
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing droplet-based microbiological assays: optofluidic detection meets multiplexed droplet generation†
Ashkan
Samimi
ab,
Sundar
Hengoju
a,
Karin
Martin
a and
Miriam A.
Rosenbaum
 *abc
*abc
aLeibniz Institute for Natural Product Research and Infection Biology – Hans-Knöll-Institute, Jena, Germany. E-mail: miriam.rosenbaum@leibniz-hki.de
bFaculty of Biological Sciences, Friedrich Schiller University, Jena, Germany
cCluster of Excellence Balance of the Microverse, Friedrich Schiller University Jena, Jena, Germany
First published on 11th June 2025
Abstract
Microbiological assays are crucial in understanding microbial ecology and developing new bioproducts. Given the significance of these assays, there is a growing interest in developing high throughput experimentation methods capable of assay multiplexing to enhance the accuracy and efficiency. In this study, we integrate a multiplexed droplet generation set-up into an optofluidic detection chip to facilitate rapid and high throughput analysis of microbiological assays. The optofluidic detection set-up at the same time enables fast and sensitive assessment of droplet condition and content, providing analysis scalability in a high throughput manner. Employing the integration, we produced unique fluorescence barcoded droplets containing defined concentrations of various carbon sources, allowing the simultaneous investigation of microbial growth and metabolic capacity under different experimental conditions. We successfully validated the robustness of the established setup in analyzing and distinguishing different fluorescence barcodes. Our findings highlight the potential of the integrated platform for a broader range of applications in high throughput drug screening, environmental monitoring, and microbiology research.
Introduction
Microbiological assays evaluate the metabolic or ecological capacities of microorganisms and are essential in developing applications in various research fields, including clinical diagnostics, environmental monitoring, and biotechnology. For instance, antibiotic susceptibility can be realized by agar diffusion1,2 and micro broth dilution,3 enabling the evaluation of pathogen susceptibility and minimum inhibitory concentrations. In environmental studies, microbiological assays have been applied to monitor the soil and water quality to detect potential contaminants or for biodiversity studies.4,5 They also enable researchers to study microbial communities from different natural habitats for their responses to various experimental conditions.6,7 Finally, these assays are utilized in biotechnology to advance fermentation and biochemical production8–10 or search for new enzymatic biocatalysts, emphasizing their central role in advancing our understanding of these research fields.Traditional microbiological assays are often limited in scalability and throughput, as shake flasks and microtiter plates are primarily used for cultivation, making these assays labor-intensive and time-consuming. To tackle these challenges, the demand is growing for high throughput multiplexed methods that facilitate large-scale, multi-factor testing. Droplet microfluidics is a technology that harnesses the immiscibility of two fluids to produce picolitre droplets in micro-scale channel sizes.11,12 This technology offers several advantages, including lower reagent consumption,13–15 faster reaction times,16 and high throughput droplet generation rates.17,18 Most importantly, this technology, for the first time, brings the scale and throughput of microbial cultivation to match the scale and diversity of microbial functions. Various microbiological assays have benefited from these unique features already.19–24 Droplets have been employed for disease identification from clinical samples23 and antibiotic susceptibility tests.20,25 The technology has also been utilized in the directed evolution of enzymes where genetic diversity is systematically created, and droplets are screened to pick the best-performing mutant.21 In these assays, droplets are analyzed through microscopy imaging or laser-induced fluorescence/absorbance measurements (using detectors installed on the microscope or optical fibers integrated into the microfluidic chip). The former provides excellent spatial resolution; however, the number of analyzed droplets is limited to a few thousand, and the image acquisition protocol is usually time-consuming. On the other hand, despite providing throughput in analysis, laser-induced fluorescence/absorbance measurements do not provide any spatial information (e.g., microbial biomass within droplets). Thus, many droplet-based assays often compromise on scalability due to limited analysis throughput.23,25 Therefore, by combining laser-induced fluorescence measurement with image acquisition, one would harness the advantages of both methods while minimizing their downsides.
Moreover, droplet-based microbial investigations, such as those on antibiotic resistance or discovery of suitable cultivation conditions (i.e., different media compositions) for microbial communities of different habitats, require droplets with multiple experimental conditions. However, due to lack of multiplexing techniques, current droplet-based approaches are limited to a single experimental condition requiring multiple experimental runs.26,27
Introducing several experimental conditions in droplet populations requires reliable tracking of the respective droplet population content since droplets lose their order during handling operations or reinjection into the microfluidic chip, and without specific encoding, it is impossible to track back the original experimental conditions. In our recent development,28 we have established a multiplexing platform that enables the production of various preset experimental conditions to assess the phenotypic characteristics of individual droplet populations in one experiment. Employing the platform, fluorescently barcoded droplet populations are generated and processed depending on the experimental design, while effective barcode analysis pipelines enable the tracking of droplet contents. In our previous study, the assay outcome for the library of droplets was assessed utilizing microscopy imaging. Despite providing high-resolution phenotypic information within droplets, this technique lacks throughput and requires high-level image processing algorithms to extract meaningful information. Moreover, the sub-sampled droplets cannot be easily recovered after imaging for further incubation and subsequent analysis. On the other hand, our group independently developed an optofluidic chip design with laser-induced fluorescence capability, enabling simultaneous detection of multiwavelength fluorescence signals from droplets when they pass through an interrogation zone in the chip.29 This technology empowers real-time, high throughput data acquisitions and processing of droplet contents. Using a single sensor, the technology eliminates the complex optical designs requiring multiple detectors on a microfluidic chip30 while providing simple and highly sensitive measurements.
In this study, we introduce an integrated workflow that combines our recently developed multiplexing platform for producing a color-coded droplet library of multiple experimental conditions with our optofluidic droplet analysis chip. We also introduced a simple in flow bright field imaging setting on the chip and evaluated the correlation of in flow imaging to microscopy by assessing the growth-promoting conditions of a model E. coli strain under the influence of different carbon sources. Our integrated workflow leverages the unique advantages of each method that provides reliable droplet library production, high throughput sensitive laser-induced fluorescence measurement, and phenotypic information through in flow imaging comparable to microscopy.
Materials and methods
Bacterial sample preparation
The Escherichia coli 081 strain was used, which is a transformation of the E. coli K-12 strain RV308 (ATCC 31608) carrying the plasmid pMK3c2GFP for constitutive green fluorescent protein (GFP) expression.31 As an overnight culture, E. coli 081 from a cryostock was inoculated in M9 medium with 1% glucose and incubated at 37 °C for around 20 hours. Bacterial cells from this culture were inoculated at an OD600 of about 0.2 in M9 medium with 1% glucose and incubated at 37 °C for about 3 hours until the OD600 of the solution reached around 0.5. Kanamycin at a final concentration of 20 μg mL−1 was added to both cultures. Next, bacterial cells were centrifuged at 5000 rpm for 5 minutes and washed three times with PBS. Then, the bacterial cell pellet was diluted in the M9 medium without any carbon sources, and an OD600 of 0.2 was prepared to load as culture inoculum into the sample well stripe of the multiplexing platform.Carbon source solution preparation
Carbon sources (alanine, arabinose, citric acid, galactose, glucose, histidine, maltose, mannose, serine, threonine, and xylose) were diluted in sterile water at a concentration of 100 mg mL−1 and stored at 4 °C for later use. Before the experiment, all solutions were filter sterilized for multiplexing sample preparation. The final concentration of carbon sources in droplet populations is 20 mg mL−1.Dye solution preparation for droplet encoding
Three dyes at two different stock concentrations per dye were used to characterize the color code measurement by the optofluidic set-up. Alexa-flour 488 (Thermo Fischer), at 8 μg mL−1 and 40 μg mL−1, Alexa-flour 647 (Thermo Fischer) at 6.4 μg mL−1 and 32 μg mL−1, and DY557 (Dyomics) at 0.8 mg mL−1 and 4.4 mg mL−1 were used for color coding purposes. Red and far-red dyes were used for color coding, as we used a green fluorescence bacterial strain for our validation experiments. Depending on the defined color code for each carbon source, all droplet generation sample plugs were prepared with the desired dye concentration.Conditions multiplexing and droplet generation
The multiplexing platform employs a commercial liquid handler (MitosDropix, Dolomite) equipped with a sample well stripe and a sample hook to move between wells and take the desired plug volumes from each well.28 To produce the multiplexed samples, we put the carbon sources and bacterial cell solutions mixed with color-coding dyes into the wells of the multiplexing platform. Using the dedicated software, defined volumes of each carbon source and bacterial cell solution are withdrawn and merged passively through transitions in the tubings of the multiplexing platform.28 The final sample is then well-mixed by creating an oscillatory fluid flow within the platform tubing and sent for droplet production.28 A syringe pump (neMESYS, cetoni) was used for fluidic operations, and for droplet generation, Novec oil (HFE7500, 3M) with 0.5% fluorinated surfactant (FluoSurf, emulseo) was utilized. All microfluidic chips have been fabricated using UV and soft lithography, as described in our previous report.32,33Microscopy imaging
Droplet populations were imaged right after generation and after 24 hours of incubation using a 10× magnification. An inverted microscope (Axio Observer Z1, Zeiss) with the standard halogen lamp was used as the light source for brightfield images. Colibri 5 Zeiss was used as the light source for fluorescence imaging. The microfluidic observation chip has a 2 mm wall-to-wall length with 150 μm height. In each field of view using 10× magnification, about 300 droplets are imaged.Optofluidic set-up and measurements
As previously reported, after droplet incubation, droplets were reinjected into a custom optofluidic chip (Fig. S1†) for fluorescence measurement.29,34 In summary, the chip includes three inlets for droplet reinjection and spacing oil flow. There are three fiber guide structures with air-PDMS micro-lenses. The bare ends of optical fibers are integrated into the chip through fiber guide structures, and the other ends are connected to the light source or detectors. A light beam from the optical fiber is collimated and focused on the microfluidic channels through micro-lenses. The contents of each droplet are analyzed as they move through the optofluidic interrogation zone. The spacing between droplets was ensured using the oil inlets by Novec-7500 oil (3M) with 0.5% flour-surfactant (Emulseo). As shown in Fig. S1,† the oil inlets 1 and 2 were set at 120 and 50 nL s−1, respectively. Droplets were injected at 10 nL s−1. Three lasers at 405, 561, and 639 nm (LASOS) wavelengths were used to simultaneously excite the content of every individual droplet. The lasers have a nominal power of 20 mW, adjusted using a custom-written LabVIEW software by controlling the input voltage (0–5 V), and for all experiments, the input voltage was set at 0.5 V. Downstream from the interrogation zone, a camera (Basler, acA1920-155uc) is positioned to capture images of these droplets. An inverted microscope (Axio Observer Z1, Zeiss) with the standard halogen lamp was used. The intensity of the microscope's halogen lamp was set at 6 V, and the camera exposure setting was at 150 μs. Using a custom-written LabVIEW software, a trigger signal was generated with a delay time (such that the droplet moves and reaches the camera interrogation zone) to capture images with an image size of 500 by 500 pixels. A time series fluorescence signal is also recorded for offline analysis.Fiber data analysis
For fiber-based analysis, peaks and the corresponding peak widths were identified from the recorded time series data using “find_peaks” and “peak_width” from the scipy-signal in Python. Next, initial data filtering was done by plotting the histogram of peak widths in one coding fluorescence channel. A normal distribution was fitted to this histogram, and data (i.e., droplet information) within full width at the tenth maximum was used for further analysis. Using this strategy, extremely small or big droplets were eliminated from the analysis pipeline. Hierarchical density-based clustering for application with noise (HDBSCAN) then analyzed filtered data to identify different populations.28Image analysis
Droplets in the microscopy images were identified using the Hough circle detection algorithm. Using the circle information from each droplet, regions of interest are created. Next, average intensities in different fluorescence channels for every droplet were calculated. The fluorescence data from the coding channels were used to determine the different populations utilizing HDBSCAN. For the growth analysis of the microscopy bright field images, we used the previously reported image analysis pipeline.28 For the binary classification (i.e., growth/no growth) of the microscopy images, the images of droplets directly after generation are utilized to quantify the growth within droplets, and the normalized mean growth value is used as a threshold.For in flow droplet imaging, single images of all droplets are recorded during measurements. For each image, the droplet was also identified using the Hough circle detection algorithm. A background image was calculated using 500 images, considering the median pixel value. For every identified droplet, the region of interest defined by the droplet area is subtracted from the background image. Next, an adaptive histogram equalization (CLAHE) from the OpenCV library is employed to improve the image's contrast. Then, a Sobel edge detection and thresholding followed by a dilating operator are applied to build the final binary image. These steps are performed to exclude minor intensity variations in the background subtracted image and connect the small gaps in the identified edges. Using the binary image, the growth is quantified as the ratio of white pixels (i.e., the area of the bacterial cells occupying the droplet) to the area of the region of interest (i.e., the droplet area). The quantified growth of individual conditions is then corrected using the average value of the two control conditions and is min-max normalized (see supplementary data).
Results and discussion
Workflow of the integrated platform
The integrated workflow is carried out in three stages. In stage I, a droplet library of different experimental conditions is produced utilizing a multiplexing platform28 by automatically configuring the content of samples for droplet generation (Fig. 1, left circle). This stage is performed in three main steps. In the first step, reagents are loaded into a sample well used by the platform to produce various small plugs of different reagents to create a final 10 μL experimental sample. The 10 μL sample is well mixed in the tubing utilizing oscillatory fluid flow created by the syringe pump to ensure a homogenous solution. These plugs are then injected into droplet generation chip (step 2). The droplet generation yields about 45–60 thousand droplets per condition. To distinguish different droplet populations within the droplet library, fluorescence dyes at specific concentrations are also diluted into the reagents and used to color code different experimental conditions. In our proposed workflow, up to three fluorescence dyes can be used for coding purposes: red, green, and far-red. However, only red and far-red dyes were used for color-coding in our current experiments as the model strain expresses GFP fluorescence. Using a pairwise combination and a single shade of these two dyes, theoretically, 90 fluorescence barcodes can be realized that can be utilized for a microbiological assay as described in our previous study.28 In step 3, droplets from all conditions are collected. In the second stage (stage II), generated droplets are incubated for a microbiological assay before measuring the desired experimental variables (growth or functional output). In our case, color-coded picolitre droplets were dynamically incubated35 for 24 hours to establish the growth of the inoculum in droplets. Subsequently, in the third stage of our workflow (stage III), the multiplexed droplet populations were reinjected in an optofluidic chip to analyze individual droplets regarding their experimental code and the selected experimental variable employing laser-induced fluorescence technology in a high throughput manner.29,34 In short, each droplet can be simultaneously excited with three different lasers, and multiplexed emission light is concurrently collected by optical fibers as they pass through an interrogation zone in the microfluidic chip. Thereby, different fluorophore signals are recorded separately using a frequency demultiplexing lock-in amplifier. The fluorophore signals are also utilized as a real-time prompt corresponding for each droplet to send a trigger signal to a camera downstream of the laser interrogation zone. The camera then captures an in flow bright field image, which is utilized for bacterial growth quantification (Fig. 1, stage III, step 3). | ||
| Fig. 1 The integrated workflow is carried out in three stages. The first stage is the generation of the multiplexed droplet library (I. Multiplexed droplet library generation). A multiplexed droplet library is produced and collected in three main steps. Initially, small plugs of different reagents, including microbial samples, are extracted from the sample wells of the multiplexing platform,28 merged, and thoroughly mixed to have a homogenous solution. The mixing of the final merged solution is achieved by employing oscillatory motion within the tubing. These samples are then sent to the microfluidic chip for droplet generation and collection in steps 2 and 3. The second stage includes the incubation of the collected droplets (II. Incubation). The droplet library is then dynamically incubated31 to ensure optimal growth of the microbial samples for the functional analysis. The third stage of the integrated workflow includes the optofluidic measurement and triggered in flow imaging. The droplet library is reinjected into an optofluidic chip after the incubation period in a three-step measurement sequence. First, the content of every droplet is excited when passing through an interrogation zone on the optofluidic chip equipped with optical fibers.29 The resulting fluorescence signal is then collected by another fiber utilized for real-time decision-making and recorded for offline data analysis. Finally, the collected fluorescence signal is utilized as a prompt to send a trigger signal to a camera downstream of the interrogation zone to capture an image of the corresponding droplet. The images of individual droplets are recorded for offline data analysis to quantify the bacterial growth within different experimental conditions. | ||
Fluorescence barcode characterization and in flow imaging using the optofluidic setup
As described previously,28 we established 24 color codes for every pairwise combination of two fluorescent dyes at two stock and three sub-concentration levels for a fixed final sample volume. Previously, we imaged every six-code matrix of each combination in separate fluorescence microscopy channels. Raw data from individual images were then analyzed to identify different clusters.To ensure the compatibility of the multiplexing coding strategy with the optofluidic measurements, we generated droplets with fluorescence barcodes of pairwise combinations of all three dyes. The droplets are then re-injected into the optofluidic chip described in Fig. 1, stage III, and droplet fluorescence signal is recorded for offline data analysis to identify the color codes.
During microbiological experiments, color-coded droplets typically undergo a period of incubation at a specific temperature, depending on the microbiological assay. Droplet size shrinkage is a common observation during incubation due to the growth of microorganisms,36 induced changes in osmolarity, and the thermal conditions of incubation. Moreover, droplet handling during transport and reinjection to the chip can introduce population size variability. Also, as the bacterial cell encapsulation follows Poisson distribution, there are empty and filled droplets depending on the initial cell concentration, which can induce great droplet size variability after incubation periods.36 Unlike microscopy image analysis, where an image undergoes a series of analytical algorithms and extremely small or big droplets can just be ignored, data from the optofluidic measurements of such a population can be challenging to analyze. To account for these changes in droplet populations, we developed a simple filtering strategy in the analysis pipeline. First, we identified the signal's peaks and corresponding widths (Fig. S2†). Then, by fitting a normal distribution over the peak widths, we only considered the peaks within the full width at the tenth maximum of the distribution for later clustering analysis. This criterion can effectively handle the small polydispersity in the population by eliminating the extremely small or large droplets.
Fig. 2 demonstrates the scatter plot of identified clusters in every pairwise combination where the filtering strategy has been applied to the data. Compared to microscopy image analysis, the optofluidic measurements indicate more localized and confined clusters (Fig. 2 and S3†). This is possibly because the photomultiplier tube has already recorded the measured fluorescence for each droplet as an average value. Also, during analysis, only the peak value from a droplet is considered as its color-coded identification value. Using two dyes, the multiplexing platform offers a decent number of 90 color codes that can be reliably identified through optofluidic measurement. Also, the coding space provides a robust coding selection depending on the microbiological assay at hand. Importantly, the color codes must still be distinguishable when introducing microbial samples to droplets after long-term incubations. To investigate the impact of droplet shrinkage, we devised an experiment with twelve experimental conditions and analyzed the droplets using microscopy imaging. We used an E. coli strain and red and far-red combinations to produce color-coded experimental conditions. As indicated in Fig. S3,† five of the 12 clusters within the library contained cell-laden droplets and were expected to exhibit growth over 24-hour incubation. The bright field images from microscopy were then analyzed to quantify the growth within droplet populations. As shown in Fig. S3,† results indicate that color codes are still detectable after 24 hours of incubation. In general, there is an overall shift in intensity since all droplets have reduced size. However, droplet populations with expected growth have more strongly shifted fluorescence intensities due to additional size shrinkage caused by bacterial growth.
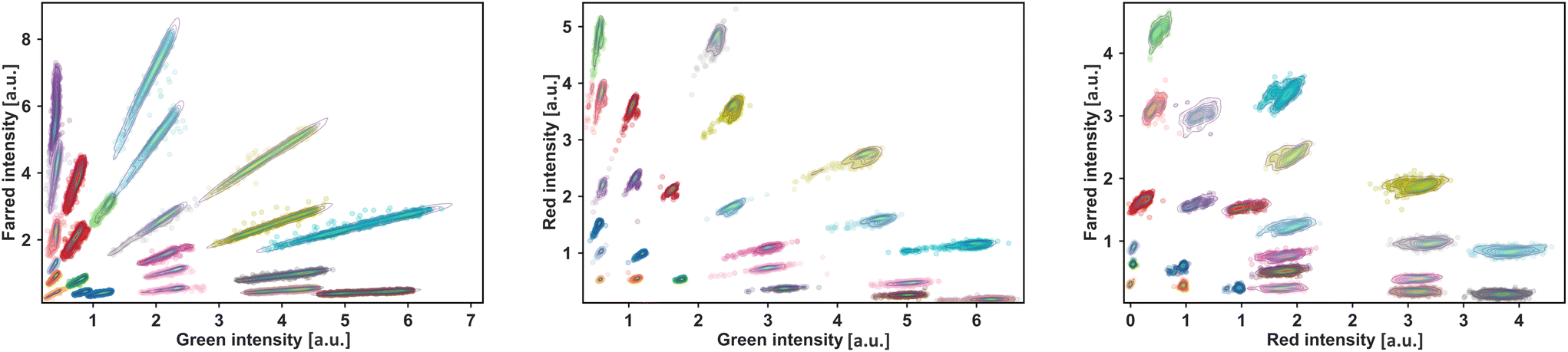 | ||
| Fig. 2 Scatter plots of the identified color codes generated by pairwise combinations of three dyes and measured by the optofluidic set-up. 24 color codes can be produced for each pairwise dye combination28 as shown in each scatter plot. Pairwise combinations of red, farred, and green fluorescent dyes were used to produce the color-coded droplets. Droplets of each combination are then reinjected into the optofluidic chip for simultaneous signal acquisition of three fluorescence channels, and a time series signal is recorded for offline analysis. Different fluorescence codes are then identified by analyzing the time series data using hierarchical density-based spatial clustering of applications with noise. The integrated workflow allows the selection and reliable identification of a total number of 72 pairwise color codes. Moreover, single shades of each dye yield six more color codes that expand the coding space to 90 color codes to choose for a biological assay. | ||
The shift in fluorescence intensity with droplet shrinkage can affect the offline classification analysis. This may increase the likelihood of identifying and counting a portion of these droplets in other coding clusters. Therefore, we also investigated the possibility of misclassification errors in identifying the droplet populations. In our test, as we expected a clear growth from inoculated droplet populations, we defined a threshold for brightfield image analysis for binary decision-making to separate growth and no-growth droplets. We used the growth histogram of all populations (Fig. S4†) to define a threshold value (equal to 0.02, normalized to maximum growth in brightfield image analysis; please check Materials and Methods for detailed information) to separate the empty droplets from growth-exhibiting ones. Using this value, we assessed non-inoculated droplet populations and looked for possible errors in analysis. This assessment revealed an error of around 2% misclassification.
In our new integrated analysis platform, we have utilized a camera to observe a section of the microfluidic chip downstream of the optofluidic interrogation zones (Fig. 1, 3B, and S1†). With droplets passing the fiber interrogation zone (see Material and Methods), a trigger signal is sent to the camera (Fig. 3A), and a bright field image is recorded for later image analysis. Alongside the image, the optofluidic fluorescence measurements are also recorded and manually matched to the images during offline analysis. Image-to-fluorescence data synchronization is achieved offline by analyzing the trigger signal time spacing in the recorded data and matching the image indexes to them. Droplet images then undergo a series of image analysis steps to quantify the growth. First, droplets are identified in the image and masked out to have a region of interest. To improve bacterial cell identification and thereby enhance the signal-to-noise ratio, a background image is calculated using the median value of pixels using 500 recorded images. The background image is then subtracted from the masked image to obtain the foreground data (Fig. 3B.III and C.III). The sharp intensity changes in the resulting image (i.e., bacterial cells) are then identified through Sobel edge detection, Sobel thresholding, and dilation operators (see Materials and Methods for detailed information). Through these operations, a final binary image is obtained to quantify the bacterial growth within droplets (Fig. 3C). The quantified growth represents the droplet area covered by the bacterial biomass. In our experimental design, all droplets are inoculated with bacterial cells, and the overall growth within individual experimental droplet populations is assessed. Therefore, there is no threshold for binary decision-making on droplet growth.
 | ||
| Fig. 3 In-flow image acquisition and analysis. The content of each droplet is excited using two lasers (Fig. 1, stage III, step 1), and the resulting multiplexed fluorescence intensity is measured using the collection fiber on the chip. The collected signal is then demultiplexed using a lock-in amplifier, and a time signal is recorded for each fluorescence channel (here, red and farred). Then, by observing the real-time signal values for each channel, a threshold is defined using one of the intensity channels (in this case 0.2 V in red channel). Whenever the signal is higher than this threshold (i.e., a droplet passed by the fiber interrogation zone), a trigger signal is generated and sent with a delay to the camera to capture an image of the corresponding droplet. An example signal of a droplet and the trigger signal is shown in A. The trigger signal is delayed since the camera's observation window is at a further distance. Part B represents the in flow droplet images of two droplets with different growth amount in I and II, and in III, the calculated background image is shown. The background image is later used in the image analysis pipeline as it will help signify the bacterial edges. In part C, the droplets shown in part B – I and II are used as example inputs for the image analysis steps. Droplets are first identified within the frame (step II). Then, the background image is subtracted to obtain the foreground (step III); next, a Sobel edge detection (step IV) followed by thresholding (step V) and dilating (step VI) is utilized to quantify the growth within each droplet. The growth is defined as the ratio of the white pixel in the binary image (i.e., bacterial biomass) to the droplet area. | ||
Comparison of the proposed workflow with microscopy imaging technique
The integrated platform enables rapid, sensitive, and high throughput assessment of multiplexed picolitre droplets. This empowers the simultaneous investigation of microbial samples in response to different environmental conditions, such as carbon sources, in a single experimental run. Also, integrating in flow brightfield imaging with the optofluidic measurement setup allows visualization of individual droplet reactors, providing detailed information on microbial sample growth when analyzed by proper image processing algorithms. To demonstrate these capabilities of the integrated platform and compare it to a conventional microscopy technique, we have devised an experiment to simultaneously evaluate the growth of a model bacterial strain across a panel of 11 carbon sources in the same background medium. We have used E. coli 081 as our model strain.31 We used red and far-red dyes to color-code the experimental conditions since our model bacterium carries a GFP label, which is utilized in other applications of strain and could be quantified in the green channel. To show the versatility of the coding space and exclude any bias regarding different color codes, we produced two populations of each carbon source in the same experiment with two different color codes. Alongside the carbon source populations, two control populations without any carbon sources build a 24-member multiplexed droplet library. The optical density of cells at inoculation was set to yield an average of 10 bacterial cells within droplets at timepoint zero (i.e., no empty or uninoculated droplets). The library of droplet populations was dynamically incubated at 37 °C for 24 hours. A subpopulation of droplets was used for microscopy image analysis, as established in previous work.28 Another sub-sample of droplets was analyzed at an average frequency of nearly 60 Hz using the chip for the optofluidic measurements. Using the fluorescence signal from a coding channel, a threshold is defined to trigger the camera to capture assay droplet images. Droplet fluorescence signals and images are then analyzed to assign the respective carbon source and assess the growth of the bacterium. The throughput scale of microscopy is comparably limited (∼6000 droplets analyzed in about 8 minutes). This time includes the loading and resting time in the observation chamber, as droplets need to be stationary during the microscopy image capture.24 In contrast, over ten thousand droplets were analyzed using the optofluidic set-up and in a reasonable time (about 5 minutes, including loading time in tubing). This throughput becomes more pronounced when a considerable number of droplets (e.g., hundreds of thousands to a million) need to be processed. Moreover, after optofluidic measurements, droplets can be recovered for further downstream processing, e.g., further incubation. In contrast, recovering droplets from a microscopy observation chamber will disrupt the droplet integrity. Despite providing higher measurement throughput and recovery of droplets, it should be noted that static microscopy imaging provides a higher lateral resolution. This is because in flow imaging requires a shorter exposure time to avoid motion blur in the captured image. On the contrary, as droplets are static during microscopy imaging, more light can be collected from the samples (i.e., higher exposure times), therefore improving the resolution and signal-to-noise ratio.Fig. 4A shows the 24 experimental conditions identified from optofluidic measurements. The quantitative growth of individual populations can be determined using the proposed integration and analysis pipeline from Fig. 3, with results for this dataset shown in Fig. 4B. The results indicate that our model E. coli strain does not sufficiently grow on amino acids as the sole carbon source substrates; however, different sugars promote the growth of our model strain at different levels, with xylose and maltose demonstrating the lowest and highest biomass production, respectively (Fig. 4B). This is expected as different carbon sources require different transporters and functional enzymes for metabolism,37 and therefore, their energy yield could be different, affecting the growth of our model strain. As shown in Fig. 4B, the optofluidic measurement and analysis of the two technical replicates demonstrated minimal variability between replicates. Glucose is typically the preferred substrate for E. coli for faster growth and higher biomass formation due to the regulatory dominance of the catabolite repression system promoting efficient pathways for metabolizing and transporting glucose.37–39 However, the excess glucose concentrations in this investigation (20 mg mL−1) can potentially result in an overflow metabolism where acetate is produced, reducing the growth rate and biomass yield in E. coli.40,41 On the other hand, maltose is a glucose polymer of two units, and there are differences in transport mechanisms and energy requirements for maltose uptake into the cell42 that potentially slow down the process and reduce the chance of overflow metabolism. Therefore, this might explain the lower biomass level observed with glucose compared to maltose at an equal provided mass of glucose equivalents, demonstrating the highest biomass productivity for our model E. coli strain. These observations are consistent for both measurement approaches, as shown in Fig. 4B.
 | ||
Fig. 4 Carbon source utilization assay to compare the performance of the in flow imaging to static bright field microscopy. After 24 hours of incubation at 37 °C, droplets are reinjected into the optofluidic chip for fluorescence measurement and triggered in flow imaging. Red and farred dyes were used to color code different experimental conditions. Part A demonstrates the identified color codes using hierarchical density-based spatial clustering of applications with noise. The error bars in red and far-red intensity are one standard deviation. Each carbon source is coded in two different color codes to ensure no bias toward growth quantification related to coding dyes and sample preparation steps of different conditions in stage I of the integrated workflow (Fig. 1). Part B shows the heat map of the normalized quantified growth [a.u.] for each carbon source for both in flow (Optofluidics) and static bright field microscopy images (Brightfield) using the model E. coli strain (ec). Correlation investigations of the two measurement approaches is shown in C to determine the comparability of them. Pearson correlation (ρ) and Spearman's rank correlation (ρs) are calculated to identify the linearity and order of each experimental condition for both measurement approaches. The diagonal histograms demonstrate the data distribution frequencies for each method. The scatter plots show the normalized growth value [a.u.] derived from the analysis of each measurement approach. The proposed integrated workflow and data analysis for carbon source utilization of E. coli (ec) demonstrates a strong correlation to static microscopy imaging (n = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980 for optofluidic and n = 6371 for microscopy). 980 for optofluidic and n = 6371 for microscopy). | ||
To further explore and validate the similarity of the two approaches, we studied two correlation methods (Pearson and Spearman's). The Pearson correlation evaluates the linear strength and direction of the two analytical approaches, assuming a normalized data distribution. On the other hand, Spearman's correlation addresses the monotonic direction and strength of the approaches. Examining both measures offers a comprehensive view of the data, as they have different statistical assumptions, which in turn enhances the confidence in the findings. As shown in Fig. 4C, Pearson and Spearman's correlation investigations demonstrate a strong correlation between the two approaches (Pearson correlation = 0.94, Spearman's rank correlation = 0.95 Fig. 4C). This result underscores the sensitivity of the optofluidic integration in identifying growth-promoting and inhibiting conditions in this experiment. The high throughput and sensitive characteristics, in flow bright field imaging, and multiplexed fluorescence signal acquisition of the optofluidic measurement enable the proposed workflow to be applied to a broader range of microbiological applications, specifically in natural product discovery.
In this study, the droplet library was processed at 60 Hz, which included a fluorescence signal and triggered image acquisition. Despite achieving a fast throughput compared to microscopy imaging, higher optofluidic measurement frequencies in the kilohertz range can be achieved without triggered image acquisition in our current settings. This is because camera readout and exposure time limit the throughput of droplet measurement. Increasing the rate of measurement (i.e., increasing droplet flow) increases the chance of missing saved images due to camera readout time and also introduces motion blur in the captured images as the droplet flow gets faster than the camera exposure time. Therefore, to improve the droplet measurement throughput while capturing images without motion blur, further technical developments, such as fast cameras with high data transfer capability and data processing on a field-programmable gate array (FPGA), are necessary. Moreover, FPGA data processing enables high-throughput real-time analysis.43
Conclusions
Droplet microfluidics has been extensively employed for microbiological assays, offering significant advantages such as lower reagent consumption, faster reaction times, and high throughput experimentations. The proposed workflow expands upon these advantages, empowering high throughput multiplexed droplet generation and analysis. Here, we have demonstrated the compatibility of integrating a multiplexing platform for producing color-coded droplet populations with different experimental conditions with optofluidic measurements and in flow imaging for comprehensive analysis of droplet contents. The multiplexed droplet library generation (Fig. 1, stage I) brings time efficiency and reduced experimental variability by automizing sample preparation and minimizing manual processing. At the same time, optofluidic measurement and in flow imaging (Fig. 1, stage III) provide sensitive fluorescence detection. In addition, the in flow bright field image acquisition and offline analysis of the droplet library yield comparable results to microscopy imaging. Moreover, unlike microscopy measurements, droplets are easily recovered after optofluidic processing, which is of great importance in microbiological screening campaigns. We have validated the applicability of coupling these technologies by simultaneously screening for growth-promoting carbon sources for a model bacterial strain. The results demonstrated that amino acids provide a poor condition for bacterial growth. In contrast, sugars demonstrated growth-promoting behavior, with xylose and maltose stipulating the lowest and highest biomass produced within droplets, respectively, and glucose performing as the second-best carbon source. The correlation evaluations of the in flow image acquisition with microscopy as the gold standard measurement further validated our integrated optofluidic approach, underscoring its robustness.The proposed workflow poses a significant potential in various microbiological applications. In applications such as antibiotic susceptibility assays, combinatorial drug screening, enzymatic activity screening, and culturomics, multiple experimental conditions should be studied simultaneously. Considering culturomics as an example, fluorescence codes can be used for different media components, while image data can be used for growth quantification as a label free approach. This would allow differentiation of fast growers from slow growing microorganisms and identification of relevant media components. Specifically, the coupled set-up can be employed to discover novel natural products from environmental samples.32 Using the multiplexing droplet library generation (Fig. 1, stage I), multiple environmental conditions (i.e., droplet composition) can be simultaneously investigated. Utilizing the optofluidic measurement and in flow imaging (Fig. 1, stage III), the microbial growth of different droplet populations can be assessed. Furthermore, these droplets can be recovered for further processing (e.g., extended incubation period) without disruption, unlike microscopy. This lowers the chance of losing potential rare hits producing valuable natural products or the uncultivated microbial taxa. Moreover, our optofluidic chip is equipped with electrodes for fluorescence-activated droplet sorting (Fig S1†). Using the fluorescence data, which indicates different color codes (i.e., droplets of different experimental conditions), different droplet populations can be isolated for microbial DNA extraction and further investigations at the genomic level using sequencing approaches. Thus, the optofluidic analysis setup facilitates the identification of diversity-promoting conditions from an environmental sample. Employing the optofluidic setup enables high throughput and sensitive detection of rare hits of potentially valuable natural products for downstream analysis. Therefore, the proposed integration of methodologies empowers the ability to explore microbial diversity in microbiological screening campaigns.
Author contributions
AS: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft. SH: conceptualization, methodology, writing – review and editing. KM: methodology, writing – review and editing. MAR: conceptualization, methodology, project administration, resources, supervision, writing – review and editing.Data availability
The data that supports the findings of this study is available from the corresponding author upon reasonable request. Example data from two experiments are provided as ESI CSV files to follow data analysis.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jill Winkler for excellent technical and microbiological assistance. This work has been funded by the Leibniz ScienceCampus InfectoOptics No. SAS-2018-HKI-LWC (Project VersaDrop), and the German Federal Ministry of Education and Research (BMBF) within the funding program Photonics Research Germany, Leibniz Center for Photonics in Infection Research (LPI), subproject LPI-BT2, contract number 13N15705. This work was also supported by the Cluster of Excellence ‘Balance of the Microverse’ under Germanýs Excellence Strategy (EXC 2051 – project ID 390713860) and the German Center for Infection Research (DZIF) in the research area Novel Antibiotics (projects 9.826). Ashkan Samimi was funded by Deutscher Akademischer Austauschdienst (DAAD). Fig. 1 contents was created with BioRender.com.References
- B. Bonev, J. Hooper and J. Parisot, J. Antimicrob. Chemother., 2008, 61, 1295–1301 CrossRef CAS PubMed.
- G. Vedel, J. Antimicrob. Chemother., 2005, 56, 657–664 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- R. Ghaju Shrestha, Y. Tanaka, B. Malla, D. Bhandari, S. Tandukar, D. Inoue, K. Sei, J. B. Sherchand and E. Haramoto, Sci. Total Environ., 2017, 601–602, 278–284 CrossRef CAS PubMed.
- C. Djemiel, S. Dequiedt, B. Karimi, A. Cottin, W. Horrigue, A. Bailly, A. Boutaleb, S. Sadet-Bourgeteau, P. A. Maron, N. Chemidlin Prévost-Bouré, L. Ranjard and S. Terrat, Front. Microbiol., 2022, 13, 889788 CrossRef PubMed.
- Z. L. Li, R. Cheng, F. Chen, X. Q. Lin, X. J. Yao, B. Liang, C. Huang, K. Sun and A. J. Wang, J. Hazard. Mater., 2021, 405, 124366 CrossRef CAS PubMed.
- L. J. Pinnell and J. W. Turner, Front. Microbiol., 2019, 10, 1252 CrossRef PubMed.
- J. E. Kufs, C. Reimer, E. Steyer, V. Valiante, F. Hillmann and L. Regestein, Microb. Cell Fact., 2022, 21, 1–10 CrossRef PubMed.
- L. Regestein, T. Klement, P. Grande, D. Kreyenschulte, B. Heyman, T. Maßmann, A. Eggert, R. Sengpiel, Y. Wang, N. Wierckx, L. M. Blank, A. Spiess, W. Leitner, C. Bolm, M. Wessling, A. Jupke, M. Rosenbaum and J. Büchs, Biotechnol. Biofuels, 2018, 11, 1–11 CrossRef PubMed.
- S. Weiser, T. Tiso, K. Willing, B. Bardl, L. Eichhorn, L. M. Blank and L. Regestein, Discover Chem. Eng., 2022, 2, 8 CrossRef CAS.
- S. Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- Y. Ding, P. D. Howes and A. J. Demello, Anal. Chem., 2020, 92, 132–149 CrossRef CAS PubMed.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- T. S. Kaminski, S. Jakiela, M. A. Czekalska, W. Postek and P. Garstecki, Lab Chip, 2012, 12, 3995–4002 RSC.
- H. Zec, T. D. Rane and T. H. Wang, Lab Chip, 2012, 12, 3055–3062 RSC.
- A. Fallah-Araghi, K. Meguellati, J. C. Baret, A. El Harrak, T. Mangeat, M. Karplus, S. Ladame, C. M. Marques and A. D. Griffiths, Phys. Rev. Lett., 2014, 112, 1–5 CrossRef PubMed.
- A. M. Kaushik, K. Hsieh, L. Chen, D. J. Shin, J. C. Liao and T. H. Wang, Biosens. Bioelectron., 2017, 97, 260–266 CrossRef CAS PubMed.
- S. S. Terekhov, I. V. Smirnov, A. V. Stepanova, T. V. Bobik, Y. A. Mokrushina, N. A. Ponomarenko, A. A. Belogurov, M. P. Rubtsova, O. V. Kartseva, M. O. Gomzikova, A. A. Moskovtsev, A. S. Bukatin, M. V. Dubina, E. S. Kostryukova, V. V. Babenko, M. T. Vakhitova, A. I. Manolov, M. V. Malakhova, M. A. Kornienko, A. V. Tyakht, A. A. Vanyushkina, E. N. Ilina, P. Masson, A. G. Gabibov and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2550–2555 CrossRef CAS PubMed.
- N. Nuti, P. Rottmann, A. Stucki, P. Koch, S. Panke and P. S. Dittrich, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202114632.
- P. Zhang, A. M. Kaushik, K. Hsieh, S. Li, S. Lewis, K. E. Mach, J. C. Liao, K. C. Carroll and T. Wang, Small Methods, 2022, 6, 1–12 Search PubMed.
- R. G. Rosenthal, X. Diana Zhang, K. I. Đurđić, J. J. Collins and D. A. Weitz, Angew. Chem., 2023, 135 DOI:10.1002/ange.202303112.
- E. J. Medcalf, M. Gantz, T. S. Kaminski and F. Hollfelder, Anal. Chem., 2023, 95, 4597–4604 CrossRef CAS PubMed.
- D. Cai, Y. Wang, J. Zou, Z. Li, E. Huang, X. Ouyang, Z. Que, Y. Luo, Z. Chen, Y. Jiang, G. Zhang, H. Wu and D. Liu, Adv. Sci., 2023, 2205863, 1–11 Search PubMed.
- L. Mahler, S. P. Niehs, K. Martin, T. Weber, K. Scherlach, C. Hertweck, M. Roth and M. A. Rosenbaum, eLife, 2021, 10, 1–23 CrossRef PubMed.
- H. Li, P. Zhang, K. Hsieh and T.-H. Wang, Lab Chip, 2022, 22, 621–631 RSC.
- A. Kulesa, J. Kehe, J. E. Hurtado, P. Tawde and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6685–6690 CrossRef.
- J. Kehe, A. Kulesa, A. Ortiz, C. M. Ackerman, S. G. Thakku, D. Sellers, S. Kuehn, J. Gore, J. Friedman and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12804–12809 CrossRef CAS PubMed.
- A. Samimi, S. Hengoju and M. A. Rosenbaum, Sens. Actuators, B, 2024, 417, 136162 CrossRef CAS.
- M. Tovar, S. Hengoju, T. Weber, L. Mahler, M. Choudhary, T. Becker and M. Roth, Anal. Chem., 2019, 91, 3055–3061 CrossRef CAS PubMed.
- J. Panwar, A. Autour and C. A. Merten, Nat. Protoc., 2023, 18, 1090–1136 CrossRef CAS PubMed.
- C. Lüdecke, J. Bossert, M. Roth and K. D. Jandt, Appl. Surf. Sci., 2013, 280, 578–589 CrossRef.
- L. Mahler, S. P. Niehs, K. Martin, T. Weber, K. Scherlach, C. Hertweck, M. Roth and M. A. Rosenbaum, eLife, 2021, 10 DOI:10.7554/ELIFE.64774.
- M. Tovar, T. Weber, S. Hengoju, A. Lovera, A. S. Munser, O. Shvydkiv and M. Roth, Biomicrofluidics, 2018, 12 DOI:10.1063/1.5013325.
- S. Hengoju, S. Wohlfeil, A. S. Munser, S. Boehme, E. Beckert, O. Shvydkiv, M. Tovar, M. Roth and M. A. Rosenbaum, Biomicrofluidics, 2020, 14, 024109 CrossRef CAS PubMed.
- L. Mahler, M. Tovar, T. Weber, S. Brandes, M. M. Rudolph, J. Ehgartner, T. Mayr, M. T. Figge, M. Roth and E. Zang, RSC Adv., 2015, 5, 101871–101878 RSC.
- É. Geersens, S. Vuilleumier and M. Ryckelynck, ACS Omega, 2022, 7, 12039–12047 CrossRef.
- Y. Luo, T. Zhang and H. Wu, Biotechnol. Adv., 2014, 32, 905–919 CrossRef CAS.
- A. Bren, J. O. Park, B. D. Towbin, E. Dekel, J. D. Rabinowitz and U. Alon, Sci. Rep., 2016, 6, 24834 CrossRef CAS PubMed.
- J. Phue, S. B. Noronha, R. Hattacharyya, A. J. Wolfe and J. Shiloach, Biotechnol. Bioeng., 2005, 90, 805–820 CrossRef CAS PubMed.
- N. Paczia, A. Nilgen, T. Lehmann, J. Gätgens, W. Wiechert and S. Noack, Microb. Cell Fact., 2012, 11, 122 CrossRef CAS PubMed.
- R. Kutscha and S. Pflügl, Int. J. Mater. Sci., 2020, 21, 8777 CAS.
- W. Boos and H. Shuman, Microbiol. Mol. Biol. Rev., 1998, 62, 204–229 CrossRef CAS PubMed.
- J. Zhou, L. Mei, M. Yu, X. Ma, D. Hou, Z. Yin, X. Liu, Y. Ding, K. Yang, R. Xiao, X. Yuan, Y. Weng, M. Long, T. Hu, J. Hou, Y. Xu, L. Tao, S. Mei, H. Shen, Y. Yalikun, F. Zhou, L. Wang, D. Wang, S. Liu and C. Lei, Light:Sci. Appl., 2025, 14, 76 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00130g |
| This journal is © The Royal Society of Chemistry 2025 |