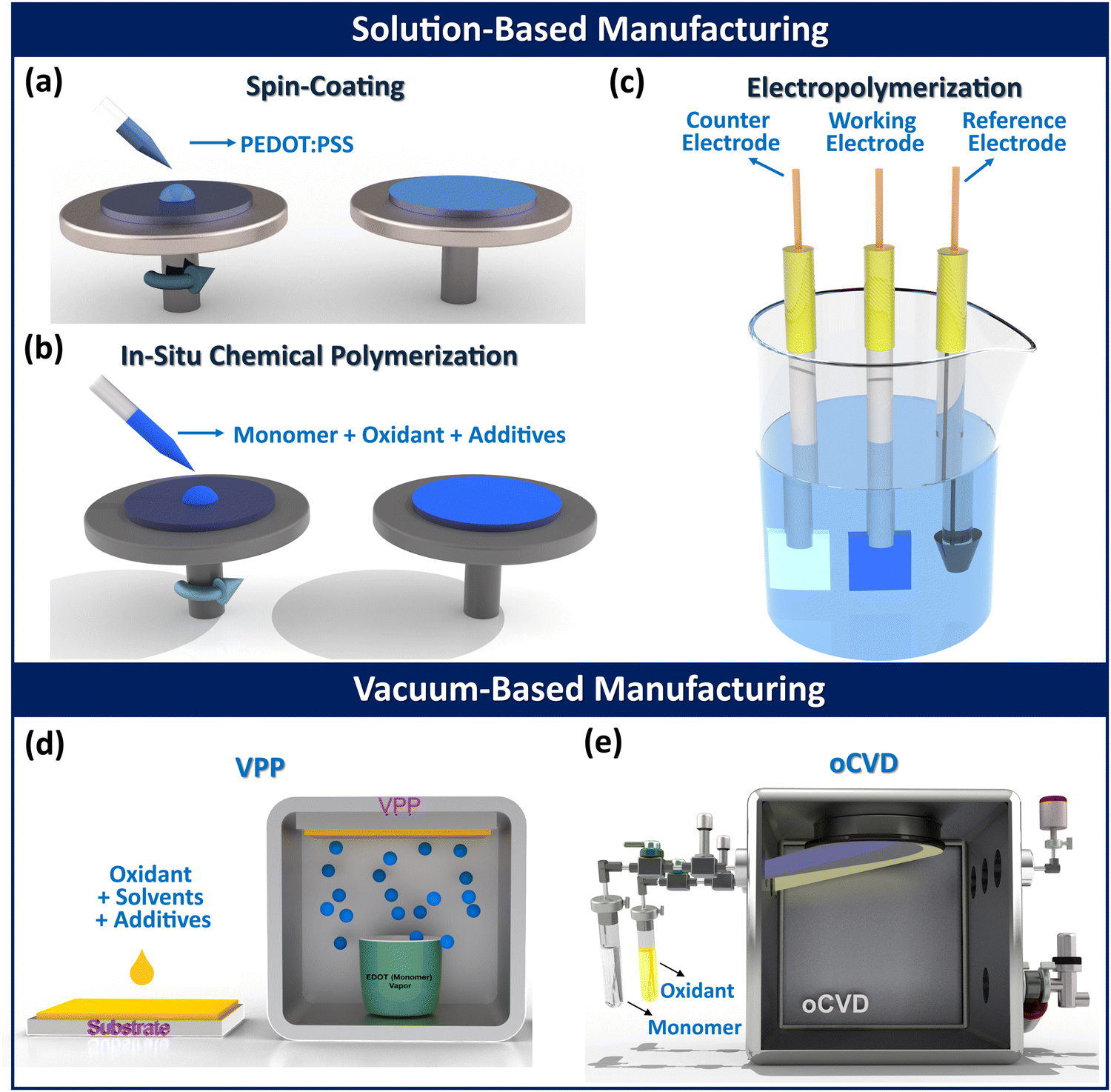 | ||
| Fig. 1 Various methods commonly used for fabricating conjugated conducting polymers. Solution-based methods include (a) spin-coating,2 (b) in situ chemical polymerization (ICP), and (c) electrochemical polymerization (ECP). Vapor-based methods include (d) vapor phase polymerization (VPP), and (e) oxidative chemical vapor deposition (oCVD).105 (a) Reproduced with permission.2 Copyright 2019, Wiley. (e) Reproduced with permission.105 Copyright 2019, Science. | ||
2.1. Solution-based fabrication methods for conducting polymers
The electrical conductivity of conducting polymer thin films fabricated by spin coating can be enhanced by adding organic polar solvents with high boiling points, such as dimethyl sulfoxide (DMSO), N,N-dimethyl formamide (DMF), glycerol, ethylene glycol (EG), N,N-dimethyl acetamide (DMAc), sorbitol, and dichloromethane (DMC).8,29,108–110 The main advantages of the spin coating method are its quickness and low cost. However, its major drawbacks include: (i) inability to create ultrathin polymeric films (thickness <10 nm), (ii) inability to provide conformal coatings, (iii) restriction to planar substrates, as the technique is ineffective for coating non-flat or complex surfaces, (iv) utilization of only a small fraction (2–5%) of the polymer solution, and (v) low quality and presence of contaminants in the fabricated thin films.2,106,111,112
Numerous oxidants are commonly used in the ICP method, including copper(II) chloride (CuCl2),114,115 copper(II) perchlorate (Cu(ClO4)2),116 iron p-toluene sulfonic acid (Fe(pTS)),117,118 iron chloride (FeCl3),117,118 iron(III) para-tosylate (Fe(Tos)3), and ammonium peroxydisulfate (APS) with p-toluene sulfonic acid (PTSA) as the oxidant and dopant, respectively.119 Various organic solvents, such as dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), acetonitrile–chloroform, and n-butanol, are used for fabricating different conducting polymers, including PEDOT, poly(ethylene terephthalate) (PET) fiber, and graphite/PMMA nanocomposites.120–123 Additionally, different additives such as base inhibitors (e.g., imidazole),124 polymeric surfactants (e.g., PEG–PPG–PEG), and high boiling point cosolvents (e.g., NMP) are employed to modify the morphology and other physicochemical characteristics of the fabricated polymer thin films through the ICP method.113
The main benefits of the ICP technique over spin-coating are (i) a simpler and faster bottom-up approach and (ii) effective dispersion of nanoparticles with less aggregation. However, the ICP method has disadvantages compared to vapor phase methods, including (i) inability to provide conformal coatings due to surface tension effects, (ii) unsuitability for solvent-sensitive substrates, and (iii) potential for flocculant formation during polymerization.
Selecting the appropriate solvent and electrolyte is critical for the electropolymerization method, as they must remain stable at the monomer's oxidation potential while providing ionic conductivity.82 The solvent type (aqueous, non-aqueous, ionic liquid, protic, aprotic) and electrolyte properties (pH, composition) significantly influence the film properties and polymerization rate.82,126,127 Organic solvents like DMSO, acetonitrile, dichloromethane, propylene carbonate, nitrobenzene, and methyl benzoate are commonly used in the electropolymerization method for fabricating conducting polymer thin films.128,129 Aqueous solvents are preferred in the electropolymerization method due to advantages like easy handling, environmental safety, lower cost, and the availability of a wide range of possible counterions for incorporation.130 Room temperature ionic liquids are also used as solvents in electrochemical polymerization method due to their non-volatility, wide electrochemical stability window, and environmental friendliness.82
The electropolymerization process is favored over other solution-based techniques for its excellent substrate adhesion, better control over film thickness and morphology, and prevention of extra chemical species accumulation (from oxidant or catalyst) on the polymeric film.131 However, the electropolymerization method has a drawback compared to other solution-based methods, as it requires conductive substrates, limiting the choice of materials.
2.2. Vapor-based fabrication methods for conducting polymers
Common oxidants used in VPP method include iron(III) chloride (FeCl3),91 iron(III) tosylate [Fe(OTs)3],136 iron(III) trifluoromethanesulfonate [Fe(OTf)3],137 and copper(II) chloride (CuCl2).138 These oxidants are typically dissolved in organic solvents like methanol, ethanol, butanol, or water to prepare spin-coatable precursors. The polymerization rate can be regulated using different solvents such as DMSO, NMP, DMF, and chelating ligands like EDTA.8,29,139 The utilization of solvents and additives in the VPP method introduces potential coordination molecules into the oxidant solution, forming metal complexes and altering ligand coordination, thereby changing the oxidant's overall reactivity. Base inhibitors such as imidazole, polyethyleneimine (PEI), or pyridine, when dissolved with the oxidant, can enhance the film's conductivity by increasing the pH and inhibiting side reactions under acidic conditions, and reducing the polymerization rate.92,93,140 The surfactant poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEG–PPG–PEG) has been reported to improve electrical conductivity of conjugated conducting polymer thin films by lowering the polymerization rate and preventing oxidant crystallization.141
The polymerization rate and characteristics of fabricated conducting polymers in VPP method can be controlled by adjusting parameters like exposure time of the substrate with the monomer vapor,92 pressure of vacuum chamber,86 substrate temperature,2,86,90,142 relative humidity inside the polymerization chamber,87 acidity level of oxidant,143 and post-deposition acidic rinsing treatment.92 The VPP method offers several key advantages for fabricating conducting polymers, including compatibility with various substrates, operation in a moderate vacuum environment for fabricating good quality thin films, fabrication of smooth thin films, and the ability to deposit on flexible or delicate substrates.90,92,133 The drawbacks of the VPP method include the complexity of tuning interconnected process parameters, the initial formation of oxidant as a solid thin film in the absence of monomer preventing fabrication of ultrathin highly conformal coatings, and the risk of substrate surface damage from solution casting, making it incompatible with solution and temperature-sensitive substrates.
Common solid oxidants include iron(III) chloride (FeCl3)30,95,98,148,149 and copper(II) chloride (CuCl2),2,97 while recently reported liquid oxidants include vanadium oxytrichloride (VOCl3)9,30,105,150 and antimony pentachloride (SbCl5).10,30,151,152 Liquid oxidants are often more effective than solid oxidants due to their higher volatility, better flow rate control, and elimination of post-treatment acidic rinsing, as they leave negligible oxidant byproducts on the as-deposited thin films.2,30,105 The characteristics of conducting polymers fabricated using the oCVD method can be controlled by tuning process parameters such as oxidant saturation ratio (OSR),105,152 deposition temperature,10,153,154 chamber pressure,2,30,146 incorporation of water-assisted growth techniques,30,152,155 and choice of oxidant.105 The most important parameters for oxidants used in oCVD method are volatility, thermal stability, and the ability to drive the reaction forward.30,156
The oCVD method enables the fabrication of conducting polymers on any substrate type (planar/non-planar, organic/inorganic, porous/dense, conductor/insulator) and produces robust, patterned thin films via covalent grafting between the polymer film and substrate without requiring any linker molecule.2 Given that majority of conjugated conducting polymers are insoluble in solvents, solvent-free and dry methods like vapor-based manufacturing (VPP, and oCVD) are highly desired.2,30,157 The most significant advantages of oCVD over other techniques include its ability to form highly conformal coatings, independence from substrate type, compatibility with large-scale roll-to-roll (R2R) production, solvent-free single-step processing, and capability to create uniform thin films at low deposition temperatures.30,144,146,148
The Fabrication methods section provided an overview of various techniques used to fabricate conjugated conducting and semiconducting polymers. These fabrication methods often rely on specific oxidants to facilitate polymerization. The commonly explored oxidants employed in various fabrication processes are summarized in Table 1.
| Fabrication method | Common explored oxidants |
|---|---|
| Spin coating | Polymer is typically dissolved in a solution, with FeCl3 occasionally added as oxidant |
| Electropolymerization | Oxidation of a monomer is induced by voltage or current |
| In situ chemical polymerization (ICP) | FeCl3, CuCl2, Cu(ClO4)2, Fe(pTS), Fe(Tos)3, ammonium peroxydisulfate (APS) |
| Vapor phase polymerization (VPP) | FeCl3, CuCl2, Fe(OTs)3, Fe(OTf)3 |
| Oxidative chemical vapor deposition (oCVD) | FeCl3, CuCl2, VOCl3, SbCl5 |
3. Specific characteristics of conjugated conducting polymers
3.1. Charge transport mechanisms in conjugated conducting polymers
Organic polymers are typically insulators due to their large band gap, which is the significant energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels.8 However, certain types of organic polymers, known as conjugated polymers, can exhibit semiconducting behavior due to the presence of alternating single (σ) and double (π) bonds along their polymer chain (Fig. 2a), creating an overlap of unbonded pz orbitals that allows π-electrons to flow along the polymer backbone skeleton.3,8,29,158 The doping process in conjugated polymers reduces the bandgap, thereby increasing their conductivity from an insulator to a conductive level.8,29 The bonding (π) and anti-bonding (π*) orbitals of carbons in the polymer chain eventually create the π (HOMO) and π* (LUMO) bands (Fig. 2b).8 The difference between the HOMO and LUMO energy levels, known as the bandgap, dictates the optical and optoelectronic characteristics of the polymer.8,152,159 As the length of polymer chain increases, the bonding and antibonding orbitals get rearranged with different energy levels and practically reducing the bandgap which enhances conductivity. | ||
| Fig. 2 Charge transport in conjugated conducting polymers. (a) Schematic illustration of sigma and pi bonds in a conjugated polymer.8 (b) Change in bandgap as a consequence of bonding and antibonding orbitals in conjugated polymers.8 Schematic illustration of charge carrier transport through (c) intra-chain coupling,8 and (d) inter-chain coupling.8 (a)–(d) Reproduced with permission.8 Copyright 2020, Elsevier. | ||
There are two distinct pathways for the movement of charge carriers inside the conjugated polymer matrix: intrachain and interchain coupling (Fig. 2c and d).8 Intrachain coupling allows charge carriers to move between adjacent localized polymer segments along the same polymer backbone, whereas interchain coupling facilitates charge transport through hopping between individual chains or within a folded chain. Charge carrier mobility is generally much faster along the conjugated backbone (intrachain transport) compared to interchain transport.8,29,30,105,160,161 Interchain transport is a rate-limiting factor, and reducing the π–π stacking distance is an effective method to enhance carrier mobility and overall electrical conductivity in various conjugated conducting polymers, including PEDOT,10,105,152,162–165 P3HT,166,167 and diketopyrrolopyrrole (DPP)-based donor–acceptor (D–A) polymers.168 While intrachain coupling provides increased carrier mobility, a high packing order in chain orientation enhances interchain coupling, reducing charge localization caused by defects and amorphous regions.8
The incorporation of dopants in the polymer results in the oxidation or reduction of the polymer backbone, forming local excitons or quasiparticles (polaron, bipolaron, and soliton) along the polymer chain.8,159 These positively or negatively charged quasiparticles act as charge carriers along the conjugated backbone chains. Based on the polarity of the dominant charge carriers, conducting polymers can be classified as p-type (hole transport), n-type (electron transport), or ambipolar.8,29
The most widely studied and applicable p-type conjugated polymers include PEDOT, PANI, PPy, and PTh, as illustrated in Fig. 3a.29 On the other hand, the most frequently reported n-type conjugated polymers are diketopyrrolopyrrole (DPP), naphthalene diimide (NDI), benzodifurandione-based oligo(p-phenylene vinylene) (BDOPV), isoindigo (IID), and benzothiadiazole (BT), shown in Fig. 3b.29 Notably, n-type (reduced) conducting polymers exhibit less stability compared to p-type (oxidized) ones, making oxidized conducting polymers (p-type conducting polymers) more widely used in practical applications.3,8
 | ||
| Fig. 3 Chemical structures of the most relevant p-type and n-type conjugated conducting polymers. (a) Chemical structures of p-type conjugated polymers, including poly(3,4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI), and polythiophene (PTh).29 (b) Chemical structures of significant n-type polymers, such as isoindigo (IID), naphthalene diimide (NDI), benzothiadiazole (BT), diketopyrrolopyrrole (DPP), and benzodifurandione-based oligo(p-phenylene vinylene) (BDOPV).29 (a) and (b) Reproduced with permission.29 Copyright 2022, MDPI. | ||
3.2. Influence of nanostructure on electrical conductivity of conjugated conducting polymers
The electrical conductivity of conjugated conducting polymers can vary significantly depending on their semicrystalline characteristics such as crystallite size, semicrystalline orientation, and π–π stacking distance.8,29,152,169–171 With respect to the substrate surface, crystallites in conducting polymer thin films can exhibit a face-on orientations, edge-on orientations, end-on orientations, and a mixture of different orientations (isotropic).8 The conjugated backbone lies parallel to the plane of substrate in face-on orientation but the π–π stacking is perpendicular; whereas for edge-on, both the conjugated backbone and π–π stacking lie parallel to the plane of substrate (Fig. 4a).2,152,155 Two of the most widely used conjugated conducting polymers, PEDOT and P3HT, exhibit a semicrystalline nature and assemble in an orthorhombic unit cell structure (Fig. 4b and c).8 The conjugated polymer chains stack along the b-axis lattice parameter to form face-to-face layers, with π–π stacking corresponding to half of the b-axis lattice parameter. In both highly edge-on and highly face-on orientations, the conjugated backbone chains lie in the plane of conjugated polymer thin films, enabling high in-plane electrical conductivity. However, a mixture of face-on and edge-on orientations reduces unperturbed percolation pathways, leading to reduced electrical conductivity.8,152 | ||
| Fig. 4 Texture and nanostructure of conjugated conducting polymers. (a) Schematic illustration of face-on and edge-on orientation along with the π–π stacking distance, which can significantly affect the overall electrical conductivity.152 Orthorhombic unit cell structure of (b) P3HT,8 and (c) PEDOT.152Charge carrier transportation pathways between different ordered crystallite regions in conjugated conducting polymers with (d) highly face-on orientation,105 and (e) highly edge-on orientation.8 (a) and (c) Reproduced with permission.152 Copyright 2020, Wiley. (b) and (e) Reproduced with permission.8 Copyright 2020, Elsevier. (d) Reproduced with permission.105 Copyright 2019, Science. | ||
The possible charge carrier transport pathways in highly face-on and highly edge-on crystallite orientations in conjugated polymers are illustrated in Fig. 4d and e, respectively. The electrical conductivity in heavily doped conjugated conducting polymers with highly face-on (or highly edge-on orientation) and short π–π stacking distance is quasi-1D.8,105,152 In the quasi-1D conductivity, charge transport parallel to the substrate primarily occurs (i) along polymer chains that traverse multiple crystallites and bridge amorphous regions, and (ii) between chains in the crystalline regions.8,171 Interchain transport is higher in the crystallite domains and slower in the less dense amorphous regions.105 Reducing the π–π stacking distance in conjugated conducting and semiconducting polymers increases both the charge transfer integral and carrier mobility and yields the electrical conductivity enhancement.8,105,152,155
Fig. 5 displays the timeline of gradual increment in the reported conductivity of a specific conducting polymer, PEDOT. The highest electrical conductivity achieved in PEDOT thin films to date is 7520 S cm−1, obtained using the water-assisted oCVD method (W-A oCVD) with SbCl5 as the oxidant.152 Kim et al.172 reported a conductivity of 1418 S cm−1 in PEDOT thin films with EG as an additive. In 2014, Kim et al.173 achieved a conductivity of 4380 S cm−1 by using different concentrations of H2SO4 as a post-treatment, which induced crystallinity and structural improvement. Worfolk et al.174 used solution shearing during deposition that yielded PEDOT:PSS thin film with a conductivity of 4600 S cm−1. Gueye et al.175 utilized the ICP method to deposit PEDOT thin films with Fe(OTf)3 as the oxidant, and N-methyl pyrrolidone (NMP) as the polymerization rate controller, achieving a conductivity of 5400 S cm−1. Using the oCVD method with solid FeCl3 oxidant and HBr rinsing, PEDOT thin films exhibited a conductivity of 6259 S cm−1.153 Moreover, using liquid SbCl5 as oxidant, the oCVD method yielded PEDOT thin films with a conductivity of 5602 S cm−1.152 Post-deposition acidic rinsing steps, such as using HCl, HBr, or H2SO4, have been shown to significantly enhance the electrical conductivity of conducting polymer thin films. Post-deposition acidic treatments improve conductivity by removing insulating impurities and inducing structural rearrangements that promote better charge transport.2,29,153,176 For instance, H2SO4 post-treatment has been reported to increase the crystallinity and overall electrical performance of PEDOT films.8,153,173
 | ||
| Fig. 5 Timeline of electrical conductivity for PEDOT thin films. The evolution of electrical conductivity enhancement in PEDOT thin films fabricated by various solution-based and vapor-based methods.30 Reproduced with permission.30 Copyright 2024, Springer. | ||
3.3. Molecular weight and glass transition temperature of conjugated conducting polymers
Conjugated conducting polymers exhibit both expansion and contraction in response to electrochemical reactions, where redox processes induce elasticity. The inclusion or exclusion of anions from the polymer matrix leads to straining.9,25,26 The extent of strain depends on the amount of anionic redox species entering or departing from the polymer. For instance, applying a positive voltage along with a suitable electrolyte causes oxidation, leading to electron loss and anion entry into the polymer matrix, resulting in polymer expansion.9 Conversely, reduction causes contraction. These electrochemical deformations are useful in applications such as artificial muscles. Semi-crystalline polymers typically exhibit superior mechanical properties compared to amorphous polymers.177The mechanical behavior of conjugated polymers depends on microscopic changes in molecular mobility, which are influenced by factors such as molecular weight, morphology, nanostructure, working temperature, and applied pressure.152 Molecular weight is commonly characterized using the number-average molecular weight  or the weight-average molecular weight
or the weight-average molecular weight  where i is the degree of polymerization, Mi and Ni are the weight and number of molecules with degree of polymerization i.178,179 Higher molecular weight in conjugated polymers enhances electrical conductivity, strength, and toughness due to increased density and better entanglement. Lower molecular weight polymers are brittle and cannot withstand high strain rates, whereas higher molecular weight polymers exhibit significant ductility and elongation.179 The mechanical properties of conjugated conducting polymer thin films can be assessed using the strain-induced elastic buckling-based method.180 The modulus of elasticity for conjugated polymer thin films (Ef) is determined using the substrate's modulus of elasticity (Es), Poisson ratios of both the thin film (ϑf) and substrate (ϑs), buckling wavelength (λb), and thin film thickness (df) by using the following equation:180
where i is the degree of polymerization, Mi and Ni are the weight and number of molecules with degree of polymerization i.178,179 Higher molecular weight in conjugated polymers enhances electrical conductivity, strength, and toughness due to increased density and better entanglement. Lower molecular weight polymers are brittle and cannot withstand high strain rates, whereas higher molecular weight polymers exhibit significant ductility and elongation.179 The mechanical properties of conjugated conducting polymer thin films can be assessed using the strain-induced elastic buckling-based method.180 The modulus of elasticity for conjugated polymer thin films (Ef) is determined using the substrate's modulus of elasticity (Es), Poisson ratios of both the thin film (ϑf) and substrate (ϑs), buckling wavelength (λb), and thin film thickness (df) by using the following equation:180
 | (1) |
Reaching the glass transition temperature (Tg) transforms the polymer from a glassy to a rubbery state, altering its mechanical properties.179 The glass transition temperature represents a second-order phase transition and describes the thermally activated reorganization of polymer chains. Below Tg the polymer is glassy, and above Tg becomes elastomeric. Tg influences the morphological stability of the polymer's solid state, affecting thermal stability, dewetting, and delamination of thin films, and is critical for the mechanical properties needed in flexible optoelectronic applications.179 The glass transition temperature is a critical processing parameter that dictates the kinetics of molecular organization in organic conjugated polymers during solidification,179 or vacuum-based deposition methods.10,152,162 In the oCVD method for fabricating PEDOT, when the deposition temperature reaches or exceeds the glass transition temperature of PEDOT (approximately 100 °C), the crystallites reorient from mixed face-on and edge-on orientation to predominantly face-on orientation.10,30,152,162 The reorientation at around glass transition temperature aims to minimize interfacial energy between the substrate and the a–c plane of the PEDOT crystallite face, resulting in a highly face-on orientation, which is essential for enhanced electrical conductivity.8,30,162
3.4. Reversible doping/de-doping in conjugated conducting polymers
Conducting polymers can undergo p-doping with counter anions upon oxidation and n-doping with counter cations upon reduction. The simplified equations for these charging processes are as follows:20| Cp → Cn+p(A−)n + ne− (p-doping) | (2) |
| Cp + ne− → Cn−p(C+)n (n-doping) | (3) |
The type of doping (p- or n-type) is typically determined from the required potential for the doping and the nature of conducting polymers, so the selection of appropriate conducting polymers based on the potential window is essential for optimal electrochemical device performance.181 N-Type doping can sometimes result in undesirable properties such as high impedance, poor processability, chemical instability, and lower capacitance and conductivity due to the need for higher negative potentials than the electrolyte solvent breakdown potential.33,181,182
Doping in conducting polymers causes volumetric changes (swelling) that revert during de-doping (shrinking), which corresponds to the oxidation or reduction reaction.10,33,183 Continuous electrochemical cycling causes large dimensional changes during charging and discharging, leading to material breakdown and degradation of the polymer structure, ultimately reducing electrochemical performance.184 The doping process in conducting polymers results in the formation of charged particles such as polarons, bipolarons, and solitons along the polymer backbone.8 During de-doping in p-type conducting polymers, bipolarons and polarons are reduced to polarons and neutral segments, respectively and positively charged ions from the electrolyte entering the film to balance the overall charge.8,10,19 Inversely, positively charged ions of electrolyte are pushed out from the p-type conducting polymers during the next cycle of doping (charging) and the initial counterions again oxidize the polaron and neutral states to the bipolaron and polaron segments, respectively.10,183
Cyclic voltammetry (CV) is a fundamental technique for studying the doping/de-doping mechanism in conducting polymers. Applying a reversible potential at a fixed scan rate in CV causes conducting polymers to donate or accept electrons and ions from the electrolyte to move in and out of the polymer structure. The surface coverage (Γ) is defined as the ratio of adsorbed molecules numbers on a specific surface to the number of molecules required to form a complete monolayer on that surface. The peak current (ip) of a reversible electrochemical system as a function of Γ, can be measured by the following equation:182
 | (4) |
485 C mol−1), A indicates the electrode surface area in cm2, Γ shows the surface coverage in mol cm−2, ϑ specifies the scan rate in mV s−1, R is the universal gas constant (8.314 J mol−1 K−1), T stands for temperature in K, and  where (E − E0) is the difference between cell potential and standard potential.
where (E − E0) is the difference between cell potential and standard potential.
In general, when the redox reaction involves larger dopant ions which are sluggish to move, the reaction becomes diffusion-controlled and it was found that ip is proportional to the square root of scan rate (ϑ1/2).185 For reversible, single-electron doping/de-doping reactions, CVs show symmetric redox peak. However, for redox reactions involving multi-electron transfers, CVs exhibit several peaks for oxidation and reduction. Moreover, the peak-to-peak distance is also observed to increase with the scan rate.186 In cyclic voltammetry, the experimental timescale is controlled by the scan rate (ϑ), and the relationship between the observed current at any specific potential (i(V)) can reveal whether the redox reaction is diffusion-controlled or surface-controlled. The general relationship between the observed current and scan rate for any material is described by the following equation:187
| i(V) = k1ϑ1/2 + k2ϑ | (5) |
The inner electrode surface stores charge via intercalation, while the outer surface, in contact with the electrolyte, stores charge via adsorption. Charge storage on the outer surface is instant and scan rate-independent, whereas storage on the inner surface is diffusion-controlled and scan rate-dependent.188 The relationship between the inner surface (diffusion-controlled) and outer surface (surface-controlled) of an electrode to the total charges (QT) measured using CV is expressed as follow:188,189
| QT = Qi + Qo | (6) |
Furthermore, the relationship between the total charges or capacity (Q) as a function of ϑ can be described through the following equation:58,189
| Q(ϑ) = Q∞ + kϑ−1/2 | (7) |
At ϑ = 0, the value of Q represents the total capacity (QT), encompassing contributions from capacities associated with both inner (Qi) and outer (Qo) surfaces, while at ϑ → ∞, Q signifies the capacity specific to outer surfaces (Qo) exclusively.188
3.5. Pseudocapacitance characteristics of conjugated conducting polymers
In electrochemical double-layer capacitors (EDLCs), charge storage occurs at the electrode–electrolyte interface through electrostatic (non-faradaic) interactions, as shown in Fig. 6a. The innermost layer, made up of solvent dipoles adsorbed onto the electrode surface, is known as the inner Helmholtz or Stern layer. Immediately next to this is the outer Helmholtz layer, which consists of an adsorbed layer of solvated ions. Beyond the outer Helmholtz layer lies the diffuse layer, where solvated ions and solvent molecules are randomly distributed into the bulk medium.190 | ||
| Fig. 6 Schematic illustration of various types of pseudocapacitance. Schematic illustration of (a) electrochemical double-layer capacitance (EDLC), (b) underpotential deposition, (c) redox pseudocapacitance, and (d) intercalation pseudocapacitance. Intercalation pseudocapacitance utilizes redox-active intercalation chemistry in electrode materials by incorporating surface redox pseudocapacitance and electrostatic pseudocapacitance. Conjugated conducting polymers exhibit redox pseudocapacitance due to their ability for bulk intercalation, which enables ions to penetrate the polymer structure volumetrically. | ||
Activated carbon is the most widely recognized electrode material for EDLCs, offering a modest capacitance of up to 200 F cm−2, constrained by its specific surface area limitations. Typical pseudocapacitance also originates from the electrode surfaces but involves an associated faradaic charge-storage reaction, leading to significantly higher capacitance. While EDLCs depend exclusively on a physical charge storage mechanism,3 pseudocapacitance describes a material's ability to replicate the electrochemical properties of capacitive electrodes (linear relationship between charge and potential) while utilizing a faradaic electron transfer mechanism for charge storage.58 Utilizing redox reactions for energy storage, instead of electrostatic ion accumulation as seen in EDLCs, leads to battery-like behavior in pseudocapacitors during charging and discharging processes.20,182
Pseudocapacitors store energy through faradaic interactions, allowing them to achieve higher specific capacity and energy density compared to EDLCs.181 The charge storage mechanisms in pseudocapacitors are categorized into three distinct faradaic processes: (i) underpotential deposition (also referred to as monolayer adsorption pseudocapacitance), (ii) surface redox pseudocapacitance, and (iii) intercalation pseudocapacitance (Fig. 6b–d). In underpotential deposition, charge storage is achieved via the adsorption of a monolayer of cations from a specific metal onto the surface of another metal at potentials more positive than their equilibrium potential (Fig. 6b). The underpotential deposition yields high capacitance but limited energy density due to a narrow potential window. Underpotential deposition can be exemplified by the deposition of lead (Pb) on a gold (Au) electrode surface. In this case, Pb is more readily deposited onto Au than onto itself due to the stronger interaction between Pb and Au compared to the Pb–Pb interaction in crystalline Pb metal.191
In surface redox pseudocapacitance, ions are electrochemically adsorbed on or near the electrode surface, with energy stored through a faradaic charge transfer mechanism (Fig. 6c). Conjugated conducting polymers demonstrate redox pseudocapacitance because they can undergo bulk intercalation, allowing ions to penetrate the entire polymer matrix, as illustrated in Fig. 6c. An example of redox pseudocapacitance can be observed in systems containing Ruthenium(IV) oxide (RuO2) nanoclusters.192
Intercalation pseudocapacitance occurs when the intercalation and deintercalation processes are not hindered by the slow solid-state diffusion of ions within the crystal structure of the electrode materials.58 It is noteworthy that intercalation pseudocapacitance allows ions to enter the layers of redox-active materials without significant crystallographic phase changes, accompanied by a faradaic charge transfer process (Fig. 6d). Intercalation pseudocapacitance differs from battery systems because, unlike batteries where charge–discharge cycles often involve phase transformations of the host material, there are no phase changes in intercalation pseudocapacitance systems.193 The intercalation pseudocapacitance is observed in systems where lithium ions intercalate into mesoporous and nanocrystalline films of orthorhombic niobium pentoxide (Nb2O5) without any phase transformation.194
In conducting polymers, pseudocapacitive behavior stems from their reversible redox capabilities. During charging (or doping), the polymer chain oxidizes and releases electrons, while during discharging (or de-doping), the chain reduces and accepts electrons. Pseudocapacitance is measured by integrating the charging/discharging current (I) over the entire potential window (E = Vf − Vi) using the following equation:10,188
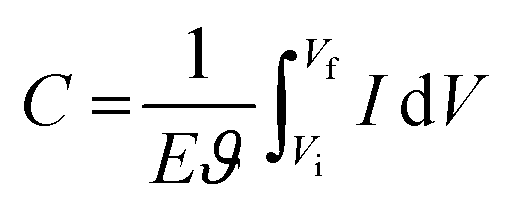 | (8) |
The gravimetric specific capacitance (Csp), which indicates the charge storage capacity and is directly related to the energy density of electrodes, can be calculated using the following equation:10,195
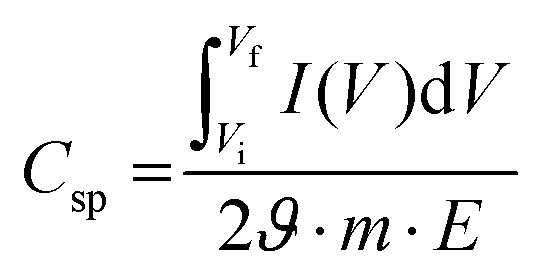 | (9) |
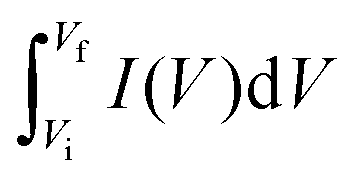 denotes the total area of the CV curve, ϑ specifies the scan rate, m represents the mass of the electrode, and E specifies the difference between the initial and final potential of sweep (E = Vf − Vi).
denotes the total area of the CV curve, ϑ specifies the scan rate, m represents the mass of the electrode, and E specifies the difference between the initial and final potential of sweep (E = Vf − Vi).
An additional common method to evaluate the behavior of electrochemical capacitors is the galvanostatic charge–discharge (GCD) test, where the specific capacitance (C) can be calculated using the following equation:190
 | (10) |
P-Type conjugated polymers are doped by oxidation, achievable through chemical reaction with an oxidant. A partially doped polymer chain for conjugated conducting polymer of poly(pyrrole) (PPy, A = N–H), poly(thiophene) (PT, A = S), and poly(selenophene) (PSe, A = Se), showing benzoid and quinoid bonding (Fig. 7a).3 Charge carriers include polarons (radical cations) and bipolarons (dications).8 In doped p-type conjugated conducting polymers, polycationic chains maintain charge neutrality with negatively charged counterions. This charge neutrality links anion migration during doping/de-doping to electron mobility. Partial charge transfer with some molecular counterions forms charge transfer complexes between the polymer and dopant.3 The doping process can be driven electrochemically by applying an external electric field to a conjugated polymer film in contact with an electrolyte.
 | ||
| Fig. 7 Schematic illustration of conjugated conducting polymer structure during electrochemical reaction. (a) The chemical structure of a partially oxidized (doped) generic conjugated chain, having neutral segments in between localized bonding states corresponding to polaronic and bipolaronic charge carriers.3 The neutral segments are in the benzoid state, while the oxidized segments display quinoid bonding. (b) Schematic illustration of conjugated polymer chain adjacent to an electrode in the (b) partially oxidized state,3 and (c) fully reduced state.3 (a)–(c) Reproduced with permission.3 Copyright 2021, Walter de Gruyter. | ||
In the doped state, polymer chains often extend to reduce repulsions between similar charges (Fig. 7b).3 Lowering the magnitude of external electric field reduces the polycationic polymer chain, expelling counterions into the electrolyte and resulting in dedoping. Reduced polymer chains typically adopt a compact conformation (Fig. 7c). The water solubility of PANI (polyaniline) makes it widely studied. Fully reduced PANI (leucoemeraldine) is yellow, while fully oxidized PANI (pernigraniline) is blue, with maximum ionic conductivity at 50% doping. Ionic conductivity is low at very low or very high doping levels, slowing electrochemical initiation unless pre-doped. This supports the hypothesis of ion transport between occupied and unoccupied states, with the rate proportional to the product of their concentrations.
Key aspects contributing to the complex ionic transport in conjugated conducting polymers include:
(1) Ion concentration dynamics: driven by chemical potential gradients from electrical potential and ionic concentration differences, leading to ion diffusion and drift.
(2) Redox reactions: occur between ions and the polymer, altering the charge state of the conjugated backbone.
(3) Electronic states: new states like polaronic and bipolaronic states emerge in the density of state diagram.8,11,19
(4) Backbone reorganization: structural changes, such as from benzoid to quinoid configurations, occur upon doping (as shown in Fig. 7a).
Intrachain migration: localized charge carrier states, situated within specific molecular structure, can migrate along the backbone via intrachain hopping, while there is a different migration rates for polarons and bipolarons.8,196
(5) Interchain transfer: electron transfer between chains via hopping, which is a rate-limiting step.29,105,152 Redox reactions between chains can also create interchain complexes in the solid conducting polymer matrix during charging, which relax during discharging.19
(6) Electrostatic forces: increase with more ion intercalation into the polymer film.
(7) Polymer conformation: the polymer chain extends to minimize electrostatic repulsion, altering its overall conformation (as illustrated in Fig. 7b)
(8) Morphology changes: conformation changes in the chains can affect the semicrystalline polymer's morphology. Also, electrochemical processes may differ in the ordered and disordered regions of the semicrystalline conjugated polymer.8–10,19,28
(9) Swelling: the electrolyte solution swells the polymer due to intercalation of redox-active components and solvent molecules.
3.6. Bulk intercalation and volumetric change in conjugated conducting polymers
The pseudocapacitive characteristics of conjugated conducting polymers differ from those of double-layer capacitors due to their capability for bulk intercalation, allowing ions to volumetrically penetrate the polymer structure, as exhibited in Fig. 6c (redox pseudocapacitance mechanism). Since the bulk of the conjugated conducting polymer participates in the redox reaction, rather than just the surface or interfacial layer between the electrode and electrolyte, pseudocapacitors typically provide higher specific capacitance and energy density than EDLCs.3,33 The insertion and extraction of redox-active species generally lead to significant changes in the dimensions of the polymer. The total volumetric change in conducting polymers can be attributed to two distinct components: (i) changes in the bond length and conformation of the polymer chain, and (ii) osmotic expansion of the polymer phase, which is the structural change associated with the movement of solvent ions.184 The concentration of ionic species within the polymer structure is determined by Donnan equilibrium,197 and controlled by fulfilling two requirements: maintaining the electroneutrality of the polymer and preserving electrochemical equilibrium between the electrolyte and the polymer, as described by the following equations:184 | (11) |
Consequently, the maintenance of electrochemical equilibrium between the electrolyte and the polymer is governed by the following equation:184
![[small mu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cd.gif) pi = Ii pi = Ii | (12) |
pi and Ii denotes the electrochemical potentials of the polymer and electrolyte phases, respectively. The subscript i for both cases refer to the various ionic species present in the system.
The swelling and shrinkage of polymer structures during doping and de-doping processes make conducting polymers prone to rapid degradation with repetitive cycles of charging and discharging, leading to diminished capacity retention and cycling stability. To improve mechanical stability of conducting polymers during doping/de-doping process, researchers have developed composite conducting polymer nanostructures,22 utilized advanced fabrication techniques like oCVD method to create rigid, durable polymeric thin films.9,10
3.7. Ionic conductivity in conjugated conducting polymers
A notable electrochemical property of conducting polymers is their ability to facilitate hybrid electrical and ionic conductivity by transferring both ions and electrons. The intercalation of ions within the conducting polymer matrix enables these materials to engage their full or partial volume during electrochemical redox reactions (doping/de-doping processes).8 The ability of conducting polymers to facilitate the intercalation of ions from the electrolyte into their matrix effectively broadens the region of active electron transfer beyond the electrode surface or the electrode/electrolyte interface, thereby enhancing their pseudocapacitance characteristics.9,10,18Ionic conduction in mixed ionic and electronic conductivity (MIEC) materials is typically described by the Nernst–Planck drift-diffusion model, which quantifies the ionic flux  as the number of moles of ions passing through a unit area per unit time (mol cm−2 s−1).8 The ionic flux is influenced by both the electrochemical gradient potential
as the number of moles of ions passing through a unit area per unit time (mol cm−2 s−1).8 The ionic flux is influenced by both the electrochemical gradient potential 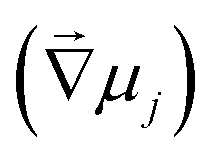 and the convective velocity
and the convective velocity  as described by the following equation:8,198,199
as described by the following equation:8,198,199
 | (13) |
The electrochemical driving force induces both the concentration gradient of species j and the gradient in the applied electrostatic potential  , as expressed by: (ref. 3 and 8)
, as expressed by: (ref. 3 and 8)
 | (14) |
In the absence of convective flow 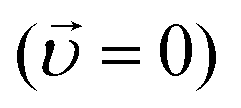 and an applied electric field
and an applied electric field  the Nernst–Planck equation simplifies to Fick's law of diffusion. The diffusivity (Dj) and mobility (μj) of an ion are assumed to be interconnected via the Nernst–Einstein equation:8
the Nernst–Planck equation simplifies to Fick's law of diffusion. The diffusivity (Dj) and mobility (μj) of an ion are assumed to be interconnected via the Nernst–Einstein equation:8
 | (15) |
The order and orientation of semicrystalline conjugated conducting polymers are expected to affect ionic transport.10,31 Ion diffusivity likely varies between crystalline domains and the surrounding amorphous regions.8,10,19 The influence of tie-chains on ionic transport has received less attention, though noted in electrical conductivity. The orientation of crystallites relative to the substrate is anticipated to cause anisotropy in ionic diffusivity.3 Variations in dopant levels, which alter electrostatic forces and polymer chain conformation, also impact ionic diffusivity.
The effects of counterion size,10,31 and semicrystalline orientation10,25,200 on ionic transport in these polymers are reported. The size and nature of the counterion dopant are crucial factors and significantly influencing the electrochemical performance of conjugated conducting polymers.8,10,29 Conducting polymers can be doped with various counterions, ranging from small monoatomic anions like chloride (Cl−) to large macromolecular polyanions such as poly(styrene sulfonate) (PSS).2,201 The chemical structures of several negatively charged molecular counterions used as dopants for PEDOT conducting polymer, including PSS, tosylate, and hexafluorophosphate, are shown in Fig. 8a–c. Additionally, the molecular structure of the dopant 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ), commonly used as a counterion dopant with P3HT and other conjugated polymers, is presented in Fig. 8d. Using small counterion dopants, such as chloride (Cl−), in conjugated conducting polymer thin films facilitates redox exchange with the electrolyte and exhibits a wider electrochemical potential window compared to larger macromolecular dopants, providing a significant advantage for electrochemical energy storage devices.8,10,30,31
 | ||
| Fig. 8 Impact of counterion size and semicrystalline orientation on electrochemical properties of conjugated polymers. Chemical structure of counterion dopants used in conjugated polymer: (a) PSS, (b) tosylate, (c) hexafluorophosphate, and (d) F4TCNQ. (e) PEDOT thin film with edge-on semicrystalline orientation shows higher pseudocapacitance and specific capacitance compared to amorphous and face-on oriented counterparts.10 (e) Reproduced with permission.10 Copyright 2024, Wiley. | ||
Minimizing π–π stacking distance is crucial for enhancing charge carrier mobility and electron conductivity, which is achievable through a more ordered structure. Conversely, facile ion migration between the electrolyte and polymer matrix, leading to increased ionic conductivity, necessitates a more relaxed structure. Optimizing the structure of conducting polymers to maximize both in-plane electrical conductivity and ionic conductivity presents a significant challenge due to the inherent trade-off between these parameters.3,7,29 Notably, the edge-on orientation of crystallites in conjugated conducting polymers has been reported to enhance pseudocapacitance due to the inherent anisotropy in the direction of the conjugated backbone (Fig. 8e).10 The edge-on orientation offers higher out-of-plane electrical conductivity, enabling faster ion exchange with the electrolyte and resulting in increased pseudocapacitance in conducting polymers.10,25,31
The ionic conductivity of a conducting polymer thin film can be measured by utilizing electrochemical impedance spectroscopy (EIS), where the conducting polymer film is used as the working electrode under various configurations.202–207 The obtained impedance response from the input containing sinusoidal perturbation is comprised of double-layer formation, diffusion and migration of ions, transport of electrons, and transfer of charges.204 In addition, electrochemical impedance models for conducting polymers describe various impedances as distributed parameters across the porous polymer thin film's thickness, rather than treating them as localized variables.208,209 This approach considers the polymer film as a non-homogeneous structure, with distributed properties instead of a macroscopic boundary between the polymer and the electrolyte. The ionic resistance (Rion) and electronic resistance (Re) of the polymer phase and the interfacial faradaic impedance (Zf) are considered as distributed circuit elements in equivalent circuit model. Assuming rapid interfacial charge transfer, the real component of the impedance (Zreal) can be defined by the following equation:204,205
 | (16) |
The value of Zreal can be obtained by extrapolating the low-frequency line to meet the real axis. Finally, the ionic conductivity (σion) of the conducting polymer film can be measured by the following equation:206,207
 | (17) |
4. Applications of conducting polymers in electrochemical energy storage devices
Energy storage systems must accommodate various demands depending on their application, whether providing high energy output over short durations (as seen in electric vehicles during acceleration) or delivering continuous energy over extended periods (as required for grid applications). Among the diverse available energy storage technologies, electrochemical energy storage devices have quickly become prominent across multiple applications due to their superior attributes, including high coulombic efficiency, elevated energy and power densities, scalability, modularity, and rapid response capabilities. Transition metal oxides and conjugated conducting polymers have attracted significant attention in the field of electrochemical energy storage devices. Conjugated conducting polymers offer key advantages for commercial electrochemical energy storage, such as inherent flexibility, unique pseudocapacitive behavior, dual ionic/electronic conduction, fast doping/de-doping dynamics, bulk ionic intercalation, high specific capacity, and excellent structural and thermal stability. Moreover, the commercialization of electrochemical energy storage devices requires large-scale production, and the CVD process is a robust manufacturing method well-suited for large-scale production via roll-to-roll (R2R) processing. The R2R CVD technique is already widely employed in the large-scale fabrication of semiconductors,144 and high-temperature superconductors.210–216 Similarly, the commercialization of conjugated conducting polymers can be achieved through the R2R oCVD process, which is currently in the early stages of research and development.2,144,146,201,2174.1. Lithium-based batteries
The large-scale adoption of electric vehicles (EVs), electronics, and drone applications has created an urgent demand for high-energy, durable, and cost-effective lithium–ion batteries (LIBs).32,218 Lithium-based (Li–ion and Li–metal) batteries have become a cornerstone of modern electrochemical energy storage technology, offering superior energy density, higher efficiency, and extended longevity. Moreover, lithium–ion batteries (LiBs) are the most advanced secondary (rechargeable) batteries currently available for industrial-scale production. The outstanding attributes of Li–ion batteries include higher energy and power densities, greater thermal stability, low self-discharge rates, and extended cycle life.219 A typical Li–ion battery comprises several key components, including a positive electrode (cathode), a negative electrode (anode), electrolytes, a separator, current collectors, electrolyte additives, and binders.The active materials used as cathodes in Li–ion batteries typically consist of lithium transition metal layered oxides, such as lithium cobalt oxide (LCO), lithium manganese oxide (LMO), lithium iron phosphate (LFP), lithium nickel cobalt aluminum oxide (LNCA), and lithium nickel manganese cobalt oxide (LNMC). The atomic percentages of the constituent elements in the cathode can be adjusted to develop desirable characteristics. The most commonly used material for anodes in Li–ion batteries is graphite, which is typically coated on copper foil. However, graphite is susceptible to low usable specific capacity due to the limited intercalation of Li between adjacent graphene planes.220 To develop anodes with higher energy and power densities, researchers are exploring materials such as silicon (Si), phosphorus (P), and lithium (Li), as well as their composites with graphite.220
The electrolytes in Li–ion batteries typically contain lithium salts such as lithium hexafluorophosphate (LiPF6), lithium perchlorate (LiClO4), and lithium hexafluoroarsenate (LiAsF6), which are dissolved in organic solvents like ethyl methyl carbonate, dimethyl carbonate, diethyl carbonate, propylene carbonate, and ethylene carbonate.221 To prevent unwanted side reactions and decomposition of the electrolytes, a variety of additives have been utilized, including vinylene carbonate, fluoroethylene carbonate, ammonium perfluoro-caprylate, and 4-propyl-[1,3,2]dioxathiolane-2,2-dioxide (PDTD).222,223
The separator, serving as a safety barrier between the anode and cathode, is electronically insulating but permeable to Li–ions and is typically made from polyethylene and polypropylene. Aluminum foils are commonly used as current collectors for the cathode, while copper foils are employed for the anode, both of which are ultimately connected to the battery terminals. Conductive additives like graphene are often incorporated into cathodes. The common binder supporting matrices include carboxymethyl cellulose (CMC), and polyvinylidene fluoride (PVDF) with N-methyl-2-pyrrolidone (NMP) as the solvent.224
The working principle of Li–ion batteries relies on the shuttling of lithium ions between the cathode (positive electrode) and anode (negative electrode) through the electrolyte.221 During charging, Li+ ions de-intercalate from the cathode, diffuse through the electrolyte, pass through the nano-porous separator, and intercalate into the anode. Concurrently, electrons travel through the external circuit to maintain electrical neutrality. During discharge, lithium atoms in the anode release electrons, becoming Li+ ions that migrate back to the cathode. Simultaneously, electrons flow from the anode to the cathode via the external circuit, providing electrical energy. The movement of Li+ ions during battery operation generates significant heat due to Joule heating and the chemical energy involved in the charge/discharge processes. While heat generation is an intrinsic aspect of battery function, insufficient dissipation pathways in certain charge/discharge states can lead to excessive heating, posing a serious risk to battery safety.225
A current major focus in Li–ion batteries is enabling fast charging while maintaining rate capacity, which is a crucial requirement for the widespread adoption of electric vehicles (EVs).226,227 The target benchmark for EV batteries is to achieve a fast charging rate that allows for 80% battery capacity to be replenished in 10 minutes or less. The current rate (C-rate) is defined as the charging or discharging current density required to achieve or utilize the theoretical capacity of a battery within 1 C−1 hour−1.228 The C-rate metric measures the rate at which a battery is charged or discharged, normalized against its maximum capacity. Despite the challenges, achieving fast charging (high C-rate charging) remains a key requirement for advancing Li–ion battery technology. However, several issues associated with fast charging, such as structural instability, poor charge transport kinetics, and accelerated degradation, need to be addressed.229
The SEI layer is a passivation layer that forms on the anode at the electrode–electrolyte interface when the redox potential of the electrode falls outside the electrochemical stability window of the electrolyte.238 The SEI layer results from the reaction between a thermodynamically unstable electrode and the electrolyte during the initial cycles, comprising decomposition products of the electrolyte. Due to its complex microstructure and chemical composition, the ionic conduction within the SEI involves multiple pathways: (i) interstitial and vacancy mechanisms in the inorganic regions, (ii) inter- and intrachain hopping in the polymeric regions, (iii) conduction through grain boundaries, and (iv) surface diffusion in highly porous SEIs. Ion diffusion in the inorganic component of the SEI typically serves as the rate-limiting step.
Optimizing the anode surface area is crucial for enhancing both energy and power density in Li–ion batteries with regard to the stable SEI formation. Minimizing the anode surface area is advantageous for energy density, as it reduces lithium consumption for SEI formation; however, it is detrimental to power density due to solid-state diffusion limitations. Conversely, increasing the anode surface area improves power density but consumes more Li for SEI formation, thereby reducing energy density.35 Introducing an artificial SEI layer is a sophisticated approach to enhancing overall battery performance by stabilizing the Li–metal anode. This surface engineering method, which utilizes fluorides, phosphates, and polymers, has been shown to improve electrode stability, increase electronic and ionic conductivity, and suppress the dissolution of transition-metal elements from the cathode.239
The artificial SEI film acts as a protective layer on the surface of the Li metal electrode, replicating the functionalities of the SEI layer. By regulating Li+ deposition and mechanically suppressing Li dendrite growth, the ionic conductive polymeric thin film effectively enhances the electrochemical performance of Li–metal batteries. Polymer materials with excellent ionic conductivity, superior processability, and high flexibility are considered ideal for artificial SEI films.239 Conducting polymer thin films, such as polypyrrole (PPy), can function as an artificial SEI layer to achieve a stable, dendrite-free Li metal anode by allowing Li ions to interact with the pyrrole ring through cation–π interactions, resulting in the dispersion of Li salt within the PPy thin film.36 PEDOT-based copolymers, PEDOT co-poly(ethylene glycol) (PEDOT-co-PEG), when deposited on the surface of a lithium anode, have been reported to act as protective layers, forming a stable and less resistive SEI layer while suppressing the growth of Li dendrites (Fig. 9a).240 The utilization of a layered conductive polyaniline (LCP) coating on Si nanoparticles, via a trimethoxysilyl propyl aniline (TMSPA) bridge, forms an LCP-integrated hybrid SEI layer with high conductivity and mechanical stability, enhancing cycling stability and maintaining high coulombic efficiency for Si anodes (Fig. 9b).241
 | ||
| Fig. 9 Artificial solid–electrolyte interface (SEI) with conducting polymers for Li-based batteries. (a) Schematic representation of a Li–S battery featuring a lithium sheet anode protected by a PEDOT-co-PEG layer.240 (b) Schematic representation of Si nanoparticles (SiNPs) modification through in situ polymerization to form a layered conductive polyaniline (LCP) coating.241 (a) Reproduced with permission.240 Copyright 2014, Royal Society of Chemistry. (b) Reproduced with permission.241 Copyright 2022, Wiley. | ||
Addressing the significant volumetric expansion of Si anodes requires the development of a stable SEI to prevent interfacial side reactions. Applying conducting polymer coatings to porous Si anodes significantly improves electrochemical performance by forming a conformal surface coating that stabilizes the SEI layer, enhances electronic conduction, improves rate capability and cycling stability, increases volumetric tolerance, enhances electrolyte uptake, and facilitates electron transport.12,37,38,246,247
Yu et al.12 implemented an n-doped, porous biphenyl-polyoxadiazole (bPOD) conducting polymer layer on a Si anode, observing significant improvement in its rate capacity due to the hybrid ionic–electronic conductivity of the bPOD porous layer and better control of SEI formation by inhibiting direct exposure between the Si and electrolyte (Fig. 10a). Furthermore, the Si-bPOD electrode exhibited higher coulombic efficiency and specific capacity compared to its pristine counterparts, with excellent capacity retention of 85% and 80% after 500 and 800 cycles, respectively (Fig. 10b).12 The construction of conducting polymer hydrogels made of PANI over Si nanoparticles (SiNPs) exhibited an enhancement in current density, with approximately two orders of magnitude higher performance than the dried PANI hydrogels (Fig. 10c and d).246 The utilization of a PEDOT coating on Si nanowires (SiNWs) improves cycling stability (the capacity retention after 100 cycles increased from 30% to 80%) and maintains structural integrity, preventing the cracks commonly found in non-coated SiNWs after cycling (Fig. 10e).247 The utilization of a poly[3-(potassium-4-butanoate)thiophene] (PPBT) conducting polymer layer on a Si anode significantly enhanced rate capability and cycling stability by increasing tolerance of volumetric changes, improving electrolyte uptake, facilitating electron transport, and stabilizing the SEI layer (Fig. 10f).38
 | ||
| Fig. 10 Utilization of conducting polymers in anodes for Li–ion batteries. (a) Schematic illustration of fabricating a porous bPOD layer on a Si electrode.12 (b) Long-term cycling performance comparison between pristine Si and Si-bPOD electrodes.12 (c) Schematic illustration of a PANI hydrogel 3D network encapsulating Si nanoparticles.246 (d) CV diagram for SiNP-PANI hydrogel and PANI hydrogel within the potential range of 0.01–1 V vs. Li/Li+.246 (e) Morphology and schematic illustration of PEDOT-coated Si nanowires (SiNWs).247 (f) Schematic illustration of procedure for fabricating PPBT conducting polymer on Si anode.38 (a) and (b) Reproduced with permission.12 Copyright 2023, Wiley. (c) and (d) Reproduced with permission.246 Copyright 2013, Nature. (e) Reproduced with permission.247 Copyright 2012, Royal Society of Chemistry. (f) Reproduced with permission.38 Copyright 2012, American Chemistry Society. | ||
A few other promising anode materials, such as Germanium (Ge), manganese-based oxides (MnxOy), and aligned carbon nanotubes (ACNTs), have also been combined with conducting polymers to mitigate their inherent limitations and enhance electrochemical performance.248–250 Ge has significantly higher conductivity and Li–ion diffusivity (about 400 times higher) than Si at room temperature, but it has a lower specific capacity (1600 mA h g−1) and also undergoes large volume changes and rapid capacity fading.251 The deposition of PEDOT:PSS conducting polymer on Ge anodes has demonstrated enhanced rate capability and cycling stability, indicating improved structural integrity and reduced pulverization and exfoliation of the Ge shell.248 The deposition of PEDOT on Mn2O3 nanowires has been shown to reduce surface resistance, prevent the pulverization of surface electron channels, and enhance cycling stability, achieving a reversible capacity of 1450 mA h g−1 after 200 cycles.249
Conducting polymers have been effectively utilized as coating layers on various cathode surfaces in lithium–ion batteries, acting as barriers between the cathode and electrolyte to prevent undesirable side reactions, provide thermal and structural stability, enhance long-term cycling stability, and improve high-rate performance.15,39,253–255 The application of a conformal PEDOT coating on the cathode surface has been shown to decrease cell impedance, reduce interfacial charge transfer resistance between the electrode and electrolyte, and improve electron transport to the current collectors.2,256 The favorable chemical and coordination bonds (between oxygen, sulfur (S) in the PEDOT structure, and manganese (Mn) in the cathode structure) stabilize the cathode surface, inhibiting Mn dissolution and preventing SEI growth, which are primary aging mechanisms.256,257
Uniformly coating NCM811 with conducting polymers is challenging due to poor bonding between the materials. The oCVD method is a versatile manufacturing technique for fabricating highly conformal, ultrathin coatings of conducting polymers on various cathode materials in Li–ion batteries, such as NMC and LMO, resulting in enhanced stability and electrochemical performance.2,8,15,255,256 These improvements are attributed to oCVD's ability to conformally coat individual particles beyond the surface layer, significantly enhancing crystal and interfacial stability for all particles by providing a stable cathode–electrolyte interface, suppressing microcrack formations, and preventing irreversible reactions.
Gan et al.253 demonstrated that a polyvinylpyrrolidone (PVP)-induced PANI coating on NCM811 cathode surfaces enhanced stability and rate performance, achieving the highest capacities at various scan rates by preventing side reactions and facilitating fast diffusion channels for Li–ions (Fig. 11a). The surface of NCM811 was modified with a PANI–PEG hybrid layer to simultaneously utilize PANI's high electronic conductivity and PEG's exceptional ionic conductivity, creating a facile diffusion pathway for Li ions and resulting in improved capacity and cycling performance, particularly at high C-rates (Fig. 11b).254 Applying a highly conformal oCVD PEDOT layer on various NCM cathodes enhanced thermal and structural stability, improving capacity retention by suppressing undesired structural evolution during charge–discharge cycles (layered to spinel structure), as shown in Fig. 11c.15
 | ||
| Fig. 11 Utilization of conducting polymer coatings on various Ni-rich NCM cathode surfaces. (a) Schematic illustration showing the mechanism of developing the PANI layer on the surface of NCM811 cathode using PVP surfactant.253 (b) TEM images of NCM811 coated with PANI–PEG copolymer.254 (c) STEM-HAADF images with corresponding EDS mappings of the secondary particles surface (left) and inner primary particles surface (right) of oCVD PEDOT-coated NCM.15 Ptychographic X-ray computed tomography and corresponding SEM images showing particles after cycling between 2.8 and 4.6 V for 200 cycles of (d) pristine NMC83 cathode and (e) oCVD PEDOT coated NMC83 cathode.32 (a) Reproduced with permission.253 Copyright 2019, American Chemistry Society. (b) Reproduced with permission.254 Copyright 2018, American Chemistry Society. (c) Reproduced with permission.15 Copyright 2019, Nature. (d) and (e) Reproduced with permission.32 Copyright 2022, American Chemical Society. | ||
The thermal stability of NMC can be assessed by comparing oxygen release profiles for highly charged pristine and PEDOT-coated NMC, to identify side reactions on the coated cathode surface.258 Previous studies have shown that oxygen species (i.e., O2−, O−, O22− and O2) are released during the phase transition of charged NMC upon heating, which can cause thermal runaway by reacting with the flammable electrolyte.258 The deposition of a conformal oCVD PEDOT thin film on the single-crystal Ni-rich NMC cathode (NMC83: LiNi0.83Mn0.1Co0.07O2) significantly improved capacity retention, achieving 85.3% after 100 cycles at a 0.5C rate, compared to 59.6% for the pristine electrode.255 The post-cycled microstructure of the pristine NMC83 cathode exhibited substantial crack formation due to heterogeneous phase transitions from layered to spinel and rock-salt structures, leading to internal strain and capacity loss; whereas PEDOT-coated NMC83 retained its initial structure (Fig. 11d and e).32
Another cathode material for next-generation Li–ion batteries, the Li-rich cathode (Li1.2Ni0.2Mn0.6O2), demonstrates a higher reversible capacity (∼250 mA h g−1) compared to traditional cathodes (LiCoO2, LiFePO4, and LiMn2O4) with capacities around 170 mA h g−1.259,260 However, side reactions at high voltages lead to undesired phase transitions, and the insulating nature of Li2MnO3 formed during cycling results in poor rate capability and low cycling performance.259 To address these issues, depositing PEDOT:PSS with optimized thickness on the Li-rich cathode improves discharge capacity and rate capability due to the conductive nature of PEDOT and its role as a barrier between the cathode and electrolyte, preventing undesired reactions.259 Su et al.261 deposited various polymeric materials, including PEDOT, on the LiCoO2 (LCO) cathode surface and observed significant enhancements in Li+ kinetics, resulting in increased rate capability at C-rates >5C and enhanced cycle life at high voltage of 4.5 V (Fig. 12a and b).
 | ||
| Fig. 12 Utilization of conducting polymer coatings on various sulfur cathodes in Li–S batteries. (a) Comparison of cell capacities for various polymer-coated LCO cathodes.261 (b) Capacity retention of different LCO cathodes at C/2 within 3.0–4.5 V.261 (c) SEM images of oCVD PEDOT–coated sulfur cathode.55 (d) Cycling performance of Li–S pouch cell with the PEDOT–coated sulfur (PE), and pristine sulfur (P2).55 (e) Schematic illustration showing PEDOT:PSS coating on CMK-3/S composite electrode, blocking polysulfide dissolution.262 (f) Illustration of the synthesis process for fabricating a PPy layer on sulfur cathodes to form S/PPy composites via in situ polymerization.56 (a) and (b) Reproduced with permission.261 Copyright 2021, American Chemical Society. (c) and (d) Reproduced with permission.55 Copyright 2023, Elsevier. (e) Reproduced with permission.262 Copyright 2011, American Chemical Society. (f) Reproduced with permission.56 Copyright 2016, Elsevier. | ||
The lithium–sulfur (Li–S) battery is considered one of the most promising Li–metal-based battery technologies, offering a high gravimetric energy density of ∼2600 W h kg−1 and serving as a low-cost energy storage method. However, the practical utilization of Li–S batteries faces several challenges: (i) poor rate capability and limited electrode rechargeability due to the insulating nature of S8 and Li2S, (ii) rapid capacity degradation resulting from the loss or dissolution of active material, as soluble lithium polysulfides (Li2Sn, where 3 ≤ n ≤ 6) migrate from the cathode to the electrolyte and anode, causing electrolyte degradation and a non-uniform anode microstructure, and (iii) significant volume increase (∼80%) during charging, as S8 converts to low-density Li2S.33,50–52 To enhance active material utilization and cycling stability of the sulfur cathode, various advanced sulfur composites have been employed, including dispersing sulfur in conductive mediums such as carbon-based materials,53,54 metal oxides/carbides, and conducting polymers like PANI, PPy, and PEDOT.55,56,262,263 Coating the sulfur cathode in Li–S batteries with conjugated conducting polymers provides substantial benefits, such as enhanced capacity retention and improved overall capacity by restricting the shuttling of polysulfide intermediates. Additionally, conducting polymers help mitigate electrode degradation caused by the volume expansion of sulfur-based cathode materials due to their self-healing properties.55,57
Zhang et al.55 demonstrated a practical Li–S application using pouch cells by depositing a highly conformal oCVD PEDOT layer on sulfur particles, achieving notable capacity retention of 85% after 300 cycles at 0.5C due to the restriction of polysulfide shuttling in the cathode (Fig. 12c and d). Yang et al.262 deposited a layer of PEDOT:PSS across the surface of a mesoporous carbon/sulfur composite (CMK-3/S) cathode and observed that the polymer layer effectively confined the shuttling of polysulfide intermediates and prevented dissolution during cycling, resulting in enhanced capacity retention and coulombic efficiency (Fig. 12e). To encapsulate sulfur and its polysulfide compounds within the cathode, PANI nanotubes are synthesized with sulfur to form a 3D, cross-linked sulfur–PANI composite, leading to improved cycling stability and rate capability.263 Xie et al.56 deposited an approximately 60-nm PPy layer on sulfur cathodes via in situ polymerization, forming S/PPy composites with a core–shell structure, which prevented sulfur particle aggregation and resulted in faster ion transfer and enhanced C-rate performance (Fig. 12f).
Additionally, conducting polymers and their composites can serve as binder materials for Li–ion battery electrodes because they provide flexible electronic contacts, maintain adhesive strength during volumetric changes, and act as both binder and conductive additive, enabling high active material loading.267–269 Zhong et al.268 used a water-soluble conductive composite binder with carboxymethyl chitosan (CCTS) as the binder and PEDOT:PSS as the conductive agent for lithium iron phosphate (LFP) cathodes, and achieved homogeneous, continuous conducting bridges and improved rate performance. Das et al.267 demonstrated a carbon black-free LFP electrode in which PEDOT:PSS served as both binder and conducting additive, resulting in enhanced rate capacity and cycling stability. Combining carboxymethyl cellulose (CMC) with PEDOT:PSS as a water-soluble composite binder for a Si anode results in superior cycling and rate capacity, as well as faster kinetics, compared to the acetylene black/CMC binder.269
4.2. Utilization of conducting polymer coatings in supercapacitors
Capacitance refers to a material's ability to store electric charge, and devices that store energy in the form of charges are commonly known as capacitors. Supercapacitors (also referred to as ultracapacitors or electrochemical capacitors) are energy storage devices that offer significantly higher capacitance compared to traditional dielectric capacitors. Supercapacitor devices are engineered to fill the gap between conventional capacitors and rechargeable secondary batteries and balancing specific power with energy density, as depicted in the Ragone plot in Fig. 13a. Supercapacitors have gained considerable attention for their exceptional capacitance (100–1000× higher than a conventional capacitors), high power density, rapid charge/discharge times (1–10 second), and extended cycling life (>30000 h), making them ideal for applications such as flexible electronics, electric vehicles, and renewable energy.7,20,58,270 Supercapacitors have found applications in several crucial areas, including electric vehicles, power grids, and aerospace technology.2,271
 | ||
| Fig. 13 Utilization of PEDOT conducting polymer coatings in supercapacitors. (a) Ragone plot illustrating the relative positions of EDLC and pseudocapacitors, showing how supercapacitors bridge the gap between batteries and traditional capacitors in terms of energy and power density. (b) Specific capacitance values for carbon cloth (CC) and oCVD PEDOT-coated carbon cloth (PEDOT-CC) electrodes. The oCVD PEDOT thin films fabricated at different deposition temperatures.10 (c) Nyquist plots for pristine carbon cloth and PEDOT-coated carbon cloths fabricated at different deposition temperatures.10 (d) Magnified SEM image showing an individual carbon fiber with a highly conformal oCVD PEDOT coating.10 (e) Schematic illustrations of the intermediate and final configurations of a vapor-printed solid-state supercapacitor, where gray for porous substrate, gold for current collector, blue for PEDOT, and green for ion gel electrolyte.272 (b)–(d) Reproduced with permission.10 Copyright 2024, Wiley. (e) Reproduced with permission.272 Copyright 2017, Wiley. | ||
Supercapacitors typically consist of a current collector, an electrode active material (which may be submerged in a liquid electrolyte or exist as a solid-state in solid-state capacitors), and a porous dielectric separator between the two electrodes.2 Two key performance metrics for supercapacitors are energy density (or specific energy) and power density (or specific power), which are evaluated in relation to parameters such as mass, volume, or area. The energy density (ED,X) and power density (PD,X) in supercapacitors are defined as follows:181
 | (18) |
 | (19) |
Depending on the energy storage mechanism, supercapacitors can be classified into three categories: (i) electrochemical double layer capacitors (EDLCs), which store charge electrostatically by forming a non-faradaic double layer at the electrode surface, (ii) pseudocapacitors, which store charge electrochemically through faradaic electron transfer via fast redox reactions, and (iii) hybrid supercapacitors, which combine both EDLC and pseudocapacitor charge storage mechanisms, and utilizing hybrid or asymmetric electrodes.273
In EDLCs, energy storage is facilitated by a non-faradaic process, where charge accumulates at the electrode–electrolyte interface, forming an electrochemical double layer that is responsible for the capacitance. In supercapacitors, capacitance increases with larger electrode surface areas, leading to the exploration of high-surface–area materials like carbon nanotubes (CNTs) and other nanoporous structures as potential electrodes.2 Carbon-based materials with high surface areas, such as activated carbon, CNTs, and graphene are widely used as electrode materials in EDLCs. The specific capacitance of EDLCs is determined by the exposed surface area and charge storage capacity of the carbon-based electrode materials, but it is inherently limited by the non-faradaic charge storage mechanism. In contrast, pseudocapacitors offer 10 to 100 times higher specific capacitance due to the faradaic (redox-reaction) charge storage mechanism that occurs within the bulk of the active materials.3 EDLCs offer higher power density but lower energy density compared to rechargeable batteries. Pseudocapacitors generally exhibit significantly higher specific capacitances compared to carbon materials utilizing the EDLC mechanism, as the entire bulk of the pseudocapacitive material, rather than just the surface layer, participates in redox reactions.10,20 Pseudocapacitors, on the other hand, typically achieve significantly greater energy density than EDLCs, positioning them in an intermediate specific energy range between EDLCs and rechargeable batteries.2
Recently, various flexible supercapacitor designs have been explored, including wire-shaped supercapacitors,274 fiber-shaped coaxial supercapacitors,275 carbon based textiles,10 and highly stretchable fiber-shaped supercapacitors.276 High surface area carbon-based materials like activated carbon (AC), carbon nanotubes (CNTs), graphene, graphene oxide (GO), carbon cloths (CCs), and carbon fibers (CFs) are commonly employed as active electrode materials in non-faradaic EDLCs. In contrast, conjugated conducting polymers and transition metal compounds (TMCs) such as metal oxides (e.g., RuO2, MnO2, V2O5, and Fe2O3), metal sulfides, metal nitrides, and metal carbides are commonly and effectively employed as electrode materials for faradaic pseudocapacitors.2 The mechanical stability of conjugated conducting polymers is effectively utilized in the fabrication of flexible supercapacitors due to their simple synthesis, redox properties, and electrical conductivity.2,20
Conducting polymers, in particular, have shown great potential as supercapacitor electrode materials due to their unique combination of hybrid ion–electron transfer capabilities, fast doping/de-doping mechanisms, excellent conductivity, mechanical flexibility, and high environmental stability.2,10 Conducting polymers are promising pseudocapacitor materials for flexible supercapacitors, advancing energy storage devices for future flexible electronics due to their high redox capacitance and inherent elasticity.
The most commonly utilized conducting polymers in supercapacitor applications include PANI, PEDOT, PPy, and PT, which are typically fabricated using solution-based or vapor-based techniques. PANI stands out as a leading candidate among conjugated conducting polymers for supercapacitors due to its excellent pseudocapacitive properties, as demonstrated by PANI nanowire arrays achieving a high specific capacitance of up to 950 F g−1.277 Typically, conjugated conducting polymers possess multiple redox states, allowing for the precise control of electrical conductivity across the entire spectrum from insulator to metal, making them suitable for use in supercapacitors operating via a pseudocapacitance mechanism.2,8,278 Conducting polymers generally exhibit high specific capacitances, such as 1284 F g−1 for PANI,59 480 F g−1 for PPy,60 and 210 F g−1 for PEDOT.10,61
PEDOT is a widely used conducting polymers in supercapacitor devices due to its desirable properties such as possessing a wide potential electrochemical window over which the capacitance is high, excellent thermal and chemical stability, high charge mobility and electrical conductivity that induces fast electrochemical kinetics, and excellent cycling stability.2,20,279 However, PEDOT has a disadvantage of relatively high molecular weight which leads to the approximately low specific capacitance, especially compared to PANI which exhibits the highest theoretical specific capacitance among conjugated conducting polymers.2
To address the cycle stability limitations of polyaniline, it is commonly composited with carbonaceous materials such as graphene, activated carbon, or graphite. Carbonaceous materials such as CNTs, graphene, and graphite hold significant potential across various fields due to their exceptional structural, mechanical, and electrical properties. A significant drawback of polyaniline–carbon based composites is their tendency to aggregate during synthesis, which reduces active sites and impairs ion mobility, ultimately compromising supercapacitor performance. To address this issue, researchers have shifted their focus to 3D porous carbon structures.
Supercapacitor devices composed entirely of conducting polymers can be configured in three main types:2,20,280
(1) Type I (symmetric): utilizes the same p-type conducting polymer for both electrodes.
(2) Type II (asymmetric): employs two different p-type conducting polymers with distinct electroactivity ranges for the electrodes.
(3) Type III (symmetric): uses the same conducting polymer for both electrodes, but the p-doped form serves as the positive electrode and the n-doped serving as the negative electrode.
Additionally, hybrid (or asymmetric) devices can be constructed with a conducting polymer as the positive electrode and a carbon or lithium-based material as the negative electrode. Among these configurations, the Type III conducting polymer-based supercapacitors offer the most promising theoretical performance due to their ability to maintain both electrodes in highly conductive states, allowing operation over a broader potential range and achieving a significantly higher maximum cell potential (Vmax,c). In Type I and II conducting polymer-based supercapacitors, the positive electrode remains in a fully doped (conductive) state when fully charged, while the negative electrode stays in a de-doped (neutral) state. During discharge, both electrodes shift to partially doped states, which limits the achievable maximum cell voltage. In contrast, Type III conducting polymer-based supercapacitors have both electrodes in highly conductive doped states (p-doped and n-doped) when fully charged, and these can transition to partially p-doped or n-doped states upon discharge, depending on the potential difference between the electrodes. This configuration enables the device to operate within a broader potential window, leading to a significantly higher Vmax,c7,20 Conjugated conducting polymers can be positively charged (p-doped) or negatively charged (n-doped) through redox reaction and ion insertion from the electrolyte into their matrix to stabilize the charge.2 However, the practical application of Type III is limited by the challenges associated with fabricating stable n-type conducting polymers.2,20,33
Carbon cloth possesses desirable properties for use as flexible electrodes in various energy storage devices but suffer from limited wettability and low specific capacitance.10 The oCVD PEDOT-coated carbon cloth, fabricated at various deposition temperatures, displayed highly conformal coatings and significantly enhanced specific capacitance, with the highest value of 170.94 F g−1 achieved in a sample fabricated at 80 °C, representing a 2.3-fold enhancement over the pristine carbon cloth (Fig. 13b–d).10 PEDOT-coated carbon cloth samples exhibit lower charge transfer resistance compared to the pristine carbon cloth (Fig. 13c), and the optimal performance of oCVD PEDOT-coated carbon cloths at 80 °C is due to a higher ionic dopant concentration and edge-on orientation, enhancing out-of-plane electrical conductivity.10 Liu et al.272 fabricated the highly conformal oCVD PEDOT thin film on both side of a flexible porous substrate to integrate all components of a supercapacitor into a flexible, porous substrate with a sandwich-like structure (Fig. 13e). This single-step process precisely controlled vapor penetration, enabling the substrate's interior to function as both a separator and solid-state electrolyte reservoir, resulting in a supercapacitor with high volumetric specific capacitance (11.3 F cm−3), power density (0.42 W cm−3), and energy density (2.98 mW h cm−3).272
Conducting polymers can be fabricated into diverse morphologies such as thin films, hydrogels, rods, particles, and conformal nanostructures, each exhibiting unique optical, electrical, mechanical, and electrochemical properties. Numerous studies have indicated a significant correlation between the morphology of conducting polymers and their electrochemical performance. Dubal et al.281 synthesized PPy nanostructures with different morphology (nanobelts, nanobricks, and nanosheets) on steel substrates via electrochemical polymerization, finding that PPy nanosheets achieved a higher specific capacitance of 586 F g−1 at the scan rate of 2 mV s−1, compared to nanobricks (357 F g−1) and nanobelts with the value of 296 F g−1 (Fig. 14a).
 | ||
| Fig. 14 Utilization of conducting polymer coatings in supercapacitors. (a) Schematic illustration of different PPy nanostructures made of nanobelts, nanobricks, and nanosheets, which are fabricated by the electropolymerization, which yields different specific capacitance.281 (b) Schematic illustration of graphene/MnO2/CNT (GMC) and graphene/MnO2/conducting polymer (GMP) systems, showing GM nanostructures wrapped with CNTs or conducting polymers (black: graphene; rose: MnO2; yellow: CNTs; blue: conducting polymer).282 (c) Schematic illustration of graphene composites formed through the polymerization of PANI nanowires.271 (d) Cross-sectional SEM images of PANI-coated Mo2C-CDC electrodes (left) and capacitance comparison of pristine and PANI-coated Mo2C-CDC electrodes across various scan rates (right).283 (a) Reproduced with permission.281 Copyright 2012, Royal Society of Chemistry. (b) Reproduced with permission.282 Copyright 2011, American Chemical Society. (c) Reproduced with permission.271 Copyright 2013, Wiley. (d) Reproduced with permission.283 Copyright 2017, Wiley. | ||
MnO2 is a promising pseudocapacitive material for high-performance supercapacitors due to its high theoretical specific capacitance, low cost, environmental friendliness, and natural abundance, but its performance is often hindered by poor electronic and ionic conductivities, limiting power density and cycling stability.282 The incorporation of PEDOT on MnO2 has been reported to improve cycling stability and increase capacitance.282 The graphene/MnO2/PEDOT:PSS (GMP) ternary composite, demonstrating exceptional cycling stability with over 95% capacitance retention after 3000 cycles and high capacitance, attributed to the significant pseudocapacitance of PEDOT:PSS (Fig. 14b).282
The integration of PANI nanowires on a flexible, free-standing graphene film with a 3D interconnected porous structure (3D-RGO film) enhances the specific capacitance to 385 F g−1, with a rate retention of 94% (Fig. 14c).271 The fabrication of a three-dimensional (3D) PANi–graphene nanoribbon (GNR)–carbon nanotube (CNT) composite (PANI–GNR–CNT composite) via in situ polymerization enhances PANI's cycle life, resulting in 89% capacity retention after 1000 cycles.284 Smolin et al.283 integrated a nanometer-thin film of oCVD PANI onto carbide-derived carbon (CDC) with a bimodal (micro/mesoporous) pore size distribution, enabling PANI to penetrate pores as small as 1.7 nm. The resulting oCVD PANI-integrated CDC supercapacitors achieved a gravimetric capacitance of 136 F g−1, more than double that of uncoated CDC electrodes (60 F g−1) at a scan rate of 10 mV s−1, and demonstrated excellent cyclability, with only a 10% decrease in capacitance after 10000 cycles (Fig. 14d).283
4.3. Utilization of conducting polymer coatings in redox flow battery batteries
Redox flow batteries (RFBs) are highly suitable for large-scale, cost-effective electrochemical energy storage due to their simple and modular design, ease of scalability, long-duration energy storage capabilities, rapid response to load demand, high efficiency, enhanced safety compared to conventional sealed battery systems, and long cycle life.9,62,63 Unlike conventional solid-state batteries, redox flow batteries store redox-active materials in external reservoirs, circulating them to electrodes, such as carbon-based electrodes, for energy storage and discharge instead of confining them within solid electrolytes. The overall power density of a redox flow battery system is determined by the size of its electrode stack, while the total energy density depends on the amount of electrolyte stored in the reservoirs.285,286 To enhance the energy density of redox flow batteries (RFBs), two primary strategies have been employed: increasing the solubility of the molecular reactants and maximizing the redox potential difference between the catholyte and anolyte.A redox flow battery system comprises three key components: (i) separate storage reservoirs for two different electrolytes (anolyte (which flows through the anode), and catholyte (which flows through the cathode)); (ii) stacks of electrodes (anodes and cathodes) separated by a porous, ion-selective membrane; and (iii) a flow system assembly (Fig. 15a). During operation, electrolytes containing dissolved electroactive species are pumped into their respective electrode compartments, where they undergo oxidation or reduction by releasing or accepting electrons at the electrode surface. When the RFB system discharges, the anolyte is oxidized at the anode, releasing electrons that travel through the current collectors to the external load and accumulate at the cathode, where the catholyte accepts the electrons and is reduced.65 Additionally, charged species or supporting ions pass through the ion exchange membrane to complete the reaction and maintain electroneutrality.62 During charging, the reactions reverse: the catholyte is oxidized and releases electrons, while the anolyte accepts electrons and is reduced. In conventional redox flow batteries, no phase changes occur in the reactants, whereas hybrid redox flow battery systems involve phase changes in at least one of the electrode reactions.287
 | ||
| Fig. 15 Utilization of conducting polymers in redox flow batteries. (a) Schematic illustration of redox flow battery system. (b) Schematic illustration exhibits the highly conformal coating of oCVD PEDOT around each carbon fiber, highlighting the penetration of ionic species from the electrolyte through the PEDOT layer and ultimately reaching to the underlying carbon fiber substrate.10 (c) Comparison of cell voltage versus capacity for blank graphite paper, PEDOT:Tos, and PEDOT:PSS.16 (b) Reproduced with permission.10 Copyright 2024, Wiley. (c) Reproduced with permission.16 Copyright 2020, Wiley. | ||
Inorganic redox couples, such as Fe3+/2+/Cr2+/3+, Cr2+/Cr3+, Ti3+/Ti4+, Zn–Br, Sn2+/Sn4+, and V2+/3+/V4+/5+, have been used for energy storage in RFBs. Despite the availability of various redox molecules, rechargeable has shifted toward organic molecules due to the abundance of their constituent elements and the flexibility of organic synthesis for chemical design.288 Additionally, organic redox couples like viologen, anthraquinone, and 2,2,6,6-tetramethylpiperidine 1-oxyl (commonly known as TEMPO), have been investigated in RFBs. However, only a few have been commercialized, including all-vanadium redox flow batteries (VRFB),63 Fe-based and Mn-based RFBs,289,290 and Zn-based hybrid flow battery systems.287,291 As an alternative to the high-cost all-vanadium RFB, RFBs using the same electroactive species with various oxidation states, such as all-chromium, all-iron, all-lead, and all-copper have been proposed.292 Notably, all-iron RFBs have gained significant attention due to their low toxicity, material abundance, and low cost.292,293 The substantial initial expense limits the widespread adoption of RFBs, with recent reports indicating that the cell stack alone accounts for about 31% of the total cost.293 Given that electrodes and membranes form the cell stack, optimizing these components is essential for cost reduction. Electrodes in RFBs must ensure strong electrochemical interactions with various redox couples and maintain stability in both acidic and alkaline environments.
Common electrode materials used in RFBs include low-cost porous carbon options such as carbon fiber paper (CFP), carbon cloth (CC), graphite felt (GF), and their composites with other materials.9,64,65 Carbon cloth holds significant promise for RFB electrodes, but it suffers from low specific capacitance, limited specific surface area, and poor wettability due to its hydrophobic nature.9,10,66 Similarly, graphite felt faces challenges such as low electrochemical activity, limited specific surface area, poor wettability, and suboptimal chemical kinetics, leading to low mass transfer.67,294,295 To address these issues and enhance the performance of carbon-based electrodes, various modification techniques have been explored, including wet chemical oxidation,68,69 electrochemical oxidation,70,71 thermal treatment,72,73 plasma enhancement,74,75 and utilization of conducting polymer coatings.9,10
Depositing thin layers of conducting polymers and their composites on carbon electrodes has emerged as a promising technique to enhance the electrochemical performance of carbon-based electrodes.9,10,16,76,296,297 Organic conjugated conducting polymers are gaining significant attention for RFBs due to their cost-effectiveness, abundance, non-toxicity, minimal environmental impact, and ease of chemical design.9,10,298,299 PEDOT, in particular, can be chemically engineered to control the diffusion of electroactive species through its volume, making it suitable as an ion-selective material.16 As the charge density or doping level of PEDOT thin films increases, electroneutrality is maintained by either expelling cations or inserting anions, depending on the mobility of the counterion dopants.19 For large, immobile counterions such as PSS, cations are expelled from the conjugated polymer chain, and the access of anions from the electrolyte is impeded due to the Donnan exclusion effect.197 Conversely, if the dopant is small and exchangeable (such as chloride or tosylate), it facilitates anion insertion from the electrolyte into the conducting polymer matrix to balance the charge transfer.9,10,19 The Donnan exclusion effect (also known as Gibbs–Donnan effect), describes the distribution of charged ions near a permeable membrane and has been applied to explain the low permeability of vanadium in anion exchange membranes (AEMs) for redox flow batteries.300
PEDOT conducting polymers exhibit notable inherent molecular-level porosity, setting them apart from conventional heterogeneous electrocatalysts.8,25,301 During the electrochemical reaction, the de-doping of PEDOT thin films involves expelling counterion dopants and incorporating anions from the electrolyte into the polymer matrix.10,19 The fabrication of oCVD PEDOT thin films with small, easily exchangeable primary counterions, such as chloride, enhances electrochemical reactions by facilitating the transport of ionic species from the electrolyte through the PEDOT pseudocapacitive layer (Fig. 15b).10 Vagin et al.16 demonstrated congruent ionic charge transport from the electrolyte (4,5-dibenzoquinone-1,3-benzenedisulfonate (tiron)) along with mobile Tos counterion dopants in PEDOT:Tos, whereas such behavior was suppressed in PEDOT:PSS due to the immobile polyanion PSS counterion dopant. This congruent ionic transport in PEDOT:Tos significantly improved the charge and discharge capacity of the flow cell compared to PEDOT:PSS (Fig. 15c).16 Generally, incorporating smaller counterions within the PEDOT polymer chain enhances doping/de-doping capability, promotes ion exchange with the electrolyte, and results in superior overall electrochemical performance.2,10
The application of a highly conformal oCVD PEDOT layer over porous carbon cloth electrodes in redox flow batteries reduces ohmic, kinetic, and mass transport resistances compared to pristine carbon cloth electrodes, leading to increased current density (Fig. 16a–c).9 Compared to unmodified carbon cloth electrodes, oCVD PEDOT-coated carbon cloth electrodes significantly increase the maximum current density at an applied overpotential of 350 mV, achieving 6.7-fold and 3.7-fold enhancements in iron sulfate and iron chloride, respectively (Fig. 16c).9 Oh et al.17 developed a symmetric, metal-free, all-organic redox flow battery using polythiophene microparticles as the redox couple in a non-aqueous electrolyte. This innovative design achieved a high cell potential of 2.5 V, stable cycling performance, and an energy efficiency of 60.9% (Fig. 16d). The performance of carbon cloth electrodes was enhanced by coating them with a TiO2 layer followed by the application of a PEDOT thin film (CC@TiO2-PEDOT) and exhibited high reversibility for the V(II)/V(III) redox reaction (Fig. 16e).302
 | ||
| Fig. 16 Utilization of conducting polymers in redox flow battery electrodes. (a) Photograph of oCVD PEDOT-coated carbon cloth electrodes integrated into the redox flow battery.9 (b) SEM images showcasing the highly conformal coating of oCVD PEDOT on carbon cloths electrodes.9 (c) Cell polarization data at 50% state-of-charge (0.25 M Fe2+/0.25 M Fe3+), comparing the performance of pristine and oCVD PEDOT-coated carbon cloth electrodes in different electrolytes with a flow rate of 15 mL min−1.9 (d) Schematic illustration of a redox flow battery utilizing polythiophene microparticles as both the cathodic and anodic redox-active species.17 (e) CV graphs of carbon cloth electrodes coated with TiO2 and TiO2-PEDOT.302 (a)–(c) Reproduced with permission.9 Copyright 2020, Wiley. (d) Reproduced with permission.17 Copyright 2014, Royal Society of Chemical. (e) Reproduced with permission.302 Copyright 2024, Elsevier. | ||
5. Conclusion and outlook
The synergy between scientific understanding and technological objectives has led to significant advancements in enhancing the electronic, optical, electrochemical, and thermal properties of conjugated conducting polymers. Moreover, their chemical reactivity, biocompatibility, magnetic properties, and mechanical flexibility make them highly attractive for a wide range of applications. In order to have a real-world impact, conjugated conducting polymers must be stable, cost-effective, scalable to large areas, and present minimal environmental, health, and safety (EHS) risks. To meet these demands, advanced manufacturing techniques are required that can economically fabricate thin films of conjugated conducting polymers over large areas while achieving the desired nanostructure within microscale layers. A crucial aspect of harnessing the full capabilities of semi-crystalline conjugated conducting polymers lies in controlling and leveraging their nanoscale order and orientation.Engineering the texture and nanostructure of conjugated conducting polymers is crucial for advancing their utilization in applications like electrochemical energy storage devices, flexible optoelectronics, and wearable devices. Key factors such as semi-crystalline orientation, π–π stacking, crystallite size, and inter-chain and inter-crystallite couplings must be precisely controlled during synthesis to optimize charge carrier mobility. Improving the pseudocapacitive properties of conducting polymers can be achieved by enhancing their semi-crystalline structure and extending polymer chain length. Ion diffusivity varies between crystalline and amorphous regions, and edge-on oriented conducting polymer thin films showing greater charge storage capacity during cycling. The size and type of counterion dopants are crucial for the electrochemical performance of conjugated conducting polymers. Small counterion dopants like chloride enhance redox exchange and expand the electrochemical potential window, benefiting electrochemical energy storage devices. Directly fabricating ultrathin, highly conformal coatings of conducting polymers via vapor phase deposition onto flexible, porous substrates has proven promising for creating flexible supercapacitors with high specific capacitance, as it preserves the substrate's high surface area, providing more active sites for redox reactions.
N-Type conjugated conducting polymers are crucial for many devices, including organic energy storage, electrochemical transistors, and spintronic devices. However, their development has lagged behind p-type counterparts. For air-stable n-type polymers, the LUMO level must be below −4.0 eV, which also improves n-type doping efficiency. Porous materials, with their reduced resistance to mass transport, are ideal for applications requiring molecular diffusion, such as adsorption, separation, sensing, and catalysis. In electrochemical energy storage, hierarchical pore structures improve access to redox-active sites and help accommodate mechanical stresses from doping and de-doping. Porous conjugated conducting polymers often have a high surface area (>1000 m2 g−1) without relying on heavy metals, using only earth-abundant elements like hydrogen, carbon, oxygen, nitrogen, and sulfur, making them suitable for large-area electrochemical energy storage devices.
Conducting polymers have become promising materials in Li–ion batteries, addressing limitations of traditional electrodes with their mixed ionic and electrical conductivity, flexibility, rapid doping/de-doping, stability, and reversible redox capabilities. The tunable nanostructure of conjugated conducting polymers allows for the design of materials with enhanced energy density, cycle stability, and rate performance. Additionally, incorporating conducting polymers improves electrode–electrolyte interfaces, mitigating issues like SEI formation and dendrite growth. As research progresses, optimizing conjugated polymer nanostructures, doping levels, and hybridization with other materials will be key to advancing Li–ion battery technology, leading to more efficient, durable, and sustainable energy storage solutions.
Research and development of conjugated conducting polymer-based flexible supercapacitors have progressed rapidly, with substantial efforts aimed at positioning these supercapacitors as a new platform for flexible energy storage devices. Interestingly, the direct growth of ultrathin, highly conformal coatings of conjugated conducting polymers via vapor phase deposition onto flexible, porous substrates has emerged as a promising approach for achieving flexible supercapacitors with high specific capacitance. This method offers a significant advantage by preserving the high surface area of the porous, flexible substrate, thereby facilitating more active sites for redox reactions. To fully realize the potential of conjugated conducting polymers in flexible supercapacitors, further advancements are crucial, particularly in optimizing specific power and energy density by exploiting the broader potential window provided by eco-friendly ionic liquid electrolyte systems. Achieving large-scale commercial applications will require not only innovative strategies but also a deeper and more refined understanding of the effects of in situ doping and the nanostructure of conjugated polymer architectures.
Conducting polymers hold significant promise for enhancing the performance of redox flow batteries. Traditional carbon-based electrode materials, such as carbon cloth and graphite felt, often face limitations like low specific capacitance, limited surface area, and poor wettability, which hinder their efficiency. However, the application of ultrathin, highly conformal coatings of conducting polymers on carbon-based electrodes has emerged as an effective strategy to overcome these challenges. Conjugated conducting polymers can be chemically engineered to improve the diffusion of electroactive species, enhance ion selectivity, and ultimately boost the overall electrochemical performance of redox flow battery electrodes, paving the way for more efficient and durable energy storage solutions.
Data availability
No software or code have been included as part of this review article.Conflicts of interest
Authors declare no competing interests.Acknowledgements
M. H. G. acknowledges the Faculty Startup Support from the San Diego State University (SDSU).References
- N. Wunderling, M. Willeit, J. F. Donges and R. Winkelmann, Nat. Commun., 2020, 11, 5177 CrossRef.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1801564 CrossRef.
- K. K. Gleason and M. H. Gharahcheshmeh, Conjugated Polymers at Nanoscale: Engineering Orientation, Nanostructure, and Properties, Walter de Gruyter GmbH & Co KG, 2021 Search PubMed.
- A. Varyani, M. Salehi and M. Heydari Gharahcheshmeh, Energies, 2024, 17, 2865 CrossRef.
- S. Ould Amrouche, D. Rekioua, T. Rekioua and S. Bacha, Int. J. Hydrogen Energy, 2016, 41, 20914–20927 CrossRef CAS.
- I. Tantis, S. Talande, V. Tzitzios, G. Basina, V. Shrivastav, A. Bakandritsos and R. Zboril, Adv. Funct. Mater., 2023, 33, 2209360 CrossRef CAS.
- A. M. Bryan, L. M. Santino, Y. Lu, S. Acharya and J. M. D’Arcy, Chem. Mater., 2016, 28, 5989–5998 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. Gleason, Mater. Today Adv., 2020, 8, 100086 CrossRef.
- M. Heydari Gharahcheshmeh, C. T. C. Wan, Y. Ashraf Gandomi, K. V. Greco, A. Forner-Cuenca, Y. M. Chiang, F. R. Brushett and K. K. Gleason, Adv. Mater. Interfaces, 2020, 7, 2000855 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. Chowdhury, Adv. Mater. Interfaces, 2024, 11, 2400118 CrossRef CAS.
- J. G. Ibanez, M. E. Rincón, S. Gutierrez-Granados, M. H. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731–4816 CrossRef CAS.
- Y. Yu, C. Yang, Y. Jiang, J. Zhu, Y. Zhao, S. Liang, K. Wang, Y. Zhou, Y. Liu and J. Zhang, Small, 2023, 19, 2303779 CrossRef CAS.
- B. Liu, K. Zhu, K. Ye, J. Yan, G. Wang and D. Cao, J. Power Sources, 2022, 552, 232226 CrossRef CAS.
- L. M. Zhu, A. W. Lei, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2013, 49, 567–569 RSC.
- G.-L. Xu, Q. Liu, K. K. S. Lau, Y. Liu, X. Liu, H. Gao, X. Zhou, M. Zhuang, Y. Ren and J. Li, Nat. Energy, 2019, 4, 484–494 CrossRef CAS.
- M. Vagin, C. Che, V. Gueskine, M. Berggren and X. Crispin, Adv. Funct. Mater., 2020, 30, 2007009 CrossRef CAS.
- S. H. Oh, C. W. Lee, D. H. Chun, J. D. Jeon, J. Shim, K. H. Shin and J. H. Yang, J. Mater. Chem. A, 2014, 2, 19994–19998 RSC.
- S.-M. Kim, C.-H. Kim, Y. Kim, N. Kim, W.-J. Lee, E.-H. Lee, D. Kim, S. Park, K. Lee, J. Rivnay and M.-H. Yoon, Nat. Commun., 2018, 9, 3858 CrossRef PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- C. Bauer, M. Kirchner and A. Krueger, Energy Adv., 2024, 3, 1422–1430 RSC.
- Y. Ma, K. Xu, X. Liu, S. Yao, X. Li, Y. Si and X. Li, Chem. Eng. J., 2024, 154348 CrossRef CAS.
- M. G. Tadesse, N. Simon and J. F. Lübben, Adv. Energy Sustainability Res., 2024, 2400006 CrossRef CAS.
- K. K. Gleason, F. Brushett, C. Wan, A. Forner-Cuenca, M. H. Gharahcheshmeh and Y. A. Gandomi, U.S. Pat., 11677078, 2023 Search PubMed.
- I. E. Jacobs, G. d’Avino, V. Lemaur, Y. Lin, Y. Huang, C. Chen, T. F. Harrelson, W. Wood, L. J. Spalek and T. Mustafa, J. Am. Chem. Soc., 2022, 144, 3005–3019 CrossRef CAS.
- F. Schauer, J. Appl. Phys., 2020, 128, 150902 CrossRef CAS.
- S. P. Arnold, J. K. Harris, B. Neelamraju, M. Rudolph and E. L. Ratcliff, Synth. Met., 2019, 253, 26–33 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Energies, 2022, 15, 3661 CrossRef CAS.
- M. Heydari Gharahcheshmeh, MRS Commun., 2024 DOI:10.1557/s43579-024-00593-w.
- P. Moni, J. Lau, A. C. Mohr, T. C. Lin, S. H. Tolbert, B. Dunn and K. K. Gleason, ACS Appl. Energy Mater., 2018, 1, 7093–7105 CrossRef CAS.
- Q. Liu, Y.-T. Liu, C. Zhao, Q.-S. Weng, J. Deng, I. Hwang, Y. Jiang, C. Sun, T. Li and W. Xu, ACS Nano, 2022, 16, 14527–14538 CrossRef CAS.
- M. E. Abdelhamid, A. P. O'Mullane and G. A. Snook, RSC Adv., 2015, 5, 11611–11626 RSC.
- J. Rehmen, T. Pathirana, L. Garcia-Quintana, R. Kerr, P. C. Howlett, K. Zuber, C. Pozo-Gonzalo and D. R. Evans, ACS Appl. Nano Mater., 2020, 3, 3820–3828 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- Z. Wen, Y. Peng, J. Cong, H. Hua, Y. Lin, J. Xiong, J. Zeng and J. Zhao, Nano Res., 2019, 12, 2535–2542 CrossRef CAS.
- S. Li, J. Huang, J. Wang and K. Han, Mater. Lett., 2021, 293, 129712 CrossRef CAS.
- R. Na, K. Minnici, G. Zhang, N. Lu, M. A. González, G. Wang and E. Reichmanis, ACS Appl. Mater. Interfaces, 2019, 11, 40034–40042 CrossRef CAS.
- T. Li, X.-Z. Yuan, L. Zhang, D. Song, K. Shi and C. Bock, Electrochem. Energy Rev., 2020, 3, 43–80 CrossRef.
- B. Zhang, J. Shen, Q. Wang, C. Hu, B. Luo, Y. Liu, Z. Xiao and X. Ou, Energy Environ. Mater., 2023, 6, e12270 CrossRef CAS.
- Y. Li, X. Liu, D. Ren, H. Hsu, G.-L. Xu, J. Hou, L. Wang, X. Feng, L. Lu and W. Xu, Nano Energy, 2020, 71, 104643 CrossRef CAS.
- Q. Xie, Z. Hu, C. Zhao, S. Zhang and K. Liu, RSC Adv., 2015, 5, 50859–50864 RSC.
- Z. Du, W. Peng, Z. Wang, H. Guo, Q. Hu and X. Li, Ionics, 2018, 24, 3717–3724 CrossRef CAS.
- Y.-H. Du, H. Sheng, X.-H. Meng, X.-D. Zhang, Y.-G. Zou, J.-Y. Liang, M. Fan, F. Wang, J. Tang and F.-F. Cao, Nano Energy, 2022, 94, 106901 CrossRef CAS.
- X. Ding, Y.-X. Li, S. Wang, J.-M. Dong, A. Yasmin, Q. Hu, Z.-Y. Wen and C.-H. Chen, Nano Energy, 2019, 61, 411–419 CrossRef CAS.
- L. Li, M. Xu, Q. Yao, Z. Chen, L. Song, Z. Zhang, C. Gao, P. Wang, Z. Yu and Y. Lai, ACS Appl. Mater. Interfaces, 2016, 8, 30879–30889 CrossRef CAS.
- T. He, Y. Lu, Y. Su, L. Bao, J. Tan, L. Chen, Q. Zhang, W. Li, S. Chen and F. Wu, ChemSusChem, 2018, 11, 1639–1648 CrossRef CAS PubMed.
- Q. Gan, N. Qin, Y. Zhu, Z. Huang, F. Zhang, S. Gu, J. Xie, K. Zhang, L. Lu and Z. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 12594–12604 CrossRef CAS.
- G.-L. Xu, Q. Liu, K. K. Lau, Y. Liu, X. Liu, H. Gao, X. Zhou, M. Zhuang, Y. Ren and J. Li, Nat. Energy, 2019, 4, 484–494 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- H. Xiang, N. Deng, H. Zhao, X. Wang, L. Wei, M. Wang, B. Cheng and W. Kang, J. Energy Chem., 2021, 58, 523–556 CrossRef CAS.
- R. Fang, S. Zhao, Z. Sun, D.-W. Wang, H.-M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- G. Zhou, D.-W. Wang, F. Li, P.-X. Hou, L. Yin, C. Liu, G. Q. M. Lu, I. R. Gentle and H.-M. Cheng, Energy Environ. Sci., 2012, 5, 8901–8906 RSC.
- Y. Zhang, H. W. Song, K. R. Crompton, X. Yang, K. Zhao and S. Lee, Nano Energy, 2023, 115, 108756 CrossRef CAS.
- Y. Xie, H. Zhao, H. Cheng, C. Hu, W. Fang, J. Fang, J. Xu and Z. Chen, Appl. Energy, 2016, 175, 522–528 CrossRef CAS.
- M. D. Hager, P. Greil, C. Leyens, S. Van Der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- C. Peng, D. Hu and G. Z. Chen, Chem. Commun., 2011, 47, 4105–4107 RSC.
- J. Zang, S.-J. Bao, C. M. Li, H. Bian, X. Cui, Q. Bao, C. Q. Sun, J. Guo and K. Lian, J. Phys. Chem. C, 2008, 112, 14843–14847 CrossRef CAS.
- H. Randriamahazaka, C. Plesse, D. Teyssie and C. Chevrot, Electrochim. Acta, 2005, 50, 1515–1522 CrossRef CAS.
- E. Sánchez-Díez, E. Ventosa, M. Guarnieri, A. Trovò, C. Flox, R. Marcilla, F. Soavi, P. Mazur, E. Aranzabe and R. Ferret, J. Power Sources, 2021, 481, 228804 CrossRef.
- M. Gencten and Y. Sahin, Int. J. Energy Res., 2020, 44, 7903–7923 CrossRef CAS.
- A. Forner-Cuenca, E. E. Penn, A. M. Oliveira and F. R. Brushett, J. Electrochem. Soc., 2019, 166, A2230–A2241 CrossRef CAS.
- J. Li, M. Al-Yasiri, H. Pham and J. Park, in Advanced Materials for Battery Separators, ed. S. Thomas, D. Rouxel, N. Kalarikkal, B. Kottathodi and H. J. Maria, Elsevier, 2024, pp. 327–347 DOI:10.1016/B978-0-12-817507-1.00003-X.
- M. Maleki, G. A. El-Nagar, D. Bernsmeier, J. Schneider and C. Roth, Sci. Rep., 2020, 10, 11153 CrossRef CAS.
- I. Azpitarte, U. Eletxigerra, A. Barros, E. Aranzabe and R. Cid, Batteries, 2023, 9, 39 CrossRef CAS.
- L. Yue, W. Li, F. Sun, L. Zhao and L. Xing, Carbon, 2010, 48, 3079–3090 CrossRef CAS.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS PubMed.
- X.-G. Li, K.-L. Huang, S.-Q. Liu, N. Tan and L.-Q. Chen, Trans. Nonferrous Met. Soc. China, 2007, 17, 195–199 CrossRef CAS.
- R. Wang and Y. Li, Energy Storage Mater., 2020, 31, 230–251 CrossRef.
- P. C. Ghimire, R. Schweiss, G. G. Scherer, T. M. Lim, N. Wai, A. Bhattarai and Q. Yan, Carbon, 2019, 155, 176–185 CrossRef CAS.
- Y.-J. Gu, W. Wen and J.-M. Wu, J. Mater. Chem. A, 2018, 6, 21078–21086 RSC.
- B. Ouyang, Y. Zhang, Y. Wang, Z. Zhang, H. J. Fan and R. S. Rawat, J. Mater. Chem. A, 2016, 4, 17801–17808 RSC.
- D. Dixon, D. J. Babu, J. Langner, M. Bruns, L. Pfaffmann, A. Bhaskar, J. J. Schneider, F. Scheiba and H. Ehrenberg, J. Power Sources, 2016, 332, 240–248 CrossRef CAS.
- M. G. Sumdani, M. R. Islam, A. N. A. Yahaya and S. I. Safie, Polym. Eng. Sci., 2022, 62, 269–303 CrossRef CAS.
- K. Choudhary, A. X. Chen, G. M. Pitch, R. Runser, A. Urbina, T. J. Dunn, M. Kodur, A. T. Kleinschmidt, B. G. Wang, J. A. Bunch, D. P. Fenning, A. L. Ayzner and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2021, 13, 51436–51446 CrossRef CAS.
- H. A. M. Mustafa and D. A. Jameel, J. Appl. Sci. Technol. Trends, 2021, 2, 91–95 Search PubMed.
- K. R. Thines, E. C. Abdullah, N. M. Mubarak and M. Ruthiraan, Biomass Bioenergy, 2017, 98, 95–111 CrossRef CAS.
- Y. Yan, J. Ju, S. Dong, Y. Wang, L. Huang, L. Cui, F. Jiang, Q. Wang, Y. Zhang and G. Cui, Adv. Sci., 2021, 8, 2003887 CrossRef CAS PubMed.
- Z. Yang, L. Ding, P. Wu, Y. Liu, F. Nie and F. Huang, Chem. Eng. J., 2015, 268, 60–66 CrossRef CAS.
- S. Cosnier and A. Karyakin, Electropolymerization: concepts, materials and applications, John Wiley & Sons, 2011 Search PubMed.
- G. Sabouraud, S. Sadki and N. Brodie, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- C. Zhao, Z. Chen, W. Wang, P. Xiong, B. Li, M. Li, J. Yang and Y. Xu, Angew. Chem., Int. Ed., 2020, 59, 11992–11998 CrossRef CAS PubMed.
- C. Friebe, M. D. Hager, A. Winter and U. S. Schubert, Adv. Mater., 2012, 24, 332–345 CrossRef CAS PubMed.
- M. Fabretto, J.-P. Autere, D. Hoglinger, S. Field and P. Murphy, Thin Solid Films, 2011, 519, 2544–2549 CrossRef CAS.
- M. Fabretto, K. Zuber, C. Hall and P. Murphy, Macromol. Rapid Commun., 2008, 29, 1403–1409 CrossRef CAS.
- A. T. Lawal and G. G. Wallace, Talanta, 2014, 119, 133–143 CrossRef CAS.
- A. Mohammadi, M. A. Hasan, B. Liedberg, I. Lundström and W. R. Salaneck, Synth. Met., 1986, 14, 189–197 CrossRef CAS.
- D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1329–1351 CrossRef CAS.
- J. Kim, E. Kim, Y. Won, H. Lee and K. Suh, Synth. Met., 2003, 139, 485–489 CrossRef CAS.
- P. A. Levermore, L. Chen, X. Wang, R. Das and D. D. C. Bradley, Adv. Mater., 2007, 19, 2379–2385 CrossRef CAS.
- B. Winther-Jensen and K. West, Macromolecules, 2004, 37, 4538–4543 CrossRef CAS.
- T. Bashir, M. Ali, S.-W. Cho, N.-K. Persson and M. Skrifvars, Polym. Adv. Technol., 2013, 24, 210–219 CrossRef CAS.
- T. Bashir, M. Skrifvars and N.-K. Persson, Polym. Adv. Technol., 2012, 23, 611–617 CrossRef CAS.
- K. K. Gleason, CVD Polym., 2015, 1–11, DOI:10.1002/9783527690275.ch1.
- S. G. Im, D. Kusters, W. Choi, S. H. Baxamusa, M. C. M. de Sanden and K. K. Gleason, ACS Nano, 2008, 2, 1959–1967 CrossRef CAS.
- S. Lee, D. C. Paine and K. K. Gleason, Adv. Funct. Mater., 2014, 24, 7187–7196 CrossRef CAS.
- F. Muralter, A. M. Coclite and K. K. S. Lau, Adv. Electron. Mater., 2021, 7, 2000871 CrossRef CAS.
- M. Rajesh, C. J. Raj, R. Manikandan, B. C. Kim, S. Y. Park and K. H. Yu, Mater. Today Energy, 2017, 6, 96–104 CrossRef.
- A. G. MacDiarmid, W. E. Jones Jr, I. D. Norris, J. Gao, A. T. Johnson Jr, N. J. Pinto, J. Hone, B. Han, F. K. Ko and H. Okuzaki, Synth. Met., 2001, 119, 27–30 CrossRef CAS.
- Q. Tang, J. Wu, X. Sun, Q. Li and J. Lin, Langmuir, 2009, 25, 5253–5257 CrossRef CAS PubMed.
- Y. Song, J.-B. Fan and S. Wang, Mater. Chem. Front., 2017, 1, 1028–1040 RSC.
- C.-S. Park, D. H. Kim, B. J. Shin, D. Y. Kim, H.-K. Lee and H.-S. Tae, Materials, 2016, 9, 812 CrossRef PubMed.
- M. Heydari Gharahcheshmeh, M. M. Tavakoli, E. F. Gleason, M. T. Robinson, J. Kong and K. K. Gleason, Sci. Adv., 2019, 5, eaay0414 CrossRef.
- R. Das and A. Chanda, Nano-size polymers: Preparation, properties, applications, 2016, pp. 283–306 Search PubMed.
- K. Norrman, A. Ghanbari-Siahkali and N. B. Larsen, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2005, 101, 174–201 RSC.
- H. Yan and H. Okuzaki, Synth. Met., 2009, 159, 2225–2228 CrossRef CAS.
- G.-F. Wang, X.-M. Tao, J. H. Xin and B. Fei, Nanoscale Res. Lett., 2009, 4, 613–617 CrossRef CAS.
- C. Gong, H. B. Yang, Q. L. Song, Z. S. Lu and C. M. Li, Sol. Energy Mater. Sol. Cells, 2012, 100, 115–119 CrossRef CAS.
- M. D. Tyona, Adv. Mater. Res., 2013, 2, 195 CrossRef.
- A. Boudrioua, M. Chakaroun and A. Fischer, in Organic Lasers, ed. A. Boudrioua, M. Chakaroun and A. Fischer, Elsevier, 2017, pp. 49–93 DOI:10.1016/B978-1-78548-158-1.50002-X.
- Y. Jiang, T. Liu and Y. Zhou, Adv. Funct. Mater., 2020, 30, 2006213 CrossRef CAS.
- V. Goel, R. Tanwar and U. Mandal, J. Chem. Technol. Biotechnol., 2021, 96, 502–513 CrossRef CAS.
- M. S. Cho, S. Y. Kim, J. D. Nam and Y. Lee, Synth. Met., 2008, 158, 865–869 CrossRef CAS.
- A. Seijas-Da Silva, J. Noguera-Gomez, P. J. Rodríguez-Canto, J. F. Sanchez-Royo, A. Cantarero, C. M. Gómez and R. Abargues, J. Phys. Chem. C, 2020, 124, 22884–22892 CrossRef CAS.
- K. H. Hong, K. W. Oh and T. J. Kang, J. Appl. Polym. Sci., 2005, 97, 1326–1332 CrossRef CAS.
- X. Wang, S. Meng, W. Ma, J. Pionteck, M. Gnanaseelan, Z. Zhou, B. Sun, Z. Qin and M. Zhu, React. Funct. Polym., 2017, 112, 74–80 CrossRef CAS.
- M. Ferenets and A. Harlin, Thin Solid Films, 2007, 515, 5324–5328 CrossRef CAS.
- L. A. A. Pettersson, F. Carlsson, O. Inganäs and H. Arwin, Thin Solid Films, 1998, 313–314, 356–361 CrossRef CAS.
- I. S. Tsagkalias, T. K. Manios and D. S. Achilias, Polymers, 2017, 9, 432 CrossRef PubMed.
- J. Wang, Z. Shi, Y. Ge, Y. Wang, J. Fan and J. Yin, Mater. Chem. Phys., 2012, 136, 43–50 CrossRef CAS.
- M. K. Erdogan, M. Karakisla and M. Sacak, J. Text. Inst., 2019, 110, 282–288 CrossRef CAS.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- C. Friebe, M. D. Hager, A. Winter and U. S. Schubert, Adv. Mater., 2012, 24, 332–345 CrossRef CAS.
- Y. Seki, M. Takahashi and M. Takashiri, RSC Adv., 2019, 9, 15957–15965 RSC.
- A. Yazdanpanah, A. Ramedani, A. Abrishamkar, P. B. Milan, Z. S. Moghadan, N. P. S. Chauhan, F. Sefat and M. Mozafari, in Fundamentals and Emerging Applications of Polyaniline, ed. M. Mozafari and N. P. S. Chauhan, Elsevier, 2019, pp. 105–119 DOI:10.1016/B978-0-12-817915-4.00006-3.
- J. Yano, Y. Ota and A. Kitani, Mater. Lett., 2004, 58, 1934–1937 CrossRef CAS.
- E. Schab-Balcerzak, Electropolymerization, BoD–Books on Demand, 2011 Search PubMed.
- M. M. Gvozdenović, B. Jugović, J. S. Stevanović and B. Grgur, Hem. Ind., 2014, 68, 673–684 CrossRef.
- S. Sadki, P. Schottland, N. Brodie and G. Sabouraud, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- E. Stussi, R. Stella and D. De Rossi, Sens. Actuators, B, 1997, 43, 180–185 CrossRef CAS.
- A. T. Lawal and G. G. Wallace, Talanta, 2014, 119, 133–143 CrossRef CAS PubMed.
- B. Winther-Jensen, J. Chen, K. West and G. Wallace, Macromolecules, 2004, 37, 5930–5935 CrossRef CAS.
- D. Evans, M. Fabretto, M. Mueller, K. Zuber, R. Short and P. Murphy, J. Mater. Chem., 2012, 22, 14889–14895 RSC.
- T. Y. Kim, C. M. Park, J. E. Kim and K. S. Suh, Synth. Met., 2005, 149, 169–174 CrossRef CAS.
- N. Massonnet, A. Carella, A. De Geyer, J. Faure-Vincent and J.-P. Simonato, Chem. Sci., 2015, 6, 412–417 RSC.
- M. S. Cho, S. Y. Kim, J. D. Nam and Y. Lee, Synth. Met., 2008, 158, 865–869 CrossRef CAS.
- P. Hojati-Talemi, C. Bächler, M. Fabretto, P. Murphy and D. Evans, ACS Appl. Mater. Interfaces, 2013, 5, 11654–11660 CrossRef CAS.
- B. Li, D. Ni, J. Fan and J. Zhou, Chem. Eng. J., 2022, 445, 136818 CrossRef CAS.
- M. Fabretto, M. Müller, K. Zuber and P. Murphy, Macromol. Rapid Commun., 2009, 30, 1846–1851 CrossRef CAS PubMed.
- D. Wu, J. Zhang, W. Dong, H. Chen, X. Huang, B. Sun and L. Chen, Synth. Met., 2013, 176, 86–91 CrossRef CAS.
- B. Winther-Jensen, D. W. Breiby and K. West, Synth. Met., 2005, 152, 1–4 CrossRef CAS.
- L. Sun, G. Yuan, L. Gao, J. Yang, M. Chhowalla, M. H. Gharahcheshmeh, K. K. Gleason, Y. S. Choi, B. H. Hong and Z. Liu, Nat. Rev. Methods Primers, 2021, 1, 5 CrossRef CAS.
- M. T. Robinson, J. Tung, M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Funct. Mater., 2021, 31, 2101310 CrossRef CAS.
- K. K. Gleason, CVD polymers: fabrication of organic surfaces and devices, John Wiley & Sons, 2015 Search PubMed.
- J. P. Lock, S. G. Im and K. K. Gleason, Macromolecules, 2006, 39, 5326–5329 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1970016 CrossRef.
- M. K. Charyton, J. Crêpellière, K. Kotwica, M. Gora, W. Le, G. Frache, P. Grysan, J. Guillot, K. Baba and N. D. Boscher, J. Mater. Chem. C, 2024, 12, 11368–11377 RSC.
- M. M. Tavakoli, M. H. Gharahcheshmeh, N. Moody, M. G. Bawendi, K. K. Gleason and J. Kong, Adv. Mater. Interfaces, 2020, 7, 2000498 CrossRef CAS.
- S. Kaviani, M. Mohammadi Ghaleni, E. Tavakoli and S. Nejati, ACS Appl. Polym. Mater., 2019, 1, 552–560 CrossRef CAS.
- M. Heydari Gharahcheshmeh, M. T. Robinson, E. F. Gleason and K. K. Gleason, Adv. Funct. Mater., 2021, 31, 2008712 CrossRef CAS.
- X. Wang, X. Zhang, L. Sun, D. Lee, S. Lee, M. Wang, J. Zhao, Y. Shao-Horn, M. Dincă and T. Palacios, Sci. Adv., 2018, 4, eaat5780 CrossRef CAS.
- G. Drewelow, H. W. Song, Z.-T. Jiang and S. Lee, Appl. Surf. Sci., 2020, 501, 144105 CrossRef CAS.
- M. Heydari Gharahcheshmeh, M. T. Robinson, E. F. Gleason and K. K. Gleason, Adv. Funct. Mater., 2021, 31, 2170097 CrossRef CAS.
- W. E. Tenhaeff and K. K. Gleason, Adv. Funct. Mater., 2008, 18, 979–992 CrossRef CAS.
- K. Zuber, H. Shere, J. Rehmen, V. Wheaton, M. Fabretto, P. J. Murphy and D. R. Evans, ACS Omega, 2018, 3, 12679–12687 CrossRef CAS.
- A. H. Malik, F. Habib, M. J. Qazi, M. A. Ganayee, Z. Ahmad and M. A. Yatoo, J. Polym. Res., 2023, 30, 115 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- R. J. Hoofman, M. P. De Haas, L. D. Siebbeles and J. M. Warman, Nature, 1998, 392, 54–56 CrossRef CAS.
- H. Sirringhaus, P. Brown, R. Friend, M. M. Nielsen, K. Bechgaard, B. Langeveld-Voss, A. Spiering, R. A. Janssen, E. Meijer and P. Herwig, Nature, 1999, 401, 685–688 CrossRef CAS.
- K. Chowdhury, S. K. Behura, M. Rahimi and M. Heydari Gharahcheshmeh, ACS Appl. Energy Mater., 2024, 7, 1068–1079 CrossRef CAS.
- S. Kee, N. Kim, B. S. Kim, S. Park, Y. H. Jang, S. H. Lee, J. Kim, J. Kim, S. Kwon and K. Lee, Adv. Mater., 2016, 28, 8625–8631 CrossRef CAS PubMed.
- C. M. Palumbiny, F. Liu, T. P. Russell, A. Hexemer, C. Wang and P. Müller-Buschbaum, Adv. Mater., 2015, 27, 3391–3397 CrossRef CAS PubMed.
- N. Kim, B. H. Lee, D. Choi, G. Kim, H. Kim, J.-R. Kim, J. Lee, Y. H. Kahng and K. Lee, Phys. Rev. Lett., 2012, 109, 106405 CrossRef.
- S. Y. Son, Y. Kim, J. Lee, G.-Y. Lee, W.-T. Park, Y.-Y. Noh, C. E. Park and T. Park, J. Am. Chem. Soc., 2016, 138, 8096–8103 CrossRef CAS PubMed.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. Fréchet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS.
- E. Collini and G. D. Scholes, Science, 2009, 323, 369–373 CrossRef CAS PubMed.
- S. P. Arnold, J. K. Harris, B. Neelamraju, M. Rudolph and E. L. Ratcliff, Synth. Met., 2019, 253, 26–33 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS.
- M. H. Gharahcheshmeh and K. K. Gleason, Conjugated Polymers for Next-Generation Applications, Elsevier, 2022, ch. 9, vol. 2, pp. 283–311 Search PubMed.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- N. Kim, S. Kee, S. H. Lee, B. H. Lee, Y. H. Kahng, Y.-R. Jo, B.-J. Kim and K. Lee, Adv. Mater., 2014, 26, 2268–2272 CrossRef CAS PubMed.
- B. J. Worfolk, S. C. Andrews, S. Park, J. Reinspach, N. Liu, M. F. Toney, S. C. Mannsfeld and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14138–14143 CrossRef CAS.
- M. N. Gueye, A. Carella, N. Massonnet, E. Yvenou, S. Brenet, J. Faure-Vincent, S. Pouget, F. Rieutord, H. Okuno, A. Benayad, R. Demadrille and J.-P. Simonato, Chem. Mater., 2016, 28, 3462–3468 CrossRef CAS.
- R. M. Howden, E. D. McVay and K. K. Gleason, J. Mater. Chem. A, 2013, 1, 1334–1340 RSC.
- Y. Lin, E. Bilotti, C. W. Bastiaansen and T. Peijs, Polym. Eng. Sci., 2020, 60, 2351–2376 CrossRef CAS.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- C. Müller, Chem. Mater., 2015, 27, 2740–2754 CrossRef.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. VanLandingham, H.-C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545–550 CrossRef CAS.
- Z. Wang, M. Zhu, Z. Pei, Q. Xue, H. Li, Y. Huang and C. Zhi, Mater. Sci. Eng., R, 2020, 139, 100520 CrossRef.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- G. Rebetez, O. Bardagot, J. Affolter, J. Réhault and N. Banerji, Adv. Funct. Mater., 2022, 32, 2105821 CrossRef CAS.
- L. Bay, T. Jacobsen, S. Skaarup and K. West, J. Phys. Chem. B, 2001, 105, 8492–8497 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Edu., 2018, 95, 197–206 CrossRef CAS.
- J.-J. Han, N. Zhang, D.-L. Liu, H. Ma, T. Han and D.-D. Sun, Ionics, 2020, 26, 1029–1038 CrossRef CAS.
- T. C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, J. Electrochem. Soc., 1998, 145, 1882 CrossRef CAS.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- K. C. S. Lakshmi and B. Vedhanarayanan, Batteries, 2023, 9, 202 CrossRef CAS.
- B.-Y. Chang, E. Ahn and S.-M. Park, J. Phys. Chem. C, 2008, 112, 16902–16909 CrossRef CAS.
- D. Rochefort and A.-L. Pont, Electrochem. Commun., 2006, 8, 1539–1543 CrossRef CAS.
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- S. Vijayakumar, S.-H. Lee and K.-S. Ryu, Electrochim. Acta, 2015, 182, 979–986 CrossRef CAS.
- M. G. Voss, J. R. Challa, D. T. Scholes, P. Y. Yee, E. C. Wu, X. Liu, S. J. Park, O. León Ruiz, S. Subramaniyan and M. Chen, Adv. Mater., 2021, 33, 2000228 CrossRef CAS.
- F. G. Donnan, Z. Electrochem., 1911, 17, 572–581 CAS.
- X. Jin, R. E. White and K. Huang, J. Electrochem. Soc., 2016, 163, A2702 CrossRef CAS.
- T. Sokalski, P. Lingenfelter and A. Lewenstam, J. Phys. Chem. B, 2003, 107, 2443–2452 CrossRef CAS.
- P. Moni, A. Al-Obeidi and K. K. Gleason, Beilstein J. Nanotechnol., 2017, 8, 723–735 CrossRef CAS PubMed.
- K. K. Gleason, Org. Mater., 2022, 4, 261–267 CrossRef CAS.
- N. A. Kukhta, A. Marks and C. K. Luscombe, Chem. Rev., 2022, 122, 4325–4355 CrossRef CAS PubMed.
- B. X. Dong, C. Nowak, J. W. Onorato, J. Strzalka, F. A. Escobedo, C. K. Luscombe, P. F. Nealey and S. N. Patel, Chem. Mater., 2019, 31, 1418–1429 CrossRef CAS.
- B. Zayat, P. Das, B. C. Thompson and S. R. Narayan, J. Phys. Chem. C, 2021, 125, 7533–7541 CrossRef CAS.
- P. Das, B. Zayat, Q. Wei, C. Z. Salamat, I.-B. Magdău, R. Elizalde-Segovia, D. Rawlings, D. Lee, G. Pace, A. Irshad, L. Ye, A. Schmitt, R. A. Segalman, T. F. Miller III, S. H. Tolbert, B. S. Dunn, S. R. Narayan and B. C. Thompson, Chem. Mater., 2020, 32, 9176–9189 CrossRef CAS.
- Y. Dobashi, A. Fannir, M. Farajollahi, A. Mahmoudzadeh, A. Usgaocar, D. Yao, G. T. M. Nguyen, C. Plesse, F. Vidal and J. D. W. Madden, Electrochim. Acta, 2017, 247, 149–162 CrossRef CAS.
- K. Leš and C.-S. Jordan, RSC Adv., 2020, 10, 41296–41304 RSC.
- G. Garcia-Belmonte, J. Bisquert, E. C. Pereira and F. Fabregat-Santiago, J. Electroanal. Chem., 2001, 508, 48–58 CrossRef CAS.
- J. Bisquert, G. G. Belmonte, F. F. Santiago, N. S. Ferriols, M. Yamashita and E. C. Pereira, Electrochem. Commun., 2000, 2, 601–605 CrossRef CAS.
- M. H. Gharahcheshmeh, E. Galstyan, A. Xu, J. Kukunuru, R. Katta, Y. Zhang, G. Majkic, X. Li and V. Selvamanickam, Supercond. Sci. Technol., 2016, 30, 015016 CrossRef.
- V. Selvamanickam, R. Mallick, X. Tao, Y. Yao, M. H. Gharahcheshmeh, A. Xu, Y. Zhang, E. Galstyan and G. Majkic, Supercond. Sci. Technol., 2016, 29, 085016 CrossRef.
- A. Xu, Y. Zhang, M. H. Gharahcheshmeh, L. Delgado, N. Khatri, Y. Liu, E. Galstyan and V. Selvamanickam, IEEE Trans. Appl. Supercond., 2017, 27, 1–5 Search PubMed.
- M. H. Gharahcheshmeh, E. Galstyan, J. Kukunuru, R. Katta, G. Majkic, X.-F. Li and V. Selvamanickam, IEEE Trans. Appl. Supercond., 2017, 27, 1–5 Search PubMed.
- V. Selvamanickam, M. Heydari Gharahcheshmeh, A. Xu, E. Galstyan, L. Delgado and C. Cantoni, Appl. Phys. Lett., 2015, 106, 032601 CrossRef.
- M. H. Gharahcheshmeh, G. Majkic, E. Galstyan, A. Xu, Y. Zhang, X. Li and V. Selvamanickam, Phys. C, 2018, 553, 26–32 CrossRef.
- E. Galstyan, M. H. Gharahcheshmeh, L. Delgado, A. Xu, G. Majkic and V. Selvamanickam, IEEE Trans. Appl. Supercond., 2014, 25, 1–5 Search PubMed.
- P. Kovacik, G. del Hierro, W. Livernois and K. K. Gleason, Mater. Horiz., 2015, 2, 221–227 RSC.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- C. Wang, C. Yang and Z. Zheng, Adv. Sci., 2022, 9, 2105213 CrossRef CAS.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- S. Park, S. Y. Jeong, T. K. Lee, M. W. Park, H. Y. Lim, J. Sung, J. Cho, S. K. Kwak, S. Y. Hong and N.-S. Choi, Nat. Commun., 2021, 12, 838 CrossRef CAS.
- Y. Lin, M. Xu, S. Wu, Y. Tian, Z. Cao, L. Xing and W. Li, ACS Appl. Mater. Interfaces, 2018, 10, 16400–16409 CrossRef CAS.
- D. Das, S. Manna and S. Puravankara, Batteries, 2023, 9, 193 CrossRef CAS.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- J. Deng, C. Bae, A. Denlinger and T. Miller, Joule, 2020, 4, 511–515 CrossRef.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- L.-L. Lu, Y.-Y. Lu, Z.-X. Zhu, J.-X. Shao, H.-B. Yao, S. Wang, T.-W. Zhang, Y. Ni, X.-X. Wang and S.-H. Yu, Sci. Adv., 2022, 8, eabm6624 CrossRef CAS PubMed.
- Y. Zhang, J. C. Kim, H. W. Song and S. Lee, Nanoscale, 2023, 15, 4195–4218 RSC.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS.
- Y. Han, B. Liu, Z. Xiao, W. Zhang, X. Wang, G. Pan, Y. Xia, X. Xia and J. Tu, InfoMat, 2021, 3, 155–174 CrossRef CAS.
- Z. Luo, X. Qiu, C. Liu, S. Li, C. Wang, G. Zou, H. Hou and X. Ji, Nano Energy, 2021, 79, 105507 CrossRef CAS.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef CAS.
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- W. Lyu, X. Yu, Y. Lv, A. M. Rao, J. Zhou and B. Lu, Adv. Mater., 2024, 36, 2305795 CrossRef.
- Y. Lu, L. Li, Q. Zhang, Z. Niu and J. Chen, Joule, 2018, 2, 1747–1770 CrossRef CAS.
- X. Yi, H. Fu, A. M. Rao, Y. Zhang, J. Zhou, C. Wang and B. Lu, Nat. Sustainability, 2024, 7, 326–337 CrossRef.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- S. Gao, F. Sun, N. Liu, H. Yang and P.-F. Cao, Mater. Today, 2020, 40, 140–159 CrossRef CAS.
- G. Ma, Z. Wen, Q. Wang, C. Shen, J. Jin and X. Wu, J. Mater. Chem. A, 2014, 2, 19355–19359 RSC.
- S. Pan, J. Han, Y. Wang, Z. Li, F. Chen, Y. Guo, Z. Han, K. Xiao, Z. Yu and M. Yu, Adv. Mater., 2022, 34, 2203617 CrossRef CAS.
- S. Ahn, C. Zor, S. Yang, M. Lagnoni, D. Dewar, T. Nimmo, C. Chau, M. Jenkins, A. J. Kibler, A. Pateman, G. J. Rees, X. Gao, P. Adamson, N. Grobert, A. Bertei, L. R. Johnson and P. G. Bruce, Nat. Chem., 2023, 15, 1022–1029 CrossRef CAS PubMed.
- P. Li, G. Zhao, X. Zheng, X. Xu, C. Yao, W. Sun and S. X. Dou, Energy Storage Mater., 2018, 15, 422–446 CrossRef.
- M. Salah, P. Murphy, C. Hall, C. Francis, R. Kerr and M. Fabretto, J. Power Sources, 2019, 414, 48–67 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 CrossRef.
- Y. Yao, N. Liu, M. T. McDowell, M. Pasta and Y. Cui, Energy Environ. Sci., 2012, 5, 7927–7930 RSC.
- S. Bai, Y. Ma, X. Jiang, Q. Li, Z. Yang, Q. Liu and D. He, Surf. Interfaces, 2017, 8, 214–218 CrossRef CAS.
- I.-H. Ko, S.-J. Kim, J. Lim, S.-H. Yu, J. Ahn, J.-K. Lee and Y.-E. Sung, Electrochim. Acta, 2016, 187, 340–347 CrossRef CAS.
- J. Chen, Y. Liu, A. I. Minett, C. Lynam, J. Wang and G. G. Wallace, Chem. Mater., 2007, 19, 3595–3597 CrossRef CAS.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O’Dwyer and K. M. Ryan, Nano Lett., 2014, 14, 716–723 CrossRef CAS PubMed.
- W. Bao, G. Qian, L. Zhao, Y. Yu, L. Su, X. Cai, H. Zhao, Y. Zuo, Y. Zhang and H. Li, Nano Lett., 2020, 20, 8832–8840 CrossRef CAS.
- Q. Gan, N. Qin, Y. Zhu, Z. Huang, F. Zhang, S. Gu, J. Xie, K. Zhang, L. Lu and Z. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 12594–12604 CrossRef CAS.
- Y. Cao, X. Qi, K. Hu, Y. Wang, Z. Gan, Y. Li, G. Hu, Z. Peng and K. Du, ACS Appl. Mater. Interfaces, 2018, 10, 18270–18280 CrossRef CAS.
- Q. Liu, Y.-T. Liu, C. Zhao, Q.-S. Weng, J. Deng, I. Hwang, Y. Jiang, C. Sun, T. Li, W. Xu, K. Du, A. Daali, G.-L. Xu, K. Amine and G. Chen, ACS Nano, 2022, 16, 14527–14538 CrossRef CAS.
- L. Su, P. M. Smith, P. Anand and B. Reeja-Jayan, ACS Appl. Mater. Interfaces, 2018, 10, 27063–27073 CrossRef CAS.
- R. Xiong, Y. Pan, W. Shen, H. Li and F. Sun, Renewable Sustainable Energy Rev., 2020, 131, 110048 CrossRef CAS.
- S.-M. Bak, K.-W. Nam, W. Chang, X. Yu, E. Hu, S. Hwang, E. A. Stach, K.-B. Kim, K. Y. Chung and X.-Q. Yang, Chem. Mater., 2013, 25, 337–351 CrossRef CAS.
- F. Wu, J. Liu, L. Li, X. Zhang, R. Luo, Y. Ye and R. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 23095–23104 CrossRef CAS.
- P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2015, 162, A2490 CrossRef CAS.
- L. Su, J. L. Weaver, M. Groenenboom, N. Nakamura, E. Rus, P. Anand, S. K. Jha, J. S. Okasinski, J. A. Dura and B. Reeja-Jayan, ACS Appl. Mater. Interfaces, 2021, 13, 9919–9931 CrossRef CAS PubMed.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS.
- Z. Wang, R. Pan, C. Xu, C. Ruan, K. Edström, M. Strømme and L. Nyholm, Energy Storage Mater., 2018, 13, 283–292 CrossRef.
- W. Liu, D. Lin, A. Pei and Y. Cui, J. Am. Chem. Soc., 2016, 138, 15443–15450 CrossRef CAS PubMed.
- H. Zhang, Y. Cao, H. Yang, S. Lu and X. Ai, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1487–1493 CrossRef CAS.
- P. R. Das, L. Komsiyska, O. Osters and G. Wittstock, J. Electrochem. Soc., 2015, 162, A674 CrossRef CAS.
- H. Zhong, A. He, J. Lu, M. Sun, J. He and L. Zhang, J. Power Sources, 2016, 336, 107–114 CrossRef CAS.
- D. Shao, H. Zhong and L. Zhang, ChemElectroChem, 2014, 1, 1679–1687 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- Y. Meng, K. Wang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 6985–6990 CrossRef CAS PubMed.
- A. Liu, P. Kovacik, N. Peard, W. Tian, H. Goktas, J. Lau, B. Dunn and K. K. Gleason, Adv. Mater., 2017, 29, 1606091 CrossRef PubMed.
- M. I. Ahamed, R. Boddula and T. Altalhi, Flexible Supercapacitor Nanoarchitectonics, John Wiley & Sons, 2021 Search PubMed.
- J. A. Lee, M. K. Shin, S. H. Kim, H. U. Cho, G. M. Spinks, G. G. Wallace, M. D. Lima, X. Lepró, M. E. Kozlov and R. H. Baughman, Nat. Commun., 2013, 4, 1970 CrossRef PubMed.
- V. T. Le, H. Kim, A. Ghosh, J. Kim, J. Chang, Q. A. Vu, D. T. Pham, J.-H. Lee, S.-W. Kim and Y. H. Lee, ACS Nano, 2013, 7, 5940–5947 CrossRef CAS PubMed.
- Z. Yang, J. Deng, X. Chen, J. Ren and H. Peng, Angew. Chem., Int. Ed., 2013, 52, 13453–13457 CrossRef CAS PubMed.
- J. Huang, K. Wang and Z. Wei, J. Mater. Chem., 2010, 20, 1117–1121 RSC.
- A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8475–8491 CrossRef CAS.
- D. Villers, D. Jobin, C. Soucy, D. Cossement, R. Chahine, L. Breau and D. Bélanger, J. Electrochem. Soc., 2003, 150, A747 CrossRef CAS.
- S. Hashmi and H. M. Upadhyaya, Solid State Ionics, 2002, 152, 883–889 CrossRef.
- D. P. Dubal, S. H. Lee, J. G. Kim, W. B. Kim and C. D. Lokhande, J. Mater. Chem., 2012, 22, 3044–3052 RSC.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS PubMed.
- Y. Y. Smolin, K. L. V. Aken, M. Boota, M. Soroush, Y. Gogotsi and K. K. S. Lau, Adv. Mater. Interfaces, 2017, 4, 1601201 CrossRef.
- M. Liu, Y.-E. Miao, C. Zhang, W. W. Tjiu, Z. Yang, H. Peng and T. Liu, Nanoscale, 2013, 5, 7312–7320 RSC.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- A. Khor, P. Leung, M. R. Mohamed, C. Flox, Q. Xu, L. An, R. G. A. Wills, J. R. Morante and A. A. Shah, Mater. Today Energy, 2018, 8, 80–108 CrossRef.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- A. Dinesh, S. Olivera, K. Venkatesh, M. S. Santosh, M. G. Priya, I. Inamuddin, A. M. Asiri and H. B. Muralidhara, Environ. Chem. Lett., 2018, 16, 683–694 CrossRef CAS.
- X. Wang, M. Ding and C. Jia, J. Phys. D: Appl. Phys., 2024, 57, 423002 CrossRef.
- A. Ashok and A. Kumar, Green Chem. Lett. Rev., 2024, 17, 2302834 CrossRef.
- K. Gong, F. Xu, J. B. Grunewald, X. Ma, Y. Zhao, S. Gu and Y. Yan, ACS Energy Lett., 2016, 1, 89–93 CrossRef CAS.
- H. Zhang and C. Sun, J. Power Sources, 2021, 493, 229445 CrossRef CAS.
- R. Wang, Y. Li, Y. Wang and Z. Fang, Appl. Energy, 2020, 261, 114369 CrossRef CAS.
- R. Wang, Y. Li and Y.-L. He, J. Mater. Chem. A, 2019, 7, 10962–10970 RSC.
- H. Wang, J. Lin and Z. X. Shen, J. Sci.: Adv. Mater. Devices, 2016, 1, 225–255 Search PubMed.
- X. Lu, W. Zhang, C. Wang, T.-C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671–712 CrossRef CAS.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chem. Soc. Rev., 2018, 47, 69–103 RSC.
- L. Zeng, T. S. Zhao, L. Wei, H. R. Jiang and M. C. Wu, Appl. Energy, 2019, 233–234, 622–643 CrossRef CAS.
- M. Vagin, C. Che, V. Gueskine, M. Berggren and X. Crispin, Adv. Funct. Mater., 2020, 30, 2007009 CrossRef CAS.
- E. K. Radwan, R. A. Omar, A. M. Ali, A. S. Elsayed and E. N. El Sawy, Sustainable Mater. Technol., 2024, e01069 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |