
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in noble-metal-free bifunctional oxygen electrode catalysts
Hengqi Liua,
Rui Xiongb,
Shengyu Maa,
Ran Wangc,
Zhiguo Liua,
Tai Yaoc and
Bo Song
 *acdefgh
*acdefgh
aSchool of Physics, Harbin Institute of Technology, Harbin 150001, China. E-mail: songbo@hit.edu.cn
bSchool of Future Technology, Harbin Institute of Technology, Harbin 150001, China
cNational Key Laboratory of Science and Technology on Advanced Composites in Special Environments, Harbin Institute of Technology, Harbin 150001, China
dNational Key Laboratory of Laser Spatial Information, Harbin Institute of Technology, Harbin 150001, China
eNational Key Laboratory of Space Environment and Matter Behaviors, Harbin Institute of Technology, Harbin 150001, China
fLaboratory for Space Environment and Physical Sciences, Harbin Institute of Technology, Harbin 150001, China
gZhengzou Research Institute, Harbin Institute of Technology, Zhengzhou 450046, China
hFrontiers Science Center for Matter Behave in Space Environment, Harbin Institute of Technology, Harbin 150001, China
First published on 18th November 2024
Abstract
The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are crucial reactions in energy storage. However, the sluggish rate of these oxidation electrode reactions and the strong dependence of these technologies on precious metal-based electrocatalysts has greatly restricted further progress. In response to this challenge, researchers have widely investigated the preparation of high-performance ORR and OER electrocatalysts using non-precious metals, reporting substantial advancements in the last ten years. This article provides a concise overview of the latest advancements in oxygen electrocatalysts that are not based on precious metals. The review focuses on the benefits and drawbacks of carbon materials, transition metal compounds, and their composite structures. Moreover, the inherent sources of activity in these materials, techniques for enhancing the density and usage of active sites, and novel design approaches and regulation methods rooted in response mechanisms are examined. Then, a statistical examination of documented bifunctional electrocatalysts is carried out to reveal the correlation between composition, structure, and performance. This report provides a comprehensive analysis of catalyst preparation, element selection, and future directions, delivering significant insights to guide future research endeavors.
 Bo Song | Bo Song is a professor in the National Key Laboratory of Science and Technology on Advanced Composites in Special Environments, Harbin Institute of Technology. He received his BS degree (2003) and MS degree (2005) from Wuhan University of Technology, and PhD degree (2008) from the Institute of Physics, Chinese Academy of Sciences. In 2013, he was promoted to full professor in Harbin Institute of Technology. His current research focuses on the electrocatalysis and energy storage, and third-generation semiconductor optoelectronic devices. |
1. Introduction
Due to rapid societal development and population growth, global energy demand has sharply increased, making energy supply and utilization critical factors in economic and social development.1,2 Electrical energy is an important source of energy that has become a major force in modern society because it is clean, efficient, and can be changed into many different forms of energy.3,4 Efficiently storing electricity remains a significant technological challenge, and developing effective electricity storage solutions will be necessary for the renewable energy applications and the stabilization of grid loads.5 Among the various energy storage technologies, electrochemical devices stand out due to their flexibility and high energy density. Thus, electrochemical technology has emerged as a core solution to these challenges.6–8 Consequently, the development of efficient electrochemical energy storage systems is increasingly urgent. Off-grid applications are becoming more common, especially with the rise of portable electronic devices. In this context, energy storage technologies play a vital role. They will significantly impact the future of energy consumption and the trajectory of sustainable development.After decades of development, lithium-ion batteries have become widely utilized.9,10 However, these batteries increasingly struggle to meet the growing demands for higher energy density, longer lifespan, and lower costs, and they also face challenges such as limited resources, high production expenses, and safety concerns.11,12 In contrast, rechargeable zinc–air batteries (ZABs) are considered a promising alternative due to their high energy density (1086 W h kg−1), low cost, and intrinsic safety.13,14 Nevertheless, the kinetic limitations of the oxygen electrode during charge and discharge processes remain a significant barrier to the widespread adoption of ZABs.15 As a result, improving the stability and efficiency of oxygen electrode catalysts has become a central research focus. Given the complex mechanisms of ORR and OER, different catalysts are typically required for each process.16,17 Precious metal catalysts such as platinum–carbon (Pt/C), iridium oxide (IrO2), and ruthenium oxide (RuO2) exhibit exceptional half-reaction catalytic efficiencies but are insufficient for simultaneously catalyzing both ORR and OER in rechargeable ZABs.18,19 While it is possible to create bifunctional catalysts by combining ORR and OER active components, simply mixing these components together often does not result in achieving accurate catalytic selectivity for both reactions.20 Moreover, the practical implementation of ZABs is hindered by considerable hurdles such as high costs, limited catalytic activity, and poor durability.21 Recently, catalysts based on transition metals and carbon materials have attracted considerable attention as cost-effective alternatives to precious metals.22,23 Strategies such as surface functionalization,24 structural engineering,25 heteroatom doping,26 and defect engineering27 have been explored to enhance bifunctional activity and stability of these catalysts. However, non-precious metal bifunctional electrocatalysts still require substantial improvements to surpass the performance benchmarks set by precious metals. Addressing this critical challenge is urgent. It requires a comprehensive review of current advancements in ORR/OER electrocatalysts. This review should summarize existing progress and guide future research directions.
This paper provides a comprehensive review of the recent advancements in non-precious metal bifunctional oxygen electrocatalysts, focusing on the benefits and drawbacks of carbon materials, transition metal compounds, and their composite structures in catalytic processes. The review focuses on the inherent sources of activity in these materials, techniques to increase the density and usage of active sites, and design principles along with regulation approaches based on reaction processes. The regulation outcomes are displayed through linear sweep voltammetry (LSV) curves, which allow for a systematic evaluation of the ORR activity (E1/2, the potential at which the current density reaches half of its maximum value), OER activity (Ej10, the potential at 10 mA cm−2), and key bifunctional performance (ΔE, the difference between E1/2 and Ej10). Additionally, the paper examines catalyst preparation processes, element selection, the application scope of single-component versus composite structures, and future development prospects. The goal of this review is to provide readers with a comprehensive perspective on advancing bifunctional oxygen electrocatalysts.
2. An introduction to rechargeable zinc–air batteries
2.1. Overview of rechargeable zinc–air batteries
Among the metal–air batteries, lithium–air batteries and ZABs are regarded as the most promising.28,29 Fig. 1(a) depicts the theoretical energy density of lithium–air batteries, which can reach 5928 W h kg−1. However, despite their promising performance, these batteries pose a serious safety risk. Lithium metal is extremely reactive, prone to explosive reactions when exposed to oxygen or water, and rapidly ignites flammable organic electrolytes. Moreover, lithium is expensive (about $60 per pound) and in scarce supply, limiting the further development of lithium–air batteries.30 In contrast, ZABs show considerable application potential. On one hand, the use of non-flammable aqueous electrolytes improves safety; on the other hand, zinc metal is inexpensive (about $0.90 per pound) and abundant. While the theoretical energy density of zinc–air batteries is only 1084 W h kg−1, which is lower than that of lithium–air batteries, this energy density is still around four times higher than that of modern lithium-ion batteries.31 ZABs show excellent promise for meeting increasing energy demand due to their consistent discharge voltage, extended lifespan, and environmental friendliness.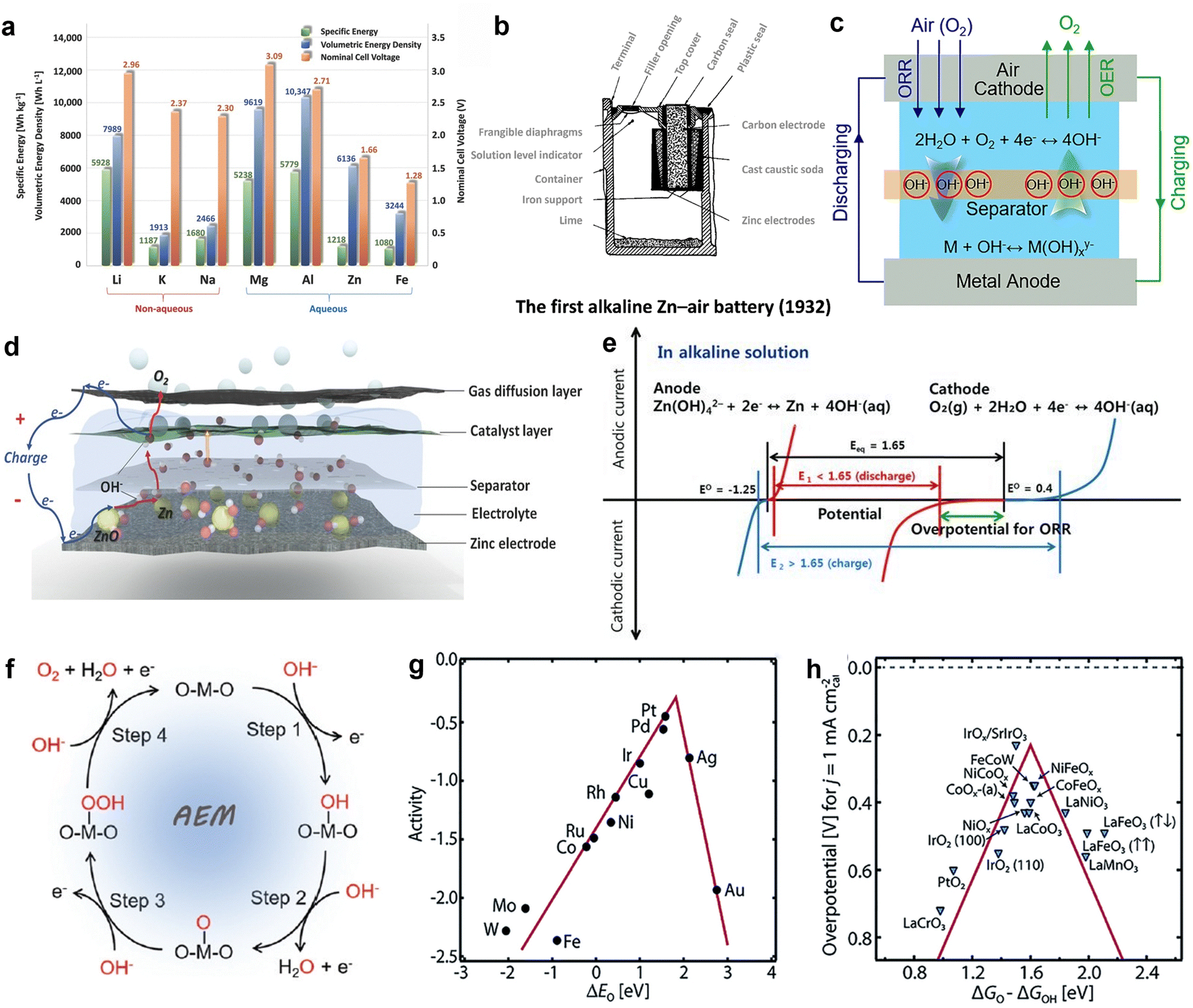 | ||
| Fig. 1 (a) The OER in metal–air batteries. Reprinted with permission.31 Copyright 2016, Wiley-VCH. (b) Theoretical specific energies, volumetric energy densities, and nominal cell voltages for various metal anodes in aqueous and non-aqueous metal–air batteries. Reprinted with permission.32 Copyright 2022, Royal Society of Chemistry. (c) Schematic illustration of the first alkaline zinc–air battery. In 1932, George W. Heise and Erwin A. Schumacher developed the first commercial alkaline zinc–air battery product. The battery employed a novel configuration, and utilized caustic soda as the electrolyte to prevent performance degradation. Reprinted with permission.33 Copyright 2022, The Author(s). (d) Schematic of an aqueous rechargeable zinc–air battery at charging status. Reprinted with permission.31 Copyright 2016, Wiley-VCH. (e) Schematic polarization curves of zinc–air cell. The equilibrium potential of the zinc–air cell (black line) is 1.65 V, but the practical voltage (red line) in discharge is lower than 1.65 V due to the sluggish ORR. A large potential is needed to charge zinc–air battery, higher than the equilibrium potential (blue line). Reprinted with permission.13 Copyright 2021, Wiley-VCH. (f) Schematic illustration of the proposed AEM pathway of OER in alkaline media on an active metal site. Reprinted with permission. Reprinted with permission.34 Copyright 2022, Royal Society of Chemistry. (g) Relationship between oxygen binding energy and catalytic activity of metals. (h) Relationship between catalytic activity and ΔGO/ΔGOH of metal oxides. Reprinted with permission.35 Copyright 2021, Elsevier Ltd. | ||
In contrast to many recent academic discoveries, the development of ZABs has taken nearly 140 years.32 This voyage began with the discovery of electrochemical principles in the early nineteenth century. In 1878, French engineer L. Maiche created the first zinc–air battery. In 1932, George W. Heise and Erwin A. Schumacher developed an improved alkaline zinc–air battery with enhanced performance, laying the groundwork for future technological developments in this field (Fig. 1(b)).36 From 1960 to 1990, ZABs became widely utilized in the U.S. space program.37 Advances in manufacturing processes and materials science further accelerated the development of these batteries.38 Nevertheless, between 1990 and 2010, further advancements were paused due to technological obstacles such as cathode catalyst instability, anode corrosion, and the rise of lithium-ion batteries. Consequently, research efforts shifted towards exploring alternative battery technologies.39,40 Since 2010, a resurgence of interest in ZABs has emerged due to breakthroughs in electrochemical theory and materials research.39 ZABs are extensively utilized in navigation equipment, signal lighting, maritime exploration, and railway signaling owing to their distinctive operating principles (Fig. 1(c)),33 high energy density, good safety, and environmental friendliness.
The primary components of ZABs include a zinc metal anode, an air electrode serving as the cathode (which consists of a gas diffusion layer, current collector, and catalyst layer), a separator, and an electrolyte (Fig. 1(d)).31 During the discharge process, the zinc anode suffers oxidation due to its interaction with hydroxide ions (OH−). This interaction to produces Zn(OH)42−, which then decomposes into ZnO. At the same time, oxygen from the air passes through the gas diffusion layer and attaches to the catalyst. Oxygen molecules undergo reduction at the three-phase interface by accepting electrons from the zinc anode, resulting in the formation of OH− ions. Subsequently, the hydroxide ions (OH−) are released from catalyst and move through the electrolyte to reach the zinc anode. As shown in Fig. 1(e), the polarization curves of ZABs demonstrate that the electrocatalysts responsible for ORR and OER in the air cathode play a crucial role in determining overall battery performance.13 However, most current catalysts do not meet the requirements for sustained operation, resulting in a substantial deficit in the actual energy density of zinc–air batteries compared to their theoretical value. Disappointingly, these batteries only achieve 40% to 50% of the anticipated energy density.41 Hence, to prepare ZABs with good performance and consistent stability, effective bifunctional electrocatalysts that can enhance the performance of the cathode must be developed.
2.2. ORR and OER mechanisms
ORR is a crucial step in the cathodic discharge process. In 1976, Wroblowa and colleagues proposed a mechanistic ORR model involving two primary pathways: a direct four-electron process and a stepwise two-electron process.42The direct four-electron pathway by the following reaction:
| O2 + 2H2O + 4e− → 4OH− | (2-1) |
In contrast, the stepwise two-electron process proceeds in two stages:
| O2 + H2O + 2e− → HO2− + OH− | (2-2) |
| HO2− + H2O + 2e− → 3OH− | (2-3) |
Under real-world conditions, the oxygen reduction process is more intricate than the simplified reactions shown in eqn (2-1)–(2-3) because both pathways can occur simultaneously. Preferably, Optimal ORR processes will preferably proceed via the more efficient four-electron pathway, which exhibits lower kinetic barriers. In contrast, the stepwise two-electron approach inhibits the total activity. The ORR process is initiated by the adsorption of oxygen molecules onto the electrocatalyst. This is followed by a sequence of reactions occurring on the surface or at the interface. These reactions involve the transfer of electrons, rearrangement of molecules, breaking of chemical bonds, and production of products.43 Ultimately, the reaction products are released from the surface of the catalyst. These complex reaction steps mean that the reaction process requires additional energy.
Additionally, because OER and ORR require different operating potentials, the charge states of the same catalyst or intermediate vary, leading to differences in electron transfer numbers for the same reversible steps. To design efficient ORR electrocatalysts, the binding energy of intermediates must be understood and controlled. Volcano plots, which are based on the Sabatier principle, are commonly used to illustrate the relationship between ORR activity and material properties despite their limitations in specific reactions.44 The oxygen binding energy (ΔEo) is a key descriptor for ORR processes, and an ideal catalyst will be located at the peak of the volcano plot.34 According to ORR volcano plot shown in Fig. 1(g), platinum (Pt) shows the best ORR performance. Although the equilibrium potentials for ORR and OER are both 1.23 V, different materials are typically selected as electrocatalysts for these reactions.45 While platinum shows exceptional ORR activity, studies have indicated that platinum has poor OER catalytic efficiency, suggesting different reaction mechanisms for these processes on platinum surfaces. To enhance OER activity, the free energy difference between O and OH (ΔGO − ΔGOH) must be optimized toward the peak position on the volcano plot shown in Fig. 1(h).46 Metal oxides are commonly used in OER electrocatalysts, and optimal activity is observed when ΔGO − ΔGOH is around 1.6 eV. Excessively high ΔGO − ΔGOH values lead to significant overpotential during O desorption, while low chemisorption energy results in a high overpotential during OH desorption.47 Thus, achieving both efficient bifunctional activity in a single catalyst remains a challenge.
During the charging process in ZABs, OER occurs at the air electrode.35 ZABs utilize alkaline solutions as electrolytes, and the OER in these batteries is a multi-step process that involves several electron transfer steps and the generation of intermediate. Each individual step has a significant influence on the total reaction efficiency. First, hydroxide ions (OH−) attach to the electrode catalyst, creating hydroxy intermediates (OH*). Subsequently, these intermediate undergo a reaction with additional hydroxide ions (OH−) to generate hydroperoxide intermediates (OOH*). Next, O–O bonds are generated. This step has significant influence on the overall reaction efficiency and determines the reaction rate. The hydroperoxide intermediates subsequently undergo decomposition, resulting in the release of oxygen molecules (O2). Consequently, active sites become available for further involvement in the OER cycle. The regenerated OH* intermediates initiate the cycle again, ensuring that the OER process proceeds uninterrupted. Thus, effective oxygen generation is promoted during battery recharging (Fig. 1(f)).48
2.3. Future trends
Developing electrocatalysts with high activity is essential for overcoming developmental challenges of ZABs and achieving large-scale commercialization. Currently, Pt/C and RuO2 are the most effective single-function catalysts. However, both of these catalyst materials are costly, and they exhibit poor stability. While combining these single-function catalysts can achieve bifunctional catalysis, this strategy increases both the cost of the battery and the complexity of the preparation process. Thermodynamically, the main challenge limiting bifunctional catalytic activity for both reactions is the interdependent adsorption energies (adsorption energy is a physical quantity that describes the strength of the interaction between molecules or atoms and a solid surface, quantifying the change in free energy during the adsorption process) of OH− and O2 molecules. To achieve bifunctional catalytic activity, the structure, composition, and electronic states of the catalyst materials must be designed to balance the binding energies of OH and OOH intermediates. Thus, identifying efficient, stable, and environmentally friendly bifunctional catalysts is crucial for enhancing the performance and advancing the commercial viability of ZABs.3. Design and research status of carbon-based catalysts
Carbon-based catalysts are characterized by their simple structure and ability to simultaneously catalyze both ORR and OER.49 These catalysts are straightforward in terms of material composition and chemical synthesis, and they demonstrate exceptional bifunctional performance.50,51 This section discusses the applications and advancements of heteroatom-doped carbon catalysts, defective carbon catalysts, and single-atom catalysts (SACs). As fundamental model systems, these catalysts offer valuable insights into the impact of atomic-level structures on performance. They are expected to serve as a basis for the advancement of more sophisticated dual-functional electrocatalysts.3.1. Heteroatom-doped carbon catalysts
Nitrogen, which is an adjacent element to carbon, has been extensively investigated as a dopant for carbon frameworks.52 Nitrogen has a high electronegativity of 3.04 and a similar atomic radius to carbon (the electronegativity of carbon is 2.55). Thus, nitrogen dopants do not damage the structural integrity of carbon frameworks. The high electronegativity of nitrogen induces the redistribution of electrons within the carbon matrix, enhancing local charge displacement and creating active sites. Additionally, nitrogen doping enhances the electrical conductivity of carbon materials by adding more electrons, resulting in lower resistance and the promotion of electrocatalytic activity. Nitrogen-doped carbon materials are regarded as eco-friendly electrocatalysts due to their abundance, sustainability, and lack of metals.53 These catalysts exhibit significant promise and research value for improving the stability of electrocatalysts.In 2009, the ability of vertically aligned nitrogen-doped carbon nanotube arrays (VA-NCNTs) to perform the ORR under alkaline conditions was reported. Since then, nitrogen-doped carbon has garnered significant attention as a single-component bifunctional electrocatalyst. Adding nitrogen atoms changes how oxygen (O2) adsorbs onto carbon nanotubes. In pure carbon nanotubes (CCNTs), oxygen adsorbs end-on. In nitrogen-doped carbon nanotubes (NCNTs), it adsorbs side-on.54 The parallel diatomic adsorption mode efficiently weakens the oxygen–oxygen (O–O) bond, which promotes the ORR process on the surface of NCNTs. Subsequently, Wang et al. developed nitrogen-doped carbon spheres (N-CS) with a microporous structure via polymerization and pyrolysis methods. Using ZnCl2 and a foaming agent greatly enhanced the specific surface area of N-CS. Compared to Pt/C, N-CS-1 exhibited exceptional performance in ZABs.55 Meanwhile, Sun and colleagues doped pyridine nitrogen at graphene edge sites with precision. They developed a new nitrogen-doped graphene material. This material exhibited significantly improved bifunctional electrocatalytic performance.56
The chemical configuration and concentration of nitrogen atoms, including pyridine nitrogen, pyrrolic nitrogen, graphitic nitrogen, and oxidized nitrogen, have significant effects on the activity of nitrogen-doped carbon-based bifunctional electrocatalysts. Scientists have thoroughly investigated the catalytic activity associated with these various forms of doped nitrogen. Guo et al. used model catalyst construction and density functional theory (DFT) calculations to prove that carbon atoms adjacent to pyridine nitrogen serve as active sites for ORR. This finding provides theoretical support for the development of nitrogen-doped carbon catalysts with high activities.57 In addition to nitrogen-doped carbon materials, various heteroatoms can be integrated into carbon matrices to induce charge redistribution. Carbon-based substances doped with boron, sulfur, and phosphorus have been successfully produced and utilized as non-metallic bifunctional electrocatalysts. For instance, Huang et al. designed a porous electrocatalyst (mf-pClNC) without any metal components. Both chlorine and nitrogen were introduced into this electrocatalyst. As shown in Fig. 2(a), this modification greatly improved the catalytic activity for ORR. The addition of chlorine enhanced the proportion of sp3-hybridized carbon atoms and improved the adsorption and desorption properties of the involved substrates and intermediates.58 Subsequently, a range of non-metallic bifunctional carbon-based electrocatalysts with dual or multiple dopants has been synthesized, including binary (e.g., N–S,59,60 N–P,61 N–B62) and multiple (e.g., N–S–P,63 N–S–B64) dopant carbon materials. These studies demonstrate that secondary doping is an effective strategy for enhancing oxygen electrode catalytic activity.
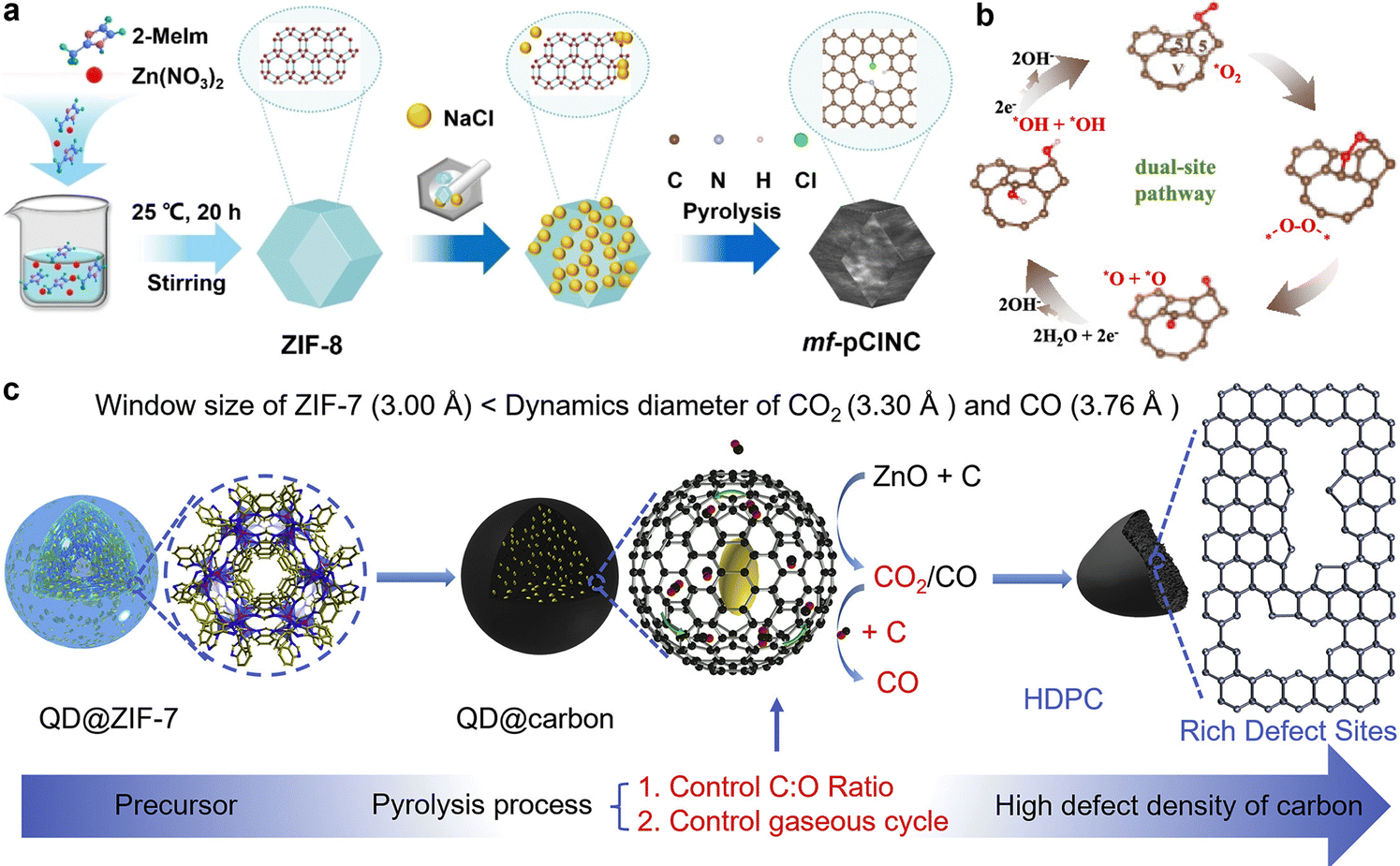 | ||
| Fig. 2 (a) The fabrication process of the metal-free porous electrocatalyst doped with Cl (mf-pClNC) electrocatalysts. Reprinted with permission.58 Copyright 2023, Wiley-VCH. (b) Schematic of the ORR mechanism for the model of the carbon vacancy coupling adjacent pentagons (VP) sites. Reprinted with permission.65 Copyright 2023, American Chemical Society. (c) Design and fabrication schematic diagram of high-defect density porous carbon (HDPC). Reprinted with permission.66 Copyright 2022, Elsevier. | ||
3.2. Defective carbon-based catalysts
Carbon atoms that are sp2 hybridized form three σ bonds and one π bond, which creates a stable planar hexagonal structure. However, pure carbon materials exhibit limited catalytic activity toward OER and ORR because their electronic structure is primarily governed by π-electrons, which lack unpaired electrons or d-orbitals for electron donation or acceptance. Consequently, pure carbon materials do not have sufficient active sites for the effective adsorption and activation of reactant molecules.67 Advances in research have revealed that defective carbon materials can show surprising catalytic activity. Atom vacancies or lattice distortions in carbon nanostructures lead to defects that disturb the symmetry of electron holes. Defective carbon materials have the ability to alter the local π-electron density.68 Therefore, these carbon materials are strong potential candidates for use as bifunctional oxygen electrode catalysts.To enhance the catalytic activity of carbon-based materials, researchers have extensively explored various methods for introducing defects. Xia et al. fabricated two-dimensional carbon nanosheet (VP/CN) electrocatalysts with a high concentration of defects utilizing a template-assisted method.65 NaCl and Zn(NO3)2·6H2O served as templates, and after heat treatment and acid washing, a material with adjacent pentagon structures and carbon vacancies was obtained. The combined effect of these structures facilitated a two-site pathway during the ORR process, as depicted in Fig. 2(b). Compared to the single-site pathway, this dual-site pathway avoided the formation of the OOH intermediate, which lowered the reaction energy barrier. Additionally, the synergistic interaction between carbon vacancies and pentagon sites further facilitated O–O bond cleavage. As a result, this catalyst achieved a E1/2 of 0.86 V for ORR, nearly matching the 0.87 V of Pt/C. To the template method, substrate corrosion is another effective approach for introducing defects. Wu et al. investigated the diffusion and transformation behavior of CO2 in carbon cavities using finite element methods (FEM) and in situ thermogravimetric-mass spectrometry (TG-MS). High-density porous carbon (HDPC) was successfully synthesized by controlling defect density through carrier gas flow adjustment to provide the carbon cavities.66 This interfacial self-corrosion strategy significantly increased ORR active sites, offering an solution for enhancing the catalytic activity of carbon-based nanomaterials. The synthesized HDPC exhibited a E1/2 of 0.90 V. This research provides a promising and versatile strategy for developing a new generation of electrocatalysts.
The combination of defects with doping can significantly amplify the activity of metal-free carbon-based materials. Jiang et al. employed a spontaneous gas foaming method to synthesize ultrathin nitrogen-doped carbon nanosheets (NCNs).69 By optimizing the carbonization temperature and precursor mass ratio, they precisely controlled the morphology, resulting in a rich porous structure that enabled simultaneous ORR, OER, and hydrogen evolution reaction (HER) catalysis. The active sites were primarily located at the armchair edges near carbon atoms doped with graphitic nitrogen. The NCN-1000-5 sample exhibited trifunctional activity. As an air electrode for ZABs, NCN-1000-5 demonstrated a higher performance.
In summary, the introduction of defects in carbon-based materials increases the number of active sites. This provides bifunctional catalytic activity, even in carbon materials not modified with any additional elements. The discovery of these catalysts has enhanced our comprehension of edge chemistry and defect chemistry. However, accurately synthesizing and characterizing carbon defect structures at the atomic level continues to be a difficult challenge. Furthermore, because defects are frequently generated during heteroatom doping, it is crucial to differentiate between the impacts of defects and doping in order to comprehend fundamental chemical principles. This is particularly important when determining active structures such as dopant type, location, and distribution as well as the specific catalytic functions of these active structures. Additionally, the ORR activity of these catalysts still falls short of that seen in transition metal-containing catalysts, especially in acidic electrolytes, which continues to limit their practical application in energy conversion devices.
3.3. Single-atom catalysts
Heteroatom doping in carbon-based materials introduces localized polarity, which allows these materials to catalyze both the ORR and OER. In addition to non-metal doping, the use of transition metal elements to create M–N–C sites (where M represents a transition metal atom) has been demonstrated to greatly improve electrocatalytic activity, offering a promising alternative to precious metal catalysts.70 Within these structures, individual transition metal atoms are uniformly distributed and bound to non-metal atoms in the same plane (often nitrogen or carbon).71 This arrangement forms unique single-atom M–N–C combinations. M–Nx (M = Fe, Co, Mn, Ni, Cu, Zn, etc.) sites are recognized as the primary active components, where the coordinated nitrogen atoms are predominantly pyridinic or pyrrolic nitrogen. The diversity in coordination numbers (x) results in the formation of M–Nx sites with great structural variety.72 At these active sites, the d orbitals of metal atoms interact with the p orbitals of oxygen molecules. In ORR, the d orbitals donate electrons to the p orbitals of oxygen molecules, which activates and reduces the oxygen. In the OER, the d orbitals at the metal centers accept electrons, which facilitates electron loss from adsorbed water molecules or OH−. This leads to the formation of intermediates and promotes the formation of O–O bonds, ultimately leading to oxygen release.73 Therefore, M–N–C electrocatalysts are considered promising candidates for bifunctional applications.Fe–N–C electrocatalysts display superior ORR performance but show limited OER activity. In comparison to other M–N–C catalysts, Co–N–C exhibits improved OER performance and maintains robust ORR activity, as demonstrated by the theoretical calculations reported by Xu et al.74 Experimentally, Co–N–C has been extensively explored as a bifunctional catalyst. Sun et al. synthesized a Co–N–C with porous structure (Co–N–C/AC) by simultaneously heating a metal–organic framework (MOF) precursor and ammonium chloride (AC).75 The ammonium chloride enhanced the active surface area and enabled the precise control of active sites by adjusting the precursor ratio and temperature. Thus, pyridinic nitrogen sites were optimized and new cobalt species were generated. The resulting Co–N–C/AC catalyst exhibited excellent bifunctional performance, with a low potential difference of 0.72 V. Zhang et al. successfully synthesized oxygen-rich cobalt–nitrogen–carbon (O–Co–N/C) porous nanosheets using template-guided CoO@ZIF-8 nanosheet growth and high-temperature thermal treatment (Fig. 3(a)).76 The O–Co–N/C electrocatalyst, which contained Co–N–C and Co–O–C active centers and an ultra-thin porous carbon structure, exhibited an E1/2 of 0.85 V (ORR) and an OER overpotential of 0.29 V. Liang et al. first reported an onion-like carbon-coated cobalt and nitrogen-doped carbon material (OLC/Co–N–C) prepared by including surfactant micelles in the precursor, which resulted in a multilayer high-curvature nanocatalyst with a mesoporous structure (Fig. 3(b) and (c)).77 This OLC/Co–N–C catalyst demonstrated effective bifunctional ORR/OER activity due to its combined electrical and structural effects. Furthermore, the presence of the curved graphitic carbon significantly increased the reactivity of metal–carbon atoms in the vicinity of the graphitic nitrogen as well as that of ortho/meta carbon atoms near pyridinic nitrogen (Fig. 3(d)).
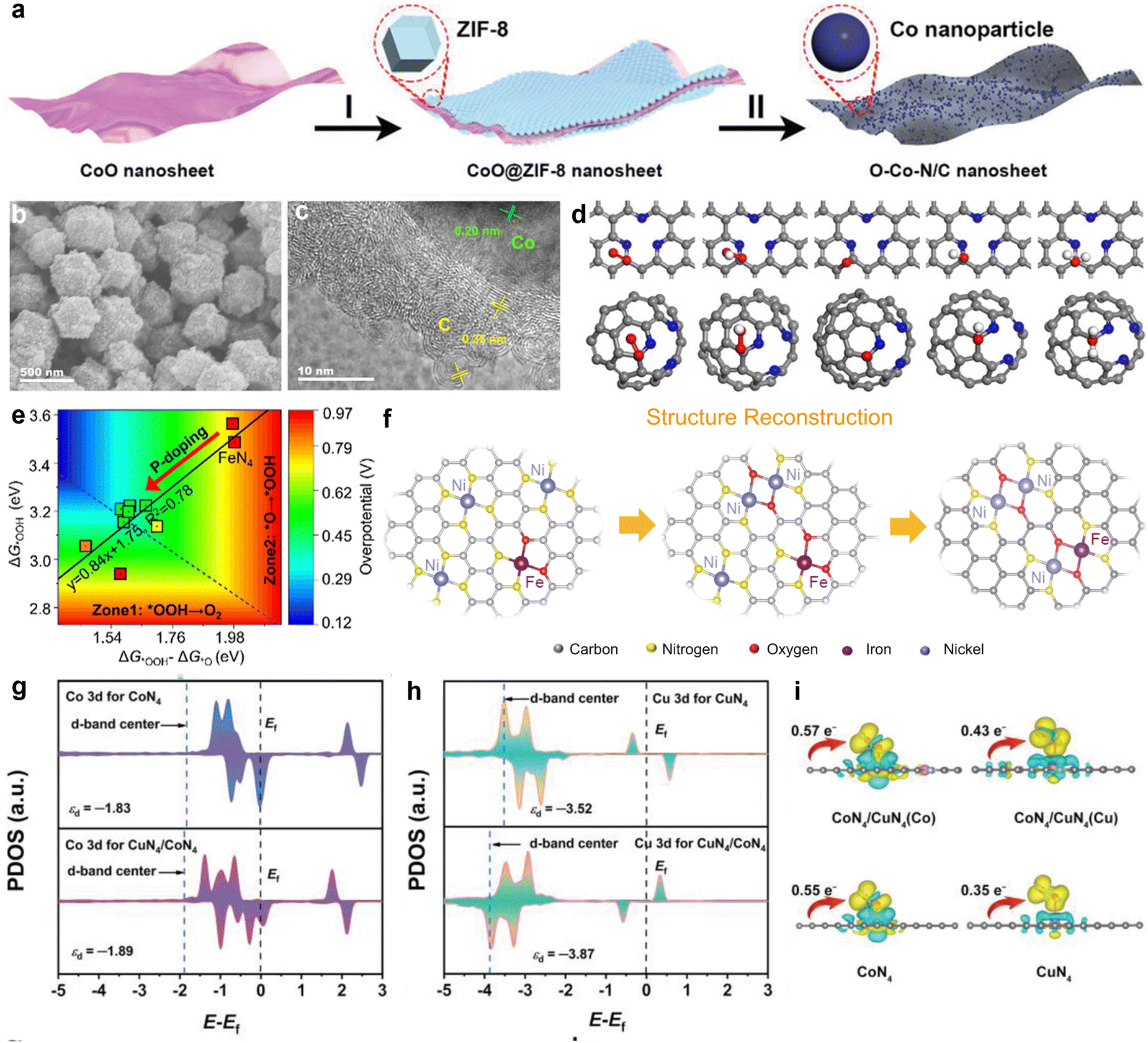 | ||
| Fig. 3 (a) Illustration of the preparation steps of O–Co–N/C nanosheets. Reprinted with permission.76 Copyright 2022, Wiley-VCH. (b) SEM image and (c) HRTEM image of OLC/Co–N–C. (d) Structural reconstruction of NiFe-CNG during OER from separate Ni and Fe centers to Ni–O–Fe/Ni moieties. Reprinted with permission.77 Copyright 2021, Wiley-VCH. (e) Contour plot of OER overpotential as a function of Gibbs adsorption energies ΔG*OOH − ΔG*O along the x-axis and ΔG*OOH along the y-axis. The inset is the scaling relation ΔG*OOH = 0.84 (ΔG*OOH − ΔG*O) + 1.75. Reprinted with permission.78 Copyright 2023, The Author(s). (f) Structural reconstruction of NiFe-CNG during OER from separate Ni and Fe centers to Ni–O–Fe/Ni moieties. Reprinted with permission.79 Copyright 2021, The Author(s). Projected density of states of Co 3d and Cu 3d orbitals in (g) CoN4 and CuN4/CoN4, (h) CuN4 and CuN4/CoN4, and the corresponding d-band centers. (i) Charge density difference and Bader charge analysis of O2 adsorbed on CuN4/CoN4 (Co), CuN4/CoN4 (Cu), CuN4, and CoN4. Reprinted with permission.80 Copyright 2022, Wiley-VCH. | ||
Aside from Co–N–C, other M–N–C systems have shown considerable promise as bifunctional oxygen electrode catalysts. Liang et al. presented design guidelines for SACs based on graphene, using theoretical models and tests to confirm the influence of the local structure and chemical environment of Ni–N–C centers on catalytic activity.81 Jin and his coworkers created a catalyst made from a biomass aerogel precursor that is atomically distributed and doped with Mn and N in carbon aerogel.82 This catalyst demonstrated exceptional efficacy, indicating its potential for application in ZABs. Mei and colleagues developed a core–shell nanostructured catalyst consisting of MnO@Cu–N–C.83 Due to the combined influence of MnO and Cu–N–C, this catalyst demonstrated effective electron transport properties and a smaller overpotential. As a result, greatly improved ZAB performance was achieved. However, despite the significant advancements reported in the development of bifunctional oxygen electrode catalysts based on M–N–C materials, simultaneously enhancing ORR and OER activity within the same reaction mechanism is still a notable challenge. Rossmeisl et al. used theoretical calculations to predict the efficiency of M–N4 active sites.84 Their volcano plots indicated that selecting an active site requires an optimal balance between the adsorption energies of different reaction intermediates. However, because the optimal Gibbs free energy values differ for ORR and OER, optimizing one reaction often compromises the activity of the other reaction. Thus, simultaneously achieving high efficiency for both reactions is difficult.
One of the most effective solutions for reducing the energy requirements of the OER pathways is adjusting the coordination environment of metal atoms. Key measures include modifying the coordination number of metal atoms in the first coordination shell and introducing dopants or structural defects in the second coordination shell. For instance, Li et al. precisely tuned the coordination environment of iron SACs by employing a polymer coating, wet chemical adsorption, ammonia treatment, and calcination strategy.7 This approach resulted in the preparation of an atomically dispersed catalyst with an Fe1N4O1 structure. By adjusting the coordination environment of the iron atoms, the electronic metal-support interaction was optimized, enhancing the adsorption and desorption capabilities of this catalyst for oxygen intermediates. Fe1N4O1 demonstrated excellent ORR activity and a low OER overpotential across the entire pH range, outperforming the Pt/C + RuO2 benchmark in terms of ZAB performance and stability. In addition, the sluggish oxygen–oxygen coupling process in Fe–N–C materials leads to low OER activity. Chen et al. synthesized a phosphorus-containing Fe–N–C catalyst (P/Fe–N–C) by carbonizing iron-doped zeolitic imidazolate frameworks.78 Spectroscopic characterization revealed that an FeN4 structure was embedded in graphite carbon. The P/Fe–N–C catalyst exhibited enhanced OER activity, and excellent ORR performance was achieved. OER activity across can be described by Gibbs adsorption energy functions (ΔGOOH − ΔGO and ΔGOOH), where ΔGOOH − ΔGO represents the O–O coupling process (O → OOH), and ΔGOOH corresponds to the O2 release process (OOH → O2). The original Fe–N–C material exhibited an OER as high overpotential, indicating poor OER activity. In contrast, the P/Fe–N–C model displayed improved OER performance due to an increase in ΔGOOH and a decrease in ΔGOOH − ΔGO. Therefore, introducing phosphorus into the second coordination shell optimized the adsorption properties of *OOH and *O, achieving improved OER performance. To further regulate catalytic activity by generating defects in the second coordination shell, Li's group developed a multifunctional molten salt-assisted pyrolysis strategy for the construction of ultrathin porous carbon nanosheets loaded with Co SACs.85 The molten salt induced the formation of Co single atoms and porous graphene-like carbon, which ensured the full exposure of active centers and endowed the Co SACs with abundant defective Co–N4 structures. The prepared Co SACs exhibited excellent bifunctional activity and stability. DFT calculations showed that defects in the second coordination shell of the Co SACs facilitated the desorption of OH intermediates in ORR and the deprotonation of OH in OER. Therefore, these defects served as effective active centers for bifunctional oxygen catalysis.
Constructing dual-atom catalysts is an effective strategy for addressing these challenges. During OER, the synergistic effects of two metal elements can enhance the adsorption and reconstruction of reaction intermediates. Wan et al. proposed a molecular design strategy that facilitated the rational design of both single-center and dual-center catalysts.79 With this strategy, a catalyst featuring single Fe sites demonstrated excellent ORR activity, achieving a E1/2 of 0.89 V. During OER process, the coordination environment around the Ni atoms underwent reconstruction, forming Ni–O–Ni/Fe bonds (Fig. 3(f)). These newly formed Ni–O–Fe bonds provided a spin channel for electron transfer, which led to improved OER activity. A dual-site water oxidation pathway was enabled at Ni–O–Fe bridge sites, where *OH deprotonation at the Ni and Fe sites led to the generation of O2. Compared to single-atom catalysts, dual-atom catalysts retain the benefits of SACs while achieving multifunctional synergistic catalysis through geometric and electronic ligand effects. Catalysts with uniformly structured and flexible active sites show better performance in bifunctional catalysis. Tang et al. developed a Janus-type dual-metal active site catalyst (FeCo–N3O3@C). In this catalyst, the Fe and Co atoms were respectively coordinated with N and O atoms, and the atoms were connected through bridging N and O atoms.86 This unique coordination environment (Fe–N3 and Co–O3) allowed for the precise regulation of bifunctional active sites, optimizing the adsorption and desorption of oxidation intermediates and enhancing reaction kinetics.
Recently, Li's team proposed a method for constructing Cu–Co dual-atom site electrocatalysts on a highly porous nitrogen-doped carbon matrix (Cu–Co/NC).80 The CuN4/CoN4, CuN4/CoN4 system exhibited improved electronic conductivity, with a negative shift in the d-band center of Co leading to the increased occupancy of anti-bonding orbitals and promotion of molecular oxygen dissociation (Fig. 3(g)). Simultaneously, the larger negative shift in the d-band center of Cu may have weakened its binding affinity with molecular oxygen, affecting O2 activation (Fig. 3(h)). The O2 species adsorbed at the Co sites in the CuN4/CoN4 system acquired more charge (0.57 e−) compared to other models, demonstrating higher charge transfer efficiency (Fig. 3(i)). Thus, the synergistic effect of the Cu–Co bimetallic sites resulted in asymmetric charge distribution of oxygen intermediates, leading to moderate adsorption and desorption behavior. By optimizing the coordination environment of the metal active sites, the adsorption and desorption efficiency of oxidation intermediates was significantly improved, enhancing the catalytic performance and reaction kinetics. This catalyst shows excellent potential for application in bifunctional oxygen electrodes.
Overall, SACs can be used to effectively incorporate highly active atomic-level sites within conductive carbon frameworks, showing great promise as candidates for bifunctional oxygen electrocatalysis. The intrinsic activity of M–N–C sites surpasses that of most carbon-based active sites, such as those with non-metal dopants or defects. From the perspective of atomic utilization efficiency, atomically dispersed active sites represent the forefront of technology, approaching theoretical limits. Looking ahead, the well-defined chemical structures of these catalysts provide valuable opportunities for molecular design and activity optimization, which will further highlight their distinct advantages in bifunctional oxygen electrocatalysis. However, the stability of SACs under operational conditions remains a challenge. Therefore, efforts must focus on simplifying the synthesis process, further enhancing activity (particularly OER activity), improving stability, and increasing the content and utilization of active sites.
4. Design and current research status of transition metal compound catalysts
Transition metal compound catalysts offer multiple active sites due to their diverse electronic structures and rich oxidation states.87,88 Additionally, doping and tuning nanostructures can optimize the physicochemical properties (e.g., conductivity and chemical stability) of these compounds, leading to a further enhancement in catalytic performance.89 Transition metal compounds are abundant and low-cost, indicating their significant potential for widespread usage. This section will provide an overview of the present application status of transition metal oxides (TMOs), transition metal nitrides (TMNs), transition metal sulfides (TMSs), transition metal phosphides (TMPs), and other transition metal compounds in bifunctional oxygen electrodes.4.1. Transition metal oxides
Co3O4 is a promising catalyst for both ORR and OER due to its plentiful oxygen vacancies and changing valence states, which enable efficient adsorption and desorption of oxygen intermediates. Moreover, Co3O4 demonstrates excellent chemical and mechanical stability in alkaline media, further enhancing its potential application range. However, the sluggish oxygen diffusion at the three-phase interface of Co3O4 catalysts results in inadequate durability and energy conversion efficiency when used in ZABs. Wu and colleagues addressed this issue systematically. They took inspiration from diving flies. They grew Co3O4 nanosheets on conductive carbon cloth. Then, they coated it with PTFE, annealed it, and activated it electrochemically to create a hydrophobic surface.90 Compared to untreated samples (Fig. 4(a), top), this hydrophobized Co3O4 NSs/CC cathode showed significantly improved oxygen diffusion properties in an oxygen-saturated alkaline electrolyte and contained more three-phase reaction interfaces (Fig. 4(a), bottom). Therefore, the kinetics of oxygen-related electrocatalytic reactions were accelerated. However, using pure Co3O4 as a bifunctional oxygen catalyst still faces some challenges, such as limited conductivity and insufficient active sites. Catalytic activity is primarily influenced by internal charge transfer resistance and the adsorption energy of surface intermediates. The electronic structure of surface atoms determines catalyst activity. Hence, modifying the electronic configuration of catalysts is anticipated to improve their catalytic performance. Zheng et al. introduced Co metal defects in Co3O4 via chlorine plasma treatment.91 The presence of these flaws improved the performance of Co(OH) sites during the OER by enabling the rapid absorption of oxygen from water in the early stage of OER and expediting the release of oxygen molecules from active sites in the final stage of OER. This promoted a seamless transition between successive OER cycles. In addition to introducing defects, doping is another technique used to manipulate the electronic structure of Co3O4. Huang et al. employed DFT to examine the impact of manganese doping on the electronic structure of Co3O4 (MCO) catalysts and enhance the interactions between oxygen species and neighboring Co sites.92 The Bader charge distributions and oxygen vacancy formation energies on the Co3O4 (001) and MCO (001) surfaces are displayed in Fig. 4(b)–(i). The probable oxygen vacancy spots are indicated by green dashed circles. The Mn and adjacent O had a stronger interaction, which extended the adjacent Co–O bond from 1.881 Å to 1.892 Å. The ΔGads values of O*, OH*, and OOH* at Co1 sites were relatively low. In contrast, the Co2 and Mn sites exhibited a higher affinity for OH groups, suggesting that oxygen species were more readily adsorbed and activated at the Mn sites. Overall, manganese doping optimized the interaction between oxygen species, leading to improved performance without the introduction of additional electronic states. Yue's team achieved superior ORR and OER activities by disrupting the structural symmetry of Co3O4.93 Consequently, the nanorods was modulated and the electron occupancy of octahedral Co3+ sites (approximately 1.010) in the e.g. orbital was optimized, leading to enhanced performance compared to Co3O4 and even Pt–IrO2. When this V-doped Co3O4 was used as an air electrode in rechargeable aqueous and flexible ZABs, a narrower charge–discharge voltage gap and higher capacity were achieved compared to pure Co3O4 and Pt/C + IrO2-based batteries (Fig. 4(i)–(n)). In addition to tuning oxygen diffusion rates at the three-phase interface and enhancing the bifunctional properties of Co3O4 through defect engineering and doping, the combination of Co3O4 with other oxides to form heterostructures represents another effective strategy for achieving improved performance. This heterostructure formation strategy will be discussed in Section 5.1.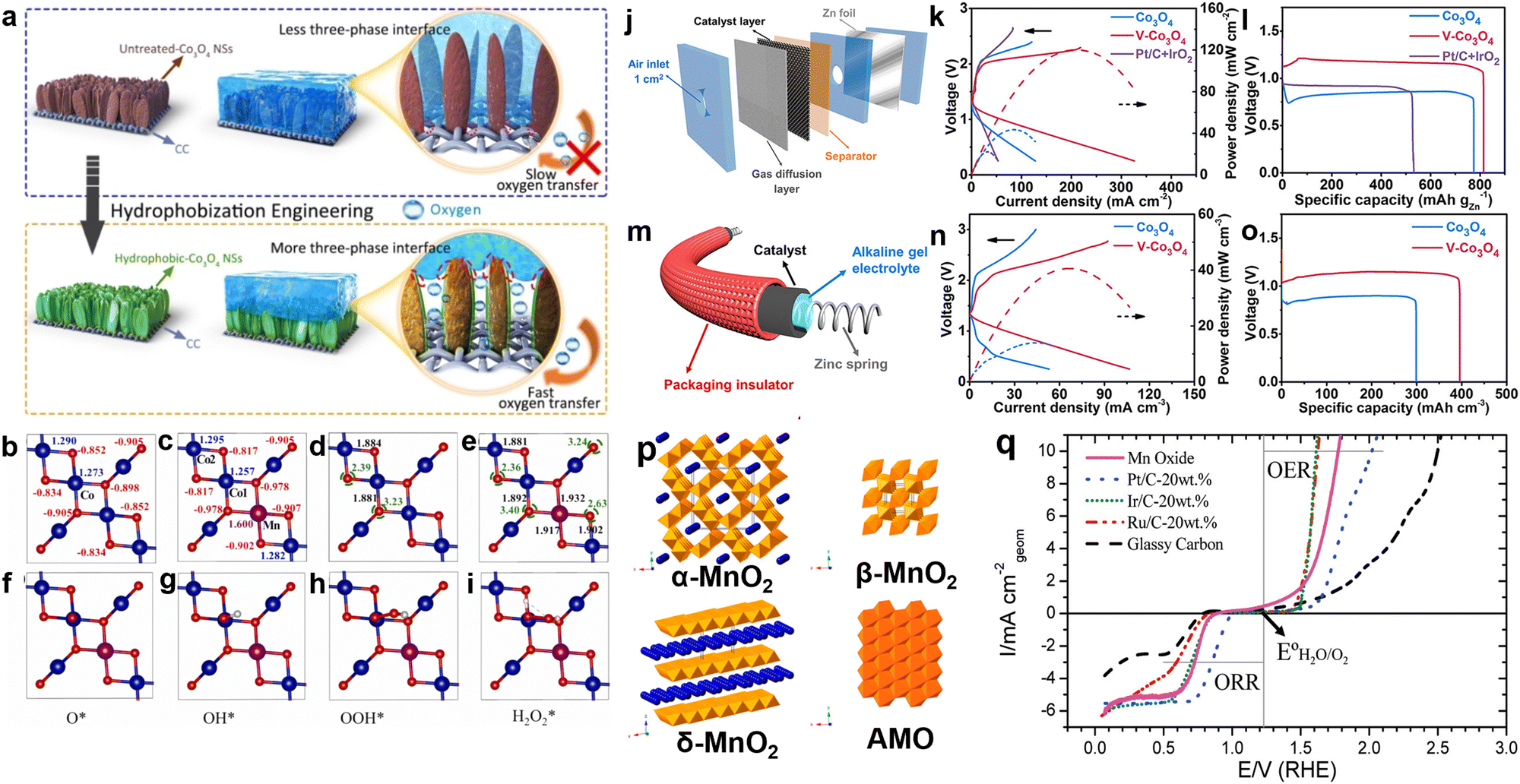 | ||
| Fig. 4 (a) Schematic depiction of the hydrophobization engineering of Co3O4 NSs to enable the formation of more three-phase reaction interfaces and promoted oxygen diffusion on the obtained hydroophobic-Co3O4 NSs/CC cathode impregnated with electrolyte, in comparison with untreated-Co3O4 NSs/CC cathode. Reprinted with permission.90 Copyright 2022, Wiley-VCH. (b) and (c) Bader charges of Co (in blue), Mn (in magenta) and O (in red) of Co3O4 (001) and MCO (001) surfaces; (d) and (e) major bond lengths (in black) and formation energies of oxygen vacancy (in green); (f)–(i) top view of the pristine MCO (001) surface as well as the adsorption states of O*, OH*, OOH*, and H2O2* over Co1 site, respectively. Note: the blue, magenta and red balls represent Co, Mn and O elements, and the green dashed circles are the potential position for oxygen vacancies. Reprinted with permission.92 Copyright 2022, Elsevier. (j) Schematic illustration of the liquid-state Zn–air battery device. (k) Polarization curves and power density profiles, and (l) voltage vs. specific capacity curves at a constant current density of 5 mA cm−2 of the liquid-state rechargeable Zn–air batteries. (m) Hierarchical structure of the cable-type Zn–air battery. (n) Polarization curves and power density profiles, and (o) voltage vs. specific capacity curves at a constant current density of 7 mA cm−3 of the cable-type all-solid-state Zn–air batteries. Reprinted with permission.93 Copyright 2021, American Chemical Society. (p) Structures of manganese oxide reported here in this study: α-MnO2 (2 × 2 tunnel), β-MnO2 (1 × 1 tunnel, pyrolusite), δ-MnO2 (layered, birnessite), and amorphous manganese oxides (AMO). Reprinted with permission.94 Copyright 2014, American Chemical Society. (q) Oxygen electrode activities of the nanostructured Mn oxide thin film, nanoparticles of Pt, Ir, and Ru, and the GC substrate. The Mn oxide thin film shows excellent activity for both the ORR and the OER. Reprinted with permission.95 Copyright 2010, American Chemical Society. | ||
Single metal oxides typically exhibit high OER catalytic activity. However, ORR performance is impeded by the considerably weaker bond between oxygen atoms and the surfaces of TMOs compared to transition metals. This challenge is further exacerbated by the powerful electric field effect on the TMO, which inhibits the breaking of O–O bonds. Nørskov and colleagues screened 7798 TMO compositions for ORR catalysts, reporting that manganese-based oxides show great potential for achieving high activity.96 The catalytic properties of manganese oxides vary based on the structural connectivity of [MnO6] units, which can be linked by shared corners or edges. One common manganese oxide structure is layered δ-MnO2, where MnO6 units share edges and the interlayer spaces are occupied. One-dimensional MnO2 materials, form various configurations based on tunnel size. Manganese oxides with tunnel structures include β-MnO2, γ-MnO2, α-MnO2, OMS-1 (3 × 3), and OMS-5 (2 × 4). Among these, α-MnO2 has been extensively studied due to its superior adsorption, catalytic, and oxidation properties.
Meng's research team facilely synthesized MnO2 nanomaterials with different crystal structures (Fig. 4(p)) and systematically analyzed the structure–performance relationship of these materials as OER and ORR catalysts in alkaline media.94 The exceptional performance of α-MnO2 was attributed to its abundant bridging oxygen bonds, mixed valence states (average oxidation state = 3.7), and low charge transfer resistance (91.8 Ω). Compared to cobalt oxides, manganese oxides exhibit a broader range of valence states, with the presence and ratio of Mn4+ and Mn3+ playing a critical role in determining catalytic activity. Inspired by the oxygen-evolving center (OEC) of Photosystem II, Jaramillo and colleagues mimicked the structure of the active site CaMn4Ox to develop a manganese oxide film with coexisting Mn4+ and Mn3+, as shown in Fig. 4(q).95 This film demonstrated high ORR and OER activity, surpassing Ru and Ir nanoparticles in terms of ORR activity. In terms of OER activity, the manganese oxide film significantly outperformed Pt and approached the activity of Ir and Ru. Beyond crystal structure and valence state, manganese-based oxides can be prepared to selectively expose different crystal facets, which also exhibit varying catalytic activity. He and colleagues found that the MnO (100) facet showed significantly enhanced electrocatalytic performance due to its higher oxygen species adsorption energy.97 They synthesized MnO nanoflowers and multi-legged structures with specific orientations using a thermal decomposition method with oleic acid as a free ligand. The unique MnO exhibited a high mass activity.
Perovskite oxides are materials that can be described by the generic formula ABO3. The A-site ions of perovskites are usually larger alkaline earth or rare earth metals such as calcium, strontium, barium, or lanthanum. These ions are arranged in a twelve-fold polyhedral environment. The B-site ions are often smaller transition metals such as titanium, manganese, iron, or cobalt that are situated in an octahedral coordination environment, where they create a three-dimensional network of BO6 octahedra. Perovskite oxides offer a diverse platform for creating efficient electrocatalysts due to the variety of A-site and B-site ions, which enables the modification and control of oxygen vacancies and modify crystal structure.
Peng's group used oxygen vacancy-rich porous CaMnO3 nanofibers (V-CMO/rGO) as air electrode catalysts for low-temperature wearable ZABs (Fig. 5(a)).98 The synergistic effect of the metal atoms and oxygen vacancies combined with the improved kinetics, conductivity, and mass transfer inside the 3D rGO-coated nanofibers resulted in dramatically improved catalytic activity. The regulated electrical filling encouraged the desorption of ORR intermediates. Thus, V-CMO/rGO enabled the operation of a flexible ZABs at −40 °C, yielding a power density of 56 mW cm−2 and a cycling life of over 80 h. This novel and dynamically active catalyst shows good promise for ZAB applications in hostile settings (Fig. 5(b)).
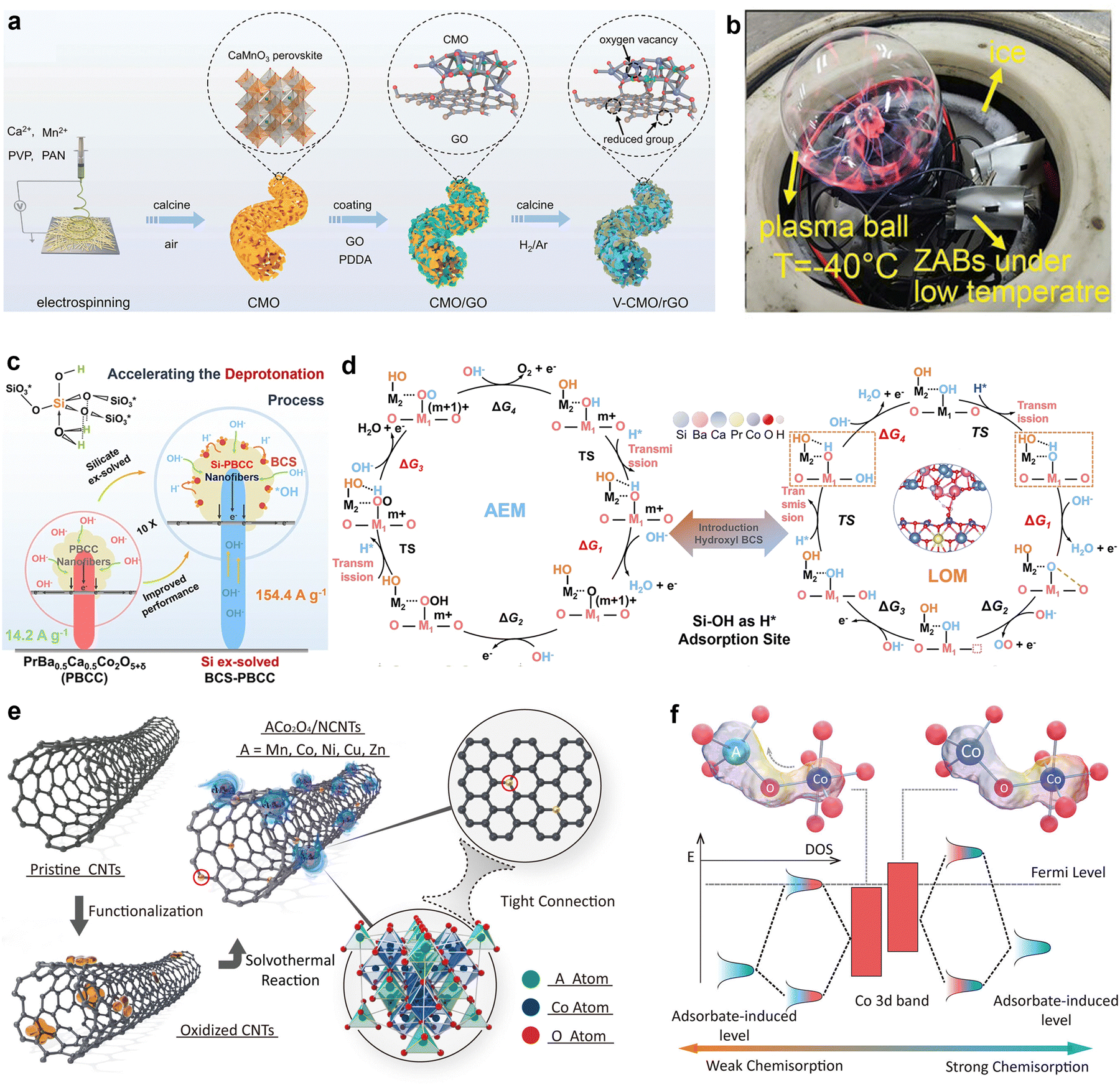 | ||
| Fig. 5 (a) Schematic diagram of the synthesis process of the vacancy-rich, reduced graphene oxidecoated porous CaMnO3 (V-CMO/rGO). (b) Photograph of a plasma ball powered by three flexible sandwich-type ZABs under −40 °C. Reprinted with permission.98 Copyright 2023, Wiley-VCH. (c) Chemical and electrochemical characterization of the solid-hydroxy silicon/perovskite oxide. A structural formula of the hydroxy silicon and a schematic diagram showing the deprotonation process on the PBCC surface. (d) DFT calculations and the electrocatalysis mechanism. Proposed OER mechanisms, including a AEM and b LOM. M1 (Co) and M2 (Si) are the adsorption centers of the AEM and LOM in the OER pathway. Other intermediates are also labeled. The empty square represents an oxygen vacancy. Reprinted with permission.99 Copyright 2023, The Author(s). (e) Schematic illustration of the formation process of ACo2O4/NCNTs (A = Mn, Co, Ni, Cu, Zn). (f) Schematic diagram of tetrahedral and octahedral interaction. Reprinted with permission.100 Copyright 2022, Wiley-VCH. | ||
The OER activity of perovskite oxides primarily depends on the charge transfer energy during the proton-coupled electron transfer (PCET) process. However, the limited energy of lone-pair electrons surrounding the oxygen atoms constrains the pace at which deprotonation occurs. Moreover, simultaneously enhancing OER and ORR is a significant challenge if both reactions follow the adsorbed-electron mechanism (AEM). To address this, Wang et al. introduced hydroxyl-containing solid base BaCaSiO4 (BCS) nanoparticles onto the surface of perovskite oxide nanofibers using a one-step calcination and desolation strategy.99 They investigated the role of Si doping in improving the physicochemical properties and OER activity of this material. During the formation of the BCS proton acceptors, the protons acted as Lewis acids, binding with electron donors. For instance, Ca2+/Ba2+ and SiO2 formed multi-anionic [SiO44−] silicates, where alkaline O2− ions were transferred from weakly acidic Ca2+/Ba2+ to strongly acidic Si4+. This increased the oxygen atom electron density, enhanced hydrogen bond formation, and stabilized the coordinated H![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(Si), causing the Si–O–Si bond angle to deviate from 180°. The resulting hydrogen bonds stabilized structures such as OH–Si–O–Si–OH and OH–Si–O–Ca2+/Ba2+–Si–OH, which more readily accepted protons from OH* intermediates. Consequently, a tenfold increase in OER performance was achieved (Fig. 5(c)). Additionally, DFT was used to study the reaction mechanism and proton transfer pathways in the BCS-PBCC system during OER (Fig. 5(d)). DFT analysis revealed that the surface-functionalized Lewis base acted as an electron pair donor, enabling effective coordination with protons and a reduction in the energy barrier of OOH* deprotonation. Thus, kinetic limitations in proton transfer were overcome and the lattice oxygen mechanism (LOM) was enabled, significantly enhancing OER performance. The lattice oxygen mechanism refers to the process in which oxygen atoms in the catalyst lattice participate in oxidation-reduction reactions by accepting or releasing electrons to facilitate chemical reactions.
O(Si), causing the Si–O–Si bond angle to deviate from 180°. The resulting hydrogen bonds stabilized structures such as OH–Si–O–Si–OH and OH–Si–O–Ca2+/Ba2+–Si–OH, which more readily accepted protons from OH* intermediates. Consequently, a tenfold increase in OER performance was achieved (Fig. 5(c)). Additionally, DFT was used to study the reaction mechanism and proton transfer pathways in the BCS-PBCC system during OER (Fig. 5(d)). DFT analysis revealed that the surface-functionalized Lewis base acted as an electron pair donor, enabling effective coordination with protons and a reduction in the energy barrier of OOH* deprotonation. Thus, kinetic limitations in proton transfer were overcome and the lattice oxygen mechanism (LOM) was enabled, significantly enhancing OER performance. The lattice oxygen mechanism refers to the process in which oxygen atoms in the catalyst lattice participate in oxidation-reduction reactions by accepting or releasing electrons to facilitate chemical reactions.
As bifunctional catalysts, the active sites of perovskite oxides mainly consist of surface oxygen vacancies, cationic defects, and transition metal centers. Crucially active locations such as surface oxygen vacancies can be used to catalyze both ORR and OER. Cationic flaws change the electronic structure of perovskite oxides, which improves activity. Moreover, important catalytic sites in perovskites that accelerate electrocatalytic processes via interactions with reactants include transition metal centers (e.g., Fe, Co, Mn). Thus, research on perovskite catalysts has concentrated on techniques for optimizing their crystal structure, cation control, anion management, nanostructure design, surface engineering, and electrocatalytic performance.
Transition metal oxides with spinel structures (i.e., those based on nickel, cobalt, and manganese) have attracted considerable interest as electrocatalysts, which is mainly due to their unique d-electron configuration and crystal structure. These materials usually adhere to the standard formula AB2O4, with A and B denoting distinct metal ions. The A-site metal ions typically occupy tetrahedral sites, while the B-site metal ions are located in octahedral sites, creating a three-dimensional network. The origins of the active sites and regulation techniques in perovskites are comparable to those in spinel-structured transition metal oxides. The metal ions, oxygen vacancies, and surface electronic structures of spinel oxides make them promising candidates as bifunctional oxygen catalysts.
For example, the spinel-structured catalyst CoFe2O4 exhibit bifunctional catalytic activity, which is often achieved through electronic structure regulation. Li's research group introduced La to adjust the electronic structure within a spinel, increasing the electron density at Fe sites. La acted as an “electron pump,” transferring electrons from Co sites to the catalytically active Fe sites. This optimized the adsorption process and enhanced bifunctional oxygen catalytic performance.101 Consequently, La0.2CoFe1.8O4/3D-G demonstrated outstanding ORR catalytic performance, with an E1/2 = 0.86 V. Moreover, this catalyst also showed a low potential and overpotential in OER (η = 320 mV), reflecting its excellent bifunctional performance (ΔE = 0.69 V). Tang et al. synthesized hollow spinel-type NiCo2O4 nanomaterials using a CTAB-assisted solvothermal method with ZIF-67 as a precursor.102 Highly active three-dimensional porous NiOOH–NiCo2O4 structures were formed on the surface of these catalysts via electrochemical reconstruction, which enhanced ORR and zinc–air battery performance. In situ Raman analysis revealed that during the OER process, this electrochemical reconstruction significantly increased pseudocapacitance compared to ORR. Theoretically, substituting atoms at the tetrahedral sites of AB2O4 spinel oxides optimizes charge distribution at the octahedral sites through ATd–O–BOh interactions. Zhao et al. proposed straightforward solvothermal process to control the structure of spinel oxides and used this method to prepare composite materials (ACo2O4/NCNTs, where A = Mn, Co, Ni, Cu, Zn) for use as oxygen electrocatalysis (Fig. 5(e)).100 The optimized MnCo2O4/NCNTs exhibited high ORR/OER activity and excellent stability. Fig. 5(f) illustrates the ATd–O–Co3+Oh and CoTd–O–Co3+Oh configurations in the spinel structure, where tetrahedral A ions were bonded to octahedral Co ions via a shared oxygen atom. The spin states of ATd and Co3+Oh are also depicted. Metal substitution adjusted the Co3+/Co2+ ratio, modulating the electronic structure. Compared to the other prepared catalysts, Co3+Oh in MnCo2O4 demonstrated more favorable binding with oxygen-containing species, which led to significantly enhanced oxygen electrocatalytic performance. Moreover, combining spinel oxides with other materials (e.g., carbon nanotubes or alloys) can markedly improve conductivity and catalytic activity. Alternatively, a one-step pyrolysis reduction strategy can be employed to prepare spinel oxide-modified alloy composites with superior catalytic performance.
4.2. Transition metal nitrides
Metal nitrides possess high melting points, hardness, excellent corrosion resistance, chemical stability, and favorable electrical and thermal conductivity. Transition metal nitrides (TMNs) have drawn particular interest in ORR/OER research due to their physical properties, which resemble those of noble metals, as well as their unique electronic structures.103 Studies have shown that the incorporation of nitrogen can enhance the electron density in the d-band of transition metals while compressing the d-band, which aligns their electronic properties more closely with those of platinum (Pt). This shift also causes the Fermi level to intersect, making TMNs promising candidates for use as efficient oxygen electrode catalysts and potential replacements for noble metals. Yang's team proposed a catalyst design concept featuring a bimetallic oxide active layer (SOAL).104 By selecting appropriate transition metal nitrides and introducing Mo as a second metal, the oxygen evolution activity was enhanced at Co sites through an “electron pump” effect. At high oxidation potentials in alkaline electrolytes, an amorphous oxide layer rich in cobalt and oxygen were generated on the Co3Mo3N catalyst. This layer, referred to as SOAL, further optimized the band structure and enhanced the OER activity. The η-carbide structure of Co3Mo3N exhibited a high d-orbital density at the Fermi level, contributing to its excellent oxygen reduction reaction (ORR) performance. Similarly, Saad et al. developed a three-dimensional Ni3FeN that demonstrated an overpotential of 259 mV in alkaline solutions, outperforming IrO2 and RuO2 catalysts.105 The superior OER performance of Ni3FeN was attributed to its hierarchical porosity of the bimetallic system. Additionally, guest metal doping and the formation of alloys or solid solutions can optimize electronic structures and modulate lattice strain. Ban and colleagues studied (CoxFe1−x)3N@NPC, 0 ≤ x < 0.5, demonstrating that phase transformation and electronic spin synergistic effects can enhance bifunctional oxygen catalytic performance (Fig. 6(a)).106 Intermediate phase transformation promoted the formation of a highly dispersed ternary nitride–carbon shell structure, which effectively suppressed grain coarsening. By adjusting the Co content, they optimized O2 adsorption and OH hydrogenation, leading to enhanced catalytic performance. Liquid and solid-state ZABs assembled from (Co0.17Fe0.83)3N@NPC exhibited a significant energy density. Given the complexity of synthesizing transition metal nitrides, refining synthesis methods and exploring cost-effective, environmentally friendly raw materials and process routes is necessary. | ||
| Fig. 6 (a) Synthetic procedure selection for (CoxFe1−x)3N@NPC catalysts. Reprinted with permission.106 Copyright 2023, Wiley-VCH. (b) TEM images of the Ni–Co–Mn–P (NCMP). (c) Comparison of the overpotentials to drive a current density of 10 mA cm−2 for the NCMP catalyst with some other transition metal phosphide-based electrocatalysts namely, (A) Co2P/Co-, N-, and P-doped carbons; (B) CoFeO@black phosphorus; (C) CoP@P, N co-doped carbon; (D) CoP N-doped carbon@CNT; (E) CoP NFs; (F) CoP/T3C2; (G) Mo–Ni3S2/NixPy/NF; (H) (Fe0.1Ni0.9)2P(O)/NF; and (I) NF@Fe2–Ni2P/C. (d) ZABs performance comparison. Reprinted with permission.107 Copyright 2024, Royal Society of Chemistry. (e) and (f) SEM images, (g) HR-TEM images, and (h) HAADF-STEM image of Co3S4/FeS@CoFe/NC. (i) The real-space distributions of spin density (the first row, the blue and orange colors of the electron cloud indicate two different spin orientations, respectively) and charge density difference (the second row, the charge accumulation and consumption regions are represented by yellow and blue, respectively) by DFT calculations for O adsorbed on Co3S4/FeS@CoFe/NC, Co3S4@Co/NC, FeS@Fe/NC, Co3S4/FeS. Dark blue, orange, yellow, blue gray, brown, red and white spheres represent cobalt, iron, sulfur, nitrogen, carbon, oxygen and hydrogen atoms, respectively. Reprinted with permission.108 Copyright 2024, Science Press and Dalian Institute of Chemical Physics. | ||
4.3. Transition metal phosphides
Transition metal phosphides (TMPs, TM = Fe, Co, Cu, Ni, W, and Mn) are widely recognized as alternatives to noble metal electrocatalysts. However, TMP electrocatalysts with single active sites typically do not exhibit remarkable bifunctional activity due to the different reaction routes of ORR and OER. Thus, modifying the surface electronic structure of TMPs through the incorporation of heteroatoms, organic molecules, or carbon supports is necessary to enhance their activity. TMPs typically exist in various crystalline phases, such as the cobalt phosphides CoP and Co2P, which are both considered active for ORR and OER. However, which of these phases is superior for specific reactions is not yet clear. Scott M. Geyer and colleagues reported that among CoP and Co2P nanocrystals synthesized using trioctylphosphine as the phosphorus source, Co2P exhibited superior OER performance, while CoP was more suitable for ORR/HER.109 Liu et al. utilized DFT to determine that within the CoxP phase (0 < x < 2), Co2P shows the highest electrocatalytic activity.110 Despite ongoing debates regarding the specific reactivity of TMPs, optimizing the stoichiometric ratio of transition metals to phosphorus is crucial for enhancing their bifunctional catalytic performance in ZABs. Effective strategies are needed to control the synthesis of TMPs with different crystalline phases and surface structures. Liu's research group proposed a cost-effective and straightforward in situ nitridation to prepare porous nitrogen–phosphorus-doped carbon-confined heterostructure composite material (CoP@Co2P) that exhibited excellent electrochemical performance.111 Using Co/Zn-ZIF as a template, this bifunctional electrocatalyst was synthesized via in situ phosphorylation and carbonization. The catalyst consisted of nanostructured cobalt phosphide heterostructure particles (Co2P and CoP) (Fig. 6(b) and (c)). The optimized charge redistribution mechanism of CoP@Co2P facilitated oxygen intermediate adsorption, leading to outstanding electrocatalytic performance in alkaline media. Recently, Khodayar et al. reported a reductive electrochemical synthesis method to prepare a Ni–Co–Mn phosphide (NCMP) electrocatalyst.107 This catalyst showed industrial-scale applicability. The trimetallic structure containing Ni, Co, and Mn offered abundant active catalytic sites, leading to excellent OER (ηj=50 = 218 mV), ORR (E1/2 = 0.74 V) performance, and higher energy density of ZABs (Fig. 6(d)).4.4. Transition metal sulfides
Transition metal sulfides (TMS) have emerged as promising electrocatalysts due to their versatile crystal structures, tunable electronic properties, and rich surface chemistry. Compared to transition metal oxides, TMS offer a broader range of active sites, higher conductivity, and more easily adjustable surface characteristics, making them particularly effective as bifunctional catalysts. Among the various TMS materials, theoretical studies have highlighted cobalt-based sulfides due to their high oxygen electrode activity, which can be attributed to kinetics that closely resemble those of noble metals. However, optimizing the OER performance of these electrocatalysts in practical applications remains a challenge. To address this, Wang et al. developed an FeCoNiSx with a high active site density.112 With low charging voltages, this catalyst showed high suitability for fast-charging zinc–air batteries operating at 100–400 mA cm−2. Additionally, quasi-solid-state ZABs fabricated with this catalyst demonstrated impressive lifespans. Detailed characterization and DFT calculations revealed that this amorphous multi-metallic sulfide was reconstructed into a defect-rich crystalline/amorphous ternary metal (oxy)hydroxide. The electronic coupling effects among the multiple active sites coupled with the migration of O* from Ni sites to Fe sites lowered the theoretical OER overpotential to as low as 170 mV. However, despite the high catalytic activity predicted by theoretical calculations, sulfides often suffer from poor stability, with many sulfides experiencing structural degradation during OER and ORR processes. To mitigate this, Li et al. successfully synthesized (FeCoNiMn2)S2 dual-phase medium-entropy metal sulfides (dp-MEMS) by regulating the Mn content to induce an interlocked structure at the microscopic scale.113 This design effectively prevented structural collapse during OER, leading to an enhancement bifunctional performance. Modifying the Mn content helped overcome kinetic barriers and accelerated the reaction rates. Additionally, tuning the electronic structure in these medium-entropy metal sulfides further enhanced their electrocatalytic performance. Thus, ZABs prepared using (FeCoNiMn2)S2 dp-MEMS demonstrated superior performance compared to those prepared using commercial noble metal catalysts. In addition to amorphous structures and medium-entropy material designs, combining different sulfides into composite materials is another key strategy. Cheng et al. developed a superhydrophobic carbon-supported metal sulfide/CoFe single-atom composite air electrode (Co3S4/FeS@CoFe/NC) (Fig. 6(e)–(h)). The CoNx/FeNx atomic sites on this electrode provided excellent ORR activity, while the Co3S4/FeS heterostructure acted as an OER catalyst.108 The multiphase coupling of this electrode regulated the charge spatial distribution, which reduced the spin density on adsorbed oxygen. Thus, the adsorption energy of oxygen intermediates was optimized and the electrocatalytic process was accelerated (Fig. 6(i)). Moreover, the superhydrophobic surface of the electrode enabled the formation of an efficient “gas–liquid–solid” three-phase interface, enhancing the electrochemical reaction efficiency and leading to outstanding bifunctional electrocatalytic activity and zinc–air battery performance.In conclusion, transition metal compounds, including oxides, sulfides, nitrides, and phosphides, offer significant advantages as bifunctional oxygen electrode catalysts. The use of these compounds shows substantial potential in electrochemical storage systems. To traditional noble metal, transition metal compounds are abundant, cost-effective, and exhibit excellent chemical and structural tunability. The unfilled d orbitals of transition metals serve as primary active sites, allowing for kinetic optimization between the OER and ORR processes through the modulation of the adsorbed intermediate bonding energies. The coordination environment and chemical bonding characteristics, such as M–O or M–S bonds, strongly influence the electronic structure and catalytic performance of these materials.
Enhancing active site density, improving electrical conductivity, and boosting catalyst stability and resistance to poisoning can be achieved through strategies such as heteroatom doping, the introduction of lattice defects, the regulation of exposed crystal facets, nanostructure optimization, and compositing with conductive carbon materials. Elemental doping optimizes reaction barriers by adjusting the electron density around metal centers, while the introduction of lattice defects and nanostructuring strategies increase the density and exposure of active sites. However, significant challenges remain, including maintaining long-term efficiency and stability under practical conditions, further reducing catalyst costs, and improving scalability. Therefore, in addition to focusing on the discovery of novel transition metal compounds, future research should also seek to gain a deeper understanding of active site mechanisms, reaction pathways, and key rate-limiting steps. This will require a combined approach involving both theoretical calculations and experimental characterization to develop more precise structure–performance tuning strategies. Additionally, validating the performance and durability of these catalysts in full-cell devices is a crucial next step for enabling large-scale commercial applications.
5. Design and current research status of composite catalysts
Composite bifunctional electrocatalysts are referred to as “composites” because they consist of two or more components. Combining the advantages of each constituent in these electrocatalysts enables them to catalyze both OER and ORR, thus achieving dual functionality. These catalysts can be prepared with many different structures, including core–shell,114 multilayer,115 and nano-composite structures,116 which offer a wider selection of active sites due to their complex designs. By addressing the limitations of single-component catalysts, composite electrocatalysts provide further enhanced bifunctional performance. However, the multi-component nature and complex structures of these catalysts mean that their preparation is more challenging and can result in poor batch-to-batch consistency. During long-term operation, some of the components of composite catalysts may dissolve, agglomerate, or degrade, which will adversely impact their overall electrochemical performance. To mitigate these issues, researchers are developing more controllable and reproducible synthesis methods and evaluating catalyst longevity through long-term stability tests. In addition, high-throughput screening and computational simulations are being utilized to accelerate the development and optimization of new materials. Given these advantages, the development of high-performance composite bifunctional electrocatalysts has become a central focus of current research. The following discussion will highlight transition metal compound composites and transition metal/compound/carbon composites, with particular emphasis on the selection, integration, and synergistic effects of active sites.5.1. Transition metal compound composites
Transition metal compound composites primarily consist of metal compounds, and these composites often exhibit distinct active sites for catalyzing both OER and ORR. By rationally designing composite structures, catalytic performance can be optimized. Transition metal compounds have commonalities in terms of crystal structure and possible synthesis techniques, facilitating their easy combination to prepare composites. These composite materials display a range of micro and nanostructures, including nanoparticles, nanowires, nanosheets, core–shell structures, and layered structures. Such diverse structures can effectively utilize their inherent reactivity, and increase the number and accessibility of active sites, thereby achieving superior bifunctional catalytic performance.Research on transition metal compound composites frequently focuses on combining various transition metal oxides and hydroxides. Yu et al. developed a CuCo2O4/NiFe LDH catalyst by adjusting the metal ion ratios and valence states within Ni–Fe–Co oxides/hydroxides.117 This approach facilitated the reconstruction of the catalyst surface via charge transfer between Ni, Fe, and Co, which optimized the catalytic performance. Experimental results indicated that ZABs prepared using CuCo2O4/NiFe LDH electrodes demonstrated higher efficiency compared to those prepared using a single CuCo2O4 electrode. Specifically, the charge/discharge voltage gap decreased from 1.02 V to 0.78 V, while the charge/discharge efficiency increased from 54% to 60%. Amorphous materials, which are characterized by a higher number of non-coordinated atoms and defect structures, provide more active sites for electrochemical processes compared to crystalline materials. Moreover, combining amorphous and crystalline phases in heterostructures can leverage the advantages of both states, leading to superior electrocatalytic performance. Zhang's research group drew inspiration from the atomic diffusion and reordering processes that occur when Co(OH)2 is heat-treated to form CoO.118 As shown in Fig. 7(a), a volcano plot was employed to illustrate the thermodynamic limitation potentials (UL) related to ΔGOOH* (for ORR) and ΔGOH* (for OER). At these UL potentials, the free energy diagrams for all fundamental reactions indicated that they were thermodynamically favorable. Ideally, ORR and OER should occur close to these thermodynamic potentials. The observed catalytic activity trend for ORR and OER was as follows: O@sur-1Ov > vac@sur-1Ov > CoO (111) > sub-1Ov. This trend demonstrates that the existence of surface vacancies significantly enhanced the intrinsic ORR/OER activity of CoO. Internal oxygen vacancies improved the performance by changing the electronic structure to facilitate electron transfer. Consequently, the precise control of oxygen defect concentrations and the formation of amorphous/crystalline heterostructures through vacuum calcination (Fig. 7(b) and (c)) effectively enhanced the electrocatalytic performance of CoO. The OER overpotential and ORR half-wave potential difference (ΔE) for this crystalline/amorphous heterostructure were 0.745 V. Additionally, during the OER process, the catalyst surface often reconstructed to form a hydroxide layer that acted as an active site. The synergistic combination of this restructured surface with the original structure provided enhanced catalytic performance.
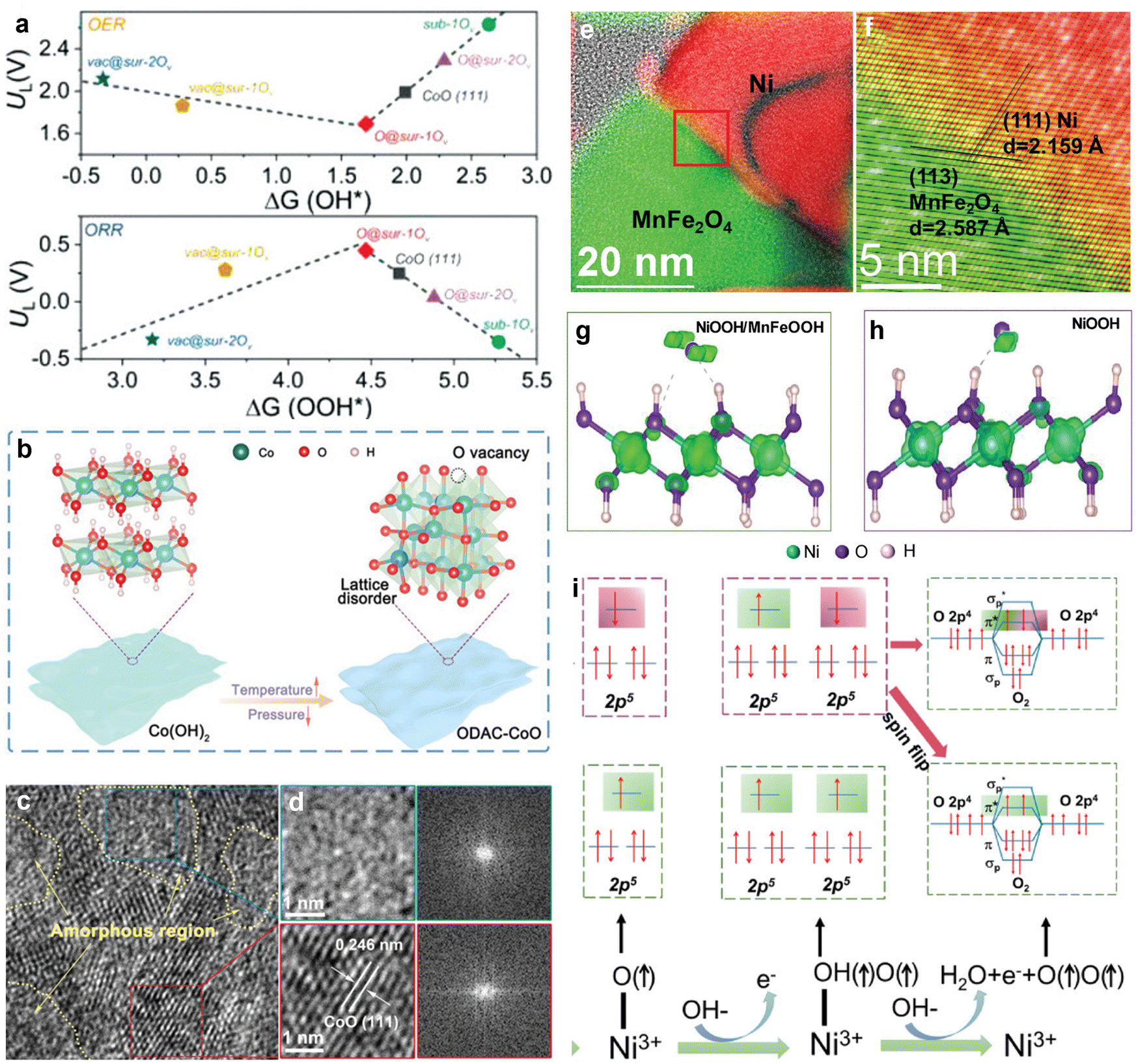 | ||
| Fig. 7 (a) Calculated UL ORR (vs. ΔGOOH*) and UL OER (vs. ΔGOH*) of different catalyst models. (b) Schematic illustration for the preparation of ODAC-CoO nanosheets. (c) Enlarged images of the selected regions in (d) and the corresponding FFT patterns in oxygen-defective amorphous-crystalline (ODAC-CoO-30). Reprinted with permission.118 Copyright 2021, Wiley-VCH. (e) and (f) Bragg-filtered image of the (111) planes of Ni and (113) planes of MnFe2O4 at the Ni35%/MnFe2O4 interface. The yellow color arising at the interface and the black curved line in the Ni particle in panel e is an artifact generated by the delocalized signal of the Bragg-filtered (111) planes. Spin polarization difference diagram of NiOOH/MnFeOOH (g) and NiOOH (h). The green contour around the atoms represents spin-up electrons. (i) OER process with and without the spin-aligned process. Reprinted with permission.119 Copyright 2024, Wiley-VCH. | ||
Yang's team constructed a Ni/MnFe2O4 heterojunction for use in rechargeable ZABs. HAADF-STEM images and EELS elemental distribution maps (Fig. 7(e)) revealed a uniform distribution of both phases, which resulted in the presence of numerous Ni/MnFe2O4 heterojunctions.119 The Bragg-filtered image, shown in the magnified red box region in Fig. 7(f), displays the relationship between the Ni (red) (111) plane and the MnFe2O4 (green) (113) plane at an angle of 66.18°. It was observed that these two crystal domains frequently grew in this relative orientation, reducing the geometric strain to 3%. During OER, the surface of this electrocatalyst reconstructed to NiOOH/MnFeOOH, and the high-spin Ni effectively tuned the spin of oxygen intermediates (Fig. 7(g) and (h)). The NiOOH/MnFeOOH electrocatalyst exhibited an overpotential of 261 mV and showed good stability for over 1000 cycles. Additionally, rechargeable ZABs prepared using this catalyst demonstrated an exceptional stability. The optimal spin state of Ni active sites with oxygen intermediates facilitated spin-selective charge transfer (Fig. 7(i)), optimizing reaction kinetics and reducing the energy barrier for oxygen evolution.
In addition to transition metal oxide and hydroxide composites, the combination of transition metal oxides with other transition metal compounds can also integrate different types of ORR and OER active sites. Xi's team reported NiFe2O4/FeNi2S4 heterostructure nanosheets (HNSs), which were used as efficient catalysts to significantly enhance the recharging performance of neutral aqueous zinc–air batteries.120 The NiFe2O4/FeNi2S4 HNSs featured a surface rich in oxide/sulfide interfaces, which effectively modulated the oxygen binding ability and activity of catalyst. In a 0.2 M PBS, NiFe2O4/FeNi2S4 HNSs exhibited excellent activity and stability. However, most transition metal sulfides, phosphides, and nitrides are typically used in combination with carbon-based materials, which will be discussed in Section 5.4.
In summary, transition metal compound composites offer notable advantages due to their tunable structures. These composites have the potential to achieve synergistic effects, show exceptional stability. However, compared to state-of-the-art bifunctional electrocatalysts, the performance remains limited. This limitation is primarily due to factors such as inadequate conductivity, and excessively high intermediate adsorption free energies during ORR. Consequently, the development of transition metal compound composites is still in its early stages, and further research and optimization are required.
5.2. Transition metal/alloy clusters and carbon
Small clusters of transition metals or their alloys (typically consisting of a few to several dozen atoms) can be combined with carbon-based materials to prepare composite materials that offer large surface areas, excellent electrical conductivity, tunable electronic structures, and multifunctionality. The metal clusters provide abundant active sites, while the carbon materials ensure rapid electron transport and offer mechanical support. Thus, higher catalytic activity is obtained. In the synthesis of metal–nitrogen–carbon catalysts, residual metal clusters work synergistically with single-atom catalytic sites to further optimize performance by modulating the electronic structure. Additionally, compared to precious metal catalysts, these composite materials offer advantages such as lower cost, abundant resources, and suitability for large-scale applications. Therefore, transition metal/alloy cluster-carbon composites hold significant potential for bifunctional electrocatalysis, offering an efficient and cost-effective alternative to precious metal.Optimizing the morphology can enhance the accessibility of active sites, while doping heteroatoms into carbon substrates can further optimize conductivity and electronic structure. Ren et al. reported a pyrolysis method utilizing dual MOFs. Specifically, FeNiM@ZnCoZ was utilized as a precursor to synthesize FeNiCo@NC-P anchored onto carbon nanotubes (CNTs).121 Compared to carbon materials derived from single MOFs, this synthesized catalyst incorporated N dopants as well as three metals (Fe, Co, and Ni). Additionally, the two MOF precursors successfully yielded carbon materials with microporous/mesoporous structures while preserving a one-dimensional carbon nanorod morphology during high-temperature pyrolysis. These features provided FeNiCo@NC-P with abundant active sites. Consequently, exceptional bifunctional electrocatalytic activity and outstanding ZAB performance were achieved. The Fe–N sites on the carbon framework exhibited excellent ORR activity, while cobalt nanoparticles significantly enhanced the OER process. The synergistic effect of these features further improved the ORR and OER performance of this catalyst. Xie et al. employed MOFs to synthesize a series of carbon-based catalysts featuring cobalt nanoparticles and highly dispersed iron species with a controlled doping strategy.122 Using ZIF-8 as the precursor, iron and cobalt sources were introduced through in situ doping and a dual-solvent method, which led to the successful anchoring of highly dispersed iron species and the confined growth of cobalt nanoparticles. The optimized catalyst (CoNP@FeNC-0.05) demonstrated a high E1/2 and a low η10 in alkaline electrolytes.
In conventional synthesis of M–N–C structures, the presence of metal particles is unavoidable. These metal particles have incomplete d-orbitals and can readily accept electrons from oxygen reaction intermediates. Thus, activation energy barriers are lowered. However, a significant issue with Fe–N–C materials is their highly symmetrical electronic structure, resulting in sluggish reaction kinetics. Therefore, electronic modulation and orbital engineering are essential for the precise design of advanced Fe–N–C. Zhang et al. prepared a composite (FeSA/AC@HNC) by utilizing a dual-ligand zinc-based zeolite framework to anchor monodisperse iron atoms and adjacent iron clusters within nitrogen-doped layered porous hollow carbon.123 The introduction of iron clusters lowered the electron density of Fe–N4 and increased the effective magnetic moment of single-atom active sites. The FeSA/AC@HNC composite demonstrated excellent ORR activity. The interaction between iron clusters and single atoms also enhanced the stability and performance of this composite in rechargeable zinc–air batteries. A persistent challenge in metal–nitrogen–carbon systems is increasing the metal loading of SACs (>5 wt%). Xu's team employed a dual-melamine pyrolysis method and a layered g-C3N4 sacrificial template to co-anchor Co single atoms and Co clusters on nitrogen-rich porous graphene, achieving a Co single-atom loading of up to 14.0 wt%.124 Due to the modulated electronic structures and optimized chemical bonding of this composite material, catalytic activity was significantly improved. Peng et al. first synthesized iron-based MOFs with a nanotube structure via co-precipitation.125 On this basis, Fe-ACSA@NC was obtained by polymerization and pyrolysis (Fig. 8(a)). The optimized electron redistribution of these materials facilitated the easier desorption of intermediates, resulting in superior ORR activity compared to nanoparticle-modified materials. A metal–air battery was assembled with Fe-ACSA@NC, and this battery demonstrated exceptional power density and capacity.
 | ||
| Fig. 8 (a) Synthesis procedure and morphology characterizations of Fe-ACSA@NC. Reprinted with permission.125 Copyright 2021, Wiley-VCH. (b) Charge density difference between the metal cluster and metal-NC of Fe4@Fe–N4, N4@Ni–N4 and Fe4@Ni/Fe–N4 (Fe4 on the Ni–N4 site). X-ray absorption to determine the chemical state and atomic local structure. DSAs: dual single atoms; DSAs/NCs: dual single atoms and nanoclusters. Reprinted with permission.126 Copyright 2024, Royal Society of Chemistry. (c) Experimental and fitted FT-EXAFS curves at the Ni Kedge. (d) Experimental and fitted FT-EXAFS curves at the Fe K-edge. Insets are the corresponding atom configurations. Reprinted with permission. (e) Cycling performance of the ZABs made from three types of composite catalysts (5 mA cm−2; 4 h duration per cycle). Reprinted with permission.127 Copyright 2023, American Chemical Society. | ||
Dual-atom catalysts (DACs), which contain multiple atomic combinations arranged at local sites, address some of the limitations of SACs. As an extension of SACs, DACs retain the advantages of SACs while also exhibiting the geometric ensemble effects and electronic ligand effects typically observed in nanoparticle catalysts. DACs offer diverse and unique local electronic structures at surface sites. This can enhance the adsorption and activation of various intermediates, enabling multifunctional and synergistic catalysis while maximizing atomic utilization. Consequently, DACs represent a more promising strategy for the preparation of bifunctional oxygen electrode catalysts.
Xu's group employed a ZIF phase strategy transformation combined with thermal fixation to prepare nitrogen-doped carbon nanorods containing embedded bimetallic CoFe single atoms and clusters (CoFe–N–C).128 The unique one-dimensional structure of these nanorods and the synergistic of the FeCo system led to enhanced performance. Meng et al. developed a bimetallic NiFe–N–C containing Fe nanoclusters via the simple pyrolysis of metal phthalocyanine and a nitrogen-doped carbon precursor.126 As shown in Fig. 8(b), the charge transfer between the Fe nanoclusters in Fe4@Ni/Fe–N4 and the active sites was significantly stronger than that in Fe4@Fe–N4 and Ni4@Ni–N4. The synergistic between Fe and Ni–N4 and Fe–N4 sites optimized the electronic structure, leading to excellent catalytic activity. Wang et al. proposed a one-step, solvent-free annealing method to construct active centers consisting of dual atoms and clusters in scalable activated carbon by leveraging the strong anchoring effect of phenanthroline on metal ions.129
Li et al. developed an efficient isolation-confinement strategy to synthesize porous carbon nanofibers (β-FeCo-PCNF) loaded with Fe–Co atomic pairs and FeCo alloy clusters.130 Fan's team also achieved the synthesis of bimetallic single atoms and atomic clusters.127 They prepared Ni, Fe-DSAs/NCs by coordinating Ni atoms are coordinated with four N atoms to form a Ni–N4 structure while neighboring Ni atoms were coordinated with three additional Ni atoms to form Ni4 clusters (Fig. 8(c)). A similar configuration was observed for Fe (Fig. 8(d)), which formed both Fe–N4 and Fe4 clusters. In contrast, other samples exhibited only single Ni–N4 or Fe–N4 structures. EXAFS and FT-EXAFS analyses ruled out the possibility of NiFe alloy clusters, indicating that the Fe and Ni in Ni, Fe-DSAs/NCs were able to coexist as stable M–N4 and M4 nanoclusters. In long-term operation tests, a composite catalyst was used to fabricate ZABs. The performance of these ZABs remained stable, with minimal degradation observed after 500 h of cycling (Fig. 8(e)).
5.3. Transition metal oxides/hydroxides and carbon
Transition metal oxides/hydroxides and carbon-based materials represent two prominent families of electrocatalysts that are valued due to their high OER activity, ease of synthesis, low cost, and excellent stability under electrocatalytic conditions. Combining these materials can produce highly effective bifunctional oxygen electrode catalysts. Typically, carbon-based materials show good ORR activity, while oxides and hydroxides contribute to OER activity. This implies that ORR and OER active sites can be selectively designed and integrated into various carbon and transition metal oxides/hydroxides, which can then be assembled into composite electrocatalysts. However, carbon and transition metal oxides/hydroxides often exhibit suboptimal interfacial compatibility and bonding strength, which can potentially affect overall electrode performance. Additionally, there are inherent conflicts between these materials, such as conductivity versus insulation and hydrophilicity versus hydrophobicity. Therefore, the rational selection and efficient integration of active sites are crucial for optimizing catalytic performance.Heteroatom-doped and defect-rich carbon-based catalysts exhibit high ORR activity, particularly single- or dual-atom catalysts. However, these materials generally show lower OER activity. Consequently, combining these carbon materials with transition metal oxides can effectively achieve bifunctional catalysis. Zhao et al. reported a nitrogen-doped bimetallic oxide-coated carbon nanotube material (FeNiO@NCNT) derived from MOFs using a straightforward pyrolysis strategy.131 FeNiO@NCNT demonstrated excellent activity. Notably, the lattice oxygen and OH− in FeNiO provide additional OER active sites beyond the metal centers, resulting in outstanding OER performance (Ej10 = 205 mV). The ultimate goal is to be able to combine transition metal oxides with carbon materials by selecting appropriate elements and employing simple preparation methods. Wang et al. developed a nickel–iron oxide material supported on foam carbon with bamboo-like carbon nanotubes.132 During high-temperature synthesis, the edges of the carbon sheets in the foam carbon structure folded inward, forming short carbon nanotubes. Raman spectroscopy revealed that the addition of bimetallic elements enhanced the graphitization (Fig. 9(a)), providing a good conductivity. The supported NiFe2O4 phase improved the ORR activity of this electrocatalyst by anchoring additional N atoms, while the high-valent nickel oxide facilitated O–O bond formation, enhancing OER performance. Shao's team employed an in situ growth strategy to prepare a nitrogen-doped graphene (NG)-supported Fe–Co bimetallic oxide (FeCoOx) with a 3D-on-2D stacked structure (Fig. 9(b) and (c)) and investigated the effect of catalytic performance.133 The optimal FeCoOx@NG composite prepared with a 20% FeCoOx loading demonstrated exceptional ORR and OER electrocatalytic performance. Yoo's group successfully synthesized a NiCoMoO4 (NCMO) catalyst with a hierarchical nanosheet structure on a rGO substrate.134 The selective substitution of Mo at specific sites was used to modulate the electrochemical environment of the active centers (Fig. 9(d)). The introduction of Mo significantly enhanced the redox activity of NiCoMoO4 (Fig. 9(e)). Further analysis revealed that the hierarchical nanostructured NiCoMoO4 supported on reduced graphene oxide demonstrated excellent bifunctional performance in alkaline solutions, with a low overpotential gap of only 0.75 V for ORR and OER. A ZABs fabricated with this catalyst exhibited good cycling stability and a higher power density of up to 125.1 mW cm−2. However, despite the inherent advantages of oxide/hydroxide and carbon-based materials, their bifunctional performance and active site utilization are limited. Rational structural design to effectively integrate oxides/hydroxides with carbon materials is crucial for maximizing their complementary properties and achieving synergistic effects.
 | ||
| Fig. 9 (a) Raman spectroscopy of Fe/NCF, Ni/NCF and FeNi/TNCF-2 samples. Reprinted with permission.132 Copyright 2022, Wiley-VCH. (b) SEM images of FeCoOx@NG. (c) Colorized TEM image of FeCoOx@NG. Reprinted with permission.133 Copyright 2022, The Author(s). (d) Typical experimental setup for an in situ XAS analysis (NiCoMoO4). Reprinted with permission. (e) The setup for in situ XRD analysis and the occurrence of the X-ray diffraction peak took place in the Zn–air battery. Reprinted with permission.134 Copyright 2023, Wiley-VCH. Reprinted with permission. Data-driven analysis of bifunctional oxygen electrocatalysts (f) performance distribution of the reported bifunctional electrocatalysts within the category of M–N–C electrocatalyst (red label), transition metal hydroxides (blue label), and other types of electrocatalysts (gray label). (g) Performance distribution of the specific active sites in “M–N–C” and “Hydroxides” subcategory. The dash lines refer to the requested overpotential to achieve DE < 0.63 V for OER and ORR, respectively. (h) The diagram for bifunctional electrocatalytic activity comparison among FeNC@LDH, Pt/C + Ir/C, and other reported bifunctional electrocatalysts tested in 0.10 M KOH (marked as the green points). Reprinted with permission.135 Copyright 2024, Elsevier. | ||
Under hydrothermal conditions, a porous, ultrathin nitrogen-doped carbon material (NCM) with enlarged interlayer spacing was prepared via self-assembly. Although this material exhibited excellent activity for ORR, its OER catalytic performance remained limited. To design a bifunctional catalyst, Wang et al. proposed embedding nitrogen-doped nanocarbon materials into Ni–Co layered double hydroxide (NiCo-LDH) nanosheets.136 The integration of these two materials provided NiCo-LDH with enhanced electrical conductivity a larger electroactive surface area, and significantly improved oxygen vacancy formation, which greatly boosted its bifunctional catalytic activity. At 50 mA cm−2, OER overpotential of NiCo-LDH was as low as 0.352 V, and ORR performance was also improved. Therefore, the combination of layered double hydroxides (LDH) with heteroatom-doped carbon materials offers unique advantages for improving both OER and ORR performance.
Achieving record-breaking bifunctional oxygen electrocatalytic activity is a key goal in the development of next-generation ZABs. Historically, the ΔE metric for bifunctional electrocatalytic activity has stagnated at ΔE > 0.60 V. Zhang's team designed and synthesized a composite catalyst with record-breaking bifunctional activity (ΔE = 0.57 V).135 Based on data-driven analysis (Fig. 9(f) and (g)), Zhang's team identified a rational pathway for integrating ORR and OER active sites. They selected Fe–N–C and NiFeCe-LDH as the ORR and OER active centers, respectively, and a composite catalyst (Fe–N–C@LDH) containing both of these active sites was prepared. This catalyst exhibited a groundbreaking ΔE of 0.57 V (Fig. 9(h)). A peak power density of 176 mW cm−2 and a discharge rate of 50 mA cm−2 were achieved in rechargeable ZABs, confirming the exceptional electrocatalytic performance of Fe–N–C@LDH. This catalyst also showed stable operation under harsher conditions. Furthermore, a practical ampere-hour-level ZAB with a unit capacity of 6.4 A h was constructed, demonstrating 20 stable cycles at 1.0 A and 1.0 A h. This work set a new benchmark for electrocatalytic activity and advanced the development of ampere-hour-level ZAB devices for practical applications. Additionally, the study introduced a new material design methodology through data-driven analysis, offering a broadly applicable research paradigm.
In summary, composite bifunctional electrocatalysts combining oxides/hydroxides with carbon materials are extensively studied catalyst materials for OER and ORR. These composites typically demonstrate excellent catalytic performance. Oxides/hydroxides and carbon materials possess high intrinsic activity, providing a natural advantage for bifunctional catalysis. Moreover, the distinct properties of these materials offer diverse opportunities for structural design, enabling the optimization of their interactions and desirable synergistic effects. As a result, composite materials based on oxides/hydroxides and carbon are widely considered as promising alternatives.
5.4. Transition metal sulfides/nitrides/phosphides and carbon
In addition to carbon-oxide/hydroxide composites, transition metal sulfide-based bifunctional electrocatalysts have also been extensively studied. In these composites, the carbon and sulfide components typically provide separate contributions toward ORR and OER electrocatalytic activity. Krishna et al. developed a carbon-coated cobalt tin sulfide nanopore material (CoSnOH/S@C NPs) through a rational design process (Fig. 10(a)).137 They investigated the influence of metal elements on the morphology of hydrothermally prepared CoSnOH nanosheet materials. The optimized CoSnOH/S@C NPs exhibited excellent catalytic performance due to their superior nanostructure, unique porous network, and high surface area. Yu's group synthesized a sulfur-doped graphene nanosheet-wrapped urchin-like NiCo2S4 microsphere composite (S-GNS/NiCo2S4).138 This composite demonstrated improved conductivity, mechanical stability, and thermal stability compared to bimetallic oxide precursors. Due to their unique layered structure, the urchin-like NiCo2S4 microspheres provided effective electron/ion transport pathways and reduced the diffusion distance of electrolyte ions. Additionally, sulfur doping enhanced the catalytic activity and significantly improved the cycling stability. When used as a catalyst in ZABs, this material achieved a higher power density (216.3 mW cm−2), a low overpotential of 1.09 V, and stable performance with no significant degradation over 100 h. Sun et al. employed Co9S8/Co1−xS heterostructures modified with carbon nanosheets (Co9S8/Co1−xS@NSC).139 By adjusting the precursor components, the formation of heterostructures was effectively controlled. A KCl salt template was used to introduce surface wrinkles into the nanosheet morphology, which maximized the exposure of nitrogen-containing functional groups and helped anchor the highly dispersed Co9S8/Co1−xS structures. The presence of the Co9S8/Co1−xS heterojunction efficiently optimized the behavior of intermediates, leading to enhanced bifunctional activity. Among the cobalt sulfide-based electrocatalysts, Co9S8/Co1−xS @NSC achieved one of the lowest Ej=10 values (1.52 V) and the highest E1/2 values (0.86 V), resulting in the smallest ΔE value (0.66 V) (Fig. 10(b)).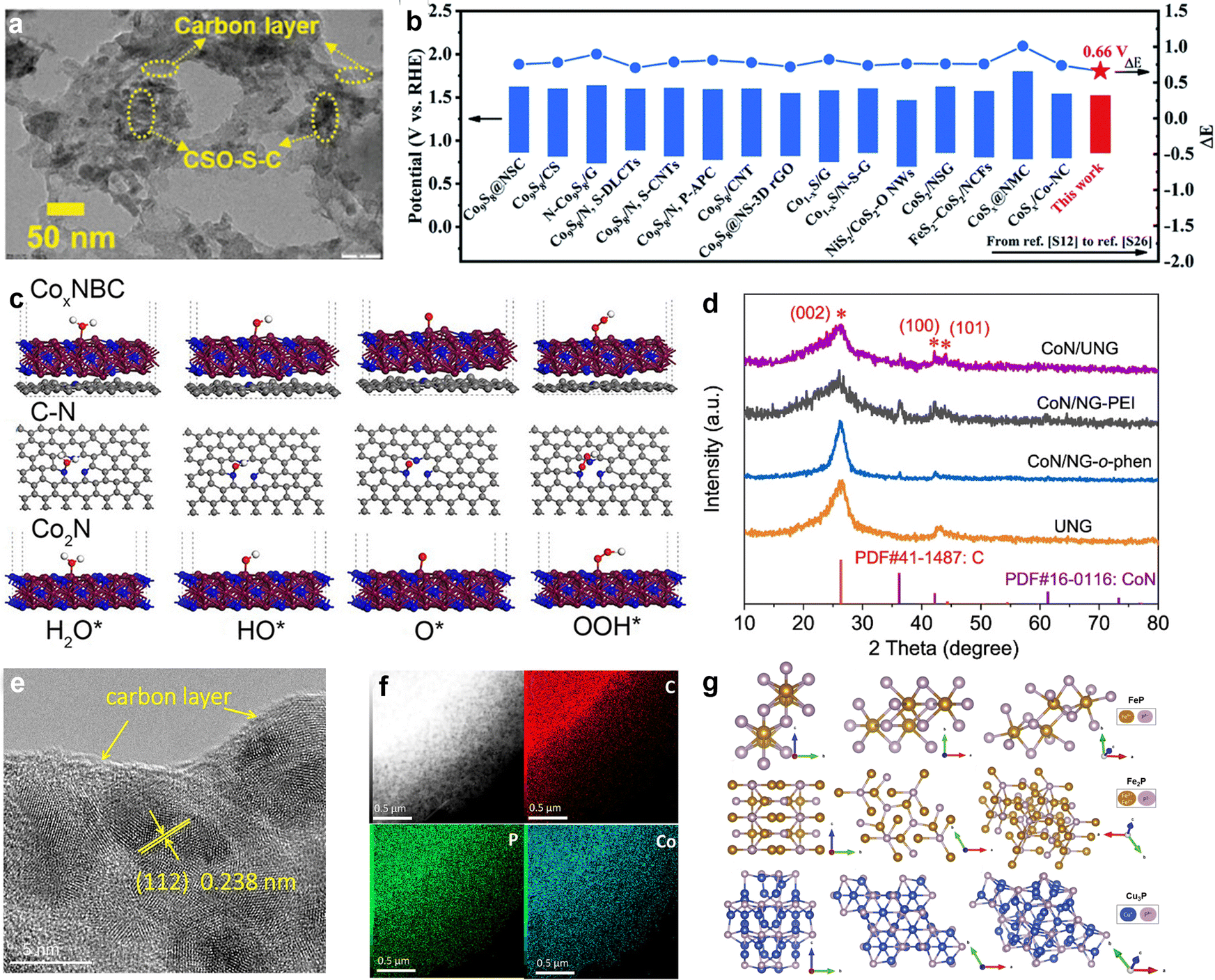 | ||
| Fig. 10 (a) TEM images of the CoSnOH/S NPs. Reprinted with permission.137 Copyright 2024, Wiley-VCH. (b) Bifunctional oxygen electrocatalytic performance comparison of some reported cobalt sulfide-based electrocatalysts. Reprinted with permission.139 Copyright 2021, Royal Society of Chemistry. (c) The process structures of ORR on C–N, Co2N, and Co2N/C–N. Reprinted with permission. Reprinted with permission.140 Copyright 2021, Zhengzhou University. (d) XRD patterns of CoN/UNG, CoN/NG-PEI and CoN/NG-o-phen and UNG. Reprinted with permission. Reprinted with permission.141 Copyright 2022, Elsevier. (e) HRTEM and (f) STEM images of the selected area for EDS elemental mapping images of C, P, and Co in the CoP@C/CNSs. Reprinted with permission.142 Copyright 2023, American Chemical Society. (g) The crystal structures of FeP, Fe2P, Cu3P in Fe–P/Cu3P-NPC. Reprinted with permission.143 Copyright 2023, Wiley-VCH. | ||
Transition metal nitrides and phosphides often exhibit excellent OER activity and possess metallic properties that provide superior electronic conductivity. By leveraging these characteristics, high-performance bifunctional electrocatalysts can be developed by combining OER-active transition metal nitrides and phosphides with carbon materials that demonstrate ORR activity. Research on nitride–carbon composite catalysts highlights the significant advantages of cobalt-based nitrides. Liu et al. proposed a strategy that utilized the electronic interactions between transition metal nitrides and biomass-derived carbon to design high-performance catalysts.140 They prepared composites consisting of Co2N nanoparticles (NPs) anchored to the ends of nitrogen-doped carbon chains. The ORR reaction pathways for C–N, Co2N, and CoxNBC were structurally similar, with variations in CoxNBC activity attributed to differences in the size and dispersion of Co2N. The electronic states of Co2N were notably enhanced in the nitrogen-doped carbon layers near the Fermi level. The synergistic effect between the Co2N NPs and nitrogen atoms increased the charge density of intermediates. This improved adsorption and charge transfer, leading to significantly enhanced ORR activity. Combined with the controllable preparation method used to prepare these Co2N composites, this innovative catalytic performance offers a promising approach for developing metal-containing catalytic carbon from biomass resources.
Wang's team reported a novel dual construction method that utilized cobalt chloride as a template to prepare a two-dimensional structure containing cobalt single-atom active sites.144 This method enabled the scalable production of CoN3 single atoms dispersed on nitrogen-doped graphene nanosheets, resulting in a bifunctional catalyst (CoSA/NCs). During synthesis, cobalt chloride served as the cobalt source, template, porogen, and graphite catalyst, and this cobalt chloride was able to be recycled. The presence of cobalt chloride endowed the CoSA/NCs with multiple active sites (CoN3 and abundant nitrogen-doped species), large graphite interlayer spacing, a high specific surface area, nanoporosity, and high graphite crystallinity. Controlling the size and the thickness during catalyst preparation is often challenging. Xu et al. synthesized CoN anchored on ultrathin nitrogen-doped graphene using a strategy that combined 1,10-phenanthroline coordination with polyethyleneimine intercalation effects.141 Polyethyleneimine was intercalated between graphene layers, which prevented graphene stacking and resulted in an ultrathin nanosheet structure. The nitrogen lone pairs in 1,10-phenanthroline coordinated with Co2+ to form Co–N bonds, which inhibited Co aggregation. XRD and SEM analyses confirmed the successful anchoring of smaller CoN nanoparticles on the ultrathin nitrogen-doped graphene. CoN/UNG exhibited excellent ORR electrochemical performance, with a E1/2 of 0.87 V and high stability. A zinc–air battery prepared using CoN/UNG demonstrated a power density of 149.3 mW cm−2.
In addition to transition metal nitrides, transition metal phosphides are commonly employed in bifunctional electrocatalysts due to their excellent electronic conductivity and inherent OER activity. Sun et al. developed a carbon dot (CD)-assisted method to fabricate CoP/C nanocomposites with a refined microstructure (Fig. 10(e) and (f)).142 In this process, the CDs acted as a stabilizing platform, adsorbing and immobilizing Co precursors while preventing the agglomeration of CoP nanoparticles. This resulted in a well-structured distribution of carbon-coated CoP particles on carbon nanosheets (CoP@C/CNSs). ZABs prepared using CoP@C/CNSs as the electrocatalyst exhibited excellent performance. Yang et al. utilized ZIF-8 as a sacrificial template and employed a nontoxic phytic acid “in situ etching-adsorption-phosphidation” process to construct a series of carbon-supported metal phosphides with hollow structures and “wall-sharing” features (e.g., Mn2P-NPC, FeP/Fe2P-NPC, CoP-NPC, Ni2P-NPC, Cu3P-NPC, Fe–P/Cu3P-NPC).143 Among these metal phosphides, Fe–P/Cu3P-NPC exhibited the best bifunctional catalytic activity. The electronic interaction between Fe and Cu enhanced the intrinsic catalytic activity of Fe–P/Cu3P-NPC, while the heterogeneous interfaces of FeP, Fe2P, and Cu3P (Fig. 10(g)) increased the number of active sites. Moreover, the ultrathin NPC framework facilitated mass transport. Liquid ZABs prepared using Fe–P/Cu3P-NPC achieved a peak power density of 158.5 mW cm−2. Additionally, excellent cycling stability was achieved in flexible ZABs, which showed stable operation for 81 h at 2 mA cm−2 without bending and for 26 h at various bending angles.
In summary, carbon–sulfide composite bifunctional electrocatalysts can be prepared using a wide variety of material combinations, and these materials demonstrate exceptional electrocatalytic performance. Enhancing the overall bifunctional activity of these electrocatalysts can be achieved by optimizing their intrinsic properties or structure. However, sulfides generally exhibit poor stability under operational conditions, leading to their conversion into oxides, hydroxides, or hydrosulfides. Currently, composite materials that combine transition metal nitrides or phosphides with carbon are receiving increasing attention. Innovations in modulating the activity of these components are emerging as key advancements. Research is evolving from a sole focus on nitrides and phosphides to exploring multi-cationic and multi-anionic compounds, as well as heterogeneous interfaces. Similar to sulfides, nitrides and phosphides also exhibit poor stability in oxidative environments. Understanding the chemical structure evolution of these materials will be crucial.
6. Summary and outlook
Highly efficient bifunctional oxygen electrocatalysts will be crucial for advancing the next generation of rechargeable aqueous metal–air batteries and other key energy technologies. The application as bifunctional ORR/OER electrocatalysts in the ZAB are summarized in Table 1. These catalysts must demonstrate high catalytic efficiency for both ORR and OER while maintaining balanced performance in both reactions. This requirement places stringent demands on the design, structural control, and synthesis of these materials. Consequently, a major focus of current research is precisely tuning the chemical structure and electronic properties of active sites at the molecular level. Various single-component and composite oxygen electrocatalysts have been developed. Single-component catalysts can achieve effective ORR and OER performance within a single material by modulating their intrinsic activity. Due to their well-defined active sites, these catalysts are often considered ideal models for studying reaction mechanisms and fundamental catalytic principles, offering theoretical insights for the development of new catalysts. In contrast, research on composite catalysts has focused on structural design and facilitate efficient conduction within constrained electron and ion transport pathways. This approach emphasizes overall performance optimization and is better suited for practical applications. Despite significant advancements, key issues such as identifying the operational mechanisms and improving the stability of bifunctional oxygen electrocatalysts remain under investigation. Given these opportunities and challenges, this paper forecasts the development prospects of advanced bifunctional oxygen electrocatalysts and outlines key directions for future research.| Catalysts | E1/2 (V vs. RHE) | η@10 mA cm−2 (V) | ΔE (V) | Power density (mW cm−2) | Cycling stability (h) | Ref. |
|---|---|---|---|---|---|---|
| N-HC@G-900 | 0.85 | 1.58 | 0.73 | — | — | 56 |
| mf-pClNC | 0.91 | — | — | 277 | 200 | 58 |
| VP/CNs | 0.86 | — | — | 155 | 250 | 65 |
| NCN-1000-5 | 0.82 | 1.55 | 0.73 | 207 | 330 | 69 |
| Co–N–C/AC | 0.87 | 1.59 | 0.72 | 203 | 72 | 75 |
| OLC/Co–N–C | 0.85 | 1.58 | 0.73 | 238 | 100 | 77 |
| Ni7Fe3-CNG | 0.89 | 1.50 | 0.61 | — | — | 79 |
| FeCo–N3O3@C | 0.93 | 1.52 | 0.59 | 143 | 200 | 86 |
| Co3O4-CP-3 | 0.69 | 1.57 | 0.88 | — | 600 | 91 |
| V-CMO/rGO | 0.86 | — | — | 145 | 40 | 98 |
| Porous NiCo2O4 | 0.79+ | 1.61 | 0.82 | 124 | 160 | 102 |
| Co3Mo3N | 0.75 | 1.52 | 0.77 | — | 240 | 104 |
| (Co0.17Fe0.83)3N@NPC | 0.88 | 1.56 | 0.68 | 126 | 160 | 106 |
| NiCoMnP | 0.72 | 1.53 | 0.81 | 148 | 1000 | 107 |
| FeCoNiSx | — | 1.43 | — | 257 | 200 | 112 |
| FeNiCo@NC-P | 0.84 | 1.54 | 0.70 | 112 | 120 | 121 |
| Fe-ACSA@NC | 0.90 | — | — | 140 | 35 | 125 |
| Fe4@Fe–N4 | 0.89 | 1.68 | 0.78 | 217 | 60 | 127 |
| FeNC@LDH | 0.88 | 1.45 | 0.57 | 176 | — | 135 |
6.1. Theoretical predictions
Designing bifunctional oxygen electrocatalysts requires strict criteria for element selection and combination. Among the many candidate elements, machine learning (Fig. 11(a) and (b)) has proven to be highly advantageous, particularly for accelerating material screening, managing complex multidimensional data, reducing experimental costs, and identifying potential material combinations.145,146 As a powerful data processing tool, machine learning provides new avenues for overcoming research bottlenecks. However, several optimization issues still need to be addressed. First, the performance of current machine learning models is limited by the quantity and quality of data, and discrepancies between theoretical simulations and experimental results often arise due to constraints in the exploration space and methods. Therefore, developing methods for data quality assessment and establishing high-quality multiscale databases will be essential. Future research should focus on leveraging data to discover new materials or reaction patterns. Techniques such as transfer learning and generative adversarial networks can integrate existing knowledge and data to generate new data or use unsupervised learning to identify patterns, thereby facilitating machine-driven decision-making and offering insights beyond traditional analytical methods. To ensure the reliability of these insights, integrating domain knowledge into descriptor definitions and model construction will be crucial. In addition to machine learning applications in catalyst design, the theoretical model for oxygen electrocatalysis developed by the Nørskov team offers new opportunities for predicting catalytic activity and guiding catalyst design. While significant progress has been made, traditional theoretical simulations still face limitations. Most theoretical calculations are conducted under vacuum and at absolute zero, conditions that markedly differ from real operating environments (e.g., 298 K and in the presence of a solvent). Entropy corrections, solvent effect adjustments, and interface adsorption corrections are promising methods for better aligning simulations with real-world scenarios. Advances in continuous solvent models, large-scale descriptions, and kinetic methods are also noteworthy. Furthermore, electrocatalysis simulations typically consider only one or a few active sites, which overlooks the complexity of actual structures. Finally, current theoretical calculations mainly rely on the thermodynamic analysis of intermediates rather than kinetic assessments, and purely thermodynamic considerations may not always be sufficient or reliable. Therefore, the precise prediction of catalyst performance remains a significant area for exploration and improvement. | ||
Fig. 11 (a) Diffusivities simulated by molecular dynamics (MD) at high temperatures and by learning-on-the-fly (LOTF-MD) at intermediate temperatures and corresponding LOTF-MD snapshots. Reprinted with permission.145 Copyright 2020, American Chemical Society. (b) Schematic diagram of geometric health indicators (solid lines denote the selected data; dash lines denote the fitted circle). Reprinted with permission.146 Copyright 2022, Elsevier. (c) Schematic diagram of phase transformation process during pyrolysis. Reprinted with permission.147 Copyright 2024, The Author(s). (d) Compared to furnace annealing, our method shows a time reduction by at least ∼5 orders of magnitude, a size reduction from 27 to 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm, and an increase in electrochemical surface area (ECSA) of 3.8 times. Reprinted with permission. (e) The general synthesis of ultrasmall nanoparticles by coordinated CTS beyond delicate metal–organic framework (MOF) chemistry. Reprinted with permission.148 Copyright 2023, The Author(s). (f) HAADFSTEM images of the Pt = N2 = Fe ABA catalyst. The structure evolution identified by in situ XAFS. (g) The Pt L3-edge and (h) XANES spectra are the corresponding local magnifications of the white-line peaks. (i) 800–1300 cm−1 range of in situ SR-FTIR characterizations for Pt = N2 = Fe ABA. Reprinted with permission.148 Copyright 2022, The Author(s). nm, and an increase in electrochemical surface area (ECSA) of 3.8 times. Reprinted with permission. (e) The general synthesis of ultrasmall nanoparticles by coordinated CTS beyond delicate metal–organic framework (MOF) chemistry. Reprinted with permission.148 Copyright 2023, The Author(s). (f) HAADFSTEM images of the Pt = N2 = Fe ABA catalyst. The structure evolution identified by in situ XAFS. (g) The Pt L3-edge and (h) XANES spectra are the corresponding local magnifications of the white-line peaks. (i) 800–1300 cm−1 range of in situ SR-FTIR characterizations for Pt = N2 = Fe ABA. Reprinted with permission.148 Copyright 2022, The Author(s). | ||
6.2. Synthesis methods
The primary synthesis methods for bifunctional oxygen electrocatalysts that exhibit both ORR and OER activities include pyrolysis, hydrothermal/solvothermal methods, precipitation, electrodeposition, and atomic layer deposition (ALD). Significant advancements have been made in the preparation of carbon materials and metal compounds using these techniques (Fig. 11(c)–(e)).147,148 For instance, pyrolysis can be used to effectively synthesize nitrogen-doped carbon-based materials, while hydrothermal methods are commonly used to produce metal oxides and metal–carbon composites. Precipitation and electrodeposition are useful for fine-tuning the structures of metal compounds, and ALD provides precise control over the distribution. However, despite the wide application of these synthesis methods, several limitations persist. Pyrolysis often requires high temperatures, which can lead to the collapse of the pore structure or the loss of active sites in carbon materials. While hydrothermal and solvothermal methods can be used to effectively synthesize nanostructures, these methods suffer from difficulties in precisely controlling the reaction conditions, leading to variability in product stability. Precipitation and electrodeposition face challenges in terms of uniformity and reproducibility, especially for the synthesis of multi-component catalysts where phase separation or compositional inhomogeneity can occur. Although ALD offers atomic-level control, high equipment costs and complex processes pose challenges for large-scale production. Future synthesis methods must address these challenges by advancing toward more precise structural control, lower costs, and higher scalability. Accurate synthesis techniques should manage the multi-level structures of catalyst materials, including their pore structures, active site distributions, and interfacial interactions. Consequently, overall catalyst performance can be optimized rather than merely enhancing individual active sites. The development of multifunctional synthesis platforms will facilitate the integration of different types of materials, such as composites that effectively couple carbon materials with metal compounds within the same reaction system to achieve synergistic effects. Additionally, leveraging machine learning and high-throughput screening technologies may enable the automated optimization of synthesis processes through intelligent design and autonomous experimental platforms, improving catalyst reproducibility and stability. These advancements will lay a solid foundation for developing the next generation of high-performance bifunctional oxygen electrocatalysts.6.3. Advanced characterization techniques
Characterization techniques are essential for revealing the active sites, coordination environments, and reaction mechanisms of bifunctional oxygen electrocatalysts. Despite the availability of various advanced characterization methods, challenges remain in precisely locating and dynamically observing active sites, probing complex coordination environments, and elucidating reaction mechanisms (Fig. 11(f)–(i)).149 First, identifying active sites and their coordination environments can be challenging. Traditional techniques such as X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), and Raman spectroscopy provide valuable elemental and structural information but have limitations in describing active site and coordination environment changes during reactions. This can hinder the accurate identification of the dominant active sites during ORR and OER, affecting catalyst design and optimization. For example, failure to differentiate between surface and bulk active sites can lead to performance prediction errors and missed opportunities for developing highly efficient catalysts. Second, traditional characterization methods often rely on static sampling and analysis. Thus, key information cannot be easily captured during dynamic processes. This limitation can result in an incomplete understanding of reaction pathways, interfacial phenomena, and the behavior of intermediates, impacting catalyst performance enhancement and durability understanding. Consequently, researchers may design catalysts based on insufficient data or incorrect assumptions. In situ and online characterization techniques are crucial for overcoming these challenges. Techniques such as in situ XAS, in situ TEM, and in situ Raman spectroscopy enable the real-time monitoring of catalyst structural evolution and surface reaction processes under realistic reaction conditions, offering more intuitive information on active sites and reaction pathways.Additionally, in situ nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) techniques are valuable for detecting molecular-level changes in electronic structures and coordination environments, enabling the further elucidation of complex reaction mechanisms. Integrating these techniques with multi-scale theoretical simulations and data analysis shows promise for providing a more comprehensive understanding of catalytic processes and better guidance for more precise catalyst design. Future advancements should focus on enhancing the spatial, temporal, and energy resolution of in situ techniques to enable the detailed analysis of catalysts under conditions closer to those used during practical operation. Moreover, developing comprehensive analytical platforms that integrate multiple characterization methods will significantly enhance the understanding of bifunctional oxygen electrocatalysts, reduce research biases, and provide critical insights for designing efficient and stable catalysts.
In summary, this review offers a comprehensive analysis of recent advancements in non-precious metal bifunctional oxygen electrode catalysts, with a particular emphasis on the advantages and limitations of carbon materials, transition metal compounds, and their composite structures in catalytic processes. It explores the sources of catalytic activity, including methods for increasing active site density, as well as design principles and regulation strategies based on reaction mechanisms. Through statistical analysis of reported bifunctional electrocatalysts, the review reveals correlations between composition, structure, and performance, offering new insights into how different materials influence electrocatalytic efficiency. Looking to the future, further advancements in this field are expected to pave new pathways for the development of next-generation metal–air batteries and other energy storage and conversion technologies, contributing to a greener and more sustainable future.
Author contributions
Hengqi Liu: conceptualization, writing draft, creating the graphics, investigation; Rui Xiong: writing draft and editing; Shengyu Ma: writing draft, creating the graphics, and editing; Ran Wang: reviewing and editing; Zhiguo Liu: reviewing and editing; Tai Yao: reviewing and editing; Bo Song: conceptualization, supervision, creating the graphics, reviewing and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review. All data supporting the conclusions of this review are available from the cited articles, which can be accessed through publicly available databases or journal repositories.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Space Environment Simulation Research Infrastructure for help in our experiment. We acknowledge the support from the National Science Fund for Distinguished Young Scholars (grant no. 52225201), the National Natural Science Foundation of China (grant no. 52072085, 52271207), the National Key Research and Development Program of China (grant no. 2023YFE0201000). This research is supported by the Science Foundation of National Key Laboratory of Science and Technology on Advanced Composites in Special Environments. We acknowledge the financial support from the Fundamental Research Funds for the Central Universities (grant no. HIT.DZJJ.2024013).References
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, 873 CAS.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- S. Gao, X. Wang, X. Liu, C. Guo, Q. Liu and G. Hu, Mater. Chem. Front., 2024, 8, 485–506 RSC.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- T. Bashir, S. Zhou, S. Yang, S. A. Ismail, T. Ali, H. Wang, J. Zhao and L. Gao, Electrochem. Energy Rev., 2023, 6, 5 CrossRef CAS.
- C. D. Quilty, D. Wu, W. Li, D. C. Bock, L. Wang, L. M. Housel, A. Abraham, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Chem. Rev., 2023, 123, 1327–1363 CrossRef CAS PubMed.
- M. Zhao, S. Li, X. Wu and L. Sun, iScience, 2024, 27, 110926 CrossRef CAS PubMed.
- X. Chang, J. Jian, G. Cai, R. Wu and J. Li, Electron. Mater. Lett., 2016, 12, 237–242 CrossRef CAS.
- Z. Ren, L. Duan, S. Xu, L. Chen, Q. Yi and D. Zhang, J. Synth. Cryst, 2023, 52, 654 CAS.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- W. Cai, Y.-X. Yao, G.-L. Zhu, C. Yan, L.-L. Jiang, C. He, J.-Q. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- I. Temprano, T. Liu, E. Petrucco, J. H. J. Ellison, G. Kim, E. Jónsson and C. P. Grey, Joule, 2020, 4, 2501–2520 CrossRef CAS.
- J. S. Lee, S. Tai Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2010, 1, 34–50 CrossRef.
- X. Deng, Z. Li, Q. Yao, S. Yin, Z. Xie, Y. Liang and F. Liu, J. Synth. Cryst, 2023, 52, 345 CAS.
- H. J. Qiu, P. Du, K. Hu, J. Gao, H. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. Chen, Adv. Mater., 2019, 31, e1900843 CrossRef PubMed.
- C. Qin, S. Chen, H. Gomaa, M. Shenashen, S. El-Safty, Q. Liu, C. An, X. Liu, Q. Deng and N. Hu, Acta Phys.-Chim. Sin, 2024, 40, 2307059 Search PubMed.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Adv. Mater., 2020, 32, e1907399 CrossRef PubMed.
- H. Liu, D. Hua, R. Wang, Z. Liu, J. Li, X. Wang and B. Song, J. Phys. D: Appl. Phys., 2023, 57, 099501 CrossRef.
- Y. Liu and X. Wu, Nano Energy, 2024, 127, 109809 CrossRef CAS.
- X. Zhang, X. Han, Z. Jiang, J. Xu, L. Chen, Y. Xue, A. Nie, Z. Xie, Q. Kuang and L. Zheng, Nano Energy, 2020, 71, 104547 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- J. Diao, F. Wang and X. Liu, J. Synth. Cryst., 2020, 49, 665 CAS.
- M. Wu, X. Yang, X. Cui, N. Chen, L. Du, M. Cherif, F.-K. Chiang, Y. Wen, A. Hassanpour, F. Vidal, S. Omanovic, Y. Yang, S. Sun and G. Zhang, Nano-Micro Lett., 2023, 15, 232 CrossRef CAS PubMed.
- W. Wang, J.-Q. Chen, Y.-R. Tao, S.-N. Zhu, Y.-X. Zhang and X.-C. Wu, ACS Catal., 2019, 9, 3498–3510 CrossRef CAS.
- S. Lu, J. Jiang, H. Yang, Y.-J. Zhang, D.-N. Pei, J.-J. Chen and Y. Yu, ACS Nano, 2020, 14, 10438–10451 CrossRef CAS PubMed.
- Y. Chen, Z. Li, Y. Zhu, D. Sun, X. Liu, L. Xu and Y. Tang, Adv. Mater., 2019, 31, e1806312 CrossRef.
- L. Su, P. Wang, X. Ma, J. Wang and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 21261–21266 CrossRef CAS PubMed.
- T. Wang, Q. Zhang, K. Lian, G. Qi, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 655, 176–186 CrossRef PubMed.
- X. Shi, B. He, L. Zhao, Y. Gong, R. Wang and H. Wang, J. Power Sources, 2021, 482, 228955 CrossRef CAS.
- X. Ren and Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS PubMed.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- J.-N. Liu, C.-X. Zhao, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542–4553 RSC.
- J. Li, Nano-Micro Lett., 2022, 14, 112 CrossRef CAS PubMed.
- J. R. J. K. Nørskov, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- G. Yasin, S. Ali, S. Ibraheem, A. Kumar, M. Tabish, M. A. Mushtaq, S. Ajmal, M. Arif, M. A. Khan, A. Saad, L. Qiao and W. Zhao, ACS Catal., 2023, 13, 2313–2325 CrossRef CAS.
- G. W. Heise and E. A. Schumacher, Trans. Electrochem. Soc., 1932, 62, 383–391 CrossRef.
- A. D. S. Tantram, Energy Policy, 1974, 2, 55–66 CrossRef.
- J. Goldstein, J. Power Sources, 1999, 80, 171–179 CrossRef CAS.
- M. Wang, X. Liu and X. Wu, Nano Energy, 2023, 114, 108681 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219191 CrossRef CAS PubMed.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- Q. Zhang, Y. Zhang, S. Zhang and G. Gao, J. Phys. Chem. C, 2023, 127, 16346–16356 CrossRef CAS.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846–854 CrossRef PubMed.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- S. Wang, S. Chen, L. Ma and J. A. Zapien, Mater. Today Energy, 2021, 20, 100659 CrossRef CAS.
- H. F. Wang, C. Tang and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1803329 CrossRef.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. Zhao, Y. Li and D. Wang, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- Y. Huang, Y. Wang, C. Tang, J. Wang, Q. Zhang, Y. Wang and J. Zhang, Adv. Mater., 2018, 31, 1803800 CrossRef PubMed.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- C. Hu and L. Dai, Adv. Mater., 2018, 31, 1804672 CrossRef PubMed.
- J. Li, Y. Zhang, X. Zhang, J. Han, Y. Wang, L. Gu, Z. Zhang, X. Wang, J. Jian, P. Xu and B. Song, ACS Appl. Mater. Interfaces, 2015, 7, 19626–19634 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- J. Li, S. Yi, R. Rajagopalan, Z. Zhang, Y. Tang and H. Wang, Green Energy Environ., 2023, 8, 308–317 CrossRef CAS.
- J. Sun, S. E. Lowe, L. Zhang, Y. Wang, K. Pang, Y. Wang, Y. Zhong, P. Liu, K. Zhao, Z. Tang and H. Zhao, Angew. Chem., Int. Ed., 2018, 57, 16511–16515 CrossRef CAS PubMed.
- R. S. D. H. Guo, C. Akiba, S. Saji, T. Kondo and J. J. Nakamura, Science, 2016, 351, 361–365 CrossRef PubMed.
- X. Feng, G. Chen, Z. Cui, R. Qin, W. Jiao, Z. Huang, Z. Shang, C. Ma, X. Zheng, Y. Han and W. Huang, Angew. Chem., Int. Ed., 2023, 63, e202316314 CrossRef PubMed.
- C. Hu and L. Dai, Adv. Mater., 2016, 29, 1604942 CrossRef.
- S. Cao, W. Shang, G.-L. Li, Z.-F. Lu, X. Wang, Y. Yan, C. Hao, S. Wang and G. Sun, Carbon, 2021, 184, 127–135 CrossRef CAS.
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H. C. Chen, Z. Zhuang, Y. Zhang, W. C. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. M. Chen, C. Chen and Y. Li, Adv. Mater., 2022, 34, 2203621 CrossRef CAS PubMed.
- P. Wei, X. Li, Z. He, X. Sun, Q. Liang, Z. Wang, C. Fang, Q. Li, H. Yang, J. Han and Y. Huang, Chem. Eng. J., 2021, 422, 130134 CrossRef CAS.
- X. Guo, J. Shi, M. Li, J. Zhang, X. Zheng, Y. Liu, B. Xi, X. An, Z. Duan, Q. Fan, F. Gao and S. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202314124 CrossRef CAS PubMed.
- S. Liu, G. Li, Y. Gao, Z. Xiao, J. Zhang, Q. Wang, X. Zhang and L. Wang, Catal. Sci. Technol., 2017, 7, 4007–4016 RSC.
- H. Xia, R. Pang, X. Dong, Q. Liu, J. Chen, E. Wang and J. Li, J. Am. Chem. Soc., 2023, 145, 25695–25704 CrossRef CAS PubMed.
- Q. Wu, Y. Jia, Q. Liu, X. Mao, Q. Guo, X. Yan, J. Zhao, F. Liu, A. Du and X. Yao, Chem, 2022, 8, 2715–2733 CAS.
- L. Lin, Y. Ni, L. Shang, H. Sun, Q. Zhang, W. Zhang, Z. Yan, Q. Zhao and J. Chen, ACS Catal., 2022, 12, 7531–7540 CrossRef CAS.
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- D. Zhang, Z. Wang, F. Liu, P. Yi, L. Peng, Y. Chen, L. Wei and H. Li, J. Am. Chem. Soc., 2024, 146, 3210–3219 CrossRef CAS PubMed.
- P. Li, Y. Jiao, Y. Ruan, H. Fei, Y. Men, C. Guo, Y. Wu and S. Chen, Nat. Commun., 2023, 14, 6936 CrossRef CAS PubMed.
- T. Patniboon and H. A. Hansen, ACS Catal., 2021, 11, 13102–13118 CrossRef CAS.
- Y. Zhang, Z.-W. Chen, X. Liu, Z. Wen, C. V. Singh, C. C. Yang and Q. Jiang, Nano Lett., 2024, 24, 4291–4299 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- X. Li, X. Lv, P. Sun and X. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 29979–29990 CrossRef CAS PubMed.
- W. Zhang, C. H. Xu, H. Zheng, R. Li and K. Zhou, Adv. Funct. Mater., 2022, 32, 2200763 CrossRef CAS.
- Z. Liang, N. Kong, C. Yang, W. Zhang, H. Zheng, H. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS PubMed.
- Z. Li, S. Ji, C. Wang, H. Liu, L. Leng, L. Du, J. Gao, M. Qiao, J. H. Horton and Y. Wang, Adv. Mater., 2023, 35, 2300905 CrossRef CAS PubMed.
- Z. Liang, M. Luo, M. Chen, X. Qi, J. Liu, C. Liu, S. G. Peera and T. Liang, J. Mater. Chem. A, 2020, 8, 20453–20462 RSC.
- Z. Yang and W. Jin, J. Mater. Sci. Mater. Electron., 2024, 35, 554 CrossRef CAS.
- Y. Ruan, H. Lei, W. Xue, T. Wang, S. Song, H. Xu, Y. Yu, G.-R. Zhang and D. Mei, J. Alloys Compd., 2023, 934, 167781 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- K. Wang, Z. Lu, J. Lei, Z. Liu, Y. Li and Y. Cao, ACS Nano, 2022, 16, 11944–11956 CrossRef CAS PubMed.
- B. Tang, Y. Zhou, Q. Ji, Z. Zhuang, L. Zhang, C. Wang, H. Hu, H. Wang, B. Mei, F. Song, S. Yang, B. M. Weckhuysen, H. Tan, D. Wang and W. Yan, Nat. Synth., 2024, 3, 878–890 CrossRef.
- Q. Du, Y. Gong, M. A. Khan, D. Ye, J. Fang, H. Zhao and J. Zhang, Green Energy Environ., 2022, 7, 16–34 CrossRef CAS.
- Y. Ran, C. Xu, D. Ji, H. Zhao, L. Li and Y. Lei, Nano Res. Energy, 2024, 3, 2790–8119 Search PubMed.
- W. Liu, D. Zheng, T. Deng, Q. Chen, C. Zhu, C. Pei, H. Li, F. Wu, W. Shi, S. W. Yang, Y. Zhu and X. Cao, Angew. Chem., Int. Ed., 2021, 60, 10614–10619 CrossRef CAS PubMed.
- K. Tang, H. Hu, Y. Xiong, L. Chen, J. Zhang, C. Yuan and M. Wu, Angew. Chem., Int. Ed., 2022, 61, e202202671 CrossRef CAS PubMed.
- J. Zheng, D. Meng, J. Guo, X. Liu, L. Zhou and Z. Wang, Adv. Mater., 2024, 36, 2405129 CrossRef CAS PubMed.
- C. Huang, Y. Zhang, X. Li, H. Cao, Y. Guo and C. Zhang, Appl. Catal., B, 2022, 319, 121909 CrossRef CAS.
- Y. Rao, S. Chen, Q. Yue and Y. Kang, ACS Catal., 2021, 11, 8097–8103 CrossRef CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- Y. Gorlin, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- H. Li, S. Kelly, D. Guevarra, Z. Wang, Y. Wang, J. A. Haber, M. Anand, G. T. K. K. Gunasooriya, C. S. Abraham, S. Vijay, J. M. Gregoire and J. K. Nørskov, Nat. Catal., 2021, 4, 463–468 CrossRef CAS.
- C.-H. Kuo, I. M. Mosa, S. Thanneeru, V. Sharma, L. Zhang, S. Biswas, M. Aindow, S. Pamir Alpay, J. F. Rusling, S. L. Suib and J. He, Chem. Commun., 2015, 51, 5951–5954 RSC.
- H. Huang, A. Huang, D. Liu, W. Han, C. H. Kuo, H. Y. Chen, L. Li, H. Pan and S. Peng, Adv. Mater., 2023, 35, 2303109 CrossRef CAS PubMed.
- Y. Wang, X. Ge, Q. Lu, W. Bai, C. Ye, Z. Shao and Y. Bu, Nat. Commun., 2023, 14, 6968 CrossRef CAS PubMed.
- Z. Wang, J. Huang, L. Wang, Y. Liu, W. Liu, S. Zhao and Z. Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114696 CrossRef CAS PubMed.
- Y. Sun, T. Zhang, P. Sun, J. Wang, W. Duan, Y. Zhuang, L. Wang and Z. Li, J. Energy Chem., 2024, 94, 778–788 CrossRef CAS.
- W. Tang, J. Tang, K. Liao and Z. Shao, Chem. Eng. J., 2023, 455, 140855 CrossRef CAS.
- Y. Zhang, Y. Jiang, G. Jiang, T. Or, R. Gao, H. Zhang, Z. Bai, N. Chen, Y.-P. Deng and Z. Chen, Nano Energy, 2023, 115, 108672 CrossRef CAS.
- Y. Yuan, S. Adimi, T. Thomas, J. Wang, H. Guo, J. Chen, J. P. Attfield, F. J. DiSalvo and M. Yang, Innovation, 2021, 2, 100096 CAS.
- A. Saad, H. Shen, Z. Cheng, R. Arbi, B. Guo, L. S. Hui, K. Liang, S. Liu, J. P. Attfield, A. Turak, J. Wang and M. Yang, Nano-Micro Lett., 2020, 12, 2311–6706 Search PubMed.
- J. Ban, H. Xu, G. Cao, Y. Fan, W. K. Pang, G. Shao and J. Hu, Adv. Funct. Mater., 2023, 33, 2300623 CrossRef CAS.
- N. Khodayar, A. Noori, M. S. Rahmanifar, M. Moloudi, N. Hassani, M. Neek-Amal, M. F. El-Kady, N. B. Mohamed, X. Xia, Y. Zhang, R. B. Kaner and M. F. Mousavi, Energy Environ. Sci., 2024, 17, 5200–5215 RSC.
- Y. Tan, Y. Wang, A. Li, X. Jiang, Y. Zhang and C. Cheng, J. Energy Chem., 2024, 96, 568–577 CrossRef CAS.
- H. Li, Q. Li, P. Wen, T. B. Williams, S. Adhikari, C. Dun, C. Lu, D. Itanze, L. Jiang, D. L. Carroll, G. L. Donati, P. M. Lundin, Y. Qiu and S. M. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef PubMed.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- Z. Li, Y. Zeng, D. Xiong, L. Zhou, J. Zhou, Y. Yang, F. Zhan, K. Wang, Y. Du and Y. Liu, Inorg. Chem. Front., 2024, 11, 549–561 RSC.
- A. Wang, X. Zhang, S. Gao, C. Zhao, S. Kuang, S. Lu, J. Niu, G. Wang, W. Li, D. Chen, H. Zhang, X. Zhou, S. Zhang, B. Zhang and W. Wang, Adv. Mater., 2022, 34, 2204247 CrossRef CAS PubMed.
- F. Li, H. Wu, S. Lv, Y. Ma, B. Wang, Y. Ren, C. Wang, Y. Shi, H. Ji, J. Gu, S. Tang and X. Meng, Small, 2023, 20, 2309025 CrossRef PubMed.
- T. Lu, Q. Sun, J. He, R. Li and C. Huang, Small, 2023, 20, 2309255 CrossRef PubMed.
- Y. Pan, J. Gao, Y. Li, E. Lv, U. Khan, X. Yang, J. Yao, A. Nairan and Q. Zhang, Small, 2023, 20, 2304594 CrossRef PubMed.
- Z. Shao, Q. Zhu, Y. Sun, Y. Zhang, Y. Jiang, S. Deng, W. Zhang, K. Huang and S. Feng, Adv. Mater., 2022, 34, 2110172 CrossRef CAS PubMed.
- X. Guo, X. Zhang, Y. Wu, Y. Xin, D. Li, Y. Zhang and P. Yu, Dalton Trans., 2023, 52, 4315–4322 RSC.
- Y. Tian, X. Liu, L. Xu, D. Yuan, Y. Dou, J. Qiu, H. Li, J. Ma, Y. Wang, D. Su and S. Zhang, Adv. Funct. Mater., 2021, 31, 2101239 CrossRef CAS.
- L. Yang, R. He, M. Botifoll, Y. Zhang, Y. Ding, C. Di, C. He, Y. Xu, L. Balcells, J. Arbiol, Y. Zhou and A. Cabot, Adv. Mater., 2024, 36, 2400572 CrossRef CAS PubMed.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- D. Ren, J. Ying, M. Xiao, Y. P. Deng, J. Ou, J. Zhu, G. Liu, Y. Pei, S. Li, A. M. Jauhar, H. Jin, S. Wang, D. Su, A. Yu and Z. Chen, Adv. Funct. Mater., 2019, 30, 1908167 CrossRef.
- Y. Xue, Y. Guo, Q. Zhang, Z. Xie, J. Wei and Z. Zhou, Nano-Micro Lett., 2022, 14, 162 CrossRef CAS PubMed.
- H. Zhang, H. C. Chen, S. Feizpoor, L. Li, X. Zhang, X. Xu, Z. Zhuang, Z. Li, W. Hu, R. Snyders, D. Wang and C. Wang, Adv. Mater., 2024, 36, 2400523 CrossRef CAS PubMed.
- M. Zhang, H. Li, J. Chen, F. X. Ma, L. Zhen, Z. Wen and C. Y. Xu, Adv. Funct. Mater., 2022, 33, 2209726 CrossRef.
- H. Huang, D. Yu, F. Hu, S. C. Huang, J. Song, H. Y. Chen, L. L. Li and S. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CrossRef CAS PubMed.
- H. Meng, B. Wu, D. Zhang, X. Zhu, S. Luo, Y. You, K. Chen, J. Long, J. Zhu, L. Liu, S. Xi, T. Petit, D. Wang, X.-M. Zhang, Z. J. Xu and L. Mai, Energy Environ. Sci., 2024, 17, 704–716 RSC.
- Z. Wang, X. Jin, R. Xu, Z. Yang, S. Ma, T. Yan, C. Zhu, J. Fang, Y. Liu, S.-J. Hwang, Z. Pan and H. J. Fan, ACS Nano, 2023, 17, 8622–8633 CrossRef CAS PubMed.
- H.-S. Fan, F.-X. Ma, Z.-H. Liu, W.-H. Wang, Z.-Q. Liu, X.-Y. Liang, Y. Du, Y.-Y. Li, L. Zhen and C.-Y. Xu, Rare Met., 2024, 43, 5769–5780 CrossRef CAS.
- Y. Wang, N. Katyal, Y. Tang, H. Li, K. Shin, W. Liu, R. He, M. Xu, G. Henkelman and S. J. Bao, Small, 2023, 20, 2306504 CrossRef PubMed.
- P. Li, F. Qiang, X. Tan, Z. Li, J. Shi, S. Liu, M. Huang, J. Chen, W. Tian, J. Wu, W. Hu and H. Wang, Appl. Catal., B, 2024, 340, 123231 CrossRef CAS.
- T. Zhao, Y.-H. Wu, Z.-R. Song, X. Wang, R.-L. Yin, H. Xu, H. Cui, X.-H. Cao and J.-K. Gao, Rare Met., 2023, 42, 3326–3336 CrossRef CAS.
- M. Wang, S. Ji, H. Wang, X. Wang, V. Linkov and R. Wang, Small, 2022, 18, 2204474 CrossRef CAS PubMed.
- Z. Zheng, C. Wang, P. Mao, Y. Zhu, R. Ran, W. Zhou, K. Liao and Z. Shao, Carbon Energy, 2022, 5, e274 CrossRef.
- R. S. Kumar, P. Mannu, S. Prabhakaran, T. T. T. Nga, Y. Kim, D. H. Kim, J. L. Chen, C. L. Dong and D. J. Yoo, Adv. Sci., 2023, 10, 2303525 CrossRef CAS PubMed.
- J.-N. Liu, C.-X. Zhao, J. Wang, X.-Q. Fang, C.-X. Bi, B.-Q. Li and Q. Zhang, Joule, 2024, 8, 1–16 CrossRef.
- Z. Jia, J. Shang, K. Xue, X. Yang, S. Wang, C. Xu and Q. Wang, ChemCatChem, 2023, 15, e202201469 CrossRef CAS.
- B. N. V. Krishna, O. R. Ankinapalli, A. R. Reddy and J. S. Yu, Small, 2024, 20, 2311176 CrossRef CAS PubMed.
- W. Liu, J. Zhang, Z. Bai, G. Jiang, M. Li, K. Feng, L. Yang, Y. Ding, T. Yu, Z. Chen and A. Yu, Adv. Funct. Mater., 2018, 28, 1706675 CrossRef.
- C. Sun, J. Ding, H. Wang, J. Liu, X. Han, Y. Deng, C. Zhong and W. Hu, J. Mater. Chem. A, 2021, 9, 13926–13935 RSC.
- X. Wu, G. Han, H. Wen, Y. Liu, L. Han, X. Cui, J. Kou, B. Li and J. Jiang, Energy Environ. Mater., 2021, 5, 935–943 CrossRef.
- S. Wu, D. Deng, E. Zhang, H. Li and L. Xu, Carbon, 2022, 196, 347–353 CrossRef CAS.
- M. Liu, X. Lv, Z. Mi, S. Chen, J. Li, J. Wu, T. Sun, L. Zhang and J. Zhang, ACS Appl. Energy Mater., 2023, 6, 7317–7322 CrossRef CAS.
- X. Yang, F. Wang, Z. Jing, M. Chen, B. Wang, L. Wang, G. Qu, Y. Kong and L. Xu, Small, 2023, 19, 2301985 CrossRef CAS PubMed.
- P. Li, H. Wang, X. Tan, W. Hu, M. Huang, J. Shi, J. Chen, S. Liu, Z. Shi and Z. Li, Appl. Catal., B, 2022, 316, 121674 CrossRef CAS.
- C. Wang, K. Aoyagi, P. Wisesa and T. Mueller, Chem. Mater., 2020, 32, 3741–3752 CrossRef CAS.
- Y. Zhou, G. Dong, Q. Tan, X. Han, C. Chen and J. Wei, Energy, 2023, 262, 125514 CrossRef CAS.
- S. Yin, H. Yi, M. Liu, J. Yang, S. Yang, B. Zhang, L. Chen, X. Cheng, H. Huang, R. Huang, Y. Jiang, H. Liao and S. Sun, Nat. Commun., 2024, 15, 6229 CrossRef CAS PubMed.
- W. Shi, Z. Li, Z. Gong, Z. Liang, H. Liu, Y. Han, H. Niu, B. Song, X. Chi, J. Zhou, H. Wang, B. Xia, Y. Yao and Z. Tian, Nat. Commun., 2023, 14, 2294 CrossRef CAS PubMed.
- W. Zhou, H. Su, W. Cheng, Y. Li, J. Jiang, M. Liu, F. Yu, W. Wang, S. Wei and Q. Liu, Nat. Commun., 2022, 13, 6414 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |